

Abstract
Cri du chat syndrome (CdCS) and primary ciliary dyskinesia (PCD) are rare diseases that present with frequent respiratory symptoms. PCD can be caused by hemizygous DNAH5 mutation in combination with a 5p segmental deletion attributable to CdCS on the opposite chromosome. Chronic oto-sino-pulmonary symptoms or organ laterality defects in CdCS should prompt an evaluation for PCD.
Cri du chat syndrome (CdCS) is a rare (1:15 000 to 1:50 000 live births)1 autosomal-dominant syndrome mainly caused by segmental deletions of chromosome 5p.2,3 Respiratory issues are common in CdCS, with some infants having neonatal respiratory distress,1 and pneumonia, bronchitis, and otitis media develop in approximately 30% of children with this syndrome.4,5 Despite the prevalence of respiratory issues in CdCS, many caregivers do not aggressively investigate other respiratory diseases because they assume these issues are secondary to chronic aspiration and hypotonia associated with CdCS.
Primary ciliary dyskinesia (PCD) is a similarly rare (1:15 000 live births) genetic disease in which dysmotile respiratory cilia result in chronic sinusitis, chronic cough, frequent otitis media, recurrent pneumonia, neonatal respiratory distress, and infertility.6 Approximately 50% of patients with PCD also have situs inversus totalis, or mirror image organ arrangement (Figure 1, A and B), and at least 12% have situs ambiguus, or organ arrangement that falls somewhere between normal and mirror image arrangements.7 PCD is a polygenic disease, with PCD-causing mutations known in 30 genes, where biallelic mutations are inherited in an autosomal-recessive pattern. Mutations in the dynein axonemal heavy chain 5 gene (DNAH5) cause outer dynein arm ciliary defects and account for approximately 30% of cases of PCD.8 This gene resides in the same chromosome 5p region affected in CdCS. We report 2 patients with CdCS who also have PCD through hemizygous DNAH5 mutations.
Figure 1.
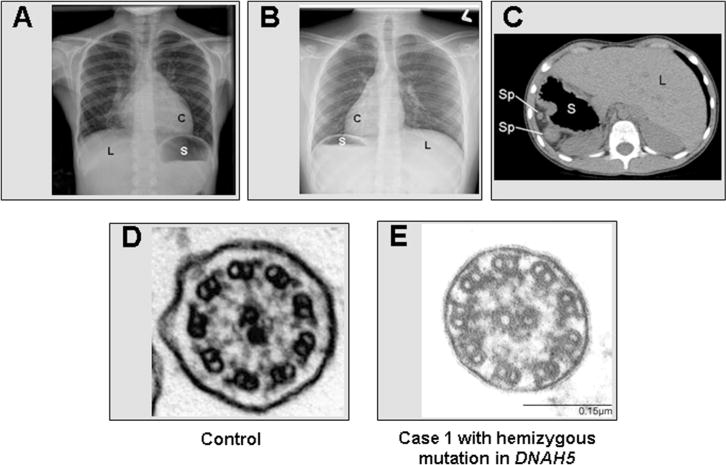
Ciliary ultrastructure and laterality defects in PCD. A, Chest radiograph of a sample patient with situs solitus or normal organ arrangement, with a left cardiac apex (C), right sided liver (L), and a left-sided stomach (S). B, Chest radiograph of sample patient with situs inversus totalis or mirror-image organ arrangement. C, CT scan of case 1 with situs ambiguus showing polysplenia (Sp), dextrogastria (S), and a left-sided liver (L). This patient also had dextrocardia, left lung isomerism, and a left superior vena cava. D, Normal ciliary ultrastructure from healthy subject. E, Individual (case 1) with apparent hemizygous mutation in DNAH5 with absent/shortened outer dynein arms.
Case 1
Case 1 is a 6-year-old white, nonconsanguineous, boy with facial dysmorphism, developmental delay, autism-spectrum disorder, and hypotonia who was diagnosed with CdCS in infancy. He also has situs ambiguus with dextrocardia, left lung isomerism, left superior vena cava, dextrogastria, polysplenia, and a left-sided liver (Figure 1, C). Despite an uncomplicated term birth, the child had neonatal respiratory distress and required supplemental oxygen for 14 days. He had 5–10 episodes of otitis media in the first 2 years of life and required pressure equalization tubes at 7 months of age. He has year-round nasal congestion and year-round wet cough, which started around 6 months of age. He has had 4 episodes of pneumonia that required hospitalization. The child has chronic right middle lobe changes on chest radiography with bronchiectasis on chest computed tomography (CT) scan.
Nasal nitric oxide screening was attempted; however, because of poor patient cooperation, a reliable measurement could not be obtained. Electron microscopy (EM) of nasal cilia confirmed the absence of the outer dynein arms, consistent with the diagnosis of PCD (Figure 1, D and E). Ciliary beat pattern was not examined by high-speed videomicroscopy. DNAH5 mutation profiling was performed by Sanger sequencing for all 79 exons and splice junctions as previously described.8 An apparently homozygous previously known frameshift mutation (c.3905delT [p.Leu1302Argfs*19]) in exon 25 of DNAH5 was identified (Figure 2, A). Multiplex ligation-dependent probe amplification (MLPA) analysis identified deletion of one copy of DNAH5, confirming presence of a hemizygous c.3905delT mutation accompanied by the deletion of DNAH5 from the trans allele due to the presence of Cri du chat (5p deletion) (Figure 2, B).9 Parental DNA was not available for segregation analysis.
Figure 2.
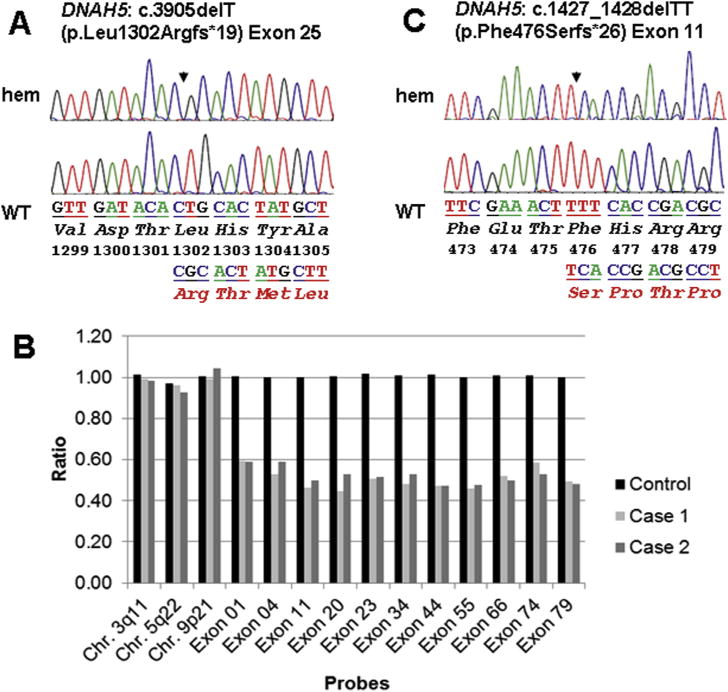
Location of PCD causing mutation in DNAH5. A, An apparent hemizygous (hem) mutation in affected (top) from case 1 and corresponding wild-type (WT) sequences in control (bottom) for exon 25 mutation c.3905delT (p.Leu1302Argfs*19). Mutations are designated by a filled arrow. B, MLPA is a technique to determine relative ploidy.9 It was carried out using SALSA MLPA P238 DNAH5 probe mix as per the manufacturer’s instructions (MRC-Holland, Amsterdam, The Netherlands). This commercial kit comprised probes for 23 of the 79 coding exons of DNAH5, including coding exons 1 and 79 harboring start and stop codons, respectively. In addition, 19 reference probes are included that defect different autosomal chromosomal locations. The dosage quotient, that is a ratio of peak area of the affected sample compared with a control sample, is shown for 3 reference probes specific for regions in no proximity to the DNAH5 locus and 11 probes specific to exons spanning the complete DNAH5 gene including start and stop codons. The bar diagram shows that the relative peak areas for DNAH5-specific probes for both cases were nearly one-half compared with the control, exhibiting Cri du chat segmental deletion of Chr. 5p includes DNAH5. C, An apparent hemizygous (hem) mutation in affected (top) from case 2 and corresponding WT sequences in control (bottom) for exon 11 mutation c.1427_1428delTT (p.Phe476Serfs*26).
Case 2
Case 2 is a 12-year-old girl of nonconsanguineous French Canadian origin who was genetically diagnosed with CdCS (46,XX,del(5)(p13)) at 3 months of age during failure-to-thrive investigations. A paternal uncle carries a diagnosis of PCD. She did not have neonatal respiratory distress, but she has had daily, year-round nasal congestion and daily, year-round wet cough since birth. She also has chronic sinusitis. She had more than 10 episodes of otitis media in the first 2 years of life and required pressure equalization tubes at 6 months of age and chronic use of hearing aids. She requires hospitalization 4 times annually since birth for pneumonia. Radiology studies show situs inversus totalis, with otherwise-normal cardiac and abdominal ultrasounds (Figure 1, B). She never had a chest CT scan, and nasal nitric oxide screening was not conducted. Genetic scanning via high-resolution melting curve analysis on a LightScanner instrument (BioFire Diagnostics, Inc, Salt Lake City, Utah) and sequencing of DNAH5 revealed a hemizygous frameshift mutation c.1427_1428delTT (p.Phe476Serfs*26) in exon 11, resulting in a premature translation termination signal codon, which is previously described in PCD (Figure 2, C).8 MLPA analysis identified deletion of one copy of DNAH5, confirming presence of a hemizygous c.1427_1428delTT mutation, as in case 1 (Figure 2, B). Ciliary biopsy was not performed in this case.
Discussion
PCD from mutations in DNAH5 and CdCS share a common genetic link through changes on chromosome 5p. Patients with CdCS who are carriers of a mutation in DNAH5 from only one parent are at risk for PCD if DNAH5 on the opposite 5p chromosome is affected by the deletion causing CdCS. With a large amount of patients with CdCS exhibiting recurrent respiratory issues, PCD may be present in a sizeable portion of these cases. Consequently, choosing when to screen patients with CdCS for PCD may be difficult. Certainly, any patients with situs inversus or situs ambiguus should have PCD testing, yet certain congenital malformations seen in situs ambiguus with PCD also can occur in CdCS (such as congenital heart disease10 and intestinal malrotation5). Therefore, any organ laterality defect in CdCS, including isolated lesions such as cardiac septal defects, interrupted inferior vena cava, intestinal malrotation, or polysplenia, could be associated with PCD.7 In addition, more than 90% of patients with CdCS harbor a gene deletion involving the DNAH5 region (5p15.2).3 Thus, PCD screening including DNAH5 gene dosage analysis should be offered to all patients with CdCS with chronic oto-sino-pulmonary symptoms or organ laterality defects.
Commercial genetic testing for PCD is available, but the polygenic nature of PCD makes genetic testing expensive, and only 50%–60% of PCD mutations currently are detected. In the past, the gold standard test for the diagnosis of PCD was EM, to investigate ciliary ultrastructural changes. However, up to 30% of patients with PCD may have normal findings on EM studies.6 Nasal nitric oxide measurement is a noninvasive, sensitive screening test for PCD, but it is unavailable outside of research centers in the US.11 High-speed videomicroscopy also is used as a diagnostic test in a limited number of centers, and in CdCS with DNAH5 mutations causing outer dynein arm defects, one would expect largely immotile or severely dysmotile ciliary motion.12 Current diagnostic strategies often rely on several testing modalities to make a definitive PCD diagnosis. If one suspects PCD in a patient with CdCS, ciliary biopsy for EM looking for outer dynein arm defects is a logical first step investigation, followed by referral to a PCD center of excellence for confirmation. Because hemizygous DNAH5 mutation with 5p alterations from CdCS deletions in trans should be the most probable genetic mechanism causing PCD in CdCS, targeted genetic analysis of DNAH5 may help limit costs and invasive biopsies.
In conclusion, PCD and CdCS share a common genetic link through mutations on chromosome 5p with hemizygous DNAH5 gene mutations. Physicians who care for children with CdCS should screen for clinical symptoms and organ laterality defects. If PCD is suspected, screening including nasal nitric oxide measurement, ciliary biopsy for EM analysis and high-speed videomicroscopy, and genetic tests are suggested to confirm the diagnosis of PCD. Follow-up in a specialized PCD center is highly recommended.
Acknowledgments
We thank patients with PCD and their families for their participation. We are grateful to Bruno Maranda, MD (Université de Sherbrooke), for his assistance in collecting clinical information for this report; Heymut Omran, MD (University of Münster), for assistance with DNAH5 gene sequencing; Whitney Wolf and Adriana Lori (University of North Carolina) for their assistance with genetic testing; Sharon McNamara, Liz Cochrane, Molly Elliott, Jennifer Soper, Robert Johnson (Seattle Children’s Hospital) for assistance with clinical data gathering; and Jennifer Guimbellot (University of North Carolina) for assistance with clinical data collection.
Supported by the National Institutes of Health (NIH)/Office of Rare Diseases Research/National Heart, Lung, and Blood Institute (NHLBI) (5US54HL096458-06 to A.S., M.R., M.Z., M.K., and M.L.), National Institutes of Health/NHLBI (5R01HL071798 to M.K. and M.Z.), and National Institutes of Health/National Center for Advancing Translational Sciences (UL1TR000083 [to University of North Carolina, Chapel Hill], UL1TR000423 [to M.R.], UL1RR02574 [to K.W.]) The Genetic Disorders of Mucociliary Clearance Consortium (5U54HL096458) is a part of the NIH Rare Disease Clinical Research Network. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
Glossary
- CdCS
Cri du chat syndrome
- CT
Computed tomography
- EM
Electron microscopy
- MLPA
Multiple ligation-dependent probe amplification
- PCD
Primary ciliary dyskinesia
Footnotes
The authors declare no conflicts of interest.
References
- 1.Cerruti MP. Cri du chat syndrome. Orphanet J Rare Dis. 2006;1:33. doi: 10.1186/1750-1172-1-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niebuhr E. Review—The Cri du Chat syndrome: epidemiology, cytogenetics, and clinical features. Hum Genet. 1978;44:227–75. doi: 10.1007/BF00394291. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X, Snijders A, Segraves R, Zhang X, Niebuhr A, Albertson D, et al. High-resolution mapping of genotype-phenotype relationships in cri du chat syndrome using array comparative genomic hybridization. Am J Hum Genet. 2005;76:312–26. doi: 10.1086/427762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mainardi PC, Pastore G, Castronovo C, Godi M, Guala A, Tamiazzo S, et al. The natural history of Cri du Chat Syndrome. A report from the Italian Register. Eur J Med Genet. 2006;49:363–83. doi: 10.1016/j.ejmg.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 5.Wilkins LE, Brown JA, Nance WE, Wolf B. Clinical heterogeneity in 80 home-reared children with cri du chat syndrome. J Pediatr. 1983;102:528–33. doi: 10.1016/s0022-3476(83)80179-6. [DOI] [PubMed] [Google Scholar]
- 6.Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med. 2013;188:913–22. doi: 10.1164/rccm.201301-0059CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shapiro AJ, Davis SD, Ferkol TF, Dell SD, Rosenfeld M, Olivier K, et al. Laterality defects other than situs inversus totalis in primary ciliary dyskinesia: insights into situs ambiguus and heterotaxy. Chest. doi: 10.1378/chest.13-1704. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hornef N, Olbrich H, Horvath J, Zariwala MA, Fliegauf M, Loges NT, et al. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am J Respir Crit Care Med. 2006;174:120–6. doi: 10.1164/rccm.200601-084OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hills C, Moller JH, Finkelstein M, Lohr J, Schimmenti L. Cri du chat syndrome and congenital heart disease: a review of previously reported cases and presentation of an additional 21 cases from the Pediatric Cardiac Care Consortium. Pediatrics. 2006;117:e924–7. doi: 10.1542/peds.2005-1012. [DOI] [PubMed] [Google Scholar]
- 11.Leigh MW, Hazucha MJ, Chawla KK, Baker BR, Shapiro AJ, Brown DE, et al. Standardizing nasal nitric oxide measurement as a test for primary ciliary dyskinesia. Ann Am Thorac Soc. 2013;10:574–81. doi: 10.1513/AnnalsATS.201305-110OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chilvers MA, Rutman A, O’Callaghan C. Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J Allergy Clin Immunol. 2003;112:518–24. doi: 10.1016/S0091-6749(03)01799-8. [DOI] [PMC free article] [PubMed] [Google Scholar]