

Abstract
The bacterial leucine transporter LeuT retains significant secondary structure similarities to the human monoamine transporters (MAT) such as the dopamine and serotonin reuptake proteins. The primary method of computational study of the MATs has been through the use of the crystallized LeuT structure. Different conformations of LeuT can give insight into mechanistic details of the MAT family. A conformational sampling performed through accelerated molecular dynamics (aMD) simulations testing different combinations of the leucine substrate and bound sodium ions revealed seven distinct conformational clusters. Further analysis has been performed to target salt-bridge residues R30–D404, Y108–F253, and R5–D369 and transmembrane domains on both the seven isolated structures and the total trajectories. In addition, solvent accessibility of LeuT and its substrate binding pockets has been analyzed using a program for calculating channel radii. Occupation of the Na2 site stabilizes the outward conformation and should bind to the open outward conformation before the leucine and Na1 sodium while two possible pathways were found to be available for intracellular transport.
Keywords: monoamine transporter, molecular dynamics, principal component analysis, HOLE, APC, FIRL
Introduction
LeuT is a bacterial transmembrane protein which functions to transport leucine across the cellular membrane and into the cell. LeuT is one of many sodium symporters which utilize a concentration gradient of sodium to help facilitate the uptake of leucine by simultaneously transporting two sodium ions along with the leucine (or symporter specific) substrate1. The monoamine transporters—e.g. dopamine, serotonin, and norepinephrine—are homologues of LeuT and are thought to have similar transport mechanisms. Even though several x-ray structures of LeuT in different conformational states are available, a complete dynamical figure has not been determined2–8.
The primary focus of LeuT experimental and computational studies has been to elucidate information relative to the monoamine transporters (MATs) as crystal structures of these proteins did not exist until recently9. MATs are of particular interest in pharmacological sciences as they include the eukaryotic reuptake transporters for dopamine (DAT), serotonin (SERT), and norepinephrine (NET) which function to remove neurotransmitter molecules from the synaptic cleft to terminate the intercellular signaling process10. Certain disease states including but not limited to addiction, Parkinson's, major depressive disorder, and anxiety have been linked to dysregulation of one or more MATs10–12. MATs are also targets for medications such as the SNRI's (serotonin–norepinephrine reuptake inhibitors) and SSRI's (selective serotonin reuptake inhibitors)13;14.
It is believed that targets for new drug therapies could be found by finding different conformational states and by understanding the macromolecular movements involved in the transport mechanism. There are two competing mechanisms to describe the transport of a solute—the alternating access model and the rocking bundle1;15. The alternating access model refers to a common conception of transport proteins: open outward, substrate binds, occluded, open inward, substrate translocation occurs1. The rocking bundle mechanism assumes that bundle 1 consisting of TM1 through TM5 “rocks” and takes the same conformation as bundle 2 consisting of TM6 through TM10 while bundle 2 takes the same conformation as bundle 115. In other words, bundle 1 and bundle 2 flip conformations, and in the process, a channel through which the substrate and potentially solvent should form.
The basis for the rocking bundle mechanism lies in the fact that the TM1 through TM5 bundle shares an inverted symmetry with the TM6 through TM10 bundle1;15. This inverted repeat has been seen in many other sodium symporters and is named the five transmembranehelix inverted–topology repeat LeuT–like (FIRL) fold which is now considered the hallmark feature of the solute carrier 6 (SLC6) subfamily of the APC transporter superfamily as well as many proteins in the rest of the superfamily16–18. Examples of transporters involved in other biological processes which share the FIRL structure as well as mechanistic dynamics with LeuT include: the sodium/galactose symporter vSGLT19; the betaine transporter BetP20; the bacterial proton pump AdiC21; the sodium–benzylhydantoin transporter Mhp122; the GABA transporters GAT-1 and GAT-223;24; and the monoamine transporters DAT, NET, and SERT2;25.
The vSGLT and BetP proteins seem to function not only as solute transporters but also in an osmotic fashion by allowing permeation of solvent molecules19;20. If the rocking bundle model holds true, then LeuT would cotransport water in this osmotic fashion; however it currently appears that LeuT does not function to cotransport the solvent. Studying LeuT could show what the selectivity is between the osmotic functioning structures and non– osmotic functioning proteins.
To further understand the MATs transport mechanism, as well as learn other processes of the APC superfamily, we took a thermally equilibrated structure of LeuT embedded in a solvated and ionized membrane system and generated seven simulations each run under accelerated molecular dynamics (aMD) for a minimum of 250 ns4. The simulations were created by modifying the presence of the three bound substrates of the LeuT structure (one leucine and two sodium ions). The initial results analyzed through PCA were presented in a previous publication and seven unique structures were found with only three of the seven structures corresponding to already published data4. Here, we present an in depth analysis of the four isolated structures, channel analysis through the binding site, and other analyses throughout the trajectories.
Materials and Methods
Starting Structure Generation
The original LeuT occluded crystal structure from 2005 (RCSB code 2A65)1 was used as the base structure. Missing residues and side chains were added using psfgen from the NAMD suite of molecular dynamics tools26. A POPE membrane was generated around the completed LeuT structure, and a physiological system was generated around the membrane and protein using the membrane, solvate, and autoionize plugins available in VMD27. The final system contained 9,527 TIP3 water molecules with a concentration of 0.2 M NaCl surrounding the protein with bound leucine substrate and two bound Na+ embedded in a 125 lipid bilayer for a total of 52,495 atoms. This system underwent classic molecular dynamics thermal equilibration for 30 ns using NAMD26. For more information regarding this generation or minimization please see the original articles3;4.
Accelerated Molecular Dynamics
The 30 ns thermally equilibrated membrane and 2A65 system was used to generate seven systems for simulation by removing the bound substrates. The seven simulations are summarized in Table I. The seven simulations underwent 5 ns of equilibration using standard molecular dynamics in AMBER928 to prevent induction of false conformations from both changing the system as well as slight differences between the CHARMM and AMBER force-fields which could have been magnified by immediate acceleration. The 5 ns equilibration was followed by 250 ns of simulation with aMD being performed on each system28;29. The acceleration was applied solely to the atoms of the protein by the addition of a dihedral boost bias potential29 while the rest of the system underwent standard MD. Finally, an analysis of the weight of each generated structure was performed by reweighting the results of the simulations. The reweighting of the simulation structures was a necessary analysis because the increase in simulation speed could have skewed the sampling distribution of conformational states which would not normally have occurred during standard thermodynamic conditions. The reweighting was a process by which a score was calculated to determine how much the structure contributes to the overall dynamics and attempts to correct this bias4;29. For more details about the simulation generation, aMD theory, aMD methods utilized, or the reweighting results and methodology, refer to the supplementary information in the 2012 article by Thomas et al.4.
Table I.
Summary showing the modifications to the bound substrates in each simulation from the original publication4. The + indicates the presence of that bound ligand while the - indicates the absence of the substrate. The convention for sodium binding pocket names conforms to the original Gouaux model1;40. The column for color corresponds to the simulation's color in Supplementary Figure S1 as well as most of the graphs in this publication.
| Simulation | Leu | Na1 | Na2 | Color | Time (ns) |
|---|---|---|---|---|---|
| A | + | + | + | red | 250 |
| B | + | + | − | orange | 280 |
| C | − | + | + | yellow | 250 |
| D | − | − | + | green | 250 |
| E | − | + | − | blue | 300 |
| F | + | − | + | sky blue | 250 |
| G | − | − | − | purple | 250 |
During the initial stages of analysis, we decided to extend the simulations of systems B and E beyond the 250 ns reported in the original publication4 because the trajectories appeared to be approaching a new conformational cluster in the PCA. After the extended simulation time, the structures settled into the same PCA clusters as the original frames of their respective simulations. RMSD to the crystallized inward structure 3TT3 in Supplementary Figure S5 also shows the systems were near equilibrium4;30. The simulations B and E extended lengths are reported in Table I and are used in all analysis in this publication.
Principal Component Analysis
Principal Component Analysis (PCA) was performed as an analytical method of clustering the simulation data as well as to discriminate between unique conformational structures. The Bio3D package was used for its powerful protein and simulation handling functions such as PCA calculation, atom selection, structural alignment, and homology analysis31;32. In our original publication, PCA on all of the published LeuT crystal structures available from the RCSB protein data bank at the time the analysis was performed4. The PCA revealed that the greatest discrimination between unique structures occurred when the PCA was performed on the Cα coordinates for the residues in TMs 1b and 6a resulting in three conformational clusters4;15;33;34. This TM1b-TM6a combination was then used for the PCA on all of the simulation trajectories combined. The seven structures determined in the PCA and reported earlier were subjected to further analysis and are discussed below4;35. For further details of the results of the x-ray structure PCA as well as comparisons between the x-ray structures and the trajectories, please refer to the Thomas et al. 2012 article4.
Channel and Path Analysis
The presence of an open accessible water channel or wire was tested using the program HOLE36. HOLE is a program designed to calculate the radius of the channel as well as visualize the channel through a protein with a channel such as aquaporin or an ion channel. HOLE functions by randomly moving from point to point relatively along an axis and calculating the distance from that point to the closest clash with an atom's van der Waals radius36. Since it does check for bad contacts while randomly selecting a path, it was hypothesized that it may be possible to have HOLE calculate a path through a membrane transporter as opposed to a channel protein. The use of HOLE for transmembrane transporters has previously been reported37;38. HOLE was run on each step of the combined trajectory taken at every 200 ps with Leu substrates removed if present in a frame. The axis of sampling was selected to be the axis along which the central core of LeuT resided after aligning all simulations to their respective first frames by using the Cα coordinates of the TM domains as the fitting parameter. A single point was selected which HOLE must sample during its calculation along the axis36. The point was defined as the geometric center of F253, Y108, S256, and A22 which should correspond to a position in the Leu substrate primary binding pocket1. The alignment of the trajectories to their first frame was performed both for visualization purposes as well as to make sure that the axis for the channel axis remained constant. The first frame of all simulations had the LeuT central structure along the z-axis which made it an ideal fitting parameter since the protein and membrane could have had displacements due to the periodic boundary conditions.
Results and Discussion
The seven simulations produced not only the seven static isolated structures4 but also an analysis of their dynamic behavior. While transport of either the leucine substrate or the sodium ions was not observed, the results of the analysis reveal a transition to an outward conformation as well as a transitioning path toward an open inward conformation even though a fully open inward structure was not sampled4.
In the original article, PCA was utilized because the RMSD (root mean square deviation) values of the published crystal structures in the RCSB protein bank at the time of analysis1;6;39–43 had very close RMSD values, and it was hypothesized that the PCA would be a better discriminator for unique structural changes over the standard convention of RMSD4;35;44;45. Performing residue by residue RMSD calculations for LeuT over all of the trajectories revealed that the reason for the lowered utility of RMSD is that the core regions of the transmembrane domains move very little throughout the simulations and that the primary source of fluctuation is in the extracellular loop regions as seen in Figure 1. It appears that the core structure is largely unchanged and that most changes in structure between conformations occurs at the extreme ends of the TM domains. This most directly conflicts with the theory of an entire TM1-5 and TM6-10 symmetrical conformation “rocks” which is known as the “rocking bundle” 15.
Figure 1.
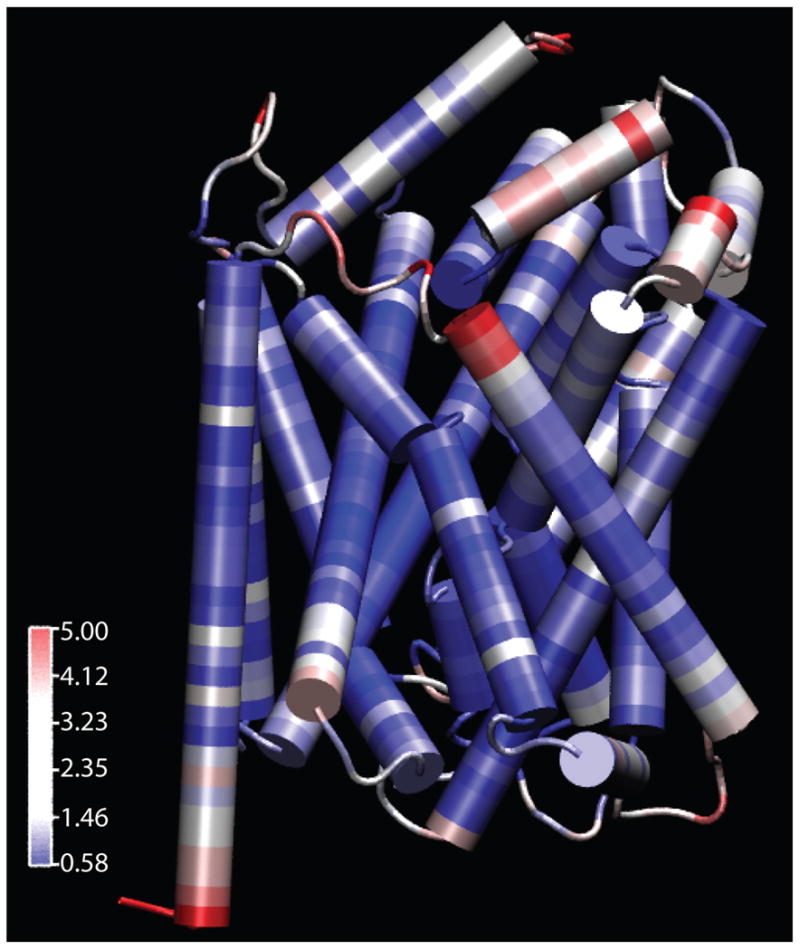
LeuT colored by RMSD of the individual residues throughout all seven simulations. Blue represents the least movement while red represents the most variations. The reference structure is the thermally equilibrated 2A65 with the 2 bound sodium ions and leucine substrate present1.
The use of the PCA revealed a second utility that was not necessarily anticipated. The PCA result showed not only a split between the absence and presence of Na2 binding4 but also happened to illustrate a possible timeline of each simulation through transport. An updated PCA is found in Supplementary Figure S1, and the mechanistic timeline of conformational changes is discussed later after combining the analysis with RMSDs of the seven isolated clusters4, the PCA, and channel analysis from HOLE36.
Analysis of Select Distances and Key Residue Interactions
Specific residues were selected for analysis based upon hypothesized implications of functions in other publications especially residues proposed to be involved in the extracellular and intracellular gating mechanisms1;6;40;41. The residues were analyzed for their interactions and relative positions by measuring the distances between them. All of the residues addressed in this section are matched to their original presenting articles in Table II and are visually represented in Figure 2 with lines drawn between the residues to highlight the specific distances.
Table II.
Table of analyzed residues and their proposed interaction functions to the structure of LeuT listed along with the original publication that proposed the interaction and/or function.
| Residue(s) | Proposed Function | Original Reference |
|---|---|---|
| R30–D404 | Extracellular Salt Bridge Gate | 1 |
| Y108–F253 | Extracellular Aromatic Gate | 1 |
| R5–D369 | Intracellular Salt Bridge Gate | 55 |
| N21–S256 | Hydrogen Bond Interaction between TM1 and TM6 | 56 |
| D401–A319 | Distance to changes to close access to extracellular vestibule. Possible hydrogen bond. | 30 |
| R5–Y268 | Intracellular cation–π interaction to stabilize a growing distance between R5–D369. | 5;30 |
Figure 2.

The highlighted residues in the distance graphs. A) Intracellular salt bridge R5–D369. B) Intracellular Interaction N21–S256. C) Extracellular Vestibule TM10–EL4 A319–D401 interaction. D) Extracellular gate system: the Y108–F253 aromatic gate and the R30–D404 salt bridge.
The Extracellular Gates of R30–D404 and Y108–F253
The R30–D404 ionic salt bridge has been implicated in substrate binding and occlusion of the primary substrate binding pocket in many studies and is generally considered a key component of profiling the S1 (primary substrate binding site) vs S2 (proposed secondary binding site) binding pockets1;6;40;41. Figure 3 shows the behavior of the R30–D404 salt bridge for the different simulations. It has been proposed that the R30 and D404 residues have two functional conformations once sodium has been bound—an open conformation which could serve as a secondary binding site and/or ionic attractant for the leucine substrate as well as a closed conformation to lock in the ligand46.
Figure 3.

Distances observed in the R30–D404 extracellular salt gate1;5;41 over time for each simulation. The averages plot is the average after 150 ns in order to prevent sampling error from early post minimization dynamics.
Our data suggests that there are two primary distances between R30–D404 leading to an open conformation with interresidue distance around 7 Å. The second is an occluded conformation with distance around 4 Å. As expected, the simulation of apo-LeuT results with a fully open conformation to facilitate the binding of substrate and sodiums. However, it appears that the presence of a sodium in the Na1 site alone shifts the conformation to close the distance but fails to maintain the closed salt bridge. In all simulations with the Na2 site occupied —except the simulation with Leu and both sodiums (Simulation A)—this ionic gate is mostly stabilized in the closed conformation.
This suggests that the first substrate to bind to LeuT is a sodium ion to the Na1 site followed by a sodium to the Na2 site and ending with LeuT closing after binding a Leu, but the bound substrate simulation analysis (Simulation A) reveals that, in this simulation, the gate is destabilized from the closed conformation and frequently adopts an open conformation. Regardless of the opening of the R30–D404 gate, the substrate did not diffuse out of the binding pocket and the rest of the protein remained in the occluded conformation (see Supplementary Figure S1)4. This behavior is only noticed in the simulation with leucine and both sodiums and not in the simulations with leucine and only one sodium suggesting that the R30 and D404 residues function as substrate attractants46 before initial Leu binding and could also serve the role a second time before transport for a second substrate in a secondary substrate binding site known as S26;40;41;47.
In contrast, the Y108–F253 aromatic gate which rests below the R30–D404 gate1;41 remains largely unchanged in most simulations as seen in Figure 4. The gate remains stable and closed around 6 Å, but in the simulation of the apo-LeuT structure, an open conformation is achieved and maintained throughout the simulation with about 9 Å between the Y108 and F253 residues measured from the center of their respective aromatic rings. In addition, toward the end of the simulation with both Na1 and Na2 sites occupied (Simulation C), the Y108–F253 gate opens. This suggests that the binding of the sodiums appears to close the aromatic extracellular gate like it does with the R30–D404 gate.
Figure 4.

Distances observed in the extracellular aromatic interaction between Y108 and F253. This is proposed to be an aromatic extracellular gate mechanism paired with the R30–D404 gate1;30;41. The averages plot is the average after 150 ns in order to prevent sampling error.
The distances of the residues involved in both “gating mechanisms” with any combination of bound sodiums starting from an open conformation were not considered. This stable closed gate result of both R30–D404 and Y108–F253 in the presence of the bound sodium ions may in fact be an artifact of starting all simulations from an occluded LeuT conformation with both gates starting in closed positions1.
The Extracellular Vestibule TM10–EL4 Interaction
The residue A319 of extracellular loop 4 (EL4) and D401 of TM10 have been proposed to interact or at least move toward each other in the occluded conformation in order to occlude the extracellular vestibule30. As seen in Supplementary Figure S3, no significant interaction or even closer distance was observed. The largest fluctuation that occurred in the distance between the carbon in the carboxylic acid group of D401 and the Cα of A319 was an increase of approximately 2 Å from the occluded conformation in Simulation G (apo-LeuT) as well as Simulation F (Leu bound and Na2 occupied). This data suggests that an open conformation has a greater distance between TM10 and EL4 which agrees that TM10 and EL4 move slightly closer during an occluded and closed conformation30. However, a major coordinated and functional movement has not been observed.
The Intracellular Gate of R5–D369
Arginine 5 and Aspartate 369 make a salt bridge interaction in LeuT between TM1a and TM8 which has been proposed to be an intracellular gating mechanism for substrate translocation5;30;48. The interaction distance between these two residues was analyzed over the course of the trajectories and plotted in Figure 5. The results show that this intracellular interaction remains stable with little increase in the distance between the residues in the simulation with all bound substrates.
Figure 5.

Graphs depicting the distances in the proposed intracellular ionic gate of R5–D3695;30. The averages plot is the average after 150 ns.
In fact, the largest distances between R5–D369 were observed at an average of 7 Å in the simulations with Na2 occupied but without the leucine substrate (Simulations C and D). It should also be mentioned that a large fluctuation occurs between this resting 7 Å and a larger than 12 Å distance in Simulation C. Overall, this result suggests that binding of the Leu substrate actually stabilizes a closer intracellular distance between TM1a and TM8. According to other hypotheses, this R5–D369 intracellular salt bridge, if it acted as a gate5;30;48, should open upon binding of the substrate and sodiums, but the opposite occurs. With the leucine substrate present, the salt bridge is more stabilized in closed conformations while only occasionally sampling the larger distances indicative of an open configuration. The systems with the smallest average distances were simulations A and B which both have a bound leucine. While simulation A which also has both sodium sites occupied appears to sample a distance of 14 Å, the data suggests that having two sodiums and a leucine substrate bound stabilizes the closed R5–D369 gate more than any other of the simulations because that large 14 Å distance is only sampled for a few sporadic points while simulation B has a prolonged time of a continuous 12 Å distance sampled for over 20 ns and has repeated instances of sampling the larger distances throughout the rest of the simulation.
The analysis of these residues over the simulations suggests that the R5–D369 interaction may not actually be a translocation gate but could be indicative of another non-gating role in transport such as substrate electrostatic attraction. This hypothesis can be supported with the idea that there could exist a different exit pathway for the Leu substrate through the transporter7. The article by Merchant et al. presents two main paths of transport of the Leu substrate through the intracellular region of the transporter–one path along a TM1a and TM6b path, and another along TM6b and TM87. The proposed TM6b–TM8 pathway may be more plausible than the TM1a–TM6b given the stability of the closed TM 1a–TM8 interaction in our simulations.
Channel Analysis
The analysis with HOLE proved to be a reliable tool for the visualization of a channel through even a transmembrane transporter36–38. In the resultant channels from HOLE (refer to Figure 6), a fully open channel was not observed which opposes the “rocking bundle” hypothesis for a full TM1–TM5 and TM6–TM10 symmetrical exchange15. The idea of a coordinated flip in with the inverted repeat symmetries of both bundles take switch conformations should cause a small channel to form. Symmetrical changes between the bundles may still occur as can be seen between TM1b and 6a4;34, but the lack of a channel suggests that the coordinated actions necessary for the bundle rocking do not occur. If water permeation were to occur, it would need to take a water wire approach37 since a straight channel had not formed even though an opening inward conformation was sampled4. The water wire method of transport has been observed in other related membrane transport families containing Mhp1 and vSGLT49.
Figure 6.
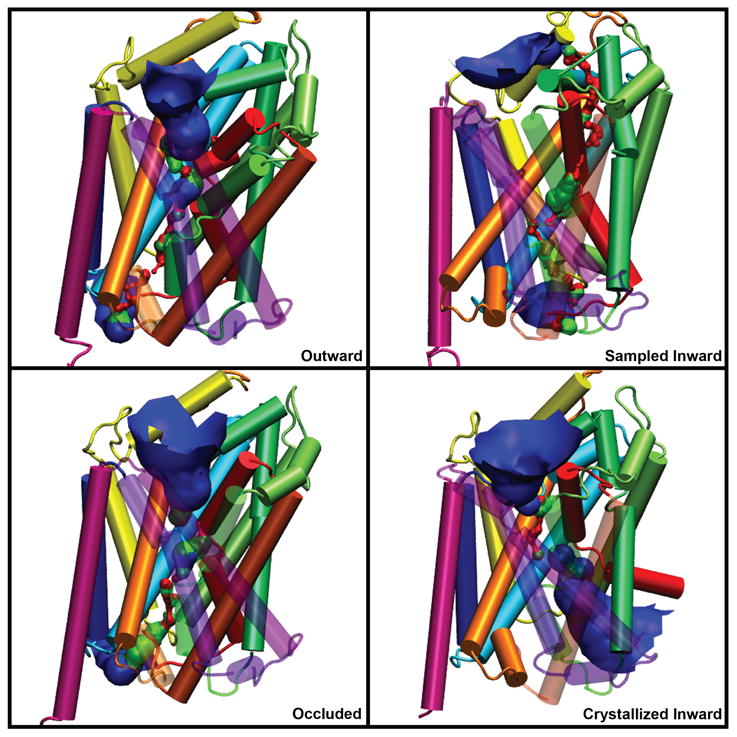
HOLE36 results as seen in the three conceptions of transport — open outward, occluded, and opening inward. The 2012 crystallized open inward structure (pdb code: 3TT3)30 is shown in the lower right protein with a calculated HOLE channel. The channels are represented as an isosurface and colored according to radius: blue is > 2.3Å, green is > 1.2Å, and red is < 1.2Å(inaccessible to water).
The channels from HOLE revealed a nearly constant path between the substrate binding pocket and the extracellular vestibule which is a similar result for a substrate exit path found from a random accelerated molecular dynamics (RaMD) publication on both LeuT and DAT7. The RaMD on LeuT and DAT also revealed that there were two possible substrate translocation paths between the substrate binding pocket and intracellular transport7. After HOLE was used to calculate the channel in each frame of the LeuT simulations, three pathways were primarily sampled, and two of the pathways were the TM1a–TM6b and the TM6b–TM8 paths sampled in the RaMD article7. When the frequency of sampling of each path was taken, the TM6b–TM8 path was sampled over 55% of the time as seen in Figure 7. Most studies propose transport down along TM1a which corresponds to the TM 1a– TM6b path5;30;50. Following the sampling frequencies in both this analysis and the RaMD paper7, it appears that the TM6b–TM8 path is preferred from the occluded position. The TM6b–TM8 preference is supported by the data from Figure 5 which compared the Arg5 and Asp369 interaction and found that the salt bridge interaction is more stable in the presence of the leucine substrate. It is unknown whether the existence of two possible paths represents options for simultaneous transport or perhaps one path is for a sodium transport and the other is for the substrate, such as the single sodium cotransport seen in the RaMD simulations7. Regardless of the implications of the two possible paths, the data demonstrates that the TM6b–TM8 path is initially preferred from the occluded conformation.
Figure 7.

Frequencies of sampling of the three primary channels and/or substrate paths calculated through HOLE36. Each graph is labeled according to the simulation from which it came. Due to nature of this analysis, only the channels for the three most diverging simulations— the ones that sampled outward, occluded, and opening inward characteristics—were utilized.
Proposed Transport Cycle
Combining structural similarities and differences from the PCA, RMSD, and HOLE36, the seven structures isolated in the original publication4 were organized into a proposed timeline during a transport cycle of LeuT. The timeline is represented in Figure 8, and the coordinate files are available online as supplementary information. It was possible to generate a linear arrangement of transitions between the first five structures from a sampled outward conformation through two transition steps beyond the standard occluded structure.
Figure 8.
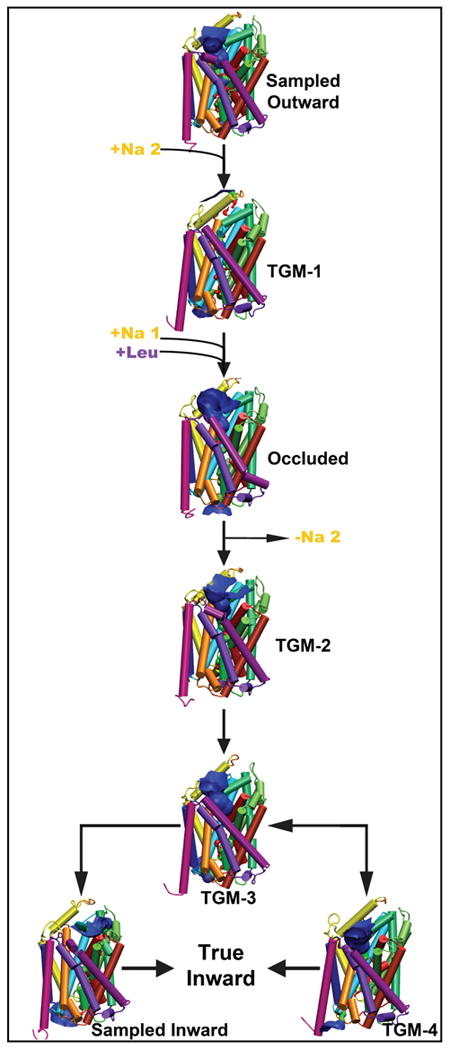
The seven sampled conformational structures isolated from the Principal Component Analysis4 arranged into a proposed timeline of involvement in the transport cycle. Each structure is represented in cartoon colored according to the convention from the original LeuT crystal paper1 and with a channel from HOLE36.
Given the data from HOLE and the distance graphs from before, it appears that the outward structure will transition to TGM-1 upon the binding of a sodium ion to the Na2 pocket of LeuT. During the change to TGM-1, there is a push of TM7 away from the core of the protein and out toward the membrane which is facilitated with a similar translation of TM5 out into the membrane. TM6a shifts in toward the binding pocket closing a small occupancy of the extracellular vestibule while TM1b remains in a more open position. Interestingly, EL2 and EL4 appear to play a role in the occlusion of the extracellular vestibule and/or generation of a tight secondary substrate binding pocket (S2)1;6;30;;33;51. Both EL2 and EL4 have a small translation toward the vestibule to slightly occlude some extracellular access.
TGM-1 appears to switch to the standard occluded structure upon the binding of the Leucine substrate to the S1 site and a sodium ion to the Na1 site, but it is currently unclear if the sodium ion binds to Na1 before the leucine binds to S1 or if the sodium ion binds to Na1 after being coordinated to the leucine substrate. EL2 continues to move a little more toward the vestibule, but EL4 appears to take a slight shift away from the vestibule to conserve the extracellular volume. In addition, both TM1b and TM6a adopt a conformation closer to the vestibule to occlude access to the substrate binding pocket. This is in agreement with the proposed mechanisms of movement for TM1b and 6a, however the movement is not a symmetrically conserved and coordinated translation1;15;30.
TGM-2 was the first transition step from the occluded structure toward the inward conformations. TM6a angles out toward TM11 while the angle between EL4a and EL4b increases a small amount. These two movements appear to open some access of the extracellular vestibule and possibly help shape the S2 pocket. TM4 and TM8 push away from the core of the protein and out into the membrane space to facilitate volume and occupancy for future internal protein conformational movements.
TGM-3 was found to occur after TGM-2 and is considered to be a transition toward the inward facing model. Both TM1b and TM6a move closer relative to each other and in toward the core of the structure occupying some of the space of the extracellular vestibule. TM10 adopts an angular rotation where its intracellular portion opens and the extracellular portion moves in toward the core of the protein. IL1 moves in and occupies part of a developing pocket between TM8 and TM2. In addition, TM1a and TM6b swing slightly outward from the core of the structure to facilitate solvent access30.
The remaining two structures each appeared to be transitions close to TGM-3. At the current time, we propose that TGM-3 transitions to our sampled opening inward conformation directly. This conformational change occurs with many transmembrane domain movements. Both TM1b and TM6a continue to move toward the core of the protein and close more of the extracellular vestibule. The intracellular portion of TM3 with IL1 held close moves outward increasing the size of the pocket between TM8 and TM2. In accordance with the theory of two substrate translocation or solvent access paths7, TM1a and TM6b move outward along with a widening of this pocket caused by an outward movement of the intracellular side of TM7.
The structure TGM-4 appeared to be a different conformation which was closest to TGM-3 but was not related enough to the sampled opening inward structure. TGM-4 could represent an alternate conformation/method of transport, a transition state from the true inward structure switching back toward an open outward conformation, or an induced conformation by having only the Na1 site occupied. For the transition from TGM-3 to TGM-4, there is a coordinated translation of TM1b and TM6a as they angle toward TM11, and TM10's extracellular portion closes in toward the core structure. Interestingly, there is a movement of TM5 and TM7 toward the core which is the only time a reversal of their pushing into the membrane between the outward conformation and TGM-1 was observed.
TGM-4 is most likely an induced conformation since all inward-like structures were isolated in simulations with the Na2 site unoccupied and the RaMD simulations published in 2012 had Na1 usually co-transport with the leucine substrate7. Even though this conformational structure's place in transport or existence is ambiguous, it gives insight into the timeline because it exists only in the presence of a sodium ion to Na1. It has become recognized that the binding of a sodium ion to the Na2 site is capable of stabilizing the open outward conformation and that dissociation from the Na2 site can contribute to the transition to the open inward conformation48;52;53. Therefore, we propose that the sodium ion in Na2 should be transported intracellularly first followed by the leucine substrate and the sodium in Na1 together. This two step transport may be facilitated by the two path intracellular vestibule model. The Na2 sodium ion could be transported along one of the intracellular paths while the coordinated substrates could be transported along the other path.
Conclusion
The LeuT structure, a homologous structure to the monoamine transporters and possibly a foundational template for the study of many more classes of transporters, retains mysteries even after its many years of study since its crystallization in 20051. After a cumulative 1.83 μs of accelerated dynamics simulations on LeuT in a membrane system, many insights were found including a proposed order of substrate binding, macro shifts in transmembrane domains during transitions from outward toward an opening inward conformation, and select gating residues involved in transporter conformation. Specifically, the results of this analysis suggest a two pathway intracellular vestibule model as well as a timeline of transport— a sodium binds to Na2, a Leucine binds to S1 with a sodium to Na1, then Na2 is transported, and finally Na1 and Leu are released into the cytoplasm. The fact that substrate transport did not occur over the course of the simulation only reaffirms the principle that there exists some remaining energy barrier. This barrier could be due to the lack of a proper sodium gradient on either side of the membrane due to periodic boundary conditions, the binding of a second substrate to the S2 site6;40;41;;47;54, or even some other energy factor not yet considered. Further studies on these potential barriers to transport are currently underway and could provide information not only for the monoamine transporter families but also for general understandings of what is currently thought of as passive symporter systems.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health, National Science Foundation, Department of Defense, and The U.S. Department of Education under awards 5R01DA27806-2, CHE-1005145(REU/ASSURE), CHE-0723109(MRI), and P116Z080180. We would like to thank Barry Grant for aid with PCA use in Bio3D and analysis, and also Eliana Asciutto Asciutto for her aid in analysis of the trajectories.
References
- 1.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−dependent neurotransmitter transporters. Nature. 2005;437:215–23. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 2.Indarte M, Madura JD, Surratt CK. Dopamine transporter comparative molecular modeling and binding site prediction using the LeuT(Aa) leucine transporter as a template. Proteins. 2008;70:1033–46. doi: 10.1002/prot.21598. [DOI] [PubMed] [Google Scholar]
- 3.Gedeon PC, Indarte M, Surratt CK, Madura JD. Molecular dynamics of leucine and dopamine transporter proteins in a model cell membrane lipid bilayer. Proteins. 2010;78:797–811. doi: 10.1002/prot.22601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas JR, Gedeon PC, Grant BJ, Madura JD. LeuT Conformational Sampling Utilizing Accelerated Molecular Dynamics and Principal Component Analysis. Biophysical journal. 2012;103:L1–3. doi: 10.1016/j.bpj.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao Y, Terry D, Shi L, Weinstein H, Blanchard SC, Javitch Ja. Single-molecule dynamics of gating in a neurotransmitter transporter homologue. Nature. 2010;465:188–93. doi: 10.1038/nature09057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quick M, Winther AML, Shi L, Nissen P, Weinstein H, Javitch Ja. Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5563–8. doi: 10.1073/pnas.0811322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merchant Ba, Madura JD. Insights from molecular dynamics: The binding site of cocaine in the dopamine transporter and permeation pathways of substrates in the leucine and dopamine transporters. Journal of molecular graphics & modelling. 2012;38C:1–12. doi: 10.1016/j.jmgm.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noskov SY, Roux B. Control of ion selectivity in LeuT: two Na+ binding sites with two different mechanisms. Journal of molecular biology. 2008;377:804–18. doi: 10.1016/j.jmb.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Penmatsa A, Wang KH, Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature. 2013:1–7. doi: 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Surratt C, Ukairo O, Ramanujapuram S. Recognition of psychostimulants, antidepressants, and other inhibitors of synaptic neurotransmitter uptake by the plasma membrane monoamine transporters. The AAPS Journal. 2005;7:739–751. doi: 10.1208/aapsj070374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ritz M, Lamb R, et al. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 12.Haapaniemi TH, Ahonen A, Torniainen P, Sotaniemi KA, Myllylä VV. [123I] beta-CIT SPECT demonstrates decreased brain dopamine and serotonin transporter levels in untreated parkinsonian patients. Movement disorders : official journal of the Movement Disorder Society. 2001;16:124–30. doi: 10.1002/1531-8257(200101)16:1<124::aid-mds1007>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 13.Meyer JH, Wilson AA, Ginovart N, Goulding V, Hussey D, Hood K, Houle S. Occupancy of serotonin transporters by paroxetine and citalopram during treatment of depression: a [(11)C]DASB PET imaging study. The American journal of psychiatry. 2001;158:1843–9. doi: 10.1176/appi.ajp.158.11.1843. [DOI] [PubMed] [Google Scholar]
- 14.Kirino E. Escitalopram for the management of major depressive disorder: a review of its efficacy, safety, and patient acceptability. Patient preference and adherence. 2012;6:853–61. doi: 10.2147/PPA.S22495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forrest LR, Rudnick G. The rocking bundle: a mechanism for ion-coupled solute flux by symmetrical transporters. Physiology (Bethesda, Md) 2009;24:377–86. doi: 10.1152/physiol.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jack DL, Paulsen IT, Saier MH. The amino acid/polyamine/organocation (APC) super-family of transporters specific for amino acids, polyamines and organocations. Microbiology (Reading, England) 2000;146(Pt 8):1797–814. doi: 10.1099/00221287-146-8-1797. [DOI] [PubMed] [Google Scholar]
- 17.Schlessinger A, Wittwer MB, Dahlin A, Khuri N, Bonomi M, Fan H, Giacomini KM, Sali A. High Selectivity of the γ-Aminobutyric Acid Transporter 2 (GAT-2, SLC6A13) Revealed by Structure-based Approach. The Journal of biological chemistry. 2012;287(37):745–56. doi: 10.1074/jbc.M112.388157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khafizov K, Perez C, Koshy C, Quick M, Fendler K, Ziegler C, Forrest LR. Investigation of the sodium-binding sites in the sodium-coupled betaine transporter BetP. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E3035–44. doi: 10.1073/pnas.1209039109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faham S, Watanabe A, Besserer GM, Cascio D, Specht A, Hirayama Ba, Wright EM, Abramson J. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science (New York, N Y) 2008;321:810–4. doi: 10.1126/science.1160406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ressl S, Terwisscha van Scheltinga AC, Vonrhein C, Ott V, Ziegler C. Molecular basis of transport and regulation in the Na(+)/betaine symporter BetP. Nature. 2009;458:47–52. doi: 10.1038/nature07819. [DOI] [PubMed] [Google Scholar]
- 21.Fang Y, Jayaram H, Shane T, Kolmakova-Partensky L, Wu F, Williams C, Xiong Y, Miller C. Structure of a prokaryotic virtual proton pump at 3.2 A resolution. Nature. 2009;460:1040–3. doi: 10.1038/nature08201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimamura T, Weyand S, Beckstein O, Rutherford NG, Hadden JM, Sharples D, Sansom MSP, Iwata S, Henderson PJF, Cameron AD. Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science (New York, N Y) 2010;328:470–3. doi: 10.1126/science.1186303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben-Yona A, Kanner BI. An acidic amino acid transmembrane helix 10 residue conserved in the neurotransmitter:sodium:symporters is essential for the formation of the extracellular gate of the γ-aminobutyric acid (GABA) transporter GAT-1. The Journal of biological chemistry. 2012;287:7159–68. doi: 10.1074/jbc.M111.323634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang X, Loo DDF, Hirayama Ba, Wright EM. The Importance of Being Aromatic: π Interactions in Sodium Symporters. Biochemistry. 2012;51:9480–7. doi: 10.1021/bi301329w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beuming T, Shi L, Javitch JA, Weinstein H. A comprehensive structure-based alignment of prokaryotic and eukaryotic neurotransmitter/Na+ symporters (NSS) aids in the use of the LeuT structure to probe NSS structure and function. Molecular pharmacology. 2006;70:1630–42. doi: 10.1124/mol.106.026120. [DOI] [PubMed] [Google Scholar]
- 26.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. Journal of computational chemistry. 2005;26:1781–802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Humphrey W, Dalke A, Schulten K. VMD- Visual Molecular Dynamics. Journal of molecular graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 28.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, et al. AMBER 9. University of California; San Francisco: 2006. [Google Scholar]
- 29.Hamelberg D, Mongan J, McCammon JA. Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. The Journal of chemical physics. 2004;120:11919–29. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 30.Krishnamurthy H, Gouaux E. X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature. 2012;25 doi: 10.1038/nature10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grant BJ, Rodrigues APC, ElSawy KM, McCammon JA, Caves LSD. Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics (Oxford, England) 2006;22:2695–6. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- 32.Development Core Team. R: A Language and Environment for Statistical Computing. 2011 [Google Scholar]
- 33.Shan J, Javitch Ja, Shi L, Weinstein H. The substrate-driven transition to an inward-facing conformation in the functional mechanism of the dopamine transporter. PloS one. 2011;6:e16–350. doi: 10.1371/journal.pone.0016350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao C, Stolzenberg S, Gracia L, Weinstein H, Noskov S, Shi L. Ion-controlled conformational dynamics in the outward-open transition from an occluded state of LeuT. Biophysical journal. 2012;103:878–88. doi: 10.1016/j.bpj.2012.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schulze BG, Evanseck JD. Cooperative Role of Arg45 and His64 in the Spectroscopic A 3 State of Carbonmonoxy Myoglobin: Molecular Dynamics Simulations, Multivariate Analysis, and Quantum Mechanical Computations. Journal of the American Chemical Society. 1999;121:6444–6454. [Google Scholar]
- 36.Smart OS, Neduvelil JG, Wang X, Wallace Ba, Sansom MS. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. Journal of molecular graphics. 1996;14:354–60. doi: 10.1016/s0263-7855(97)00009-x. 376. [DOI] [PubMed] [Google Scholar]
- 37.Vanni S, Campomanes P, Marcia M, Rothlisberger U. Ion Binding and Internal Hydration in the Multidrug Resistance Secondary Active Transporter NorM Investigated by Molecular Dynamics Simulations. Biochemistry. 2012 doi: 10.1021/bi2015184. [DOI] [PubMed] [Google Scholar]
- 38.Shaikh SA, Tajkhorshid E. Modeling and dynamics of the inward-facing state of a Na+/Cl- dependent neurotransmitter transporter homologue. PLoS computational biology. 2010;6 doi: 10.1371/journal.pcbi.1000905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith MEa, Wang DN. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science (New York, N Y) 2007;317:1390–3. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448:952–6. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- 41.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science (New York, N Y) 2008;322:1655–61. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou Z, Zhen J, Karpowich NK, Law CJ, Reith MEA, Wang Dn. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nature structural & molecular biology. 2009;16:652–7. doi: 10.1038/nsmb.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kroncke BM, Horanyi PS, Columbus L. Structural origins of nitroxide side chain dynamics on membrane protein α-helical sites. Biochemistry. 2010;49(10):045–60. doi: 10.1021/bi101148w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stein S, Loccisano A, Firestine S, Evanseck J. Chapter 13 Principal Components Analysis: A Review of its Application on Molecular Dynamics Data. Annual Reports in Computational Chemistry. 2006;2:233–261. [Google Scholar]
- 45.Maisuradze GG, Liwo A, Scheraga HA. Principal component analysis for protein folding dynamics. Journal of molecular biology. 2009;385:312–29. doi: 10.1016/j.jmb.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piscitelli CL, Gouaux E. Insights into transport mechanism from LeuT engineered to transport tryptophan. The EMBO journal. 2011;31:228–235. doi: 10.1038/emboj.2011.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y, Terry DS, Shi L, Quick M, Weinstein H, Blanchard SC, Javitch JA. Substrate-modulated gating dynamics in a Na+-coupled neurotransmitter transporter homologue. Nature. 2011;474:109–13. doi: 10.1038/nature09971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao C, Noskov SY. The molecular mechanism of ion-dependent gating in secondary transporters. PLoS computational biology. 2013;9:e1003–296. doi: 10.1371/journal.pcbi.1003296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J, Shaikh Sa, Enkavi G, Wen PC, Huang Z, Tajkhorshid E. Transient formation of water-conducting states in membrane transporters. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:7696–701. doi: 10.1073/pnas.1218986110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang H, Elferich J, Gouaux E. Structures of LeuT in bicelles define conformation and substrate binding in a membrane-like context. Nature structural & molecular biology. 2012;19:212–9. doi: 10.1038/nsmb.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quick M, Shi L, Zehnpfennig B, Weinstein H, Javitch Ja. Experimental conditions can obscure the second high-affinity site in LeuT. Nature structural & molecular biology. 2012:1–6. doi: 10.1038/nsmb.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao C, Noskov SY. The role of local hydration and hydrogen-bonding dynamics in ion and solute release from ion-coupled secondary transporters. Biochemistry. 2011;50:1848–56. doi: 10.1021/bi101454f. [DOI] [PubMed] [Google Scholar]
- 53.Koldsø H, Noer P, Grouleff J, Autzen HE, Sinning S, Schiø tt B. Unbiased simulations reveal the inward-facing conformation of the human serotonin transporter and Na(+) ion release. PLoS computational biology. 2011;7:e1002–246. doi: 10.1371/journal.pcbi.1002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng MH, Bahar I. Coupled global and local changes direct substrate translocation by neurotransmitter-sodium symporter ortholog LeuT. Biophysical journal. 2013;105:630–9. doi: 10.1016/j.bpj.2013.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Forrest L, Zhang Y, Jacobs M, Gesmonde J, Xie L, Honig B, Rudnick G. Mechanism for alternating access in neurotransmitter transporters. Proceedings of the National Academy of Sciences. 2008;105(10):338. doi: 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jø rgensen AM, Tagmose L, Jø rgensen AMM, Bø gesø KP, Peters GH. Molecular dynamics simulations of Na+/Cl(-)-dependent neurotransmitter transporters in a membrane-aqueous system. ChemMedChem. 2007;2:827–40. doi: 10.1002/cmdc.200600243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.