

Abstract
The compstatin family of complement inhibitors has shown promise in various immuno-inflammatory disorders. Although recent analogues show beneficial pharmacokinetics, further extension of the plasma half-life is expected to benefit systemic application of these peptidic inhibitors. We therefore synthesized conjugates of compstatin analogues and albumin-binding molecules (ABM) to increase circulatory residence. Equilibrium dialysis in complement-depleted serum showed a marked increase in plasma protein binding from <8% to >99% for a resulting chimera (ABM2-Cp20). Further analysis confirmed interaction with albumin from different species, primarily via site II. Importantly, ABM2-Cp20 bound 20-fold stronger to its target protein C3b (KD=150 pm) than the parent peptide. Kinetic and in silico analysis suggested that ABM2 occupies a secondary site on C3b and improves the dissociation rate via additional contacts. Addition of an ABM modifier thereby not only improved plasma protein binding but also produced the most potent compstatin analogue to date with potential implications for the treatment of systemic complement-related diseases.
Keywords: albumin binding, complement inhibitor, compstatin, conjugation, peptide drugs
Inappropriate or excessive activation of the human complement system is implicated in many clinical disorders.[1] Compstatin, a 13-residue cyclic peptide originally discovered by phage-display library screening, interacts with the complement component C3 and its activation fragment C3b and broadly inhibits complement activation.[2,3] The central role of C3 in complement initiation and amplification pathways renders C3 inhibitors an attractive option for the treatment of a wide range of complement-related conditions, and compstatin analogues have shown promise in disorders ranging from sepsis and bio-material-induced thromboinflammation to transplantation.[3–6] Whereas an early analogue of compstatin (POT-4, Potentia Pharmaceuticals) is in clinical development for the local treatment of age-related macular degeneration, the pharmacokinetic profile of this analogue may limit systemic applications. New generations of compstatin derivatives with enhanced inhibitory activity and plasma residence have therefore been developed.[7, 8] Backbone N-methylation resulted in analogue Cp20 (Ac-Ile-[Cys-Val-Trp(Me)-Gln-Asp-Trp-Sar-Ala-His-Arg-Cys]-mIle-NH2) that showed 10-fold improved affinity.[8] Introducing an additional amino acid at the N terminus of Cp20, thereby extending the target binding site, produced the lead analogue Cp40 ((d)Tyr-Ile-[Cys-Val-Trp(Me)-Gln-Asp-Trp-Sar-Ala-His-Arg-Cys]-mIle-NH2) with sub-nanomolar binding affinity for C3 (KD= 0.5 nm). Importantly, pharmacokinetic evaluation in non-human primates (NHP) revealed that these next-generation compstatin analogues follow target-driven elimination kinetics and feature half-life values of up to 12 h, thereby exceeding those typically reported for peptidic drugs.[7] Cp40 has shown promise in preclinical models of paroxysmal nocturnal hemo-globinuria and periodontal disease,[9, 10] and is currently developed for a variety of systemic disorders (Amanden, Amyndas Pharmaceuticals).[11] Whereas suitable inhibitor levels for chronic treatment could be achieved via subcutaneous application of Cp40,[9] further extension of its plasma residence is considered beneficial through a decrease in dose intervals.
Among the various strategies to improve the half-life of pep-tidic drugs, the coupling to albumin-binding tags appears particularly promising.[12] Albumin constitutes ∼60% of the total plasma protein pool and has a long circulation residence (t½ ∼ 20 d); binding to serum albumin has therefore been recognized as an attractive route to extend the plasma residence of biopharmaceuticals.[12, 13] Alongside direct coupling ap-proaches,[13, 14] several affinity tags based on albumin-binding peptides or molecules (ABP and ABM, respectively) have been developed that allow noncovalent interaction with circulating albumin.[15–19] Chimeras of a compstatin derivative with an ABP have been successfully constructed,[20] yet their synthesis is demanding, given the involvement of two cyclic peptides. In the present study, we therefore evaluated conjugation to a low-molecular-weight ABM tag as potential strategy to improve the half-life of compstatin analogues. For this purpose, we based our approach on two previously described naphthalene acylsulfonamide[17] and diphenylcyclohexanol phosphate ester[18, 21] tags that were shown to improve the plasma half-life of therapeutic peptides.[18] One of these tags is being used clinically in the case of MS-325 (gadofosveset trisodium; Ablavar, Lanteus Medical Imaging), a rationally designed magnetic resonance imaging (MRI) contrast agent with prolonged intravascular half-life (18.5±3 h in humans).[21] Conjugation of advanced compstatin analogues such as Cp20 and Cp40 to these established ABM therefore holds promise to result in a safe and cost-effective way of improving the plasma residence of compstatin and extend its use in systemic indications.
Previous analysis of the co-crystal structure of an early compstatin analogue with the target protein fragment C3c revealed that both termini of the cyclic peptide were minimally engaged in binding site contacts and may be amenable for modification.[22] For initial studies, two previously described albumin-binding tags (ABM1 and ABM2; each extended with a carboxylic acid group for conjugation) were coupled to the N terminus of compstatin analogue Cp20 through an amide linkage (Scheme 1).[8,17,18] Following cleavage from the resin, each peptide was oxidized with hydrogen peroxide to form the intramolecular disulfide bond. All peptides were purified by reversed-phase high-performance liquid chromatography (RP-HPLC), resulting in average overall yields of 9–16% after lyophilization.
Scheme 1.
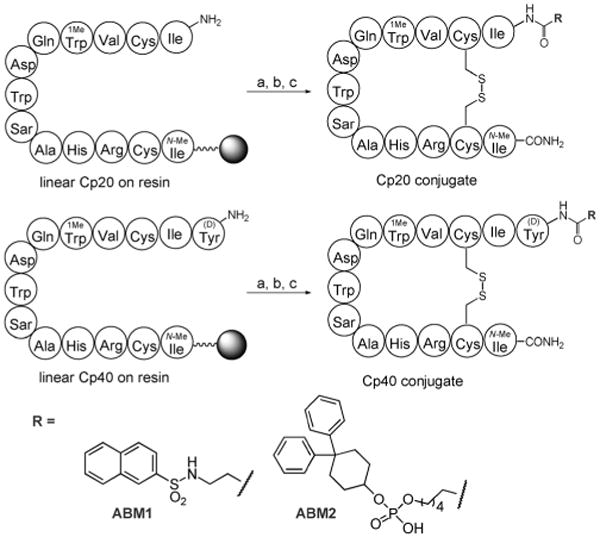
Synthesis of conjugates between compstatin analogues Cp20 and Cp40 with albumin-binding molecules. Reagents and conditions: a) coupling of RCOOH: DIPEA, HATU, DMF (R=ABM1), or DIPEA, PyBOP, NMP, CH2Cl2 (R=ABM2);[17, 18] b) resin cleavage with 90% TFA, 5% thioanisole, 3% EDT, 2% anisole; c) cyclization with H2O2. (Detailed procedures are given in the Supporting Information.)
To confirm maintenance of target binding affinity after addition of the albumin-binding tags, the synthesized compstatin conjugates were assayed using surface plasmon resonance (SPR) by injecting them over site-specifically immobilized C3b.[7, 23] Due to the slow dissociation rate of these compounds, a single-cycle kinetics approach was used to improve the assay efficiency.[24] The SPR responses of all peptides were fitted to a 1:1 Langmuir binding model to obtain kinetic association and dissociation rate constants (ka, kd) and binding affinities (KD; Table 1). As expected, conjugation of ABM1 to the N terminus of Cp20 fully maintained the target binding affinity; the slight improvement over the parent peptide may be explained by additional hydrophobic contacts of the new N terminus with the binding site of C3, as had been observed with Cp40.[7] Surprisingly, however, conjugation with ABM2 led to a 20-fold improvement in binding affinity (KD=150 pm), rendering ABM2-Cp20 the most potent compstatin analogue described so far. To further explore the potential of ABM2-mediated enhancement of compstatin affinity, we conjugated ABM2 to the N terminus of the current lead compound Cp40 (Scheme 1). In contrast to Cp20, the addition of ABM2 to Cp40 led to a comparatively minor improvement in affinity (Table 1). Analysis of the compstatin binding site using a structural model of ABM2-Cp20 suggested that the alkyl linker in ABM2 may ideally align the diphenyl-cyclohexanol moiety with a shallow groove formed by macroglobulin domain 4 of the C3 β-chain (Figure 1). In the case of ABM2-Cp40, the presence of an additional amino acid at the N terminus would lead to a less preferred placement of this moiety.
Table 1.
Evaluation of C3b interaction profiles of compstatin conjugates.[a]
| Peptide | ka [106 m−1 s−1] | kd [10−3 s−1] | KD [nm] |
|---|---|---|---|
| Cp20 | 2.3±0.7 | 6.7±2.9 | 2.9±0.4 |
| ABM1-Cp20 | 1.7±0.1 | 3.5±0.1 | 2.0±0.1 |
| ABM2-Cp20 | 2.7±0.9 | 0.4±0.1 | 0.15±0.06 |
| Cp40 | 2.8±0.6 | 1.3±0.2 | 0.48±0.09 |
| ABM2-Cp40 | 1.8±0.2 | 0.5±0.2 | 0.26±0.09 |
Single-cycle kinetic analysis of compstatin conjugates was performed. Sets of five increasing concentrations were consecutively injected over a C3b surface (3000–5000 RU density) in a single cycle. The processed signals were fitted to a 1:1 binding model.
Figure 1.
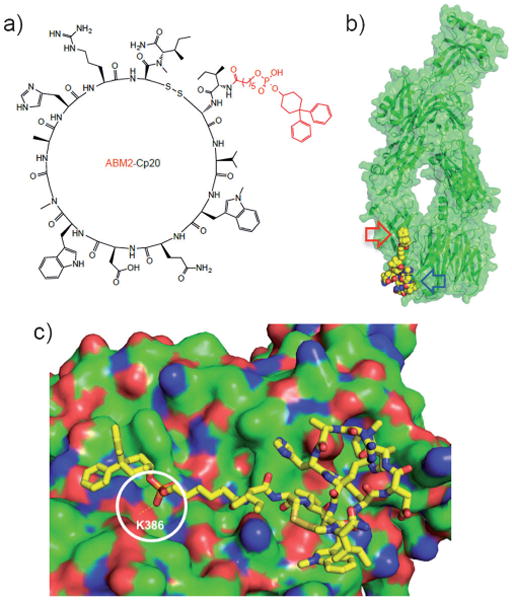
Structure and proposed binding model of ABM2-Cp20. a) Structure of ABM2-Cp20 with the ABM2 tag shown in red. b) Docking of ABM2-Cp20 (yellow spheres) into the compstatin binding site of C3c (green cartoon/surface representation; PDB code: 2QKI); the primary compstatin binding site and the proposed extended contact site for ABM2 are marked with blue and red arrows, respectively. c) Close-up of ABM2-Cp20 (stick representation) docked to C3c (green surface; positive and negative surface charges are shown in red and blue, respectively). The hydrogen bond between ABM2-Cp20 and lysine residue 386 of C3c (K386) predicted from the computational analysis is highlighted by a white circle.
As the binding of ABM–compstatin conjugates to albumin in circulation may potentially affect their complement-directed potency, we also evaluated the inhibitory activity of all pep-tides toward immune complex-induced complement activation in human plasma using an established ELISA format.[25] Compared with the parent peptides, the conjugates retain or improve the inhibitory activity (Table 2; Figure S4), thereby indicating that they are able to bind their target despite the high plasma concentration of albumin. Whether the less significant improvement in IC50 values between ABM conjugates and parent peptides relative to the KD values is caused by the presence of albumin or the limited dynamic range of the ELISA needs to be further investigated.
Table 2.
Complement inhibition potency of compstatin conjugates.[a]
| Peptide | IC50 [μm] |
|---|---|
| Cp20 | 0.26±0.12 |
| ABM1-Cp20 | 0.2±0.00 |
| ABM2-Cp20 | 0.17±0.11 |
| Cp40 | 0.14±0.05 |
| ABM2-Cp40 | 0.08±0.03 |
Complement inhibition assay based on initiation via the classical pathway.
To confirm the albumin-directed activity of the ABM–compstatin conjugates and further explore their simultaneous binding to C3 and albumin, we performed additional SPR binding studies. For this purpose, an ABM2-Cp20 derivative with C-ter-minal biotinylation was synthesized as a probe compound. In addition, biotinylated Cp40 (without an ABM2 tag) was used as control. The biotinylated peptides were immobilized on a strep-tavidin-coated SPR sensor chip, and the interactions with albumins from different species (human, baboon, bovine, rabbit, and mouse) were characterized (Table 3; Figure S2). Notably, given the narrow species specificity of compstatin for C3 from human and NHP, only the binding to human and baboon albumin is of direct importance for use in disease models. Also, limitations in the available concentration range of albumin only allows an estimation of affinity values. Nevertheless, the apparent binding affinity of ABM2-Cp20 to human serum albumin (KDapp = 97 μm; Table 3) was similar to the reported affinity of MS-325, which uses the same tag (KD = 164 μm).[26] The apparent affinities of albumin from other species to ABM2-Cp20 were in a similar range, with baboon albumin showing values nearly identical to those of the human form (Table 3). In contrast, none of the albumins bound significantly to the comp-statin control lacking the ABM2 tag (data not shown). These results demonstrate that conjugation to ABMs enables compsta-tin analogues to bind serum albumins; importantly, ABM2-Cp20 binds more tightly to C3 than to albumin (with affinities of ∼0.2 nM vs. ∼100 μm), thereby indicating minimal interference with the pharmacodynamic profile of the peptide.[18]
Table 3.
Evaluation of albumin interaction profiles to immobilized ABM2-Cp20.[a]
| Albumin Species | KD app [μm] |
|---|---|
| Human | 97±2 |
| Bovine | 208±5 |
| Mouse | 134±3 |
| Rabbit | 64±2 |
| Baboon | 106±3 |
The interactions of ABM2-Cp20 with albumins were characterized by multi-cycle analysis. Sets of five increasing concentrations of albumin from different species (6.3–100 μm) were injected over the chip surface. The processed signals were fit to a single binding site model.
To confirm the albumin binding mode of ABM2-Cp20, an SPR-based competition assay was performed. Fluorescent probe displacement studies showed that MS-325 binds primarily to site II on human serum albumin (HSA), as MS-325 can displace site II ligands rather than site I ligands (such as warfarin).[27] Ibuprofen, which binds to site II of human serum albumin (KD = 0.37 μm), was therefore chosen as a probe of site II ligands.[28,29] When HSA (50 μm) was injected to the ABM2-Cp20 sensor chip in the presence of ibuprofen, inhibition of albumin binding to the conjugate was observed (Figure S3), which suggests that ABM2-Cp20 indeed binds primarily to site II in a similar manner to MS-325.
Previous pharmacokinetic studies of Cp20 in NHP revealed a distinctive target-driven elimination profile, in which the strong binding to the abundant plasma protein C3 (∼ 1 mg mL−1) defines the slow terminal elimination of the compound; unbound peptide in excess of the plasma C3 level is excreted more rapidly[7] This strong influence of target binding on the elimination profile was further supported by the fact that the more potent Cp40 had a significantly longer half-life than Cp20 (12 vs. 9 h).[7] The ∼ 20-fold increased binding affinity of ABM2-Cp20 for C3 itself is therefore expected to contribute to an enhanced pharmacokinetic profile. In addition, however, the binding to albumin is considered important, as it would facilitate the maintenance of target-saturating inhibitor concentration. To assess the influence of and cooperation between C3 and albumin binding in the case of Cp20 and ABM2-Cp20, we performed in vitro plasma protein binding studies using rapid equilibrium dialysis (RED).[30] The free fractions of each peptide (percent free) were determined by mass spectrometry from the concentration ratios between the buffer and plasma side after 24 h of incubation at 37°C, and used to calculate the protein-bound fraction (percent bound). Two peptide concentrations (5 and 10 μm) were selected that represent inhibitor levels relevant for therapeutic complement inhibition.[7,9] When C3-depleted serum was used to assess plasma protein binding in the absence of target-mediated effects, conjugation of the ABM2 tag to Cp20 resulted in a profound increase in the bound fraction relative to the parent peptide (Table 4). Recon-stitution of the serum to a defined C3 concentration within the physiological range (5μm) leveled the difference of the two peptides to >99% bound at an equimolar target/inhibitor ratio, thereby clearly reflecting the strong influence of target binding. When 10μm peptide concentrations in excess of C3 were used, the influence of the ABM2 tag became more pronounced even in the C3-positive serum (Table 4). These studies clearly indicate that, when compared with its parent peptide, ABM2-Cp20 is more likely to reside in plasma independently of C3 due to its binding to albumin. The observed profile is expected to decrease the comparatively rapid elimination of excess peptide and facilitate the maintenance of target-saturating inhibitor levels. While the expected lower solubility of ABM2-Cp20 due to the hydrophobic ABM2 tag likely requires adjustment in the formulation for parenteral injection, it will be interesting to evaluate the pharmacokinetic profile in NHP in the future.
Table 4.
Evaluation of plasma protein binding profiles of compstatin conjugates.[a]
| Peptide (c[μm]) | Bound [%] | |
|---|---|---|
| C3-depleted serum | C3-positive serum[b] | |
| Cp20 (5) | 7±3 | >99 |
| ABM2-Cp20 (5) | >99 | >99 |
| Cp20 (10) | 3±1 | 84±1 |
| ABM2-Cp20 (10) | >99 | >99 |
Peptides were extracted from post-dialysis samples using solid phase extraction (SPE) and analyzed by reversed-phase ultra performance liquid chromatography coupled to high-definition mass spectrometry (UPLC-HDMS) as described in the Supporting Information.
A defined, physiological amount of purified C3 (5 μm) was added to C3-depleted serum.
In summary, we have developed a new series of compstatin derivatives with significant improvements regarding both potency and pharmacokinetic properties through the introduction of an albumin affinity tag. Such enhanced complement inhibitors appear particularly suitable for the systemic treatment of chronic complement-mediated diseases, as they may potentially allow sustained maintenance of therapeutic inhibitor levels at decreased dose intervals. Besides, the identification of a secondary/extended binding site for N-terminally tagged compstatin analogues is expected to facilitate the rational design of complement inhibitors with enhanced pharmacokinetic and pharmacodynamic profiles in the future.
Supplementary Material
Acknowledgments
We thank Dr. Georgios Paraskevopoulos and Dr. Hongchang Qu for their support in the synthesis and selection of albumin-binding molecules, and Dr. Melanie Loedige for critically reading the manuscript. This work was supported by National Institutes of Health grants AI030040, AI068730, AI097805, EY020633, GM097747, and DE021685, and by funding from the European Community's Seventh Framework Programme under grant agreement number 602699 (DIREKT).
Footnotes
These authors shared supervision of this study.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.201402212.
References
- 1.Ricklin D, Lambris JD. J Immunol. 2013;190:3831–3838. doi: 10.4049/jimmunol.1203487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sahu A, Kay BK, Lambris JD. J Immunol. 1996;157:884–891. [PubMed] [Google Scholar]
- 3.Ricklin D, Lambris JD. Adv Exp Med Biol. 2008;632:273–292. doi: 10.1007/978-0-387-78952-1_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kourtzelis I, Markiewski MM, Doumas M, Rafail S, Kambas K, Mitroulis I, Panagoutsos S, Passadakis P, Vargemezis V, Magotti P, Qu H, Mollnes TE, Ritis K, Lambris JD. Blood. 2010;116:631–639. doi: 10.1182/blood-2010-01-264051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silasi-Mansat R, Zhu H, Popescu NI, Peer G, Sfyroera G, Magotti P, Ivanciu L, Lupu C, Mollnes TE, Taylor FB, Kinasewitz G, Lambris JD, Lupu F. Blood. 2010;116:1002–1010. doi: 10.1182/blood-2010-02-269746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kourtzelis I, Rafail S, DeAngelis RA, Foukas PG, Ricklin D, Lambris JD. FASEB J. 2013;27:2768–2776. doi: 10.1096/fj.12-225888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qu H, Ricklin D, Bai H, Chen H, Reis ES, Maciejewski M, Tzekou A, DeAngelis RA, Resuello RRG, Lupu F, Barlow PN, Lambris JD. Immunobiology. 2013;218:496–505. doi: 10.1016/j.imbio.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qu H, Magotti P, Ricklin D, Wu EL, Kourtzelis I, Wu YQ, Kaznessis YN, Lambris JD. Mol Immunol. 2011;48:481–489. doi: 10.1016/j.molimm.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Risitano AM, Ricklin D, Huang Y, Reis ES, Chen H, Ricci P, Lin Z, Pascariello C, Raia M, Sica M, Del Vecchio L, Pane F, Lupu F, Notaro R, Resuello RRG, DeAngelis RA, Lambris JD. Blood. 2014;123:2094–2101. doi: 10.1182/blood-2013-11-536573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maekawa T, Abe T, Hajishengallis E, Hosur KB, DeAngelis RA, Ricklin D, Lambris JD, Hajishengallis G. J Immunol. 2014;192:6020–6027. doi: 10.4049/jimmunol.1400569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ricklin D, Lambris JD. J Immunol. 2013;190:3839–3847. doi: 10.4049/jimmunol.1203200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pollaro L, Heinis C. MedChemComm. 2010;1:319. [Google Scholar]
- 13.Kratz F. J Controlled Release. 2008;132:171–183. doi: 10.1016/j.jconrel.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 14.Gao ZH, Bai G, Chen JQ, Zhang Q, Pan PW, Bai F, Geng P. Biosci Biotechnol Biochem. 2009;73:688–694. doi: 10.1271/bbb.80742. [DOI] [PubMed] [Google Scholar]
- 15.Langenheim JF, Chen WY. J Endocrinol. 2009;203:375–387. doi: 10.1677/JOE-09-0211. [DOI] [PubMed] [Google Scholar]
- 16.Dennis MS, Zhang M, Meng YG, Kadkhodayan M, Kirchhofer D, Combs D, Damico LA. J Biol Chem. 2002;277:35035–35043. doi: 10.1074/jbc.M205854200. [DOI] [PubMed] [Google Scholar]
- 17.Koehler MFT, Zobel K, Beresini MH, Caris LD, Combs D, Paasch BD, Lazarus RA. Bioorg Med Chem Lett. 2002;12:2883–2886. doi: 10.1016/s0960-894x(02)00610-8. [DOI] [PubMed] [Google Scholar]
- 18.Zobel K, Koehler MFT, Beresini MH, Caris LD, Combs D. Bioorg Med Chem Lett. 2003;13:1513–1515. doi: 10.1016/s0960-894x(03)00209-9. [DOI] [PubMed] [Google Scholar]
- 19.Dumelin CE, Trüssel S, Buller F, Trachsel E, Bootz F, Zhang Y, Mannocci L, Beck SC, Drumea-Mirancea M, Seeliger MW, Baltes C, Müggler T, Kranz F, Rudin M, Melkko S, Scheuermann J, Neri D. Angew Chem Int Ed. 2008;47:3196–3201. doi: 10.1002/anie.200704936. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2008;120:3240–3245. [Google Scholar]
- 20.Qu H, Magotti P, Ricklin D, Lambris JD. In: Proceedings of the Twenty-First American Peptide Symposium. Lebl M, editor. Prompt Scientific; Bloomington: 2009. pp. 219–220. [Google Scholar]
- 21.Lauffer RB, Parmelee DJ, Dunham SU, Ouellet HS, Dolan RP, Witte S, McMurry TJ, Walovitch RC. Radiology. 1998;207:529–538. doi: 10.1148/radiology.207.2.9577506. [DOI] [PubMed] [Google Scholar]
- 22.Janssen BJC, Halff EF, Lambris JD, Gros P. J Biol Chem. 2007;282:29241–29247. doi: 10.1074/jbc.M704587200. [DOI] [PubMed] [Google Scholar]
- 23.Magotti P, Ricklin D, Qu H, Wu YQ, Kaznessis YN, Lambris JD. J Mol Recognit. 2009;22:495–505. doi: 10.1002/jmr.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karlsson R, Katsamba PS, Nordin H, Pol E, Myszka DG. Anal Biochem. 2006;349:136–147. doi: 10.1016/j.ab.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 25.Katragadda M, Magotti P, Sfyroera G, Lambris JD. J Med Chem. 2006;49:4616–4622. doi: 10.1021/jm0603419. [DOI] [PubMed] [Google Scholar]
- 26.Muller RN, Radüchel B, Laurent S, Platzek J, Piérart C, Mareski P, Vander Elst L. Eur J Inorg Chem. 1999:1949–1955. [Google Scholar]
- 27.Caravan P, Cloutier NJ, Greenfield MT, McDermid SA, Dunham SU, Bulte JWM, Amedio JC, Looby RJ, Supkowski RM, Horrocks WD, McMurry TJ, Lauffer RB. J Am Chem Soc. 2002;124:3152–3162. doi: 10.1021/ja017168k. [DOI] [PubMed] [Google Scholar]
- 28.Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. J Mol Biol. 2005;353:38–52. doi: 10.1016/j.jmb.2005.07.075. [DOI] [PubMed] [Google Scholar]
- 29.Aarons L, Grennan DM, Siddiqui M. Eur J Clin Pharmacol. 1983;25:815–818. doi: 10.1007/BF00542526. [DOI] [PubMed] [Google Scholar]
- 30.Waters NJ, Jones R, Williams G, Sohal B. J Pharm Sci. 2008;97:4586–4595. doi: 10.1002/jps.21317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.