

Abstract
Copy number variants (CNVs) are associated with many neurocognitive disorders; however, these events are typically large and the underlying causative gene is unclear. We created an expanded CNV morbidity map from 29,085 children with developmental delay versus 19,584 healthy controls, identifying 70 significant CNVs. We resequenced 26 candidate genes in 4,716 additional cases with developmental delay or autism and 2,193 controls. An integrated analysis of CNV and single-nucleotide variant (SNV) data pinpointed ten genes enriched for putative loss of function. Patient follow-up on a subset identified new clinical subtypes of pediatric disease and the genes responsible for disease-associated CNVs. This includes haploinsufficiency of SETBP1 associated with intellectual disability and loss of expressive language and truncations of ZMYND11 in patients with autism, aggression and complex neuropsychiatric features. This combined CNV and SNV approach facilitates the rapid discovery of new syndromes and neuropsychiatric disease genes despite extensive genetic heterogeneity.
Introduction
Copy number variants (CNVs) collectively have an appreciable impact on human mental health but their large size often precludes specifying the underlying genes involved in the disorder. The pathogenicity of many CNVs observed in the clinic is unknown because the typical variant is also extremely rare, requiring large surveys to achieve case–control significance1-4. Large-scale analyses of clinical microarray data from children with developmental delay (DD), intellectual disability (ID), and autism spectrum disorder (ASD) are now possible and have been used to catalogue regions of human dosage imbalance. In most cases, multiple candidate genes still underlie the smallest region of overlap. In contrast, exome sequencing studies of parent–child trios provide the necessary specificity to discover de novo truncating mutations, i.e., nonsense and frameshift indel mutations with gene-level specificity5-14. Due to the extreme locus heterogeneity of such diseases, however, relatively few recurrences have been reported because surveys of tens of thousands of exomes are still prohibitively expensive. Since large-scale deletions and truncating mutations result in the same dosage imbalance of critical genes, we reasoned that systematically integrating both classes of mutation would improve our power to discover genes associated with DD. Here, we construct one of the largest CNV morbidity maps of patients with ID/DD/ASD both as a clinical resource for pathogenic CNVs and also to identify genes potentially sensitive to dosage imbalance. We then integrate with published exome sequencing data and use next-generation sequencing methods to rapidly resequence candidate genes in patients with unexplained DD. The approach identifies pathogenic mutations in new genes with both statistical significance and clinical relevance.
Results
Construction of a CNV morbidity map
We constructed an expanded CNV morbidity map as previously described1 using array comparative genomic hybridization (CGH) data from 29,085 primarily pediatric cases with ID/ASD/DD compared to 19,584 adult population controls (Methods).The set included 13,318 previously unpublished patients and 11,255 new controls providing enhanced power to detect large-scale, potentially pathogenic deletions and duplications (Supplementary Table 1). As expected, we observe a striking patient increase for rare (<1% frequency) CNVs (p < 10-16, Peto & Peto) driven overwhelmingly by deletions (>=500 kbp deletion OR = 5.09 vs. duplication OR = 1.76). An analysis of 2,086 transmissions shows that likely deleterious CNVs are transmitted preferentially from mothers (58%, p = 0.008, binomial test) (Supplementary Figure 2D-E)15.
We identified 2,184 CNVs (1,348 deletions and 836 duplications) in 55 known autosomal genomic disorder regions, most of which (40/55) corresponded to genomic hotspots flanked by segmental duplication (Supplementary Tables 2 and 3). Among these were 19 loci (Supplementary Tables 2) that have been suspected as pathogenic and now reach nominal significance in our new screen (7 deletion loci, 7 duplication loci, 5 significant for both). This includes the 2q11.2 deletion16 as well as several reciprocal duplications of known deletion syndromes such as a 15q24 microduplication (B to C region; p = 0.027, Fisher's exact test), the reciprocal duplication of the 17q11.2 NF1 deletion (7 cases vs. 0 controls; p = 0.027, Fisher's exact test), and the 16p13.11 microduplication (p = 0.0112, Fisher's exact test).
To identify novel regions of genomic imbalance and potential candidate genes, we performed three analyses. First, we performed a gene-level (RefSeq) analysis to assess the excess of deletions or duplications in cases when compared to controls. Overall, we detected 1,945 genes enriched for deletions and 2,633 genes enriched for duplications (3,800 unique genes combined) at a nominal level of significance (p < 0.01 one-tailed Fisher's exact test; Supplementary Table 4). Since many of these are clustered within specific regions, we next computed enrichment in probands using a genomic windowing approach focused on case CNVs >250kbp (Supplementary Figure 1, Supplementary Figure 3), and a simulation-based empirical p-value. The analysis identified 14 significant regions (most are either novel or previously discussed in the context of case reports, or single gene studies 17-27).This table also includes some well-established risk loci such as NRXN1, SATB2 and MEF2C which reach genome-wide significance with additional refinement of incidence and deletion boundaries18,21,22,25,27,28. Unlike genomic hotspots (Supplementary Tables 2 and 3), most of these regions were not flanked by segmental duplications, and a smaller significant region of overlap (SRO) corresponding to a few genes could be identified because of the multiple breakpoints (Table 1, Supplementary Figure 1, Supplementary Figure 4). In addition we performed a reciprocal analysis for control enrichment and identified one duplication locus on 19q13.33 enriched for KRAB C2H2 zinc finger transcription factor genes which shows a moderate protective odds ratio and nominal significance (Supplementary Note).
Table 1. New CNVs and Smallest Region of Overlap.
| Region | Chr | Start (hg18 Mbp) | End (hg18 Mbp) | Typea | State | Casesb | Controlsb | Inheritancec | Window q-valued | Simulated p-valuee | |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| de novo | Inherited | ||||||||||
|
| |||||||||||
| 1q24 (FMO Deletions and DNM3)17 | 1 | 167.00 | 172.00 | MB | Deletion | 12 | 0 | 2 | 0.0324 | 0.011 | |
| 2q33.1 (SATB2)22,25 | 2 | 199.87 | 200.22 | MB | Deletion | 13 | 0 | 1 | 0.0211 | 0.0002 | |
| 2p16.1 (NRXN1)18,28 | 2 | 50.00 | 51.11 | MB | Deletion | 30 | 9 | 4 | 8 | focal | 0.00005 |
| 2p15-16.1 proximal (PEX13 to AHSA2) | 2 | 59.50 | 63.00 | MB | Duplication | 9 | 0 | 1 | 0.285 | 0.00001 | |
| 3p25.3 (JAGN1 to TATDN2) | 3 | 9.50 | 11.00 | MB | Duplication | 10 | 0 | 1 | 3 | 0.036 | 0.00103 |
| 3p11.2 (CHMP2B to POU1F1) | 3 | 87.32 | 87.64 | MB | Deletion | 9 | 0 | 3 | 0.0489 | 0.000075 | |
| 3q13 (GAP43)19,29 | 3 | 116.72 | 117.13 | MB | Deletion | 9 | 0 | 4 | 0.0489 | 0.0003 | |
| 3q28-29 (FGF12) | 3 | 193.00 | 194.50 | MB | Deletion | 13 | 1 | 3 | focal | 0.00005 | |
| 4q21 (BMP3) | 4 | 81.00 | 83.50 | MB | Deletion | 11 | 0 | 2 | 0.0324 | 0.00025 | |
| 5q14 (MEF2C)21,27 | 5 | 88.00 | 88.26 | MB | Deletion | 10 | 0 | 2 | focal | 0.00005 | |
| 9p13 | 9 | 32.00 | 39.00 | MB | Duplication | 18 | 0 | 2 | 2f | 0.00216 | - |
| 10q1123 | 10 | 49.06 | 52.06 | HS, MB | Duplication | 10 | 0 | 5 | 0.036 | - | |
| 10q23.1 (SFTPD to GLUD1, NRG3 inclusive)24,55 | 10 | 81.68 | 88.93 | HS, MB | Deletion | 11 | 0 | 5 | 0.0211 | - | |
| 12p13 (SCNN1A to PIANP)20 | 12 | 6.34 | 6.68 | MB | Duplication | 23 | 1 | 3 | 1f | 0.00115 | - |
Hotspot (HS) or multiple breakpoint (MB) locus.
Due to complex CNV structure the case–control counts are representative of the region but may vary throughout.
De novo counts also include cases from Hehir-Kwa et al.56
Window q-value is the weighted median for unique segments in the critical region
Reported as the median simulation p-value for all genes in the region (Supplementary Table 4).
Carrier of a balanced translocation.
We next estimated the false discovery rate of our CNV calls by designing a customized microarray and independently validating a subset (39/40 or 97.5%) of the events corresponding to the 14 regions (Methods). Similarly, we assessed transmission of 61 CNVs and found that 28 were de novo and 33 were inherited (21 maternal and 12 paternal, including 3 parental balanced carriers).In several cases a single SRO was apparent, such as the 360 kbp duplication region on chromosome 12p13.3 corresponding to 19 genes (SCNN1A to PIANP) where a focal 92.6 kbp CNV highlighted five genes, including CHD4. In a few cases, a single gene was implicated (e.g., NRXN1, SATB2, or MEF2C) (Table 1). We observed a significant enrichment at GAP4329 (p = 0.0003, simulated) with four deletions arising de novo. In other cases, such as the chromosome 1q24q25 microdeletion, we observed several peaks of significance making it impossible to refine the CNVs to a single candidate gene (e.g., DNM3 vs. FMO1/2; Figure 1).
Figure 1. Truncating SETBP1 mutations and phenotypes.
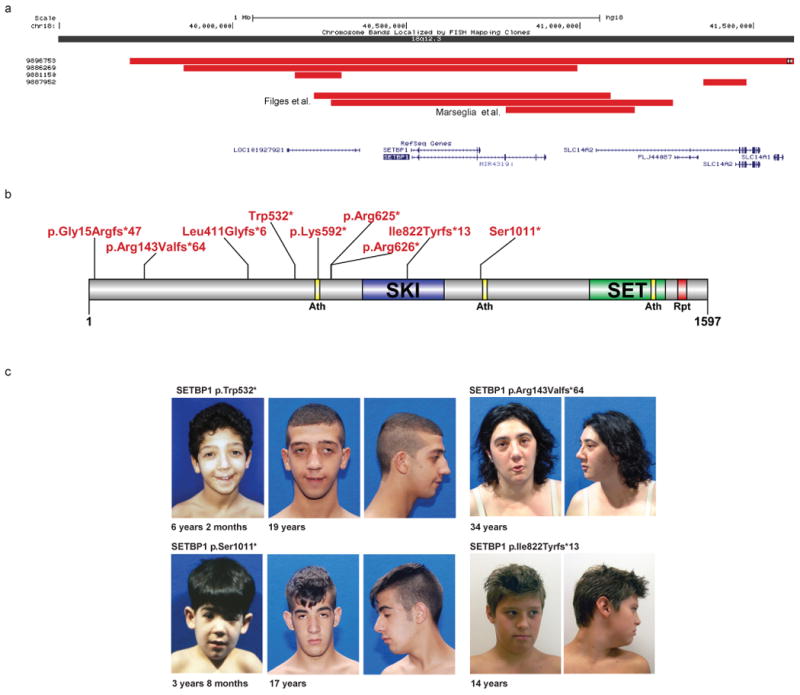
CNV data define a focal CNV region around SETBP1 (a). Combining a focal de novo deletion observed in our study (9886269) and CNVs from Filges and Marseglia et al.41,42 (red bars) highlights minimal common regions, including SETBP1 and LOC101927921. Targeted resequencing identified eight truncating variants in SETBP1 and none in controls. Integration of published exome data identified one additional case, and no truncating events in controls (b). Phenotypic assessment of these cases identified a recognizable phenotype (c-d), including IQ deficits ranging from mild to severe, impaired speech, and distinctive facial features. See the Supplementary Note for additional patient photos and write-ups. We obtained informed consent to publish the photographs.
Integration of CNV and exome sequencing data
As a final analysis to identify high-impact candidate genes, we integrated our CNV deletion data with de novo truncating mutation data identified in 1,879 probands from recently published exome sequencing studies of ASD, ID, congenital heart defects, and schizophrenia5-14. Overall, we detect deletion enrichment at 17.4% of genes with at least one truncating mutation (43/247 with CNV deletion, p < 0.05, Fisher's exact test), which is similar to the expected number of intersections by random chance (OR 1.15 (95% CI, 0.8 to 1.6) p= 1, Fisher's exact test). However, if we limit our analysis to the 21 genes with two or more truncating mutations in probands, we observe significant deletion enrichment for 33.3% of genes (7 of 21 genes, OR = 2.72, p = 0.034, Fisher's exact test) supporting the notion that integrating CNV data and exome sequencing data increases power to detect disease genes. Using a statistical framework based on a hypergeometric distribution, we computed a joint probability of putative loss of function (Methods), combining the CNV data with the single-nucleotide variant (SNV) data for 6,500 individuals from the Exome Sequencing Project (ESP) (ESP6500 controls) and published de novo loss-of-function (LoF) mutations in probands. This analysis highlighted 38 of 247 genes with nominally significant increases in loss-of-function events in cases compared to controls (19 with q-values ≤ 0.01), including 13 genes previously identified as disease-causing (OMIM) (Table 2).
Table 2. Intersection of CNV and Exome Data.
| Exome Data | Array CGH | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Gene Name | Isoform | 1,879 Published Cases LoF | 1,879 Published Cases de novo LoF (ESPAvg Read Depth >20, Dustmasked) | 6,500 ESP LoF (ESPAvg Read Depth>20, Dustmasked) | Signature Dels (n=29,085) | Control Dels (n=19,584) | Combined LoF p-value | Combined LoF q-valuee |
|
| ||||||||
| ANK2a | NM_020977.3b | 1 | 1 | 0 | 5 | 0 | 0.0171 | 0.169 |
| ARHGAP5 | NM_001030055.1 | 1 | 1 | 0 | 7 | 0 | 0.0061 | 0.0833 |
| BCL11A | NM_022893.3 | 1 | 0 | 0 | 4 | 0 | 0.0286 | 0.244 |
| CAPRIN1 | NM_005898.4 | 1 | 1 | 0 | 4 | 0 | 0.0286 | 0.244 |
| CARKD | NM_001242881.1c | 1 | 1 | 0 | 12 | 4 | 0.0363 | 0.28 |
| CHD2 a | NM_001271.3 | 3 | 3 | 0 | 0 | 0 | 0.0113 | 0.127 |
| CHD8 a | NM_001170629.1 | 3 | 3 | 0 | 2 | 0 | 0.00402 | 0.0703 |
| CSDE1 | NM_001130523.2 | 1 | 1 | 0 | 3 | 0 | 0.0479 | 0.311 |
| CUL3 a | NM_003590.4 | 2 | 2 | 0 | 5 | 0 | 0.00383 | 0.0703 |
| DLL1 | NM_005618.3 | 1 | 0 | 0 | 32 | 1 | 2.17E-07 | 2.68E-05 |
| DYRK1A a | NM_001396.3 | 2 | 2 | 0 | 11 | 0 | 1.74E-04 | 8.60E-03 |
| FAM8A1 | NM_016255.2 | 1 | 1 | 0 | 5 | 0 | 0.0171 | 0.169 |
| FOXP1 a | NM_001244810.1 | 1 | 1 | 0 | 4 | 0 | 0.0286 | 0.244 |
| GRIN2B a | NM_000834.3 | 3 | 3 | 0 | 2 | 0 | 0.00402 | 0.0703 |
| GTPBP4 | NM_012341.2 | 1 | 1 | 0 | 3 | 0 | 0.0479 | 0.311 |
| LTN1 | NM_015565.2 | 1 | 1 | 0 | 6 | 0 | 0.0102 | 0.12 |
| MBD5 a | NM_018328.4 | 1 | 1 | 0 | 16 | 6 | 0.0343 | 0.273 |
| MYT1L | NM_015025.2 | 1 | 1 | 0 | 8 | 0 | 0.00365 | 0.0703 |
| NAA15 | NM_057175.3 | 2 | 2 | 0 | 5 | 3 | 0.0296 | 0.244 |
| NCKAP1 | NM_205842.1 | 2 | 2 | 0 | 7 | 0 | 0.00137 | 0.0564 |
| NFIA | NM_001134673.3 | 1 | 1 | 0 | 3 | 0 | 0.0479 | 0.311 |
| NRXN1 a | NM_001135659.1 | 1 | 1 | 0 | 30 | 9 | 0.00427 | 0.0703 |
| NTM | NM_001144058.1 | 1 | 1 | 0 | 40 | 0 | 2.53E-10 | 6.25E-08 |
| PCOLCE | NM_002593.3 | 1 | 1 | 0 | 7 | 0 | 0.0061 | 0.0833 |
| PHF2 | NM_005392.3 | 1 | 1 | 0 | 4 | 0 | 0.0286 | 0.244 |
| RAB2A | NM_002865.2 | 1 | 1 | 0 | 3 | 0 | 0.0479 | 0.311 |
| SCN1A a | NM_001165963.1 | 4 | 4 | 0 | 10 | 1 | 7.36E-05 | 4.55E-03 |
| SCN2A a | NM_021007.2 | 6 | 5 | 0 | 10 | 0 | 7.34E-07 | 6.04E-05 |
| SLC6A1 | NM_003042.3 | 1 | 1 | 0 | 6 | 0 | 0.0102 | 0.12 |
| SRM | NM_003132.2 | 1 | 1 | 0 | 9 | 0 | 0.00218 | 0.0703 |
| STXBP1 a | NM_003165.3 | 2 | 2 | 0 | 4 | 0 | 0.00641 | 0.0833 |
| SUV420H1 | NM_016028.4d | 1 | 1 | 0 | 3 | 0 | 0.0479 | 0.31135 |
| SYNGAP1 a | NM_006772.2 | 4 | 4 | 0 | 0 | 1 | 0.00252 | 0.0703 |
| TBR1 | NM_006593.2 | 2 | 2 | 0 | 7 | 1 | 0.00522 | 0.0806 |
| UBN2 | NM_173569.3 | 1 | 1 | 0 | 5 | 0 | 0.0171 | 0.169 |
| WAC | NM_016628.4 | 1 | 1 | 0 | 3 | 0 | 0.0479 | 0.31135 |
| WDFY3 | NM_014991.4 | 1 | 1 | 0 | 8 | 0 | 0.00365 | 0.0703 |
| ZMYND11 | NM_006624.5 | 1 | 1 | 0 | 8 | 0 | 0.00365 | 0.0703 |
OMIM disease gene.
Variant (2) This is the major form of ankyrin in the adult brain.
Variant (2) This isoform and variants 3 and 4 are shorter than variant 1.
Variant (2) This isoform is shorter and has a distinct C-terminus compared to isoform 1.
Please see the Supplementary Note for discussion of the q-value in this table.
Targeted resequencing of candidate genes in ASD/ID
Based on the analyses above, we selected a set of 26 candidate genes with significant CNV enrichment, rare focal CNVs with de novo mutations from exome sequencing studies, and top candidates from targeted resequencing in ASD/ID (Table 3). For three of these regions, we selected at least two adjacent genes mapping within the SRO; we also selected six genes (GRIN2B, ARID1B, MBD5, PTEN, SCN1A and KANSL1) known to be associated with ASD/ID as positive controls30-35. We utilized molecular inversion probe (MIP)-based capture36 to sequence the 26 genes in 3,387 cases of ID/DD and 1,329 cases of ASD, totaling 4,716 patients. Putative loss-of-function single-nucleotide variation and indels were validated by Sanger sequencing and assessed in parental DNA when available to determine inheritance. Genes with significant enrichment were identified by comparison with MIP resequencing data of 2,193 unaffected siblings from the Simons Simplex Collection37 and the ESP6500. We tested each gene for combined enrichment of loss-of-function variation across CNV and SNV data (Methods) and identified 16 genes (Table 3, Supplementary Table 5) with a significant enrichment of disruptive mutations in cases. Additionally, to control for the differential effect of terminal truncating events we applied a statistical model based on predicted protein lengths for genes with truncating or splice events in the ESP6500 (Supplementary Figure 5) as this was complementary to the case–control results (Table 3).
Table 3. Combined CNV and Targeted Sequencing.
| CNV Deletions | CNV Duplications | Severe Variants (nonsense, start-loss, frameshift, splice-site) ESP Read Depth > 20, Dustmasker (FALSE/NEAR) |
Joint p-valuesb | Joint q-valuec | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||
| Gene | RefSeq | Cases | Controls | Cases | Controls | ID/DD (n=3,387) |
ASD (n=1,329) |
ESP6500 | Simons Siblings (n=2,193) |
ID/DD | ID/DD/ASD | ID/DD | ID/DD/ASD | Truncation p-value |
|
| ||||||||||||||
| ACACA | NM_198839.1 | 28 | 4 | 34 | 10 | 1 | 1 | 7 | 1 | 0.611 | 0.517 | 0.691 | 0.64 | 0.73 |
| ADNP | NM_015339.2 | 1 | 0 | 4 | 0 | 5 | 0 | 1 | 1 | 0.0138 | 0.0376 | 0.0326 | 0.0698 | 0.044 |
| ARID1Ba | NM_017519.2 | 5 | 1 | 3 | 2 | 9 | 0 | 1 | 0 | 0.0000205 | 0.000151 | 0.000183 | 0.000654 | 0.00028 |
| CHD1L | NM_004284.3 | 78 | 7 | 71 | 8 | 12 | 1 | 40 | 0 | 0.419 | 0.628 | 0.545 | 0.71 | 0.94 |
| CYFIP1 | NM_014608.2 | 230 | 69 | 175 | 98 | 0 | 1 | 1 | 0 | 2.75E-08 | 3.10E-09 | 7.15E-07 | 8.06E-08 | - |
| DIP2A | NM_015151.3 | 13 | 3 | 74 | 26 | 1 | 1 | 43 | 1 | 1 | 1 | 1 | 1 | - |
| DNM3 | NM_015569.3 | 11 | 3 | 2 | 0 | 2 | 1 | 0 | 0 | 0.0095 | 0.00524 | 0.0247 | 0.0142 | - |
| DYRK1A | NM_001396.3 | 11 | 0 | 66 | 2 | 2 | 1 | 0 | 0 | 0.00027 | 0.00015 | 0.00117 | 0.000654 | - |
| FOXP1 | NM_032682.5 | 4 | 0 | 6 | 4 | 1 | 0 | 0 | 0 | 0.0358 | 0.0449 | 0.0665 | 0.0778 | - |
| GRIN2Ba | NM_000834.3 | 2 | 0 | 17 | 1 | 2 | 2 | 0 | 0 | 0.0281 | 0.00546 | 0.0562 | 0.0142 | - |
| KANSL1a | NM_001193466.1 | 32 | 3 | 4 | 8 | 4 | 2 | 2 | 1 | 0.00251 | 0.000418 | 0.00816 | 0.00155 | - |
| MAPT | NM_016835.4 | 32 | 1 | 4 | 3 | 1 | 0 | 6 | 0 | 0.33 | 0.35 | 0.452 | 0.455 | - |
| MBD5a | NM_018328.4 | 16 | 6 | 8 | 5 | 1 | 0 | 0 | 0 | 0.0429 | 0.054 | 0.0744 | 0.0878 | 0.095 |
| NRG3 | NM_001165973.1 | 18 | 7 | 9 | 23 | 2 | 1 | 1 | 0 | 0.0468 | 0.0307 | 0.0761 | 0.0614 | - |
| NRXN1 | NM_004801.4 | 30 | 9 | 6 | 0 | 0 | 1 | 0 | 0 | 0.019 | 0.00669 | 0.0412 | 0.0158 | - |
| PTENa | NM_000314.4 | 1 | 1 | 0 | 5 | 1 | 0 | 0 | 0 | 0.235 | 0.295 | 0.339 | 0.404 | - |
| SCN1Aa | NM_006920.4 | 10 | 1 | 5 | 0 | 2 | 0 | 0 | 0 | 0.00229 | 0.00361 | 0.00816 | 0.0117 | - |
| SCN2A | NM_021007.2 | 10 | 0 | 6 | 0 | 3 | 1 | 0 | 0 | 0.000128 | 0.0000888 | 0.000666 | 0.000577 | - |
| TTC21B | NM_024753.3 | 10 | 0 | 9 | 0 | 1 | 0 | 51 | 0 | 1 | 1 | 1 | 1 | - |
| SETBP1 | NM_015559.2 | 2 | 0 | 28 | 1 | 5 | 0 | 2 | 0 | 0.0093 | 0.0262 | 0.0247 | 0.0568 | 0.011 (ID only) |
| SLC1A1 | NM_004170.5 | 33 | 3 | 26 | 1 | 0 | 0 | 0 | 0 | 0.0000221 | 0.0000221 | 0.000183 | 0.000287 | - |
| SOX5 | NM_006940.4 | 15 | 4 | 17 | 3 | 0 | 1 | 2 | 0 | 0.512 | 0.292 | 0.605 | 0.404 | - |
| TBL1XR1 | NM_024665.4 | 3 | 0 | 4 | 5 | 0 | 0 | 0 | 0 | 0.2134 | 0.2134 | 0.326 | 0.326 | - |
| TSPAN17 | NM_012171.2 | 12 | 2 | 7 | 0 | 0 | 0 | 3 | 0 | 0.64 | 0.711 | 0.693 | 0.77 | - |
| DIP2C | NM_014974.2 | 10 | 0 | 36 | 6 | 0 | 0 | 2 | 0 | 0.48 | 0.557 | 0.594 | 0.658 | - |
| ZMYND11 | NM_006624.5 | 8 | 0 | 25 | 15 | 5 | 0 | 0 | 0 | 0.0000281 | 0.0000874 | 0.000183 | 0.000577 | - |
Bolded genes represent genes passing nominal significance.
Bolded entries represent q < 0.1. Please see the Supplementary Note.
Among the positive controls, our analysis confirmed the pathogenicity of five genes using a nominal threshold of significance on the joint p-values: ARID1B (5 CNVs and 9 truncating SNVs, one of which confirmed de novo, p = 1.51×10-4, q=6.54×10-4), GRIN2B (2 CNVs—one focal disrupting the distal end of GRIN2B—and 4 new truncating variants; p = 0.00546, q=0.0142), MBD5 (p = 0.0429, q=0.0744), as well as SCN1A (p = 0.0036, q=0.0117) compared to the adjacent gene TTC21B (p = 1.00, q=1.00). Integration of SNV and CNV data confirms KANSL1 as the gene responsible for the 17q21.31 deletion syndrome32 (p = 0.000418, q=0.00155) compared to the adjacent gene MAPT (p = 0.36, q=0.455) (Table 3, Supplementary Table 5).Patient follow-up for one KANSL1 patient with a severe frameshift demonstrates a striking phenotypic resemblance to microdeletion carriers confirming this gene as the major contributor to the phenotype of 17q21.31 microdeletion (Koolen-de Vries) syndrome32,38.
An enrichment of patient loss-of-function mutations was observed for ten additional genes (ADNP, DYRK1A, NRXN1, NRG3, SETBP1, ZMYND11, DNM3, CYFIP1, FOXP1 and SCN2A) (Table 4). In one case with a de novo DYRK1A splice-site mutation (see Troina1818, Supplementary Table 5), the patient presented with severe microcephaly consistent with published autism de novo truncating mutations and earlier CNV studies36,39. Among those genes where there was no enrichment in cases versus controls, two are notable: CHD1L and ACACA—candidates for the 1q21 deletion and 17q12 deletion syndromes, respectively40. In our resequencing study of CHD1L, for example, we identified 14 likely truncating variants (Table 3), compared to nine independent truncating variants in controls, indicating that rare truncating mutations of CHD1L are not uncommon (Table 3, Supplementary Table 5). There is also no significant decrease in predicted protein sizes in cases compared to controls (p = 0.94, Log Rank).
Table 4. Brief Phenotypic Description of Patients with SETBP1 LoF Variants.
| Patient | Age at Examination |
Gender | Mutation | Inheritance | Cognitive | Hyperactive / ADHD |
Social Difficulties |
Other Behavioral Difficulties |
Speech Delay |
Motor Delay |
Facial Dysmorphism |
Seizures or EEG Abnormalities |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| DNA03-00335 | 14 yrs | M | p.Ile822Tyrfs*13 | de novo | Normal IQ | + | + | + | + | |||
| DNA-008897 | 73 yrs | M | p.Leu411Glyfs*6 | Profound ID | + | + | + | + | + | |||
| Troina 1274 | 19 yrs | M | p.Trp532* | de novo | Severe ID | + | + | + | + | - | ||
| Troina 1512 | 17 yrs | M | p.Ser1011* | de novo | Mild ID | + (3y8m) | + | + | + | + | - | |
| Troina 3097 | 34 yrs | F | p.Arg143Valfs*64 | Severe ID | + | + | + | + | ||||
| DNA11-21308Z | 36 years | F | p.Arg625* | Mild to Moderate ID | + | + | + | + | + | + | ||
| DNA11-19324Z | 9 yrs | F | p.Arg626* | 2 to 2.5 year delay at 9 yr | + | - | + | - | ||||
| DNA08-08272 | 9 yrs | M | p.Gly15Argfs*47 | Mild ID | + | + | + | + | + | + | ||
| Rauch et al | 13 yrs | F | p.Lys592* | Mild ID | + | + | + | - | + | |||
| 9886269 | 5 yrs | M | deletion | de novo | Global Delay | + | + | + | + | + | ||
| Marseglia et al | 15 yrs | M | deletion | de novo | Mild ID | + | + | + | + | + | + | + |
| Filges et al pt 1 | 7 yrs | M | deletion | de novo | Moderate ID | + | + | + | + | |||
| Filges et al pt 2 | 4 yrs | M | deletion | de novo | + | + | + | |||||
Phenotypic examination of SETBP1 and ZMYND11 truncations
Among the significant genes, we focused on SETBP1 and ZYMD11 for further phenotypic characterization. We confirmed a focal de novo deletion and five cases with truncating mutations (3 tested and confirmed de novo) in the SET binding protein 1 (SETBP1) gene. Disruptive mutations were absent in controls, with the exception of a splice-site alteration predicted to lead to the loss of an in-frame exon encoding 18 amino acids. Notably, all truncating mutations in patients occur in cohorts of ID, where we observe an enrichment of mutations (p = 0.0093, joint LoF), and decreased predicted protein sizes (p = 0.011, Log Rank) (Figure 2, Table 3, Supplementary Tables 5 and 6). Integration of our cases with two additional truncating variants found in a separate ID screen (n = 847) with the same MIPs, as well as published small deletions and de novo variants, highlights a similar phenotype12,41,42. The majority of cases demonstrate IQ and language deficits (completely absent or significantly impaired speech in 92% (12/13) of the cases).Patients also frequently exhibit impairment of fine motor skills (n=8), hyperactivity/ADHD (n = 7), and autistic features/poor social skills (n = 4). We observe a dysmorphism typified by a long face (n = 10), characteristic eyebrows, and less frequently, low-set ears (n = 4) and café-au-lait spots (n = 4) (Figure 1, Table 4 Supplementary Table 6).
Figure 2. Truncating ZMYND11 mutations and phenotypes.
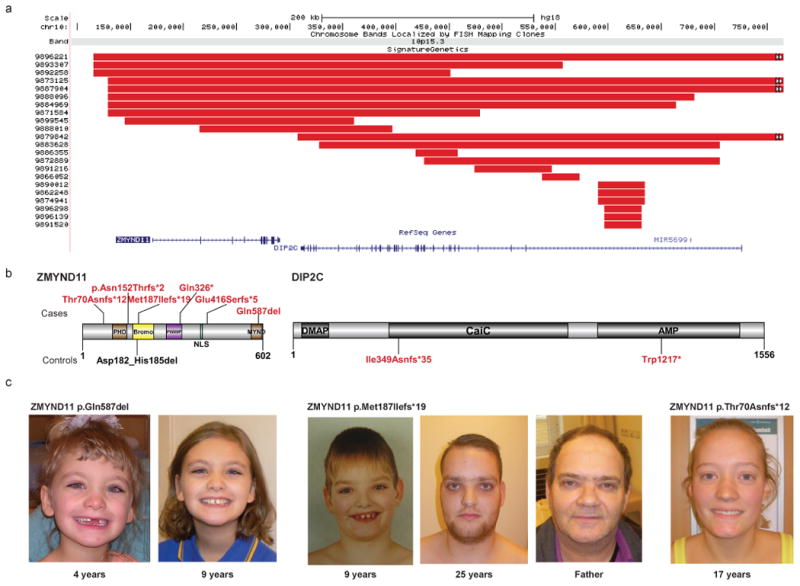
CNV data refines a focal CNV deletion region (red bars) containing two genes: ZMYND11 and DIP2C (a). Targeted resequencing identified five truncating variants and one single amino acid deletion predicted to behave as LoF variants by removing a critical binding residue in the MYND domain (Gln587) (b). Analysis of control resequencing and exome data identified no additional truncating events in ZMYND11 but highlighted two truncating mutations in DIP2C. Phenotypic assessment revealed a consistent phenotype characterized by mild ID concurrent with speech and motor delays, as well as complex neuropsychiatric behavioral and characteristic facial features (c-d). See the Supplementary Note for additional patient photos and write-ups. We obtained informed consent to publish the photographs.
The smallest region of overlap for the 10p15.3 microdeletion predicts two possible candidate genes43: ZMYND11 and DIP2C (Figure 3). We resequenced both candidates and detected five truncating variants in ZMYND11 (two confirmed de novo and one inherited from an affected father) and none in DIP2C. In contrast, concurrent examination of controls identified truncating mutations only for DIP2C (Figure 3, Table 3, Supplementary Table 5). Integration of CNV and truncating SNV data strongly support ZMYND11 (DD p = 2.81×10-5, joint LoF), as opposed to DIP2C (DD p-value = 0.48, joint LoF), as the critical gene. Comparing the ZMYND11 phenotypes of patients with truncating SNVs (Figure 2, Table 5, Supplementary Table 7) reveals a striking resemblance to the 10p15.3 microdeletion cases described previously43 and highlights a consistent set of behavioral features, mild ID and subtle facial features including hypertelorism (n = 6), ptosis (n = 3) and a wide mouth (n = 4). The most consistent features seen in all subjects were speech and motor delays, which were observed in all patients for which information was available, including CNV cases43. Interestingly, a psychiatric phenotype is apparent in 3/5 patients including aggression in 3/4 males. Three cases were accessible for parental DNA testing revealing two de novo variants and one paternally inherited variant. The paternal carrier of the p.Met187Ilefs*19 variant also had DD, including walking at 3-4 years of age, and learning problems in addition to aggression in childhood with mood swings. We also detected a de novo in-frame deletion (p.Gln587Del) in the MYND domain (Gln87), which represents a critical residue in co-repressor binding (including NCoR)44-46. Examination of this patient reveals similarities to published 10p15.3 microdeletion syndrome cases (Figure 2, Supplementary Table 7) including characteristic facial dysmorphisms, global DD, and speech delay. Taking this evidence together, we propose that ZMYND11 is the critical gene associated with the 10p15.3 microdeletion syndrome.
Table 5. Brief Phenotypic Description of Patients with ZMYND11 LoF Variants.
| Patient | Age at Examination | Gender | Mutation | Inheritance | Cognitive | Speech Delay | Social Difficulties | Behavioral Problems | Facial Dysmorphism |
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Adelaide20124 | 4 y & 9y | F | Gln587del | de novo | Global DD | + | + | + | |
| Adelaide3553 | 22y | M | p.Asn152Thrfs*26 | Global DD | + | + | |||
| DNA-017151 | 17 y | F | p.Thr70Asnfs*12 | de novo | Normal IQ | + | + | + | + |
| DNA04-02424 | 41 y | M | p.Gln326* | Mild ID | + | + | + | + | |
| DNA05-04370 | M | p.Glu416Serfs*5 | Severe ID | + | + | + | |||
| DNA-013587 | 25 y | M | p.Met187Ilefs*19 | inherited | Global DD | + | + | + | + |
| Fa of DNA-013587 | M | p.Met187Ilefs*19 | carrier | DD | + | ||||
Discussion
In this study, we leverage the large sample size of patients available from CNV clinical microarrays and the precision of next-generation sequencing to identify specific genes associated with neurodevelopmental disease. The expanded CNV morbidity map offers clinical utility as a resource to assess pathogenic significance of rare events as well as a research tool to prioritize genes discovered from exome sequencing studies that are currently too underpowered to achieve statistical significance5-14,36. It is important to note that the large sample size (nearly 50,000 patients and controls) has begun to highlight regions that map outside of recurrent CNVs mediated by segmental duplications. The sample size is, thus, sufficient to survey the background level of CNVs, identifying critical regions outside of regions with elevated mutation rates (Table 2). In addition, the sample size identifies various recurrent duplications (Supplementary Tables 2 and 3) that are neither necessary nor sufficient to cause disease but are more likely to act as genetic modifiers or risk factors similar to the 15q11.2 microdeletion47. It is possible that copy number polymorphisms >1% may also contribute as weaker risk factors but such events are typically smaller and have not been sufficiently assayed by microarrays. We identify, for example, the 16p13.11 microduplication among 68 cases compared to 27 controls, giving a likelihood ratio (LR) of 1.7 (95% CI 1.13-2.56). Exploring these high-impact risk factors will be important in understanding the genetic architecture of ASDand DD and its relationship to other neuropsychiatric features.
Under the assumption that different classes of genetic mutation (microdeletion and truncating SNVs and indels) will expose the same genic haploinsufficiency, we developed a joint probability statistic to identify 38 specific genes (Table 3) with a higher prior of disease involvement. Although we have not explored it here, a similar approach may be useful in assessing microduplications and hypermorphic missense mutations. While it is clear that not all CNVs are monogenic and will be amenable to this integrated strategy, forward resequencing of 23 candidate regions (including 6 controls) identifies eleven genes where there is an excess of deletions and truncating mutations in cases when compared to controls (Table 3). Targeted resequencing, in particular, allows discrimination of adjacent genes within an SRO (i.e.,SCN1A vs. TTC21B, KANSL1 vs. MAPT or ZMYND11 vs. DIP2C). A comparison of the frequency of truncating mutations in cases and controls also reduces the likelihood that specific genes highlighted by case reports of atypical CNVs are pathogenic (e.g., ACACA and CHD1L)40.
Patient follow-up and phenotypic evaluation provides the most compelling evidence that we have identified likely genes underlying CNV haploinsufficiency. Studies of microdeletion and translocation patients originally narrowed a 1 Mbp deletion region on chromosome 18q12.3 to a 372 kbp critical region spanning three genes: SETBP1, SLC14A2 and MIR431941,42. We identified five truncating mutations (3/3 tested and confirmed de novo) in SETBP1 among patients with moderate to severe ID. The phenotypic similarity among microdeletion patients and truncating SNVs and indels, including ID, craniofacial dysmorphism, and the almost complete absence of expressive language (92% of cases), strongly suggests that loss of function of this gene underlies this condition. Interestingly, gain-of-function mutations result in a completely different phenotype known as the Schinzel-Giedionsyndrome. In contrast to the likely loss-of-function, mutations, gain-of-function mutations cluster within a 12 amino domain and result in a more severe DD with multiple congenital abnormalities and death in infancy48,49. In addition, identical somatic mutations in this hotspot region have recently also been reported in a variety of myeloid malignancies50,51.
Similarly, a study of 19 unrelated DD patients with submicroscopic deletions in chromosome 10p15.3 (as well as data from the CNV morbidity map in this study, which has six shared samples) narrowed the critical region to two genes: DIP2C and ZMYND1143. Our targeted sequencing identified truncating ZMYND11 mutations exclusively in cases but none in DIP2C. ZMYND11 (zinc finger MYND domain 11) encodes a tumor suppressor gene that recognizes H3K36 trimethylated DNA and regulates RNA polymerase II elongation52. It is associated with highly expressed genes and may be an important transcriptional co-repressor early in development. Additionally, ZMYND11 has been demonstrated to play an inhibitory role in neuronal differentiation53. Patients with truncating mutations show borderline IQ and a mild dysmorphism similar to microdeletion patients. Interestingly, both females studied have been described with autistic tendencies, while the three males in this study have been identified with aggressive behaviors, temper tantrums and rage. The oldest male patient in this study (45 years of age) has had, in fact, differing psychiatric diagnoses including borderline personality disorder, bipolar disorder, psychosis, depression, low frustration tolerance leading to aggression, and ADHD. In this regard, it is noteworthy that Frommer and colleagues recently reported a de novo frameshift mutation of ZMYND11 in a patient with schizophrenia54. We suggest that truncating mutations in ZMYND11 are likely to be associated with other more complex neuropsychiatric disorders as children age. Early diagnoses of such carriers as children may be critical to improving their prognosis and outcome.
In conclusion, we have demonstrated that a genotype-first approach combining copy number and mutation screening across a broad range of neurodevelopmental phenotypes has the potential to discover new syndromes and to identify the critical genes underlying pathogenic CNVs. Given the large number of exome sequencing studies that are projected and the locus heterogeneity underlying neurocognitive disease, this CNV-SNV integrated approach in conjunction with forward resequencing in large cohorts will serve to identify additional high-impact genes and pathways important in neurodevelopment.
URLs
Exome Variant Server, http://evs.gs.washington.edu/EVS/
Wellcome Trust Case Control Consortium 2, http://www.wtccc.org.uk/ccc2/
Online Methods
Microarray platforms and samples
We combined the 15,767 cases previously published in Cooper et al.1 with 13,318 new cases with ID/DD and related phenotypes that were submitted to Signature Genomic Laboratories, LLC, for clinical microarray-based CGH. Array CGH was performed on nine different CGH platforms (Supplementary Table 9). All arrays were reanalyzed from the underlying raw data for CNVs (Supplementary Note). The majority of samples were profiled on a 135,000 probe or higher array (64%) with increased density in regions associated with known disorders1,57. Initial CNV calls were generated as previously described57. Cases were filtered by the following criteria: First, CNVs were filtered for absolute log2 ratio >0.3. Second, to account for excess segmentation, CNVs were manually inspected for potential merging when two CNVs of the same state were within 10% of the larger CNV's size of each other. Cases were also filtered based on the following criteria: σ > 0.29722, or excess CNVs (Q3 + 3xIQR per array platform). Cases with >3 large (500+ kbp) subtelomeric (initiating in the first 1.5 subtelomeric Mbp of the p or q arm) events, or over 11 CNVs (1.5 IQR across all cases), were manually inspected to account for wave artifacts in low-quality samples. Finally, we inspected CNVs completely contained in the following regions prone to low-ratio CNVs due to wave artifacts (Supplementary Table 10). CNVs highlighting new regions of interest were validated on a custom 8-plex Agilent array (Supplementary Note). In addition 5,531 cases previously published by Vulto-Van Silfhout were screened for de novo CNVs overlapping regions of interest4.
We constructed a CNV atlas map from combining 8,329 controls from Cooper et al.1 (dbVar study accession nsdt54) with 11,255 new controls profiled on Affymetrix SNP6 arrays from the Wellcome Trust Case Control Consortium 2 58C and NBS cohorts (http://www.wtccc.org.uk/ccc2/), as well as the Atherosclerosis Risk in Communities (ARIC) Community Surveillance Cohort (dbGaP: phs000090v1) (Supplementary Table 1). All CNV calling for the ARIC and WTCCC2 58C cohorts was performed using GTC4.1 with default parameters, except for the minimum CNV size and minimum number of probes, which were set to 10 and 20 kbp, respectively. One array batch with very low ratio responses (log2 ratios at most 16.8% of expected) were removed from the ARIC study due to poor CNV calling. Additional filtering was applied to remove cases with excessive CNV counts, and a threshold of >72 CNVs per case was established using an outlier detection method for skewed data58. Finally, we trimmed CNV calls that falsely extended across centromeric gaps due to small polymorphisms on both arms.
A total of 29,415 rare autosomal CNV calls in cases and 741,729 (289,359 new) control CNVs were detected (Supplementary Table 1) and deposited into dbVar (dbVar study accession nstd100). Patient informed consent was obtained to publish clinical information and photographs and to further characterize the CNVs present in the individuals with detailed information presented in this paper using a protocol approved by the Signature Genomic Laboratories, LLC, Institutional Review Board - Spokane. Controls were not ascertained specifically for neurological disorders, but all controls were obtained from adult samples providing informed consent, so severe developmental phenotypes should be exceedingly rare in this group.
Statistical analysis
CNV burden was compared between cases and controls for rare CNVs (<1%), using CNV length excluding gaps and regions annotated as segmental duplications (hg18). The distribution of these CNVs is indicated in Supplementary Figure 6. Burden was defined using only the largest CNV to account for the large number of bases encompassed in small CNVs and the significant difference in array resolutions between cases and controls. Statistical comparisons utilized the Peto & Peto modification of the Gehan-Wilcoxon test (due to non-proportional hazard ratios) to assess overall burden. For significance at specific thresholds we utilized the Fisher's exact test. Significance for CNV enrichment was enumerated for all RefSeq genes (NCBI36). All isoforms for each gene were combined into a single entry representing all possible coding bases. Rare CNVs from cases and all control CNVs were then enumerated for only cases where the CNV intersects an exon. The resulting counts were then compared using the one-tailed Fisher's exact test. Likelihood ratios were calculated as per standard formulae, and confidence bounds were estimated by using the binomial confidence interval for case and control counts calculated by the Clopper–Pearson exact tail area method as described in Rosenfeld et al59. Additionally, we calculated an empirical p-value for genes affected by rare CNVs. To do so we first excluded CNVs residing in regions with elevated mutation rates or unreliable CNV detection. These regions include subtelomeric CNVs initiating in the first 1.5 Mbp of each chromosome, over 75% of bases intersecting hotspots (145.1 Mbp across 58 sites) and segmental duplications (130.4 Mbp across 7,264 sites), initiating or terminating in a centromere gap region. All CNVs under 10 Mbp were then randomly shuffled (chromosome selection was weighted by the number of bases not filtered) under these constraints for cases and controls and Fisher's exact tests were calculated for deletions and duplications of each gene 20,000 times. The empirical p-value was defined as the number of simulations more significant than observed plus one divided by the number of simulations plus one. CNV burden for regions was also enumerated using a windowed analysis of rare case CNVs over 250 kbp. Window starts/ends were defined based on all unique breakpoints in the signature array. Breakpoint pairs under 50 kbp were then filtered as these represent the uncertainty in edges of Signature calls. Counts for p-values are based on 40% coverage of each window by cases (over 250 kbp) or controls (all CNVS). Significance was calculated using the one-tailed Fisher's exact test, and Supplementary Figure 2 shows the negative logarithm of the p-value. In many cases the critical region may represent multiple subregions that individually reach significance. Here, we report the larger region where smaller subregions are indicated by a number of additional CNVs over the background preventing refinement to a single candidate gene. Due to high prior probability of pathogenicity for large CNVs, the lack of independence between genes disrupted by CNVs, and the high odds ratio for most pathogenic loci, we have chosen to report nominal significance in all cases in addition to the Benjamini-Hochberg q-value, which represents an overestimate of the false discovery rate in our analyses60. Please see the Supplementary Note for details on our interpretation of q-values in this study.
Joint CNV and SNV haploinsufficient mutation probabilities
We developed a model based on the hypergeometric distribution for event counts to calculate the probability of gene enrichment by integration of truncating SNV mutations and CNV deletions. For each gene we enumerated the total number of LoF events observed: cases with and without deletion CNVs (a and b); controls with and without deletion CNVs (c and d); cases with and without truncating SNV and indel mutations (a2 and b2); and controls with and without truncating SNV and indel mutations (c2 and d2). We computed the observed frequency (Z) of LoF events (CNVs and SNVs) (equation 1). We assume that mutations and CNVs are independent (supported by the rare nature of these events); however, in cases with more frequent observations the interaction term could be included in the calculation of Z. This threshold was applied to calculate probabilities as per equation 2. When CNV or truncating SNV and indel mutation counts are 0 for both cases and controls, the p-value reduces to the equivalent of the one-tailed Fisher's exact test for the assay with counts. This method also has the benefit of allowing negative observations from one assay to decrease the significance of a gene. For example, a gene with no CNVs in controls, but many truncating SNV mutations, will be negatively impacted by those events.
| (1) |
| (2) |
Truncation p-values
For genes with truncating mutations in controls we also compared the effect on protein lengths (in the context of retained wild-type amino acids) in cases and controls based on annotated isoforms. Although early stop gains may lead to either nonsense mediated decay or truncated proteins, this model does not discriminate between these outcomes since both result in proteins without wild-type function. For splice-site mutations we extracted the most likely lost exon and determined the likely protein effect (in-frame loss or introduction of a frameshift or stop codon). Predicted protein lengths for ESP6500 and cases were compared using the log-rank test.
MIP sequencing and sample cohorts
Targeted sequencing of candidate genes was accomplished using the MIP resequencing method as described in O'Roak et al36. In total, we successfully targeted the coding sequence and splice-donor/acceptor sites of 26 genes with 1,388 MIPs. MIP sequences, their concentrations in the assay, and relative performances are detailed in Supplementary Table 8. 192 samples were barcoded and sequenced per Illumina HiSeq lane, and all analyses were performed as described in O'Roak et al.36. 192 samples were included in each Illumina HiSeq 2000 lane with 1,388 MIP probes covering 26 genes. Details on the MIP probes used, their individual performance, and concentrations in the pool are detailed in Supplementary Table 9.
In order to compare data between exome and MIP sequencing, we calculated statistics only for sites (case and control) with an average read depth >20 in the ESP6500, and no intersection with low complexity repeat sequence (as defined by Dustmasker).
In total, we screened 8,060 unique samples including 5,633 probands and 2,427 unaffected siblings from the Simons Simplex Collection (SSC). In addition to variant-level filtering, samples were filtered by QC based on the percentage of MIPs with at least 20 reads (our minimum for variant calling). Probands were required to have sufficient coverage for 75% of targets, while control samples were required to have 90% of targets covered. This resulted in the inclusion of 2,193/2,427 controls and 4,716/5,633 cases in the final analysis (Supplementary Figure 7).
Patients were consented for resequencing and recontact for inheritance testing. Patient samples were acquired from the Autism Phenome Project (David Amaral, UC Davis), Leuven (Hilde Peeters, University Hospitals Leuven), Murdoch (Ingrid E. Scheffer, Murdoch Children's Research Institute), Adelaide (Jozef Gécz, University of Adelaide), Nijmegen (Bert B.A. de Vries, Radboud University Medical Center), SAGE (Raphael Bernier, University of Washington), and Troina (Corrado Romano, Associazione Oasi Maria Santissima) (Supplementary Table 11).
Supplementary Material
Acknowledgments
We thank F. Hormozdiari, M. Dennis, and T. Brown for useful discussions and for editing the manuscript. B.P.C. is supported by a fellowship from the Canadian Institutes of Health Research. This study makes use of data generated by the Wellcome Trust Case Control Consortium. A full list of the investigators who contributed to the generation of the data is available from http://www.wtccc.org.uk/. J.A.R. and B.T. are employees of Signature Genomic Laboratories, LLC, a subsidiary of PerkinElmer, Inc. This work was supported by U.S. National Institutes of Mental Health MH101221 to E.E.E. E.E.E. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Accession Codes: CNV for the combined cases and new controls have been deposited into dbVar under accession nstd100.
Author Contributions: B.P.C. and E.E.E. designed the study. B.P.C. performed the data analysis. B.P.B., K.W., and C.B. performed array CGH, MIP sequencing, and Sanger validations. J.A.R. and B.S.T. supervised array CGH experiments and coordinated clinical data collection at Signature Genomics. B.W.M.v.B., A.T.V-v.S., P.B., K.L.F., S.B., L.E.L.M.V., J.H.S-H., A.H., D.L., D.A., N.B., P.J.L., I.E.S., A.A., R.P., R.T., N.d.L., M.R.F.R., H.P., M.F., H.C.M., E.H., C.R., J.G., and B.B.A.d.V. provided clinical samples for resequencing, clinical reports, and inheritance testing. J.Y.H-K. and N.d.L. curated the Nijmegen de novo CNV calls. B.P.B., K.W., C.B., B.O., J.S., and E.E.E. designed the MIP gene panel. G.L.C. and H.C.M. identified two SETBP1 variants in an independent screen. N.K. curated published de novo mutations. B.P.C. and E.E.E. wrote the manuscript. All authors have read and approved the final version of the manuscript.
References
- 1.Cooper GM, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–46. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaminsky EB, et al. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. 2011;13:777–84. doi: 10.1097/GIM.0b013e31822c79f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moreno-De-Luca D, et al. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol Psychiatry. 2013;18:1090–5. doi: 10.1038/mp.2012.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vulto-van Silfhout AT, et al. Clinical significance of de novo and inherited copy-number variation. Hum Mutat. 2013;34:1679–87. doi: 10.1002/humu.22442. [DOI] [PubMed] [Google Scholar]
- 5.Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–21. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Ligt J, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–9. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 7.Gulsuner S, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154:518–29. doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iossifov I, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–99. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang YH, et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet. 2013;93:249–63. doi: 10.1016/j.ajhg.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–5. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O'Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–50. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rauch A, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–82. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 13.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–41. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaidi S, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–3. doi: 10.1038/nature12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacquemont S, et al. A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am J Hum Genet. 2014;94:415–25. doi: 10.1016/j.ajhg.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rudd MK, et al. Segmental duplications mediate novel, clinically relevant chromosome rearrangements. Hum Mol Genet. 2009;18:2957–62. doi: 10.1093/hmg/ddp233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burkardt DD, et al. Distinctive phenotype in 9 patients with deletion of chromosome 1q24-q25. Am J Med Genet A. 2011;155A:1336–51. doi: 10.1002/ajmg.a.34049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dabell MP, et al. Investigation of NRXN1 deletions: clinical and molecular characterization. Am J Med Genet A. 2013;161A:717–31. doi: 10.1002/ajmg.a.35780. [DOI] [PubMed] [Google Scholar]
- 19.Gimelli S, et al. A rare 3q13.31 microdeletion including GAP43 and LSAMP genes. Mol Cytogenet. 2013;6:52. doi: 10.1186/1755-8166-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madrigal I, Martinez M, Rodriguez-Revenga L, Carrio A, Mila M. 12p13 rearrangements: 6 Mb deletion responsible for ID/MCA and reciprocal duplication without clinical responsibility. Am J Med Genet A. 2012;158A:1071–6. doi: 10.1002/ajmg.a.35287. [DOI] [PubMed] [Google Scholar]
- 21.Paciorkowski AR, et al. MEF2C Haploinsufficiency features consistent hyperkinesis, variable epilepsy, and has a role in dorsal and ventral neuronal developmental pathways. Neurogenetics. 2013;14:99–111. doi: 10.1007/s10048-013-0356-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenfeld JA, et al. Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome. PLoS One. 2009;4:e6568. doi: 10.1371/journal.pone.0006568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stankiewicz P, et al. Recurrent deletions and reciprocal duplications of 10q11.21q11.23 including CHAT and SLC18A3 are likely mediated by complex low-copy repeats. Hum Mutat. 2012;33:165–79. doi: 10.1002/humu.21614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Bon BW, et al. The phenotype of recurrent 10q22q23 deletions and duplications. Eur J Hum Genet. 2011;19:400–8. doi: 10.1038/ejhg.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Docker D, et al. Further delineation of the SATB2 phenotype. Eur J Hum Genet. 2013 doi: 10.1038/ejhg.2013.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thorsson T, et al. Chromosomal Imbalances in Patients with Congenital Cardiac Defects: A Meta-analysis Reveals Novel Potential Critical Regions Involved in Heart Development. Congenit Heart Dis. 2014 doi: 10.1111/chd.12179. [DOI] [PubMed] [Google Scholar]
- 27.Le Meur N, et al. MEF2C haploinsufficiency caused by either microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. J Med Genet. 2010;47:22–9. doi: 10.1136/jmg.2009.069732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ching MS, et al. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:937–47. doi: 10.1002/ajmg.b.31063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shuvarikov A, et al. Recurrent HERV-H-mediated 3q13.2-q13.31 deletions cause a syndrome of hypotonia and motor, language, and cognitive delays. Hum Mutat. 2013;34:1415–23. doi: 10.1002/humu.22384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Endele S, et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet. 2010;42:1021–6. doi: 10.1038/ng.677. [DOI] [PubMed] [Google Scholar]
- 31.Goffin A, Hoefsloot LH, Bosgoed E, Swillen A, Fryns JP. PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet. 2001;105:521–4. doi: 10.1002/ajmg.1477. [DOI] [PubMed] [Google Scholar]
- 32.Koolen DA, et al. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat Genet. 2012;44:639–41. doi: 10.1038/ng.2262. [DOI] [PubMed] [Google Scholar]
- 33.Lossin C. A catalog of SCN1A variants. Brain Dev. 2009;31:114–30. doi: 10.1016/j.braindev.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 34.Santen GW, et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet. 2012;44:379–80. doi: 10.1038/ng.2217. [DOI] [PubMed] [Google Scholar]
- 35.Talkowski ME, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet. 2011;89:551–63. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Roak BJ, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–22. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fischbach GD, Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–5. doi: 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Sharp AJ, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Bon BW, et al. Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clin Genet. 2011;79:296–9. doi: 10.1111/j.1399-0004.2010.01544.x. [DOI] [PubMed] [Google Scholar]
- 40.Girirajan S, et al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am J Hum Genet. 2013;92:221–37. doi: 10.1016/j.ajhg.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Filges I, et al. Reduced expression by SETBP1 haploinsufficiency causes developmental and expressive language delay indicating a phenotype distinct from Schinzel-Giedion syndrome. J Med Genet. 2011;48:117–22. doi: 10.1136/jmg.2010.084582. [DOI] [PubMed] [Google Scholar]
- 42.Marseglia G, et al. 372 kb microdeletion in 18q12.3 causing SETBP1 haploinsufficiency associated with mild mental retardation and expressive speech impairment. Eur J Med Genet. 2012;55:216–21. doi: 10.1016/j.ejmg.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 43.Descipio C, et al. Subtelomeric deletion of chromosome 10p15.3: Clinical findings and molecular cytogenetic characterization. Am J Med Genet A. 2012;158A:2152–61. doi: 10.1002/ajmg.a.35574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ansieau S, Leutz A. The conserved Mynd domain of BS69 binds cellular and oncoviral proteins through a common PXLXP motif. J Biol Chem. 2002;277:4906–10. doi: 10.1074/jbc.M110078200. [DOI] [PubMed] [Google Scholar]
- 45.Kateb F, et al. Structural and functional analysis of the DEAF-1 and BS69 MYND domains. PLoS One. 2013;8:e54715. doi: 10.1371/journal.pone.0054715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masselink H, Bernards R. The adenovirus E1A binding protein BS69 is a corepressor of transcription through recruitment of N-CoR. Oncogene. 2000;19:1538–46. doi: 10.1038/sj.onc.1203421. [DOI] [PubMed] [Google Scholar]
- 47.Stefansson H, et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature. 2014;505:361–6. doi: 10.1038/nature12818. [DOI] [PubMed] [Google Scholar]
- 48.Hoischen A, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet. 2010;42:483–5. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 49.Schinzel A, Giedion A. A syndrome of severe midface retraction, multiple skull anomalies, clubfeet, and cardiac and renal malformations in sibs. Am J Med Genet. 1978;1:361–75. doi: 10.1002/ajmg.1320010402. [DOI] [PubMed] [Google Scholar]
- 50.Makishima H, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45:942–6. doi: 10.1038/ng.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piazza R, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. 2013;45:18–24. doi: 10.1038/ng.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wen H, et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 2014 doi: 10.1038/nature13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu B, et al. BS69 undergoes SUMO modification and plays an inhibitory role in muscle and neuronal differentiation. Exp Cell Res. 2009;315:3543–53. doi: 10.1016/j.yexcr.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 54.Fromer M, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–84. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alliman S, et al. Clinical and molecular characterization of individuals with recurrent genomic disorder at 10q22.3q23.2. Clin Genet. 2010;78:162–8. doi: 10.1111/j.1399-0004.2010.01373.x. [DOI] [PubMed] [Google Scholar]
- 56.Hehir-Kwa JY, et al. De novo copy number variants associated with intellectual disability have a paternal origin and age bias. J Med Genet. 2011;48:776–8. doi: 10.1136/jmedgenet-2011-100147. [DOI] [PubMed] [Google Scholar]
- 57.Duker AL, et al. Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur J Hum Genet. 2010;18:1196–201. doi: 10.1038/ejhg.2010.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.M H, S VdV. Outlier detection for skewed data. J Chemometr. 2008;22:235–246. [Google Scholar]
- 59.Rosenfeld JA, Coe BP, Eichler EE, Cuckle H, Shaffer LG. Estimates of penetrance for recurrent pathogenic copy-number variations. Genet Med. 2013;15:478–81. doi: 10.1038/gim.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc. 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.