

Abstract
Psychiatric disorders are complex multifactorial illnesses involving chronic alterations in neural circuit structure and function. While genetic factors are important in the etiology of disorders such as depression and addiction, relatively high rates of discordance among identical twins clearly indicate the importance of additional mechanisms. Environmental factors such as stress or prior drug exposure are known to play a role in the onset of these illnesses. Such exposure to environmental insults induces stable changes in gene expression, neural circuit function, and ultimately behavior, and these maladaptations appear distinct between developmental and adult exposures. Increasing evidence indicates that these sustained abnormalities are maintained by epigenetic modifications in specific brain regions. Indeed, transcriptional dysregulation and associated aberrant epigenetic regulation is a unifying theme in psychiatric disorders. Aspects of depression and addiction can be modeled in animals by inducing disease-like states through environmental manipulations (e.g., chronic-stress, drug administration). Understanding how environmental factors recruit the epigenetic machinery in animal models is revealing new insight into disease mechanisms in humans.
INTRODUCTION
Psychiatric disorders impose an ever-increasing burden on society. Major depression and drug addiction are complex phenomena resulting from the interaction of several factors including neurobiological, genetic, cultural, and life experience. Both depression and addiction are characterized by functional and transcriptional alterations in several limbic brain regions implicated in regulating stress responses and reward1; 2; 3; 4; 5. Advances in the last decade have identified epigenetic mechanisms as important effectors in psychiatric conditions. Indeed, being at the foundation of gene regulation, epigenetic mechanisms are ideal candidates for the study of depression and addiction. Epigenetic mechanisms refer to the highly complex organization of DNA. This includes primarily many types of histone modifications6 and nucleotide modifications such as DNA methylation7 and hydroxymethylation8. More recently, non-coding RNAs such as microRNAs (miRNA) have emerged as a related mechanism although not epigenetic per se. Mounting evidence has identified epigenetic regulation in the context of stress-induced depression and drug-induced addiction. These epigenetic alterations are site-specific, largely in post-mitotic cells, and appear to be de novo rather than inherited. The majority of these findings come from animal models although interesting insights in humans are starting to accumulate.
Early development marks a time of rapid brain development and enhanced susceptibility to environmental insults. Epigenetic mechanisms of gene regulation are a particularly attractive explanation for how early life exposures to stress or drugs exert life-long effects on neuropsychiatric phenomena. Developmental exposures to stress or drugs may have broader impact on epigenetic states and brain circuits than similar exposure later in life. Research to date has focused on relatively distinct neural circuits in exploring the consequences of developmental versus adult exposures.
The present review brings together findings relating to epigenetic mechanisms in depression and addiction from both adult and developmental studies and elaborates the potential offered by epigenetic analyses to better understand these complex disorders.
EPIGENETICS AND DEPRESSION
Depression is a complex and heterogeneous disorder. Stressful life events represent a major factor in vulnerability to depression. However, the difficulty in defining specific subsets of depression has made clear identification of its multiple etiologies very difficult. Animal models offer a useful approach to study depression. Indeed, the development of chronic stress paradigms over the last decade, combined with the ability to objectively measure anhedonia and stress susceptibility in rodents, have helped clarify the neural circuitry and neuroadaptations underlying aspects of depression.
Adult onset of depression
Our understanding of the role of epigenetics in adult depression comes primarily from studies of animals exposed to stress. While acute stress paradigms are designed to evaluate an animal’s initial coping response, chronic stress paradigms involve prolonged exposure to either physical9 or psychological stressors, such as social subordination10. Such chronic stress paradigms successfully recapitulate certain behavioral features of human depression. For instance, chronic stressors produce anhedonia-like symptoms, characterized by a decrease in reward-related behaviors such as reduced preference for sucrose and social interaction10; 11 that are rarely seen following acute stress. Additionally, some behavioral alterations induced by chronic stress are long-lasting and effectively reversed by chronic but not acute treatment with existing antidepressant medications10; 11, a treatment course comparable to that required in humans. Together, these findings suggest that chronic stress paradigms are more effective at modeling at least certain features or subtypes of the human depression syndrome, while acute studies may provide insight into neuronal adaptations that regulate short-lived responses to stressful events.
Histone modifications in depression
Histone tails may be posttranslationally modified by acetylation, methylation, phosphorylation, poly(ADP-ribosyl)ation, and ubiquitination, among others. The combinations of various marks at a multitude of residues are seemingly endless and ongoing research is attempting to translate the effect of this “histone code” to transcriptional regulation. Meanwhile, study of some better understood histone modifications are providing important insights into the pathophysiology of depression12.
Histone Acetylation
The potential importance of histone acetylation in depression was initially suggested by observations that histone deacetylase (HDAC) inhibition alone or in combination with antidepressant treatment ameliorated depression-like behavior in rodents13; 14; 15; 16; 17,18; 19; 20. In mice, chronic social defeat stress induces a transient decrease of H3K14 acetylation in nucleus accumbens (NAc; see Figure 1B; Figure 2), a key brain reward region, associated with a persistent reduction of HDAC214. Both findings are seen in the NAc of depressed humans as well. The observation that intra-NAc infusion of MS275, a specific inhibitor of class I HDACs, yields antidepressant-like effects suggests that the persistent increase in histone acetylation in NAc may facilitate adaptation to chronic stress14. In support of this, in a chronic unpredictable stress paradigm, overexpression of an Hdac2 dominant-negative in NAc was antidepressant, while expression of a form of Hdac2 with increased chromatin binding affinity was pro-depressant20. However, expression of Hdac5, a class II HDAC, is decreased in mice susceptible to social defeat stress and increased by chronic imipramine treatment suggesting a pro-resiliency effect of this HDAC. This is further supported by the heightened susceptibility to social defeat of Hdac5 knock-out mice21. The opposing effects of HDAC2 and HDAC5 are difficult to reconcile with a simplistic role of histone acetylation in depression and stress adaptation. However, it is conceivable that HDAC2 and HDAC5 may regulate distinct populations of genes. Additionally, it is important to note that HDAC5 could also regulate non-histone targets due to its cytoplasmic as well as nuclear localization. Genome-wide studies of NAc gene expression in defeated mice treated with fluoxetine or intra-NAc infusion of MS275 demonstrated that both treatments are able to reverse a large proportion of defeat-induced differential gene expression. Although each treatment regulated subsets of unique gene expression changes, there was also a degree of overlap in regulated targets, suggesting that antidepressant effects of fluoxetine may in part be mediated by regulation of histone acetylation14.
Figure 1. Epigenetic dysregulation in the HPA-axis and reward circuitry is implicated in psychiatric disorders.
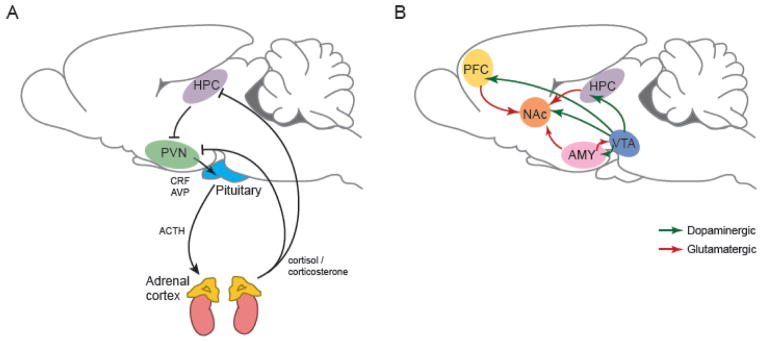
A majority of research on altered epigenetic regulation in depression and other stress-related disorders has focused on changes within the hypothalamic-pituitary-adrenal (HPA) axis (A) and the brain’s reward circuitry (B), depicted here in rodent brain. Studies examining effects of early life manipulations on epigenetic regulation of behavior have focused on changes within the HPA axis, in contrast to adult studies, which have concentrated on epigenetic alterations in reward circuitry.
A) Main components of the HPA axis: corticotropin releasing factor (CRF) and vasopressin (AVP) from the paraventricular nucleus of the hypothalamus (PVN) stimulates adrenocorticotropic hormone (ACTH) release from the anterior pituitary, which induces glucocorticoid (cortisol [human] or corticosterone [rodent]) release from the adrenal cortex. Glucocorticoid receptors (GRs) in the hippocampus (HPC) and other brain regions mediate negative feedback to reduce the stress response. B) Depicted are the major components of the limbic-reward circuitry: dopaminergic neurons (green) project from ventral tegmental area (VTA) to nucleus accumbens (NAc), prefrontal cortex (PFC), amygdala (AMY), and HPC. NAc receives excitatory glutamatergic innervation (red) from HPC, PFC, and AMY.
Figure 2. Examples of chromatin modifications regulated by stress or antidepressant treatment.

Illustration (top) indicates histone octamers (pink) in heterochromatin (left) and euchromatin (right), along with associated proteins and histone tail/DNA modifications. Table (bottom) lists histone tail modifications of specific residues—depicted on the expanded histone tail illustration (left)—that are regulated by various stress paradigms or antidepressant treatments within the indicated brain regions. Arrows indicate an increase (green) or decrease (blue) in specific modifications. Abbreviations: A, acetylation; P, phosphorylation; M (in a square), histone methylation; M (in a circle), DNA methylation; AMY, amygdala; HAT, histone acetyltransferase; HDAC, histone deacetylase; HPC, hippocampus; HMT, histone methyltransferase; HR and LR, high responding and low responding, respectively (with respect to baseline locomotor activity); PFC, prefrontal cortex; NAc, nucleus accumbens; pol II, RNA polymerase II. Modified from Ref 12, with permission.
Chronic stress paradigms also robustly regulate histone acetylation in the hippocampus (HPC; see Figure 1A&B), a brain region that is both highly sensitive to the effects of stress and implicated in the regulation of stress responses. In contrast to effects in the NAc, chronic social defeat stress transiently increases then persistently decreases global H3K14 acetylation and this decrease is reversed by chronic imipramine22 (see Figure 2). Imipramine also increases H3 acetylation at the brain-derived neurotrophic factor (Bdnf) promoter and this increase correlates with increased hippocampal Bdnf expression13. However, the initial stress-induced decrease in Bdnf after defeat may be mediated by increased H3K27 methylation at the Bdnf promoter rather than histone acetylation mechanisms. Intra-hippocampal HDAC inhibition by MS275 restores normal sucrose preference in defeated mice but alone does not alter social avoidance. However, MS275 treatment combined with social housing reversed social interaction deficits suggesting that histone acetylation in HPC may serve to facilitate adaptation and that defeat-induced reduction in H3K14 acetylation post-defeat is maladaptive. Whereas Hdac5 expression in NAc is antidepressant, in HPC Hdac5 is downregulated by imipramine in defeated mice and hippocampal overexpression of Hdac5 blocks the antidepressant actions of imipramine.
In a genetic rat model of stress susceptibility, high responders (HR), which exhibit basal reductions in anxiety and increased sucrose preference relative to low responders (LR), have increased CREB-binding protein (a histone acetyl transferase or HAT), lower HDAC3, and higher global H3 and H2B acetylation levels in hippocampus23. However, HR rats are more susceptible to chronic social defeat than LR rats and exhibit reduced sucrose preference post-defeat. After chronic social defat, HR rats have decreased hippocampal global H3 and H2B acetylation, whereas LR rats have increased H3 acetylation. Both groups showed equivalent decreases in H4 acetylation. Repeated electroconvulsive seizures (ECS), which induce a robust antidepressant effect, regulates H3 and H4 acetylation at Bdnf, c-Fos, and Creb promoters in a time-dependent manner that correlates with gene expression changes. Downregulation of c-Fos was linked to reduced H4 acetylation whereas sustained induction of Bdnf was linked to increased H3 acetylation24. Potentially H3 and H4 acetylation differentially modulate depression-like states in HPC further highlighting the complexity of histone mechanisms in depression.
Histone acetylation in the amygdala (AMY; see Figure 1B, Figure 2) is also implicated in the expression of depressive-like behaviors after chronic social defeat. H3K14 acetylation is transiently increased in AMY after social defeat but is not persistently changed25. HDAC inhibition by intra-AMY MS275 reverses social avoidance but not sucrose preference deficits, suggesting that AMY and HPC histone acetylation may regulate different aspects of depression-like behavior. Chronic unpredictable stress in rats reduced Hdac5 expression in the AMY central nucleus26 and acute, but not chronic, defeat transiently decreased AMY H3 acetylation27. Clearly more work is needed to fully understand the significance of AMY histone acetylation in depression.
Data on the role of histone acetylation in depression in prefrontal cortex (PFC; see Figure 1B) is similarly scarce. Although some studies report no global change in acetylation levels25; 27, conflicting reports suggest that social defeat stress increases global H3 acetylation in PFC28 although this study did not differentiate between mice that are susceptible versus resilient to defeat stress making the significance of this regulation difficult to interpret. A recent study found that rats less resilient to chronic social defeat stress had increased levels of H3K18 acetylation in PFC29. At this point, the lack of data documenting effects of manipulating histone acetylation in PFC prohibits a clearer understanding of the importance of potential stress-induced acetylation changes in depression.
In summary, histone acetylation is robustly regulated by chronic stress in multiple brain regions however the significance of these changes is region-specific. Existing data suggest that, in general, increased histone acetylation, which would favor an open chromatin conformation and increased transcription, is antidepressant. More work is needed to understand how histone acetylation changes regulate expression of specific gene targets implicated in depression.
Histone Methylation
Chronic social defeat stress robustly decreases global levels of H3K9me2 in NAc, a repressive histone modification (see Figure 2), with the coincident downregulation of the histone methyltransferases G9a and G9a-like protein which catalyze this mark22. Overexpression of G9a in NAc is antidepressant22 and increased H3K9me2 at specific gene promoters is implicated in the antidepressant effect of fluoxetine30. Indeed, chronic exposure to fluoxetine reduces Camkii expression in NAc by reducing H3Ac and increasing H3K3me2 levels at the Camkiia promoter in NAc. Interestingly, these effects are found in the NAc of depressed humans exposed to antidepressants suggesting that the stress-induced loss of repressive methylation is maladaptive and that the therapeutic effects of antidepressant drugs may act via the reinstatement of these marks at specific gene loci. One gene which illustrates this mode of regulation is Ras. Reduced H3K9me2 at this gene in the NAc of susceptible mice results in increased Ras expression, induction of ERK signaling and, ultimately, CREB activation which induces depression-like behavior22.
Another repressive histone mark, H3K27me3, is increased upstream to the promoter of the Rac1 gene in susceptible mice and this is associated with a sustained reduction in transcript expression that influences characteristic dendritic spine changes in defeated mice31. These findings are corroborated in humans as H3K27me3 levels are inversely correlated with Rac1 expression, which is also decreased in NAc of depression patients31. As well, susceptible mice exhibit decreased expression of Mll and Lsd1, the methyltransferase and demethylase of H3K4, respectively, although no global changes in H3K4 methylation are apparent22.
The observed decreases in H3K9me2 in NAc would be expected to mediate a more permissive transcriptional state similar to the effect of the global increases in H3 acetylation described above. Curiously, however, manipulations that decrease repressive methylation induce susceptibility whereas manipulations that increase acetylation induce resilience. To reconcile and integrate these findings, genome-wide approaches are required to examine regulation of histone modifications at specific gene loci to understand the precise coordinated regulation of depression-related target genes. ChIP-chip analysis (chromatin immunoprecipitation followed by genome-wide promoter microarrays) examined stress-induced redistribution of H3K9me2 and H3K27me2 in NAc of mice subjected to chronic social defeat or protracted social isolation. Significant and dynamic changes in repressive histone methylation were observed in upstream regulatory regions in both models with more genes evidencing increased H3 methylation. Interestingly approximately 20% of genes were similarly regulated in both models32. The genome-wide finding of increased H3 methylation opposes the finding of reduced global levels of H3K9me2 in NAc of susceptible mice22, further highlighting the requirement for more refined analytical approaches that target histone modifications at specific genomic loci.
Stress also regulates histone methylation in the HPC in a complex time dependent manner33. For instance, acute and sub-chronic restraint stress increases global H3K27me3 levels, an effect that returns to basal levels with chronic restraint stress. Moreover, H3K9me1 is decreased after acute stress and these changes are not evident after more prolonged stress. In addition, sub-chronic stress increases global H3K4me3 levels whereas chronic stress decreases levels of this same mark and this decrease is reversed by antidepressant treatment. Potentially these temporal patterns of histone methylation may reflect different processes of initial stress adaptation subsiding into eventual maladaptation with sustained stress. However, experimental manipulations of such modifications are needed to interpret the functional consequences of these adaptations. Acute stress also increases H3K9me3 levels at transposable elements which may be important in limiting potential genomic instability34. Whole forebrain overexpression of Setdb1, a histone methyltransferase that catalyzes H3K9me3, reduced depression-like behavior35, suggesting that the increase in H3K9me3 after acute stress may represent an adaptive response.
Aside from the few examples cited above, human postmortem studies examining histone modifications in depression are sparse. Elevated levels of H3K4me3 were reported at the synapsin gene family. These changes associate with higher expression of SYN2 in the PFC36. In addition, contrasting chromatin profiles were found in brains from bipolar disorder cases suggesting that these changes may be specific to major depression. Similar changes have been described in the promoter of the polyamine gene OAZ where higher levels of H3K4me3 are associated with higher expression in the PFC of suicide completers37.
Consistent with evidence from animal models, postmortem studies in human brains suggest that antidepressants promote open chromatin structure by decreasing H3K27me3 levels at certain BDNF promoters in PFC of a depression sample38. Follow up studies in peripheral blood revealed higher peripheral BDNF expression in treatment responders compared to non-responders, with H3K27me3 levels being inversely correlated with both BDNF IV expression levels and with symptom severity39. Decreased expression of the tyrosine receptor kinase B (TRKB), which is activated by BDNF, in the PFC of depressed cases is also associated with an enrichment of H3K27me3 levels in the promoter of both TRKB and its astrocytic variant, TRKB.T140; 41. The elevated H3K27me3 levels associate with changes in DNA methylation in the promoter suggesting the presence of dual epigenetic control over TRKB.T1 expression, as reported for many genes in simpler systems. In addition, mice overexpressing TRKB.T1 are more susceptible to chronic social stress than wild type mice42 suggesting that epigenetic changes at the TRKB.T1 promoter could define the vulnerability to chronic social stress and the development of depression.
Chromatin remodeling in depression
Very little is known concerning the role of chromatin remodeling complexes in depression. Chromatin remodeling complexes use the energy of ATP hydrolysis to alter the packing state of chromatin and work in concert with chromatin modifying enzymes to direct nucleosomal dynamics. Preliminary data suggest that chronic social defeat regulates the expression levels of several families of chromatin remodelers in the NAc, including the ISWI family, and enhancing ISWI remodeling complexes in NAc controls susceptibility to social defeat43. As our understanding of these molecules as well as their functional role evolves, more work will be needed to uncover the precise nature of their impact on behavioral regulation induced by stress in the context of depression.
DNA methylation
In addition to the chromatin modifications described above, a growing body of evidence supports a role for DNA methylation in mediating the impact of stress. DNA methylation is a relatively stable epigenetic mark and thus an interesting candidate mechanism for sustained stress-induced susceptibility to depression. Several of these alterations have been described in different animal models and more recently in human brain.
Chronic social defeat stress increases transcript levels of the de novo DNA methyltransferase Dnmt3a in NAc. Overexpressing Dnmt3a in NAc increases depression-like behavior after sub-maximal social defeat and intra-NAc infusion of a DNMT inhibitor, RG108, reverses defeat induced social avoidance44. DNMT3a activity is generally associated with transcriptional repression suggesting that susceptibility may associate with downregulation of transcriptional expression in NAc. Genome-wide analysis of DNA methylation will be important in establishing the precise mechanisms of this epigenetic modification in defeat-induced susceptibility. DNA methylation in NAc may play a role in regulation of glial cell-derived neurotrophic factor (Gdnf)20. Chronic unpredictable stress increases Gdnf expression in NAc of a stress-sensitive mouse line (BALB/C) but increases Gdnf expression in a more resilient line (C57BL/6). Although DNA methylation and MeCP2 (methyl CpG binding protein 2) binding was increased at the Gdnf promoter in NAc of both mouse lines, MeCP2 complexes with different proteins in the two lines. In the susceptible mouse line, MeCP2 reportedly interacts with HDAC2 to decrease H3 acetylation and repress Gdnf transcription, while in the resilient mouse line MeCP2 associates with the transcriptional activator CREB to facilitate transcription. The authors suggest that these differences are mediated by differing patterns of methylation at the Gdnf promoter although more work is required to confirm this and elucidate its underlying mechanisms.
DNA methylation is implicated in the regulation of corticotropin releasing factor (CRF) in the paraventricular nucleus of the hypothalamus (PVN)26; 45 (see Figure 1A). CRF is a critical regulator of the hypothalamic-pituitary-adrenal (HPA)-axis activation and other stress actions in the brain. CRF is increased in the PVN of mice that are susceptible to social defeat and this is accompanied by decreased DNA methylation at the Crf promoter. Both effects are reversed by chronic imipramine treatment45. DNA methylation is also increased at the Crf promoter in the PVN of female rats subjected to chronic unpredictable stress, suggesting that DNA methylation may play a role in determining sex-specific regulation of HPA-axis function26. Knockout of Mecp2 in PVN results in an exaggerated physiological stress response, however, the precise mechanism of MeCP2 action remains to be fully elucidated46.
Flinders Sensitive Line rats, a model of genetic susceptibility to depression, exhibit elevated DNA methylation at the P11 promoter which is reduced by escitalopram treatment47. The significance of these findings lies in the fact that P11 is known to interact with serotonin receptors and decreased levels of P11 in certain brain regions are associated with depression-like behavior in mice and humans48; 49; 50. Additionally, chromatin remodeling factor SMARCA3 is a target of the p11 complex and is required for neurogenesis and behavioral responses induced by fluoxetine treatment, indicating interactions between epigenetic states51.
In addition to the effects mentioned above, exposure to aggressive mothers and traumatic stress in rats decrease the expression of Bdnf transcripts III and IV in the HPC and PFC52; 53. These effects are associated with alteration of DNA methylation patterns in distinct regions of the HPC53.
The studies elaborated above highlight epigenetic modifications targeted toward specific genes frequently associated with behavioral alterations. However, it is clear that stress effects are not restricted to a small number of candidate genes. Genome-wide studies mapping DNA methylation alterations induced by stress are lacking in animals. A series of recent genome-wide studies addressing this issue in humans in the context of early life adversity are discussed below. Furthermore, analysis of PFC of psychotic and bipolar cases found numerous sites of differential DNA methylation that were enriched in various functions such as glutamatergic and GABAergic neurotransmission, brain development, and response to stress54. Importantly, these studies compared different tissues (blood versus brain) and brain regions (HPC versus PFC) and globally suggest that stress-induced epigenetic adaptations are region-specific but also cell-type specific, consistent with the emerging notion of epigenetic heterogeneity across tissues55 and cell types56; 57.
In summary, experiments to date have identified stress-induced increases in DNA methylation at a small number of genic loci. Based on the current literature, chronic stress would appear to favor increased DNA methylation and transcriptional repression. However, genome-wide approaches will be very important in identifying the global DNA-methylation “foot-print” and the transcriptional consequences in stress models in animals and in human depression.
Non-coding RNAs in depression
A relatively novel area of research concerns the regulation of miRNAs by stress and antidepressant treatments. miRNAs are posttranscriptional regulators that bind to complementary sequences in the 3′ UTR of their target mRNAs to repress translation or alter mRNA stability58. While still in its infancy, this area of research has the promise to identify important mechanisms through which stress and antidepressants exert their effects.
Microarray analysis of PFC and HPC following acute or repeated restraint stress in mice revealed stress induced changes in miRNA expression profiles. Following acute restraint, let-7a, miR-9, and miR 26-a/b expression was increased in the PFC but not in the HPC, while no change was reported after repeated restraint59. Contrasting results showed elevated expression of let-7a-1 in AMY after acute and chronic stress. In addition, miR-376b and miR-208 were both increased following acute or chronic stress, whereas miR-9-1 decreased in both conditions in HPC60. Other reports implicate miRNAs in the post-transcriptional regulation of Gr (glucocorticoid receptor; Nr3c1). Compared to Sprague-Dawley rats, F344 stress-sensitive rats have lower GR protein but not mRNA levels in the PVN and corresponding increases in miR-18a levels. Overexpression of miR-18a downregulates GR protein levels in cultured neurons61 suggesting that the exaggerated HPA stress response in F344 rats may be mediated by miR-18a regulation of GR translation. Interestingly, in AMY, acute stress induced expression of several miRNAs is implicated in regulation of anxiety through effects on mineralocorticoid receptor and corticotropin-releasing factor receptor 1 (Crfr1) expression62; 63. A significant downregulation of several miRNAs has been reported in HPC of rats that do not develop learned helplessness (LH) following inescapable foot shock compared to LH rats64. LH is a model that is often used to mimic certain symptoms of depression65. These effects coincide with drastic changes in HPC gene expression66 and are believed to represent a potential coping mechanism that allows the development of physiological responses to overcome the effects of stress.
miRNAs may also be therapeutic targets for mood stabilizers. A screening for miRNAs targeted by lithium and valproic acid in rat HPC identified several candidates, among which nine were common to both treatments and are predicted to target genes frequently associated with bipolar disorder. Moreover, lithium was shown to regulate the immobilization stress-induced alteration in HPC miR-34c and AMY miR-15a levels but only in a stress-specific fashion67. Chronic fluoxetine treatment in mice increases miR-16 levels in serotonergic raphe nuclei which downregulates serotonin transporter (Sert) levels, thus altering 5-HT activity in the raphe68. Intra-raphe administration of fluoxetine induced the release of S100β, a known inhibitor of miR-16. Via the reciprocal connections between dorsal raphe and locus coeruleus (LC), S100β is believed to migrate to noradrenergic neurons in the LC and decrease expression of miR-16. By decreasing miR-16 in the LC, S100β turned on the expression of serotonergic functions in noradrenergic neurons. Moreover, intra-raphe and -LC miR-16 administration alleviated the behavioral deficits induced by 6 weeks of chronic unpredictable stress suggesting that the therapeutic effects of fluoxetine may be mediated via its actions on miRNA expression in the brain68.
Recent findings also support the involvement of miRNA in human depression. For instance, 21 miRNAs were downregulated in PFC of depressed suicide cases69. The miRNAs identified in this study were predicted to bind several targets among which DNMT3B was shown to be highly upregulated, an effect consistent with findings from previous postmortem studies in depression70 and with several reports of stress-induced hypermethylation in the context of depression (see below). Interestingly, the authors suggest the existence of a highly interconnected network of miRNAs specific to depression which may underlie PFC hypoactivity. Microarray screening of known miRNAs in brain found significant enrichment of miR-185 and miR-491-3p in the brains of depressed suicide cases71. miR-185 is predicted to bind 5 sites in the 3′ UTR of TRKB.T1, the astrocytic variant of TRKB and miR-185 levels were inversely correlated with TRKB.T1, with expression decreased in the PFC of depressed suicide cases40; 71. Luciferase assays confirmed the functional relationship of these two genes.
The above findings suggest the involvement of miRNAs in the pathophysiology of depressive disorders. However, studies are still in their infancy and one key obstacle is the lack of powerful prediction tools for miRNA targets. Indeed, most miRNAs are predicted to, and most likely do, bind several genes, making it difficult to predict the impact of one miRNA on stress-induced behavioral alterations.
Epigenetics and developmental vulnerability to depression
Early life exposures to stress may have life-long impact on neuropsychiatric states and behavior via epigenetic mechanisms. Adults who experienced childhood stress or maltreatment are at significantly greater lifetime risk of a range of mood or other psychiatric disorders72; 73; 74; 75. DNA methylation, post-translational histone modifications, and non-coding RNAs may alter expression of key genes, or may alter their ability to be induced or repressed in response to subsequent environmental perturbations, enhancing vulnerability to psychiatric disorders. This section will highlight research on epigenetic alterations resulting from developmental (in utero, early post-natal, and peri-adolescent) exposures to stress, with consequences for depression and and related disorders.
Early life adversity has been modeled in rodents and non-human primates using maternal separation (MS) or maternal deprivation. These paradigms involve removing offspring from the mother for 2–8 hours per day prior to weaning, and lead to robust stress responses among offspring. Natural variations in maternal care likewise associate with differential stress responses among adult offspring. Rats reared by dams that display low frequencies of offspring licking and grooming (LG) exhibit heightened stress sensitivity and elevated HPA activity in adulthood compared to those reared by dams that groom their offspring with high frequency76. Research in the last decade has shown that epigenetic alterations play a role in the enduring effects of early life stress on adult mood disorders.
Histone Modifications
Prenatal Stress
Few studies have explored the effects of prenatal stress on post-translational histone modifications directly. Treatment of adult mice exposed to prenatal stress with valproic acid—which is a non-specific HDAC inhibitor among several additional actions—ameliorate locomotor hyperactivity, deficits in social interaction, prepulse inhibition, and fear conditioning77. However, more work is needed to understand the role of chromatin modifications and chromatin modifying enzymes in both the initial response to, and lasting effects of, prenatal stress.
Postnatal Stress
Postnatal adversity in the form of maternal separation in rodents is associated with broad changes in histone modifying enzymes and post-translational histone modifications. Adult male rats that underwent MS displayed reduced levels of Hdac1 mRNA in PFC, although this molecular phenotype was not observed in infancy or adolescence78. Among Balb/c mice that display enhanced susceptibility to stress (but not among C57Bl/6 mice), MS reduced levels of Hdac 1, 3, 7, 8, and 10 in the forebrain in adulthood, and increased acetylation of histone H4, particularly at H4K1279. Elevated H3 and H4 acetylation was also found in the HPC of juvenile mice immediately after MS80. Adolescent fluoxetine treatment potentiated effects of MS on histone modifications, and co-administration of fluoxetine with an HDAC inhibitor ameliorated both HDAC and behavioral effects of MS79. Not surprisingly, treatment during adolescence with theophylline—which can activate HDACs in addition to its better described action as a phosphodiesterase inhibitor—exacerbated the behavioral effects of MS79. These findings suggest that adolescence may be a relevant period for pharmacological intervention and that it may be possible to erase at least some of the epigenetic signature of early life stress.
Chromatin modifications additionally mediate some of the effects of maternal LG on rat stress responses. Low maternal LG associates with decreased hippocampal H3K9-acetylation at the Gr exon 17 promoter17; 81; 82; 83. These modifications co-localize with changes in DNA methylation and span across large regions of the genome as evidenced by microarray analyses82. The changes are also associated with depressive-like symptoms, reduced gene expression, with promoter hypermethylation. Treatment with the HDAC inhibitor TSA, infused either intracerebroventricularly (ICV) or intra-HPC, reversed the effects of low maternal care on adult H3K9-Ac levels and anxiety-like behavior17; 84.
DNA methylation
Prenatal Stress
Activation of the HPA-axis (see Figure 1A) leads to release of CRF and vasopressin (AVP) from the hypothalamic PVN, release of adrenocorticotropic hormone (ACTH) from the anterior pituitary, and release of glucocorticoids (cortisol in primates and corticosterone in rodents) from the adrenal gland. Under normal conditions, acute stress is shunted by negative feedback from neurons expressing GR in the brain, such as in the HPC, while chronic stress has been associated with impaired GR feedback and elevated HPA-activity85. The developing fetus is largely protected from maternal glucocorticoids by the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), which converts active glucocorticoids to their inactive form. However, 10–20% of maternal cortisol is estimated to pass through the placenta to the fetus86. Evidence paradoxically suggests that maternal adversity during pregnancy leads to a suppression of 11β-HSD2 levels and enzymatic activity87; 88 and heightened stress-responses among offspring89; 90. DNA hypermethylation of Hsd11b2 in the placenta and hypomethylation in the fetal hypothalamus consequent to prenatal maternal stress may contribute to long-term alterations in offspring stress programming91.
Persistent changes in DNA methylation have been identified in the brains of adult mice exposed to early prenatal stress92. Within the hypothalamus, early prenatal stress associates with elevated DNA methylation at CpG sites in the NGF1-A binding region of the Gr promoter exon 17, and decreased methylation at CpG sites within the Crf promoter, with no changes in Bdnf methylation92. These alterations occur at specific CpG sites, in specific genes, and in specific brain regions, highlighting both the difficulties in using peripheral tissues to predict meaningful changes within the brain, as well as the need for more precise tools to manipulate epigenetic gene regulation in functional pre-clinical studies.
Hypermethylation in the GR promoter 1F was likewise found in infant cord blood from mothers who experienced depression during the third trimester of pregnancy93; 94, and from mothers reporting intimate partner violence during their pregnancy95. The effects were however not reversed by antidepressant treatment. This suggests that maternal prenatal stress induces specific long-lasting epigenetic alterations affecting GR expression. Altered DNA methylation at the serotonin transporter (SERT, SLC6A4) in offspring cord blood is likewise associated with prenatal maternal depression in the context of a genetic variant within methylenetetrahydrofolate reductase, an enzyme required for folate metabolism and generation of methyl groups96. Contrary to these reports, a recent study examined the relationship of maternal depression or other psychiatric diagnosis with DNA methylation across the genome in offspring cord blood and found no association between maternal depression and altered DNA methylation at any site examined97. Modest differences in methylation at CpG sites in two genes (TNFRSF21 and CHRNA2) were associated with maternal antidepressant use.
In addition, elevated levels of Dnmt3a mRNA were found in the placenta (but not hypothalamus or cortex) of rats exposed to in utero stress, while elevated Dnmt1 mRNA was found within the cortex of offspring at gestational day 2091. Mice exposed to prenatal stress had elevated levels of Dnmt3a and Dnmt1 mRNA in the PFC and HPC at birth, and these relative differences persisted at postnatal (PN) day 7, 14, and 6077. Furthermore, prenatal stress induced increased binding of DNMT1 and MeCP2, along with increased 5-methylcytosine and 5-hydroxymethylcytosine, within CpG-righ regions of the Reelin and Gad67 promoters, at both PN1 and PN6077.
In sum, existing evidence points to a role of prenatal stress in altering adult vulnerability to depression via altered DNA methylation at specific genes. However, work to date is limited by its focus on a small number of candidate genes. Genome-wide analyses of DNA methylation changes in rodent models of prenatal stress will facilitate comparisons of potential epigenetic biomarkers in peripheral tissue such as cord blood and placenta with epigenetic and gene expression changes in specific brain regions implicated in stress regulation and mood.
Postnatal Stress
Variations in maternal care alter DNA methylation levels in genes thought to be critically involved in behavioral stress responses. The offspring of low LG mothers, as opposed to those raised by high LG dams, exhibit lower hippocampal expression of several variants of GR, including the hippocampal specific variant Gr 1717; 82. This is associated with higher DNA methylation levels in promoter regions, including those overlapping with binding sites for transcription factors (i.e. NGF1A) known to regulate GR expression. However, the impact of maternal care on the establishment of DNA methylation profiles is not targeted to specific genes but rather spreads across large genomic regions. Indeed, microarray analysis of a 6.5 million base-pair region centered on the Gr locus showed that low maternal care induces hundreds of parallel DNA methylation changes co-localized with other chromatin modifications98. These adaptations preferentially affect promoters, as evidenced by the cluster at protocadherin genes, and follow a non-random, discontinuous pattern across large genomic regions98. While it is known that DNA methylation recruits cofactors carrying enzymatic activity toward chromatin structure99, it is still unclear how both DNA methylation and chromatin conformation are co-regulated in the context of stress.
Similar alterations have been reported in the HPC of suicide completers with a history of child abuse. Abused suicide completers exhibit lower expression levels of GR1B, 1C, and 1F compared to non-abused suicides and controls100; 101. These changes are associated with changes in DNA methylation within respective promoters, may interfere with transcription factor binding as evidenced by functional luciferase assays. Furthermore, DNA methylation levels at the GR 1F promoter are positively correlated with childhood sexual abuse severity and number of distinct maltreatment conditions in individuals with major depressive disorders102; 103. Importantly, these alterations appear to be specific to early-life adversity as GR transcriptional modifications found in the brains of depressed patients do not associate with changes in DNA methylation104.
Low maternal care in rats also affects Gad1 and Grm1 expression in the HPC81; 83. Pups raised by low LG mothers show lower Gad1 and Grm1 HPC expression levels associated with promoter hypermethylation and lower levels of H3K9Ac compared to pups raised by high LG dams. This is accompanied by elevated HPC levels of Dnmt1 similar to the elevated levels of Dnmt3a in mice susceptible to chronic social defeat. It is possible that the hypermethylated state found in gene promoters following stress originates from the increase in DNMT levels. Human data support this possibility. For instance, levels of DNMT1 in the brain of schizophrenia and bipolar disorder cases correlates with promoter hypermethylation and lower expression of REELIN and GAD1 genes105; 106; 107; 108; 109. In addition, the expression of all three DNMTs (DNMT1, 3A, and 3B) are altered in limbic and brain stem regions in depressed suicide completers70.
Interactions between early attachment patterns and the methylation state of the SERT promoter in peripheral blood mononuclear cells in Rhesus macaques has also been reported110, suggesting that increased methylation in this genomic region may associate with increased reactivity to stress in maternally deprived, but not in mother-reared, infants. Interestingly, in humans, a significant association has been reported between sexual abuse and overall DNA methylation in different regions of the SERT gene, in transformed lymphoblast cell lines derived from subjects recruited through the Iowa Adoption Study111; 112. Furthermore, DNA methylation patterns in SERT have been associated with the emergence of antisocial personality disorder in adulthood.
Early maternal separation also altered DNA methylation in a known enhancer region for Avp expression in PVN113. Consistent with the observed overexpression of Avp, a sustained hypomethylation state within the Avp enhancer region was characterized in the PVN of stressed mice 6 weeks, 3 months, and 1 year following the stress regimen113. A similar overexpression was also reported in stressed mice at 10 days, although no methylation differences were observed in the Avp enhancer, revealing a dual regulatory mode depending on the timing of the stress. These findings point to the existence of complex mechanisms of transcriptional regulation by DNA methylation in non-promoter regions, highlighting the importance of investigating such mechanisms beyond the traditional promoter-centric focus.
Stress beyond the early neonatal period also leaves an epigenetic mark. Three weeks of adolescent isolation stress in a Disc1 mutant mouse induced mood-related behavioral alterations accompanied by hypermethylation of the tyrosine hydroxylase (Th) gene promoter in the VTA of mice114. However, Th promoter hypermethylation was observed in response to both Disc1 mutations and adolescent isolation stress, and these effects were additive although only in VTA dopamine cells projecting to frontal cortex and not in dopamine cells projecting to NAc114. Hypermethylation was observed at the end of the three-week adolescent isolation, and sustained 12 weeks later in the absence of further stress. Th promoter hypermethylation was rescued by treatment with the GR antagonist RU38486, suggesting that GR-mediation of the stress response in this chronic adolescent stress paradigm underlies stress-induced alterations in the mesocortical reward pathway114.
DNA methylation is also altered by extreme childhood adversity in the form of abuse. Experience of maternal maltreatment in rats (tramping, dragging, rough handling) leads to chronic hypomethylation at the Bdnf exon IX gene promoter in the PFC52. These effects are at least partially rescued by ICV treatment for seven days with zebularine, a DNA methylation inhibitor. One recent human study assessed the impact of child abuse on genome-wide DNA methylation signatures in gene promoters115. HPC DNA methylation patterns were compared between suicide completers with a severe history of child abuse (sexual or physical) and healthy controls, and hundreds of differentially methylated sites were identified. Interestingly, DNA methylation levels in gene promoters were inversely correlated with gene expression at a genome-wide level, supporting the globally repressive role of DNA methylation at promoters, as reported by other groups54; 116. Similar observations have been made in suicide completers117. The impact of abuse becomes obvious when assessing the gene functions enriched with differential methylation: differential methylation in the abused suicide group is enriched in genes related to cellular plasticity, while learning and memory genes were particularly affected in suicide. This suggests that intense early-life adversity may induce long-lasting alterations that may not be found in the brain of suicide completers not exposed to early-life adversity. Importantly, the changes in DNA methylation levels reported in these studies occurred in specific cell types as most of the methylation changes were found exclusively in neuronal DNA.
In line with the previous results, these studies suggest that the experience of stress, whether during early-life or adulthood, have profound, genome-wide epigenetic consequences in the brain and peripheral tissues. Indeed, modifications of DNA methylation signatures in different regions of the brain are a plausible mechanism to explain how stress can induce behavioral alterations. Methylation signatures in peripheral tissues may provide a biomarker of stress exposure and vulnerability, however, it is highly implausible that the same genes regulated within a particular brain region will be similar affected in blood. It will be interesting in future studies to better correlate regulation of DNA methylation genome-wide between blood and brain and across numerous brain regions of animal models and to translate such findings to humans.
Non-coding RNAs in depression
Prenatal Stress
Several miRNAs were down- (miR-145, miR-151, miR-425) or up- (miR-103, miR-219-2-3p, miR-98, miR-323) regulated in whole brain tissue of neonatal rats exposed to gestational stress from E12-18118. Targets of these miRNAs include genes implicated in neurotransmission, stress response, and disorders such as schizophrenia and bipolar disorder. For example, prenatal stress increases levels of GPM6A, a neuronal glycoprotein involved in filopodium extension, in the HPC and PFC of male mice at both PN28 and PN60119. This is accompanied by enhanced miR-133b, Dnmt3a, and Mecp2 levels and with alterations in methylation patterns within two CpG islands of the Gpm6a gene119. Furthermore, miR-133b downregulated Gpm6a levels and reduced neurite extensions in cultured HPC neurons, demonstrating functional consequences of miR-133b dysregulation by prenatal stress.
Altered miRNA expression was also found in the second generation of animals exposed to prenatal stress, promoting miRNAs as one potential epigenetic mechanism of trans-generational stress effects. First generation male mice exposed to first trimester gestational stress have a demasculinized stress response, and their second generation male offspring have reduced levels of miR-322, miR-574, and miR-873 that are similar to patterns among control females120. Similarly, altered microRNA patterns were observed after neonatal treatment with the aromatase inhibitor formasetane, implicating testosterone in the regulation of miRNAs and organization of sexually dimorphic brain and behavior120. Similar effects of prenatal stress on miRNA patterns have yet to be observed in peripheral or central human tissue.
Postnatal stress
Maternal separation in rats is known to induce the expression of the repressor element-1 silencing transcription factor 4 (Rest4)121 in PFC, the expression of which is believed to influence the processing of several miRNAs including miR--9-1, -9-3, -212, -29a, 124-1, and -132122. Among these miRNAs, miR-132 and -124 have CREB binding sites within their respective promoters and their expression can be induced by BDNF123. Overexpressing Bdnf in cultured neurons increases miR-132, which upregulates several glutamate receptors subunits (NMDA NA2A and 2B subunits and AMPA GluA1 subunit) and neurite outgrowth, potentially by decreasing levels of the GTPase-activating protein, p250GAP124; 125. CREB can also regulate miR-124 at the synapse through 5-HT-induced long-term facilitation and CREB de-repression123. Interestingly, treatment with the MAPK/ERK pathway inhibitor U0126, suppressed BDNF induction of miR-132, suggesting that MAPK/ERK may represent another pathway besides CREB by which BDNF upregulates miR-132. Moreover, BDNF increases the expression of DICER, one of the key members of the miRNA processing machinery, increasing mature miRNA levels and inducing RNA processing bodies in neurons126. Together, these findings suggest that miRNAs may be affected by stress via altered BDNF signaling.
EPIGENETICS AND ADDICTION
In rodents, addiction-relevant transcriptional regulation is studied by exposing animals to drugs of abuse. Most commonly this is experimenter-administered drug exposure, and it should be noted that, although many effects are recapitulated in self-administration models, clearly some mechanisms are distinct. More work is needed to examine the generalizability of epigenetic mechanisms revealed by passive drug exposure paradigms to more rigorous drug self administration models. The following sections will review epigenetic regulation in response to exposure to several common drugs of abuse.
Histone modifications in addiction
Histone acetylation
Global H3 and H4 acetylation levels in the NAc, a brain region critical for drug reward as noted above, are increased after a single exposure to cocaine and remain elevated with chronic administration127; 128; 129. The specific K residues at which cocaine induces acetylation have not been documented. The functional significance of elevated histone acetylation in regulating the rewarding effects of cocaine is complex and manipulations of acetylation exert variable effects on drug responses. While manipulations that acutely increase histone acetylation generally increase behavioral responses to cocaine, more sustained increases in acetylation attenuate cocaine’s rewarding effects. The opposing effect of long-term increases in histone acetylation may be explained by compensatory increases in repressive histone methylation130. Cocaine regulation of the HAT CREB binding protein (CBP) and HDAC5 are implicated in HDAC regulation of cocaine’s behavioral effects21; 131; 132; 133. CBP heterozygous knockout mice exhibit attenuated locomotor and reward-related responses to cocaine131; 132, while homozygous HDAC5 knockout mice are hypersensitive to cocaine and overexpression of HDAC4 or HDAC5 in NAc attenuates cocaine-elicited behaviors21; 127; 134. In contrast, NAc-specific deletion of HDAC1 (but not HDAC2 or 3) attenuates cocaine responses via induction of G9a and H3K9me2130.
In many instances, cocaine or other stimulant-induced alterations in histone acetylation at candidate gene loci in NAc correlate with gene expression. Studies of candidate genes suggest that with the progression from acute to chronic drug exposure, the gene targets associated with acetylation changes shift from immediate early genes to a later enrichment of genes implicated in long-term plasticity (e.g. Cdk5, Bdnf, Camkiia). Acute, but not chronic, cocaine increases H4 acetylation at the c-Fos promoter127; 135. In contrast, chronic, but not acute, cocaine increased H3 acetylation at the Bdnf, Cdk5, and Camkiia promoters in NAc30; 127; 134. Genome-wide studies of total H3 and H4 acetylation using ChIP-chip found both hyper- and hypo-acetylation at many gene promoter regions that were largely non-overlapping between the marks. Interestingly, although altered histone acetylation associated with expected regulation of mRNA for many genes, expression levels of most genes did not correlate with acetylation changes. This highlights the importance of considering individual histone modifications within the context of broader chromatin regulation136. Two interesting targets identified by this genome-wide approach are Sirt1 and Sirt2, class III HDACs, which are induced in NAc by chronic cocaine. Overexpression of Sirt1 or Sirt2 in NAc enhances cocaine reward and NAc-specific knockdown of Sirt1 has the opposite effect. These actions of SIRT1 are mediated via the regulation of numerous synaptic proteins and the associated induction of dendritic spines137.
In contrast to cocaine, far less is known about the role of histone acetylation in mediating the rewarding effects of other drugs of abuse. Opiate regulation of several target genes correlates with altered histone acetylation138; 139 and inhibition of HDAC activity in NAc potentiates the rewarding effects of opiates140. Similar to cocaine, opiates induce SIRT1, although not SIRT2, in NAc and overexpression of either Sirt1 or Sirt2 in this region increases opiate reward137. Ethanol increases global H3 and H4 acetylation in PFC and AMY, increases CBP in AMY, and decreases HDAC activity in brain and these effects are associated with increased ethanol reward141; 142; 143; 144. However, ethanol withdrawal is associated with increased HDAC activity and decreased H3 and H4 acetylation, which was rescued by systemic treatment with the HDAC inhibitor TSA and also prevented withdrawal-associated anxiety144. Much further work is needed to characterize the actions of these other drugs, most importantly genome-wide assessments of histone acetylation.
Histone methylation
Much of our understanding of the role of histone methylation in drug addiction is informed by recent studies of two repressive histone methylation signatures, H3K9me2 and H3K9me3, both of which are decreased by cocaine and opiates in mouse NAc, effects recapitulated in human addicts145; 140. Both cocaine and morphine decrease expression of G9a and GLP, two HMTs that catalyze H3K9me2. Ethanol also decreases G9a in cultured cortical neurons146. Manipulations of G9a in NAc bi-directionally control behavioral responses to cocaine and opiates. Increasing G9a function inhibits and decreasing G9a enhances drug-elicited behaviors145; 147. Cocaine-induced downregulation of G9a plays a central role in the characteristic increases in dendritic arborizations and synaptic protein expression that is associated with cocaine exposure145.
Regulation of G9a and associated alterations in H3K9me2 levels appear especially relevant to understanding the prolonged effects of cocaine. Chronic cocaine followed by one month withdrawal decreases H3K9me2 at the Fosb gene and this is associated with increased inducibility of the Fosb gene148. Prior history of cocaine exposure increases susceptibility to subsequent stress and reduction of G9a in NAc by cocaine mediates this cross sensitization22. G9a may play a critical role in homeostatic control of epigenetic regulatory mechanisms. Extended HDAC inhibition in NAc attenuates cocaine effects, as stated above, and this is mediated via G9a induction130 (see Figure 3). G9a exerts this effect via complex regulation of certain GABAA subunits in this brain region. Thus, while chronic cocaine or extended HDAC inhibition individually increases expression of GABAA subunits in NAc, chronic cocaine in combination with extended HDAC inhibition paradoxically attenuates the behavioral effects of cocaine and inhibits expression of these subunits. The mechanism of this suppression involves induction of G9a and subsequent increased H3K9me2 binding at specific GABAA subunit genes. In this instance, G9a induction may be triggered in response to excessive hyperacetylation induced by combined HDAC inhibition and cocaine as a homeostatic brake130.
Figure 3. Regulation of GABAA receptor subunit gene expression in NAc through crosstalk between histone acetylation and repressive methylation.
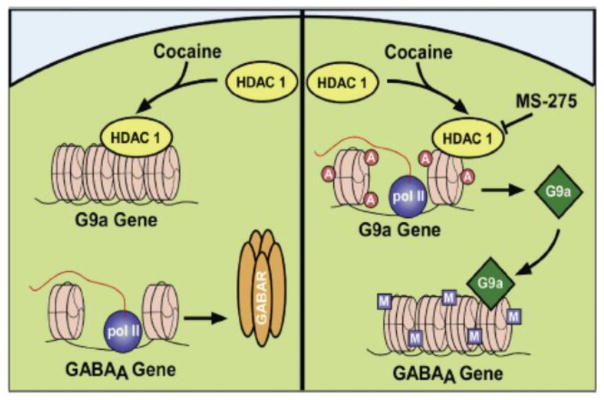
Repeated cocaine targets HDAC1 to the G9a/GLP (G9a-like protein) promoters, leading to decreased G9a/GLP gene expression and decreased binding of these histone methyltransferases (HMTs) at the promoters of certain GABAA receptor subunit genes. The resulting decreased repressive histone methylation (reduced H3K9me2) allows for increased transcription of the GABAA receptor subunits and increased inhibitory tone in the NAc. Chronic cocaine plus chronic intra-NAc infusion of MS275, by inhibiting HDAC1, promotes excessive histone acetylation at several genes, including G9a/GLP, and leads to the induction of G9a/GLP gene expression. These HMTs then catalyze increased H3K9me2 at GABAA receptor subunit gene promoters to block cocaine-induced transcriptional activation of the GABAA subunits and increased inhibitory tone. From Ref 130, with permission.
Genome-wide analysis of H3K9me2 expression by ChIP-chip or more sensitive ChIP-seq (ChIP followed by next generation sequencing) suggests that, although patterns of expression after cocaine or opiates are associated with changes in gene expression, this single mark—as with measures of histone acetylation noted earlier—is not predictive of gene expression changes135; 147; 149. ChIP-seq of H3K9me3 in NAc shows that this mark is almost exclusively located in non-genic regions, with such binding reduced by chronic cocaine at many sites. Of particular interest, cocaine reduces H3K9me3 binding at several repetitive elements, including long-interspersed nuclear element-1 (Line1), the expression of which is decreased by cocaine150. The next important challenge will be to understand cell-type specific regulation. Recent findings suggest that cocaine regulation of H3K9me2 and H3K9me3 follows a cell-type specific time course151. Interestingly, although cocaine represses G9a expression in both D1 and D2 medium spiny neurons (MSNs) in NAc, cell-type specific manipulation suggest that G9a exerts opposing effects152. Developmental knockdown of G9a in D1 MSNs suppresses behavioral responses to cocaine, conversely, G9a knockdown in D2 MSNs enhances cocaine’s effects. Moreover, overexpression of G9a in D2 MSNs of adult mice attenuates cocaine reward but has no effect in D1 MSNs, suggesting that G9a may regulate cocaine reward primarily through effects on D2 MSNs.
Finally, advances in designer-transcription factors are allowing for unprecedented precision in dissecting the regulatory influence of specific histone marks. Recent work using zinc finger proteins to specifically target G9a to the FosB promoter establishes a direct causal link between increased H3K9me2 at this specific locus and transcriptional repression of the FosB as well as attenuated locomotor responses to cocaine. In contrast, targeting of the transcriptional-activator p65 to the same locus increases H3 acetylation, increases FosB transcription, and enhances cocaine responses153.
Other histone and related mechanisms
Far less is known about other histone modifications and their role in drug reward. Chronic cocaine alters histone phosphorylation154; 155 and arginine methylation156. A recent paper established a critical role for poly(ADP-ribosyl)ation. Chronic cocaine induces poly(ADP-ribose) polymerase-1 (PARP-1) in the NAc and this upregulation potentiates behavioral responses to cocaine. ChIP-seq revealed cocaine-induced increases in PARP-1 binding across the genome and this correlated with increased gene expression measured by RNA-seq157.
As with depression, very little is known concerning the role of chromatin remodeling complexes in addiction. Chromatin remodeling likely contributes to cocaine-induced expansion of euchromatin. Recent work suggests that cocaine, like chronic social defeat, regulates the expression of the ISWI family of chromatin remodelers and that this upregulation may be important for altered nucleosome spacing156.
Studies of drug-induced regulation of histone post-translational modifications have enriched our understanding of the complex interplay of several different types of modifications149. Exposure to drugs of abuse may initially increase histone acetylation, favoring transcriptional activation. With sustained drug exposure, increases in histone acetylation act to induce certain histone methyltransferases and subsequently increase repressive histone methylation, curtailing the initial period of excessive transcriptional activation. Thus, it is critical to identify the interactions among diverse histone modifications to fully characterize the transcriptional alterations induced by drugs of abuse.
DNA methylation in addiction
In comparison to histone methylation, far less is known concerning how DNA methylation influences molecular and behavioral adaptations in addiction. Acute exposure to cocaine inhibits the expression of both maintenance and de novo DNMTs, Dnmt1 and Dnmt3a/b, in the NAc44; 158. However, Dnmt3a expression is actually increased after chronic cocaine exposure with extended withdrawal. NAc-specific deletion or pharmacological inhibition of DNMT3a in NAc, which reduces global DNA methylation levels, potentiates the rewarding effects of cocaine suggesting that DNMT3a and DNA methylation negatively regulated cocaine reward. Accordingly, viral-mediated overexpression of Dnmt3a, but not Dnmt1, in NAc reduces cocaine conditioned place preference. Paradoxically, Dnmt3a overexpression induced increases in thin spines comparable to increases observed after cocaine44. This finding suggests that DNMT3a may not simply repress gene transcription. Consistent with this, inhibition of DNMT activity has been reported to block histone acetylation159.
Chronic extended access cocaine self-administration increases methyl-CpG binding protein in the dorsal striatum160. Knockdown of MeCP2 in the dorsal striatum suppresses cocaine self-administration specifically in an extended availability paradigm. In contrast, MeCP2 knockdown in NAc enhances behavioral responses to amphetamine, while MeCP2 overexpression attenuates amphetamine reward161. More work is needed to understand the complexities of MeCP2 regulation in addiction models. It is important to note that MeCP2 and DNMTs may not exclusively mediate their reported effects on drug reward via DNA methylation, such as through recruitment of HDACs162 and H3K9 methylation163. Several studies have reported cocaine-induced DNA methylation changes at candidate genes158; 164; 165, however, genome-wide studies of DNA methylation are needed to more fully and directly assess the role of DNA methylation in addiction.
Recent work suggests that cocaine also decreases expression of Tet1 in NAc. TET1 is an enzyme that catalyzes 5-methyl cytosine oxidation to 5-hydroxymethylcytosine (5hmc), which has been viewed as a transient epigenetic state between methylated and unmethylated cytosines. Overexpression of Tet1 attenuates behavioral responses to cocaine. Genome-wide 5hmc capture and deep sequencing combined with RNA-seq found robust regulation of 5hmc at distal enhancer regions and within coding regions of genes that are induced in response to a cocaine challenge149. These findings suggest that 5hmc may be a stable epigenetic mark induced by cocaine although more work is needed.
Non-coding RNAs in addiction
Many studies report up- and downregulation of several miRNAs by drugs of abuse. Next generation sequencing recently identified tens of miRNAs that are altered in NAc whole extracts and in purified striatal postsynaptic densities after chronic cocaine166. Among these, cocaine increased expression of miR-181a and decreased expression of miR-124 and let-7d in rat striatum126; 167; 168; 169; 170. Moreover manipulations that recapitulate these effects also enhanced behavioral responses to cocaine. miR212 is induced in the rat dorsal striatum by cocaine self-administration and this induction suppresses cocaine intake171. The antagonistic effect of miR212 on cocaine self-administration appears to be mediated through indirect activation of the transcription factor CREB, known to antagonize cocaine reward5. Finally, chronic ethanol suppressed BK channel expression through induction of miR9 in both the striatum and preoptic area of the hypothalamus172. Knockout of argonaut-2 (Ago2), which is required for miRNA processing and mRNA silencing, from D2 neurons in striatum reduced cocaine self-administration, further supporting the importance of miRNAs in cocaine action169. This is a promising field of research and more work is needed to fully explore the population of miRNAs regulated by drugs of abuse and to define mRNA targets and functional consequences for each.
Epigenetics and developmental vulnerability to addiction
Developmental exposure to drugs of abuse results in altered epigenetic states in the brain. In some cases, clear links to addiction vulnerability exist, while in other cases the ultimate effect on addiction or other psychiatric disorders remains to be elucidated.
Histone Modifications
Prenatal Exposure
Fetal alcohol exposure can result in impaired motor coordination and balance and has been linked to alterations in cerebellar development. Guo and colleagues173 identified reduced acetylation of H3 and H4 in the cerebellum of rats exposed to ethanol in the third trimester-equivalent of human pregnancy (rat PN2-12). Reduced histone acetylation may be mediated by reduced CBP in this region after developmental ethanol173. More research is needed to understand whether deficits in H3 and H4 acetylation similarly contribute to deficits in learning, cognition, judgment, attention, and social behavior associated with fetal alcohol exposure.
Developmental Δ9-tetrahydrocannabinol (THC) exposure enhances rat behavioral preference for THC in adulthood174, which may be mediated by altered dopamine receptor levels. Reduced D2 (DRD2) expression is observed in adult drug abusers, raising the question of causality or consequence. Human fetal exposure to cannabis also decreased D2 expression levels in NAc, suggesting that downregulated D2 levels may be both a consequence of exposure and predate drug abuse vulnerability174. Rats prenatally exposed to THC show D2 downregulation in association with increased H3K9me2 and decreased H3K4me3 binding at the Drd2 gene, consistent with the change in receptor expression174.
Postnatal Exposure
Altered histone modifications may underlie the enhanced vulnerability to alcohol and other drugs found during the peri-adolescent period. Binge-like intermittent adolescent alcohol exposure alters acetylation of H3 and H4 in PFC, NAc, and dorsal striatum of rats and is associated with downregulation of D2 receptors in the PFC175. ChIP has not been used to confirm reduced H3 and H4 at the Drd2 gene locus specifically.s Interestingly, these findings were specific for adolescent, but not adult, ethanol exposure175, indicating enhanced vulnerability to chromatin-modifying effects of alcohol during development.
Cocaine administered to adolescent rats in ascending doses to mimic patterns of drug binging observed in human adolescents resulted in abnormally rapid attention shifting and altered gene expression in the PFC176. Decreased H3K4me3 and H3K27me3 levels accompanied downregulated expression of transcription factors including Egr1/2, Homer1, and Jmjd1a in PFC one day after final cocaine administration176. However, array data also showed upregulation of genes associated with cell adhesion and extracellular matrix not explained directly by altered H3 methylation. Interestingly, expression of fewer genes was altered 24 days after final cocaine administration indicating that persistent behavioral effects may be due to organizational changes and altered connectivity that no longer require sustained changes in gene expression or epigenetic gene regulation.
Adolescent THC exposure has been shown to enhance heroin self-administration and dysregulation of the endogenous opiate system. Adolescent rats exposed to THC have elevated levels of proenkephalin (Penk) mRNA in the NAc, which is associated with decreased H3K9me2 and increased H3K4me3 at the Penk promoter177. Furthermore, overexpression of Penk in the striatum of THC-exposed rats potentiated heroin self-administration, while knockdown of Penk attenuated self-administration. These studies elegantly demonstrate gene-specific regulation by posttranslational histone modifications in the development of drug addiction, rather than global dysregulation of histone modifications or histone modifying enzymes.
DNA methylation
Prenatal Exposure
Altered DNA methylation patterns are implicated in long-term neurological alterations associated with fetal alcohol exposure. Sufficient folate and methyl donors are essential for proper DNA methylation in rapidly dividing cells during embryonic development. Recent evidence in humans shows that chronic alcohol consumption during pregnancy impairs folate transfer from the mother across the placenta to the developing fetus178, although additional research is needed to determine whether alcohol actively downregulates placental folate receptors and transporters. Alcohol also has a direct effect on developing neurons: neuronal stem cells cultured with alcohol for just six hours showed altered patterns of 5-methylcytosine and DNMT1 immunostaining, as well as inhibited differentiation, growth, and migration179. Alcohol-induced alterations in DNA methylation persist past embryonic development, and DNMT activity was increased at weaning180 and alterations in CpG methylation were observed into adulthood in the whole brain of male mice181. These studies are an excellent beginning to understanding the effects of fetal alcohol exposure on gene regulation by DNA methylation. Future studies would benefit from examining more specific genes or gene networks that may be affected by methylation changes, as well as how these patterns change from embryonic development through adulthood, and whether a diet rich in methyl donors (either during fetal development or later in life) is able to rescue some of the effects of fetal alcohol exposure.
Prenatal exposure to cigarette smoke has a small but significant effect on methylation within repetitive DNA elements in human placenta182; 183, buccal epithelium184, and blood samples185. Among the genes aberrantly methylated, BDNF exon 5 was hypermethylated in blood samples taken during adolescence185. Further studies in animal models of prenatal or neonatal cigarette smoke exposure should explore whether DNA methylation at specific genes in these peripheral tissues are predictive of methylation patterns (even if at different genes) within brain regions relevant for cognitive, emotional, social, or rewarding behaviors.
Fetal cocaine exposure during the second and third trimesters of mouse gestation significantly affected CpG methylation in HPC pyramidal neurons of both neonatal (PN3) and juvenile (PN30) offspring186. Interestingly, the patterns of aberrant methylation observed at PN3 were somewhat dissimilar to the patterns observed at PN30 in response to fetal cocaine exposure, which highlights the necessity of longitudinal, tissue-specific studies to better understand long-term epigenetic consequences of in utero drug exposure. However, the relevance of these changes to later addiction liability is unknown.
Postnatal Exposure
Surprisingly little is known about methylation patterns resulting from postnatal exposure to various drugs of abuse. Among weight-restored anorexic patients, smoking (but not malnutrition) was associated with decreased POMC promoter methylation within peripheral monocytes187. However, the degree to which this is relevant for POMC expression or epigenetic regulation within the brain is unknown.
The broad epigenetic changes induced by early life stress may also be relevant to our understanding of sensitivity to drugs of abuse in adulthood. Maternal stress was recently shown to paradoxically increase Dnmt1, Dnmt3a, and Dnmt3b mRNA levels and decrease global DNA methylation levels in the adult NAc, although methylation was increased at promoters of genes potentially relevant to stress or drug behavior including protein phosphatase 1 catalytic subunit (Pp1c) and adenosine A2A receptor (A2ar)188. Maternal separation-induced DNA methylation changes in NAc may underlie enhanced locomotor responses to acute cocaine treatment in adulthood188. Further studies are needed to understand the role of epigenetics in the cross-sensitization of early life stress and drug sensitivity.
Non-coding RNAs
Prenatal Exposure
Persistent effects of developmental drug exposure are additionally mediated by miRNAs. Levels of miRNAs including miR-140-3 were reduced by ethanol and dose-dependently upregulated by nicotine exposure in neural progenitor cells derived from fetal mouse cortex189. Nicotinic acetylcholine receptor subunits α4 and β2 (Nchar-a2 and b2) were altered in the same direction as miR140-3 indicating a potential mechanism for long-term developmental effects of prenatal alcohol or nicotine on neurotransmission. Microarray analysis identified dramatic changes in miRNA patterns in whole brains of mice exposed to alcohol during gestation181; 190. Targets of these dysregulated miRNAs converged on several genes relevant to fetal alcohol spectrum disorders, including Pten, Nmnat1, Slitrk2, Otx2, and Hoxa1181. Co-incubation of alcohol-exposed mouse embryos with folate was able to block upregulation of miR-10a and -10b and downregulation of a target gene, Hoxa1190, indicating that these miRNAs are themselves regulated epigenetically by DNA methylation, and that dietary supplements may be able to mitigate some of the effects of fetal alcohol exposure.
Prenatal cocaine exposure in zebrafish altered mRNA levels of Drd1, Drd2a, Drd2b, and Drd3, Pitx3, and Th, genes known to be relevant for drug taking behavior191. These expression changes were accompanied by decreased miR-133b, in both encephalon and in the periphery191. It will be interesting to validate these findings in mammalian systems and test whether miR-133b may be a useful biomarker for changes occurring within the developing brain reward circuitry.
Postnatal Exposure
Altered miRNA expression was observed after chronic nicotine exposure in an atypical C. elegans model, from post-embryonic stage L1-L4 (juvenile)192. In both human placental tissue and C. elegans exposed to nicotine, miRNA variation was proportional to nicotine dose192; 193. However, it is unknown as of yet whether developmental nicotine exposure results in chronic alterations in miRNAs within mammalian brain.
LIMITATIONS, FUTURE DIRECTIONS, AND CONCLUDING REMARKS
A wealth of data from animal models and evolving evidence from postmortem human samples has established the important role of a diverse array of epigenetic regulatory mechanisms in mediating the transcriptional abnormalities that underlie depression and addiction. The studies in depression and addiction discussed here illustrate approaches that are being used to understand other psychiatric and neurodevelopmental disorders, including schizophrenia, bipolar disorder, autism spectrum disorders, and more. An important challenge in the field of psychiatric epigenetics is to develop an integrated understanding of how these various mechanisms, including histone modifications, DNA methylation, and non-coding RNAs, interact to ultimately orchestrate the characteristic abnormalities in gene expression. Integrating data from multiple levels of epigenetic modulation is not trivial but will progress with increasing collaboration between biology and bioinformatics. An interesting question to be addressed by future research is the extent to which altered epigenetic states observed in addiction and depression reflect adaptive or maladaptive responses to environmental insults. Potentially, epigenetic and gene expression changes could help prime the individual to cope with environmental challenges. Alternatively, stress and drug exposure could impair the ability of neurons to dynamically regulate epigenetic states in response to ongoing environmental influences by hijacking the epigenetic machinery. Another important challenge is to determine the cell-type specificity of observed epigenetic alterations, with virtually all studies carried out to date being performed on crude brain extracts. Initial studies suggest that epigenetic modifications are highly cell-type specific even at the level of neuronal subtype, let alone in neuronal versus non-neuronal populations.
Although existing studies document how early developmental exposures lead to life-long changes in gene expression and behavior, to fully understand these altered trajectories, it will be important to profile the epigenetic landscape across development from initial exposure to adulthood. For instance, certain epigenetic modifications could conceivably be altered proximal to the time of stress or drug exposure, whereas other epigenetic modifications may incubate (build) over time but may be more stably maintained. A more precise understanding of such temporal dynamics obtained from genome-wide studies will facilitate attempts to identify critical windows for therapeutic intervention. Additionally, while several studies have described developmental stress- or drug-induced alterations in chromatin modifications and enzymes regulating epigenetic marks, few have identified specific genomic loci at which these changes exert a functional effect. Future research with ChIP and ChIP-seq will shed light on specific genes targeted by these changes. Another significant limitation to our current understanding of developmental vulnerability to stress and addictive disorders is the lack of targeted manipulations of epigenetic states. While many studies examining developmental stress or drug exposure report consequent epigenetic modifications, very few studies have examined the behavioral or molecular consequences of altered epigenetic modifications for psychiatric vulnerability. The few studies which have attempted to assess this important question employed manipulations during adulthood that broadly disrupt histone acetylation or methylation genome-wide and with many known side-effects, leaving it unclear how altered epigenetic regulation at any specific genic loci may relate to behavioral outcomes.
Access to human brain tissue is extremely limited and, increasingly, researchers have attempted to understand epigenetic consequences of early life stress or drug exposure by examining peripheral tissues (e.g., placenta, blood, buccal epithelium). While certain epigenetic modifications may emerge as reliable biomarkers for concurrent alterations in brain, few studies have directly examined this. At best, results from studies investigating both peripheral and central tissue suggest that any correlations are limited and highly specific to brain region, gene, and epigenetic modification. Although animal models can be powerful tools to study complex disorders, translational studies are critical to validate the relevance of these findings to human depression and addiction. The rarity of the tissue, combined with the inability to directly study mechanisms in humans, has been a significant limitation of the field. However, as shown in specific cases13; 19; 31, combining findings from animal and human studies can provide great insights into the functional mechanisms of psychiatric disease.
There is an urgent demand for increased specificity in studies attempting to understand the interplay of developmental stress or drug exposure, epigenetics, and adult neuropsychiatric behavior. Recent research efforts have begun at long last to generate significant data sets profiling genome-wide alterations in methylation and other epigenetic patterns after stress or drug exposure. The current challenge is now to refine these analyses to understand how epigenetic regulation of specific genes and gene networks relate to depression- and addiction-related outcomes. Technology is emerging that enables brain region-specific, gene-specific targeting of epigenetic modifiers to determine whether epigenetic regulation at a specific locus is responsible for life-long perturbations in psychiatric disorders. This same technology may offer therapeutic promise in the future.
Highlights.
Discussion of current evidence for epigenetic dysregulation in depression and addiction
Insights from both animal and human studies
Special focus on findings from developmental vs. adult studies
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cisler JM, James GA, Tripathi S, Mletzko T, Heim C, Hu XP, Mayberg HS, Nemeroff CB, Kilts CD. Differential functional connectivity within an emotion regulation neural network among individuals resilient and susceptible to the depressogenic effects of early life stress. Psychol Med. 2013;43:507–18. doi: 10.1017/S0033291712001390. [DOI] [PubMed] [Google Scholar]
- 2.Hamani C, Mayberg H, Stone S, Laxton A, Haber S, Lozano AM. The subcallosal cingulate gyrus in the context of major depression. Biological Psychiatry. 2011;69:301–8. doi: 10.1016/j.biopsych.2010.09.034. [DOI] [PubMed] [Google Scholar]
- 3.Sequeira A, Klempan T, Canetti L, ffrench-Mullen J, Benkelfat C, Rouleau GA, Turecki G. Patterns of gene expression in the limbic system of suicides with and without major depression. Mol Psychiatry. 2007;12:640–55. doi: 10.1038/sj.mp.4001969. [DOI] [PubMed] [Google Scholar]
- 4.Sequeira A, Mamdani F, Ernst C, Vawter MP, Bunney WE, Lebel V, Rehal S, Klempan T, Gratton A, Benkelfat C, Rouleau GA, Mechawar N, Turecki G. Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PLoS ONE. 2009;4:e6585. doi: 10.1371/journal.pone.0006585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–37. doi: 10.1038/nrn3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–76. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 8.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–30. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aapola U, Kawasaki K, Scott HS, Ollila J, Vihinen M, Heino M, Shintani A, Minoshima S, Krohn K, Antonarakis SE, Shimizu N, Kudoh J, Peterson P. Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics. 2000;65:293–8. doi: 10.1006/geno.2000.6168. [DOI] [PubMed] [Google Scholar]
- 10.Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–8. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 11.Wallace DL, Han MH, Graham DL, Green TA, Vialou V, Iniguez SD, Cao JL, Kirk A, Chakravarty S, Kumar A, Krishnan V, Neve RL, Cooper DC, Bolanos CA, Barrot M, McClung CA, Nestler EJ. CREB regulation of nucleus accumbens excitability mediates social isolation-induced behavioral deficits. Nature Neuroscience. 2009;12:200–9. doi: 10.1038/nn.2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vialou V, Feng J, Robison AJ, Nestler EJ. Epigenetic mechanisms of depression and antidepressant action. Annu Rev Pharmacol Toxicol. 2013;53:59–87. doi: 10.1146/annurev-pharmtox-010611-134540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nature Neuroscience. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 14.Covington HE, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ. Antidepressant actions of histone deacetylase inhibitors. Journal of Neuroscience. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biological Psychiatry. 2007;62:55–64. doi: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- 16.Semba J, Kuroda Y, Takahashi R. Potential antidepressant properties of subchronic GABA transaminase inhibitors in the forced swimming test in mice. Neuropsychobiology. 1989;21:152–156. doi: 10.1159/000118569. [DOI] [PubMed] [Google Scholar]
- 17.Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nature Neuroscience. 2004;7:847–54. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- 18.Yamawaki Y, Fuchikami M, Morinobu S, Segawa M, Matsumoto T, Yamawaki S. Antidepressant-like effect of sodium butyrate (HDAC inhibitor) and its molecular mechanism of action in the rat hippocampus. World J Biol Psychiatry. 2012;13:458–467. doi: 10.3109/15622975.2011.585663. [DOI] [PubMed] [Google Scholar]
- 19.Zhu H, Huang Q, Xu H, Niu L, Zhou JN. Antidepressant-like effects of sodium butyrate in combination with estrogen in rat forced swimming test: involvement of 5-HT(1A) receptors. Behavioural Brain Research. 2009;196:200–206. doi: 10.1016/j.bbr.2008.08.039. [DOI] [PubMed] [Google Scholar]
- 20.Uchida S, Hara K, Kobayashi A, Otsuki K, Yamagata H, Hobara T, Suzuki T, Miyata N, Watanabe Y. Epigenetic status of Gdnf in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron. 2011;69:359–72. doi: 10.1016/j.neuron.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 21.Renthal W, Maze I, Krishnan V, Covington HE, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone Deacetylase 5 Epigenetically Controls Behavioral Adaptations to Chronic Emotional Stimuli. Neuron. 2007;56:13–13. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 22.Covington HE, Maze I, Sun H, Bomze HM, DeMaio KD, Wu EY, Dietz DM, Lobo MK, Ghose S, Mouzon E, Neve RL, Tamminga CA, Nestler EJ. A role for repressive histone methylation in cocaine-induced vulnerability to stress. Neuron. 2011;71:656–670. doi: 10.1016/j.neuron.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hollis F, Duclot F, Gunjan A, Kabbaj M. Individual differences in the effect of social defeat on anhedonia and histone acetylation in the rat hippocampus. Hormones and Behavior. 2011;59:331–337. doi: 10.1016/j.yhbeh.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsankova NM, Kumar A, Nestler EJ. Histone Modifications at Gene Promoter Regions in Rat Hippocampus after Acute and Chronic Electroconvulsive Seizures. Journal of Neuroscience. 2004;24:5603–5610. doi: 10.1523/JNEUROSCI.0589-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Covington HE, Vialou VF, Laplant Q, Ohnishi YN, Nestler EJ. Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neuroscience Letters. 2011;493:122–126. doi: 10.1016/j.neulet.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sterrenburg L, Gaszner B, Boerrigter J, Santbergen L, Bramini M, Elliott E, Chen A, Peeters BWMM, Roubos EW, Kozicz T. Chronic stress induces sex-specific alterations in methylation and expression of corticotropin-releasing factor gene in the rat. PLoS ONE. 2011;6:e28128. doi: 10.1371/journal.pone.0028128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hollis F, Wang H, Dietz D, Gunjan A, Kabbaj M. The effects of repeated social defeat on long-term depressive-like behavior and short-term histone modifications in the hippocampus in male Sprague-Dawley rats. Psychopharmacology. 2010;211:69–77. doi: 10.1007/s00213-010-1869-9. [DOI] [PubMed] [Google Scholar]
- 28.Hinwood M, Tynan RJ, Day TA, Walker FR. Repeated social defeat selectively increases δ FosB expression and histone H3 acetylation in the infralimbic medial prefrontal cortex. Cerebral Cortex. 2011;21:262–271. doi: 10.1093/cercor/bhq080. [DOI] [PubMed] [Google Scholar]
- 29.Kenworthy CA, Sengupta A, Luz SM, Ver Hoeve ES, Meda K, Bhatnagar S, Abel T. Social defeat induces changes in histone acetylation and expression of histone modifying enzymes in the ventral hippocampus, prefrontal cortex, and dorsal raphe nucleus. Neuroscience. 2013 doi: 10.1016/j.neuroscience.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 30.Robison AJ, Vialou V, Sun H-S, Labonté BA, Golden S, Dias C, Turecki G, Tamminga C, Russo S, Mazei-Robison M, Nestler EJ. Fluoxetine Epigenetically Alters the CaMKII α Promoter in Nucleus Accumbens to Regulate Δ FosB Binding and Antidepressant Effects. Neuropsychopharmacology. 2013;5:1178–86. doi: 10.1038/npp.2013.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golden SA, Christoffel DJ, Heshmati M, Hodes GE, Magida J, Davis K, Cahill ME, Dias C, Ribeiro E, Ables JL, Kennedy PJ, Robison AJ, Gonzalez-Maeso J, Neve RL, Turecki G, Ghose S, Tamminga CA, Russo SJ. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nature Medicine. 2013;19:337–344. doi: 10.1038/nm.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilkinson MB, Xiao G, Kumar A, Laplant Q, Renthal W, Sikder D, Kodadek TJ, Nestler EJ. Imipramine Treatment and Resiliency Exhibit Similar Chromatin Regulation in the Mouse Nucleus Accumbens in Depression Models. Journal of Neuroscience. 2009;29:7820–7832. doi: 10.1523/JNEUROSCI.0932-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hunter RG, McCarthy KJ, Milne TA, Pfaff DW, Mcewen BS. Regulation of hippocampal H3 histone methylation by acute and chronic stress. Proc Natl Acad Sci U S A. 2009;106:20912–20917. doi: 10.1073/pnas.0911143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunter RG, Murakami G, Dewell S, Seligsohn Maa, Baker MER, Datson NA, Mcewen BS, Pfaff DW. Acute stress and hippocampal histone H3 lysine 9 trimethylation, a retrotransposon silencing response. Proc Natl Acad Sci U S A. 2012;109:17657–17662. doi: 10.1073/pnas.1215810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neuroscience. 2008;9:42–42. doi: 10.1186/1471-2202-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cruceanu C, Alda M, Nagy C, Freemantle E, Rouleau GA, Turecki G. H3K4 tri-methylation in synapsin genes leads to different expression patterns in bipolar disorder and major depression. Int J Neuropsychopharmacol. 2013;16:289–99. doi: 10.1017/S1461145712000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fiori LM, Gross JA, Turecki G. Effects of histone modifications on increased expression of polyamine biosynthetic genes in suicide. Int J Neuropsychopharmacol. 2012;15:1161–6. doi: 10.1017/S1461145711001520. [DOI] [PubMed] [Google Scholar]
- 38.Chen ES, Ernst C, Turecki G. The epigenetic effects of antidepressant treatment on human prefrontal cortex BDNF expression. Int J Neuropsychopharmacol. 2011;14:427–9. doi: 10.1017/S1461145710001422. [DOI] [PubMed] [Google Scholar]
- 39.Lopez JP, Mamdani F, Labonte B, Beaulieu MM, Yang JP, Berlim MT, Ernst C, Turecki G. Epigenetic regulation of BDNF expression according to antidepressant response. Mol Psychiatry. 2013;18:398–9. doi: 10.1038/mp.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ernst C, Chen ES, Turecki G. Histone methylation and decreased expression of TrkB.T1 in orbital frontal cortex of suicide completers. Mol Psychiatry. 2009;14:830–2. doi: 10.1038/mp.2009.35. [DOI] [PubMed] [Google Scholar]
- 41.Ernst C, Deleva V, Deng X, Sequeira A, Pomarenski A, Klempan T, Ernst N, Quirion R, Gratton A, Szyf M, Turecki G. Alternative splicing, methylation state, and expression profile of tropomyosin-related kinase B in the frontal cortex of suicide completers. Arch Gen Psychiatry. 2009;66:22–32. doi: 10.1001/archpsyc.66.1.22. [DOI] [PubMed] [Google Scholar]
- 42.Razzoli M, Domenici E, Carboni L, Rantamaki T, Lindholm J, Castren E, Arban R. A role for BDNF/TrkB signaling in behavioral and physiological consequences of social defeat stress. Genes Brain Behav. 2011 doi: 10.1111/j.1601-183X.2011.00681.x. [DOI] [PubMed] [Google Scholar]
- 43.Sun H, Damez-Werno D, Scobie K, Shao N, Wright K, Dias C, Koo J, Maze I, Kennedy P, Mouzon E, Dietz D, Neve R, Allis DC, Turecki G, Tamminga C, Kabbaj M, Varga-Weisz P, Shen L, Nestler EJ. Role of chromatin remodeling in the mouse nucleus accumbens in preclinical models of depression and cocaine addiction. Abstract, Society for Neuroscience; San Diego, CA: 2013. [Google Scholar]
- 44.LaPlant Q, Vialou V, Covington HE, Dumitriu D, Feng J, Warren BL, Maze I, Dietz DM, Watts EL, Iñiguez SD, Koo JW, Mouzon E, Renthal W, Hollis F, Wang H, Noonan MA, Ren Y, Eisch AJ, Bolaños CA, Kabbaj M, Xiao G, Neve RL, Hurd YL, Oosting RS, Fan G, Morrison JH, Nestler EJ. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nature Neuroscience. 2010;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elliott EE, Ezra-Nevo GG, Regev LL, Neufeld-Cohen AA, Chen AA. Resilience to social stress coincides with functional DNA methylation of the Crf gene in adult mice. Nature Neuroscience. 2010;13:1351–1353. doi: 10.1038/nn.2642. [DOI] [PubMed] [Google Scholar]
- 46.Fyffe SL, Neul JL, Samaco RC, Chao HT, Ben-Shachar S, Moretti P, McGill BE, Goulding EH, Sullivan E, Tecott LH, Zoghbi HY. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron. 2008;59:947–958. doi: 10.1016/j.neuron.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melas PA, Rogdaki M, Lennartsson A, Björk K, Qi H, Witasp A, Werme M, Wegener G, Mathé AA, Svenningsson P, Lavebratt C. Antidepressant treatment is associated with epigenetic alterations in the promoter of P11 in a genetic model of depression. Int J Neuropsychopharmacol. 2012;15:669–679. doi: 10.1017/S1461145711000940. [DOI] [PubMed] [Google Scholar]
- 48.Anisman H, Gibb J, Hayley S. Influence of continuous infusion of interleukin-1beta on depression-related processes in mice: corticosterone, circulating cytokines, brain monoamines, and cytokine mRNA expression. Psychopharmacology. 2008;199:231–44. doi: 10.1007/s00213-008-1166-z. [DOI] [PubMed] [Google Scholar]
- 49.Alexander B, Warner-Schmidt J, Eriksson T, Tamminga C, Arango-Lievano M, Ghose S, Vernov M, Stavarache M, Musatov S, Flajolet M, Svenningsson P, Greengard P, Kaplitt MG. Reversal of depressed behaviors in mice by p11 gene therapy in the nucleus accumbens. Sci Transl Med. 2010;2:54ra76. doi: 10.1126/scitranslmed.3001079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Svenningsson P, Chergui K, Rachleff I, Flajolet M, Zhang X, El Yacoubi M, Vaugeois JM, Nomikos GG, Greengard P. Alterations in 5-HT1B receptor function by p11 in depression-like states. Science. 2006;311:77–80. doi: 10.1126/science.1117571. [DOI] [PubMed] [Google Scholar]
- 51.Oh YS, Gao P, Lee KW, Ceglia I, Seo JS, Zhang X, Ahn JH, Chait BT, Patel DJ, Kim Y, Greengard P. SMARCA3, a chromatin-remodeling factor, is required for p11-dependent antidepressant action. Cell. 2013;152:831–43. doi: 10.1016/j.cell.2013.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting Epigenetic Influence of Early-Life Adversity on the BDNF Gene. Biological Psychiatry. 2009;65:10–10. doi: 10.1016/j.biopsych.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roth TL, Zoladz PR, Sweatt JD, Diamond DM. Epigenetic modification of hippocampal Bdnf DNA in adult rats in an animal model of post-traumatic stress disorder. Journal of Psychiatric Research. 2011;45:919–926. doi: 10.1016/j.jpsychires.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, Jia P, Assadzadeh A, Flanagan J, Schumacher A, Wang SC, Petronis A. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82:696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ladd-Acosta C, Pevsner J, Sabunciyan S, Yolken RH, Webster MJ, Dinkins T, Callinan PA, Fan JB, Potash JB, Feinberg AP. DNA methylation signatures within the human brain. Am J Hum Genet. 2007;81:1304–15. doi: 10.1086/524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deaton AM, Webb S, Kerr AR, Illingworth RS, Guy J, Andrews R, Bird A. Cell type-specific DNA methylation at intragenic CpG islands in the immune system. Genome Res. 2011;21:1074–86. doi: 10.1101/gr.118703.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iwamoto K, Bundo M, Ueda J, Oldham MC, Ukai W, Hashimoto E, Saito T, Geschwind DH, Kato T. Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. 2011;21:688–96. doi: 10.1101/gr.112755.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schratt G. microRNAs at the synapse. Nat Rev Neurosci. 2009;10:842–9. doi: 10.1038/nrn2763. [DOI] [PubMed] [Google Scholar]
- 59.Rinaldi A, Vincenti S, De Vito F, Bozzoni I, Oliverio A, Presutti C, Fragapane P, Mele A. Stress induces region specific alterations in microRNAs expression in mice. Behavioural Brain Research. 2010;208:265–9. doi: 10.1016/j.bbr.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 60.Meerson A, Cacheaux L, Goosens KA, Sapolsky RM, Soreq H, Kaufer D. Changes in brain MicroRNAs contribute to cholinergic stress reactions. J Mol Neurosci. 2010;40:47–55. doi: 10.1007/s12031-009-9252-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uchida S, Nishida A, Hara K, Kamemoto T, Suetsugi M, Fujimoto M, Watanuki T, Wakabayashi Y, Otsuki K, McEwen BS, Watanabe Y. Characterization of the vulnerability to repeated stress in Fischer 344 rats: possible involvement of microRNA-mediated down-regulation of the glucocorticoid receptor. Eur J Neurosci. 2008;27:2250–61. doi: 10.1111/j.1460-9568.2008.06218.x. [DOI] [PubMed] [Google Scholar]
- 62.Mannironi C, Camon J, De Vito F, Biundo A, De Stefano ME, Persiconi I, Bozzoni I, Fragapane P, Mele A, Presutti C. Acute stress alters amygdala microRNA miR-135a and miR-124 expression: inferences for corticosteroid dependent stress response. PLoS ONE. 2013;8:e73385. doi: 10.1371/journal.pone.0073385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haramati S, Navon I, Issler O, Ezra-Nevo G, Gil S, Zwang R, Hornstein E, Chen A. MicroRNA as repressors of stress-induced anxiety: the case of amygdalar miR-34. Journal of Neuroscience. 2011;31:14191–14203. doi: 10.1523/JNEUROSCI.1673-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smalheiser NR, Lugli G, Rizavi HS, Zhang H, Torvik VI, Pandey GN, Davis JM, Dwivedi Y. MicroRNA expression in rat brain exposed to repeated inescapable shock: differential alterations in learned helplessness vs. non-learned helplessness. Int J Neuropsychopharmacol. 2011;14:1315–25. doi: 10.1017/S1461145710001628. [DOI] [PubMed] [Google Scholar]
- 65.Willner P. Animal models of depression: validity and applications. Adv Biochem Psychopharmacol. 1995;49:19–41. [PubMed] [Google Scholar]
- 66.Kohen R, Kirov S, Navaja GP, Happe HK, Hamblin MW, Snoddy JR, Neumaier JF, Petty F. Gene expression profiling in the hippocampus of learned helpless and nonhelpless rats. Pharmacogenomics J. 2005;5:278–91. doi: 10.1038/sj.tpj.6500322. [DOI] [PubMed] [Google Scholar]
- 67.O’Connor RM, Dinan TG, Cryan JF. Little things on which happiness depends: microRNAs as novel therapeutic targets for the treatment of anxiety and depression. Mol Psychiatry. 2012;17:359–76. doi: 10.1038/mp.2011.162. [DOI] [PubMed] [Google Scholar]
- 68.Baudry A, Mouillet-Richard S, Schneider B, Launay JM, Kellermann O. miR-16 targets the serotonin transporter: a new facet for adaptive responses to antidepressants. Science. 2010;329:1537–1541. doi: 10.1126/science.1193692. [DOI] [PubMed] [Google Scholar]
- 69.Smalheiser NR, Lugli G, Rizavi HS, Torvik VI, Turecki G, Dwivedi Y. MicroRNA expression is down-regulated and reorganized in prefrontal cortex of depressed suicide subjects. PLoS ONE. 2012;7:e33201. doi: 10.1371/journal.pone.0033201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Poulter MO, Du L, Weaver IC, Palkovits M, Faludi G, Merali Z, Szyf M, Anisman H. GABAA receptor promoter hypermethylation in suicide brain: implications for the involvement of epigenetic processes. Biological Psychiatry. 2008;64:645–52. doi: 10.1016/j.biopsych.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 71.Maussion G, Yang J, Yerko V, Barker P, Mechawar N, Ernst C, Turecki G. Regulation of a truncated form of tropomyosin-related kinase B (TrkB) by Hsa-miR-185* in frontal cortex of suicide completers. PLoS ONE. 2012;7:e39301. doi: 10.1371/journal.pone.0039301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heim C, Nemeroff CB. The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biological Psychiatry. 2001;49:1023–39. doi: 10.1016/s0006-3223(01)01157-x. [DOI] [PubMed] [Google Scholar]
- 73.Holmes SJ, Robins LN. The influence of childhood disciplinary experience on the development of alcoholism and depression. J Child Psychol Psychiatry. 1987;28:399–415. doi: 10.1111/j.1469-7610.1987.tb01762.x. [DOI] [PubMed] [Google Scholar]
- 74.Enoch MA. The role of early life stress as a predictor for alcohol and drug dependence. Psychopharmacology. 2011;214:17–31. doi: 10.1007/s00213-010-1916-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Riggs S, Alario AJ. Adolescent substance use and the role of the primary care provider. R I Med J. 1990;73:253–7. [PubMed] [Google Scholar]
- 76.Meaney MJ. Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu Rev Neurosci. 2001;24:1161–92. doi: 10.1146/annurev.neuro.24.1.1161. [DOI] [PubMed] [Google Scholar]
- 77.Matrisciano F, Tueting P, Dalal I, Kadriu B, Grayson DR, Davis JM, Nicoletti F, Guidotti A. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology. 2013;68:184–194. doi: 10.1016/j.neuropharm.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blaze J, Roth TL. Exposure to caregiver maltreatment alters expression levels of epigenetic regulators in the medial prefrontal cortex. Int J Devl Neuroscience. 2013;31:804–810. doi: 10.1016/j.ijdevneu.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Levine A, Worrell TR, Zimnisky R, Schmauss C. Early life stress triggers sustained changes in histone deacetylase expression and histone H4 modifications that alter responsiveness to adolescent antidepressant treatment. Neurobiology of disease. 2012;45:488–498. doi: 10.1016/j.nbd.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xie L, Korkmaz KS, Braun K, Bock J. Early life stress-induced histone acetylations correlate with activation of the synaptic plasticity genes Arc and Egr1 in the mouse hippocampus. J Neurochem. 2013;125:457–464. doi: 10.1111/jnc.12210. [DOI] [PubMed] [Google Scholar]
- 81.Bagot RC, Zhang TY, Wen X, Nguyen TT, Nguyen HB, Diorio J, Wong TP, Meaney MJ. Variations in postnatal maternal care and the epigenetic regulation of metabotropic glutamate receptor 1 expression and hippocampal function in the rat. Proc Natl Acad Sci U S A. 2012;109:17200–7. doi: 10.1073/pnas.1204599109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mcgowan PO, Suderman M, Sasaki A, Huang TCT, Hallett M, Meaney MJ, Szyf M, Sirigu A. Broad Epigenetic Signature of Maternal Care in the Brain of Adult Rats. PLoS ONE. 2011;6:e14739. doi: 10.1371/journal.pone.0014739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang TY, Hellstrom IC, Bagot RC, Wen X, Diorio J, Meaney MJ. Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. Journal of Neuroscience. 2010;30:13130–7. doi: 10.1523/JNEUROSCI.1039-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weaver ICG, Meaney MJ, Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci U S A. 2006;103:3480–3485. doi: 10.1073/pnas.0507526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meaney MJ, Viau V, Bhatnagar S, Betito K, Iny LJ, O’Donnell D, Mitchell JB. Cellular mechanisms underlying the development and expression of individual differences in the hypothalamic-pituitary-adrenal stress response. J Steroid Biochem Mol Biol. 1991;39:265–274. doi: 10.1016/0960-0760(91)90072-d. [DOI] [PubMed] [Google Scholar]
- 86.Gitau R, Cameron A, Fisk NM, Glover V. Fetal exposure to maternal cortisol. Lancet. 1998;352:707–708. doi: 10.1016/S0140-6736(05)60824-0. [DOI] [PubMed] [Google Scholar]
- 87.Mairesse J, Lesage J, Breton C, Bréant B, Hahn T, Darnaudéry M, Dickson SL, Seckl J, Blondeau B, Vieau D, Maccari S, Viltart O. Maternal stress alters endocrine function of the feto-placental unit in rats. Am J Physiol Endo Metab. 2007;292:E1526–33. doi: 10.1152/ajpendo.00574.2006. [DOI] [PubMed] [Google Scholar]
- 88.O’Donnell KJ, Bugge Jensen A, Freeman L, Khalife N, O'Connor TG, Glover V. Maternal prenatal anxiety and downregulation of placental 11β-HSD2. Psychoneuroendocrinology. 2012;37:818–826. doi: 10.1016/j.psyneuen.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 89.Holmes MC, Abrahamsen CT, French KL, Paterson JM, Mullins JJ, Seckl JR. The mother or the fetus? 11beta-hydroxysteroid dehydrogenase type 2 null mice provide evidence for direct fetal programming of behavior by endogenous glucocorticoids. Journal of Neuroscience. 2006;26:3840–3844. doi: 10.1523/JNEUROSCI.4464-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Welberg LA, Seckl JR, Holmes MC. Inhibition of 11beta-hydroxysteroid dehydrogenase, the foeto-placental barrier to maternal glucocorticoids, permanently programs amygdala GR mRNA expression and anxiety-like behaviour in the offspring. Eur J Neurosci. 2000;12:1047–1054. doi: 10.1046/j.1460-9568.2000.00958.x. [DOI] [PubMed] [Google Scholar]
- 91.Jensen Peña C, Monk C, Champagne FA. Epigenetic effects of prenatal stress on 11β-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS ONE. 2012;7:e39791–e39791. doi: 10.1371/journal.pone.0039791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mueller BR, Bale TL. Sex-specific programming of offspring emotionality after stress early in pregnancy. Journal of Neuroscience. 2008;28:9055–9065. doi: 10.1523/JNEUROSCI.1424-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106. doi: 10.4161/epi.3.2.6034. [DOI] [PubMed] [Google Scholar]
- 94.Hompes T, Izzi B, Gellens E, Morreels M, Fieuws S, Pexsters A, Schops G, Dom M, Van Bree R, Freson K, Verhaeghe J, Spitz B, Demyttenaere K, Glover V, Van den Bergh B, Allegaert K, Claes S. Investigating the influence of maternal cortisol and emotional state during pregnancy on the DNA methylation status of the glucocorticoid receptor gene (NR3C1) promoter region in cord blood. Journal of Psychiatric Research. 2013;47:880–891. doi: 10.1016/j.jpsychires.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 95.Radtke KM, Ruf M, Gunter HM, Dohrmann K, Schauer M, Meyer A, Elbert T. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Translational Psychiatry. 2011;1:1–6. doi: 10.1038/tp.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Devlin AM, Brain U, Austin J, Oberlander TF. Prenatal exposure to maternal depressed mood and the MTHFR C677T variant affect SLC6A4 methylation in infants at birth. PLoS ONE. 2010;5:e12201. doi: 10.1371/journal.pone.0012201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schroeder J, Smith A, Brennan P, Conneely K, Kilaru V, Knight B, Newport D, Cubells J, Stowe Z. DNA methylation in neonates born to women receiving psychiatric care. Epigenetics. 2012;7:409–414. doi: 10.4161/epi.19551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Suderman M, McGowan PO, Sasaki A, Huang TC, Hallett MT, Meaney MJ, Turecki G, Szyf M. Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc Natl Acad Sci U S A. 2012;109:17266–72. doi: 10.1073/pnas.1121260109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 100.Labonte B, Yerko V, Gross J, Mechawar N, Meaney MJ, Szyf M, Turecki G. Differential glucocorticoid receptor exon 1(B), 1(C), and 1(H) expression and methylation in suicide completers with a history of childhood abuse. Biological Psychiatry. 2012;72:41–8. doi: 10.1016/j.biopsych.2012.01.034. [DOI] [PubMed] [Google Scholar]
- 101.Mcgowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonté B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nature Neuroscience. 2009;12:342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Perroud N, Paoloni-Giacobino A, Prada P, Olié E, Salzmann A, Nicastro R, Guillaume S, Mouthon D, Stouder C, Dieben K, Huguelet P, Courtet P, Malafosse A. Increased methylation of glucocorticoid receptor gen (NR3C1) in adults with a history of childhood maltreatment: a link with the severity and type of trauma. Translational Psychiatry. 2011;1 doi: 10.1038/tp.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tyrka AR, Price LH, Marsit C, Walters OC, Carpenter LL. Childhood adversity and epigenetic modulation of the leukocyte glucocorticoid receptor: preliminary findings in healthy adults. PLoS ONE. 2012;7:e30148–e30148. doi: 10.1371/journal.pone.0030148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Alt SR, Turner JD, Klok MD, Meijer OC, Lakke EA, Derijk RH, Muller CP. Differential expression of glucocorticoid receptor transcripts in major depressive disorder is not epigenetically programmed. Psychoneuroendocrinology. 2010;35:544–56. doi: 10.1016/j.psyneuen.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 105.Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, Impagnatiello F, Pandey G, Pesold C, Sharma R, Uzunov D, Costa E. Decrease in reelin and glutamic acid decarboxylase 67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57:1061–9. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- 106.Veldic M, Caruncho HJ, Liu WS, Davis J, Satta R, Grayson DR, Guidotti A, Costa E. DNA-methyltransferase 1 mRNA is selectively overexpressed in telencephalic GABAergic interneurons of schizophrenia brains. Proc Natl Acad Sci U S A. 2004;101:348–53. doi: 10.1073/pnas.2637013100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kundakovic M, Chen Y, Costa E, Grayson DR. DNA methyltransferase inhibitors coordinately induce expression of the human reelin and glutamic acid decarboxylase 67 genes. Mol Pharmacol. 2007;71:644–53. doi: 10.1124/mol.106.030635. [DOI] [PubMed] [Google Scholar]
- 108.Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, Costa E. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A. 2005;102:9341–6. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tamura Y, Kunugi H, Ohashi J, Hohjoh H. Epigenetic aberration of the human REELIN gene in psychiatric disorders. Mol Psychiatry. 2007;12:519, 593–600. doi: 10.1038/sj.mp.4002014. [DOI] [PubMed] [Google Scholar]
- 110.Capitanio JP, Mendoza SP, Mason WA, Maninger N. Rearing environment and hypothalamic-pituitary-adrenal regulation in young rhesus monkeys (Macaca mulatta) Dev Psychobiol. 2005;46:318–30. doi: 10.1002/dev.20067. [DOI] [PubMed] [Google Scholar]
- 111.Beach SR, Brody GH, Todorov AA, Gunter TD, Philibert RA. Methylation at 5HTT mediates the impact of child sex abuse on women’s antisocial behavior: an examination of the Iowa adoptee sample. Psychosomatic Medicine. 2011;73:83–7. doi: 10.1097/PSY.0b013e3181fdd074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vijayendran M, Beach SR, Plume JM, Brody GH, Philibert RA. Effects of genotype and child abuse on DNA methylation and gene expression at the serotonin transporter. Frontiers in Psychiatry. 2012;3:55. doi: 10.3389/fpsyt.2012.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Murgatroyd C, Patchev AV, Wu Y, Micale V, Bockmühl Y, Fischer D, Holsboer F, Wotjak CT, Almeida OFX, Spengler D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nature Neuroscience. 2009;12:1559–1566. doi: 10.1038/nn.2436. [DOI] [PubMed] [Google Scholar]
- 114.Niwa M, Jaaro-Peled H, Tankou S, Seshadri S, Hikida T, Matsumoto Y, Cascella NG, Kano Si, Ozaki N, Nabeshima T, Sawa A. Adolescent Stress-Induced Epigenetic Control of Dopaminergic Neurons via Glucocorticoids. Science. 2013;339:335–339. doi: 10.1126/science.1226931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Labonte B, Suderman M, Maussion G, Navaro L, Yerko V, Mahar I, Bureau A, Mechawar N, Szyf M, Meaney MJ, Turecki G. Genome-wide epigenetic regulation by early-life trauma. Arch Gen Psychiatry. 2012;69:722–31. doi: 10.1001/archgenpsychiatry.2011.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Uddin M, Aiello AE, Wildman DE, Koenen KC, Pawelec G, de Los Santos R, Goldmann E, Galea S. Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc Natl Acad Sci U S A. 2010;107:9470–5. doi: 10.1073/pnas.0910794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Labonte B, Suderman M, Maussion G, Lopez JP, Navarro-Sanchez L, Yerko V, Mechawar N, Szyf M, Meaney MJ, Turecki G. Genome-wide methylation changes in the brains of suicide completers. Am J Psychiatry. 2013;170:511–20. doi: 10.1176/appi.ajp.2012.12050627. [DOI] [PubMed] [Google Scholar]
- 118.Zucchi FCR, Yao Y, Ward ID, Ilnytskyy Y, Olson DM, Benzies K, Kovalchuk I, Kovalchuk O, Metz GAS. Maternal stress induces epigenetic signatures of psychiatric and neurological diseases in the offspring. PLoS ONE. 2013;8:e56967–e56967. doi: 10.1371/journal.pone.0056967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Monteleone MC, Adrover E, Pallarés ME, Antonelli MC, Frasch AC, Brocco MA. Prenatal stress changes the glycoprotein GPM6A gene expression and induces epigenetic changes in rat offspring brain. Epigenetics. 2013:9. doi: 10.4161/epi.25925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Morgan CP, Bale TL. Early prenatal stress epigenetically programs dysmasculinization in second-generation offspring via the paternal lineage. Journal of Neuroscience. 2011;31:11748–11755. doi: 10.1523/JNEUROSCI.1887-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Uchida S, Hara K, Kobayashi A, Funato H, Hobara T, Otsuki K, Yamagata H, McEwen BS, Watanabe Y. Early life stress enhances behavioral vulnerability to stress through the activation of REST4-mediated gene transcription in the medial prefrontal cortex of rodents. Journal of Neuroscience. 2010;30:15007–18. doi: 10.1523/JNEUROSCI.1436-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Conaco C, Otto S, Han JJ, Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc Natl Acad Sci U S A. 2006;103:2422–7. doi: 10.1073/pnas.0511041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rajasethupathy P, Fiumara F, Sheridan R, Betel D, Puthanveettil SV, Russo JJ, Sander C, Tuschl T, Kandel E. Characterization of small RNAs in Aplysia reveals a role for miR-124 in constraining synaptic plasticity through CREB. Neuron. 2009;63:803–17. doi: 10.1016/j.neuron.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kawashima H, Numakawa T, Kumamaru E, Adachi N, Mizuno H, Ninomiya M, Kunugi H, Hashido K. Glucocorticoid attenuates brain-derived neurotrophic factor-dependent upregulation of glutamate receptors via the suppression of microRNA-132 expression. Neuroscience. 2010;165:1301–11. doi: 10.1016/j.neuroscience.2009.11.057. [DOI] [PubMed] [Google Scholar]
- 125.Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, Impey S. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci U S A. 2005;102:16426–31. doi: 10.1073/pnas.0508448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Huang YW, Ruiz CR, Eyler EC, Lin K, Meffert MK. Dual regulation of miRNA biogenesis generates target specificity in neurotrophin-induced protein synthesis. Cell. 2012;148:933–46. doi: 10.1016/j.cell.2012.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DEH, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 128.Schroeder FA, Penta KL, Matevossian A, Jones SR, Konradi C, Tapper AR, Akbarian S. Drug-induced activation of dopamine D(1) receptor signaling and inhibition of class I/II histone deacetylase induce chromatin remodeling in reward circuitry and modulate cocaine-related behaviors. Neuropsychopharmacology. 2008;33:2981–2992. doi: 10.1038/npp.2008.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shen HY, Kalda A, Yu L, Ferrara J, Zhu J, Chen JF. Additive effects of histone deacetylase inhibitors and amphetamine on histone H4 acetylation, cAMP responsive element binding protein phosphorylation and DeltaFosB expression in the striatum and locomotor sensitization in mice. Neuroscience. 2008;157:644–655. doi: 10.1016/j.neuroscience.2008.09.019. [DOI] [PubMed] [Google Scholar]
- 130.Kennedy PJ, Feng J, Robison AJ, Maze I, Badimon A, Mouzon E, Chaudhury D, Damez-Werno DM, Haggarty SJ, Han MH, Bassel-Duby R, Olson EN, Nestler EJ. Class I HDAC inhibition blocks cocaine-induced plasticity by targeted changes in histone methylation. Nature Neuroscience. 2013;16:434–440. doi: 10.1038/nn.3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Levine AA, Guan Z, Barco A, Xu S, Kandel ER, Schwartz JH. CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum. Proc Natl Acad Sci U S A. 2005;102:19186–19191. doi: 10.1073/pnas.0509735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Malvaez M, Mhillaj E, Matheos DP, Palmery M, Wood MA. CBP in the nucleus accumbens regulates cocaine-induced histone acetylation and is critical for cocaine-associated behaviors. Journal of Neuroscience. 2011;31:16941–16948. doi: 10.1523/JNEUROSCI.2747-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Taniguchi M, Carreira MB, Smith LN, Zirlin BC, Neve RL, Cowan CW. Histone deacetylase 5 limits cocaine reward through cAMP-induced nuclear import. Neuron. 2012;73:108–120. doi: 10.1016/j.neuron.2011.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang L, Lv Z, Hu Z, Sheng J, Hui B, Sun J, Ma L. Chronic cocaine-induced H3 acetylation and transcriptional activation of CaMKIIalpha in the nucleus accumbens is critical for motivation for drug reinforcement. Neuropsychopharmacology. 2010;35:913–928. doi: 10.1038/npp.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Renthal W, Carle TL, Maze I, Covington HE, Truong HT, Alibhai I, Kumar A, Montgomery RL, Olson EN, Nestler EJ. Delta FosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. Journal of Neuroscience. 2008;28:7344–7349. doi: 10.1523/JNEUROSCI.1043-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Renthal W, Kumar A, Xiao G, Wilkinson M, Covington HE, Maze I, Sikder D, Robison AJ, Laplant Q, Dietz DM, Russo SJ, Vialou V, Chakravarty S, Kodadek TJ, Stack A, Kabbaj M, Nestler EJ. Genome-wide analysis of chromatin regulation by cocaine reveals a role for sirtuins. Neuron. 2009;62:335–348. doi: 10.1016/j.neuron.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ferguson D, Koo JW, Feng J, Heller E, Rabkin J, Heshmati M, Renthal W, Neve R, Liu X, Shao N, Sartorelli V, Shen L, Nestler EJ. Essential Role of SIRT1 Signaling in the Nucleus Accumbens in Cocaine and Morphine Action. Journal of Neuroscience. 2013;33:16088–16098. doi: 10.1523/JNEUROSCI.1284-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hwang CK, Song KY, Kim CS, Choi HS, Guo XH, Law PY, Wei LN, Loh HH. Epigenetic programming of mu-opioid receptor gene in mouse brain is regulated by MeCP2 and Brg1 chromatin remodelling factor. J Cellular Mol Med. 2009;13:3591–3615. doi: 10.1111/j.1582-4934.2008.00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mazei-Robison MS, Koo JW, Friedman AK, Lansink CS, Robison AJ, Vinish M, Krishnan V, Kim S, Siuta MA, Galli A, Niswender KD, Appasani R, Horvath MC, Neve RL, Worley PF, Snyder SH, Hurd YL, Cheer JF, Han MH, Russo SJ, Nestler EJ. Role for mTOR Signaling and Neuronal Activity in Morphine-Induced Adaptations in Ventral Tegmental Area Dopamine Neurons. Neuron. 2011;72:977–990. doi: 10.1016/j.neuron.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sheng J, Lv Zg, Wang L, Zhou Y, Hui B. Histone H3 phosphoacetylation is critical for heroin-induced place preference. Neuroreport. 2011;22:575–580. doi: 10.1097/WNR.0b013e328348e6aa. [DOI] [PubMed] [Google Scholar]
- 141.Pascual M, Do Couto BR, Alfonso-Loeches S, Aguilar MA, Rodriguez-Arias M, Guerri C. Changes in histone acetylation in the prefrontal cortex of ethanol-exposed adolescent rats are associated with ethanol-induced place conditioning. Neuropharmacology. 2012;62:2309–2319. doi: 10.1016/j.neuropharm.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 142.Botia B, Legastelois R, Alaux-Cantin S, Naassila M. Expression of ethanol-induced behavioral sensitization is associated with alteration of chromatin remodeling in mice. PLoS ONE. 2012;7:e47527. doi: 10.1371/journal.pone.0047527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sakharkar AJ, Zhang H, Tang L, Shi G, Pandey SC. Histone deacetylases (HDAC)-induced histone modifications in the amygdala: a role in rapid tolerance to the anxiolytic effects of ethanol. Alcohol Clin Exp Res. 2012;36:61–71. doi: 10.1111/j.1530-0277.2011.01581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. Brain chromatin remodeling: a novel mechanism of alcoholism. Journal of Neuroscience. 2008;28:3729–3737. doi: 10.1523/JNEUROSCI.5731-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Maze I, Covington HE, Dietz DM, Laplant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren Y, Sampath SC, Hurd YL, Greengard P, Tarakhovsky A, Schaefer A, Nestler EJ. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Qiang M, Denny A, Lieu M, Carreon S, Li J. Histone H3K9 modifications are a local chromatin event involved in ethanol-induced neuroadaptation of the NR2B gene. Epigenetics. 2011;6:1095–1104. doi: 10.4161/epi.6.9.16924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sun H, Maze I, Dietz DM, Scobie KN, Kennedy PJ, Damez-Werno D, Neve RL, Zachariou V, Shen L, Nestler EJ. Morphine epigenomically regulates behavior through alterations in histone H3 lysine 9 dimethylation in the nucleus accumbens. Journal of Neuroscience. 2012;32:17454–17464. doi: 10.1523/JNEUROSCI.1357-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Damez-Werno D, Laplant Q, Sun H, Scobie KN, Dietz DM, Walker IM, Koo JW, Vialou VF, Mouzon E, Russo SJ, Nestler EJ. Drug Experience Epigenetically Primes Fosb Gene Inducibility in Rat Nucleus Accumbens. Journal of Neuroscience. 2012;32:10267–10272. doi: 10.1523/JNEUROSCI.1290-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Feng J, Wilkinson M, Liu X, Purushothaman I, Ferguson D, Vialou V, Maze I, Shao N, Kennedy P, Koo J, Dias C, Laitman B, Stockman V, LaPlant Q, Cahill M, Nestler EJ, Shen L. Chronic cocaine-regulated epigenome in mouse nucleus accumbens. Genome Biol. 2014 doi: 10.1186/gb-2014-15-4-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Maze I, Feng J, Wilkinson MB, Sun H, Shen L, Nestler EJ. Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc Natl Acad Sci U S A. 2011;108:3035–3040. doi: 10.1073/pnas.1015483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jordi E, Heiman M, Marion-Poll L, Guermonprez P, Cheng SK, Nairn AC, Greengard P, Girault JA. Differential effects of cocaine on histone posttranslational modifications in identified populations of striatal neurons. Proc Natl Acad Sci U S A. 2013;110:9511–9516. doi: 10.1073/pnas.1307116110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Maze I, Chaudhury D, Dietz DM, Von Schimmelmann M, Kennedy PJ, Lobo MK, Sillivan SE, Miller ML, Bagot RC, Sun H, Turecki G, Neve RL, Hurd YL, Shen L, Han MH, Schaefer A, Nestler EJ. G9a influences neuronal subtype specification in striatum. Nature Neuroscience. 2014 doi: 10.1038/nn.3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Heller E, Cates H, Sun H-S, Ferguson D, Neve R, Knight S, Zhang F, Zhang S, Nestler EJ. Locus specific epigenetic reprogramming: Bidirectional regulation of the FosB gene using synthetic transcription factors in vivo. Abstract Society for Neuroscience; San Diego, CA: 2013. [Google Scholar]
- 154.Brami-Cherrier K, Roze E, Girault JA, Betuing S, Caboche J. Role of the ERK/MSK1 signalling pathway in chromatin remodelling and brain responses to drugs of abuse. J Neurochem. 2009;108:1323–1335. doi: 10.1111/j.1471-4159.2009.05879.x. [DOI] [PubMed] [Google Scholar]
- 155.Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Hervé D, Valjent E, Girault JA. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. Journal of Neuroscience. 2008;28:5671–5685. doi: 10.1523/JNEUROSCI.1039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Damez-Werno D, Scobie KN, Sun H, Dias C, Cates H, Shao N, Gancarz AM, Maze I, Neve RL, Dietz DM, Shen L, Guccione E, Allis DC, Nestler EJ. Histone arginine methylation in the nucleus accumbens in response to chronic cocaine and chronic stress. Abstract, Society for Neuroscience; San Diego, CA: 2013. [Google Scholar]
- 157.Scobie KN, Damez-Werno D, Sun H, Shao N, Gancarz A, Panganiban CH, Dias C, Koo J, Caiafa P, Kaufman L, Neve RL, Dietz DM, Shen L, Nestler EJ. Essential role of poly(ADP-ribosyl)ation in cocaine action. Proc Natl Acad Sci U S A. 2014;111:2005–10. doi: 10.1073/pnas.1319703111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Anier K, Malinovskaja K, Aonurm-Helm A, Zharkovsky A, Kalda A. DNA methylation regulates cocaine-induced behavioral sensitization in mice. Neuropsychopharmacology. 2010;35:2450–2461. doi: 10.1038/npp.2010.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Miller CA, Campbell SL, Sweatt JD. DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiology of Learning and Memory. 2008;89:599–603. doi: 10.1016/j.nlm.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Im HI, Hollander JA, Bali P, Kenny PJ. MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nature Neuroscience. 2010;13:1120–1127. doi: 10.1038/nn.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Deng JV, Rodriguiz RM, Hutchinson AN, Kim IH, Wetsel WC, West AE. MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nature Neuroscience. 2010;13:1128–1136. doi: 10.1038/nn.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–91. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 163.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278:4035–40. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 164.Nielsen DA, Yuferov V, Hamon S, Jackson C, Ho A, Ott J, Kreek MJ. Increased OPRM1 DNA methylation in lymphocytes of methadone-maintained former heroin addicts. Neuropsychopharmacology. 2009;34:867–873. doi: 10.1038/npp.2008.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. Journal of Neuroscience. 2012;32:1884–97. doi: 10.1523/JNEUROSCI.3136-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Eipper-Mains JE, Kiraly DD, Palakodeti D, Mains RE, Eipper BA, Graveley BR. microRNA-Seq reveals cocaine-regulated expression of striatal microRNAs. RNA. 2011;17:1529–1543. doi: 10.1261/rna.2775511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chandrasekar V, Dreyer JL. microRNAs miR-124, let-7d and miR-181a regulate cocaine-induced plasticity. Mol Cell Neurosci. 2009;42:350–362. doi: 10.1016/j.mcn.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 168.Chandrasekar V, Dreyer JL. Regulation of MiR-124, Let-7d, and MiR-181a in the accumbens affects the expression, extinction, and reinstatement of cocaine-induced conditioned place preference. Neuropsychopharmacology. 2011;36:1149–1164. doi: 10.1038/npp.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Schaefer AA, Im HIH, Venø MTM, Fowler CDC, Min AA, Intrator AA, Kjems JJ, Kenny PJP, O'Carroll DD, Greengard PP. Argonaute 2 in dopamine 2 receptor-expressing neurons regulates cocaine addiction. Journal of Experimental Medicine. 2010;207:1843–1851. doi: 10.1084/jem.20100451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sartor GC, St Laurent G, Wahlestedt C. The Emerging Role of Non-Coding RNAs in Drug Addiction. Frontiers in Genetics. 2012;3:106. doi: 10.3389/fgene.2012.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hollander JA, Im HI, Amelio AL, Kocerha J, Bali P, Lu Q, Willoughby D, Wahlestedt C, Conkright MD, Kenny PJ. Striatal microRNA controls cocaine intake through CREB signalling. Nature. 2010;466:197–202. doi: 10.1038/nature09202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Pietrzykowski AZ, Friesen RM, Martin GE, Puig SI, Nowak CL, Wynne PM, Siegelmann HT, Treistman SN. Posttranscriptional regulation of BK channel splice variant stability by miR-9 underlies neuroadaptation to alcohol. Neuron. 2008;59:274–287. doi: 10.1016/j.neuron.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Guo W, Crossey EL, Zhang L, Zucca S, George OL, Valenzuela CF, Zhao X. Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS ONE. 2011;6:e19351–e19351. doi: 10.1371/journal.pone.0019351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.DiNieri JA, Wang X, Szutorisz H, Spano SM, Kaur J, Casaccia P, Dow-Edwards D, Hurd YL. Maternal cannabis use alters ventral striatal dopamine D2 gene regulation in the offspring. Biological Psychiatry. 2011;70:763–769. doi: 10.1016/j.biopsych.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Pascual M, Boix J, Felipo V, Guerri C. Repeated alcohol administration during adolescence causes changes in the mesolimbic dopaminergic and glutamatergic systems and promotes alcohol intake in the adult rat. J Neurochem. 2009;108:920–931. doi: 10.1111/j.1471-4159.2008.05835.x. [DOI] [PubMed] [Google Scholar]
- 176.Black YD, Maclaren FR, Naydenov AV, Carlezon WA, Baxter MG, Konradi C. Altered attention and prefrontal cortex gene expression in rats after binge-like exposure to cocaine during adolescence. Journal of Neuroscience. 2006;26:9656–9665. doi: 10.1523/JNEUROSCI.2391-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tomasiewicz HC, Jacobs MM, Wilkinson MB, Wilson SP, Nestler EJ, Hurd YL. Proenkephalin mediates the enduring effects of adolescent cannabis exposure associated with adult opiate vulnerability. Biological Psychiatry. 2012;72:803–810. doi: 10.1016/j.biopsych.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hutson JR, Stade B, Lehotay DC, Collier CP, Kapur BM. Folic acid transport to the human fetus is decreased in pregnancies with chronic alcohol exposure. PLoS ONE. 2012;7:e38057–e38057. doi: 10.1371/journal.pone.0038057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zhou FC, Balaraman Y, Teng M, Liu Y, Singh RP, Nephew KP. Alcohol alters DNA methylation patterns and inhibits neural stem cell differentiation. Alcohol Clin Exp Res. 2011;35:735–746. doi: 10.1111/j.1530-0277.2010.01391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Perkins A, Lehmann C, Lawrence RC, Kelly SJ. Alcohol exposure during development: Impact on the epigenome. Int J Devl Neuroscience. 2013;31:391–397. doi: 10.1016/j.ijdevneu.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Laufer BI, Mantha K, Kleiber ML, Diehl EJ, Addison SMF, Singh SM. Long-lasting alterations to DNA methylation and ncRNAs could underlie the effects of fetal alcohol exposure in mice. Disease Models & Mechanisms. 2013;6:977–992. doi: 10.1242/dmm.010975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wilhelm-Benartzi CS, Houseman EA, Maccani MA, Poage GM, Koestler DC, Langevin SM, Gagne LA, Banister CE, Padbury JF, Marsit CJ. In utero exposures, infant growth, and DNA methylation of repetitive elements and developmentally related genes in human placenta. Environ Health Perspect. 2012;120:296–302. doi: 10.1289/ehp.1103927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Suter M, Ma J, Harris A, Patterson L, Brown KA, Shope C, Showalter L, Abramovici A, Aagaard-Tillery KM. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics. 2011;6:1284–94. doi: 10.4161/epi.6.11.17819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med. 2009;180:462–467. doi: 10.1164/rccm.200901-0135OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Toledo-Rodriguez MM, Lotfipour SS, Leonard GG, Perron MM, Richer LL, Veillette SS, Pausova ZZ, Paus TT. Maternal smoking during pregnancy is associated with epigenetic modifications of the brain-derived neurotrophic factor-6 exon in adolescent offspring. Am J Med Genet Part B. 2010;153B:1350–1354. doi: 10.1002/ajmg.b.31109. [DOI] [PubMed] [Google Scholar]
- 186.Novikova SI, He F, Bai J, Cutrufello NJ, Lidow MS, Undieh AS. Maternal cocaine administration in mice alters DNA methylation and gene expression in hippocampal neurons of neonatal and prepubertal offspring. PLoS ONE. 2008;3:e1919–e1919. doi: 10.1371/journal.pone.0001919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ehrlich S, Walton E, Roffman JL, Weiss D, Puls I, Doehler N, Burghardt R, Lehmkuhl U, Hillemacher T, Muschler M, Frieling H. Smoking, but not malnutrition, influences promoter-specific DNA methylation of the proopiomelanocortin gene in patients with and without anorexia nervosa. Can J Psychiatry. 2012;57:168–76. doi: 10.1177/070674371205700306. [DOI] [PubMed] [Google Scholar]
- 188.Anier K, Malinovskaja K, Pruus K, Aonurm-Helm A, Zharkovsky A, Kalda A. Maternal separation is associated with DNA methylation and behavioural changes in adult rats. Eur Neuropsychopharm. 2013 doi: 10.1016/j.euroneuro.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 189.Balaraman S, Winzer-Serhan UH, Miranda RC. Opposing actions of ethanol and nicotine on microRNAs are mediated by nicotinic acetylcholine receptors in fetal cerebral cortical-derived neural progenitor cells. Alcohol Clin Exp Res. 2012;36:1669–1677. doi: 10.1111/j.1530-0277.2012.01793.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wang LL, Zhang Z, Li Q, Yang R, Pei X, Xu Y, Wang J, Zhou SF, Li Y. Ethanol exposure induces differential microRNA and target gene expression and teratogenic effects which can be suppressed by folic acid supplementation. Human Reproduction. 2009;24:562–579. doi: 10.1093/humrep/den439. [DOI] [PubMed] [Google Scholar]
- 191.Barreto-Valer K, López-Bellido R, Sánchez-Simón FM, Rodríguez RE. Modulation by Cocaine of Dopamine Receptors through miRNA-133b in Zebrafish Embryos. PLoS ONE. 2012;7:e52701–e52701. doi: 10.1371/journal.pone.0052701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Taki FA, Pan X, Zhang B. Chronic Nicotine Exposure Systemically Alters MicroRNA Expression Profiles During Post-Embryonic Stages in Caenorhabditis elegans. Journal of Cellular Physiology. 2014;229:79–89. doi: 10.1002/jcp.24419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Maccani MA, Avissar-Whiting M, Banister CE, McGonnigal B, Padbury JF, Marsit CJ. Maternal cigarette smoking during pregnancy is associated with downregulation of miR-16, miR-21, and miR-146a in the placenta. Epigenetics. 2010;5:583–9. doi: 10.4161/epi.5.7.12762. [DOI] [PMC free article] [PubMed] [Google Scholar]