

Abstract
Alcoholic liver disease is a major health problem in the United States and worldwide. Chronic alcohol consumption can cause steatosis, inflammation, fibrosis, cirrhosis and even liver cancer. Significant progress has been made to understand key events and molecular players for the onset and progression of alcoholic liver disease from both experimental and clinical alcohol studies. No successful treatments are currently available for treating alcoholic liver disease; therefore, development of novel pathophysiological-targeted therapies is urgently needed. This review summarizes the recent progress on animal models used to study alcoholic liver disease and the detrimental factors that contribute to alcoholic liver disease pathogenesis including miRNAs, S-adenosylmethionine, Zinc deficiency, cytosolic lipin-1β, IRF3-mediated apoptosis, RIP3-mediated necrosis and hepcidin. In addition, we summarize emerging adaptive protective effects induced by alcohol to attenuate alcohol-induced liver pathogenesis including FoxO3, IL-22, autophagy and nuclear lipin-1α.
Keywords: Alcoholic liver disease, Autophagy, Fatty liver, Lipin-1, FoxO3, RIP3, IL-22
Core tip: Alcoholic liver disease is a major health problem worldwide. Significant progress has been made to understand key events and molecular players for the onset and progression of alcoholic liver disease. This review summarizes the recent progress on animal models used to study alcoholic liver disease and the detrimental factors that contribute to alcoholic liver disease pathogenesis including miRNAs, S-adenosylmethionine, zinc deficiency, cytosolic lipin-1β, IRF3-mediated apoptosis, RIP3-mediated necrosis and hepcidin. In addition, we summarize emerging adaptive protective effects induced by alcohol to attenuate alcohol-induced liver pathogenesis including FoxO3, IL-22, autophagy and nuclear lipin-1α.
INTRODUCTION
Alcohol consumption and abuse are major causes of chronic liver disease, which is a significant health problem in the United States and around the world. The pathogenesis of alcoholic liver disease (ALD) in humans is characterized by steatosis (mild stage), which is an accumulation of fat in hepatocytes. In most heavy alcohol consumers, steatosis is caused by inhibiting fatty acid oxidation while increasing uptake of fat into the liver along with fatty acid and triglyceride synthesis. Approximately 8%-20% of heavy drinkers with steatosis can further develop steatohepatitis (moderate stage) and fibrosis and cirrhosis (advanced stage), and some (3%-10%) eventually develop hepatocellular carcinoma (HCC)[1,2]. Alcoholic steatohepatitis is characterized by hepatic inflammation and injury in addition to steatosis and also includes fibrotic and cirrhotic disease states. Fibrosis is a typical wound-healing response induced by liver injury that is characterized by an accumulation of extracellular matrix proteins, such as collagen, which are produced by activated hepatic stellate cells (HSCs). Continuous activation of this wound healing response leads to cirrhosis of the liver, which can eventually progress to HCC.
Most heavy alcohol consumers do not progress beyond steatosis of the liver, which suggests that other factors contribute to progression of ALD in addition to heavy alcohol consumption. There have been several factors shown to contribute to progression and severity of ALD in humans including race, sex, and comorbidities like obesity or hepatitis C virus (HCV). Genetic polymorphisms and epigenetic modifications have also been shown to have roles in ALD progression. Two of the most important factors in susceptibility to ALD progression are race and sex. African-Americans and Hispanics are more likely to progress to alcohol-induced cirrhosis than Caucasians[3,4]. In addition, women are more likely to progress to ALD with more severity than men[5-7]. Women were shown to have increased blood alcohol levels compared to men after alcohol consumption, which was likely due to females having decreased gastric alcohol dehydrogenase, contributing to a decreased first-pass metabolism and greater bioavailability of alcohol after consumption[7,8]. The greater likelihood of women to have increased risk of developing ALD may also be due to estrogen levels[9-12].
Comorbidity with other diseases, such as obesity or HCV, in addition to lifestyle factors have also been shown to play a role in ALD progression. Obesity and metabolic syndrome have been shown to have a synergistic effect on alcohol-induced liver injury[13,14], which may be due to nitrosative stress in the liver caused by type 1 macrophage activation, increased ER and mitochondrial stress, and adiponectin resistance[15]. In addition, obesity has been associated with an increased mortality rate in ALD patients[16]. HCV has also been associated with severity of ALD[17,18]. A combination of HCV and ALD has been shown to cause more liver injury than either disease alone[17-19], and the risk of developing cirrhosis was greatly increased in HCV patients that were heavy alcohol consumers[20]. Smoking has also been shown to have a role in ALD severity and progression to liver cirrhosis[21-23]. In contrast, drinking coffee has been shown to protect against ALD severity and development of alcohol-induced liver cirrhosis[22,24,25].
Finally, genetic polymorphisms and epigenetics have been shown to contribute to ALD progression and severity. For example, several polymorphisms of genes necessary for alcohol metabolism have been found including polymorphisms in alcohol dehydrogenase (ADH2 and ADH3), aldehyde dehydrogenase (ALDH2) and in the cytochrome P450 2e1 (Cyp2e1) promoter[26,27]. Polymorphisms in glutathione S-transferase (GST) genes necessary to reduce oxidative stress may also have a role in ALD progression[28]. In addition, polymorphisms in genes involved in cytokine regulation and response have been associated with ALD progression, such as a polymorphism of the TNF-α promoter[29,30] in addition to polymorphisms in IL-1β, TGF-β1, and IL-10, which all influence progression to hepatic fibrosis[31]. Recently, variations in the patatin-like phospholipase domain-containing protein 3 (PNPLA3) gene have been associated with ALD progression to cirrhosis[32-35] fibrosis and HCC[36]. In addition to genetic polymorphisms, epigenetics have also been shown to have a role in ALD progression. Alcohol has also been shown to influence epigenetics and histone modification in the GI tract and liver, which may increase progression and severity of ALD. For example, alcohol has been shown to alter expression of ADH due to histone modification. More critically, epigenetic changes induced by alcohol consumption may be transmitted to offspring, which could affect their development[37].
ALD is a substantial problem worldwide that is caused by heavy alcohol consumption in addition to other environmental and genetic factors. Significant research progress has been made in the past few years for understanding ALD pathogenesis, but a universal treatment to cure ALD is still lacking. Several mediators that result in progression of ALD that have already been thoroughly reviewed include hepatocyte apoptosis, activation of innate and adaptive immunity, and inhibition of liver regeneration[38-40]. In this review, we discuss newly available mouse models for studying ALD, new players in alcohol-induced liver pathogenesis and novel adaptive mechanisms that the liver may utilize to protect against alcohol-induced liver injury.
A PREVIEW OF CURRENT RODENT MODELS OF ALD
While many species have been used to study ALD including baboons, pigs and rats, mice have been used predominantly in current ALD research. This is due to the availability of numerous transgenic and knockout mice that can easily help scientists determine the role of a particular molecule or signaling pathway in the pathogenesis of ALD. In addition, mice have more than 85% genetic similarity to humans, and their physiology and genetics have been extensively studied. Moreover, mice can reproduce quickly and are relatively inexpensive compared to baboons and pigs. Although many different mouse models have been established to study the pathogenesis of ALD, they are only able to mimic some early pathogenic changes of ALD in humans. In fact, none of the current mouse models have been able to reproduce the exact pathogenic process in human ALD. Nevertheless, since ALD is a chronic liver disease, the understanding of these early pathogenic changes that can be seen in mouse ALD models, such as hepatic steatosis and inflammation, can still shed light on future development of therapeutics for prevention of ALD progression. Several mouse models have been used to study alcoholic liver injury that include acute oral gavage, ad libitum oral alcohol in drinking water, intragastric infusion (Tsukamoto-French model), chronic Lieber-DeCarli diet ethanol feeding and the most recent National Institute on Alcohol Abuse and Alcoholism (NIAAA) chronic-binge ethanol model, which was developed by Bin Gao’s group (hereafter referred to as Gao-Binge model). Mice administered acute oral gavage, ad libitum oral alcohol in drinking water or chronic Lieber-DeCarli diet ethanol feeding show mild elevation of serum alanine aminotransferase (ALT), which only mimics early pathogenic changes in human ALD. Aimed to establish a mouse model to better recapitulate human ALD pathogenesis, Dr. Tsukamoto and Dr. French invented the intragastric infusion model in which the mice/rats are surgically implanted with an intragastric tube to allow continuous enteral alcohol feeding, which results in blood alcohol concentrations that are much higher than ad libitum alcohol feeding[41,42]. The intragastric alcohol fed mice show marked elevation in serum ALT levels, steatosis and some mild liver fibrotic changes[43]. However, an increased mortality rate was found in long-term intragastric fed mice, and thus these mice require extensive medical care during experiments. Moreover, this model requires a technically challenging surgery that is difficult to perform for most laboratories.
Alcoholic hepatitis (AH) is a clinical syndrome among chronic alcohol drinkers who often also binge drink, resulting in hospitalization. Approximately 30%-40% of AH patients die within one month of diagnosis, and current treatment options, which are use of corticosteroids or pentoxifylline, only provide about a 50% survival benefit[44]. Therefore, there is an urgent need for development of new targeted therapies for AH. As previously mentioned, most mouse models only mimic early ALD pathogenesis. However, Dr. Gao’s laboratory recently established a novel mouse model to mimic AH conditions (Gao-binge model)[45]. In this model, mice are fed a control Lieber-DeCarli liquid diet for 5 d before switching them to a 10 d ethanol Lieber-DeCarli diet (1%-5%) followed by a one-time ethanol binge by oral gavage (31.5% v/v) on the morning of day 11 for 9 h. Control animals are fed a calorie-matched diet and receive a maltose-dextran binge (45%) on day 11. Mice were found to have a blood alcohol level of 140 mmol/L 1-2 h after ethanol binge using this model[46]. Mice treated with chronic feeding together with an acute binge showed markedly elevated ALT, aspartate aminotransferase (AST), and steatosis along with significant inflammation in mouse livers compared with either the chronic 10 d feeding or the acute binge alone-treated mice[45,47]. The advantages of this model are obvious because it is easy to perform and is more relevant to the human AH condition because it generates more liver injury and inflammation than other frequently used models. However, it should be noted that the phenotype in this model does not progress beyond liver steatosis, inflammation, and injury. Therefore, improved animal models that more accurately reflect the human condition are still needed for studying ALD progression.
NOVEL PLAYERS IN ALD PATHOGENESIS
Chronic alcohol consumption is well known to induce liver steatosis, which can eventually progress to more severe forms of liver injury, such as AH, fibrosis, and HCC as previously discussed. Owing to the active research in the past decade, many mediators of ALD pathogenesis have been identified and extensively reviewed recently[38-40], this section of the review will thus focus on novel players in ALD pathogenesis including modulation of microRNA levels, changes in methionine metabolism and S-adenosylmethionine, induction of osteopontin, hepcidin and zinc depletion, and newly discovered mechanisms of alcohol-induced cell death.
MicroRNA
MicroRNAs (miRNAs) are small noncoding RNAs that post-transcriptionally regulate gene expression via RNA cleavage and degradation or inhibition of RNA translation[48]. There have been several studies over the past 5 years investigating the role of miRNAs in ALD. A microarray analysis to determine the hepatic miRNA profile in mice fed the Lieber-DeCarli diet (4.5%) for 5 wk revealed that chronic ethanol feeding caused significant changes in miRNA levels. Specifically, chronic ethanol feeding caused down-regulation of miR-27b, miR-214, miR-199a-3p, miR-182, miR-183, miR-200a and miR-322, but caused up-regulation of miR-705 and miR-1224[49]. While these results indicate that chronic alcohol consumption can alter hepatic miRNA levels, the exact contribution of these miRNA changes in the pathogenesis of ALD is still not clear. In a separate study conducted by the same group, it was found that RAW 264.7 macrophages exposed to either lipopolysaccharide (LPS) or ethanol increased the induction of miR-155, which was important for TNFα induction. Interestingly, combined treatment of LPS with ethanol further synergistically increased the induction of miR-155 in RAW 264.7 macrophages, which also correlated with ethanol-induced TNFα production. More importantly, they further found that mice that were fed the Lieber-DeCarli diet (5%) for 4 wk had significantly increased miR-155 levels and TNFα production in isolated Kuppfer cells (KCs) when compared with pair-fed controls. Mechanistically, miR-155 increased mRNA stability of TNFα and, in turn, promoted alcohol-induced elevation of TNFα production[50]. The ethanol-induced increase in miR-155 seemed to be more robust in KCs because it was only slightly increased in hepatocytes in chronic ethanol fed mice, which is consistent with the role of miR-155 in regulating inflammation. In contrast to miR-155, miR-122 is abundant in hepatocytes. Interestingly, it was found that chronic ethanol feeding decreased hepatic miR-122 levels but significantly increased blood circulating levels of both miR-122 and miR-155. Further studies revealed that serum/plasma levels of miR-122 correlated with ALT levels whereas serum/plasma miR-155 levels correlated with inflammation induced by alcohol[51]. These results suggest that circulating miRNAs may serve as additional biomarkers for alcohol-induced liver injury and inflammation in addition to serum ALT levels.
Yin et al[52] recently demonstrated that miR-217 plays a critical role in ethanol-induced liver steatosis via down-regulation of sirtuin 1 (SIRT1). SIRT1 is a deacetylase that regulates lipid metabolism via deacetylation of genes involved in lipid synthesis and break down, such as sterol regulatory element-binding protein 1 (SREBP-1) and Peroxisome-proliferator-activated receptor gamma (PPARγ) co-activator-1α (PGC-1α). Ethanol treatment increased miR-217 levels in both ethanol-treated AML-12 cells and in mice fed a modified Lieber-DeCarli diet for 4 wk. In addition, both ethanol and miR-217 overexpression increased triglycerides (TG) in AML-12 cells, and co-treatment of AML-12 cells with both ethanol and miR-217 further increased TG levels compared to ethanol or miR-217 overexpression alone. Mechanistically, it was found that both miR-217 overexpression and alcohol treatment inhibited SIRT1 mRNA and protein expression as well as its deacetylase activity. Co-treatment of miR-217 with ethanol further exacerbated the inhibitory effects on SIRT1. Moreover, miR-217 overexpression and ethanol treatment both inhibited AMP-activated protein kinase (AMPK) activity, which has critical roles in ethanol-induced liver fat accumulation[53,54]. In line with these results, miR-217 overexpression increased mRNA expression of several genes involved in lipid synthesis with a corresponding decrease in genes involved in fatty acid oxidation. Furthermore, miR-217 and ethanol treatment both induced lipin-1, a protein that has phosphatidate phosphatase activity important for regulating lipid homeostasis (also see below discussion), to be localized in the cytoplasm, which is associated with induction of lipid synthesis and reduction of fatty acid oxidation in the liver[52]. In addition to miR-217, miR-34a was also found to down-regulate SIRT1 expression after ethanol treatment in human hepatocytes, but the effect of miR-34a on ethanol-induced steatosis was not investigated[55].
In addition to having a role in ethanol-induced inflammation and steatosis, miRNAs may also have an impact on ethanol-induced liver regeneration. Dippold et al[56] studied miRNA profiles in livers from rats fed ethanol for 5 wk compared with pair-fed controls after both groups were further subjected to partial hepatectomy (PHx). They found that expression of particular miRNAs differed between ethanol and control rats after PHx, and that some miRNAs present in control rats were absent in ethanol-treated rats or vice versa. Importantly, ethanol-treated rats had decreased expression of miR-196a and miR-196c in the early liver regenerative phase. Chip analysis revealed that these miRNAs were regulated by nuclear factor-kappaB (NF-κB), and that binding of NF-κB to the miR-196c promoter was decreased after PHx[56]. These results suggest that decreased expression of miR-196a and miR-196c is associated with impaired liver regeneration during ethanol-induced liver injury, but the exact role of miR-196a and miR-196c in ethanol-induced impairment of liver regeneration needs to be further studied. In addition to miR-196a and 196c, it should also be noted that many other miRNAs were altered during liver regeneration in chronic ethanol-fed rats. Therefore, it will be interesting to further investigate the role of other miRNAs in liver recovery and regeneration after ethanol-induced liver injury.
Ethanol-induced oxidative stress has recently been shown to involve miR-214. Rats fed with ethanol for 4 wk had increased expression of miR-214, which binds specifically to the 3’-UTR of glutathione reductase and cytochrome P450 oxidoreductase, two important antioxidant genes[57]. Binding of miR-214 to the promoters of these oxidative stress genes inhibits their expression and decreases their activity, which likely promotes alcohol-induced oxidative stress. Indeed, inhibition of miR-214 in human hepatoma cells (Bel7402) and in rat liver cells (BRL) treated with ethanol rescued the ethanol-induced reduction in expression of glutathione reductase and cytochrome P450 oxidoreductase and suppressed ethanol-induced oxidative stress[57]. These findings suggest that miR-214 may be a future therapeutic target for reducing ethanol-induced oxidative stress and liver injury.
In summary, it seems that miRNAs play roles in alcohol-induced inflammation, steatosis, liver regeneration/repair and oxidative stress. miRNAs might be novel therapeutic targets as well as diagnostic biomarkers for ALD. However, it should be noted that the miRNA profiles found in mice and rats treated with ethanol were quite different. For example, none of the microRNA alterations found in Dolganic’s mouse liver microarray analysis[49] were found in the rat microarray analysis completed by Hoek’s group[56], which could be due to species differences between rats and mice. Moreover, the miRNA profile in mice treated with the Gao-binge model has not been determined. Future work to further characterize the role of miRNAs in ALD is definitely needed.
S-adenosylmethionine
Methionine is an essential amino acid important for synthesis of cysteine and phospholipids, such as phosphatidylcholine. Methionine must be ingested through the diet because it cannot be newly synthesized by humans. Methionine metabolism occurs predominantly in the liver and results in production of the methyl donor S-adenosylmethionine (SAM), which is necessary to perform most methylation reactions. The first reaction in methionine metabolism occurs via the transmethylation cycle, which results in production of SAM from methionine by methionine adenosyltransferase (MAT). After donating its methyl group, SAM is converted to S-adenosylhomocysteine (SAH), which is then further converted to adenosine and homocysteine by S-adenosylhomocysteine hydrolase (SAHH). Homocysteine is then used to regenerate methionine or to produce glutathione (GSH), which is a well-known anti-oxidant important for prevention of liver injury. The vitamin B6 dependent transsulfuration pathway is utilized to produce GSH via reduction of homocysteine to cystathionine using cystathionine beta synthase (CβS), which can then be further metabolized to cysteine and subsequently, GSH. Regeneration of methionine from homocysteine occurs by two pathways. The folate-dependent pathway regenerates methionine from homocysteine via transfer of a methyl group from N5-methyltetrahydrofolate (MTHF) to vitamin B12, which is then subsequently transferred to homocysteine by methionine synthase (MS) to form methionine. The second pathway is folate independent and uses betaine as a substrate for methionine synthesis from homocysteine via betaine homocysteine methyltransferase (BHMT)[58-61].
It is well known that ethanol induces alterations in multiple steps of methionine metabolism, which is associated with progression of ALD (Figure 1)[58-63]. Studies in the 1960s showed that alcoholic patients had hematosuppressive effects due to alcohol’s effect on folate metabolism. In addition, ethanol-treated rat hepatocytes had increased MTHF. These studies were the earliest evidence indicating that ethanol may affect methionine metabolism[64,65]. Then, pioneer work from Lieber et al[66] showed that baboons fed chronic ethanol for 18 to 36 mo had decreased hepatic SAM levels, and SAM supplementation in these baboons significantly attenuated ethanol-induced mitochondrial damage in the liver. Studies from Dr. Tuma’s group also showed that rats that received betaine in their diets had increased hepatic SAM levels and markedly reduced liver steatosis after chronic ethanol feeding[67]. Mechanistically, it was found that chronic ethanol consumption inhibits the activity of methionine synthase, resulting in a compensatory increase of BHMT activity. However, this compensatory BHMT-mediated pathway cannot be maintained under chronic ethanol exposure conditions, which consequently results in decreased levels of SAM and increased levels of SAH and homocysteine[58,62]. A decreased SAM-to-SAH ratio and increased homocysteine leads to progression of liver injury, steatosis and ALD, which may be involved in multiple mechanisms including inflammation, oxidative stress, endoplasmic reticulum (ER) stress, accumulation of damaged protein, altered gene expression, chromatin structure modification, GSH depletion and apoptosis[60,61,63,68-71].
Figure 1.
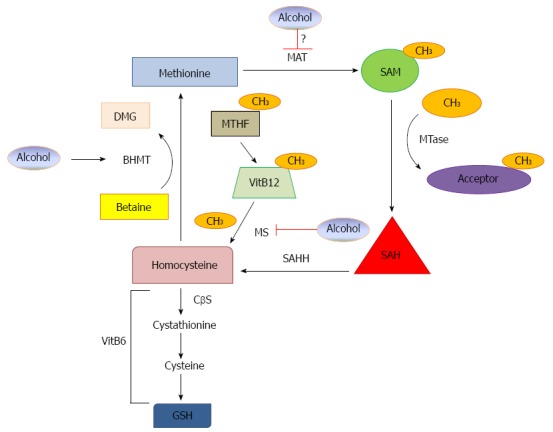
S-adenosylmethionine is produced from methionine by methionine adenosyltransferase. After donating its methyl group, S-adenosylmethionine (SAM) is converted to S-adenosylhomocysteine (SAH), which is then converted to adenosine and homocysteine by S-adenosylhomocysteine hydrolase (SAHH). Homocysteine is used to regenerate methionine or to produce glutathione (GSH). The vitamin B6 dependent transsulfuration pathway produces GSH via reduction of homocysteine to cystathionine using cystathionine beta synthase (CβS), which can then be further metabolized to cysteine and GSH. Regeneration of methionine from homocysteine occurs by two pathways. The folate-dependent pathway regenerates methionine from homocysteine via transfer of a methyl group from N5-methyltetrahydrofolate (MTHF) to vitamin B12, which is then transferred to homocysteine by methionine synthase (MS) to form methionine. The second pathway is folate independent and uses betaine as a substrate for methionine synthesis from homocysteine via betaine homocysteine methyltransferase (BHMT). Alcohol may affect methionine metabolism via three mechanisms: alcohol inhibits MS, alcohol increases BHMT as a compensatory effect and alcohol may inhibit MAT activity, but this is still controversial.
Therapeutically, SAM treatment reduced ethanol-induced inflammation, which was possibly through inhibition of TNFα production from KCs after LPS treatment[69] and repression of ethanol-induced increases in mRNA expression of toll-like receptors 2 and 4 likely via histone methylation[72]. SAM has also been shown to be important in preserving mitochondrial function and reducing oxidative stress during ethanol consumption. It was recently shown that betaine treatment increases the SAM-to-SAH ratio along with increasing GSH and its component cysteine, which allowed for protection against oxidative stress. Betaine treatment improved liver antioxidant capacity and reduced inducible nitric oxide synthase (iNOS) expression and nitric oxide production in the liver during chronic ethanol treatment[70,71]. Furthermore, betaine treatment also inhibited ethanol-induced reduction of cytochrome c oxidase and NADH dehydrogenase, two components of the electron transport chain necessary for mitochondrial respiration[70]. It is known that chronic alcohol consumption induces hyperhomocysteinemia in rats and humans, which is associated with ER stress. Interestingly, betaine treatment significantly reduced chronic ethanol-induced ER stress and ER stress-initiated hepatocyte apoptosis in intragastric alcohol-fed mice[68]. Finally, betaine and SAM supplements significantly reduced blood alcohol levels by increasing epinephrine-mediated metabolic rate and alcohol dehydrogenase-mediated alcohol oxidation[73].
Although SAM and betaine treatments have shown beneficial effects in reducing ethanol-induced liver injury and steatosis in animal models, it was also found that methionine metabolism may differ among the various alcohol models (acute vs chronic) and in different species (mouse vs rat). Chronic ethanol feeding increased plasma homocysteine in mice, but not in rats. Additionally, BHMT protein levels were increased in rat livers after ethanol feeding, but were unchanged in mice fed ethanol compared to controls. Furthermore, BHMT promoter activity was different in rat and mouse primary hepatocytes. Homocysteine treatment inhibited BHMT promoter activity in mouse hepatocytes, which was rescued by betaine treatment, but homocysteine treatment did not affect BHMT promoter activity in rat hepatocytes[74]. Therefore, more studies are definitely needed to further dissect these differences. These various changes in methionine metabolism in different animal models may also help to explain why therapeutic use of SAM in humans has not proven useful. Several studies in ALD patients have been recently performed to investigate the therapeutic potential of SAM treatment. In a clinical study where 13 ALD patients were given either SAM or placebo, no differences in ALT, AST, or bilirubin levels were found. In addition, there was no difference between the groups for histopathology[75,76]. In another patient study, French and colleagues found no differences in liver biopsy characteristics between alcoholic patients treated or not treated with SAM while abstaining from alcohol use for 24 d[75]. However, it should be noted that the sample size used in these studies was relatively small. In addition, many patients had some fibrosis at baseline, which may render injured hepatocytes non-responsive to SAM treatment. Moreover, chronic ethanol use has been associated with decreased vitamin B6[60], which is an important cofactor for production of GSH from homocysteine, as previously discussed. Therefore, it is possible that new trials using co-administration of SAM and vitamin B6 may provide more promising results in human patients. Moreover, increased patient numbers are also needed in future clinical trials.
Osteopontin
Osteopontin (OPN), also known as secreted phosphoprotein 1 (SPP1), is a pro-inflammatory protein found in extracellular matrix that binds to CD44 to initiate its transcription and to integrins on target cells to promote cell adherence and migration[77]. OPN undergoes multiple posttranslational modifications including phosphorylation, O-glycosylation and proteolytic processing[78]. Thrombin or matrix metalloproteinase (MMP) 7 cleaves OPN to yield a more active form of OPN[79]. Accumulating evidence suggests that OPN plays a role in various liver diseases including hepatic steatosis, inflammation, fibrosis and the pathogenesis of ALD[80-85]. OPN mRNA and protein levels were first found to be elevated in mice and rats after feeding them the Lieber-DeCarli diet for 6 wk[82,86,87], and OPN induction occurred mainly in biliary epithelium[86]. It is known that alcohol consumption inhibits hepatic peroxisome proliferator-activated receptor-α (PPAR-α) activity resulting in hepatic steatosis and inflammation. Interestingly, it was found that alcohol-induced down-regulation of PPAR-α was markedly suppressed in OPN knockout (KO) mice, which suggests that OPN might positively regulate PPAR-α in alcohol-treated mice. Conversely, treatment of mice with a PPAR-α agonist increased PPAR-α expression and reduced OPN expression in mice that were treated with alcohol and carbon tetrachloride (CCl4), suggesting that PPARα might also regulate OPN expression[88].
OPN has been suggested to increase alcohol-induced inflammation by acting as a chemokine for neutrophil infiltration in the liver[82,86,89], and OPN levels have been shown to correlate with liver neutrophil numbers in human patients[90]. Neutrophil infiltration was reduced when rats were treated with an OPN neutralizing antibody before treatment with LPS using a Lieber DeCarli ethanol diet + LPS ALD model[89]. These early results suggest that OPN may contribute to the pathogenesis of ALD. Indeed, OPN KO mice had reduced serum ALT levels after ethanol treatment using the Gao-Binge model compared to WT mice[83]. In addition, WT mice, but not OPN KO mice, had increased hepatic inflammatory gene expression and neutrophil infiltration after ethanol treatment with the Gao-Binge model[83]. Moreover, studies using anti-osteopontin antibodies to both osteopontin and its β3 integrin receptor have been shown to protect against non-alcoholic hepatocyte toxicity and concavalin-A induced liver injury, respectively[91,92]. In addition to playing a role in ALD, OPN has also been shown to promote liver fibrosis in mice that were treated with CCl4 and thioacetamide (TAA). It was shown that OPN promoted hepatic stellate cell activation through the phosphoinositide 3-kinase (PI3K)-Akt pathway and integrin αvβ3 engagement. More importantly, OPN KO mice showed less liver injury and fibrosis in response to CCl4 and TAA[93]. In line with the results obtained from these animal studies, ALD patients also have increased serum OPN levels when they progress beyond hepatic steatosis to hepatic inflammation, AH, and fibrosis[83,90]. Taken together, the above evidence suggests that OPN may contribute to the pathogenesis of ALD, and inhibiting OPN may be a potential therapeutic target for treating patients with alcohol hepatitis.
While most evidence supports a detrimental role of OPN in ALD, Nieto and colleagues recently found that OPN could improve the pathogenesis of ALD by targeting the gut-liver axis. Supplementation of the breast milk form of OPN protected against ethanol-induced liver inflammation, steatosis, and injury by maintaining gut permeability after mice were treated with the Lieber-DeCarli diet for 3 wk[85]. Moreover, the same group also reported that steatosis and liver injury were increased in OPN KO mice but were significantly decreased in OPN transgenic mice overexpressing OPN in hepatocytes compared with wild type mice. Mechanistically, it was proposed that OPN may block gut derived LPS and TNFα, and thus alleviate LPS/TNFα-mediated liver injury in chronic alcohol-fed mice[84]. These contradictory results could be due to multiple factors. First, it is known that OPN is post-translationally regulated and OPN in breast milk has a greater number of phosphorylated serine residues than endogenous forms of OPN in adults[77,94]. OPN can also be glycosylated, sulfated, and cleaved by proteases[95]. Second, it is also possible that OPN may play different roles in different parts of ALD pathogenesis because Morales-Ibanez et al[83] used the Gao-Binge model whereas Ge et al[84] fed mice with the Lieber-DeCarli diet for 7 wk. Therefore, future work is needed to further investigate the role of OPN in ALD. In particular, the role of various post-translational modifications of OPN in different animal ALD models should be investigated.
Hepcidin
Hepcidin is a peptide synthesized in the liver and secreted into the bloodstream to help regulate iron homeostasis[96]. Hepcidin is synthesized as an 83 amino acid protein known as prohepcidin, which is then cleaved into its 25 amino acid mature form. When searching for iron-regulated liver-expressed genes, Tomas Ganz coined the name hepcidin[97]. In addition, Ganz and others found that the hepcidin gene is highly expressed in the liver and has antimicrobial properties[97,98]. Hepcidin suppresses iron absorption from the small intestine and iron release from macrophages resulting in reduced serum iron stores[96]. Mechanistically, hepcidin regulates iron metabolism by binding to the iron exporter ferroportin (FPN1), which is expressed on macrophages and on enterocytes in the small intestine, resulting in its internalization and subsequent degradation by the lysosome. By inducing degradation of FPN1, hepcidin prevents iron overload by inducing sequestration of iron in macrophages and enterocytes, which prevents secretion of iron into the circulation[99]. Hepcidin overexpression has been shown to cause severe anemia and a lack of hepcidin gene expression causes iron overload in tissues, demonstrating that regulation of hepcidin expression is extremely important for maintaining iron homeostasis[100,101]. Hepcidin expression is regulated by several factors including iron levels, inflammation, and erythropoiesis. Hepcidin expression is increased during iron overload to induce degradation of FPN1 to decrease intestinal absorption and export of stored iron, and hepcidin expression is decreased during iron deficiency to increase iron absorption and export into the circulation. Additionally, hepcidin expression is up-regulated by the inflammatory cytokines IL-6, IL-22, IL-1α, and IL-1β. Hepcidin expression is also regulated by erythropoietic activity because hepcidin levels decrease with increased red blood cell production in order to supply iron needed for erythropoiesis[102].
Accumulating evidence suggests that iron homeostasis and hepcidin play a role in the pathogenesis of ALD. It is well known that ALD patients have excess iron accumulation in their livers[103]. While iron is required for several important processes in the body, such as red blood cell synthesis and cellular respiration, excess free iron can be toxic. Excess alcohol use is well known to cause production of reactive oxygen species (ROS) such as H2O2 and superoxide radical[104]. Free iron reacts with H2O2 via the Fenton reaction to produce hydroxyl radical (OH-), which is a potent form of ROS that causes cell damage and eventual toxicity via lipid peroxidation, DNA mutation, and breakdown of cell membranes that contribute to ALD[104]. Iron overload is thought to be a second hit in the progression of ALD because hepatic iron content in alcoholic cirrhosis patients correlated with their mortality[105]. Excess iron storage in liver KCs has been shown to increase NF-κB and TNFα expression, which can aggravate alcohol-induced liver inflammation and injury[106,107]. Iron chelation was shown to inhibit TNFα expression[108,109], alcohol-induced liver lipid peroxidation and steatosis in intragastric ethanol fed rats[110]. Conversely, co-treatment with alcohol and iron has been shown to exacerbate alcohol-induced liver injury in intragastric alcohol fed rats[111]. This evidence supports that iron overload can cause progression of alcohol-induced liver injury.
Accumulation of hepatic iron in ALD patients was thought to be due to increased hepatocellular uptake of iron via transferrin receptor 1 (TfR1) and increased intestinal absorption of iron, and it was later realized that accumulation of iron in human ALD patients was also likely due to down-regulation of hepcidin expression[112]. Indeed, accumulating evidence indicates that alcohol down-regulates hepcidin mRNA expression in both rodents and humans, which results in up-regulation of FPN1 and subsequent hepatic iron overload[113-118]. Suppression of hepcidin expression has also been shown to correlate with disease severity in ALD patients[118]. Several mechanisms have been proposed to contribute to alcohol-induced decrease of hepcidin expression. Alcohol-induced oxidative stress is thought to play a role in reducing hepcidin expression because mice treated with the antioxidants N-acetylcysteine (NAC) or vitamin E during alcohol exposure maintained hepcidin expression levels[113]. In addition, the metabolism of ethanol to acetaldehyde was shown to be required for down-regulation of hepcidin expression because co-treatment with 4-Methylpyrazole, which is an inhibitor of alcohol dehydrogenase and Cyp2e1, inhibited the alcohol-induced decrease in hepcidin expression[113]. Furthermore, alcohol reduced DNA binding activity and protein levels of CCAAT/enhancer binding protein alpha (C/EBPα), which is a transcription factor required for hepcidin synthesis[113,116,117,119]. Recent research has demonstrated a role for hypoxia-inducible transcription factor-1 alpha (HIF-1α) in alcohol-induced suppression of hepcidin via down-regulation of C/EBPα expression. HIF-1α, which is a transcription factor that is up-regulated in hypoxic conditions, has been shown to down-regulate hepcidin expression[119,120]. It was shown that HIF-1α mRNA and nuclear protein expression was increased in mice treated with ethanol using the Lieber DeCarli diet (4%) for 8 wk[117]. Interestingly, the repression of hepcidin expression by alcohol was blocked with hepatocyte loss of Arnt, which is the heterodimer binding partner needed for HIF-1α transcriptional activity. Furthermore, HIF-1α mediated suppression of hepcidin expression was mediated by a decrease in C/EBPα expression via its proteasomal degradation. Mice lacking Arnt did not have decreased C/EBPα expression after ethanol treatment, whereas overexpression of C/EBPα before ethanol treatment prevented suppression of hepcidin[119]. These findings support a role for the HIF-1α-C/EBPα axis in the regulation of hepcidin in ALD.
It should be noted that several therapeutic experimental studies have been conducted that suggest a beneficial role for targeting iron overload in ALD. For example, administration of epigallocatechin-3-gallate (EGCG) to mice treated with alcohol for 12 wk by intragastric feeding was shown to decrease serum ALT, AST, and steatosis by decreasing serum and hepatic iron in addition to increasing hepcidin mRNA levels in the liver[121]. Vitamin C has also been implicated as a therapeutic option for ALD patients to reduce iron overload. Mice given 50 mg/kg of vitamin C in addition to treatment with ethanol (20% in their drinking water) had decreased serum ALT and serum and hepatic iron levels compared to mice treated with ethanol alone. In addition, mice given vitamin C also had increased hepcidin expression compared to mice treated with ethanol alone[122]. In addition to dietary therapeutic options, others have proposed pharmacologic options for reducing iron overload in ALD. For example, minihepcidin, which is a small peptide similar to hepcidin, has been shown to reduce iron overload in mice[123]. However, use of this peptide has not been evaluated in ALD models and should be investigated further. Nevertheless, targeting hepcidin and iron overload may be a promising approach for treating ALD.
Alcohol-induced cell death
Alcohol metabolism is well known to produce ROS which then induce lipid peroxidation and GSH depletion, leading to hepatocyte injury and death via apoptosis, necrosis, or necroptosis. Apoptotic cell death is characterized by nuclear fragmentation, chromatin condensation, and cellular shrinkage and is dependent on activation of caspases. When the cell dies by apoptosis, it breaks apart into apoptotic bodies, which are membrane-enclosed particles containing intact organelles that are later degraded by phagocytosis without inducing an inflammatory response. Ethanol can induce apoptosis via the intrinsic (mitochondrial) or extrinsic (death receptor regulated) pathway[124-126]. Early studies showed that ethanol selectively depletes mitochondrial GSH and sensitizes cultured hepatocytes to TNFα-induced apoptosis[127]. Chronic ethanol treatment also increases the expression of CD95 and induces the onset of mitochondrial permeability transition to promote apoptosis in rat hepatocytes[128]. Szabo and colleagues recently discovered that ethanol-induced apoptosis requires activation of interferon regulator factor 3 (IRF3), which is a transcription factor involved in regulating innate immunity. They found that ethanol-induced ER stress caused IRF3 association with the ER adaptor stimulator of interferon genes (STING), which then activated IRF3 by phosphorylation. Interestingly, they found that IRF3 activation was required for initiation of ethanol-induced hepatocyte apoptosis in mice fed ethanol for 4 wk and that its role in apoptosis induction was independent of its role in innate immunity regulation and inflammation[129].
In addition to apoptosis, ethanol can also induce hepatocyte cell death via the necrosis pathway. Necrosis, which was initially thought of as a non-programmed cell death response, is characterized by cell swelling, membrane rupture, and release of cell contents that leads to a subsequent inflammatory response[124,130]. However, recent evidence suggests that necrosis can also be highly regulated, which involves the receptor-interacting protein kinase 1 (RIP1) and RIP3, a process also referred to as necroptosis or programmed necrosis[131-134]. Necroptosis is similar in nature to necrosis, but is a caspase-independent programmed form of cell death that requires initiation by death receptors, similar to the extrinsic apoptotic pathway. Upon TNFα binding to its receptor TNFR1, it recruits downstream factors such as TNFR-associated death domain (TRADD), RIP1, TNFR-associated factor 2 (TRAF2), and cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1/2) to form the pro-survival TNFR complex I[135,136]. In addition to the E3 ligases cIAP1/2, the linear ubiquitin assembly complex (LUBAC) is also recruited to this complex, which triggers the ubiquitination of RIP1. Ubiquitinated RIP1 then serves as a scaffold protein to recruit the IκB kinase (IKK) complex to activate the NF-κB pathway[135,136]. Depending on the cellular conditions, RIP1 can also be de-ubiquitinated by the enzyme cylindromatosis (CLYD), which then recruits TRADD, the Fas-associated protein with a death domain (FADD) and caspase-8 to form the pro-death complex II. Activated caspase-8 then further cleaves Bid to trigger the mitochondrial-apoptotic pathway (Type II cells) or directly cleaves and activates downstream caspase-3 (Type I cells) to induce apoptosis[137,138]. RIP3 can also be recruited to complex II to form complex IIb (also called the necrosome), which includes RIP1, RIP3, FADD and caspase-8. Activated caspase-8 cleaves RIP3 to inactivate RIP3, suggesting that induction of apoptosis can suppress necroptosis[139]. However, in the absence of caspase-8 activation, RIP1-RIP3 then activates necroptosis. RIP1 and RIP3 are serine/threonine kinases, and their kinase activities are necessary for the formation of the necrosome[134,139]. Xiaodong Wang’s group recently discovered that mixed lineage kinase domain-like protein (MLKL) is a downstream target of RIP1-RIP3, which is critical for TNFα-induced necroptosis[140]. They further identified that RIP3 also interacts with phosphoglycerate mutase family member 5 (PGAM5), a mitochondrial phosphoglycerate mutase, which can dephosphorylate dynamin-related protein 1 (Drp1), a critical mitochondrial fission molecule. Dephosphorylated Drp1 translocates to mitochondria to induce mitochondrial fragmentation and subsequent necroptosis[141].
Induction of RIP1-RIP3-mediated necroptosis seems to also be pathologically and physiologically relevant. For example, RIP1-RIP3-mediated necroptosis has been shown to be involved in ischemic brain injury[142], ischemic-reperfusion-induced myocardial injury[143], kidney injury[144], pancreatitis[134], skin inflammation[145] and immune response against certain viral infections[134,146]. In addition, we and others also recently reported that RIP3 and MLKL are important in acetaminophen-induced necrosis in mouse liver[147,148].
RIP3-mediated necroptosis has recently been shown to play a role in ALD[149]. Nagy and colleagues showed that RIP3 was induced by ethanol feeding in mouse livers. ALD patients had increased hepatic expression of RIP3 compared to control patients. Furthermore, ethanol-induced liver injury, steatosis, and inflammation were decreased in RIP3 KO mice compared to control mice, verifying the importance of RIP3 in mediating ethanol-induced liver injury and progression of ALD[149]. Since ethanol also induces apoptosis in hepatocytes and apoptosis has been found in human ALD, it is not clear how these two events occur during the pathogenesis of ALD because apoptosis would normally suppress necrosis by caspase-mediated cleavage of RIP3. It is possible that different cell death modes could occur in different stages of ALD pathogenesis. Perhaps apoptosis occurs in early ALD, such as in steatosis, and necrosis occurs in later stages of ALD, such as in AH. Future work is needed to further elucidate these possibilities. Moreover, it is also unclear whether RIP1 or downstream MLKL or PGAM5 also play a role in ALD. The possible switch between apoptosis and necrosis may also raise some concerns on the current ongoing clinical trial using a pan-caspase inhibitor for ALD[44]. It will be interesting to see the outcomes of this clinical trial and how the results correlate with experimental animal studies.
Zinc
As one of the most prevalent trace elements in the body, zinc plays a critical role in regulating enzyme activity, metabolism, signal transduction, and gene expression, making it an important element for maintenance of human health[150,151]. As early as the 1950s, it was observed that zinc deficiency is associated with liver disease, particularly in human ALD[152]. Decreases in serum zinc concentrations correlate with liver damage and ALD progression[153-155], and supplementation with zinc decreases ethanol-induced liver injury in rodent models[156,157]. In addition, zinc supplementation in human ALD-induced cirrhosis patients improved liver function, emphasizing the importance of maintaining proper zinc levels in ethanol-induced liver disease[158]. Another important piece of experimental evidence supporting the role of zinc in ALD is that metallothionein (MT) transgenic mice with hepatic overexpression of MT, a major protein regulating cellular zinc homeostasis, had elevated zinc levels and were resistant to ethanol-induced liver injury. In contrast, MT knockout mice, which had reduced hepatic zinc levels, were more susceptible to alcohol toxicity[159,160]. Mechanistically, supplementation of zinc can improve zinc-deficiency-induced gut permeability and improve alcoholic endotoxemia[161]. In addition, zinc deficiency allowed for a greater increase in liver neutrophil infiltration, gut permeability and up-regulation of some pro-inflammatory genes after ethanol feeding compared to ethanol feeding alone, suggesting that zinc deficiency may worsen the ethanol-induced inflammatory response[162]. Dietary Zinc deficiency exacerbated ethanol-induced liver injury and steatosis by increasing ethanol-induced inflammation, oxidative stress, and plasma leptin levels[162]. In contrast, Zinc supplementation reduced chronic ethanol-feeding induced oxidative stress via suppressing ethanol-induced Cyp2e1 activity while increasing the activity of alcohol dehydrogenase in the liver[163]. Moreover, zinc supplementation reduced ethanol-induced apoptosis possibly through inhibiting ethanol-induced serum and hepatic TNFα levels and TNF-R1 and Fas proteins in the liver[164]. Furthermore, zinc supplementation enhanced liver regeneration after ethanol treatment likely by preserving the function of nuclear factor-4α (HNF-4α), a liver-enriched, zinc-finger transcription factor[157]. However, HNF-4α is known to inhibit hepatocyte proliferation and loss of HNF-4α promotes HCC[165]. Therefore, increased HNF-4α expression during zinc-enhanced liver regeneration in chronic ethanol fed mice could reflect a compensatory effect rather than a contributory factor for liver regeneration. Zinc supplementation also attenuated alcohol-induced increases in plasma triglycerides and partially reversed decreases in gonadal adipose depot mass, which was likely due to the restoration of PPAR-α activity[156]. Moreover, zinc deficiency suppressed expression of hepatocyte growth factor (HGF), insulin-like growth factor I (IGF-I), insulin-like growth factor binding protein 1 (IGFBP1), MT, and cyclin D1 in cultured HepG2 cells[157]. It will be interesting to see whether zinc supplementation also affects these factors in ethanol fed mouse livers. Finally, Liuzzi et al[166] recently showed that zinc is necessary for autophagy induction after ethanol treatment, which has been shown to be a protective mechanism against ethanol-induced liver injury and steatosis (see a later section for further discussion). Taken together, there is no doubt that zinc deficiency contributes to both experimental and human ALD pathogenesis, but more clinical evidence is still needed to confirm the beneficial effects of zinc supplementation in attenuating ALD.
PROTECTIVE PATHWAYS IN ALD
As discussed above, owing to the progress of decades of research on ALD, the pathogenesis of ALD and mechanisms for alcohol-induced liver injury have been well studied[38-40]. However, cells, animals and humans may activate some adaptive responses against the detrimental effects caused by alcohol exposure. Indeed, in addition to the detrimental effects caused by alcohol exposure during the pathogenesis of ALD, emerging evidence now supports that alcohol exposure can also activate cellular protective pathways to alleviate these detrimental effects, including some beneficial cytokines, lipin-1, autophagy, and the FoxO3 transcription factor. Modulating these protective pathways may also offer a novel avenue for treating ALD.
Cytokines
In addition to inducing detrimental inflammatory cytokines such as TNFα and IL-1, alcohol consumption also induces IL-6, IL-10 and IL-22, which may have some beneficial effects against ALD. IL-6 is both a pro and anti-inflammatory cytokine, but it has a hepatoprotective role in ALD by inducing signal transducer and activator of transcription 3 (STAT3) activation. IL-6 induces activation of STAT3 in ALD via binding to its IL-6 receptor and gp130 subunit in hepatocytes, which leads to activation of the Janus-Kinase (JAK)-STAT3 pathway resulting in up-regulation of anti-apoptosis and anti-oxidative stress genes along with down-regulation of fatty acid synthesis genes[39,167,168]. IL-10 is an anti-inflammatory cytokine that also activates the STAT3 pathway as a protective mechanism by binding to its IL-101 and IL-102 receptors, which results in inhibition of pro-inflammatory cytokine production and innate immunity activation[39,168-170]. The roles of IL-10, IL-6, and the JAK/STAT3 pathway in ALD have been extensively reviewed elsewhere[39,167-170]. Therefore, this section will focus on the recent findings regarding the role of IL-22 in protection against alcohol-induced liver injury and steatosis.
IL-22, previously known as IL-10-related T cell inducible factor (IL-TIF)[171], is secreted by T cells (TH1, TH17, TH22, γδ, natural killer, and cytotoxic)[172,173] and shares 22% amino acid sequence identity with IL-10[171]. IL-22 initiates its signaling pathway by binding to IL-10R2 and IL-22R1 receptors as a heterodimer[174]. After binding IL-22R1, the IL-22/IL-22R1 complex binds to the IL-10R2 receptor[175-177] to initiate activation of STAT3 via tyrosine phosphorylation[178]. IL-22 has also been shown to activate STAT1, STAT5, extracellular signal-related kinase 1 and 2 (ERK1/2), c-JUN N-terminal kinase (JNK), and p38 to a lesser extent in some cell lines[178]. The IL-10R2 receptor is ubiquitously expressed on many cell types, but the IL-22R1 receptor is mainly expressed on epithelial cells in skin, kidney, pancreas, small intestine, and colon. In addition, liver hepatocytes have been shown to express IL-22R1[172]. IL-22 has been shown to promote pathogenesis of certain diseases such as rheumatoid arthritis[179,180] and psoriasis[181], but IL-22 has also been shown to be protective in various disease models such as T-cell mediated hepatitis[182], ischemia-reperfusion[183], and ALD[46,184]. It was recently shown that treatment with recombinant IL-22 protein or injection of IL-22 adenovirus reduced chronic-binge ethanol-induced liver injury and steatosis in mice[46]. This protection was due to STAT3 activation because IL-22 recombinant protein and adenovirus overexpression of IL-22 did not protect against chronic-binge ethanol-induced liver injury or steatosis in STAT3 hepatocyte-specific knockout mice. Moreover, IL-22 may protect against steatosis by decreasing the expression of fatty acid transport protein (FATP)[46]. IL-22 treatment was also protective in an acute ethanol model where mice were gavaged each day for 7 d and treated with IL-22 one hour after gavage. IL-22 treatment reduced hepatocyte apoptosis and steatosis after ethanol binge[184]. IL-22 treatment in this model resulted in decreased lipid peroxidation and glutathione depletion after ethanol binge. In addition, TNFα expression in the liver was reduced with IL-22 treatment, indicating that IL-22 may protect against ethanol-induced inflammation and injury[184].
IL-22 treatment was also shown to be protective against hepatic fibrosis. In addition to hepatocytes, Kong et al[185] recently showed that quiescent and activated HSCs express IL-10R2 and IL-22R1, and HSC IL-22R1 mRNA and protein expression levels increased after IL-22 treatment. Similar to hepatocytes, IL-22 also induced STAT3 activation in HSCs[185]. Deletion of IL-22 increased fibrosis in a CCl4 mouse model of fibrosis[186], and IL-22 protected against CCl4-induced liver fibrosis by promoting HSC senescence[185]. IL-22-induced HSC senescence required STAT3 and suppressor of cytokine signaling 3 (SOCS3) through p53- and p21-dependent pathways[185]. Unfortunately, there is not an available model for inducing significant fibrosis via alcohol treatment alone to test the anti-fibrogenic effects of IL-22. Furthermore, the role of IL-22 in protection against later stages of ALD, such as in ALD-induced liver fibrosis and cirrhosis, is unknown and still needs to be further investigated.
Lipin-1
The lipin family proteins are evolutionarily conserved proteins that play a critical role in regulating lipid metabolism and related diseases. There are three lipin proteins (lipin-1, lipin-2 and lipin-3) in mammals and orthologous of these lipin genes are also found in plants, invertebrates, and single cell eukaryotes[187,188]. The lipin1 gene (LPIN1) was originally discovered from the mouse strains BALB/cByl-fld and C3H/HeJ-fld that carry mutations in fatty liver dystrophy[189]. The fld mouse is deficient for lipin1 due to a null mutation in the LPIN1 gene, and these mutant mice lack normal adipose tissue depots throughout the body and develop peripheral neuropathy[189]. These phenotypes suggest that lipin1-deficiency impairs lipid metabolism in liver, adipose tissue and peripheral nerves. Autosomal recessive LPIN1 mutations have also been identified in humans, and these people develop severe rhabdomyolysis and recurrent acute myoglobinuria in early childhood[190,191]. The three mammalian lipins have very distinctive but overlapping tissue expression patterns. Lipin-1 is mainly expressed in adipose tissue, skeletal muscle, and testis and has lower expression levels in liver, kidney, lung, brain and heart. Lipin-2 is highly expressed in liver and brain, whereas lipin-3 exhibits high expression levels in intestine and other regions of the gastrointestinal tract[188]. All three lipin proteins exhibit Mg2+-dependent phosphatidate phosphatase (PAP) activity that catalyzes triacylglycerol synthesis by converting phosphatidic acid to diacylglycerol (DAG). However, it appears that lipin-1 has higher PAP activity than lipin-2 and lipin-3. In addition to regulating triglyceride synthesis, the mammalian lipin proteins also have a LXXIL motif that allows them to act as a transcriptional co-activator, but this transcriptional co-activator activity has only been convincingly demonstrated for mouse lipin-1[192].
Lipin-1 is a key regulator of lipid metabolism that also exists in three isoforms as a consequence of alternative splicing: lipin-1α, lipin-1β, and lipin-1γ. Lipin-1α is a mostly nuclear protein that is important for adipogenesis. Lipin-1β is mainly localized to the cytosol and is involved in lipogenesis. It is also the major isoform expressed in the liver. Lipin-1γ is mostly localized to lipid droplets and expressed in brain tissues[193,194]. Lipin-1 regulates lipid metabolism via two pathways involving nuclear and cytosolic isoforms of lipin-1. As discussed above, cytosolic lipin-1β has PAP activity and regulates triacylglycerol synthesis. Nuclear lipin-1α has transcriptional co-activator activity and regulates genes involved in β-oxidation and lipid synthesis[187]. Specifically, nuclear lipin-1α co-activates peroxisome PPARα and PGC-1α and inhibits expression of SREBP-1, which are genes important for lipid break down by β-oxidation and for lipid synthesis, respectively[192,195]. Interestingly, posttranslational modification of lipin1 seems to play a critical role in regulating the cellular localization of lipin-1. Insulin increased the phosphorylation of multiple sites on lipin-1, which was likely via downstream PI3K. Lipin-1 was dephosphorylated in response to epinephrine or oleic acid. The phosphorylation status of lipin-1 did not change the PAP activity of lipin-1, but rather regulated the cellular location of lipin-1, in which phosphorylated lipin-1 was found to translocate from cytosol to the microsomal fraction[196]. It is well known that the mammalian target of rapamycin complex I (mTORC1) is a key sensor for cellular nutrients that positively regulates the anabolic process including protein synthesis and lipogenesis. Interestingly, mTORC1 directly phosphorylates lipin-1, and inactivation of mTORC1 leads to the retention of nuclear lipin-1. Dephosphorylated nuclear lipin-1 regulates the nuclear abundance and promoter activity of SREBP, which represses SREBP-dependent transcription of lipogenesis genes[195]. Mice deficient in mTORC1 are resistant to hepatic steatosis induced by a high-fat and cholesterol diet in a lipin-1 dependent fashion[195]. In addition to phosphorylation, lipin-1 was found to be sumoylated in neuronal cells, and sumoylation promotes the nuclear localization of lipin-1, which appears to promote the transcriptional co-activator activity of liplin-1[197].
Recent studies from Min You’s group suggest that lipin-1 also plays a protective role in ALD by preventing ethanol-induced steatosis and liver injury[198-200]. They found that ethanol significantly increased the cytosolic pro-lipogenic activity of lipin-1β but decreased the nuclear entry of lipin-1α by chronic ethanol exposure in cultured hepatocytes and in mouse liver resulting in excessive liver fat accumulation[200]. Interestingly, while ethanol consumption increased PAP activity in wild type mouse livers, liver-specific lipin-1-deficient mice showed markedly greater steatosis and liver injury as well as increased hepatic pro-inflammatory cytokine production compared to pair-fed control wild type mice with a modified Lieber-DeCarli ethanol diet for 4 wk[200]. These surprising results suggest that induction of lipin-1 by ethanol in wild type mice may actually serve as a protective mechanism against alcohol-induced steatosis. Alternatively, it would suggest that other functions of lipin-1, excluding cytosolic lipin-1β form-mediated triglyceride synthesis, play more important roles in alcohol-induced steatosis. Indeed, they found that hepatic lipin-1 deficient mice showed impaired PGC1-α activation resulting in impaired hepatic fatty acid oxidation[200]. Thus, these data suggest that modulating the nuclear lipin-1α form might be a promising target to attenuate alcohol-induced steatosis by promoting PGC1-α activation.
Ethanol exposure robustly induced activity of a mouse lipin-1 promoter, promoted cytoplasmic localization of lipin-1, and caused excess lipid accumulation, both in cultured hepatic cells and in mouse livers[199]. Mechanistic studies showed that ethanol feeding reduced the sumoylation levels of lipin-1 while increasing its acetylation, resulting in reduced nuclear retention and increased cytosolic levels of lipin-1. Ethanol-induced lipin-1 gene expression was inhibited by a known activator of AMPK or overexpression of a constitutively active form of AMPK. Activation of AMPK by 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) suppressed ethanol-mediated induction of lipin-1 gene-expression, which was abolished by the overexpression of the processed nuclear form of SREBP-1c. Furthermore, ethanol exposure significantly increased the association of acetylated histone H3 with the SRE-containing region in the promoter of the lipin-1 gene[199]. As discussed, ethanol metabolism increased cellular NADH/NAD+ ratio resulting in the inhibition of Sirt1 activity. Using the ethanol Gao-Binge model, Yin et al[198] showed that liver-specific Sirt1 knockout mice had greater hepatic lipid accumulation, inflammatory cytokine production and liver injury than wild type mice. Ethanol-treated liver-specific Sirt1 KO mice also had increased levels of ER stress and nuclear factor of activated T cells c4 (NFATc4) expression, which is an important inflammatory factor. Ethanol-treated liver-specific SIRT1 KO mice had decreased mRNA and protein levels of SFRS10, which is a SR-LIKE protein family of splicing factors that favors the lipin-1β isoform and is sufficient to increase expression of lipogenic genes and induces hepatic steatosis in mice[198]. This decrease in SFRS10 was associated with an increased lipin-1β/lipin-1α ratio. Ethanol treatment significantly increased the lipin-1β/lipin-1α ratio in AML-12 cells compared to control treatment, which was blocked by overexpressing either Sirt1 or SFRS10. More importantly, AH patients were found to have decreased expression of lipin-1α and had a higher lipin-1β/lipin-1α ratio[198]. These results indicate that targeting hepatic Sirt1 may be a promising approach to attenuate ALD by altering the lipin-1 signaling pathway.
Autophagy
Macroautophagy, hereafter referred to as autophagy, is an evolutionarily conserved process that results in degradation of cellular proteins and organelles due to a cell’s “self-eating” in response to starvation and other cellular stress conditions. Autophagy involves the formation of double-membrane autophagosomes, which is tightly regulated by the autophagy-related (Atg) genes. Two ubiquitin-like conjugation systems regulate autophagy induction. The first of these systems includes Atg7 (E1) and Atg10 (E2), which promote Atg5 conjugation with Atg12. The second involves Atg7 (E1) and Atg3 (E2), which promote microtubule-associated protein 1 light chain 3 (LC3) conjugation with phosphatidylethanolamine (PE), which is critical for the elongation of pre-autophagosome structures and formation of autophagosomes. Before its conjugation with PE, LC3 is a cytosolic protein (referred to as LC3-I), and the LC3-PE conjugated form (referred to as LC3-II) targets autophagosome membranes. The level of LC3-II has been widely used as an autophagy marker to monitor autophagy activation[201,202]. Autophagosomes then engulf individual organelles, protein aggregates, or portions of cytoplasm before fusing with lysosomes to form autolysosomes where the engulfed contents are degraded[203]. Autophagy is a protective process that can be either selective or non-selective. Non-selective autophagy occurs during starvation to break down the cell’s components in order to provide a source of energy and nutrients. Selective autophagy occurs in either nutrient-rich or poor conditions as a protective mechanism by ridding the cell of protein aggregates and damaged or excess organelles[204,205].
Autophagy was originally discovered in the liver, and accumulating evidence has demonstrated that autophagy plays a critical role in regulating liver physiology and pathogenesis of various liver diseases[206-208]. Our lab and others have recently demonstrated that alcohol consumption may activate autophagy to selectively remove excess lipid droplets and damaged mitochondria and in turn attenuate alcohol-induced steatosis and liver injury in mice[209-213]. However, there has also been evidence to suggest that alcohol consumption may also suppress autophagy, particularly in chronic alcohol consumption conditions[214,215]. Several possibilities could explain these discrepancies, including animal models used to assess ALD, assays used to determine autophagy and the limitation of steady-state assessment of autophagy using one single time point in vivo. For example, it is now well known that use of LC3-II levels to monitor autophagy is troublesome because LC3-II itself is degraded in the autolysosomes during autophagy. For this reason, autophagic flux assays (assessing LC3-II levels with and without a lysosomal inhibitor) are now mandatory for assessing autophagy[201], and autophagy flux was not always evaluated when determining the effect of ethanol on autophagy. Autophagy is also a dynamic process, and autophagic activity can fluctuate during experimental conditions over time[216]. Moreover, autophagy activity can be influenced by circadian rhythm[217]. Therefore, special attention should be paid to experimental conditions when evaluating the effect of alcohol on autophagy.
Using an acute ethanol gavage model (33% v/v, 4.5 g/kg), we first demonstrated that ethanol treatment increased autophagosome numbers by electron microscopy, assessment of GFP-LC3 positive autophagosomes by confocal microscopy and detection of LC3-II protein levels by Western blot analysis in vivo and in vitro[211]. Intriguingly, we further demonstrated that acute ethanol-induced autophagy seemed to selectively remove damaged mitochondria and excess lipid droplets, but not long-lived proteins[213]. The induction of autophagy was mediated by ethanol-induced production of ROS and inhibition of mTOR, and induction of autophagy required ethanol metabolism. In addition to liver, acute ethanol treatment also increased autophagy in mouse myocardial cells and in the developing brain through inhibition of mTOR[218,219]. While autophagy was found to serve as a protective role against ethanol-induced neurotoxicity similar to the liver, it might contribute to ethanol-induced myocardial dysfunction[218,219].
In line with our findings, some recent studies also found that chronic ethanol treatment increased autophagosome content and autophagic flux in mouse livers and cultured hepatocytes[212,215]. Otsuki’s group recently showed that autophagy was protective in chronic ethanol treated rats fed the Lieber-DeCarli ethanol diet for 10 wk. This group noticed an induction of autophagosomes engulfing damaged mitochondria or lipids in addition to several lysosomes containing degraded organelles in ethanol-treated rats compared to control rats by electron microscopy. Interestingly, they found several autophagosomes that contained both mitochondria and lipid droplets, which suggest that these degradative pathways may be linked. They also saw an induction of LC3-II puncta and an increase in autophagosome-lysosome fusion after ethanol treatment compared to controls[220]. However, this study lacked an autophagy flux assay, so whether the accumulation of autophagosomes was due to the induction of autophagy by ethanol treatment was not known. Lin et al[212] also showed that chronic ethanol consumption activated autophagy using mice fed the Lieber-DeCarli diet for 4 wk. They showed that autophagy was activated in the chronic feeding model using an autophagy flux assay, where co-treatment with CQ and ethanol diet increased GFP-LC3 puncta and protein levels more than ethanol-treatment alone. However, they only treated mice with the ethanol diet for 4 wk, so the role of ethanol feeding for a longer period of time on autophagy should be more critically evaluated.
In contrast to the evidence supporting acute and chronic ethanol induction of autophagy, some other studies suggest that ethanol may suppress autophagy in liver and pancreas[214,221,222]. It is well known that chronic alcohol consumption can cause hepatomegaly and protein accumulation[214,223], which would suggest impaired autophagy in this chronic alcohol exposure scenario. However, it should be noted that alcohol consumption has been shown to inhibit hepatic proteasome activity, another important cellular catabolic pathway in addition to autophagy[210,224,225]. Moreover, there is crosstalk between the proteasome and autophagy, and proteasome inhibition can increase autophagy as a compensatory mechanism[226-228]. Therefore, chronic alcohol consumption-induced accumulation of hepatic proteins and hepatomegaly could be due to multiple factors and might not be due simply to impaired autophagy. While Cederbaum’s group recently reported that acute ethanol inhibited autophagy, their observations were only based on the observations that ethanol treatment decreased LC3-II protein and LC3 positive puncta levels, and an autophagy flux assay was not implemented[221,229]. In addition, the previously discussed effects of circadian rhythm and dynamic nature of autophagy will make it technically challenging to determine autophagic flux in a chronic ethanol consumption scenario. Despite the controversy on the autophagy status of acute versus chronic alcohol exposure conditions, all studies unanimously demonstrated a beneficial role for autophagy in protecting against alcohol-induced steatosis and hepatotoxicity. Therefore pharmacological induction of autophagy may be a promising approach for treating ALD.
In addition to hepatocytes, there are many other cell types such as HSCs and macrophages in the liver that may also play a role in the pathogenesis of ALD. Emerging evidence indicates that autophagy in other cell types in the liver may also be critical in liver physiology and pathogenesis. HSCs are one of the key factors in regulating hepatic fibrosis, and recent evidence suggests that autophagy in HSCs promotes liver fibrosis likely by providing free fatty acids as an energy source for HSCs activation through lipophagy[230]. Cre-induced specific deletion of Atg7 in HSCs attenuated CCl4-induced fibrosis in vivo[230]. The decreased fibrogenic capacity of HSCs by inhibiting autophagy was also confirmed in vitro using primary cultured HSCs and immortalized HSC cell lines[230,231]. Interestingly, a study from Friedman’s group also recently showed that autophagy was activated in HSCs in an 8-wk chronic ethanol feeding model in rats. They showed that ER stress was induced in HSCs isolated from ethanol-fed rats, and that this ER stress further induced autophagy activation and subsequent HSC activation[232]. These results imply that chronic ethanol-induced autophagy in HSCs may promote fibrosis during the pathogenesis of ALD. In contrast to HSCs, specific deletion of autophagy in macrophages was also reported to exacerbate CCl4-induced fibrosis in mouse livers by promoting HSC activation through enhanced secretion of inflammatory cytokines from macrophages[233]. Moreover, we also found that hepatocyte-specific Atg5 knockout mice had severe liver injury, and these mice develop fibrosis (Ni et al unpublished observations). While it will be technically difficult to pharmacologically target autophagy in a specific cell type in the liver, rapamycin, an autophagy inducer, showed beneficial effects against CCl4- or bile duct ligation-induced fibrosis in rat livers[234,235]. It will be interesting to determine the autophagy status in different cell types after rapamycin and CCl4 treatment in mouse livers in the future. As discussed above, because of a lack of proper animal models to study fibrosis in ALD, it is not yet clear how modulating autophagy would affect fibrosis in ALD pathogenesis.
FoxO3
FoxO3 is a member of the Forkhead box-containing protein, class O (FoxO) family of DAF-16 like transcription factors, which is ubiquitously expressed and evolutionarily conserved[236-238]. There are four FoxO proteins in mammals: FoxO1, FoxO3, FoxO4 and FoxO6[237]. FoxO1, FoxO3 and FoxO4 are ubiquitously expressed in most tissues whereas FoxO6 is mainly expressed in neurons[237,238]. The activities of FoxO family proteins are mainly regulated by multiple post-translational modifications, including phosphorylation, acetylation, methylation and ubiquitination[236,238]. FoxO3 has recently been shown to play an important role in protection against alcohol-induced steatosis and liver injury via induction of autophagy and antioxidant gene expression, which is further discussed below.
Accumulating evidence now supports that FoxO family proteins can regulate autophagy by at least three distinctive mechanisms: direct transcriptional up-regulation of autophagy-related genes[239,240], modulation of intracellular glutamine levels[241] and direct interaction with Atg7 independent of transcription activity[242]. It was first reported that FoxO3 controls the transcription of autophagy-related genes, including LC3, Atg12, Beclin 1, ULK2 and Bnip3, to promote autophagy in skeletal muscle in mice and cultured C2C12 myotubes[239,240]. Subsequently, it was found that FoxO1 and FoxO3 are also required to regulate the expression of autophagy-related genes as well as antioxidant genes in protection against ischemia/reperfusion-induced cardiomyocyte injury in mice[243]. In the liver, FoxO1 was found to directly regulate expression of Atg14 and in turn regulate hepatic autophagy to control hepatic lipid homeostasis likely via promoting lipophagy[244]. In addition to directly regulating expression of autophagy-related genes, activation of FoxO also up-regulates the expression of glutamine synthetase resulting in increased production of glutamine. Increased glutamine inhibits mTOR activity, likely via suppressing translocation of mTOR to the lysosomal membrane, to trigger autophagy for cell survival[241]. In response to oxidative stress or serum starvation, FoxO1 was acetylated by dissociation from SIRT2, a cytosolic deacetylase. Acetylated FoxO1 then bound to Atg7 to promote autophagy in several cancer cell lines, although it was not clear how the interaction of FoxO1 and Atg7 promoted autophagy[242].
Recent studies from our lab and others suggest that FoxO3 plays a role against alcohol-induced steatosis and hepatotoxicity[245-247]. Using an acute ethanol binge model, we recently demonstrated that ethanol treatment increased the expression of autophagy-related genes in mouse liver and in primary cultured mouse and human hepatocytes. More importantly, hepatic nuclear accumulation of FoxO3 was increased in ethanol-treated mouse livers and primary cultured mouse hepatocytes, which was due to decreased Akt-mediated FoxO3 phosphorylation at Ser253. Activating SIRT1 by resveratrol caused deacetylation of FoxO3, which increased ethanol-induced expression of autophagy-related genes compared to cells only treated with ethanol. More importantly, FoxO3 knockout mice had decreased expression of autophagy-related genes and had increased steatosis and liver injury compared to wild type mice after acute ethanol treatment. These results support a protective role of FoxO3-mediated autophagy against alcohol hepatotoxicity[245]. In contrast, it was reported that ethanol inhibited expression of several autophagy-related genes by promoting cytosolic FoxO3 translocation, which was reversed by globular adiponectin[248]. However, HepG2, a human hepatoma cell line, was used in this study. The cancerous origin and the lack of alcohol metabolism enzymes in HepG2 cells may explain the contradictory observations. Recent studies from Dr. Weinman’s group also support a protective role of FoxO3 in HCV and alcohol-induced liver injury model. Consistent with findings from the acute alcohol model, FoxO3 knockout mice fed the Lieber-DeCarli alcohol diet for 3 wk developed more severe steatosis, inflammation and liver injury compared to wild type mice. Intriguingly, in cultured Huh7 cells, a human hepatoma cell line that expresses alcohol dehydrogenase, combined treatment with ethanol and HCV decreased FoxO3 nuclear retention and transcriptional activity, but either ethanol or HCV infection alone increased FoxO3 transcriptional activity in HuH7 cells[246,247]. Using a capillary isoelectric focusing (IEF) approach, they were able to identify several patterns of FoxO3 posttranslational modifications to dissect the differential roles of ethanol, HCV and their combination on FoxO3 activity. A novel JNK phosphorylation site at Ser574 on FoxO3 was induced by HCV, which promoted FoxO3 nuclear retention and transcriptional activity. In contrast, ethanol treatment inhibited arginine-methylation of FOXO3, which increased FoxO3 nuclear export and degradation of the JNK phosphorylated form. Consequently, HCV and ethanol co-treatment decreased FoxO3-mediated expression of superoxide dismutase 2, which may subsequently exacerbate HCV-alcohol-induced liver injury[246,247]. While more studies are definitely needed to determine whether other FoxO family proteins are also involved in alcohol-induced liver injury, these results support a protective role of FoxO3 against alcohol-induced hepatotoxicity in the normal liver by promoting the expression of autophagy-related genes and antioxidant genes. The possible role of FoxO3 and its regulation in ALD is summarized in Figure 2.
Figure 2.
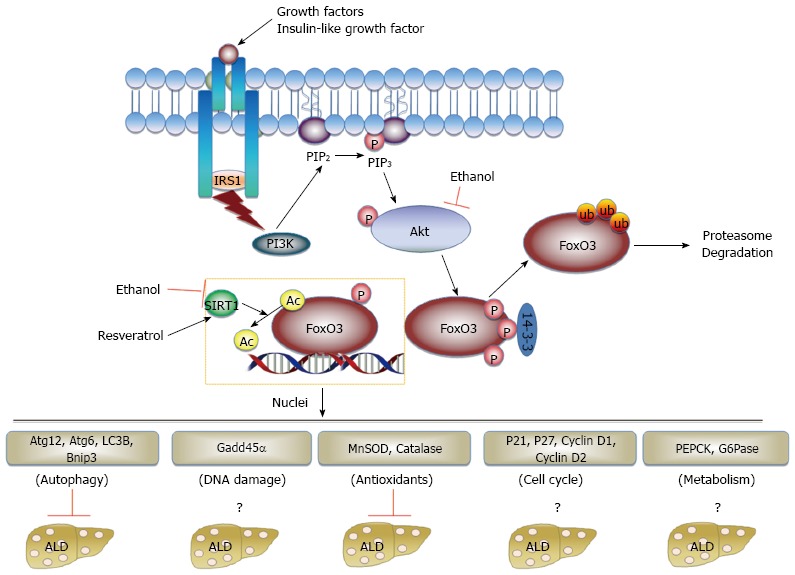
Regulation of FoxO3 and its role in alcohol-induced liver injury. Growth factors, such as insulin-like growth factor I (IGF-1), bind to their receptor and recruit insulin receptor substrate 1 (IRS-1), which further activates PI3K. Activated PI3K then converts the plasma membrane lipid phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-trisphosphate (PIP3), and PIP3 directly binds AKT to promote its activation. AKT phosphorylates FoxO3 resulting in its sequestration in the cytoplasm and interaction with 14-3-3 proteins. FoxO3 can also be ubiquitinated and degraded by the ubiquitin proteasome system, or deacetylated by SIRT1, which regulates the specificity of FoxO3 target gene expression. Dephosphorylated nuclear retained FoxO3 then binds to the promoter regions of target genes with various distinctive functions, including autophagy, DNA damage, antioxidants, cell cycle and metabolism. Acute alcohol regulates hepatic FoxO3 by inhibiting AKT, resulting in increased nuclear retention of FoxO3, which leads to increased expression of autophagy-related genes. Acute alcohol also increases acetylated levels of FoxO3, which may serve as a negative regulator of acute alcohol-induced expression of autophagy-related genes. Resveratrol activates SIRT1, which leads to FoxO3 deacetylation and enhanced expression of autophagy-related genes. Alcohol-induced activation of FoxO3 also promotes the expression of antioxidant genes. Whether alcohol affects other FoxO3-mediated target genes such as those involved in DNA damage, cell cycle and metabolism remains to be studied. Overall, alcohol-induced activation of FoxO3 protects against alcohol-induced steatosis and liver injury.
CONCLUSION
ALD is a major health problem in the United States and worldwide, which claims millions of lives each year. During the past decades, significant progress has been made to understand the key events and molecular players for the onset and progression of ALD as discussed previously. Unfortunately there are no successful treatments available for treating ALD; therefore, development of novel pathophysiological-targeted therapies is urgently needed. Recent evidence showed that alcohol consumption induces various detrimental effects including changes in miRNA expression, a decrease in SAM/SAH ratio, an increase in cytosolic lipin-1β expression, induction of IRF3-mediated apoptosis and RIP3-mediated necrosis, and decreases in hepcidin and zinc levels. Meanwhile, it was also shown that alcohol can activate several adaptive protective responses to attenuate alcohol-induced liver pathogenesis including FoxO3, IL-22, autophagy and nuclear lipin-1α. The role of osteopontin in ALD is still controversial, but it seems that milk osteopontin protects against ALD (Figure 3). Several clinical trials funded by the NIAAA are ongoing to target these detrimental effects induced by alcohol consumption including use of the antibiotics rifaximin and norfloxacin to decrease plasma endotoxin, IL-1 receptor antagonist anakinra to inhibit hepatic recruitment of inflammatory cells, pan-caspase inhibitor emricasan to block caspase-mediated apoptosis and sterile inflammation as well as farnesoid X receptor agonist obeticholic acid to improve bile acid metabolism in AH[44]. Development of new approaches to target the discussed protective mechanisms induced by alcohol consumption, such as FoxO3 and IL-22, is much anticipated. Indeed, the use of IL-22 for treating AH has recently been proposed and might be in clinical trials shortly[44]. In addition, development of new animal models that accurately represent human ALD pathogenesis is needed because current animal models of alcoholic-liver disease do not result in progression beyond mild liver injury and steatosis, which makes studying ALD pathogenesis and development of novel therapeutics challenging. In summary, it is clear that the knowledge we have gained from experimental alcohol animal models concerning detrimental and protective effects in the liver induced by alcohol consumption will lead to advances in treatment of ALD, but better animal models are needed for expanding our understanding of ALD pathogenesis and for development of novel therapeutics.
Figure 3.
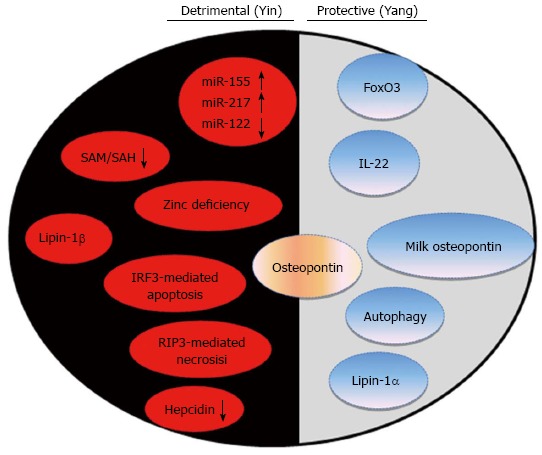
New progress in alcohol-induced detrimental (Yin) and adaptive protective (Yang) pathways in the liver. Experimental and clinical research for alcoholic liver disease (ALD) in the past several years has revealed many detrimental factors leading to alcohol-induced liver pathogenesis. In addition to detrimental factors, emerging evidence indicates that alcohol consumption can also activate cellular adaptive/compensatory protective mechanisms to attenuate alcohol-induced detrimental effects. The eventual outcomes seem to be decided by the balance between the “bad” (detrimental) and “good” (adaptive protective) factors induced by alcohol in each individual person or cell. This is more like a battle between “Yin” (the dark side of detrimental) and “Yang” (the light side of protective). The emerging detrimental factors induced by alcohol are listed on the dark side and the adaptive protective factors are listed on the light side. The role of osteopontin is still controversial. Therefore approaches to inhibit the dark side (detrimental “Yin”) or boost the light side (protective “Yang”) may provide novel therapeutic tools for treating ALD.
Footnotes
Supported by NIAAA funds R01 AA020518 and National Center for Research Resources No. 5P20RR021940 and No. T32 ES007079; Williams JA and Manley S are recipients of the Biomedical Research Training Program Fellowship from University of Kansas Medical Center
P- Reviewer: Dhanda A, Sheedfar F S- Editor: Qi Y L- Editor: A E- Editor: Zhang DN
References
- 1.Altamirano J, Bataller R. Alcoholic liver disease: pathogenesis and new targets for therapy. Nat Rev Gastroenterol Hepatol. 2011;8:491–501. doi: 10.1038/nrgastro.2011.134. [DOI] [PubMed] [Google Scholar]
- 2.Teli MR, Day CP, Burt AD, Bennett MK, James OF. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet. 1995;346:987–990. doi: 10.1016/s0140-6736(95)91685-7. [DOI] [PubMed] [Google Scholar]
- 3.Stewart SH. Racial and ethnic differences in alcohol-associated aspartate aminotransferase and gamma-glutamyltransferase elevation. Arch Intern Med. 2002;162:2236–2239. doi: 10.1001/archinte.162.19.2236. [DOI] [PubMed] [Google Scholar]
- 4.Stinson FS, Grant BF, Dufour MC. The critical dimension of ethnicity in liver cirrhosis mortality statistics. Alcohol Clin Exp Res. 2001;25:1181–1187. [PubMed] [Google Scholar]
- 5.Moshage H. Alcoholic liver disease: a matter of hormones? J Hepatol. 2001;35:130–133. doi: 10.1016/s0168-8278(01)00115-5. [DOI] [PubMed] [Google Scholar]
- 6.O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Hepatology. 2010;51:307–328. doi: 10.1002/hep.23258. [DOI] [PubMed] [Google Scholar]
- 7.Sato N, Lindros KO, Baraona E, Ikejima K, Mezey E, Järveläinen HA, Ramchandani VA. Sex difference in alcohol-related organ injury. Alcohol Clin Exp Res. 2001;25:40S–45S. doi: 10.1097/00000374-200105051-00007. [DOI] [PubMed] [Google Scholar]
- 8.Baraona E, Abittan CS, Dohmen K, Moretti M, Pozzato G, Chayes ZW, Schaefer C, Lieber CS. Gender differences in pharmacokinetics of alcohol. Alcohol Clin Exp Res. 2001;25:502–507. [PubMed] [Google Scholar]
- 9.Enomoto N, Yamashina S, Schemmer P, Rivera CA, Bradford BU, Enomoto A, Brenner DA, Thurman RG. Estriol sensitizes rat Kupffer cells via gut-derived endotoxin. Am J Physiol. 1999;277:G671–G677. doi: 10.1152/ajpgi.1999.277.3.G671. [DOI] [PubMed] [Google Scholar]
- 10.Iimuro Y, Frankenberg MV, Arteel GE, Bradford BU, Wall CA, Thurman RG. Female rats exhibit greater susceptibility to early alcohol-induced liver injury than males. Am J Physiol. 1997;272:G1186–G1194. doi: 10.1152/ajpgi.1997.272.5.G1186. [DOI] [PubMed] [Google Scholar]
- 11.Kono H, Wheeler MD, Rusyn I, Lin M, Seabra V, Rivera CA, Bradford BU, Forman DT, Thurman RG. Gender differences in early alcohol-induced liver injury: role of CD14, NF-kappaB, and TNF-alpha. Am J Physiol Gastrointest Liver Physiol. 2000;278:G652–G661. doi: 10.1152/ajpgi.2000.278.4.G652. [DOI] [PubMed] [Google Scholar]
- 12.Yin M, Ikejima K, Wheeler MD, Bradford BU, Seabra V, Forman DT, Sato N, Thurman RG. Estrogen is involved in early alcohol-induced liver injury in a rat enteral feeding model. Hepatology. 2000;31:117–123. doi: 10.1002/hep.510310119. [DOI] [PubMed] [Google Scholar]
- 13.Chiang DJ, McCullough AJ. The impact of obesity and metabolic syndrome on alcoholic liver disease. Clin Liver Dis. 2014;18:157–163. doi: 10.1016/j.cld.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naveau S, Giraud V, Borotto E, Aubert A, Capron F, Chaput JC. Excess weight risk factor for alcoholic liver disease. Hepatology. 1997;25:108–111. doi: 10.1002/hep.510250120. [DOI] [PubMed] [Google Scholar]
- 15.Xu J, Lai KK, Verlinsky A, Lugea A, French SW, Cooper MP, Ji C, Tsukamoto H. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J Hepatol. 2011;55:673–682. doi: 10.1016/j.jhep.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stepanova M, Rafiq N, Younossi ZM. Components of metabolic syndrome are independent predictors of mortality in patients with chronic liver disease: a population-based study. Gut. 2010;59:1410–1415. doi: 10.1136/gut.2010.213553. [DOI] [PubMed] [Google Scholar]
- 17.Degos F. Hepatitis C and alcohol. J Hepatol. 1999;31 Suppl 1:113–118. doi: 10.1016/s0168-8278(99)80386-9. [DOI] [PubMed] [Google Scholar]
- 18.Monto A, Patel K, Bostrom A, Pianko S, Pockros P, McHutchison JG, Wright TL. Risks of a range of alcohol intake on hepatitis C-related fibrosis. Hepatology. 2004;39:826–834. doi: 10.1002/hep.20127. [DOI] [PubMed] [Google Scholar]
- 19.Befrits R, Hedman M, Blomquist L, Allander T, Grillner L, Kinnman N, Rubio C, Hultcrantz R. Chronic hepatitis C in alcoholic patients: prevalence, genotypes, and correlation to liver disease. Scand J Gastroenterol. 1995;30:1113–1118. doi: 10.3109/00365529509101616. [DOI] [PubMed] [Google Scholar]
- 20.Harris DR, Gonin R, Alter HJ, Wright EC, Buskell ZJ, Hollinger FB, Seeff LB. The relationship of acute transfusion-associated hepatitis to the development of cirrhosis in the presence of alcohol abuse. Ann Intern Med. 2001;134:120–124. doi: 10.7326/0003-4819-134-2-200101160-00012. [DOI] [PubMed] [Google Scholar]
- 21.Altamirano J, Bataller R. Cigarette smoking and chronic liver diseases. Gut. 2010;59:1159–1162. doi: 10.1136/gut.2008.162453. [DOI] [PubMed] [Google Scholar]
- 22.Corrao G, Lepore AR, Torchio P, Valenti M, Galatola G, D’Amicis A, Aricó S, di Orio F. The effect of drinking coffee and smoking cigarettes on the risk of cirrhosis associated with alcohol consumption. A case-control study. Provincial Group for the Study of Chronic Liver Disease. Eur J Epidemiol. 1994;10:657–664. doi: 10.1007/BF01719277. [DOI] [PubMed] [Google Scholar]
- 23.Pessione F, Ramond MJ, Peters L, Pham BN, Batel P, Rueff B, Valla DC. Five-year survival predictive factors in patients with excessive alcohol intake and cirrhosis. Effect of alcoholic hepatitis, smoking and abstinence. Liver Int. 2003;23:45–53. doi: 10.1034/j.1600-0676.2003.01804.x. [DOI] [PubMed] [Google Scholar]
- 24.Danielsson J, Kangastupa P, Laatikainen T, Aalto M, Niemelä O. Dose- and gender-dependent interactions between coffee consumption and serum GGT activity in alcohol consumers. Alcohol Alcohol. 2013;48:303–307. doi: 10.1093/alcalc/agt017. [DOI] [PubMed] [Google Scholar]
- 25.Stroffolini T, Cotticelli G, Medda E, Niosi M, Del Vecchio-Blanco C, Addolorato G, Petrelli E, Salerno MT, Picardi A, Bernardi M, et al. Interaction of alcohol intake and cofactors on the risk of cirrhosis. Liver Int. 2010;30:867–870. doi: 10.1111/j.1478-3231.2010.02261.x. [DOI] [PubMed] [Google Scholar]
- 26.Monzoni A, Masutti F, Saccoccio G, Bellentani S, Tiribelli C, Giacca M. Genetic determinants of ethanol-induced liver damage. Mol Med. 2001;7:255–262. [PMC free article] [PubMed] [Google Scholar]
- 27.Zintzaras E, Stefanidis I, Santos M, Vidal F. Do alcohol-metabolizing enzyme gene polymorphisms increase the risk of alcoholism and alcoholic liver disease? Hepatology. 2006;43:352–361. doi: 10.1002/hep.21023. [DOI] [PubMed] [Google Scholar]
- 28.Ladero JM, Martínez C, García-Martin E, Fernández-Arquero M, López-Alonso G, de la Concha EG, Díaz-Rubio M, Agúndez JA. Polymorphisms of the glutathione S-transferases mu-1 (GSTM1) and theta-1 (GSTT1) and the risk of advanced alcoholic liver disease. Scand J Gastroenterol. 2005;40:348–353. doi: 10.1080/00365520510012109. [DOI] [PubMed] [Google Scholar]
- 29.Grove J, Daly AK, Bassendine MF, Day CP. Association of a tumor necrosis factor promoter polymorphism with susceptibility to alcoholic steatohepatitis. Hepatology. 1997;26:143–146. doi: 10.1002/hep.510260119. [DOI] [PubMed] [Google Scholar]
- 30.McClain CJ, Song Z, Barve SS, Hill DB, Deaciuc I. Recent advances in alcoholic liver disease. IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2004;287:G497–G502. doi: 10.1152/ajpgi.00171.2004. [DOI] [PubMed] [Google Scholar]
- 31.Bataller R, North KE, Brenner DA. Genetic polymorphisms and the progression of liver fibrosis: a critical appraisal. Hepatology. 2003;37:493–503. doi: 10.1053/jhep.2003.50127. [DOI] [PubMed] [Google Scholar]
- 32.Burza MA, Molinaro A, Attilia ML, Rotondo C, Attilia F, Ceccanti M, Ferri F, Maldarelli F, Maffongelli A, De Santis A, et al. PNPLA3 I148M (rs738409) genetic variant and age at onset of at-risk alcohol consumption are independent risk factors for alcoholic cirrhosis. Liver Int. 2014;34:514–520. doi: 10.1111/liv.12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stickel F, Buch S, Lau K, Meyer zu Schwabedissen H, Berg T, Ridinger M, Rietschel M, Schafmayer C, Braun F, Hinrichsen H, et al. Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in caucasians. Hepatology. 2011;53:86–95. doi: 10.1002/hep.24017. [DOI] [PubMed] [Google Scholar]
- 34.Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA. Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet. 2010;42:21–23. doi: 10.1038/ng.488. [DOI] [PubMed] [Google Scholar]
- 35.Trépo E, Gustot T, Degré D, Lemmers A, Verset L, Demetter P, Ouziel R, Quertinmont E, Vercruysse V, Amininejad L, et al. Common polymorphism in the PNPLA3/adiponutrin gene confers higher risk of cirrhosis and liver damage in alcoholic liver disease. J Hepatol. 2011;55:906–912. doi: 10.1016/j.jhep.2011.01.028. [DOI] [PubMed] [Google Scholar]
- 36.Singal AG, Manjunath H, Yopp AC, Beg MS, Marrero JA, Gopal P, Waljee AK. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a meta-analysis. Am J Gastroenterol. 2014;109:325–334. doi: 10.1038/ajg.2013.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shukla SD, Lim RW. Epigenetic effects of ethanol on the liver and gastrointestinal system. Alcohol Res. 2013;35:47–55. [PMC free article] [PubMed] [Google Scholar]
- 38.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–1585. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller AM, Horiguchi N, Jeong WI, Radaeva S, Gao B. Molecular mechanisms of alcoholic liver disease: innate immunity and cytokines. Alcohol Clin Exp Res. 2011;35:787–793. doi: 10.1111/j.1530-0277.2010.01399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orman ES, Odena G, Bataller R. Alcoholic liver disease: pathogenesis, management, and novel targets for therapy. J Gastroenterol Hepatol. 2013;28 Suppl 1:77–84. doi: 10.1111/jgh.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsukamoto H, French SW, Reidelberger RD, Largman C. Cyclical pattern of blood alcohol levels during continuous intragastric ethanol infusion in rats. Alcohol Clin Exp Res. 1985;9:31–37. doi: 10.1111/j.1530-0277.1985.tb05046.x. [DOI] [PubMed] [Google Scholar]
- 42.Tsukamoto H, French SW, Benson N, Delgado G, Rao GA, Larkin EC, Largman C. Severe and progressive steatosis and focal necrosis in rat liver induced by continuous intragastric infusion of ethanol and low fat diet. Hepatology. 1985;5:224–232. doi: 10.1002/hep.1840050212. [DOI] [PubMed] [Google Scholar]
- 43.Deng QG, She H, Cheng JH, French SW, Koop DR, Xiong S, Tsukamoto H. Steatohepatitis induced by intragastric overfeeding in mice. Hepatology. 2005;42:905–914. doi: 10.1002/hep.20877. [DOI] [PubMed] [Google Scholar]
- 44.Singal AK, Kamath PS, Gores GJ, Shah VH. Alcoholic hepatitis: current challenges and future directions. Clin Gastroenterol Hepatol. 2014;12:555–64; quiz e31-2. doi: 10.1016/j.cgh.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protoc. 2013;8:627–637. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ki SH, Park O, Zheng M, Morales-Ibanez O, Kolls JK, Bataller R, Gao B. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology. 2010;52:1291–1300. doi: 10.1002/hep.23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology. 2013;58:1814–1823. doi: 10.1002/hep.26419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 49.Dolganiuc A, Petrasek J, Kodys K, Catalano D, Mandrekar P, Velayudham A, Szabo G. MicroRNA expression profile in Lieber-DeCarli diet-induced alcoholic and methionine choline deficient diet-induced nonalcoholic steatohepatitis models in mice. Alcohol Clin Exp Res. 2009;33:1704–1710. doi: 10.1111/j.1530-0277.2009.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bala S, Marcos M, Kodys K, Csak T, Catalano D, Mandrekar P, Szabo G. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor {alpha} (TNF{alpha}) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem. 2011;286:1436–1444. doi: 10.1074/jbc.M110.145870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bala S, Petrasek J, Mundkur S, Catalano D, Levin I, Ward J, Alao H, Kodys K, Szabo G. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology. 2012;56:1946–1957. doi: 10.1002/hep.25873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yin H, Hu M, Zhang R, Shen Z, Flatow L, You M. MicroRNA-217 promotes ethanol-induced fat accumulation in hepatocytes by down-regulating SIRT1. J Biol Chem. 2012;287:9817–9826. doi: 10.1074/jbc.M111.333534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hou X, Xu S, Maitland-Toolan KA, Sato K, Jiang B, Ido Y, Lan F, Walsh K, Wierzbicki M, Verbeuren TJ, et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem. 2008;283:20015–20026. doi: 10.1074/jbc.M802187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127:1798–1808. doi: 10.1053/j.gastro.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 55.Meng F, Glaser SS, Francis H, Yang F, Han Y, Stokes A, Staloch D, McCarra J, Liu J, Venter J, et al. Epigenetic regulation of miR-34a expression in alcoholic liver injury. Am J Pathol. 2012;181:804–817. doi: 10.1016/j.ajpath.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dippold RP, Vadigepalli R, Gonye GE, Patra B, Hoek JB. Chronic ethanol feeding alters miRNA expression dynamics during liver regeneration. Alcohol Clin Exp Res. 2013;37 Suppl 1:E59–E69. doi: 10.1111/j.1530-0277.2012.01852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dong X, Liu H, Chen F, Li D, Zhao Y. MiR-214 promotes the alcohol-induced oxidative stress via down-regulation of glutathione reductase and cytochrome P450 oxidoreductase in liver cells. Alcohol Clin Exp Res. 2014;38:68–77. doi: 10.1111/acer.12209. [DOI] [PubMed] [Google Scholar]
- 58.Kharbanda KK. Alcoholic liver disease and methionine metabolism. Semin Liver Dis. 2009;29:155–165. doi: 10.1055/s-0029-1214371. [DOI] [PubMed] [Google Scholar]
- 59.Kharbanda KK. Methionine metabolic pathway in alcoholic liver injury. Curr Opin Clin Nutr Metab Care. 2013;16:89–95. doi: 10.1097/MCO.0b013e32835a892a. [DOI] [PubMed] [Google Scholar]
- 60.Halsted CH. B-Vitamin dependent methionine metabolism and alcoholic liver disease. Clin Chem Lab Med. 2013;51:457–465. doi: 10.1515/cclm-2012-0308. [DOI] [PubMed] [Google Scholar]
- 61.Halsted CH, Medici V. Vitamin-dependent methionine metabolism and alcoholic liver disease. Adv Nutr. 2011;2:421–427. doi: 10.3945/an.111.000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Halsted CH, Medici V. Aberrant hepatic methionine metabolism and gene methylation in the pathogenesis and treatment of alcoholic steatohepatitis. Int J Hepatol. 2012;2012:959746. doi: 10.1155/2012/959746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shukla SD, Velazquez J, French SW, Lu SC, Ticku MK, Zakhari S. Emerging role of epigenetics in the actions of alcohol. Alcohol Clin Exp Res. 2008;32:1525–1534. doi: 10.1111/j.1530-0277.2008.00729.x. [DOI] [PubMed] [Google Scholar]
- 64.Sullivan LW, Herbert V. Suppression hematopoiesis by ethanol. J Clin Invest. 1964;43:2048–2062. doi: 10.1172/JCI105079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Horne DW, Briggs WT, Wagner C. Ethanol stimulates 5-methyltetrahydrofolate accumulation in isolated rat liver cells. Biochem Pharmacol. 1978;27:2069–2074. doi: 10.1016/0006-2952(78)90071-0. [DOI] [PubMed] [Google Scholar]
- 66.Lieber CS, Casini A, DeCarli LM, Kim CI, Lowe N, Sasaki R, Leo MA. S-adenosyl-L-methionine attenuates alcohol-induced liver injury in the baboon. Hepatology. 1990;11:165–172. doi: 10.1002/hep.1840110203. [DOI] [PubMed] [Google Scholar]
- 67.Barak AJ, Beckenhauer HC, Junnila M, Tuma DJ. Dietary betaine promotes generation of hepatic S-adenosylmethionine and protects the liver from ethanol-induced fatty infiltration. Alcohol Clin Exp Res. 1993;17:552–555. doi: 10.1111/j.1530-0277.1993.tb00798.x. [DOI] [PubMed] [Google Scholar]
- 68.Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- 69.Veal N, Hsieh CL, Xiong S, Mato JM, Lu S, Tsukamoto H. Inhibition of lipopolysaccharide-stimulated TNF-alpha promoter activity by S-adenosylmethionine and 5’-methylthioadenosine. Am J Physiol Gastrointest Liver Physiol. 2004;287:G352–G362. doi: 10.1152/ajpgi.00316.2003. [DOI] [PubMed] [Google Scholar]
- 70.Kharbanda KK, Todero SL, King AL, Osna NA, McVicker BL, Tuma DJ, Wisecarver JL, Bailey SM. Betaine treatment attenuates chronic ethanol-induced hepatic steatosis and alterations to the mitochondrial respiratory chain proteome. Int J Hepatol. 2012;2012:962183. doi: 10.1155/2012/962183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jung YS, Kim SJ, Kwon do Y, Ahn CW, Kim YS, Choi DW, Kim YC. Alleviation of alcoholic liver injury by betaine involves an enhancement of antioxidant defense via regulation of sulfur amino acid metabolism. Food Chem Toxicol. 2013;62:292–298. doi: 10.1016/j.fct.2013.08.049. [DOI] [PubMed] [Google Scholar]
- 72.Oliva J, Bardag-Gorce F, Li J, French BA, French SW. S-adenosylmethionine prevents the up regulation of Toll-like receptor (TLR) signaling caused by chronic ethanol feeding in rats. Exp Mol Pathol. 2011;90:239–243. doi: 10.1016/j.yexmp.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li J, Li XM, Caudill M, Malysheva O, Bardag-Gorce F, Oliva J, French BA, Gorce E, Morgan K, Kathirvel E, et al. Betaine feeding prevents the blood alcohol cycle in rats fed alcohol continuously for 1 month using the rat intragastric tube feeding model. Exp Mol Pathol. 2011;91:540–547. doi: 10.1016/j.yexmp.2011.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shinohara M, Ji C, Kaplowitz N. Differences in betaine-homocysteine methyltransferase expression, endoplasmic reticulum stress response, and liver injury between alcohol-fed mice and rats. Hepatology. 2010;51:796–805. doi: 10.1002/hep.23391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Le MD, Enbom E, Traum PK, Medici V, Halsted CH, French SW. Alcoholic liver disease patients treated with S-adenosyl-L-methionine: an in-depth look at liver morphologic data comparing pre and post treatment liver biopsies. Exp Mol Pathol. 2013;95:187–191. doi: 10.1016/j.yexmp.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Medici V, Virata MC, Peerson JM, Stabler SP, French SW, Gregory JF, Albanese A, Bowlus CL, Devaraj S, Panacek EA, et al. S-adenosyl-L-methionine treatment for alcoholic liver disease: a double-blinded, randomized, placebo-controlled trial. Alcohol Clin Exp Res. 2011;35:1960–1965. doi: 10.1111/j.1530-0277.2011.01547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang KX, Denhardt DT. Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev. 2008;19:333–345. doi: 10.1016/j.cytogfr.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 78.Anborgh PH, Mutrie JC, Tuck AB, Chambers AF. Pre- and post-translational regulation of osteopontin in cancer. J Cell Commun Signal. 2011;5:111–122. doi: 10.1007/s12079-011-0130-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kazanecki CC, Uzwiak DJ, Denhardt DT. Control of osteopontin signaling and function by post-translational phosphorylation and protein folding. J Cell Biochem. 2007;102:912–924. doi: 10.1002/jcb.21558. [DOI] [PubMed] [Google Scholar]
- 80.Seth D, Gorrell MD, Cordoba S, McCaughan GW, Haber PS. Intrahepatic gene expression in human alcoholic hepatitis. J Hepatol. 2006;45:306–320. doi: 10.1016/j.jhep.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 81.Kiefer FW, Zeyda M, Gollinger K, Pfau B, Neuhofer A, Weichhart T, Säemann MD, Geyeregger R, Schlederer M, Kenner L, et al. Neutralization of osteopontin inhibits obesity-induced inflammation and insulin resistance. Diabetes. 2010;59:935–946. doi: 10.2337/db09-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Apte UM, Banerjee A, McRee R, Wellberg E, Ramaiah SK. Role of osteopontin in hepatic neutrophil infiltration during alcoholic steatohepatitis. Toxicol Appl Pharmacol. 2005;207:25–38. doi: 10.1016/j.taap.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 83.Morales-Ibanez O, Domínguez M, Ki SH, Marcos M, Chaves JF, Nguyen-Khac E, Houchi H, Affò S, Sancho-Bru P, Altamirano J, et al. Human and experimental evidence supporting a role for osteopontin in alcoholic hepatitis. Hepatology. 2013;58:1742–1756. doi: 10.1002/hep.26521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ge X, Leung TM, Arriazu E, Lu Y, Urtasun R, Christensen B, Fiel MI, Mochida S, Sørensen ES, Nieto N. Osteopontin binding to lipopolysaccharide lowers tumor necrosis factor-α and prevents early alcohol-induced liver injury in mice. Hepatology. 2014;59:1600–1616. doi: 10.1002/hep.26931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ge X, Lu Y, Leung TM, Sørensen ES, Nieto N. Milk osteopontin, a nutritional approach to prevent alcohol-induced liver injury. Am J Physiol Gastrointest Liver Physiol. 2013;304:G929–G939. doi: 10.1152/ajpgi.00014.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Banerjee A, Burghardt RC, Johnson GA, White FJ, Ramaiah SK. The temporal expression of osteopontin (SPP-1) in the rodent model of alcoholic steatohepatitis: a potential biomarker. Toxicol Pathol. 2006;34:373–384. doi: 10.1080/01926230600806543. [DOI] [PubMed] [Google Scholar]
- 87.Lee JH, Banerjee A, Ueno Y, Ramaiah SK. Potential relationship between hepatobiliary osteopontin and peroxisome proliferator-activated receptor alpha expression following ethanol-associated hepatic injury in vivo and in vitro. Toxicol Sci. 2008;106:290–299. doi: 10.1093/toxsci/kfn165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nan YM, Kong LB, Ren WG, Wang RQ, Du JH, Li WC, Zhao SX, Zhang YG, Wu WJ, Di HL, et al. Activation of peroxisome proliferator activated receptor alpha ameliorates ethanol mediated liver fibrosis in mice. Lipids Health Dis. 2013;12:11. doi: 10.1186/1476-511X-12-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Banerjee A, Apte UM, Smith R, Ramaiah SK. Higher neutrophil infiltration mediated by osteopontin is a likely contributing factor to the increased susceptibility of females to alcoholic liver disease. J Pathol. 2006;208:473–485. doi: 10.1002/path.1917. [DOI] [PubMed] [Google Scholar]
- 90.Patouraux S, Bonnafous S, Voican CS, Anty R, Saint-Paul MC, Rosenthal-Allieri MA, Agostini H, Njike M, Barri-Ova N, Naveau S, et al. The osteopontin level in liver, adipose tissue and serum is correlated with fibrosis in patients with alcoholic liver disease. PLoS One. 2012;7:e35612. doi: 10.1371/journal.pone.0035612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Diao H, Kon S, Iwabuchi K, Kimura C, Morimoto J, Ito D, Segawa T, Maeda M, Hamuro J, Nakayama T, Taniguchi M, Yagita H, Van Kaer L, Onóe K, Denhardt D, Rittling S, Uede T. Osteopontin as a mediator of NKT cell function in T cell-mediated liver diseases. Immunity. 2004;21:539–550. doi: 10.1016/j.immuni.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 92.Sahai A, Pan X, Paul R, Malladi P, Kohli R, Whitington PF. Roles of phosphatidylinositol 3-kinase and osteopontin in steatosis and aminotransferase release by hepatocytes treated with methionine-choline-deficient medium. Am J Physiol Gastrointest Liver Physiol. 2006;291:G55–G62. doi: 10.1152/ajpgi.00360.2005. [DOI] [PubMed] [Google Scholar]
- 93.Urtasun R, Lopategi A, George J, Leung TM, Lu Y, Wang X, Ge X, Fiel MI, Nieto N. Osteopontin, an oxidant stress sensitive cytokine, up-regulates collagen-I via integrin α(V)β(3) engagement and PI3K/pAkt/NFκB signaling. Hepatology. 2012;55:594–608. doi: 10.1002/hep.24701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Christensen B, Nielsen MS, Haselmann KF, Petersen TE, Sørensen ES. Post-translationally modified residues of native human osteopontin are located in clusters: identification of 36 phosphorylation and five O-glycosylation sites and their biological implications. Biochem J. 2005;390:285–292. doi: 10.1042/BJ20050341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nagoshi S. Osteopontin: Versatile modulator of liver diseases. Hepatol Res. 2014;44:22–30. doi: 10.1111/hepr.12166. [DOI] [PubMed] [Google Scholar]
- 96.Drakesmith H, Prentice AM. Hepcidin and the iron-infection axis. Science. 2012;338:768–772. doi: 10.1126/science.1224577. [DOI] [PubMed] [Google Scholar]
- 97.Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 98.Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loréal O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 99.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 100.Nicolas G, Bennoun M, Porteu A, Mativet S, Beaumont C, Grandchamp B, Sirito M, Sawadogo M, Kahn A, Vaulont S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci USA. 2002;99:4596–4601. doi: 10.1073/pnas.072632499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA. 2001;98:8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012;1823:1434–1443. doi: 10.1016/j.bbamcr.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Harrison-Findik DD. Role of alcohol in the regulation of iron metabolism. World J Gastroenterol. 2007;13:4925–4930. doi: 10.3748/wjg.v13.i37.4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wu D, Cederbaum AI. Oxidative stress and alcoholic liver disease. Semin Liver Dis. 2009;29:141–154. doi: 10.1055/s-0029-1214370. [DOI] [PubMed] [Google Scholar]
- 105.Ganne-Carrié N, Christidis C, Chastang C, Ziol M, Chapel F, Imbert-Bismut F, Trinchet JC, Guettier C, Beaugrand M. Liver iron is predictive of death in alcoholic cirrhosis: a multivariate study of 229 consecutive patients with alcoholic and/or hepatitis C virus cirrhosis: a prospective follow up study. Gut. 2000;46:277–282. doi: 10.1136/gut.46.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xiong S, She H, Sung CK, Tsukamoto H. Iron-dependent activation of NF-kappaB in Kupffer cells: a priming mechanism for alcoholic liver disease. Alcohol. 2003;30:107–113. doi: 10.1016/s0741-8329(03)00100-9. [DOI] [PubMed] [Google Scholar]
- 107.Xiong S, She H, Takeuchi H, Han B, Engelhardt JF, Barton CH, Zandi E, Giulivi C, Tsukamoto H. Signaling role of intracellular iron in NF-kappaB activation. J Biol Chem. 2003;278:17646–17654. doi: 10.1074/jbc.M210905200. [DOI] [PubMed] [Google Scholar]
- 108.Xiong S, She H, Tsukamoto H. Signaling role of iron in NF-kappa B activation in hepatic macrophages. Comp Hepatol. 2004;3 Suppl 1:S36. doi: 10.1186/1476-5926-2-S1-S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xiong S, She H, Zhang AS, Wang J, Mkrtchyan H, Dynnyk A, Gordeuk VR, French SW, Enns CA, Tsukamoto H. Hepatic macrophage iron aggravates experimental alcoholic steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2008;295:G512–G521. doi: 10.1152/ajpgi.90327.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sadrzadeh SM, Nanji AA, Price PL. The oral iron chelator, 1,2-dimethyl-3-hydroxypyrid-4-one reduces hepatic-free iron, lipid peroxidation and fat accumulation in chronically ethanol-fed rats. J Pharmacol Exp Ther. 1994;269:632–636. [PubMed] [Google Scholar]
- 111.Tsukamoto H, Horne W, Kamimura S, Niemelä O, Parkkila S, Ylä-Herttuala S, Brittenham GM. Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest. 1995;96:620–630. doi: 10.1172/JCI118077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kohgo Y, Ohtake T, Ikuta K, Suzuki Y, Torimoto Y, Kato J. Dysregulation of systemic iron metabolism in alcoholic liver diseases. J Gastroenterol Hepatol. 2008;23 Suppl 1:S78–S81. doi: 10.1111/j.1440-1746.2007.05290.x. [DOI] [PubMed] [Google Scholar]
- 113.Harrison-Findik DD, Schafer D, Klein E, Timchenko NA, Kulaksiz H, Clemens D, Fein E, Andriopoulos B, Pantopoulos K, Gollan J. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J Biol Chem. 2006;281:22974–22982. doi: 10.1074/jbc.M602098200. [DOI] [PubMed] [Google Scholar]
- 114.Iqbal T, Diab A, Ward DG, Brookes MJ, Tselepis C, Murray J, Elias E. Is iron overload in alcohol-related cirrhosis mediated by hepcidin? World J Gastroenterol. 2009;15:5864–5866. doi: 10.3748/wjg.15.5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Harrison-Findik DD, Klein E, Evans J, Gollan J. Regulation of liver hepcidin expression by alcohol in vivo does not involve Kupffer cell activation or TNF-alpha signaling. Am J Physiol Gastrointest Liver Physiol. 2009;296:G112–G118. doi: 10.1152/ajpgi.90550.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Harrison-Findik DD, Klein E, Crist C, Evans J, Timchenko N, Gollan J. Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology. 2007;46:1979–1985. doi: 10.1002/hep.21895. [DOI] [PubMed] [Google Scholar]
- 117.Heritage ML, Murphy TL, Bridle KR, Anderson GJ, Crawford DH, Fletcher LM. Hepcidin regulation in wild-type and Hfe knockout mice in response to alcohol consumption: evidence for an alcohol-induced hypoxic response. Alcohol Clin Exp Res. 2009;33:1391–1400. doi: 10.1111/j.1530-0277.2009.00969.x. [DOI] [PubMed] [Google Scholar]
- 118.Costa-Matos L, Batista P, Monteiro N, Simões M, Egas C, Pereira J, Pinho H, Santos N, Ribeiro J, Cipriano MA, et al. Liver hepcidin mRNA expression is inappropriately low in alcoholic patients compared with healthy controls. Eur J Gastroenterol Hepatol. 2012;24:1158–1165. doi: 10.1097/MEG.0b013e328355cfd0. [DOI] [PubMed] [Google Scholar]
- 119.Anderson ER, Taylor M, Xue X, Martin A, Moons DS, Omary MB, Shah YM. The hypoxia-inducible factor-C/EBPα axis controls ethanol-mediated hepcidin repression. Mol Cell Biol. 2012;32:4068–4077. doi: 10.1128/MCB.00723-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Peyssonnaux C, Zinkernagel AS, Schuepbach RA, Rankin E, Vaulont S, Haase VH, Nizet V, Johnson RS. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs) J Clin Invest. 2007;117:1926–1932. doi: 10.1172/JCI31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ren Y, Deng F, Zhu H, Wan W, Ye J, Luo B. Effect of epigallocatechin-3-gallate on iron overload in mice with alcoholic liver disease. Mol Biol Rep. 2011;38:879–886. doi: 10.1007/s11033-010-0180-5. [DOI] [PubMed] [Google Scholar]
- 122.Guo X, Li W, Xin Q, Ding H, Zhang C, Chang Y, Duan X. Vitamin C protective role for alcoholic liver disease in mice through regulating iron metabolism. Toxicol Ind Health. 2011;27:341–348. doi: 10.1177/0748233710387007. [DOI] [PubMed] [Google Scholar]
- 123.Ramos E, Ruchala P, Goodnough JB, Kautz L, Preza GC, Nemeth E, Ganz T. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood. 2012;120:3829–3836. doi: 10.1182/blood-2012-07-440743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev. 2010;90:1165–1194. doi: 10.1152/physrev.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Casey CA, Nanji A, Cederbaum AI, Adachi M, Takahashi T. Alcoholic liver disease and apoptosis. Alcohol Clin Exp Res. 2001;25:49S–53S. doi: 10.1097/00000374-200105051-00009. [DOI] [PubMed] [Google Scholar]
- 126.Feldstein AE, Gores GJ. Apoptosis in alcoholic and nonalcoholic steatohepatitis. Front Biosci. 2005;10:3093–3099. doi: 10.2741/1765. [DOI] [PubMed] [Google Scholar]
- 127.Colell A, García-Ruiz C, Miranda M, Ardite E, Marí M, Morales A, Corrales F, Kaplowitz N, Fernández-Checa JC. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology. 1998;115:1541–1551. doi: 10.1016/s0016-5085(98)70034-4. [DOI] [PubMed] [Google Scholar]
- 128.Miñana JB, Gómez-Cambronero L, Lloret A, Pallardó FV, Del Olmo J, Escudero A, Rodrigo JM, Pellíin A, Viña JR, Viña J, et al. Mitochondrial oxidative stress and CD95 ligand: a dual mechanism for hepatocyte apoptosis in chronic alcoholism. Hepatology. 2002;35:1205–1214. doi: 10.1053/jhep.2002.32969. [DOI] [PubMed] [Google Scholar]
- 129.Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, Fitzgerald KA, Szabo G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci USA. 2013;110:16544–16549. doi: 10.1073/pnas.1308331110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology. 2006;43:S31–S44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- 131.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 132.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 133.Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 135.Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol. 2011;12:439–452. doi: 10.1038/nrm3143. [DOI] [PubMed] [Google Scholar]
- 136.Zhou Z, Han V, Han J. New components of the necroptotic pathway. Protein Cell. 2012;3:811–817. doi: 10.1007/s13238-012-2083-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ding WX, Yin XM. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J Cell Mol Med. 2004;8:445–454. doi: 10.1111/j.1582-4934.2004.tb00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yin XM, Ding WX. Death receptor activation-induced hepatocyte apoptosis and liver injury. Curr Mol Med. 2003;3:491–508. doi: 10.2174/1566524033479555. [DOI] [PubMed] [Google Scholar]
- 139.Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- 140.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 141.Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 142.Northington FJ, Chavez-Valdez R, Graham EM, Razdan S, Gauda EB, Martin LJ. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J Cereb Blood Flow Metab. 2011;31:178–189. doi: 10.1038/jcbfm.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Smith CC, Davidson SM, Lim SY, Simpkin JC, Hothersall JS, Yellon DM. Necrostatin: a potentially novel cardioprotective agent? Cardiovasc Drugs Ther. 2007;21:227–233. doi: 10.1007/s10557-007-6035-1. [DOI] [PubMed] [Google Scholar]
- 144.Linkermann A, Bräsen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2013;110:12024–12029. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, Haase I, Pasparakis M. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity. 2011;35:572–582. doi: 10.1016/j.immuni.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 146.Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology. 2013;58:2099–2108. doi: 10.1002/hep.26547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sharma M, Gadang V, Jaeschke A. Critical role for mixed-lineage kinase 3 in acetaminophen-induced hepatotoxicity. Mol Pharmacol. 2012;82:1001–1007. doi: 10.1124/mol.112.079863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57:1773–1783. doi: 10.1002/hep.26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Maret W. Zinc biochemistry: from a single zinc enzyme to a key element of life. Adv Nutr. 2013;4:82–91. doi: 10.3945/an.112.003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Roohani N, Hurrell R, Kelishadi R, Schulin R. Zinc and its importance for human health: An integrative review. J Res Med Sci. 2013;18:144–157. [PMC free article] [PubMed] [Google Scholar]
- 152.Vallee BL, Wacker WE, Bartholomay AF, Hoch FL. Zinc metabolism in hepatic dysfunction. II. Correlation of metabolic patterns with biochemical findings. N Engl J Med. 1957;257:1055–1065. doi: 10.1056/NEJM195711282572201. [DOI] [PubMed] [Google Scholar]
- 153.Sullivan JF, Heaney RP. Zinc metabolism in alcoholic liver disease. Am J Clin Nutr. 1970;23:170–177. doi: 10.1093/ajcn/23.2.170. [DOI] [PubMed] [Google Scholar]
- 154.McClain CJ, Su LC. Zinc deficiency in the alcoholic: a review. Alcohol Clin Exp Res. 1983;7:5–10. doi: 10.1111/j.1530-0277.1983.tb05402.x. [DOI] [PubMed] [Google Scholar]
- 155.Rodríguez-Moreno F, González-Reimers E, Santolaria-Fernández F, Galindo-Martín L, Hernandez-Torres O, Batista-López N, Molina-Perez M. Zinc, copper, manganese, and iron in chronic alcoholic liver disease. Alcohol. 1997;14:39–44. doi: 10.1016/s0741-8329(96)00103-6. [DOI] [PubMed] [Google Scholar]
- 156.Kang X, Zhong W, Liu J, Song Z, McClain CJ, Kang YJ, Zhou Z. Zinc supplementation reverses alcohol-induced steatosis in mice through reactivating hepatocyte nuclear factor-4alpha and peroxisome proliferator-activated receptor-alpha. Hepatology. 2009;50:1241–1250. doi: 10.1002/hep.23090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kang X, Song Z, McClain CJ, Kang YJ, Zhou Z. Zinc supplementation enhances hepatic regeneration by preserving hepatocyte nuclear factor-4alpha in mice subjected to long-term ethanol administration. Am J Pathol. 2008;172:916–925. doi: 10.2353/ajpath.2008.070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mohammad MK, Zhou Z, Cave M, Barve A, McClain CJ. Zinc and liver disease. Nutr Clin Pract. 2012;27:8–20. doi: 10.1177/0884533611433534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhou Z, Sun X, Lambert JC, Saari JT, Kang YJ. Metallothionein-independent zinc protection from alcoholic liver injury. Am J Pathol. 2002;160:2267–2274. doi: 10.1016/S0002-9440(10)61174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhou Z, Sun X, James Kang Y. Metallothionein protection against alcoholic liver injury through inhibition of oxidative stress. Exp Biol Med (Maywood) 2002;227:214–222. doi: 10.1177/153537020222700310. [DOI] [PubMed] [Google Scholar]
- 161.Lambert JC, Zhou Z, Wang L, Song Z, McClain CJ, Kang YJ. Prevention of alterations in intestinal permeability is involved in zinc inhibition of acute ethanol-induced liver damage in mice. J Pharmacol Exp Ther. 2003;305:880–886. doi: 10.1124/jpet.102.047852. [DOI] [PubMed] [Google Scholar]
- 162.Zhong W, Zhao Y, Sun X, Song Z, McClain CJ, Zhou Z. Dietary zinc deficiency exaggerates ethanol-induced liver injury in mice: involvement of intrahepatic and extrahepatic factors. PLoS One. 2013;8:e76522. doi: 10.1371/journal.pone.0076522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhou Z, Wang L, Song Z, Saari JT, McClain CJ, Kang YJ. Zinc supplementation prevents alcoholic liver injury in mice through attenuation of oxidative stress. Am J Pathol. 2005;166:1681–1690. doi: 10.1016/S0002-9440(10)62478-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhou Z, Liu J, Song Z, McClain CJ, Kang YJ. Zinc supplementation inhibits hepatic apoptosis in mice subjected to a long-term ethanol exposure. Exp Biol Med (Maywood) 2008;233:540–548. doi: 10.3181/0710-RM-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lazarevich NL, Cheremnova OA, Varga EV, Ovchinnikov DA, Kudrjavtseva EI, Morozova OV, Fleishman DI, Engelhardt NV, Duncan SA. Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology. 2004;39:1038–1047. doi: 10.1002/hep.20155. [DOI] [PubMed] [Google Scholar]
- 166.Liuzzi JP, Yoo C. Role of zinc in the regulation of autophagy during ethanol exposure in human hepatoma cells. Biol Trace Elem Res. 2013;156:350–356. doi: 10.1007/s12011-013-9816-3. [DOI] [PubMed] [Google Scholar]
- 167.Gao B, Seki E, Brenner DA, Friedman S, Cohen JI, Nagy L, Szabo G, Zakhari S. Innate immunity in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2011;300:G516–G525. doi: 10.1152/ajpgi.00537.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.An L, Wang X, Cederbaum AI. Cytokines in alcoholic liver disease. Arch Toxicol. 2012;86:1337–1348. doi: 10.1007/s00204-012-0814-6. [DOI] [PubMed] [Google Scholar]
- 169.Dhanda AD, Lee RW, Collins PL, McCune CA. Molecular targets in the treatment of alcoholic hepatitis. World J Gastroenterol. 2012;18:5504–5513. doi: 10.3748/wjg.v18.i39.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gao B. Hepatoprotective and anti-inflammatory cytokines in alcoholic liver disease. J Gastroenterol Hepatol. 2012;27 Suppl 2:89–93. doi: 10.1111/j.1440-1746.2011.07003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Dumoutier L, Van Roost E, Ameye G, Michaux L, Renauld JC. IL-TIF/IL-22: genomic organization and mapping of the human and mouse genes. Genes Immun. 2000;1:488–494. doi: 10.1038/sj.gene.6363716. [DOI] [PubMed] [Google Scholar]
- 172.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 173.Witte E, Witte K, Warszawska K, Sabat R, Wolk K. Interleukin-22: a cytokine produced by T, NK and NKT cell subsets, with importance in the innate immune defense and tissue protection. Cytokine Growth Factor Rev. 2010;21:365–379. doi: 10.1016/j.cytogfr.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 174.Kotenko SV, Izotova LS, Mirochnitchenko OV, Esterova E, Dickensheets H, Donnelly RP, Pestka S. Identification of the functional interleukin-22 (IL-22) receptor complex: the IL-10R2 chain (IL-10Rbeta ) is a common chain of both the IL-10 and IL-22 (IL-10-related T cell-derived inducible factor, IL-TIF) receptor complexes. J Biol Chem. 2001;276:2725–2732. doi: 10.1074/jbc.M007837200. [DOI] [PubMed] [Google Scholar]
- 175.Wei CC, Ho TW, Liang WG, Chen GY, Chang MS. Cloning and characterization of mouse IL-22 binding protein. Genes Immun. 2003;4:204–211. doi: 10.1038/sj.gene.6363947. [DOI] [PubMed] [Google Scholar]
- 176.Logsdon NJ, Jones BC, Josephson K, Cook J, Walter MR. Comparison of interleukin-22 and interleukin-10 soluble receptor complexes. J Interferon Cytokine Res. 2002;22:1099–1112. doi: 10.1089/10799900260442520. [DOI] [PubMed] [Google Scholar]
- 177.Li J, Tomkinson KN, Tan XY, Wu P, Yan G, Spaulding V, Deng B, Annis-Freeman B, Heveron K, Zollner R, et al. Temporal associations between interleukin 22 and the extracellular domains of IL-22R and IL-10R2. Int Immunopharmacol. 2004;4:693–708. doi: 10.1016/j.intimp.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 178.Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Biology of interleukin-22. Semin Immunopathol. 2010;32:17–31. doi: 10.1007/s00281-009-0188-x. [DOI] [PubMed] [Google Scholar]
- 179.Colin EM, Asmawidjaja PS, van Hamburg JP, Mus AM, van Driel M, Hazes JM, van Leeuwen JP, Lubberts E. 1,25-dihydroxyvitamin D3 modulates Th17 polarization and interleukin-22 expression by memory T cells from patients with early rheumatoid arthritis. Arthritis Rheum. 2010;62:132–142. doi: 10.1002/art.25043. [DOI] [PubMed] [Google Scholar]
- 180.Ikeuchi H, Kuroiwa T, Hiramatsu N, Kaneko Y, Hiromura K, Ueki K, Nojima Y. Expression of interleukin-22 in rheumatoid arthritis: potential role as a proinflammatory cytokine. Arthritis Rheum. 2005;52:1037–1046. doi: 10.1002/art.20965. [DOI] [PubMed] [Google Scholar]
- 181.Wolk K, Witte E, Wallace E, Döcke WD, Kunz S, Asadullah K, Volk HD, Sterry W, Sabat R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36:1309–1323. doi: 10.1002/eji.200535503. [DOI] [PubMed] [Google Scholar]
- 182.Radaeva S, Sun R, Pan HN, Hong F, Gao B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology. 2004;39:1332–1342. doi: 10.1002/hep.20184. [DOI] [PubMed] [Google Scholar]
- 183.Chestovich PJ, Uchida Y, Chang W, Ajalat M, Lassman C, Sabat R, Busuttil RW, Kupiec-Weglinski JW. Interleukin-22: implications for liver ischemia-reperfusion injury. Transplantation. 2012;93:485–492. doi: 10.1097/TP.0b013e3182449136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Xing WW, Zou MJ, Liu S, Xu T, Wang JX, Xu DG. Interleukin-22 protects against acute alcohol-induced hepatotoxicity in mice. Biosci Biotechnol Biochem. 2011;75:1290–1294. doi: 10.1271/bbb.110061. [DOI] [PubMed] [Google Scholar]
- 185.Kong X, Feng D, Wang H, Hong F, Bertola A, Wang FS, Gao B. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. 2012;56:1150–1159. doi: 10.1002/hep.25744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, Cong M, Iwaisako K, Liu X, Zhang M, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 2012;143:765–76.e1-3. doi: 10.1053/j.gastro.2012.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Csaki LS, Reue K. Lipins: multifunctional lipid metabolism proteins. Annu Rev Nutr. 2010;30:257–272. doi: 10.1146/annurev.nutr.012809.104729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Donkor J, Sariahmetoglu M, Dewald J, Brindley DN, Reue K. Three mammalian lipins act as phosphatidate phosphatases with distinct tissue expression patterns. J Biol Chem. 2007;282:3450–3457. doi: 10.1074/jbc.M610745200. [DOI] [PubMed] [Google Scholar]
- 189.Péterfy M, Phan J, Xu P, Reue K. Lipodystrophy in the fld mouse results from mutation of a new gene encoding a nuclear protein, lipin. Nat Genet. 2001;27:121–124. doi: 10.1038/83685. [DOI] [PubMed] [Google Scholar]
- 190.Michot C, Hubert L, Brivet M, De Meirleir L, Valayannopoulos V, Müller-Felber W, Venkateswaran R, Ogier H, Desguerre I, Altuzarra C, et al. LPIN1 gene mutations: a major cause of severe rhabdomyolysis in early childhood. Hum Mutat. 2010;31:E1564–E1573. doi: 10.1002/humu.21282. [DOI] [PubMed] [Google Scholar]
- 191.Zeharia A, Shaag A, Houtkooper RH, Hindi T, de Lonlay P, Erez G, Hubert L, Saada A, de Keyzer Y, Eshel G, et al. Mutations in LPIN1 cause recurrent acute myoglobinuria in childhood. Am J Hum Genet. 2008;83:489–494. doi: 10.1016/j.ajhg.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC, Kelly DP. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 193.Csaki LS, Dwyer JR, Fong LG, Tontonoz P, Young SG, Reue K. Lipins, lipinopathies, and the modulation of cellular lipid storage and signaling. Prog Lipid Res. 2013;52:305–316. doi: 10.1016/j.plipres.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wang H, Zhang J, Qiu W, Han GS, Carman GM, Adeli K. Lipin-1γ isoform is a novel lipid droplet-associated protein highly expressed in the brain. FEBS Lett. 2011;585:1979–1984. doi: 10.1016/j.febslet.2011.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, Madden KL, Carpenter AE, Finck BN, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Harris TE, Huffman TA, Chi A, Shabanowitz J, Hunt DF, Kumar A, Lawrence JC. Insulin controls subcellular localization and multisite phosphorylation of the phosphatidic acid phosphatase, lipin 1. J Biol Chem. 2007;282:277–286. doi: 10.1074/jbc.M609537200. [DOI] [PubMed] [Google Scholar]
- 197.Liu GH, Gerace L. Sumoylation regulates nuclear localization of lipin-1alpha in neuronal cells. PLoS One. 2009;4:e7031. doi: 10.1371/journal.pone.0007031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R, Odena G, Stevens SM, You M. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology. 2014;146:801–811. doi: 10.1053/j.gastro.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hu M, Wang F, Li X, Rogers CQ, Liang X, Finck BN, Mitra MS, Zhang R, Mitchell DA, You M. Regulation of hepatic lipin-1 by ethanol: role of AMP-activated protein kinase/sterol regulatory element-binding protein 1 signaling in mice. Hepatology. 2012;55:437–446. doi: 10.1002/hep.24708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hu M, Yin H, Mitra MS, Liang X, Ajmo JM, Nadra K, Chrast R, Finck BN, You M. Hepatic-specific lipin-1 deficiency exacerbates experimental alcohol-induced steatohepatitis in mice. Hepatology. 2013;58:1953–1963. doi: 10.1002/hep.26589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Batoko H, Bay BH, Beau I, Bechet DM, Begley TJ, Behl C, Behrends C, Bekri S, Bellaire B, Bendall LJ, Benetti L, Berliocchi L, Bernardi H, Bernassola F, Besteiro S, Bhatia-Kissova I, Bi X, Biard-Piechaczyk M, Blum JS, Boise LH, Bonaldo P, Boone DL, Bornhauser BC, Bortoluci KR, Bossis I, Bost F, Bourquin JP, Boya P, Boyer-Guittaut M, Bozhkov PV, Brady NR, Brancolini C, Brech A, Brenman JE, Brennand A, Bresnick EH, Brest P, Bridges D, Bristol ML, Brookes PS, Brown EJ, Brumell JH, Brunetti-Pierri N, Brunk UT, Bulman DE, Bultman SJ, Bultynck G, Burbulla LF, Bursch W, Butchar JP, Buzgariu W, Bydlowski SP, Cadwell K, Cahova M, Cai D, Cai J, Cai Q, Calabretta B, Calvo-Garrido J, Camougrand N, Campanella M, Campos-Salinas J, Candi E, Cao L, Caplan AB, Carding SR, Cardoso SM, Carew JS, Carlin CR, Carmignac V, Carneiro LA, Carra S, Caruso RA, Casari G, Casas C, Castino R, Cebollero E, Cecconi F, Celli J, Chaachouay H, Chae HJ, Chai CY, Chan DC, Chan EY, Chang RC, Che CM, Chen CC, Chen GC, Chen GQ, Chen M, Chen Q, Chen SS, Chen W, Chen X, Chen X, Chen X, Chen YG, Chen Y, Chen Y, Chen YJ, Chen Z, Cheng A, Cheng CH, Cheng Y, Cheong H, Cheong JH, Cherry S, Chess-Williams R, Cheung ZH, Chevet E, Chiang HL, Chiarelli R, Chiba T, Chin LS, Chiou SH, Chisari FV, Cho CH, Cho DH, Choi AM, Choi D, Choi KS, Choi ME, Chouaib S, Choubey D, Choubey V, Chu CT, Chuang TH, Chueh SH, Chun T, Chwae YJ, Chye ML, Ciarcia R, Ciriolo MR, Clague MJ, Clark RS, Clarke PG, Clarke R, Codogno P, Coller HA, Colombo MI, Comincini S, Condello M, Condorelli F, Cookson MR, Coombs GH, Coppens I, Corbalan R, Cossart P, Costelli P, Costes S, Coto-Montes A, Couve E, Coxon FP, Cregg JM, Crespo JL, Cronje MJ, Cuervo AM, Cullen JJ, Czaja MJ, D’Amelio M, Darfeuille-Michaud A, Davids LM, Davies FE, De Felici M, de Groot JF, de Haan CA, De Martino L, De Milito A, De Tata V, Debnath J, Degterev A, Dehay B, Delbridge LM, Demarchi F, Deng YZ, Dengjel J, Dent P, Denton D, Deretic V, Desai SD, Devenish RJ, Di Gioacchino M, Di Paolo G, Di Pietro C, Diaz-Araya G, Diaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dikic I, Dinesh-Kumar SP, Ding WX, Distelhorst CW, Diwan A, Djavaheri-Mergny M, Dokudovskaya S, Dong Z, Dorsey FC, Dosenko V, Dowling JJ, Doxsey S, Dreux M, Drew ME, Duan Q, Duchosal MA, Duff K, Dugail I, Durbeej M, Duszenko M, Edelstein CL, Edinger AL, Egea G, Eichinger L, Eissa NT, Ekmekcioglu S, El-Deiry WS, Elazar Z, Elgendy M, Ellerby LM, Eng KE, Engelbrecht AM, Engelender S, Erenpreisa J, Escalante R, Esclatine A, Eskelinen EL, Espert L, Espina V, Fan H, Fan J, Fan QW, Fan Z, Fang S, Fang Y, Fanto M, Fanzani A, Farkas T, Farre JC, Faure M, Fechheimer M, Feng CG, Feng J, Feng Q, Feng Y, Fesus L, Feuer R, Figueiredo-Pereira ME, Fimia GM, Fingar DC, Finkbeiner S, Finkel T, Finley KD, Fiorito F, Fisher EA, Fisher PB, Flajolet M, Florez-McClure ML, Florio S, Fon EA, Fornai F, Fortunato F, Fotedar R, Fowler DH, Fox HS, Franco R, Frankel LB, Fransen M, Fuentes JM, Fueyo J, Fujii J, Fujisaki K, Fujita E, Fukuda M, Furukawa RH, Gaestel M, Gailly P, Gajewska M, Galliot B, Galy V, Ganesh S, Ganetzky B, Ganley IG, Gao FB, Gao GF, Gao J, Garcia L, Garcia-Manero G, Garcia-Marcos M, Garmyn M, Gartel AL, Gatti E, Gautel M, Gawriluk TR, Gegg ME, Geng J, Germain M, Gestwicki JE, Gewirtz DA, Ghavami S, Ghosh P, Giammarioli AM, Giatromanolaki AN, Gibson SB, Gilkerson RW, Ginger ML, Ginsberg HN, Golab J, Goligorsky MS, Golstein P, Gomez-Manzano C, Goncu E, Gongora C, Gonzalez CD, Gonzalez R, Gonzalez-Estevez C, Gonzalez-Polo RA, Gonzalez-Rey E, Gorbunov NV, Gorski S, Goruppi S, Gottlieb RA, Gozuacik D, Granato GE, Grant GD, Green KN, Gregorc A, Gros F, Grose C, Grunt TW, Gual P, Guan JL, Guan KL, Guichard SM, Gukovskaya AS, Gukovsky I, Gunst J, Gustafsson AB, Halayko AJ, Hale AN, Halonen SK, Hamasaki M, Han F, Han T, Hancock MK, Hansen M, Harada H, Harada M, Hardt SE, Harper JW, Harris AL, Harris J, Harris SD, Hashimoto M, Haspel JA, Hayashi S, Hazelhurst LA, He C, He YW, Hebert MJ, Heidenreich KA, Helfrich MH, Helgason GV, Henske EP, Herman B, Herman PK, Hetz C, Hilfiker S, Hill JA, Hocking LJ, Hofman P, Hofmann TG, Hohfeld J, Holyoake TL, Hong MH, Hood DA, Hotamisligil GS, Houwerzijl EJ, Hoyer-Hansen M, Hu B, Hu CA, Hu HM, Hua Y, Huang C, Huang J, Huang S, Huang WP, Huber TB, Huh WK, Hung TH, Hupp TR, Hur GM, Hurley JB, Hussain SN, Hussey PJ, Hwang JJ, Hwang S, Ichihara A, Ilkhanizadeh S, Inoki K, Into T, Iovane V, Iovanna JL, Ip NY, Isaka Y, Ishida H, Isidoro C, Isobe K, Iwasaki A, Izquierdo M, Izumi Y, Jaakkola PM, Jaattela M, Jackson GR, Jackson WT, Janji B, Jendrach M, Jeon JH, Jeung EB, Jiang H, Jiang H, Jiang JX, Jiang M, Jiang Q, Jiang X, Jiang X, Jimenez A, Jin M, Jin S, Joe CO, Johansen T, Johnson DE, Johnson GV, Jones NL, Joseph B, Joseph SK, Joubert AM, Juhasz G, Juillerat-Jeanneret L, Jung CH, Jung YK, Kaarniranta K, Kaasik A, Kabuta T, Kadowaki M, Kagedal K, Kamada Y, Kaminskyy VO, Kampinga HH, Kanamori H, Kang C, Kang KB, Kang KI, Kang R, Kang YA, Kanki T, Kanneganti TD, Kanno H, Kanthasamy AG, Kanthasamy A, Karantza V, Kaushal GP, Kaushik S, Kawazoe Y, Ke PY, Kehrl JH, Kelekar A, Kerkhoff C, Kessel DH, Khalil H, Kiel JA, Kiger AA, Kihara A, Kim DR, Kim DH, Kim DH, Kim EK, Kim HR, Kim JS, Kim JH, Kim JC, Kim JK, Kim PK, Kim SW, Kim YS, Kim Y, Kimchi A, Kimmelman AC, King JS, Kinsella TJ, Kirkin V, Kirshenbaum LA, Kitamoto K, Kitazato K, Klein L, Klimecki WT, Klucken J, Knecht E, Ko BC, Koch JC, Koga H, Koh JY, Koh YH, Koike M, Komatsu M, Kominami E, Kong HJ, Kong WJ, Korolchuk VI, Kotake Y, Koukourakis MI, Kouri Flores JB, Kovacs AL, Kraft C, Krainc D, Kramer H, Kretz-Remy C, Krichevsky AM, Kroemer G, Kruger R, Krut O, Ktistakis NT, Kuan CY, Kucharczyk R, Kumar A, Kumar R, Kumar S, Kundu M, Kung HJ, Kurz T, Kwon HJ, La Spada AR, Lafont F, Lamark T, Landry J, Lane JD, Lapaquette P, Laporte JF, Laszlo L, Lavandero S, Lavoie JN, Layfield R, Lazo PA, Le W, Le Cam L, Ledbetter DJ, Lee AJ, Lee BW, Lee GM, Lee J, Lee JH, Lee M, Lee MS, Lee SH, Leeuwenburgh C, Legembre P, Legouis R, Lehmann M, Lei HY, Lei QY, Leib DA, Leiro J, Lemasters JJ, Lemoine A, Lesniak MS, Lev D, Levenson VV, Levine B, Levy E, Li F, Li JL, Li L, Li S, Li W, Li XJ, Li YB, Li YP, Liang C, Liang Q, Liao YF, Liberski PP, Lieberman A, Lim HJ, Lim KL, Lim K, Lin CF, Lin FC, Lin J, Lin JD, Lin K, Lin WW, Lin WC, Lin YL, Linden R, Lingor P, Lippincott-Schwartz J, Lisanti MP, Liton PB, Liu B, Liu CF, Liu K, Liu L, Liu QA, Liu W, Liu YC, Liu Y, Lockshin RA, Lok CN, Lonial S, Loos B, Lopez-Berestein G, Lopez-Otin C, Lossi L, Lotze MT, Low P, Lu B, Lu B, Lu B, Lu Z, Luciano F, Lukacs NW, Lund AH, Lynch-Day MA, Ma Y, Macian F, MacKeigan JP, Macleod KF, Madeo F, Maiuri L, Maiuri MC, Malagoli D, Malicdan MC, Malorni W, Man N, Mandelkow EM, Manon S, Manov I, Mao K, Mao X, Mao Z, Marambaud P, Marazziti D, Marcel YL, Marchbank K, Marchetti P, Marciniak SJ, Marcondes M, Mardi M, Marfe G, Marino G, Markaki M, Marten MR, Martin SJ, Martinand-Mari C, Martinet W, Martinez-Vicente M, Masini M, Matarrese P, Matsuo S, Matteoni R, Mayer A, Mazure NM, McConkey DJ, McConnell MJ, McDermott C, McDonald C, McInerney GM, McKenna SL, McLaughlin B, McLean PJ, McMaster CR, McQuibban GA, Meijer AJ, Meisler MH, Melendez A, Melia TJ, Melino G, Mena MA, Menendez JA, Menna-Barreto RF, Menon MB, Menzies FM, Mercer CA, Merighi A, Merry DE, Meschini S, Meyer CG, Meyer TF, Miao CY, Miao JY, Michels PA, Michiels C, Mijaljica D, Milojkovic A, Minucci S, Miracco C, Miranti CK, Mitroulis I, Miyazawa K, Mizushima N, Mograbi B, Mohseni S, Molero X, Mollereau B, Mollinedo F, Momoi T, Monastyrska I, Monick MM, Monteiro MJ, Moore MN, Mora R, Moreau K, Moreira PI, Moriyasu Y, Moscat J, Mostowy S, Mottram JC, Motyl T, Moussa CE, Muller S, Muller S, Munger K, Munz C, Murphy LO, Murphy ME, Musaro A, Mysorekar I, Nagata E, Nagata K, Nahimana A, Nair U, Nakagawa T, Nakahira K, Nakano H, Nakatogawa H, Nanjundan M, Naqvi NI, Narendra DP, Narita M, Navarro M, Nawrocki ST, Nazarko TY, Nemchenko A, Netea MG, Neufeld TP, Ney PA, Nezis IP, Nguyen HP, Nie D, Nishino I, Nislow C, Nixon RA, Noda T, Noegel AA, Nogalska A, Noguchi S, Notterpek L, Novak I, Nozaki T, Nukina N, Nurnberger T, Nyfeler B, Obara K, Oberley TD, Oddo S, Ogawa M, Ohashi T, Okamoto K, Oleinick NL, Oliver FJ, Olsen LJ, Olsson S, Opota O, Osborne TF, Ostrander GK, Otsu K, Ou JH, Ouimet M, Overholtzer M, Ozpolat B, Paganetti P, Pagnini U, Pallet N, Palmer GE, Palumbo C, Pan T, Panaretakis T, Pandey UB, Papackova Z, Papassideri I, Paris I, Park J, Park OK, Parys JB, Parzych KR, Patschan S, Patterson C, Pattingre S, Pawelek JM, Peng J, Perlmutter DH, Perrotta I, Perry G, Pervaiz S, Peter M, Peters GJ, Petersen M, Petrovski G, Phang JM, Piacentini M, Pierre P, Pierrefite-Carle V, Pierron G, Pinkas-Kramarski R, Piras A, Piri N, Platanias LC, Poggeler S, Poirot M, Poletti A, Pous C, Pozuelo-Rubio M, Praetorius-Ibba M, Prasad A, Prescott M, Priault M, Produit-Zengaffinen N, Progulske-Fox A, Proikas-Cezanne T, Przedborski S, Przyklenk K, Puertollano R, Puyal J, Qian SB, Qin L, Qin ZH, Quaggin SE, Raben N, Rabinowich H, Rabkin SW, Rahman I, Rami A, Ramm G, Randall G, Randow F, Rao VA, Rathmell JC, Ravikumar B, Ray SK, Reed BH, Reed JC, Reggiori F, Regnier-Vigouroux A, Reichert AS, Reiners JJ, Jr., Reiter RJ, Ren J, Revuelta JL, Rhodes CJ, Ritis K, Rizzo E, Robbins J, Roberge M, Roca H, Roccheri MC, Rocchi S, Rodemann HP, Rodriguez de Cordoba S, Rohrer B, Roninson IB, Rosen K, Rost-Roszkowska MM, Rouis M, Rouschop KM, Rovetta F, Rubin BP, Rubinsztein DC, Ruckdeschel K, Rucker EB, 3rd, Rudich A, Rudolf E, Ruiz-Opazo N, Russo R, Rusten TE, Ryan KM, Ryter SW, Sabatini DM, Sadoshima J, Saha T, Saitoh T, Sakagami H, Sakai Y, Salekdeh GH, Salomoni P, Salvaterra PM, Salvesen G, Salvioli R, Sanchez AM, Sanchez-Alcazar JA, Sanchez-Prieto R, Sandri M, Sankar U, Sansanwal P, Santambrogio L, Saran S, Sarkar S, Sarwal M, Sasakawa C, Sasnauskiene A, Sass M, Sato K, Sato M, Schapira AH, Scharl M, Schatzl HM, Scheper W, Schiaffino S, Schneider C, Schneider ME, Schneider-Stock R, Schoenlein PV, Schorderet DF, Schuller C, Schwartz GK, Scorrano L, Sealy L, Seglen PO, Segura-Aguilar J, Seiliez I, Seleverstov O, Sell C, Seo JB, Separovic D, Setaluri V, Setoguchi T, Settembre C, Shacka JJ, Shanmugam M, Shapiro IM, Shaulian E, Shaw RJ, Shelhamer JH, Shen HM, Shen WC, Sheng ZH, Shi Y, Shibuya K, Shidoji Y, Shieh JJ, Shih CM, Shimada Y, Shimizu S, Shintani T, Shirihai OS, Shore GC, Sibirny AA, Sidhu SB, Sikorska B, Silva-Zacarin EC, Simmons A, Simon AK, Simon HU, Simone C, Simonsen A, Sinclair DA, Singh R, Sinha D, Sinicrope FA, Sirko A, Siu PM, Sivridis E, Skop V, Skulachev VP, Slack RS, Smaili SS, Smith DR, Soengas MS, Soldati T, Song X, Sood AK, Soong TW, Sotgia F, Spector SA, Spies CD, Springer W, Srinivasula SM, Stefanis L, Steffan JS, Stendel R, Stenmark H, Stephanou A, Stern ST, Sternberg C, Stork B, Stralfors P, Subauste CS, Sui X, Sulzer D, Sun J, Sun SY, Sun ZJ, Sung JJ, Suzuki K, Suzuki T, Swanson MS, Swanton C, Sweeney ST, Sy LK, Szabadkai G, Tabas I, Taegtmeyer H, Tafani M, Takacs-Vellai K, Takano Y, Takegawa K, Takemura G, Takeshita F, Talbot NJ, Tan KS, Tanaka K, Tanaka K, Tang D, Tang D, Tanida I, Tannous BA, Tavernarakis N, Taylor GS, Taylor GA, Taylor JP, Terada LS, Terman A, Tettamanti G, Thevissen K, Thompson CB, Thorburn A, Thumm M, Tian F, Tian Y, Tocchini-Valentini G, Tolkovsky AM, Tomino Y, Tonges L, Tooze SA, Tournier C, Tower J, Towns R, Trajkovic V, Travassos LH, Tsai TF, Tschan MP, Tsubata T, Tsung A, Turk B, Turner LS, Tyagi SC, Uchiyama Y, Ueno T, Umekawa M, Umemiya-Shirafuji R, Unni VK, Vaccaro MI, Valente EM, Van den Berghe G, van der Klei IJ, van Doorn W, van Dyk LF, van Egmond M, van Grunsven LA, Vandenabeele P, Vandenberghe WP, Vanhorebeek I, Vaquero EC, Velasco G, Vellai T, Vicencio JM, Vierstra RD, Vila M, Vindis C, Viola G, Viscomi MT, Voitsekhovskaja OV, von Haefen C, Votruba M, Wada K, Wade-Martins R, Walker CL, Walsh CM, Walter J, Wan XB, Wang A, Wang C, Wang D, Wang F, Wang F, Wang G, Wang H, Wang HG, Wang HD, Wang J, Wang K, Wang M, Wang RC, Wang X, Wang X, Wang YJ, Wang Y, Wang Z, Wang ZC, Wang Z, Wansink DG, Ward DM, Watada H, Waters SL, Webster P, Wei L, Weihl CC, Weiss WA, Welford SM, Wen LP, Whitehouse CA, Whitton JL, Whitworth AJ, Wileman T, Wiley JW, Wilkinson S, Willbold D, Williams RL, Williamson PR, Wouters BG, Wu C, Wu DC, Wu WK, Wyttenbach A, Xavier RJ, Xi Z, Xia P, Xiao G, Xie Z, Xie Z, Xu DZ, Xu J, Xu L, Xu X, Yamamoto A, Yamamoto A, Yamashina S, Yamashita M, Yan X, Yanagida M, Yang DS, Yang E, Yang JM, Yang SY, Yang W, Yang WY, Yang Z, Yao MC, Yao TP, Yeganeh B, Yen WL, Yin JJ, Yin XM, Yoo OJ, Yoon G, Yoon SY, Yorimitsu T, Yoshikawa Y, Yoshimori T, Yoshimoto K, You HJ, Youle RJ, Younes A, Yu L, Yu L, Yu SW, Yu WH, Yuan ZM, Yue Z, Yun CH, Yuzaki M, Zabirnyk O, Silva-Zacarin E, Zacks D, Zacksenhaus E, Zaffaroni N, Zakeri Z, Zeh HJ, 3rd, Zeitlin SO, Zhang H, Zhang HL, Zhang J, Zhang JP, Zhang L, Zhang L, Zhang MY, Zhang XD, Zhao M, Zhao YF, Zhao Y, Zhao ZJ, Zheng X, Zhivotovsky B, Zhong Q, Zhou CZ, Zhu C, Zhu WG, Zhu XF, Zhu X, Zhu Y, Zoladek T, Zong WX, Zorzano A, Zschocke J, Zuckerbraun B. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–216. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 203.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Reggiori F, Komatsu M, Finley K, Simonsen A. Selective types of autophagy. Int J Cell Biol. 2012;2012:156272. doi: 10.1155/2012/156272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Manley S, Williams JA, Ding WX. Role of p62/SQSTM1 in liver physiology and pathogenesis. Exp Biol Med (Maywood) 2013;238:525–538. doi: 10.1177/1535370213489446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Czaja MJ, Ding WX, Donohue TM, Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, et al. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology. 2008;47:1773–1785. doi: 10.1002/hep.22146. [DOI] [PubMed] [Google Scholar]
- 208.Ding WX. Role of autophagy in liver physiology and pathophysiology. World J Biol Chem. 2010;1:3–12. doi: 10.4331/wjbc.v1.i1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ding WX, Manley S, Ni HM. The emerging role of autophagy in alcoholic liver disease. Exp Biol Med (Maywood) 2011;236:546–556. doi: 10.1258/ebm.2011.010360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Dolganiuc A, Thomes PG, Ding WX, Lemasters JJ, Donohue TM. Autophagy in alcohol-induced liver diseases. Alcohol Clin Exp Res. 2012;36:1301–1308. doi: 10.1111/j.1530-0277.2012.01742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lin CW, Zhang H, Li M, Xiong X, Chen X, Chen X, Dong XC, Yin XM. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013;58:993–999. doi: 10.1016/j.jhep.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Ding WX, Li M, Yin XM. Selective taste of ethanol-induced autophagy for mitochondria and lipid droplets. Autophagy. 2011;7:248–249. doi: 10.4161/auto.7.2.14347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Donohue TM. Autophagy and ethanol-induced liver injury. World J Gastroenterol. 2009;15:1178–1185. doi: 10.3748/wjg.15.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Thomes PG, Ehlers RA, Trambly CS, Clemens DL, Fox HS, Tuma DJ, Donohue TM. Multilevel regulation of autophagosome content by ethanol oxidation in HepG2 cells. Autophagy. 2013;9:63–73. doi: 10.4161/auto.22490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, Ding WX. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy. 2011;7:188–204. doi: 10.4161/auto.7.2.14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Ma D, Panda S, Lin JD. Temporal orchestration of circadian autophagy rhythm by C/EBPβ. EMBO J. 2011;30:4642–4651. doi: 10.1038/emboj.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Chen G, Ke Z, Xu M, Liao M, Wang X, Qi Y, Zhang T, Frank JA, Bower KA, Shi X, et al. Autophagy is a protective response to ethanol neurotoxicity. Autophagy. 2012;8:1577–1589. doi: 10.4161/auto.21376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kandadi MR, Hu N, Ren J. ULK1 plays a critical role in AMPK-mediated myocardial autophagy and contractile dysfunction following acute alcohol challenge. Curr Pharm Des. 2013;19:4874–4887. doi: 10.2174/1381612811319270010. [DOI] [PubMed] [Google Scholar]
- 220.Eid N, Ito Y, Maemura K, Otsuki Y. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: an immunohistochemical and electron microscopic study. J Mol Histol. 2013;44:311–326. doi: 10.1007/s10735-013-9483-x. [DOI] [PubMed] [Google Scholar]
- 221.Wu D, Wang X, Zhou R, Yang L, Cederbaum AI. Alcohol steatosis and cytotoxicity: the role of cytochrome P4502E1 and autophagy. Free Radic Biol Med. 2012;53:1346–1357. doi: 10.1016/j.freeradbiomed.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Fortunato F, Bürgers H, Bergmann F, Rieger P, Büchler MW, Kroemer G, Werner J. Impaired autolysosome formation correlates with Lamp-2 depletion: role of apoptosis, autophagy, and necrosis in pancreatitis. Gastroenterology. 2009;137:350–60, 360.e1-5. doi: 10.1053/j.gastro.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 223.Baraona E, Leo MA, Borowsky SA, Lieber CS. Alcoholic hepatomegaly: accumulation of protein in the liver. Science. 1975;190:794–795. doi: 10.1126/science.1198096. [DOI] [PubMed] [Google Scholar]
- 224.Thomes PG, Trambly CS, Thiele GM, Duryee MJ, Fox HS, Haorah J, Donohue TM. Proteasome activity and autophagosome content in liver are reciprocally regulated by ethanol treatment. Biochem Biophys Res Commun. 2012;417:262–267. doi: 10.1016/j.bbrc.2011.11.097. [DOI] [PubMed] [Google Scholar]
- 225.Donohue TM, Zetterman RK, Zhang-Gouillon ZQ, French SW. Peptidase activities of the multicatalytic protease in rat liver after voluntary and intragastric ethanol administration. Hepatology. 1998;28:486–491. doi: 10.1002/hep.510280228. [DOI] [PubMed] [Google Scholar]
- 226.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:141–150. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- 228.Wu WK, Cho CH, Lee CW, Wu YC, Yu L, Li ZJ, Wong CC, Li HT, Zhang L, Ren SX, et al. Macroautophagy and ERK phosphorylation counteract the antiproliferative effect of proteasome inhibitor in gastric cancer cells. Autophagy. 2010;6:228–238. doi: 10.4161/auto.6.2.11042. [DOI] [PubMed] [Google Scholar]
- 229.Yang L, Wu D, Wang X, Cederbaum AI. Cytochrome P4502E1, oxidative stress, JNK, and autophagy in acute alcohol-induced fatty liver. Free Radic Biol Med. 2012;53:1170–1180. doi: 10.1016/j.freeradbiomed.2012.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, Friedman SL. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–946. doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Thoen LF, Guimarães EL, Dollé L, Mannaerts I, Najimi M, Sokal E, van Grunsven LA. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55:1353–1360. doi: 10.1016/j.jhep.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 232.Hernández-Gea V, Hilscher M, Rozenfeld R, Lim MP, Nieto N, Werner S, Devi LA, Friedman SL. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J Hepatol. 2013;59:98–104. doi: 10.1016/j.jhep.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Ding WX. Induction of autophagy, a promising approach for treating liver injury. Hepatology. 2014;59:340–343. doi: 10.1002/hep.26572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Bridle KR, Popa C, Morgan ML, Sobbe AL, Clouston AD, Fletcher LM, Crawford DH. Rapamycin inhibits hepatic fibrosis in rats by attenuating multiple profibrogenic pathways. Liver Transpl. 2009;15:1315–1324. doi: 10.1002/lt.21804. [DOI] [PubMed] [Google Scholar]
- 235.Zhu J, Wu J, Frizell E, Liu SL, Bashey R, Rubin R, Norton P, Zern MA. Rapamycin inhibits hepatic stellate cell proliferation in vitro and limits fibrogenesis in an in vivo model of liver fibrosis. Gastroenterology. 1999;117:1198–1204. doi: 10.1016/s0016-5085(99)70406-3. [DOI] [PubMed] [Google Scholar]
- 236.Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479–2487. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 237.Arden KC. FOXO animal models reveal a variety of diverse roles for FOXO transcription factors. Oncogene. 2008;27:2345–2350. doi: 10.1038/onc.2008.27. [DOI] [PubMed] [Google Scholar]
- 238.Tikhanovich I, Cox J, Weinman SA. Forkhead box class O transcription factors in liver function and disease. J Gastroenterol Hepatol. 2013;28 Suppl 1:125–131. doi: 10.1111/jgh.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 240.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 241.van der Vos KE, Eliasson P, Proikas-Cezanne T, Vervoort SJ, van Boxtel R, Putker M, van Zutphen IJ, Mauthe M, Zellmer S, Pals C, et al. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat Cell Biol. 2012;14:829–837. doi: 10.1038/ncb2536. [DOI] [PubMed] [Google Scholar]
- 242.Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12:665–675. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- 243.Sengupta A, Molkentin JD, Paik JH, DePinho RA, Yutzey KE. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J Biol Chem. 2011;286:7468–7478. doi: 10.1074/jbc.M110.179242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Xiong X, Tao R, DePinho RA, Dong XC. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem. 2012;287:39107–39114. doi: 10.1074/jbc.M112.412569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Ni HM, Du K, You M, Ding WX. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am J Pathol. 2013;183:1815–1825. doi: 10.1016/j.ajpath.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Tikhanovich I, Kuravi S, Campbell RV, Kharbanda KK, Artigues A, Villar MT, Weinman SA. Regulation of FOXO3 by phosphorylation and methylation in hepatitis C virus infection and alcohol exposure. Hepatology. 2014;59:58–70. doi: 10.1002/hep.26618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Tumurbaatar B, Tikhanovich I, Li Z, Ren J, Ralston R, Kuravi S, Campbell R, Chaturvedi G, Huang TT, Zhao J, et al. Hepatitis C and alcohol exacerbate liver injury by suppression of FOXO3. Am J Pathol. 2013;183:1803–1814. doi: 10.1016/j.ajpath.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Nepal S, Park PH. Activation of autophagy by globular adiponectin attenuates ethanol-induced apoptosis in HepG2 cells: involvement of AMPK/FoxO3A axis. Biochim Biophys Acta. 2013;1833:2111–2125. doi: 10.1016/j.bbamcr.2013.05.013. [DOI] [PubMed] [Google Scholar]