

Abstract
Non-alcoholic fatty liver disease (NAFLD) is the most frequent cause of liver disease in the Western world. Furthermore, it is increasing worldwide, paralleling the obesity pandemic. Though highly frequent, only about one fifth of affected subjects are at risk of developing the progressive form of the disease, non-alcoholic steatohepatitis with fibrosis. Even in the latter, liver disease is slowly progressive, though, since it is so prevalent, it is already the third cause of liver transplantation in the United States, and it is predicted to get to the top of the ranking in few years. Of relevance, fatty liver is also associated with increased overall mortality and particularly increased cardiovascular mortality. The literature and amount of published papers on NAFLD is increasing as fast as its prevalence, which makes it difficult to keep updated in this topic. This review aims to summarize the latest knowledge on NAFLD, in order to help clinicians understanding its pathogenesis and advances on diagnosis and treatment.
Keywords: Non-alcoholic fatty liver disease, Metabolic syndrome, Insulin resistance, Epidemiology, Pathogenesis, Genetics, Diagnosis, Prognosis, Management
Core tip: This is a review in non-alcoholic fatty liver disease (NAFLD) that puts the disease into context, highlights the recent advances in pathology, and gives special focus to the diagnosis and management of those patients. We present NAFLD patients in a holistic view, understanding that it many cases thinking outside the liver, namely in the cardiovascular and neoplastic risk, may have a bigger impact in the prognosis. In the era of genomics and high-throughput approaches, we also summarized the latest breakthroughs regarding the genetic associations with NAFLD.
EPIDEMIOLOGY
Non-alcoholic fatty liver disease (NAFLD) is a spectrum of liver disease characterized by the presence of ectopic fat in the liver, steatosis, which cannot be explained by alcohol consumption. It ranges from simple steatosis to non-alcoholic steatohepatitis (NASH) that can have different degrees of fibrosis and progress to liver cirrhosis and end-stage liver disease, with its associated complications, including hepatocellular carcinoma (HCC).
NAFLD is the pandemic liver disease from the twenty-first century. It has been estimated that about one billion individuals worldwide have NAFLD[1]. In fact, it is the number one cause of altered aminotransferases in the Western world[2], where one third of the population is affected[3]. In Asia, recent reports showed similar prevalence of NAFLD[4,5], whereas in the Indian continent it is similar in urban populations, although much lower, less than 10%, in rural populations[6-8]. The prevalence in Africa is not known[1], though a Nigerian study showed a prevalence of less than 10%[9].
Among patients with NAFLD, one to two in each ten patients will have NASH[3]. A recent survey showed that NAFLD-associated cirrhosis is the third indication for liver transplantation in the United States, and it is expected to be the number one indication in 2020[10].
NAFLD is strongly associated with obesity, insulin resistance (IR)/type 2 diabetes mellitus (T2DM) and the metabolic syndrome. Obesity, particularly central obesity, is highly predictive of hepatic steatosis and disease progression[11,12]. In overweight subjects, the prevalence of steatosis is at least two times more frequent than in lean subjects[13], being directly proportional to the increase of body mass index (BMI)[14]. In morbid obesity, almost all patients present steatosis and more than one third have NASH[15]. The association with T2DM is also very strong, being 5-9 times more frequent in patients with NAFLD as compared to the general population[16]. On the other hand, more than two third of patients with T2DM have NAFLD[17]. T2DM also associates with disease severity, namely NASH, fibrosis, liver cirrhosis and HCC[16]. Lastly, the metabolic syndrome is a cluster of cardiovascular risk factors that associates with IR, namely central obesity, hypertension, dyslipidemia and glucose intolerance[18]. At least one third of patients with NAFLD have the metabolic syndrome and 80% at least one of its components[19,20]. Noteworthy, the prevalence of NAFLD increases with the number of components of the metabolic syndrome[20].
There are racial differences in the susceptibility for NAFLD, being more frequent in East Asian Indians, followed by Hispanics, Asians, Caucasians and less frequent in African Americans[1,19,21,22]. Also, among patients with NAFLD, African Americans have less frequently NASH as opposed to Hispanics[23]. The racial disparities are not fully understood. A possible explanation may ensue from a higher BMI and adiposity in Hispanics[24,25] and a lower visceral adiposity in African Americans[26]. Genetic differences in metabolism and wound healing response may also have an influence. For instance, African Americans have lower fructose absorption rates than Hispanics, and fructose is considered an important driver of liver steatogenesis[27].
In this review we will summarize the latest evidence in the pathogenesis, diagnosis and management of NAFLD, with a clinical-oriented focus.
PATHOGENESIS
How do we get liver steatosis?
The hallmark of NAFLD is the presence of ectopic fat in the hepatocytes. The underlying mechanisms of this pathological fat deposition in the liver are reasonably known. The main store of lipids in the hepatocyte is in the form of triglycerides, though other lipids can be increased, such as, different free fatty acids (FFA), diacylglycerol, free cholesterol, cholesterol esters, ceramides, and phospholipids[28]. Triglycerides are synthesized from FFA. The accumulation of triglycerides depends on the presence of FFA in the liver and its disposal. There are three major sources of FFA in the liver, with the following order of relevance: 60% from plasmatic nonesterified fatty acids (NEFA’s), 25% from de novo lipogenesis and 15% from dietary fatty acids (FA) in the form of chylomicrons lipoproteins[29]. Starting with the former, the hepatic uptake of FFA is unregulated; as such, it is directly proportional to the plasmatic concentrations of NEFAs[30]. Of relevance, plasmatic NEFAs come mainly from lipolysis in the adipose tissue, which represents the source of 80% of FA in the fasting state. Postprandial, dietary FA also plays a relevant contribution, but even then, 60% of FFA derives from adipocytes[29]. Adipose tissue lipolysis is induced, during fasting, through the stimulation of beta-adrenergic receptors by catecholamines[31]. When adipose tissue storage capacity is overcome, either by energy surplus as occur in obesity, or by primary defect in adipocytes as occurs in lipodystrophies, the efflux of FFA increases. Also, with IR, there is a decreased ability of insulin to suppress adipose tissue lipolysis[32]. De novo lipogenesis, during fasting, is increased by 3 fold in patients with NAFLD as compared to those with lean liver[33]. Also, it has no diurnal variation[29]. This probably represents hyperinsulinism, which induces sterol response element-binding protein (SREBP)-1c and peroxisome proliferator-activated receptor (PPAR)-γ that in turn promote the expression of several lipogenic genes[33]. De novo lipogenesis is also important, since when activated, a key intermediate, malonyl-CoA, inhibits the oxidation of FA from any source[34]. Lastly, the contribution of dietary FA increases with high fat diet (by definition, more than 30% of total energy requirements)[29]. FFA in the liver may follow 3 different destinations: (1) oxidation, mainly in mitochondria, but also in extra-mitochondrial organelles; (2) assembly and export as very low-density lipoproteins (VLDL); and (3) production of triglycerides and storage as lipid droplets. Regarding the latter, when production of triglycerides is inhibited, it decreases steatosis, but increases liver damage by accumulation of active FA intermediates[35]. An excessive increase in FFA influx in the liver may over saturate the ability for FA oxidation. Although patients may have mitochondrial abnormalities and dysfunction, there is no evidence of decreased FA oxidation in most of the patients[36]. On the contrary, when this pathway is overactive it may promote the production of oxygen reactive species and oxidative stress[37,38]. Hyperinsulinism may decrease VLDL assembly, since insulin decreases the synthesis and stability of apolipoprotein B, one component of VLDL[39,40]. Also, NAFLD/NASH can occur in familial hypolipoproteinemia, in which there is a defect in VLDL assembly, independently of obesity and IR[41] (Figure 1).
Figure 1.
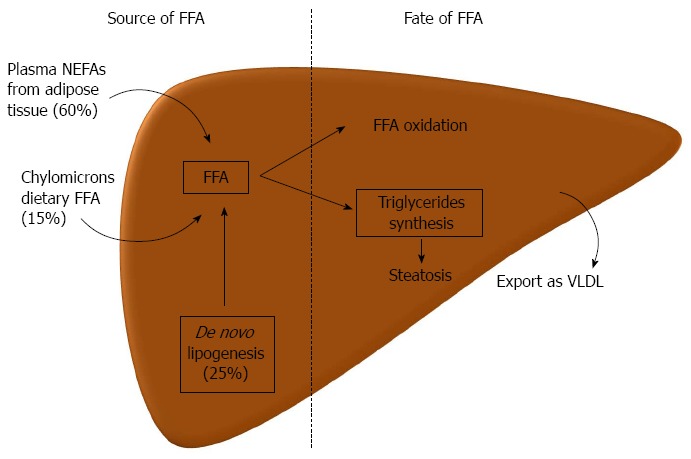
Pathogenesis of liver steatosis. Hepatic steatosis can result from an increased influx of lipids, free fatty acids (FFA), to the liver or a decreased lipid disposal. Three main sources of FFA in the liver are the plasmatic nonesterified fatty acids (NEFAs), which originate predominantly from lipolysis in the adipose tissue, from de novo lipogenesis, mainly from glucose or other carbohydrates, and from FFA that come in chylomicrons from the gut (dietary FFA). In the liver, FFA can either be oxidized, mainly in the mitochondria, beta-oxidation, or can be used to produce triglycerides. The latter can be exported as very low density lipoproteins (VLDL) to the circulation or can accumulate in lipid droplets in the hepatocyte leading to steatosis.
As described, IR has a major role in the development of steatosis. But steatosis itself also promotes IR, endorsing a self-perpetuating vicious cycle. The most accepted model is the initiation of IR peripherally in the adipose tissue[42,43]. Obesity translates an expansion in adipose tissue. That expansion may occur in inert subcutaneous tissue, being more an esthetical condition, but it may also occur in visceral, metabolically active adipose tissue. The excessive accumulation of fat in adipocytes promotes an increase in oxidative stress, which deregulates adipocytokines production[44] and promotes low grade inflammatory state in the adipose tissue, through release of interleukin (IL)-6 and monocyte chemotactic protein (MCP)-1 among others[44]. Subsequently, there is activation of macrophages, M1, and lymphocytes, Th1, promoting further release of proinflammatory cytokines, tumor necrosis factor (TNF)-α and interferon-γ. The latter also promotes IR[45] directly or through the deregulation of adipocytokines secretion, namely through inhibition of adiponectin. In fact, with obesity, adiponectin, an anti-inflammatory, insulin sensitizer adipokine, decreases, whereas leptin, a pro-inflammatory, pro-fibrogenic and satiety inducer adipokine increases. Of note, central resistance to leptin also develops, decreasing its anorexigenic effects[46]. The spillover of FFA from the adipose tissue leads to ectopic accumulation of fat in muscle and liver. In those tissues, ectopic fat induces IR by generation of lipid-derived second messengers such as diacylglycerol (DAG) and ceramides that directly interfere with the insulin receptor pathway[47]. Noteworthy, in the liver, not all insulin actions are impaired; it preserves its lipogenic actions further inducing steatosis, and its pro-mitogenic actions, which may enhance hepatocarcinogenesis[48].
Injury and inflammation leads to NASH and fibrogenesis
Lipid accumulation in the liver is linked with endoplasmic reticulum (ER) stress, oxidative stress/mitochondria stress, and impaired autophagy, resulting in the condition known as lipotoxicity[31]. In some patients, lipotoxicity leads to cell damage and cell death, which induces an inflammatory and wound healing response that can drive fibrogenesis. Why in some people lipid accumulation in the liver is inert and in others is toxic to cells is still not fully understood. It may be related to the type of fat itself, since triglycerides per se do not seem toxic[35], whereas FA, mainly saturated FA[49,50] as well as cholesterol[51] and its metabolites do seem highly toxic. The higher toxicity of saturated FA as compared to polyunsaturated FA may be in part due to a limited capacity of hepatocytes to use them to produce triglycerides[52]. Also, individual differences in lipid metabolism (e.g., in the enzymes that desaturate FA[50]) and susceptibility for cell damage, may promote NASH development (Figure 2).
Figure 2.
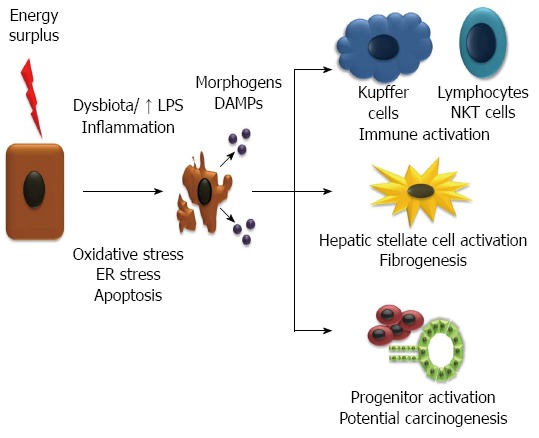
Injury and inflammation leads to non-alcoholic steatohepatitis and fibrogenesis. Energy surplus leads to fat accumulation in the hepatocytes which promote oxidative stress, endoplasmic reticulum (ER) stress and apoptosis. The injury of hepatocytes is promoted by an inflammatory state, among other factors, favored by a deregulated gut microbiota and increase in lipopolysaccharide (LPS). Injured and dying hepatocytes release damage associated molecular patterns (DAMPs) and morphogens (e.g. hedgehog and Wnt), that act on the immune system increasing inflammation, in stellate cells and progenitors cells activating them and inducing fibrogenesis and pathways of hepatocarcinogenesis. FFA: Free fatty acids; NEFA: Nonesterified fatty acids.
Cell death
Apoptosis is a form of programmed cell death that plays an important role in NAFLD[53,54]. Considerable amounts of evidence show an increase in hepatocyte apoptosis in animal models[55-58] and in human NASH[59-62]. FFA can increase the susceptibility of hepatocytes to cell death through induction of expression of cell death receptors that promote the extrinsic pathway of apoptosis, Fas and DR5, TRAIL (TNF related apoptosis inducing ligand) receptor 5[56,59,63]. FFA and free cholesterol can also promote apoptosis through dose-dependent mitochondrial toxicity, which leads not only to increased production of oxygen reactive species[64], but also through activation of pro-apoptotic proteins Bim and Bax that trigger intrinsic apoptotic pathway[36,65]. Ceramide, a sphingolipid that can also accumulate in NAFLD, can be synthesized de novo from long chain saturated FA[28]. It has been associated with apoptosis induction and inhibition of insulin pathway leading to IR[66].
FFA-promoted oxidative stress has several deleterious effects, through direct injury of DNA, proteins and lipids[52], but also by promoting cell death with the activation of stress-sensitive pathways such as NF-κB, p38 MAPK and c-Jun N terminal kinase[67] as well as promoting ER stress[68]. Lipotoxicity is, in fact, associated with an ER stress response and unfolded protein response, mainly by saturated and not polyunsaturated FA[69].
Sterile inflammation
Lobular chronic inflammatory infiltrate, in the absence of pathogens, is a hallmark of NASH. Injured cells and necrotic cells (to a lesser extent also apoptotic cells) can release molecules, termed “damage-associated molecular patterns” (DAMPs), which trigger inflammation, through the binding of several receptors[70]. Those receptors can be specific or shared with pathogen-associated molecular patterns (PAMPs) that recognize molecular patterns associated with microbial pathogens or cellular stress, such as some toll-like receptors (TLR). An important mechanism to initiate inflammation is through the inflammasomes, cytosolic multiprotein complexes, present in the liver in parenchymal and non-parenchymal cells, which respond to DAMPs and PAMPs. It requires two signals: the first, through the NF-κB pathway having upstream TNF receptor or TLRs, increases the transcription of components of the inflammasome; and the second in response to DAMPs and PAMPs with assembly of the inflammasome. The inflammasome is formed by a complex of a receptor from the family of NOD-like receptors, with pro-caspase-1 and an adaptor: apoptosis-associated speck like CARD-domain containing protein (ASC). The assembly of the complex leads to autocatalytic cleavage and activation of caspase-1 which then cleaves pro-IL-1β, pro-IL-18 activating them and IL-33 neutralizing it[71]. The first 2 cytokines are pro-inflammatory and IL-33 drives Th2 responses[72]. Kupffer cells, resident macrophages in the liver, seem to be a crucial element in detecting DAMPs, and reacting with an inflammasome response[73,74], though that can also occur in endothelial cells, stellate cells and hepatocytes[71].
This pathway seems to be important in the pathogenesis of NASH. In fact, several animal models of NASH showed an increase of IL-1β[73-77]. Also, mice deficient in IL-1β[76] or components of the inflammasome[77] were protected from NASH and fibrosis. Moreover, increased expression of inflammasome components has been shown in liver biopsies from patients with NASH[75].
The role of Kupffer cells in the inflammatory and fibrogenic response in NAFLD is starting to be highlighted by hepatologists. Studies with human biopsies showed an increase in CD68, a pan-macrophage marker in patients with NASH, as compared with simple steatosis[78,79]. Also, in vitro studies with co-cultures with Kupffer cells and hepatic stellate cells, showed increased proliferation and activation of the latter, with increased production of collagen 1a, which was inhibited by catalase[80]. Regarding studies with animal models, methionine-choline deficient diet fed mice, in which Kupffer cells were depleted by administration of clodronate-containing liposomes, developed less features of NASH[81]. In the early phases of NAFLD, the classic activation of macrophages, M1, in the liver, may promote more inflammation and IR, as well as steatosis. However, an alternative activation of macrophages, M2, which is anti-inflammatory and insulin-sensitizer, but also profibrogenic seems to play a major role. In fact, in several mouse models, ob/ob, high fat-diet and lipoatrophic diabetic A-ZIP transgenic mice with fatty liver, an M2 response, in the adipose tissue and in the liver, protects from glucose intolerance and IR, as well as hepatic steatosis[82-84]. The effect of M2 polarization in liver fibrogenesis is still not characterized with conflicting results in the literature[85]. However, there are concerns that M2 polarization may promote carcinogenesis[86].
Other inflammatory cells in the innate immune system, such as NKT cells and NK cells play a crucial role in the development of NAFLD and NASH. Also, adaptive immune system seems to have a role, having been described a Th1-polarization in the liver and peripheral blood of patients with NASH[87,88]. A more profound review is out of the scope of this article[89,90].
Microbiota
Obesity and the metabolic syndrome have been associated with disturbances in the gut microbiota. Microbiota can induce a higher extraction of energy from diet and change whole-body lipid metabolism shifting it from oxidation to de novo production[91]. Also, the diet itself can modulate the microbiota, with high-fat diets favoring a metabolically less favorable microbiome[91]. Dysbiota also seems important in the development of NAFLD and NASH. Recently, a mouse model with dysbiota, induced by defects of inflammasome in the gut, promoted NASH development. Noteworthy, when the gut microbiota from affected mice was transferred to regular mice lead to the induction of NASH per se[92]. Furthermore, NAFLD has been associated with small intestinal bacterial overgrowth[93-95] and increased intestinal permeability[96]. That can result in an increase in endotoxin, lipopolysaccharide (LPS)[97-99]. TLR-4 is a receptor for the LPS from Gram-negative bacteria, being one of the most potent activators of Kupffer cells. Rivera et al[100] found that mice under methionine-choline deficient-diet expressed higher TLR-4, which was blunted after Kupffer cells depletion with clodronate liposomes. Additionally, mice with a mutant TLR-4 were protected from steatohepatitis development[100]. Gut dysbiosis can promote NASH by other mechanisms, such as ethanol production and disturbing choline metabolism[91].
Morphogens inducing fibrogenic and progenitors response
Injured cells, such as ballooned hepatocytes and dying cells that will undergo apoptosis and necrosis, release cues to hepatic stellate cells, inflammatory cells and progenitors[101]. It is adaptive because it promotes a wound-healing response, with repair and regeneration, which could not be accomplished by the senescent hepatocytes. However, if those cues are sustained, it leads to excessive activation of stellate cells driving fibrogenesis. Sustained activation of progenitor cells may also promote hepatocarcinogenesis. Some of those cues have been proved to be morphogens such as hedgehog and Wnt ligands. In fact, ballooned hepatocytes produce sonic hedgehog[102]. Hedgehog is known to activate hepatic stellate cells[103,104] as well as progenitor cells[105]. The role of hedgehog pathway has been extensively demonstrated in NASH development and fibrogenesis in animal models[106-108] and corroborated by association studies in human NAFLD[109]. Recently, administration of a hedgehog antagonist protected from liver injury in a mouse model of diet-induced NASH[110].
Lastly, the mechanisms that promote steatosis are intricate with the pro-fibrogenic process. For instance, hyperinsulinism may be profibrogenic by itself, since insulin promotes stellate cell proliferation and activation increasing the production of collagen 1a and alpha-smooth muscle actin[111,112].
GENETICS
NAFLD development is highly dependent on the environment, with tremendous influence by the dietary patterns and a sedentary lifestyle. However, it surely requires an interaction with a genetic susceptibility. In fact, independent studies showed a strong familial aggregation[113-116]. The racial differences in the prevalence of NAFLD and NASH also highlight a genetic background. Furthermore, a study on 157 patients with familial combined hyperlipidemia and 20 spouses, showed higher NAFLD prevalence in the patients and its direct family members as compared to their spouses, which differentiates an inherited transmission rather than shared lifestyles[117]. In fact, a recent large population study suggested that the heredity of NAFLD is 26%-27%[118].
Genetic associations with NAFLD have been widely studied. The first studies looked for candidate genes with case-control designs. Recently, in the era of omics, a non-biased approach has been applied, with several genome wide associations studies (GWAS) already published in this topic[118-125].
The most consistent genetic association is with patatin-like phospholipase domain containing 3 (PNPLA3) or adiponutrin. That association was described in the first GWAS study performed in NAFLD patients[119]. Romeo et al[119] studied a multiethnic population from the Dallas Heart Study comprising 2111 subjects, including African Americans, European-Americans and Hispanics. Hepatic steatosis was assessed by proton-magnetic resonance spectroscopy (1H-MRS). They found a specific non-synonymous single nucleotide polymorphism (SNP), rs738409, with a substitution of cytosine by guanine (C>G) translating in a substitution of an isoleucine for a methionine in residue 148 (I148M), that strongly associated with hepatic fat content and serum levels of aminotransferases. It did not associate, however, with metabolic factors such as BMI, presence of diabetes, indices of insulin sensitivity or plasma lipid profile. Noteworthy, that SNP was more frequent in Hispanics, followed by European-Americans and less frequent in African Americans, resembling the racial differences in the prevalence of NAFLD. They also described another SNP, with a non-synonymous substitution of an isoleucin for methionine at residue 48 (I48M) that associated with lower hepatic fat content and was more frequent in African Americans. The association of PNPLA3 rs738409 was reproduced in different genetic backgrounds, by several other GWAS[121,122,125] and candidate gene studies[126-137]. This SNP has also been associated with severity of histological NAFLD, namely with severity of steatosis[126,129], presence of NASH[121,126,133], NAFLD Activity Score (NAS)[129] and fibrosis[131,133,136,138,139], irrespective of degree of obesity or metabolic co-morbidities. The function of adiponutrin is not clearly understood. In humans it is predominantly expressed in the liver but in mice in the adipose tissue. Its expression is positively regulated by carbohydrate-responsive element-binding protein (ChREBP), in response to glucose and SREBP-1c in response to insulin[140]. It is located in the ER and lipid droplets membrane[141]. In vitro studies have shown lipolysis properties, triglyceride hydrolase and diacylglycerol transacylase activities[142], but also possible function in lipid synthesis with lysophosphatidic acid acyltransferase activity, converting lysophosphatidic acid in phosphatidic acid[143].
Other genetic associations suggested by GWAS and confirmed by case-control candidate gene studies are: neurocan (NCAN), an adhesion molecule, with steatosis[118,136,137,144], lobular inflammation[136] and perivenular fibrosis[136]; glucokinase regulatory gene (GCKR) also not only with NAFLD[118,144-146], but also with NASH[137,146] and fibrosis[146]; lysophospholipase-like 1 (LYPLAL1), an enzyme in triglycerides breakdown, and protein phosphatase-1 regulatory subunit 3b (PPP1R3B), an enzyme in glycogen breakdown, with steatosis[118,125,137,144].
A vast amount of genetic polymorphisms have been evaluated in case-control studies on NAFLD. Most of them either were not reproduced by different groups or presented conflicting results among studies, needing more extensive validation. Four genes are, however, worthy mention. Manganese superoxide dismutase, encoded by the gene SOD2, is a mitochondrial enzyme relevant in detoxification of reactive oxygen species. It is synthesized in the cytosol and, after posttranslational changes, transported to the mitochondria. Patients with NAFLD have been shown to present lower hepatic levels of manganese superoxide dismutase[147]. A non-synonymous polymorphism in SOD2, C1183T, with substitution of cysteine for threonine at residue 47 (C47T), associates with less efficient transport of the protein to the mitochondria[148]. Four independent cohorts, from different ethnic backgrounds, Japanese[149], European[150,151], African[152], showed an association with NASH and hepatic fibrosis. Kruppel-like factor 6 (KLF6) is a transcriptional factor expressed in activated stellate cells that regulates the expression of several fibrogenic genes such as collagen 1a, transforming growth factor (TGF)-β and its receptors[153]. KLF6-IVS1-27G>A is a functional polymorphism that induces a site of alternative splicing, inactivating it[154]. A study with 415 patients[155], from 3 different cohorts, one from United Kingdom, other from Italy and a trio with 2 parents with an affected child showed an association between that SNP and steatosis, inflammation and hepatic fibrosis severity. More recently, a pediatric study also showed an association with NASH[151]. Furthermore, the renin-angiotensin system is believed to promote hepatic fibrogenesis. Variants in the angiotensin II receptor-1 (ATGR-1) were associated with NAFLD and hepatic fibrosis, in two independent cohorts[156,157]. Lastly, TNF-α is a crucial pro-inflammatory cytokine in NAFLD that also promotes IR. Two polymorphisms in the promoter of TNF gene, -308A/G and -238A/G, known to associate with increased expression of TNF-α and IR, have been evaluated in several studies as potential modifiers of disease risk, with conflicting results[158-163]. Recently, a meta-analysis on 8 studies and more than 1500 NAFLD patients and healthy controls, showed that the polymorphism -238G/A in homozygosity conferred a two-fold increased risk for NAFLD, whereas the polymorphism -308A/G was not associated with an increased risk[164].
In the recent years, there has been a change in how we think in genetics. Not only the genes are important, but also epigenetic that regulates how the genes are expressed has been shown highly relevant in the pathogenesis of NAFLD. In fact, several independent studies showed that the male offsprings of female mice fed high fat diet during pregnancy, presented a higher risk for developing advanced NASH[165-170]. Also, they presented increased genetic expression of genes in lipid metabolism, inflammation and oxidative stress[165,170], which was associated with a correspondent different pattern of DNA methylation[170]. An animal study with male Wilstar rats fed with NAFLD-inducing high fat diet, showed increased expression of genes in lipid metabolism, with correspondent decrease in the methylation status of their promoters[171]. In human NAFLD, two important studies were done with epigenetics. Ahrens et al[171] studied NAFLD associated with morbid obesity; comparing with healthy controls, patients had different methylation status in lipid metabolism and insulin signaling, that was partially reversed by bariatric surgery. Murphy et al[172] studied patients with NAFLD with mild or advanced fibrosis and found differences in the methylation status of fibrogenic genes, enzymes of one-carbon metabolism and components of inflammasome, which correlated with gene expression.
DIAGNOSIS
For better understanding the diagnosis, there is need to clarify some definitions. Fatty liver or steatosis, is considered when more than 5% of the hepatocytes present ectopic lipid droplets in a liver biopsy[173,174]. Though this is an arbitrary definition, it is corroborated by quantitative data using H1-MRS in a large healthy population, showing that 95% of subjects will have less than 5% steatosis[175]. However, it should be noticed that H1-MRS does not measure the same as a liver biopsy. In fact, it measures the amount of triglycerides in the parenchyma and not the number of positive hepatocytes. Steatohepatitis requires the presence of lobular inflammation and hepatocyte lesion, usually in the form of hepatocellular ballooning with or without Mallory-Denk bodies, besides steatosis[176]. When cirrhosis is fully developed, features of NASH may be lost, such as ballooning and even steatosis, which is called burned-out NASH. To be considered non-alcoholic, the patient must drink less than the amounts of alcohol that have been linked to increased risk for liver disease. Large prospective cohort studies done in the nineties, showed that alcohol intake of more than 2-3 drinks per day in men and 1-2 drinks per day in women increased the risk for liver-associated mortality[177-179]. Taking that into consideration, European[173] and American[174] guidelines consider the threshold to be non-alcoholic intakes lower than 210 and 140 g per week (roughly equivalent to 3 and 2 drinks per day) in men and women, respectively. The Asian guidelines are a little more restrictive, considering intakes lower than 140 and 70 g per week (two and one drink per day) in men and women, respectively[180].
The gold standard for the diagnosis is the liver biopsy, mainly for diagnosing NASH and staging fibrosis. However, for the diagnosis and quantification of steatosis, H1-MRS may be the new reference since it assesses larger volumes of liver and it detects amounts of triglycerides that may not be enough to form macrovesicles amenable of histological visualization[181]. Liver biopsy cannot be used routinely, since it is an invasive and expensive procedure, in which the low rate of complications would be significant if massively applied to all patients with NAFLD. Also, it has some limitations that should not be forgotten: it is prone to sample error, since it only assesses an insignificant volume of the liver, and this is a disease in which lesions are unevenly distributed throughout the liver, which results in wrong exclusion of NASH in one forth of cases and misclassification of fibrosis severity in one third[182]. On the other hand, it is highly dependent on the pathologist, mainly for the diagnosis of NASH. Kleiner et al[183] proposed a histological score, NAS, that combines different degrees of steatosis, hepatocellular ballooning and lobular inflammation. It was not intended to diagnose NASH, though several studies have used it with that purpose. It was designed to help monitoring the effect of interventional and therapeutic strategies. The utility of the NAS score as a prognostic marker and even as a guidance to evaluate treatment-efficacy has been questioned[184-186]. In fact, it has several limitations, including the fact that the degree of steatosis confers a huge impact on the score, although the severity of steatosis does not have proven prognostic value. On the other hand, it does not take into account the stage of fibrosis. More recently, Bedossa et al[187] proposed a different score, steatosis activity fibrosis (SAF) score that sequentially adds steatosis, ballooning and lobular inflammation for the diagnosis of NASH. It needs external validation.
Most of NAFLD patients will have an incidental diagnosis, mainly because the majority of patients are asymptomatic and symptoms, when present, are unspecific such as fatigue and abdominal discomfort. Aminotransferases are generally in normal-range values, and when altered, the increase is usually mild and fluctuant[188]. Adding to that, it is not recommended to screen the population, even in high risk groups, since a cost-effective analysis has not been done and there is no effective specific treatment for NAFLD[174]. Having said that, the first method to be used in the clinical setting for diagnosing NAFLD is abdominal ultrasound. It is non-invasive, inexpensive, widely available, and presents reasonable accuracy for the diagnosis of hepatic steatosis, with 60%-94% sensitivity and 66%-97% specificity[189]. However, its sensitivity decreases extremely for mild steatosis, being only reliable for steatosis higher than 30%[190]. Also, its specificity is compromised by the presence of fibrosis, edema, necrosis and extra-hepatic adipose tissue. Computed tomography does not add accuracy to the ultrasound, and because it is more expensive and exposes patients to radiation, should not be used with the sole purpose of diagnosing hepatic steatosis. Magnetic resonance is more sensitive than ultrasound, mainly for the diagnosis of mild steatosis[191], but it is time-consuming and expensive, so it is seldom used in clinical practice. The most accurate radiological method is H1-MRS[175], but it is costly and lacks broaden availability, which limits its use to research sets. More recently, a method that uses the equipment of transient elastography, controlled attenuation parameter (CAP), is promising, with great accuracy, not being influenced by fibrosis[192]. Though the added benefit in accuracy with ultrasound has not been properly assessed, it seems more sensitive for lower levels of steatosis[193]. There are several combined panels with clinical and laboratorial data that were designed to predict steatosis[194-197]. They are particularly relevant to large-scale epidemiological studies without availability of ultrasound and for a quick clinical suspicion of the presence of NAFLD in high-risk populations such as patients with T2DM. For large-scale epidemiological studies, fatty liver index (FLI), which incorporates BMI, waist circumference, serum levels of triglycerides and γ-glutamyltranspeptidase, can be very useful, and has been extensively validated[195]. For high-risk populations, NAFLD Liver Fat score, is a very simple score that incorporates the presence of metabolic syndrome or T2DM, fasting serum insulin levels, aspartate aminotransferases (AST) and AST/alanine aminotransferases (ALT) ratio[196]. It showed good performance in predicting NAFLD in the original publication and when reproduced by independent authors.
As important as diagnosing NAFLD, is the stratification of patients as having NASH and/or fibrosis. Again, clinical clues such as symptoms and physical examination do not help, though the presence of metabolic syndrome should raise the clinical suspicion of NASH. Aminotransferases are also not reliable, though if increased associate with a higher risk[198]. Several complex models incorporating different clinical and chemical variables have been proposed, most of them with suboptimal accuracy or lacking external validation[199-206]. The fragments of keratin 18 (CK18) is for now, the best surrogate marker for NASH, showing AUROC 0.82 in a meta-analysis of 10 studies and more than 1000 patients with NAFLD[207]. However, it is not a perfect tool, and more recent studies including a second meta-analysis showed lower accuracy with sensitivity about 60%[208,209].
Regarding estimation of liver fibrosis, several biomarkers[210-212] and composite scores have also been proposed[185,213-219]. Of those, the most robust, with more extensive evaluation by different groups and good performance is the NAFLD Fibrosis score[216]. It incorporates, in an equation, age, glycemia, BMI, platelet count, albumin and AST/ALT ratio. Values lower or equal to -1.455 and higher than 0.676, presented high accuracy in excluding and identifying advanced fibrosis, respectively. A meta-analysis of 13 studies, comprising more than 3000 patients, confirmed those results[207]. One limitation of this score is that nearly one fourth of the patients will not be classified, because will fall in between those cut-offs - intermediate risk. More recently, NAFLD Fibrosis Score (among other non-invasive scores) has been shown to be useful in predicting overall and liver-related mortality, in 3 studies with follow up ranging from 8 to 14 years[220-222]. Transient elastography (Fibroscan©), already widely used in chronic hepatitis C, also has good accuracy to predict advanced fibrosis and exclude it in NAFLD, with cutoffs higher and lower than 9.6 and 7.9, respectively[207,209,223-227], although probably not as sensitive as in hepatitis C[228]. A concern is that elastography is more prone to failure in obesity, which is not completely overcome by the XL probes, and also in the presence of ascites and narrow intercostals spaces. Also, elastography is influenced by meals, acute hepatitis, cholestasis and liver congestion. Other methods to evaluate elastography, such as real-time elastography that takes advantage of a B mode conventional mechanism, and magnetic resonance elastography that allows estimation of elasticity in whole liver, are promising. More recently, acoustic radiation force impulse (ARFI) that is integrated in conventional ultrasound systems shows very good results that need further validation[229-233].
In conclusion, most of the times, the diagnosis of NAFLD is incidental. When a patient presents in the clinic with ultrasound-diagnosed NAFLD, there is no clinical or laboratorial accurate way to stratify the patient in terms of presence of NASH and fibrosis. However, several risk factors should increase the suspicion, such as, older age and postmenopausal women, being Hispanic, obese, with IR, T2DM, hypertension or the metabolic syndrome, and increased aminotransferases levels[234]. NAFLD Fibrosis Score is a simple test with good prediction ability for fibrosis and even prognostic implications. If the score is lower than -1.4555, the risk of having fibrosis is very low. If none of the other risk factors are identified, it can be discharged for primary care follow up. If the above risk factors are present, and there is availability for performing CK18 fragments determination, it could be done, and if predictive of NASH, confirmed by liver biopsy. On the other extreme, NAFLD Fibrosis Score higher than 0.676, a liver biopsy should be performed to confirm advanced fibrosis, unless there is clinical evidence of liver cirrhosis. Lastly, patients with intermediate NAFLD Fibrosis Score values should be offered another non-invasive tool for assessing fibrosis, such as transient elastography, and if high level of suspicion of fibrosis, should be considered for liver biopsy.
NATURAL HISTORY AND PROGNOSIS
Patients with NAFLD have a decreased survival as compared to the general population[235]. The number one cause of death in patients with NAFLD is cardiovascular disease, followed by malignancies and only then liver disease[235-237]. In fact, patients with NAFLD die two fold more frequently due to cardiovascular disease than to liver disease itself[207]. Even in patients with liver cirrhosis, cardiovascular disease is still the second cause of death[207]. Importantly, patients with NAFLD have a two-fold increase in cardiovascular mortality as compared to the general population[235,237,238]. However, liver death takes the third position in the ranking of mortality in patients with NAFLD, whereas it is only the thirteenth cause of death in the general population[235]. As such, NAFLD patients die 9 times more often from liver disease than the general population[236]. Despite the fact that NASH is considered the progressive form of NAFLD[239,240], there is no difference on overall or cardiovascular mortality between patients with simple steatosis and NASH, though the latter group does have a more than 10 fold increase in liver mortality[237].
The high cardiovascular mortality may be explained by the fact that NAFLD is a surrogate marker of metabolic derangement. Moreover, NAFLD itself may contribute to the pathogenesis and development of T2DM and the metabolic syndrome. In fact, in longitudinal studies, NAFLD either diagnosed by altered liver enzymes[241], abdominal ultrasound[242-247] or non-invasive scores markers of NAFLD[248,249], showed an increased risk for developing T2DM and metabolic syndrome. That increase in T2DM risk was even higher in patients with NASH vs simple steatosis[240].
Regarding liver prognosis, NAFLD is a slowly progressive disease. Simple steatosis is believed to be a non-progressive condition, whereas NASH can develop progressive fibrosis in more than one fourth of the patients in 4 years and nearly half of the patients in 6 years[207]. Fibrosis may evolve in liver cirrhosis. Over 14 years of follow up, 13% of patients with F2 grade fibrosis and 25% with F3 grade are expected to develop liver cirrhosis[240]. Grossly, one fifth of patients with NASH will develop liver cirrhosis in the long term[1].
The main risk factors for having advanced fibrosis are older age[250,251], presence of obesity and central obesity[12], as well as T2DM[252] and hypertension[199]. A systematic review[253] of 10 studies comprising 221 patients submitted to paired biopsies apart on average from 5 years, showed that fibrosis progression, of at least one stage, occurred in a little more than one third of the patients. Overall, the mean rate of fibrosis progression was 0.03 stages per year; however, analyzing only the patients in whom fibrosis progressed, the rate was 0.41 stages per year. The stronger predictor of fibrosis progression was necroinflammation. Having any lobular inflammation increased by 2.5 fold the likelihood of developing advanced fibrosis. Steatosis severity did not correlate with fibrosis progression. Of note, ballooning was not possible to access. In fact, ballooning correlates with severity of fibrosis[184].
Patients with NAFLD can also progress to HCC, even without cirrhosis[254]. Some cases of HCC were reported in patients with simple steatosis, without NASH or fibrosis[255,256]. A recent systematic review on 17 studies showed that in non-cirrhotics, the cumulative HCC mortality rate was 3% over a 20 year follow up. In patients with cirrhosis, that rate was 2.4-12.8 after 3 to 12 years of follow up[257]. In fact, NAFLD-associated cirrhosis accounts for 15%-30% of cases of HCC[258].
MANAGEMENT
The first difficult decision is who to treat and who to discharge from a specialized assistance to a general physician. Patients who should continue to be followed by hepatologists are the ones at risk for progressive liver disease, that is, patients with NASH, particularly if they have liver fibrosis. However, non-pharmacological measurements should be applied to all patients with NAFLD, even simple steatosis, since NAFLD confers an increased risk for metabolic derangement and cardiovascular disease as well as an increased risk for several malignancies (Table 1). Screening for hepatocellular carcinoma should be offered to all patients with liver cirrhosis. In patients with NASH and even simple steatosis with no fibrosis, though there are several reports on the literature of liver cancer, there is not sufficient evidence to recommend a screening program. Though a cost-efficacy analysis has not been done in patients with NAFLD without fibrosis, the incidence of liver cancer in those patients is so low that would not warrant its application. Also, because patients with NAFLD are at increased risk of malignancies, they should be carefully monitored in the regular screening programs for colorectal, prostate, breast and cervical cancer.
Table 1.
Summary of the management of non-alcoholic fatty liver disease
| Lifestyle interventions - diet and physical exercise |
| Weight loss: 3%-5% if simple steatosis, 7%-10% if NASH |
| Accompanied by cognitive-behavior therapy program |
| Diet - hypocaloric, adjusted to the patients needs and body weight |
| Fat - prefer PUFAs, mainly ω3 - advise 2-3 oily fish meals/wk |
| ≤ 25% as MUFA’s, avoid SAF (less than 7% total energy) Cholesterol ≤ 200 mg/d |
| Carbohydrates - ≥ 50% as whole grains, avoid high fructose corn syrup |
| No need to restrict coffee |
| Mild alcohol intake - do not prohibit, do not advise; recommend against in patients with cirrhosis |
| Exercise - aerobic and restrictive, ≥ 3-4 times/wk, ≥ 400 calories per session |
| Treat risk factors when present |
| Insulin sensitizers - no clear evidence to prefer thiazolidinediones or biguanides |
| Lipid-lowering drugs - statins are safe; protect from cardiovascular risk (more than in non-NAFLD) |
| Anti-hypertensive drugs - prefer ARAII if no contraindication, mainly telmisartan |
| Specific treatment for NAFLD |
| Consider vitamin E in patients with NASH, non-diabetic and without hypertension or at risk for prostate cancer |
| Pentoxifilin - promising agent that needs more evidence from large randomized clinical trials |
| Probiotics - promising agents that need more evidence from large randomized clinical trials |
| Screening for cancer |
| Screening for hepatocellular carcinoma every 6 mo in cirrhotic patients |
| Screening program (colorectal, breast, prostate and cervical cancer) as general population |
NAFLD: Non-alcoholic fatty liver disease; NASH: Non-alcoholic steatohepatitis; PUFA: Polyunsaturated fatty acids; MUFA: Monounsaturated fatty acids; SAF: Saturated fatty acids; ARA II: Angiotensin II receptor antagonists.
Despite huge investigation on it, we still do not have available an effective treatment for NAFLD. Most of the trials done in patients with NAFLD showed some efficacy in decreasing steatosis, inflammation and even ballooning, though there is no solid evidence for a treatment being able to decrease fibrosis. This apparent failure could be either because we still did not find a good anti-fibrotic agent in NAFLD, or because the design of the trials, that usually have no more than 2 years of intervention, was unable to detect differences in fibrosis progression. Also, the desirable trials would be the ones that could show an effect on survival and major events such as liver decompensation. However, due to the slowing progressive nature of NAFLD, those trials would have needed long follow up, which makes them impractical.
Non-pharmacological approach
It is recommended a personalized approach with lifestyle intervention, focusing on diet and exercise, in order to lose weight, to all patients with NAFLD. Except for the rare patients with normal weight, it should be promoted weight loss of 3%-5% in patients with simple steatosis and 7%-10% in patients with NASH[174]. In the long term, normal weight should be the goal. Though several studies have shown a benefit of weight loss in NAFLD[259-272], the rational for those weight goals comes mainly from 3 studies, one observational[273] and two interventional[274,275]. The first is a longitudinal study, by Zelber-Sagi et al, with 7 years follow up of 147 healthy subjects[273]. The authors found that weight gain and loss associated, respectively, with de novo NAFLD and NAFLD remission. Of notice, if the weight loss was 5% of the body weight or more, the remission rate of NAFLD was 75%. The two interventional studies were both on 31 obese or overweight patients with NASH, managed either for 48 wk[274] or 36 wk[275]. What collectively they found was that weight loss of at least 5% of body weight associated with improvements in steatosis and IR, but only when weight loss was at least 7%-9% there were improvements in inflammation, hepatocellular ballooning and NAS. Of notice, compliance in lifestyle intervention programs and weight loss are difficult to accomplish, usually almost half of the patients cannot achieve the proposed goals. To improve the efficacy of those programs, it would be preferable to associate them with cognitive-behavior therapy programs[276].
The effect of weight loss is independent of the composition of the hypocaloric diet[277-280]. However, some recommendations should be made. Patients with dyslipidemia may benefit from low fat diets[277], whereas patients with IR/T2DM from low carbohydrate diets[278]. Regarding lipids composition, polyunsaturated FA should be preferred, mainly ω3 from fish oil; up to 25% of fat should be in the form of mono-unsaturated FA, in detriment of saturated FA that should account for less than 7% of total energy[281]. Cholesterol intake should be lowered to 200 mg per day[282]. Those recommendations rely on several lines of evidence. Patients with NAFLD, and even more with NASH, eat a lower polyunsaturated vs saturated FA ratio, a lower ω3 vs ω6 ratio, and a higher amount of cholesterol, as compared to the general population[283-287]. Also, saturated FA associate with IR[285], oxidative stress[288], ER stress and apoptosis[49,50], whereas polyunsaturated FA may decrease hepatic lipid content through inhibition of SREBP-1c[289]. Furthermore, several studies evaluated the effect of supplementation with ω3 polyunsaturated FA in patients with NAFLD[290-292], and a recent meta-analysis[293] suggested a benefit in decreasing liver fat and aminotransferases, starting at doses as low as 0.83 g per day. The authors could not infer the ideal dose, due to the high heterogeneity among studies. Lastly, two large-scale population-based prospective cohort studies found a positive correlation between cholesterol intake[287] and a negative correlation between ω3 polyunsaturated FA-rich fish consumption[294] and increased risk for liver cirrhosis and HCC.
Concerning carbohydrates consumption, at least half should come from whole grains. Most importantly, high fructose corn syrup, a major source of fructose in occidental diet[295], should be avoided. Actually, higher fructose consumption, in the form of soft drinks, has been associated with NAFLD[296] and fibrosis stage, inflammation as well as hepatocellular ballooning[297]. In fact, the steatogenic effect of fructose can be explained by its metabolism that bypasses the regulatory points of glycolysis and thus can act as an unregulated source of substrates for lipolysis; it regulates the expression of lipogenic genes through induction of SREBP-1c and ChREBP; it decreases fatty acid oxidation through downregulation of PPAR-α; and it promotes bacterial overgrowth and hence increases the load of endotoxin that reaches the liver[298].
Several studies have shown that exercise alone, even when weight loss is not achieved, has beneficial effects on NAFLD[268,270], and in IR[299-301]. However, the combination of exercise and diet has synergic value[302,303]. The type of exercise does not matter[302,304,305], as long as at least 400 kcal are spent per session[306]. Patients should be advised to perform moderate exercise, preferentially 3-4 times per week[307].
Regarding alcohol consumption, it is known, since the nineties, that consumption higher than 2-3 drinks per day not only increases the risk for liver cirrhosis, it also increases the risk for overall death and some malignancies[177,179,308,309]. However, there is a U relation between alcohol consumption with lower intakes decreasing overall mortality at expense of a decrease in cardiovascular events[310,311], less risk for diabetes mellitus[312] and the metabolic syndrome[313-315]. Some studies also showed a possible beneficial effect in NAFLD for very mild alcohol consumption[199,316-320]. In our view, we should not actively recommend mild alcohol consumption in patients with NAFLD since the evidence come largely from cross-sectional studies[321]. Also, the benefit of mild alcohol consumption in obese patients is not known, since obesity and alcohol are synergic in promoting NAFLD and HCC[322-325]. In patients with NASH-associated cirrhosis, however, it should be strongly recommended against, since any regular alcohol consumption, in this set of patients, increases by more than 3-fold the risk for HCC[326].
Lastly, coffee should not be recommended against, because an important amount of evidence, either by epidemiological studies either by studies in animal models[327-331], suggest a protective effect in terms of metabolic control[332,333] and NAFLD development and progression[334-339].
Pharmacological therapy to promote weight loss does not seem to confer any benefit over lifestyle intervention alone, as been shown in trials with orlistat[275,340]. Lastly, bariatric surgery may be an alternative to promote weight loss and improve metabolic profile in morbid obese patients[341]. Even in moderately obese patients, it showed beneficial effects in promoting metabolic control[342]. A meta-analysis on bariatric surgery applied to morbid obese patients with NAFLD, but without cirrhosis, and paired liver biopsies, showed improvement in steatosis in 92% of patients, steatohepatitis in 82% and fibrosis in 65%[343].
Pharmacological approach
Since there is no clear curative treatment for NAFLD, the management of these patients should rely first on the control of co-morbidities known to promote not only liver disease, but also cardiovascular disease and overall mortality, such as IR/T2DM, dyslipidemia and hypertension. The effect of some of those treatments in the liver has been evaluated. For now, the presence of NAFLD alone should not be an indication to use anti-diabetics, lipid-lowering drugs or anti-hypertensive drugs. However, in patients who also present those metabolic disturbances, some drugs may be preferred in patients with NAFLD.
Agents that target risk factors
Starting with anti-diabetics, there is no clear evidence to prefer a specific class of drugs. Thiazolidinediones have been widely studied on NAFLD, mainly rosiglitazone and pioglitazone. We will focus on the latter, since rosiglitazone has been abolished from the European market and has very restricted use in the United States, due to its association with increased risk of adverse cardiovascular events. Three well designed randomized controlled trials evaluated pioglitazone against placebo treatment, during 6, 12 and 24 mo, including 55, 74 and 247 patients with NASH, respectively[344-346]. Collectively they found pioglitazone to be better than placebo in improving glucose metabolism, decreasing aminotransferases levels and improving steatosis, ballooning and inflammation, but not in improving liver fibrosis. Thought pioglitazone, during the time-course it was administered, did not improve fibrosis, it decreased the rate of fibrosis progression, as described by two meta-analysis[347,348]. Of notice, only the smaller study included patients with glucose intolerance or T2DM, the other two were in non-diabetics, which might not be the better target for insulin-sensitizers. There are some concerns that preclude us to propose its systematic use in patients with NAFLD with glucose intolerance. First, there are some safety issues, as it associates with considerable weight gain that does not return back to baseline after stopping the drug[349]. Also, it has been associated with increased risk for congestive heart failure[350], bone fractures[351] and bladder cancer after two years of treatment[352]. Lastly, it showed no additional benefit after one year of treatment, and even more importantly, the relapse is certain after discontinuation of pioglitazone[349]. Other insulin-sensitizer, the biguanide metformin, though it has obvious benefic metabolic effects, not only in terms of glucose control, but also in increasing high density lipoprotein (HDL)-cholesterol, it promotes weight loss, which can be an advantage when starting treatment in patients with NAFLD and overweight. However, it has no proved benefit in liver histology[3,348,353].
The effect of lipid lowering agents in NAFLD is still not completely understood, though small studies suggested a mild benefit in steatosis and NAS score, without clear effect on fibrosis, for either statins[354], fibrates[355,356], probucol[357,358] and ezetimibe[359,360]. However, the benefits of statins in these patients go beyond the liver. In fact, post-hoc analysis of two large studies on the effect of statins in cardiovascular outcomes in high risk patients, showed that patients with NAFLD, as assessed by an increase in aminotransferases without other cause for liver disease, had an even greater decrease in cardiovascular events, as compared to patients with normal aminotransferases baseline[361,362]. We can then conclude that we should not be afraid of treating patients with NAFLD and elevated aminotransferases with statins, and they might even be used with a lower threshold for dyslipidemia.
Lastly, a specific class of anti-hypertensive, angiotensin II receptor antagonists, should be preferred in patients with NAFLD, regarding no contraindication is obvious. In fact, losartan has been shown to decrease liver fibrosis and hepatic stellate cells activation in a very small pilot study[363,364]. Telmisartan looks even more promising[365] since it has, unlike other drugs from the same class, anti-steatogenic and insulin-sensitizer actions through agonistic effects on PPAR-γ[366].
Agents specific for NAFLD
Vitamin E is the one with more evidence. Two large randomized controlled studies in patients with NASH, without diabetes mellitus, in adult and pediatric population, showed similar results. It had benefit over placebo in decreasing aminotransferases, and in improving liver histology, steatosis, NASH, inflammation and ballooning, but not fibrosis. Also, in patients who did not achieve weight loss, the disease seemed to progress less in the ones taking vitamin E. Again, we do not recommend its systematic use since, though it was better than placebo, more than half of the patients did not show any histological improvement. On the other hand, there is no evidence for treating patients with diabetes, nor patients with simple steatosis or liver cirrhosis, so it should not be prescribed in those cases. Lastly, several safety concerns cannot be under looked: it has been associated with increased mortality, starting at doses as low as 150 IU/d[367], increased risk for prostate cancer after 3 years of treatment[368], and increased risk for hemorrhagic stroke, though decreased for ischemic ones[369].
Ursodeoxycholic acid has been widely studied and it showed no effect in NAFLD[348,370-373], though it may improve adiponectin levels[374] and IR[373]. Pentoxifilin[375-378] and probiotics[379-383] look promising in pilot studies, but more robust evidence from large scale randomized controlled-trials is warranted.
Several anti-fibrotic, anti-apoptotic and immune therapies are in the pipeline in pre-clinical and phase II trials, and hopefully in the next decade, we will achieve a more efficient liver-specific treatment.
CONCLUSION
NAFLD is the new pandemic of the twenty first century, walking together with obesity. It can be seen as a consequence of metabolic deregulation associated with energy surplus and exceeded reservoir ability of adipose tissue to store fat/energy. However, it is becoming clearer that hepatic steatosis also drives that metabolic deregulation, promoting T2DM development and cardiovascular disease. In fact, patients with NAFLD have increased mortality, mainly at expenses of cardiovascular disease, though liver-associated mortality becomes relevant in patients with more severe disease, particularly, with NASH and fibrosis.
Though NAFLD has profound influences by environmental factors, such as obesity, western diet and a sedentary lifestyle, genetic influences play together with environment in orchestrating the pathogenesis of the disease and its spectrum. In the last 5 years, genetic susceptibility has been studied by GWAS unbiased studies, with some important genes popping out that otherwise would have not been linked to NAFLD. More recently, a new layer of complexity has been added, with the understanding that epigenetic modifications also can modulate the susceptibility for this disease.
In patients with NAFLD, the gold standard for diagnosing NASH and fibrosis is liver biopsy, but some non-invasive methods have high accuracy in predicting fibrosis, which can help us selecting patients for histological assessment.
Treatment should be aimed not only to decrease liver disease progression, but also to decrease cardiovascular events and mortality. The basis remains lifestyle education with hypocaloric diet and physical exercise promoting weight loss. Metabolic risk factors, such as IR/T2DM, hypertension and dyslipidemia should be treated aggressively. There is no approved specific treatment for the liver disease itself, although vitamin E could be used, with caution, in non-diabetic patients with NASH and fibrosis, but without cirrhosis and with no expected increased risk for prostate cancer.
Footnotes
P- Reviewer: Anty R, Inzaugarat E, Ji G, Kobyliak NK S- Editor: Ding Y L- Editor: A E- Editor: Zhang DN
References
- 1.Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol. 2013;10:686–690. doi: 10.1038/nrgastro.2013.171. [DOI] [PubMed] [Google Scholar]
- 2.Clark JM, Brancati FL, Diehl AM. The prevalence and etiology of elevated aminotransferase levels in the United States. Am J Gastroenterol. 2003;98:960–967. doi: 10.1111/j.1572-0241.2003.07486.x. [DOI] [PubMed] [Google Scholar]
- 3.Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- 4.Eguchi Y, Hyogo H, Ono M, Mizuta T, Ono N, Fujimoto K, Chayama K, Saibara T. Prevalence and associated metabolic factors of nonalcoholic fatty liver disease in the general population from 2009 to 2010 in Japan: a multicenter large retrospective study. J Gastroenterol. 2012;47:586–595. doi: 10.1007/s00535-012-0533-z. [DOI] [PubMed] [Google Scholar]
- 5.Farrell GC, Wong VW, Chitturi S. NAFLD in Asia--as common and important as in the West. Nat Rev Gastroenterol Hepatol. 2013;10:307–318. doi: 10.1038/nrgastro.2013.34. [DOI] [PubMed] [Google Scholar]
- 6.Amarapurkar D, Kamani P, Patel N, Gupte P, Kumar P, Agal S, Baijal R, Lala S, Chaudhary D, Deshpande A. Prevalence of non-alcoholic fatty liver disease: population based study. Ann Hepatol. 2007;6:161–163. [PubMed] [Google Scholar]
- 7.Singh SP, Nayak S, Swain M, Rout N, Mallik RN, Agrawal O, Meher C, Rao M. Prevalence of nonalcoholic fatty liver disease in coastal eastern India: a preliminary ultrasonographic survey. Trop Gastroenterol. 2004;25:76–79. [PubMed] [Google Scholar]
- 8.Das K, Das K, Mukherjee PS, Ghosh A, Ghosh S, Mridha AR, Dhibar T, Bhattacharya B, Bhattacharya D, Manna B, et al. Nonobese population in a developing country has a high prevalence of nonalcoholic fatty liver and significant liver disease. Hepatology. 2010;51:1593–1602. doi: 10.1002/hep.23567. [DOI] [PubMed] [Google Scholar]
- 9.Onyekwere CA, Ogbera AO, Balogun BO. Non-alcoholic fatty liver disease and the metabolic syndrome in an urban hospital serving an African community. Ann Hepatol. 2011;10:119–124. [PubMed] [Google Scholar]
- 10.Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhising RA. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology. 2011;141:1249–1253. doi: 10.1053/j.gastro.2011.06.061. [DOI] [PubMed] [Google Scholar]
- 11.Fan JG, Zhu J, Li XJ, Chen L, Lu YS, Li L, Dai F, Li F, Chen SY. Fatty liver and the metabolic syndrome among Shanghai adults. J Gastroenterol Hepatol. 2005;20:1825–1832. doi: 10.1111/j.1440-1746.2005.04058.x. [DOI] [PubMed] [Google Scholar]
- 12.Wong VW, Wong GL, Choi PC, Chan AW, Li MK, Chan HY, Chim AM, Yu J, Sung JJ, Chan HL. Disease progression of non-alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut. 2010;59:969–974. doi: 10.1136/gut.2009.205088. [DOI] [PubMed] [Google Scholar]
- 13.Bellentani S, Tiribelli C. The spectrum of liver disease in the general population: lesson from the Dionysos study. J Hepatol. 2001;35:531–537. doi: 10.1016/s0168-8278(01)00151-9. [DOI] [PubMed] [Google Scholar]
- 14.Patt CH, Yoo HY, Dibadj K, Flynn J, Thuluvath PJ. Prevalence of transaminase abnormalities in asymptomatic, healthy subjects participating in an executive health-screening program. Dig Dis Sci. 2003;48:797–801. doi: 10.1023/a:1022809430756. [DOI] [PubMed] [Google Scholar]
- 15.Machado M, Marques-Vidal P, Cortez-Pinto H. Hepatic histology in obese patients undergoing bariatric surgery. J Hepatol. 2006;45:600–606. doi: 10.1016/j.jhep.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 16.Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol. 2013;10:330–344. doi: 10.1038/nrgastro.2013.41. [DOI] [PubMed] [Google Scholar]
- 17.Targher G, Bertolini L, Rodella S, Tessari R, Zenari L, Lippi G, Arcaro G. Nonalcoholic fatty liver disease is independently associated with an increased incidence of cardiovascular events in type 2 diabetic patients. Diabetes Care. 2007;30:2119–2121. doi: 10.2337/dc07-0349. [DOI] [PubMed] [Google Scholar]
- 18.Grundy SM, Brewer HB, Cleeman JI, Smith SC, Lenfant C. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109:433–438. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- 19.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 20.Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N, Melchionda N, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–923. doi: 10.1053/jhep.2003.50161. [DOI] [PubMed] [Google Scholar]
- 21.Petersen KF, Dufour S, Feng J, Befroy D, Dziura J, Dalla Man C, Cobelli C, Shulman GI. Increased prevalence of insulin resistance and nonalcoholic fatty liver disease in Asian-Indian men. Proc Natl Acad Sci USA. 2006;103:18273–18277. doi: 10.1073/pnas.0608537103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider AL, Lazo M, Selvin E, Clark JM. Racial differences in nonalcoholic fatty liver disease in the U.S. population. Obesity (Silver Spring) 2014;22:292–299. doi: 10.1002/oby.20426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kallwitz ER, Guzman G, TenCate V, Vitello J, Layden-Almer J, Berkes J, Patel R, Layden TJ, Cotler SJ. The histologic spectrum of liver disease in African-American, non-Hispanic white, and Hispanic obesity surgery patients. Am J Gastroenterol. 2009;104:64–69. doi: 10.1038/ajg.2008.12. [DOI] [PubMed] [Google Scholar]
- 24.Flegal KM, Carroll MD, Kuczmarski RJ, Johnson CL. Overweight and obesity in the United States: prevalence and trends, 1960-1994. Int J Obes Relat Metab Disord. 1998;22:39–47. doi: 10.1038/sj.ijo.0800541. [DOI] [PubMed] [Google Scholar]
- 25.Casas YG, Schiller BC, DeSouza CA, Seals DR. Total and regional body composition across age in healthy Hispanic and white women of similar socioeconomic status. Am J Clin Nutr. 2001;73:13–18. doi: 10.1093/ajcn/73.1.13. [DOI] [PubMed] [Google Scholar]
- 26.Perry AC, Applegate EB, Jackson ML, Deprima S, Goldberg RB, Ross R, Kempner L, Feldman BB. Racial differences in visceral adipose tissue but not anthropometric markers of health-related variables. J Appl Physiol (1985) 2000;89:636–643. doi: 10.1152/jappl.2000.89.2.636. [DOI] [PubMed] [Google Scholar]
- 27.Walker RW, Lê KA, Davis J, Alderete TL, Cherry R, Lebel S, Goran MI. High rates of fructose malabsorption are associated with reduced liver fat in obese African Americans. J Am Coll Nutr. 2012;31:369–374. doi: 10.1080/07315724.2012.10720445. [DOI] [PubMed] [Google Scholar]
- 28.Alkhouri N, Dixon LJ, Feldstein AE. Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Rev Gastroenterol Hepatol. 2009;3:445–451. doi: 10.1586/egh.09.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tamura S, Shimomura I. Contribution of adipose tissue and de novo lipogenesis to nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1139–1142. doi: 10.1172/JCI24930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koo SH. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clin Mol Hepatol. 2013;19:210–215. doi: 10.3350/cmh.2013.19.3.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- 33.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes. 2002;51:7–18. doi: 10.2337/diabetes.51.1.7. [DOI] [PubMed] [Google Scholar]
- 35.Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, Bhanot S, Monia BP, Li YX, Diehl AM. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 36.Fromenty B, Robin MA, Igoudjil A, Mansouri A, Pessayre D. The ins and outs of mitochondrial dysfunction in NASH. Diabetes Metab. 2004;30:121–138. doi: 10.1016/s1262-3636(07)70098-8. [DOI] [PubMed] [Google Scholar]
- 37.Fromenty B, Pessayre D. Inhibition of mitochondrial beta-oxidation as a mechanism of hepatotoxicity. Pharmacol Ther. 1995;67:101–154. doi: 10.1016/0163-7258(95)00012-6. [DOI] [PubMed] [Google Scholar]
- 38.Seifert EL, Estey C, Xuan JY, Harper ME. Electron transport chain-dependent and -independent mechanisms of mitochondrial H2O2 emission during long-chain fatty acid oxidation. J Biol Chem. 2010;285:5748–5758. doi: 10.1074/jbc.M109.026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown AM, Gibbons GF. Insulin inhibits the maturation phase of VLDL assembly via a phosphoinositide 3-kinase-mediated event. Arterioscler Thromb Vasc Biol. 2001;21:1656–1661. doi: 10.1161/hq1001.096640. [DOI] [PubMed] [Google Scholar]
- 40.Qin B, Anderson RA, Adeli K. Tumor necrosis factor-alpha directly stimulates the overproduction of hepatic apolipoprotein B100-containing VLDL via impairment of hepatic insulin signaling. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1120–G1129. doi: 10.1152/ajpgi.00407.2007. [DOI] [PubMed] [Google Scholar]
- 41.Amaro A, Fabbrini E, Kars M, Yue P, Schechtman K, Schonfeld G, Klein S. Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology. 2010;139:149–153. doi: 10.1053/j.gastro.2010.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murdolo G, Bartolini D, Tortoioli C, Piroddi M, Iuliano L, Galli F. Lipokines and oxysterols: novel adipose-derived lipid hormones linking adipose dysfunction and insulin resistance. Free Radic Biol Med. 2013;65:811–820. doi: 10.1016/j.freeradbiomed.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 43.Lomonaco R, Ortiz-Lopez C, Orsak B, Webb A, Hardies J, Darland C, Finch J, Gastaldelli A, Harrison S, Tio F, et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology. 2012;55:1389–1397. doi: 10.1002/hep.25539. [DOI] [PubMed] [Google Scholar]
- 44.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tateya S, Kim F, Tamori Y. Recent advances in obesity-induced inflammation and insulin resistance. Front Endocrinol (Lausanne) 2013;4:93. doi: 10.3389/fendo.2013.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kwon H, Pessin JE. Adipokines mediate inflammation and insulin resistance. Front Endocrinol (Lausanne) 2013;4:71. doi: 10.3389/fendo.2013.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Machado MV, Ferreira DM, Castro RE, Silvestre AR, Evangelista T, Coutinho J, Carepa F, Costa A, Rodrigues CM, Cortez-Pinto H. Liver and muscle in morbid obesity: the interplay of fatty liver and insulin resistance. PLoS One. 2012;7:e31738. doi: 10.1371/journal.pone.0031738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marra F, Lotersztajn S. Pathophysiology of NASH: perspectives for a targeted treatment. Curr Pharm Des. 2013;19:5250–5269. doi: 10.2174/13816128113199990344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao J, Dai DL, Yao L, Yu HH, Ning B, Zhang Q, Chen J, Cheng WH, Shen W, Yang ZX. Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol Cell Biochem. 2012;364:115–129. doi: 10.1007/s11010-011-1211-9. [DOI] [PubMed] [Google Scholar]
- 50.Li ZZ, Berk M, McIntyre TM, Feldstein AE. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-CoA desaturase. J Biol Chem. 2009;284:5637–5644. doi: 10.1074/jbc.M807616200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caballero F, Fernández A, De Lacy AM, Fernández-Checa JC, Caballería J, García-Ruiz C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol. 2009;50:789–796. doi: 10.1016/j.jhep.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 52.Zámbó V, Simon-Szabó L, Szelényi P, Kereszturi E, Bánhegyi G, Csala M. Lipotoxicity in the liver. World J Hepatol. 2013;5:550–557. doi: 10.4254/wjh.v5.i10.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Machado MV, Cortez-Pinto H. Cell death and nonalcoholic steatohepatitis: where is ballooning relevant? Expert Rev Gastroenterol Hepatol. 2011;5:213–222. doi: 10.1586/egh.11.16. [DOI] [PubMed] [Google Scholar]
- 54.Feldstein AE, Gores GJ. Apoptosis in alcoholic and nonalcoholic steatohepatitis. Front Biosci. 2005;10:3093–3099. doi: 10.2741/1765. [DOI] [PubMed] [Google Scholar]
- 55.Rashid A, Wu TC, Huang CC, Chen CH, Lin HZ, Yang SQ, Lee FY, Diehl AM. Mitochondrial proteins that regulate apoptosis and necrosis are induced in mouse fatty liver. Hepatology. 1999;29:1131–1138. doi: 10.1002/hep.510290428. [DOI] [PubMed] [Google Scholar]
- 56.Feldstein AE, Canbay A, Guicciardi ME, Higuchi H, Bronk SF, Gores GJ. Diet associated hepatic steatosis sensitizes to Fas mediated liver injury in mice. J Hepatol. 2003;39:978–983. doi: 10.1016/s0168-8278(03)00460-4. [DOI] [PubMed] [Google Scholar]
- 57.Witek RP, Stone WC, Karaca FG, Syn WK, Pereira TA, Agboola KM, Omenetti A, Jung Y, Teaberry V, Choi SS, et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology. 2009;50:1421–1430. doi: 10.1002/hep.23167. [DOI] [PubMed] [Google Scholar]
- 58.Farrell GC, Larter CZ, Hou JY, Zhang RH, Yeh MM, Williams J, dela Pena A, Francisco R, Osvath SR, Brooling J, et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J Gastroenterol Hepatol. 2009;24:443–452. doi: 10.1111/j.1440-1746.2009.05785.x. [DOI] [PubMed] [Google Scholar]
- 59.Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, Gores GJ. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 60.Ribeiro PS, Cortez-Pinto H, Solá S, Castro RE, Ramalho RM, Baptista A, Moura MC, Camilo ME, Rodrigues CM. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol. 2004;99:1708–1717. doi: 10.1111/j.1572-0241.2004.40009.x. [DOI] [PubMed] [Google Scholar]
- 61.Ramalho RM, Cortez-Pinto H, Castro RE, Solá S, Costa A, Moura MC, Camilo ME, Rodrigues CM. Apoptosis and Bcl-2 expression in the livers of patients with steatohepatitis. Eur J Gastroenterol Hepatol. 2006;18:21–29. doi: 10.1097/00042737-200601000-00005. [DOI] [PubMed] [Google Scholar]
- 62.Bechmann LP, Gieseler RK, Sowa JP, Kahraman A, Erhard J, Wedemeyer I, Emons B, Jochum C, Feldkamp T, Gerken G, et al. Apoptosis is associated with CD36/fatty acid translocase upregulation in non-alcoholic steatohepatitis. Liver Int. 2010;30:850–859. doi: 10.1111/j.1478-3231.2010.02248.x. [DOI] [PubMed] [Google Scholar]
- 63.Malhi H, Barreyro FJ, Isomoto H, Bronk SF, Gores GJ. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut. 2007;56:1124–1131. doi: 10.1136/gut.2006.118059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marí M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, García-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 65.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- 66.Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 67.Ghosh J, Das J, Manna P, Sil PC. Taurine prevents arsenic-induced cardiac oxidative stress and apoptotic damage: role of NF-kappa B, p38 and JNK MAPK pathway. Toxicol Appl Pharmacol. 2009;240:73–87. doi: 10.1016/j.taap.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 68.Bánhegyi G, Margittai E, Szarka A, Mandl J, Csala M. Crosstalk and barriers between the electron carriers of the endoplasmic reticulum. Antioxid Redox Signal. 2012;16:772–780. doi: 10.1089/ars.2011.4437. [DOI] [PubMed] [Google Scholar]
- 69.Wang D, Wei Y, Pagliassotti MJ. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 2006;147:943–951. doi: 10.1210/en.2005-0570. [DOI] [PubMed] [Google Scholar]
- 70.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 71.Szabo G, Csak T. Inflammasomes in liver diseases. J Hepatol. 2012;57:642–654. doi: 10.1016/j.jhep.2012.03.035. [DOI] [PubMed] [Google Scholar]
- 72.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 73.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. 2010;139:323–34.e7. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stienstra R, Saudale F, Duval C, Keshtkar S, Groener JE, van Rooijen N, Staels B, Kersten S, Müller M. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology. 2010;51:511–522. doi: 10.1002/hep.23337. [DOI] [PubMed] [Google Scholar]
- 75.Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54:133–144. doi: 10.1002/hep.24341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, Olteanu S, Barshack I, Dotan S, Voronov E, et al. Lack of interleukin-1α or interleukin-1β inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol. 2011;55:1086–1094. doi: 10.1016/j.jhep.2011.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lefkowitch JH, Haythe JH, Regent N. Kupffer cell aggregation and perivenular distribution in steatohepatitis. Mod Pathol. 2002;15:699–704. [Google Scholar]
- 79.Park JW, Jeong G, Kim SJ, Kim MK, Park SM. Predictors reflecting the pathological severity of non-alcoholic fatty liver disease: comprehensive study of clinical and immunohistochemical findings in younger Asian patients. J Gastroenterol Hepatol. 2007;22:491–497. doi: 10.1111/j.1440-1746.2006.04758.x. [DOI] [PubMed] [Google Scholar]
- 80.Nieto N. Oxidative-stress and IL-6 mediate the fibrogenic effects of [corrected] Kupffer cells on stellate cells. Hepatology. 2006;44:1487–1501. doi: 10.1002/hep.21427. [DOI] [PubMed] [Google Scholar]
- 81.Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J Biol Chem. 2012;287:40161–40172. doi: 10.1074/jbc.M112.417014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kitade H, Sawamoto K, Nagashimada M, Inoue H, Yamamoto Y, Sai Y, Takamura T, Yamamoto H, Miyamoto K, Ginsberg HN, et al. CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes. 2012;61:1680–1690. doi: 10.2337/db11-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nio Y, Yamauchi T, Iwabu M, Okada-Iwabu M, Funata M, Yamaguchi M, Ueki K, Kadowaki T. Monocyte chemoattractant protein-1 (MCP-1) deficiency enhances alternatively activated M2 macrophages and ameliorates insulin resistance and fatty liver in lipoatrophic diabetic A-ZIP transgenic mice. Diabetologia. 2012;55:3350–3358. doi: 10.1007/s00125-012-2710-2. [DOI] [PubMed] [Google Scholar]
- 84.Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, Pecker F, Tran A, Gual P, Mallat A, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. 2014;59:130–142. doi: 10.1002/hep.26607. [DOI] [PubMed] [Google Scholar]
- 85.Yi HS, Jeong WI. Interaction of hepatic stellate cells with diverse types of immune cells: foe or friend? J Gastroenterol Hepatol. 2013;28 Suppl 1:99–104. doi: 10.1111/jgh.12017. [DOI] [PubMed] [Google Scholar]
- 86.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Inzaugarat ME, Ferreyra Solari NE, Billordo LA, Abecasis R, Gadano AC, Cherñavsky AC. Altered phenotype and functionality of circulating immune cells characterize adult patients with nonalcoholic steatohepatitis. J Clin Immunol. 2011;31:1120–1130. doi: 10.1007/s10875-011-9571-1. [DOI] [PubMed] [Google Scholar]
- 88.Sutti S, Jindal A, Locatelli I, Vacchiano M, Gigliotti L, Bozzola C, Albano E. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology. 2014;59:886–897. doi: 10.1002/hep.26749. [DOI] [PubMed] [Google Scholar]
- 89.Zhan YT, An W. Roles of liver innate immune cells in nonalcoholic fatty liver disease. World J Gastroenterol. 2010;16:4652–4660. doi: 10.3748/wjg.v16.i37.4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Farrell GC, van Rooyen D, Gan L, Chitturi S. NASH is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut Liver. 2012;6:149–171. doi: 10.5009/gnl.2012.6.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Machado MV, Cortez-Pinto H. Gut microbiota and nonalcoholic fatty liver disease. Ann Hepatol. 2012;11:440–449. [PubMed] [Google Scholar]
- 92.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wigg AJ, Roberts-Thomson IC, Dymock RB, McCarthy PJ, Grose RH, Cummins AG. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2001;48:206–211. doi: 10.1136/gut.48.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sajjad A, Mottershead M, Syn WK, Jones R, Smith S, Nwokolo CU. Ciprofloxacin suppresses bacterial overgrowth, increases fasting insulin but does not correct low acylated ghrelin concentration in non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2005;22:291–299. doi: 10.1111/j.1365-2036.2005.02562.x. [DOI] [PubMed] [Google Scholar]
- 95.Sabaté JM, Jouët P, Harnois F, Mechler C, Msika S, Grossin M, Coffin B. High prevalence of small intestinal bacterial overgrowth in patients with morbid obesity: a contributor to severe hepatic steatosis. Obes Surg. 2008;18:371–377. doi: 10.1007/s11695-007-9398-2. [DOI] [PubMed] [Google Scholar]
- 96.Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Mascianà R, Forgione A, Gabrieli ML, Perotti G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 97.Thuy S, Ladurner R, Volynets V, Wagner S, Strahl S, Königsrainer A, Maier KP, Bischoff SC, Bergheim I. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J Nutr. 2008;138:1452–1455. doi: 10.1093/jn/138.8.1452. [DOI] [PubMed] [Google Scholar]
- 98.Harte AL, da Silva NF, Creely SJ, McGee KC, Billyard T, Youssef-Elabd EM, Tripathi G, Ashour E, Abdalla MS, Sharada HM, et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm (Lond) 2010;7:15. doi: 10.1186/1476-9255-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ruiz AG, Casafont F, Crespo J, Cayón A, Mayorga M, Estebanez A, Fernadez-Escalante JC, Pons-Romero F. Lipopolysaccharide-binding protein plasma levels and liver TNF-alpha gene expression in obese patients: evidence for the potential role of endotoxin in the pathogenesis of non-alcoholic steatohepatitis. Obes Surg. 2007;17:1374–1380. doi: 10.1007/s11695-007-9243-7. [DOI] [PubMed] [Google Scholar]
- 100.Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. 2007;47:571–579. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jung Y, Witek RP, Syn WK, Choi SS, Omenetti A, Premont R, Guy CD, Diehl AM. Signals from dying hepatocytes trigger growth of liver progenitors. Gut. 2010;59:655–665. doi: 10.1136/gut.2009.204354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rangwala F, Guy CD, Lu J, Suzuki A, Burchette JL, Abdelmalek MF, Chen W, Diehl AM. Increased production of sonic hedgehog by ballooned hepatocytes. J Pathol. 2011;224:401–410. doi: 10.1002/path.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sicklick JK, Li YX, Choi SS, Qi Y, Chen W, Bustamante M, Huang J, Zdanowicz M, Camp T, Torbenson MS, et al. Role for hedgehog signaling in hepatic stellate cell activation and viability. Lab Invest. 2005;85:1368–1380. doi: 10.1038/labinvest.3700349. [DOI] [PubMed] [Google Scholar]
- 104.Choi SS, Omenetti A, Witek RP, Moylan CA, Syn WK, Jung Y, Yang L, Sudan DL, Sicklick JK, Michelotti GA, et al. Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2009;297:G1093–G1106. doi: 10.1152/ajpgi.00292.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fleig SV, Choi SS, Yang L, Jung Y, Omenetti A, VanDongen HM, Huang J, Sicklick JK, Diehl AM. Hepatic accumulation of Hedgehog-reactive progenitors increases with severity of fatty liver damage in mice. Lab Invest. 2007;87:1227–1239. doi: 10.1038/labinvest.3700689. [DOI] [PubMed] [Google Scholar]
- 106.Syn WK, Jung Y, Omenetti A, Abdelmalek M, Guy CD, Yang L, Wang J, Witek RP, Fearing CM, Pereira TA, et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology. 2009;137:1478–1488.e8. doi: 10.1053/j.gastro.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Syn WK, Choi SS, Liaskou E, Karaca GF, Agboola KM, Oo YH, Mi Z, Pereira TA, Zdanowicz M, Malladi P, et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology. 2011;53:106–115. doi: 10.1002/hep.23998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Syn WK, Agboola KM, Swiderska M, Michelotti GA, Liaskou E, Pang H, Xie G, Philips G, Chan IS, Karaca GF, et al. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut. 2012;61:1323–1329. doi: 10.1136/gutjnl-2011-301857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Guy CD, Suzuki A, Zdanowicz M, Abdelmalek MF, Burchette J, Unalp A, Diehl AM. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology. 2012;55:1711–1721. doi: 10.1002/hep.25559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hirsova P, Ibrahim SH, Bronk SF, Yagita H, Gores GJ. Vismodegib suppresses TRAIL-mediated liver injury in a mouse model of nonalcoholic steatohepatitis. PLoS One. 2013;8:e70599. doi: 10.1371/journal.pone.0070599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Svegliati-Baroni G, Ridolfi F, Di Sario A, Casini A, Marucci L, Gaggiotti G, Orlandoni P, Macarri G, Perego L, Benedetti A, et al. Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: differential effects on signal transduction pathways. Hepatology. 1999;29:1743–1751. doi: 10.1002/hep.510290632. [DOI] [PubMed] [Google Scholar]
- 112.Lin J, Zheng S, Chen A. Curcumin attenuates the effects of insulin on stimulating hepatic stellate cell activation by interrupting insulin signaling and attenuating oxidative stress. Lab Invest. 2009;89:1397–1409. doi: 10.1038/labinvest.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Struben VM, Hespenheide EE, Caldwell SH. Nonalcoholic steatohepatitis and cryptogenic cirrhosis within kindreds. Am J Med. 2000;108:9–13. doi: 10.1016/s0002-9343(99)00315-0. [DOI] [PubMed] [Google Scholar]
- 114.Willner IR, Waters B, Patil SR, Reuben A, Morelli J, Riely CA. Ninety patients with nonalcoholic steatohepatitis: insulin resistance, familial tendency, and severity of disease. Am J Gastroenterol. 2001;96:2957–2961. doi: 10.1111/j.1572-0241.2001.04667.x. [DOI] [PubMed] [Google Scholar]
- 115.Abdelmalek MF, Liu C, Shuster J, Nelson DR, Asal NR. Familial aggregation of insulin resistance in first-degree relatives of patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2006;4:1162–1169. doi: 10.1016/j.cgh.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 116.Schwimmer JB, Celedon MA, Lavine JE, Salem R, Campbell N, Schork NJ, Shiehmorteza M, Yokoo T, Chavez A, Middleton MS, et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology. 2009;136:1585–1592. doi: 10.1053/j.gastro.2009.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brouwers MC, Cantor RM, Kono N, Yoon JL, van der Kallen CJ, Bilderbeek-Beckers MA, van Greevenbroek MM, Lusis AJ, de Bruin TW. Heritability and genetic loci of fatty liver in familial combined hyperlipidemia. J Lipid Res. 2006;47:2799–2807. doi: 10.1194/jlr.M600312-JLR200. [DOI] [PubMed] [Google Scholar]
- 118.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, Gudnason V, Eiriksdottir G, Garcia ME, Launer LJ, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7:e1001324. doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chalasani N, Guo X, Loomba R, Goodarzi MO, Haritunians T, Kwon S, Cui J, Taylor KD, Wilson L, Cummings OW, et al. Genome-wide association study identifies variants associated with histologic features of nonalcoholic Fatty liver disease. Gastroenterology. 2010;139:1567–176, 1567-176. doi: 10.1053/j.gastro.2010.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kawaguchi T, Sumida Y, Umemura A, Matsuo K, Takahashi M, Takamura T, Yasui K, Saibara T, Hashimoto E, Kawanaka M, et al. Genetic polymorphisms of the human PNPLA3 gene are strongly associated with severity of non-alcoholic fatty liver disease in Japanese. PLoS One. 2012;7:e38322. doi: 10.1371/journal.pone.0038322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kitamoto T, Kitamoto A, Yoneda M, Hyogo H, Ochi H, Nakamura T, Teranishi H, Mizusawa S, Ueno T, Chayama K, et al. Genome-wide scan revealed that polymorphisms in the PNPLA3, SAMM50, and PARVB genes are associated with development and progression of nonalcoholic fatty liver disease in Japan. Hum Genet. 2013;132:783–792. doi: 10.1007/s00439-013-1294-3. [DOI] [PubMed] [Google Scholar]
- 123.Chen QR, Braun R, Hu Y, Yan C, Brunt EM, Meerzaman D, Sanyal AJ, Buetow K. Multi-SNP analysis of GWAS data identifies pathways associated with nonalcoholic fatty liver disease. PLoS One. 2013;8:e65982. doi: 10.1371/journal.pone.0065982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gerhard GS, Chu X, Wood GC, Gerhard GM, Benotti P, Petrick AT, Gabrielsen J, Strodel WE, Still CD, Argyropoulos G. Next-generation sequence analysis of genes associated with obesity and nonalcoholic fatty liver disease-related cirrhosis in extreme obesity. Hum Hered. 2013;75:144–151. doi: 10.1159/000351719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Feitosa MF, Wojczynski MK, North KE, Zhang Q, Province MA, Carr JJ, Borecki IB. The ERLIN1-CHUK-CWF19L1 gene cluster influences liver fat deposition and hepatic inflammation in the NHLBI Family Heart Study. Atherosclerosis. 2013;228:175–180. doi: 10.1016/j.atherosclerosis.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sookoian S, Castaño GO, Burgueño AL, Gianotti TF, Rosselli MS, Pirola CJ. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Res. 2009;50:2111–2116. doi: 10.1194/jlr.P900013-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kotronen A, Johansson LE, Johansson LM, Roos C, Westerbacka J, Hamsten A, Bergholm R, Arkkila P, Arola J, Kiviluoto T, et al. A common variant in PNPLA3, which encodes adiponutrin, is associated with liver fat content in humans. Diabetologia. 2009;52:1056–1060. doi: 10.1007/s00125-009-1285-z. [DOI] [PubMed] [Google Scholar]
- 128.Kantartzis K, Peter A, Machicao F, Machann J, Wagner S, Königsrainer I, Königsrainer A, Schick F, Fritsche A, Häring HU, et al. Dissociation between fatty liver and insulin resistance in humans carrying a variant of the patatin-like phospholipase 3 gene. Diabetes. 2009;58:2616–2623. doi: 10.2337/db09-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rotman Y, Koh C, Zmuda JM, Kleiner DE, Liang TJ. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology. 2010;52:894–903. doi: 10.1002/hep.23759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Speliotes EK, Butler JL, Palmer CD, Voight BF, Hirschhorn JN. PNPLA3 variants specifically confer increased risk for histologic nonalcoholic fatty liver disease but not metabolic disease. Hepatology. 2010;52:904–912. doi: 10.1002/hep.23768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hotta K, Yoneda M, Hyogo H, Ochi H, Mizusawa S, Ueno T, Chayama K, Nakajima A, Nakao K, Sekine A. Association of the rs738409 polymorphism in PNPLA3 with liver damage and the development of nonalcoholic fatty liver disease. BMC Med Genet. 2010;11:172. doi: 10.1186/1471-2350-11-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hyysalo J, Stojkovic I, Kotronen A, Hakkarainen A, Sevastianova K, Makkonen J, Lundbom N, Rissanen A, Krauss RM, Melander O, et al. Genetic variation in PNPLA3 but not APOC3 influences liver fat in non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2012;27:951–956. doi: 10.1111/j.1440-1746.2011.07045.x. [DOI] [PubMed] [Google Scholar]
- 133.Zain SM, Mohamed R, Mahadeva S, Cheah PL, Rampal S, Basu RC, Mohamed Z. A multi-ethnic study of a PNPLA3 gene variant and its association with disease severity in non-alcoholic fatty liver disease. Hum Genet. 2012;131:1145–1152. doi: 10.1007/s00439-012-1141-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li Y, Xing C, Tian Z, Ku HC. Genetic variant I148M in PNPLA3 is associated with the ultrasonography-determined steatosis degree in a Chinese population. BMC Med Genet. 2012;13:113. doi: 10.1186/1471-2350-13-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Peng XE, Wu YL, Lin SW, Lu QQ, Hu ZJ, Lin X. Genetic variants in PNPLA3 and risk of non-alcoholic fatty liver disease in a Han Chinese population. PLoS One. 2012;7:e50256. doi: 10.1371/journal.pone.0050256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gorden A, Yang R, Yerges-Armstrong LM, Ryan KA, Speliotes E, Borecki IB, Harris TB, Chu X, Wood GC, Still CD, et al. Genetic variation at NCAN locus is associated with inflammation and fibrosis in non-alcoholic fatty liver disease in morbid obesity. Hum Hered. 2013;75:34–43. doi: 10.1159/000346195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Palmer ND, Musani SK, Yerges-Armstrong LM, Feitosa MF, Bielak LF, Hernaez R, Kahali B, Carr JJ, Harris TB, Jhun MA, et al. Characterization of European ancestry nonalcoholic fatty liver disease-associated variants in individuals of African and Hispanic descent. Hepatology. 2013;58:966–975. doi: 10.1002/hep.26440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, Nobili V, Mozzi E, Roviaro G, Vanni E, et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1209–1217. doi: 10.1002/hep.23622. [DOI] [PubMed] [Google Scholar]
- 139.Ballestri S, Day CP, Daly AK. Polymorphism in the farnesyl diphosphate farnesyl transferase 1 gene and nonalcoholic fatty liver disease severity. Gastroenterology. 2011;140:1694–1695. doi: 10.1053/j.gastro.2011.01.060. [DOI] [PubMed] [Google Scholar]
- 140.Dubuquoy C, Robichon C, Lasnier F, Langlois C, Dugail I, Foufelle F, Girard J, Burnol AF, Postic C, Moldes M. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes. J Hepatol. 2011;55:145–153. doi: 10.1016/j.jhep.2010.10.024. [DOI] [PubMed] [Google Scholar]
- 141.Dubuquoy C, Burnol AF, Moldes M. PNPLA3, a genetic marker of progressive liver disease, still hiding its metabolic function? Clin Res Hepatol Gastroenterol. 2013;37:30–35. doi: 10.1016/j.clinre.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 142.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem. 2004;279:48968–48975. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- 143.Kumari M, Schoiswohl G, Chitraju C, Paar M, Cornaciu I, Rangrez AY, Wongsiriroj N, Nagy HM, Ivanova PT, Scott SA, et al. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012;15:691–702. doi: 10.1016/j.cmet.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hernaez R, McLean J, Lazo M, Brancati FL, Hirschhorn JN, Borecki IB, Harris TB, Nguyen T, Kamel IR, Bonekamp S, et al. Association between variants in or near PNPLA3, GCKR, and PPP1R3B with ultrasound-defined steatosis based on data from the third National Health and Nutrition Examination Survey. Clin Gastroenterol Hepatol. 2013;11:1183–1190.e2. doi: 10.1016/j.cgh.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yang Z, Wen J, Tao X, Lu B, Du Y, Wang M, Wang X, Zhang W, Gong W, Ling C, et al. Genetic variation in the GCKR gene is associated with non-alcoholic fatty liver disease in Chinese people. Mol Biol Rep. 2011;38:1145–1150. doi: 10.1007/s11033-010-0212-1. [DOI] [PubMed] [Google Scholar]
- 146.Tan HL, Zain SM, Mohamed R, Rampal S, Chin KF, Basu RC, Cheah PL, Mahadeva S, Mohamed Z. Association of glucokinase regulatory gene polymorphisms with risk and severity of non-alcoholic fatty liver disease: an interaction study with adiponutrin gene. J Gastroenterol. 2014;49:1056–1064. doi: 10.1007/s00535-013-0850-x. [DOI] [PubMed] [Google Scholar]
- 147.Krautbauer S, Eisinger K, Lupke M, Wanninger J, Ruemmele P, Hader Y, Weiss TS, Buechler C. Manganese superoxide dismutase is reduced in the liver of male but not female humans and rodents with non-alcoholic fatty liver disease. Exp Mol Pathol. 2013;95:330–335. doi: 10.1016/j.yexmp.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 148.Stewart SF, Leathart JB, Chen Y, Daly AK, Rolla R, Vay D, Mottaran E, Vidali M, Albano E, Day CP. Valine-alanine manganese superoxide dismutase polymorphism is not associated with alcohol-induced oxidative stress or liver fibrosis. Hepatology. 2002;36:1355–1360. doi: 10.1053/jhep.2002.36940. [DOI] [PubMed] [Google Scholar]
- 149.Namikawa C, Shu-Ping Z, Vyselaar JR, Nozaki Y, Nemoto Y, Ono M, Akisawa N, Saibara T, Hiroi M, Enzan H, et al. Polymorphisms of microsomal triglyceride transfer protein gene and manganese superoxide dismutase gene in non-alcoholic steatohepatitis. J Hepatol. 2004;40:781–786. doi: 10.1016/j.jhep.2004.01.028. [DOI] [PubMed] [Google Scholar]
- 150.Al-Serri A, Anstee QM, Valenti L, Nobili V, Leathart JB, Dongiovanni P, Patch J, Fracanzani A, Fargion S, Day CP, et al. The SOD2 C47T polymorphism influences NAFLD fibrosis severity: evidence from case-control and intra-familial allele association studies. J Hepatol. 2012;56:448–454. doi: 10.1016/j.jhep.2011.05.029. [DOI] [PubMed] [Google Scholar]
- 151.Nobili V, Donati B, Panera N, Vongsakulyanon A, Alisi A, Dallapiccola B, Valenti L. A 4-polymorphism risk score predicts steatohepatitis in children with nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr. 2014;58:632–636. doi: 10.1097/MPG.0000000000000279. [DOI] [PubMed] [Google Scholar]
- 152.El-Koofy NM, El-Karaksy HM, Mandour IM, Anwar GM, El-Raziky MS, El-Hennawy AM. Genetic polymorphisms in non-alcoholic fatty liver disease in obese Egyptian children. Saudi J Gastroenterol. 2011;17:265–270. doi: 10.4103/1319-3767.82582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ratziu V, Lalazar A, Wong L, Dang Q, Collins C, Shaulian E, Jensen S, Friedman SL. Zf9, a Kruppel-like transcription factor up-regulated in vivo during early hepatic fibrosis. Proc Natl Acad Sci USA. 1998;95:9500–9505. doi: 10.1073/pnas.95.16.9500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Narla G, DiFeo A, Yao S, Banno A, Hod E, Reeves HL, Qiao RF, Camacho-Vanegas O, Levine A, Kirschenbaum A, et al. Targeted inhibition of the KLF6 splice variant, KLF6 SV1, suppresses prostate cancer cell growth and spread. Cancer Res. 2005;65:5761–5768. doi: 10.1158/0008-5472.CAN-05-0217. [DOI] [PubMed] [Google Scholar]
- 155.Miele L, Beale G, Patman G, Nobili V, Leathart J, Grieco A, Abate M, Friedman SL, Narla G, Bugianesi E, et al. The Kruppel-like factor 6 genotype is associated with fibrosis in nonalcoholic fatty liver disease. Gastroenterology. 2008;135:282–291.e1. doi: 10.1053/j.gastro.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yoneda M, Hotta K, Nozaki Y, Endo H, Uchiyama T, Mawatari H, Iida H, Kato S, Fujita K, Takahashi H, et al. Association between angiotensin II type 1 receptor polymorphisms and the occurrence of nonalcoholic fatty liver disease. Liver Int. 2009;29:1078–1085. doi: 10.1111/j.1478-3231.2009.01988.x. [DOI] [PubMed] [Google Scholar]
- 157.Zain SM, Mohamed Z, Mahadeva S, Rampal S, Basu RC, Cheah PL, Salim A, Mohamed R. Susceptibility and gene interaction study of the angiotensin II type 1 receptor (AGTR1) gene polymorphisms with non-alcoholic fatty liver disease in a multi-ethnic population. PLoS One. 2013;8:e58538. doi: 10.1371/journal.pone.0058538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Valenti L, Fracanzani AL, Dongiovanni P, Santorelli G, Branchi A, Taioli E, Fiorelli G, Fargion S. Tumor necrosis factor alpha promoter polymorphisms and insulin resistance in nonalcoholic fatty liver disease. Gastroenterology. 2002;122:274–280. doi: 10.1053/gast.2002.31065. [DOI] [PubMed] [Google Scholar]
- 159.Tokushige K, Takakura M, Tsuchiya-Matsushita N, Taniai M, Hashimoto E, Shiratori K. Influence of TNF gene polymorphisms in Japanese patients with NASH and simple steatosis. J Hepatol. 2007;46:1104–1110. doi: 10.1016/j.jhep.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 160.Wong VW, Wong GL, Tsang SW, Hui AY, Chan AW, Choi PC, So WY, Tse AM, Chan FK, Sung JJ, et al. Genetic polymorphisms of adiponectin and tumor necrosis factor-alpha and nonalcoholic fatty liver disease in Chinese people. J Gastroenterol Hepatol. 2008;23:914–921. doi: 10.1111/j.1440-1746.2008.05344.x. [DOI] [PubMed] [Google Scholar]
- 161.Hu ZW, Luo HB, Xu YM, Guo JW, Deng XL, Tong YW, Tang X. Tumor necrosis factor--alpha gene promoter polymorphisms in Chinese patients with nonalcoholic fatty liver diseases. Acta Gastroenterol Belg. 2010;72:215–221. [PubMed] [Google Scholar]
- 162.Aller R, de Luis DA, Izaola O, González Sagrado M, Conde R, Alvarez Gago T, Pacheco D, González JM, Velasco MC. G308A polymorphism of TNF-alpha gene is associated with insulin resistance and histological changes in non alcoholic fatty liver disease patients. Ann Hepatol. 2010;9:439–444. [PubMed] [Google Scholar]
- 163.Zhou YJ, Li YY, Nie YQ, Yang H, Zhan Q, Huang J, Shi SL, Lai XB, Huang HL. Influence of polygenetic polymorphisms on the susceptibility to non-alcoholic fatty liver disease of Chinese people. J Gastroenterol Hepatol. 2010;25:772–777. doi: 10.1111/j.1440-1746.2009.06144.x. [DOI] [PubMed] [Google Scholar]
- 164.Wang JK, Feng ZW, Li YC, Li QY, Tao XY. Association of tumor necrosis factor-α gene promoter polymorphism at sites -308 and -238 with non-alcoholic fatty liver disease: a meta-analysis. J Gastroenterol Hepatol. 2012;27:670–676. doi: 10.1111/j.1440-1746.2011.06978.x. [DOI] [PubMed] [Google Scholar]
- 165.Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC, Bateman AC, Clough GF, Poston L, Hanson MA, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology. 2009;50:1796–1808. doi: 10.1002/hep.23205. [DOI] [PubMed] [Google Scholar]
- 166.Elahi MM, Cagampang FR, Mukhtar D, Anthony FW, Ohri SK, Hanson MA. Long-term maternal high-fat feeding from weaning through pregnancy and lactation predisposes offspring to hypertension, raised plasma lipids and fatty liver in mice. Br J Nutr. 2009;102:514–519. doi: 10.1017/S000711450820749X. [DOI] [PubMed] [Google Scholar]
- 167.Oben JA, Mouralidarane A, Samuelsson AM, Matthews PJ, Morgan ML, McKee C, Soeda J, Fernandez-Twinn DS, Martin-Gronert MS, Ozanne SE, et al. Maternal obesity during pregnancy and lactation programs the development of offspring non-alcoholic fatty liver disease in mice. J Hepatol. 2010;52:913–920. doi: 10.1016/j.jhep.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 168.Ashino NG, Saito KN, Souza FD, Nakutz FS, Roman EA, Velloso LA, Torsoni AS, Torsoni MA. Maternal high-fat feeding through pregnancy and lactation predisposes mouse offspring to molecular insulin resistance and fatty liver. J Nutr Biochem. 2012;23:341–348. doi: 10.1016/j.jnutbio.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 169.Dahlhoff M, Pfister S, Blutke A, Rozman J, Klingenspor M, Deutsch MJ, Rathkolb B, Fink B, Gimpfl M, Hrabě de Angelis M, et al. Peri-conceptional obesogenic exposure induces sex-specific programming of disease susceptibilities in adult mouse offspring. Biochim Biophys Acta. 2014;1842:304–317. doi: 10.1016/j.bbadis.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 170.Pruis MG, Lendvai A, Bloks VW, Zwier MV, Baller JF, de Bruin A, Groen AK, Plösch T. Maternal western diet primes non-alcoholic fatty liver disease in adult mouse offspring. Acta Physiol (Oxf) 2014;210:215–227. doi: 10.1111/apha.12197. [DOI] [PubMed] [Google Scholar]
- 171.Ahrens M, Ammerpohl O, von Schönfels W, Kolarova J, Bens S, Itzel T, Teufel A, Herrmann A, Brosch M, Hinrichsen H, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013;18:296–302. doi: 10.1016/j.cmet.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 172.Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, Abdelmalek MF, Garrett ME, Ashley-Koch A, Suzuki A, Tillmann HL, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology. 2013;145:1076–1087. doi: 10.1053/j.gastro.2013.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ratziu V, Bellentani S, Cortez-Pinto H, Day C, Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J Hepatol. 2010;53:372–384. doi: 10.1016/j.jhep.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 174.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142:1592–1609. doi: 10.1053/j.gastro.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 175.Szczepaniak LS, Nurenberg P, Leonard D, Browning JD, Reingold JS, Grundy S, Hobbs HH, Dobbins RL. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab. 2005;288:E462–E468. doi: 10.1152/ajpendo.00064.2004. [DOI] [PubMed] [Google Scholar]
- 176.Brunt EM. Nonalcoholic steatohepatitis: definition and pathology. Semin Liver Dis. 2001;21:3–16. doi: 10.1055/s-2001-12925. [DOI] [PubMed] [Google Scholar]
- 177.Fuchs CS, Stampfer MJ, Colditz GA, Giovannucci EL, Manson JE, Kawachi I, Hunter DJ, Hankinson SE, Hennekens CH, Rosner B. Alcohol consumption and mortality among women. N Engl J Med. 1995;332:1245–1250. doi: 10.1056/NEJM199505113321901. [DOI] [PubMed] [Google Scholar]
- 178.Becker U, Deis A, Sørensen TI, Grønbaek M, Borch-Johnsen K, Müller CF, Schnohr P, Jensen G. Prediction of risk of liver disease by alcohol intake, sex, and age: a prospective population study. Hepatology. 1996;23:1025–1029. doi: 10.1002/hep.510230513. [DOI] [PubMed] [Google Scholar]
- 179.Thun MJ, Peto R, Lopez AD, Monaco JH, Henley SJ, Heath CW, Doll R. Alcohol consumption and mortality among middle-aged and elderly U.S. adults. N Engl J Med. 1997;337:1705–1714. doi: 10.1056/NEJM199712113372401. [DOI] [PubMed] [Google Scholar]
- 180.Gao X, Fan JG. Diagnosis and management of non-alcoholic fatty liver disease and related metabolic disorders: consensus statement from the Study Group of Liver and Metabolism, Chinese Society of Endocrinology. J Diabetes. 2013;5:406–415. doi: 10.1111/1753-0407.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Fierbinteanu-Braticevici C, Dina I, Petrisor A, Tribus L, Negreanu L, Carstoiu C. Noninvasive investigations for non alcoholic fatty liver disease and liver fibrosis. World J Gastroenterol. 2010;16:4784–4791. doi: 10.3748/wjg.v16.i38.4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Merriman RB, Ferrell LD, Patti MG, Weston SR, Pabst MS, Aouizerat BE, Bass NM. Correlation of paired liver biopsies in morbidly obese patients with suspected nonalcoholic fatty liver disease. Hepatology. 2006;44:874–880. doi: 10.1002/hep.21346. [DOI] [PubMed] [Google Scholar]
- 183.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 184.Ekstedt M, Franzén LE, Mathiesen UL, Kechagias S. Low clinical relevance of the nonalcoholic fatty liver disease activity score (NAS) in predicting fibrosis progression. Scand J Gastroenterol. 2012;47:108–115. doi: 10.3109/00365521.2011.634024. [DOI] [PubMed] [Google Scholar]
- 185.Younossi ZM, Stepanova M, Rafiq N, Makhlouf H, Younoszai Z, Agrawal R, Goodman Z. Pathologic criteria for nonalcoholic steatohepatitis: interprotocol agreement and ability to predict liver-related mortality. Hepatology. 2011;53:1874–1882. doi: 10.1002/hep.24268. [DOI] [PubMed] [Google Scholar]
- 186.Sanyal AJ, Brunt EM, Kleiner DE, Kowdley KV, Chalasani N, Lavine JE, Ratziu V, McCullough A. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology. 2011;54:344–353. doi: 10.1002/hep.24376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bedossa P, Poitou C, Veyrie N, Bouillot JL, Basdevant A, Paradis V, Tordjman J, Clement K. Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology. 2012;56:1751–1759. doi: 10.1002/hep.25889. [DOI] [PubMed] [Google Scholar]
- 188.Fracanzani AL, Valenti L, Bugianesi E, Vanni E, Grieco A, Miele L, Consonni D, Fatta E, Lombardi R, Marchesini G, et al. Risk of nonalcoholic steatohepatitis and fibrosis in patients with nonalcoholic fatty liver disease and low visceral adiposity. J Hepatol. 2011;54:1244–1249. doi: 10.1016/j.jhep.2010.09.037. [DOI] [PubMed] [Google Scholar]
- 189.Machado MV, Cortez-Pinto H. Non-invasive diagnosis of non-alcoholic fatty liver disease. A critical appraisal. J Hepatol. 2013;58:1007–1019. doi: 10.1016/j.jhep.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 190.Ryan CK, Johnson LA, Germin BI, Marcos A. One hundred consecutive hepatic biopsies in the workup of living donors for right lobe liver transplantation. Liver Transpl. 2002;8:1114–1122. doi: 10.1053/jlts.2002.36740. [DOI] [PubMed] [Google Scholar]
- 191.Fishbein M, Castro F, Cheruku S, Jain S, Webb B, Gleason T, Stevens WR. Hepatic MRI for fat quantitation: its relationship to fat morphology, diagnosis, and ultrasound. J Clin Gastroenterol. 2005;39:619–625. doi: 10.1097/00004836-200508000-00012. [DOI] [PubMed] [Google Scholar]
- 192.de Lédinghen V, Vergniol J, Capdepont M, Chermak F, Hiriart JB, Cassinotto C, Merrouche W, Foucher J, Brigitte le B. Controlled attenuation parameter (CAP) for the diagnosis of steatosis: a prospective study of 5323 examinations. J Hepatol. 2014;60:1026–1031. doi: 10.1016/j.jhep.2013.12.018. [DOI] [PubMed] [Google Scholar]
- 193.Yilmaz Y, Ergelen R, Akin H, Imeryuz N. Noninvasive detection of hepatic steatosis in patients without ultrasonographic evidence of fatty liver using the controlled attenuation parameter evaluated with transient elastography. Eur J Gastroenterol Hepatol. 2013;25:1330–1334. doi: 10.1097/MEG.0b013e3283623a16. [DOI] [PubMed] [Google Scholar]
- 194.Poynard T, Ratziu V, Naveau S, Thabut D, Charlotte F, Messous D, Capron D, Abella A, Massard J, Ngo Y, et al. The diagnostic value of biomarkers (SteatoTest) for the prediction of liver steatosis. Comp Hepatol. 2005;4:10. doi: 10.1186/1476-5926-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Bedogni G, Bellentani S, Miglioli L, Masutti F, Passalacqua M, Castiglione A, Tiribelli C. The Fatty Liver Index: a simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 2006;6:33. doi: 10.1186/1471-230X-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kotronen A, Peltonen M, Hakkarainen A, Sevastianova K, Bergholm R, Johansson LM, Lundbom N, Rissanen A, Ridderstråle M, Groop L, et al. Prediction of non-alcoholic fatty liver disease and liver fat using metabolic and genetic factors. Gastroenterology. 2009;137:865–872. doi: 10.1053/j.gastro.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 197.Bedogni G, Kahn HS, Bellentani S, Tiribelli C. A simple index of lipid overaccumulation is a good marker of liver steatosis. BMC Gastroenterol. 2010;10:98. doi: 10.1186/1471-230X-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Verma S, Jensen D, Hart J, Mohanty SR. Predictive value of ALT levels for non-alcoholic steatohepatitis (NASH) and advanced fibrosis in non-alcoholic fatty liver disease (NAFLD) Liver Int. 2013;33:1398–1405. doi: 10.1111/liv.12226. [DOI] [PubMed] [Google Scholar]
- 199.Dixon JB, Bhathal PS, O‘Brien PE. Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology. 2001;121:91–100. doi: 10.1053/gast.2001.25540. [DOI] [PubMed] [Google Scholar]
- 200.Palekar NA, Naus R, Larson SP, Ward J, Harrison SA. Clinical model for distinguishing nonalcoholic steatohepatitis from simple steatosis in patients with nonalcoholic fatty liver disease. Liver Int. 2006;26:151–156. doi: 10.1111/j.1478-3231.2005.01209.x. [DOI] [PubMed] [Google Scholar]
- 201.Poynard T, Ratziu V, Charlotte F, Messous D, Munteanu M, Imbert-Bismut F, Massard J, Bonyhay L, Tahiri M, Thabut D, et al. Diagnostic value of biochemical markers (NashTest) for the prediction of non alcoholo steato hepatitis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2006;6:34. doi: 10.1186/1471-230X-6-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Shimada M, Kawahara H, Ozaki K, Fukura M, Yano H, Tsuchishima M, Tsutsumi M, Takase S. Usefulness of a combined evaluation of the serum adiponectin level, HOMA-IR, and serum type IV collagen 7S level to predict the early stage of nonalcoholic steatohepatitis. Am J Gastroenterol. 2007;102:1931–1938. doi: 10.1111/j.1572-0241.2007.01322.x. [DOI] [PubMed] [Google Scholar]
- 203.Anty R, Iannelli A, Patouraux S, Bonnafous S, Lavallard VJ, Senni-Buratti M, Amor IB, Staccini-Myx A, Saint-Paul MC, Berthier F, et al. A new composite model including metabolic syndrome, alanine aminotransferase and cytokeratin-18 for the diagnosis of non-alcoholic steatohepatitis in morbidly obese patients. Aliment Pharmacol Ther. 2010;32:1315–1322. doi: 10.1111/j.1365-2036.2010.04480.x. [DOI] [PubMed] [Google Scholar]
- 204.Feldstein AE, Lopez R, Tamimi TA, Yerian L, Chung YM, Berk M, Zhang R, McIntyre TM, Hazen SL. Mass spectrometric profiling of oxidized lipid products in human nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J Lipid Res. 2010;51:3046–3054. doi: 10.1194/jlr.M007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Younossi ZM, Page S, Rafiq N, Birerdinc A, Stepanova M, Hossain N, Afendy A, Younoszai Z, Goodman Z, Baranova A. A biomarker panel for non-alcoholic steatohepatitis (NASH) and NASH-related fibrosis. Obes Surg. 2011;21:431–439. doi: 10.1007/s11695-010-0204-1. [DOI] [PubMed] [Google Scholar]
- 206.Tamimi TI, Elgouhari HM, Alkhouri N, Yerian LM, Berk MP, Lopez R, Schauer PR, Zein NN, Feldstein AE. An apoptosis panel for nonalcoholic steatohepatitis diagnosis. J Hepatol. 2011;54:1224–1229. doi: 10.1016/j.jhep.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Musso G, Gambino R, Cassader M, Pagano G. Meta-analysis: natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med. 2011;43:617–649. doi: 10.3109/07853890.2010.518623. [DOI] [PubMed] [Google Scholar]
- 208.Cusi K, Chang Z, Harrison S, Lomonaco R, Bril F, Orsak B, Ortiz-Lopez C, Hecht J, Feldstein AE, Webb A, et al. Limited value of plasma cytokeratin-18 as a biomarker for NASH and fibrosis in patients with non-alcoholic fatty liver disease. J Hepatol. 2014;60:167–174. doi: 10.1016/j.jhep.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 209.Kwok R, Tse YK, Wong GL, Ha Y, Lee AU, Ngu MC, Chan HL, Wong VW. Systematic review with meta-analysis: non-invasive assessment of non-alcoholic fatty liver disease--the role of transient elastography and plasma cytokeratin-18 fragments. Aliment Pharmacol Ther. 2014;39:254–269. doi: 10.1111/apt.12569. [DOI] [PubMed] [Google Scholar]
- 210.Suzuki A, Angulo P, Lymp J, Li D, Satomura S, Lindor K. Hyaluronic acid, an accurate serum marker for severe hepatic fibrosis in patients with non-alcoholic fatty liver disease. Liver Int. 2005;25:779–786. doi: 10.1111/j.1478-3231.2005.01064.x. [DOI] [PubMed] [Google Scholar]
- 211.Yoneda M, Mawatari H, Fujita K, Yonemitsu K, Kato S, Takahashi H, Kirikoshi H, Inamori M, Nozaki Y, Abe Y, et al. Type IV collagen 7s domain is an independent clinical marker of the severity of fibrosis in patients with nonalcoholic steatohepatitis before the cirrhotic stage. J Gastroenterol. 2007;42:375–381. doi: 10.1007/s00535-007-2014-3. [DOI] [PubMed] [Google Scholar]
- 212.Yoneda M, Mawatari H, Fujita K, Iida H, Yonemitsu K, Kato S, Takahashi H, Kirikoshi H, Inamori M, Nozaki Y, et al. High-sensitivity C-reactive protein is an independent clinical feature of nonalcoholic steatohepatitis (NASH) and also of the severity of fibrosis in NASH. J Gastroenterol. 2007;42:573–582. doi: 10.1007/s00535-007-2060-x. [DOI] [PubMed] [Google Scholar]
- 213.Ratziu V, Giral P, Charlotte F, Bruckert E, Thibault V, Theodorou I, Khalil L, Turpin G, Opolon P, Poynard T. Liver fibrosis in overweight patients. Gastroenterology. 2000;118:1117–1123. doi: 10.1016/s0016-5085(00)70364-7. [DOI] [PubMed] [Google Scholar]
- 214.Rosenberg WM, Voelker M, Thiel R, Becka M, Burt A, Schuppan D, Hubscher S, Roskams T, Pinzani M, Arthur MJ. Serum markers detect the presence of liver fibrosis: a cohort study. Gastroenterology. 2004;127:1704–1713. doi: 10.1053/j.gastro.2004.08.052. [DOI] [PubMed] [Google Scholar]
- 215.Ratziu V, Massard J, Charlotte F, Messous D, Imbert-Bismut F, Bonyhay L, Tahiri M, Munteanu M, Thabut D, Cadranel JF, et al. Diagnostic value of biochemical markers (FibroTest-FibroSURE) for the prediction of liver fibrosis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2006;6:6. doi: 10.1186/1471-230X-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Angulo P, Hui JM, Marchesini G, Bugianesi E, George J, Farrell GC, Enders F, Saksena S, Burt AD, Bida JP, et al. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology. 2007;45:846–854. doi: 10.1002/hep.21496. [DOI] [PubMed] [Google Scholar]
- 217.Harrison SA, Oliver D, Arnold HL, Gogia S, Neuschwander-Tetri BA. Development and validation of a simple NAFLD clinical scoring system for identifying patients without advanced disease. Gut. 2008;57:1441–1447. doi: 10.1136/gut.2007.146019. [DOI] [PubMed] [Google Scholar]
- 218.Calès P, Lainé F, Boursier J, Deugnier Y, Moal V, Oberti F, Hunault G, Rousselet MC, Hubert I, Laafi J, et al. Comparison of blood tests for liver fibrosis specific or not to NAFLD. J Hepatol. 2009;50:165–173. doi: 10.1016/j.jhep.2008.07.035. [DOI] [PubMed] [Google Scholar]
- 219.McPherson S, Stewart SF, Henderson E, Burt AD, Day CP. Simple non-invasive fibrosis scoring systems can reliably exclude advanced fibrosis in patients with non-alcoholic fatty liver disease. Gut. 2010;59:1265–1269. doi: 10.1136/gut.2010.216077. [DOI] [PubMed] [Google Scholar]
- 220.Angulo P, Bugianesi E, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Barrera F, Haflidadottir S, Day CP, George J. Simple noninvasive systems predict long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2013;145:782–9.e4. doi: 10.1053/j.gastro.2013.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Kim D, Kim WR, Kim HJ, Therneau TM. Association between noninvasive fibrosis markers and mortality among adults with nonalcoholic fatty liver disease in the United States. Hepatology. 2013;57:1357–1365. doi: 10.1002/hep.26156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Treeprasertsuk S, Björnsson E, Enders F, Suwanwalaikorn S, Lindor KD. NAFLD fibrosis score: a prognostic predictor for mortality and liver complications among NAFLD patients. World J Gastroenterol. 2013;19:1219–1229. doi: 10.3748/wjg.v19.i8.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Yoneda M, Yoneda M, Fujita K, Inamori M, Tamano M, Hiriishi H, Nakajima A. Transient elastography in patients with non-alcoholic fatty liver disease (NAFLD) Gut. 2007;56:1330–1331. doi: 10.1136/gut.2007.126417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Yoneda M, Yoneda M, Mawatari H, Fujita K, Endo H, Iida H, Nozaki Y, Yonemitsu K, Higurashi T, Takahashi H, et al. Noninvasive assessment of liver fibrosis by measurement of stiffness in patients with nonalcoholic fatty liver disease (NAFLD) Dig Liver Dis. 2008;40:371–378. doi: 10.1016/j.dld.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 225.Wong VW, Vergniol J, Wong GL, Foucher J, Chan HL, Le Bail B, Choi PC, Kowo M, Chan AW, Merrouche W, et al. Diagnosis of fibrosis and cirrhosis using liver stiffness measurement in nonalcoholic fatty liver disease. Hepatology. 2010;51:454–462. doi: 10.1002/hep.23312. [DOI] [PubMed] [Google Scholar]
- 226.Lupsor M, Badea R, Stefanescu H, Grigorescu M, Serban A, Radu C, Crişan D, Sparchez Z, Iancu S, Maniu A. Performance of unidimensional transient elastography in staging non-alcoholic steatohepatitis. J Gastrointestin Liver Dis. 2010;19:53–60. [PubMed] [Google Scholar]
- 227.Kumar R, Rastogi A, Sharma MK, Bhatia V, Tyagi P, Sharma P, Garg H, Chandan Kumar KN, Bihari C, Sarin SK. Liver stiffness measurements in patients with different stages of nonalcoholic fatty liver disease: diagnostic performance and clinicopathological correlation. Dig Dis Sci. 2013;58:265–274. doi: 10.1007/s10620-012-2306-1. [DOI] [PubMed] [Google Scholar]
- 228.Gaia S, Carenzi S, Barilli AL, Bugianesi E, Smedile A, Brunello F, Marzano A, Rizzetto M. Reliability of transient elastography for the detection of fibrosis in non-alcoholic fatty liver disease and chronic viral hepatitis. J Hepatol. 2011;54:64–71. doi: 10.1016/j.jhep.2010.06.022. [DOI] [PubMed] [Google Scholar]
- 229.Yoneda M, Suzuki K, Kato S, Fujita K, Nozaki Y, Hosono K, Saito S, Nakajima A. Nonalcoholic fatty liver disease: US-based acoustic radiation force impulse elastography. Radiology. 2010;256:640–647. doi: 10.1148/radiol.10091662. [DOI] [PubMed] [Google Scholar]
- 230.Palmeri ML, Wang MH, Rouze NC, Abdelmalek MF, Guy CD, Moser B, Diehl AM, Nightingale KR. Noninvasive evaluation of hepatic fibrosis using acoustic radiation force-based shear stiffness in patients with nonalcoholic fatty liver disease. J Hepatol. 2011;55:666–672. doi: 10.1016/j.jhep.2010.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Friedrich-Rust M, Romen D, Vermehren J, Kriener S, Sadet D, Herrmann E, Zeuzem S, Bojunga J. Acoustic radiation force impulse-imaging and transient elastography for non-invasive assessment of liver fibrosis and steatosis in NAFLD. Eur J Radiol. 2012;81:e325–e331. doi: 10.1016/j.ejrad.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 232.Guzmán-Aroca F, Frutos-Bernal MD, Bas A, Luján-Mompeán JA, Reus M, Berná-Serna Jde D, Parrilla P. Detection of non-alcoholic steatohepatitis in patients with morbid obesity before bariatric surgery: preliminary evaluation with acoustic radiation force impulse imaging. Eur Radiol. 2012;22:2525–2532. doi: 10.1007/s00330-012-2505-3. [DOI] [PubMed] [Google Scholar]
- 233.Fierbinteanu Braticevici C, Sporea I, Panaitescu E, Tribus L. Value of acoustic radiation force impulse imaging elastography for non-invasive evaluation of patients with nonalcoholic fatty liver disease. Ultrasound Med Biol. 2013;39:1942–1950. doi: 10.1016/j.ultrasmedbio.2013.04.019. [DOI] [PubMed] [Google Scholar]
- 234.Torres DM, Harrison SA. NAFLD: Predictive value of ALT levels for NASH and advanced fibrosis. Nat Rev Gastroenterol Hepatol. 2013;10:510–511. doi: 10.1038/nrgastro.2013.138. [DOI] [PubMed] [Google Scholar]
- 235.Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 236.Ong JP, Pitts A, Younossi ZM. Increased overall mortality and liver-related mortality in non-alcoholic fatty liver disease. J Hepatol. 2008;49:608–612. doi: 10.1016/j.jhep.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 237.Rafiq N, Bai C, Fang Y, Srishord M, McCullough A, Gramlich T, Younossi ZM. Long-term follow-up of patients with nonalcoholic fatty liver. Clin Gastroenterol Hepatol. 2009;7:234–238. doi: 10.1016/j.cgh.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 238.Söderberg C, Stål P, Askling J, Glaumann H, Lindberg G, Marmur J, Hultcrantz R. Decreased survival of subjects with elevated liver function tests during a 28-year follow-up. Hepatology. 2010;51:595–602. doi: 10.1002/hep.23314. [DOI] [PubMed] [Google Scholar]
- 239.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 240.Ekstedt M, Franzén LE, Mathiesen UL, Thorelius L, Holmqvist M, Bodemar G, Kechagias S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:865–873. doi: 10.1002/hep.21327. [DOI] [PubMed] [Google Scholar]
- 241.Fraser A, Harris R, Sattar N, Ebrahim S, Davey Smith G, Lawlor DA. Alanine aminotransferase, gamma-glutamyltransferase, and incident diabetes: the British Women’s Heart and Health Study and meta-analysis. Diabetes Care. 2009;32:741–750. doi: 10.2337/dc08-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Okamoto M, Takeda Y, Yoda Y, Kobayashi K, Fujino MA, Yamagata Z. The association of fatty liver and diabetes risk. J Epidemiol. 2003;13:15–21. doi: 10.2188/jea.13.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Fan JG, Li F, Cai XB, Peng YD, Ao QH, Gao Y. Effects of nonalcoholic fatty liver disease on the development of metabolic disorders. J Gastroenterol Hepatol. 2007;22:1086–1091. doi: 10.1111/j.1440-1746.2006.04781.x. [DOI] [PubMed] [Google Scholar]
- 244.Shibata M, Kihara Y, Taguchi M, Tashiro M, Otsuki M. Nonalcoholic fatty liver disease is a risk factor for type 2 diabetes in middle-aged Japanese men. Diabetes Care. 2007;30:2940–2944. doi: 10.2337/dc07-0792. [DOI] [PubMed] [Google Scholar]
- 245.Yamada T, Fukatsu M, Suzuki S, Wada T, Yoshida T, Joh T. Fatty liver predicts impaired fasting glucose and type 2 diabetes mellitus in Japanese undergoing a health checkup. J Gastroenterol Hepatol. 2010;25:352–356. doi: 10.1111/j.1440-1746.2009.05998.x. [DOI] [PubMed] [Google Scholar]
- 246.Bae JC, Rhee EJ, Lee WY, Park SE, Park CY, Oh KW, Park SW, Kim SW. Combined effect of nonalcoholic fatty liver disease and impaired fasting glucose on the development of type 2 diabetes: a 4-year retrospective longitudinal study. Diabetes Care. 2011;34:727–729. doi: 10.2337/dc10-1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Sung KC, Kim SH. Interrelationship between fatty liver and insulin resistance in the development of type 2 diabetes. J Clin Endocrinol Metab. 2011;96:1093–1097. doi: 10.1210/jc.2010-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Balkau B, Lange C, Vol S, Fumeron F, Bonnet F. Nine-year incident diabetes is predicted by fatty liver indices: the French D.E.S.I.R. study. BMC Gastroenterol. 2010;10:56. doi: 10.1186/1471-230X-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Cicero AF, D’Addato S, Reggi A, Marchesini G, Borghi C. Gender difference in hepatic steatosis index and lipid accumulation product ability to predict incident metabolic syndrome in the historical cohort of the Brisighella Heart Study. Metab Syndr Relat Disord. 2013;11:412–416. doi: 10.1089/met.2012.0147. [DOI] [PubMed] [Google Scholar]
- 250.Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology. 1999;30:1356–1362. doi: 10.1002/hep.510300604. [DOI] [PubMed] [Google Scholar]
- 251.Noureddin M, Yates KP, Vaughn IA, Neuschwander-Tetri BA, Sanyal AJ, McCullough A, Merriman R, Hameed B, Doo E, Kleiner DE, et al. Clinical and histological determinants of nonalcoholic steatohepatitis and advanced fibrosis in elderly patients. Hepatology. 2013;58:1644–1654. doi: 10.1002/hep.26465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Nakahara T, Hyogo H, Yoneda M, Sumida Y, Eguchi Y, Fujii H, Ono M, Kawaguchi T, Imajo K, Aikata H, Tanaka S, Kanemasa K, Fujimoto K, Anzai K, Saibara T, Sata M, Nakajima A, Itoh Y, Chayama K, Okanoue T; Japan Study Group of Nonalcoholic Fatty Liver Disease (JSG-NAFLD) Type 2 diabetes mellitus is associated with the fibrosis severity in patients with nonalcoholic fatty liver disease in a large retrospective cohort of Japanese patients. J Gastroenterol. 2013:Epub ahead of print. doi: 10.1007/s00535-013-0911-1. [DOI] [PubMed] [Google Scholar]
- 253.Argo CK, Northup PG, Al-Osaimi AM, Caldwell SH. Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J Hepatol. 2009;51:371–379. doi: 10.1016/j.jhep.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 254.Yasui K, Hashimoto E, Tokushige K, Koike K, Shima T, Kanbara Y, Saibara T, Uto H, Takami S, Kawanaka M, et al. Clinical and pathological progression of non-alcoholic steatohepatitis to hepatocellular carcinoma. Hepatol Res. 2012;42:767–773. doi: 10.1111/j.1872-034X.2012.00986.x. [DOI] [PubMed] [Google Scholar]
- 255.Guzman G, Brunt EM, Petrovic LM, Chejfec G, Layden TJ, Cotler SJ. Does nonalcoholic fatty liver disease predispose patients to hepatocellular carcinoma in the absence of cirrhosis? Arch Pathol Lab Med. 2008;132:1761–1766. doi: 10.5858/132.11.1761. [DOI] [PubMed] [Google Scholar]
- 256.Paradis V, Zalinski S, Chelbi E, Guedj N, Degos F, Vilgrain V, Bedossa P, Belghiti J. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology. 2009;49:851–859. doi: 10.1002/hep.22734. [DOI] [PubMed] [Google Scholar]
- 257.White DL, Kanwal F, El-Serag HB. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol. 2012;10:1342–1359.e2. doi: 10.1016/j.cgh.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol. 2013;10:656–665. doi: 10.1038/nrgastro.2013.183. [DOI] [PubMed] [Google Scholar]
- 259.Bellentani S, Dalle Grave R, Suppini A, Marchesini G. Behavior therapy for nonalcoholic fatty liver disease: The need for a multidisciplinary approach. Hepatology. 2008;47:746–754. doi: 10.1002/hep.22009. [DOI] [PubMed] [Google Scholar]
- 260.Cowin GJ, Jonsson JR, Bauer JD, Ash S, Ali A, Osland EJ, Purdie DM, Clouston AD, Powell EE, Galloway GJ. Magnetic resonance imaging and spectroscopy for monitoring liver steatosis. J Magn Reson Imaging. 2008;28:937–945. doi: 10.1002/jmri.21542. [DOI] [PubMed] [Google Scholar]
- 261.Eckard C, Cole R, Lockwood J, Torres DM, Williams CD, Shaw JC, Harrison SA. Prospective histopathologic evaluation of lifestyle modification in nonalcoholic fatty liver disease: a randomized trial. Therap Adv Gastroenterol. 2013;6:249–259. doi: 10.1177/1756283X13484078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Hickman IJ, Jonsson JR, Prins JB, Ash S, Purdie DM, Clouston AD, Powell EE. Modest weight loss and physical activity in overweight patients with chronic liver disease results in sustained improvements in alanine aminotransferase, fasting insulin, and quality of life. Gut. 2004;53:413–419. doi: 10.1136/gut.2003.027581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Huang MA, Greenson JK, Chao C, Anderson L, Peterman D, Jacobson J, Emick D, Lok AS, Conjeevaram HS. One-year intense nutritional counseling results in histological improvement in patients with non-alcoholic steatohepatitis: a pilot study. Am J Gastroenterol. 2005;100:1072–1081. doi: 10.1111/j.1572-0241.2005.41334.x. [DOI] [PubMed] [Google Scholar]
- 264.Kantartzis K, Thamer C, Peter A, Machann J, Schick F, Schraml C, Königsrainer A, Königsrainer I, Kröber S, Niess A, et al. High cardiorespiratory fitness is an independent predictor of the reduction in liver fat during a lifestyle intervention in non-alcoholic fatty liver disease. Gut. 2009;58:1281–1288. doi: 10.1136/gut.2008.151977. [DOI] [PubMed] [Google Scholar]
- 265.Kugelmas M, Hill DB, Vivian B, Marsano L, McClain CJ. Cytokines and NASH: a pilot study of the effects of lifestyle modification and vitamin E. Hepatology. 2003;38:413–419. doi: 10.1053/jhep.2003.50316. [DOI] [PubMed] [Google Scholar]
- 266.Lazo M, Solga SF, Horska A, Bonekamp S, Diehl AM, Brancati FL, Wagenknecht LE, Pi-Sunyer FX, Kahn SE, Clark JM. Effect of a 12-month intensive lifestyle intervention on hepatic steatosis in adults with type 2 diabetes. Diabetes Care. 2010;33:2156–2163. doi: 10.2337/dc10-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 2005;54:603–608. doi: 10.2337/diabetes.54.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Sreenivasa Baba C, Alexander G, Kalyani B, Pandey R, Rastogi S, Pandey A, Choudhuri G. Effect of exercise and dietary modification on serum aminotransferase levels in patients with nonalcoholic steatohepatitis. J Gastroenterol Hepatol. 2006;21:191–198. doi: 10.1111/j.1440-1746.2005.04233.x. [DOI] [PubMed] [Google Scholar]
- 269.Suzuki A, Lindor K, St Saver J, Lymp J, Mendes F, Muto A, Okada T, Angulo P. Effect of changes on body weight and lifestyle in nonalcoholic fatty liver disease. J Hepatol. 2005;43:1060–1066. doi: 10.1016/j.jhep.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 270.Thoma C, Day CP, Trenell MI. Lifestyle interventions for the treatment of non-alcoholic fatty liver disease in adults: a systematic review. J Hepatol. 2012;56:255–266. doi: 10.1016/j.jhep.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 271.Thomas EL, Brynes AE, Hamilton G, Patel N, Spong A, Goldin RD, Frost G, Bell JD, Taylor-Robinson SD. Effect of nutritional counselling on hepatic, muscle and adipose tissue fat content and distribution in non-alcoholic fatty liver disease. World J Gastroenterol. 2006;12:5813–5819. doi: 10.3748/wjg.v12.i36.5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Wong VW, Chan RS, Wong GL, Cheung BH, Chu WC, Yeung DK, Chim AM, Lai JW, Li LS, Sea MM, et al. Community-based lifestyle modification programme for non-alcoholic fatty liver disease: a randomized controlled trial. J Hepatol. 2013;59:536–542. doi: 10.1016/j.jhep.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 273.Zelber-Sagi S, Lotan R, Shlomai A, Webb M, Harrari G, Buch A, Nitzan Kaluski D, Halpern Z, Oren R. Predictors for incidence and remission of NAFLD in the general population during a seven-year prospective follow-up. J Hepatol. 2012;56:1145–1151. doi: 10.1016/j.jhep.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 274.Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, Fava JL, Wing RR. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51:121–129. doi: 10.1002/hep.23276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Harrison SA, Fecht W, Brunt EM, Neuschwander-Tetri BA. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology. 2009;49:80–86. doi: 10.1002/hep.22575. [DOI] [PubMed] [Google Scholar]
- 276.Moscatiello S, Di Luzio R, Bugianesi E, Suppini A, Hickman IJ, Di Domizio S, Dalle Grave R, Marchesini G. Cognitive-behavioral treatment of nonalcoholic Fatty liver disease: a propensity score-adjusted observational study. Obesity (Silver Spring) 2011;19:763–770. doi: 10.1038/oby.2010.254. [DOI] [PubMed] [Google Scholar]
- 277.Haufe S, Engeli S, Kast P, Böhnke J, Utz W, Haas V, Hermsdorf M, Mähler A, Wiesner S, Birkenfeld AL, et al. Randomized comparison of reduced fat and reduced carbohydrate hypocaloric diets on intrahepatic fat in overweight and obese human subjects. Hepatology. 2011;53:1504–1514. doi: 10.1002/hep.24242. [DOI] [PubMed] [Google Scholar]
- 278.Kirk E, Reeds DN, Finck BN, Mayurranjan SM, Patterson BW, Klein S. Dietary fat and carbohydrates differentially alter insulin sensitivity during caloric restriction. Gastroenterology. 2009;136:1552–1560. doi: 10.1053/j.gastro.2009.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Westerbacka J, Lammi K, Häkkinen AM, Rissanen A, Salminen I, Aro A, Yki-Järvinen H. Dietary fat content modifies liver fat in overweight nondiabetic subjects. J Clin Endocrinol Metab. 2005;90:2804–2809. doi: 10.1210/jc.2004-1983. [DOI] [PubMed] [Google Scholar]
- 280.Browning JD, Baker JA, Rogers T, Davis J, Satapati S, Burgess SC. Short-term weight loss and hepatic triglyceride reduction: evidence of a metabolic advantage with dietary carbohydrate restriction. Am J Clin Nutr. 2011;93:1048–1052. doi: 10.3945/ajcn.110.007674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Stamler J. Diet-heart: a problematic revisit. Am J Clin Nutr. 2010;91:497–499. doi: 10.3945/ajcn.2010.29216. [DOI] [PubMed] [Google Scholar]
- 282.Conlon BA, Beasley JM, Aebersold K, Jhangiani SS, Wylie-Rosett J. Nutritional management of insulin resistance in nonalcoholic fatty liver disease (NAFLD) Nutrients. 2013;5:4093–4114. doi: 10.3390/nu5104093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Yasutake K, Nakamuta M, Shima Y, Ohyama A, Masuda K, Haruta N, Fujino T, Aoyagi Y, Fukuizumi K, Yoshimoto T, et al. Nutritional investigation of non-obese patients with non-alcoholic fatty liver disease: the significance of dietary cholesterol. Scand J Gastroenterol. 2009;44:471–477. doi: 10.1080/00365520802588133. [DOI] [PubMed] [Google Scholar]
- 284.Cortez-Pinto H, Jesus L, Barros H, Lopes C, Moura MC, Camilo ME. How different is the dietary pattern in non-alcoholic steatohepatitis patients? Clin Nutr. 2006;25:816–823. doi: 10.1016/j.clnu.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 285.Musso G, Gambino R, De Michieli F, Cassader M, Rizzetto M, Durazzo M, Fagà E, Silli B, Pagano G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology. 2003;37:909–916. doi: 10.1053/jhep.2003.50132. [DOI] [PubMed] [Google Scholar]
- 286.Toshimitsu K, Matsuura B, Ohkubo I, Niiya T, Furukawa S, Hiasa Y, Kawamura M, Ebihara K, Onji M. Dietary habits and nutrient intake in non-alcoholic steatohepatitis. Nutrition. 2007;23:46–52. doi: 10.1016/j.nut.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 287.Ioannou GN, Morrow OB, Connole ML, Lee SP. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the United States population. Hepatology. 2009;50:175–184. doi: 10.1002/hep.22941. [DOI] [PubMed] [Google Scholar]
- 288.Machado MV, Ravasco P, Jesus L, Marques-Vidal P, Oliveira CR, Proença T, Baldeiras I, Camilo ME, Cortez-Pinto H. Blood oxidative stress markers in non-alcoholic steatohepatitis and how it correlates with diet. Scand J Gastroenterol. 2008;43:95–102. doi: 10.1080/00365520701559003. [DOI] [PubMed] [Google Scholar]
- 289.Mater MK, Thelen AP, Pan DA, Jump DB. Sterol response element-binding protein 1c (SREBP1c) is involved in the polyunsaturated fatty acid suppression of hepatic S14 gene transcription. J Biol Chem. 1999;274:32725–32732. doi: 10.1074/jbc.274.46.32725. [DOI] [PubMed] [Google Scholar]
- 290.Zhu FS, Liu S, Chen XM, Huang ZG, Zhang DW. Effects of n-3 polyunsaturated fatty acids from seal oils on nonalcoholic fatty liver disease associated with hyperlipidemia. World J Gastroenterol. 2008;14:6395–6400. doi: 10.3748/wjg.14.6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Capanni M, Calella F, Biagini MR, Genise S, Raimondi L, Bedogni G, Svegliati-Baroni G, Sofi F, Milani S, Abbate R, et al. Prolonged n-3 polyunsaturated fatty acid supplementation ameliorates hepatic steatosis in patients with non-alcoholic fatty liver disease: a pilot study. Aliment Pharmacol Ther. 2006;23:1143–1151. doi: 10.1111/j.1365-2036.2006.02885.x. [DOI] [PubMed] [Google Scholar]
- 292.Hatzitolios A, Savopoulos C, Lazaraki G, Sidiropoulos I, Haritanti P, Lefkopoulos A, Karagiannopoulou G, Tzioufa V, Dimitrios K. Efficacy of omega-3 fatty acids, atorvastatin and orlistat in non-alcoholic fatty liver disease with dyslipidemia. Indian J Gastroenterol. 2004;23:131–134. [PubMed] [Google Scholar]
- 293.Parker HM, Johnson NA, Burdon CA, Cohn JS, O’Connor HT, George J. Omega-3 supplementation and non-alcoholic fatty liver disease: a systematic review and meta-analysis. J Hepatol. 2012;56:944–951. doi: 10.1016/j.jhep.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 294.Sawada N, Inoue M, Iwasaki M, Sasazuki S, Shimazu T, Yamaji T, Takachi R, Tanaka Y, Mizokami M, Tsugane S. Consumption of n-3 fatty acids and fish reduces risk of hepatocellular carcinoma. Gastroenterology. 2012;142:1468–1475. doi: 10.1053/j.gastro.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 295.Johnson RJ, Segal MS, Sautin Y, Nakagawa T, Feig DI, Kang DH, Gersch MS, Benner S, Sánchez-Lozada LG. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am J Clin Nutr. 2007;86:899–906. doi: 10.1093/ajcn/86.4.899. [DOI] [PubMed] [Google Scholar]
- 296.Abid A, Taha O, Nseir W, Farah R, Grosovski M, Assy N. Soft drink consumption is associated with fatty liver disease independent of metabolic syndrome. J Hepatol. 2009;51:918–924. doi: 10.1016/j.jhep.2009.05.033. [DOI] [PubMed] [Google Scholar]
- 297.Abdelmalek MF, Suzuki A, Guy C, Unalp-Arida A, Colvin R, Johnson RJ, Diehl AM. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1961–1971. doi: 10.1002/hep.23535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Nomura K, Yamanouchi T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J Nutr Biochem. 2012;23:203–208. doi: 10.1016/j.jnutbio.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 299.Pan XR, Li GW, Hu YH, Wang JX, Yang WY, An ZX, Hu ZX, Lin J, Xiao JZ, Cao HB, et al. Effects of diet and exercise in preventing NIDDM in people with impaired glucose tolerance. The Da Qing IGT and Diabetes Study. Diabetes Care. 1997;20:537–544. doi: 10.2337/diacare.20.4.537. [DOI] [PubMed] [Google Scholar]
- 300.Perseghin G, Price TB, Petersen KF, Roden M, Cline GW, Gerow K, Rothman DL, Shulman GI. Increased glucose transport-phosphorylation and muscle glycogen synthesis after exercise training in insulin-resistant subjects. N Engl J Med. 1996;335:1357–1362. doi: 10.1056/NEJM199610313351804. [DOI] [PubMed] [Google Scholar]
- 301.Ross R, Dagnone D, Jones PJ, Smith H, Paddags A, Hudson R, Janssen I. Reduction in obesity and related comorbid conditions after diet-induced weight loss or exercise-induced weight loss in men. A randomized, controlled trial. Ann Intern Med. 2000;133:92–103. doi: 10.7326/0003-4819-133-2-200007180-00008. [DOI] [PubMed] [Google Scholar]
- 302.Rice B, Janssen I, Hudson R, Ross R. Effects of aerobic or resistance exercise and/or diet on glucose tolerance and plasma insulin levels in obese men. Diabetes Care. 1999;22:684–691. doi: 10.2337/diacare.22.5.684. [DOI] [PubMed] [Google Scholar]
- 303.Tamura Y, Tanaka Y, Sato F, Choi JB, Watada H, Niwa M, Kinoshita J, Ooka A, Kumashiro N, Igarashi Y, et al. Effects of diet and exercise on muscle and liver intracellular lipid contents and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab. 2005;90:3191–3196. doi: 10.1210/jc.2004-1959. [DOI] [PubMed] [Google Scholar]
- 304.Bacchi E, Negri C, Targher G, Faccioli N, Lanza M, Zoppini G, Zanolin E, Schena F, Bonora E, Moghetti P. Both resistance training and aerobic training reduce hepatic fat content in type 2 diabetic subjects with nonalcoholic fatty liver disease (the RAED2 Randomized Trial) Hepatology. 2013;58:1287–1295. doi: 10.1002/hep.26393. [DOI] [PubMed] [Google Scholar]
- 305.Fenkci S, Sarsan A, Rota S, Ardic F. Effects of resistance or aerobic exercises on metabolic parameters in obese women who are not on a diet. Adv Ther. 2006;23:404–413. doi: 10.1007/BF02850161. [DOI] [PubMed] [Google Scholar]
- 306.O’Donovan G, Kearney EM, Nevill AM, Woolf-May K, Bird SR. The effects of 24 weeks of moderate- or high-intensity exercise on insulin resistance. Eur J Appl Physiol. 2005;95:522–528. doi: 10.1007/s00421-005-0040-5. [DOI] [PubMed] [Google Scholar]
- 307.Harrison SA, Day CP. Benefits of lifestyle modification in NAFLD. Gut. 2007;56:1760–1769. doi: 10.1136/gut.2006.112094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Gaziano JM, Gaziano TA, Glynn RJ, Sesso HD, Ajani UA, Stampfer MJ, Manson JE, Hennekens CH, Buring JE. Light-to-moderate alcohol consumption and mortality in the Physicians’ Health Study enrollment cohort. J Am Coll Cardiol. 2000;35:96–105. doi: 10.1016/s0735-1097(99)00531-8. [DOI] [PubMed] [Google Scholar]
- 309.Smith-Warner SA, Spiegelman D, Yaun SS, van den Brandt PA, Folsom AR, Goldbohm RA, Graham S, Holmberg L, Howe GR, Marshall JR, et al. Alcohol and breast cancer in women: a pooled analysis of cohort studies. JAMA. 1998;279:535–540. doi: 10.1001/jama.279.7.535. [DOI] [PubMed] [Google Scholar]
- 310.Di Castelnuovo A, Costanzo S, Bagnardi V, Donati MB, Iacoviello L, de Gaetano G. Alcohol dosing and total mortality in men and women: an updated meta-analysis of 34 prospective studies. Arch Intern Med. 2006;166:2437–2445. doi: 10.1001/archinte.166.22.2437. [DOI] [PubMed] [Google Scholar]
- 311.King DE, Mainous AG, Geesey ME. Adopting moderate alcohol consumption in middle age: subsequent cardiovascular events. Am J Med. 2008;121:201–206. doi: 10.1016/j.amjmed.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Howard AA, Arnsten JH, Gourevitch MN. Effect of alcohol consumption on diabetes mellitus: a systematic review. Ann Intern Med. 2004;140:211–219. doi: 10.7326/0003-4819-140-6-200403160-00011. [DOI] [PubMed] [Google Scholar]
- 313.Athyros VG, Liberopoulos EN, Mikhailidis DP, Papageorgiou AA, Ganotakis ES, Tziomalos K, Kakafika AI, Karagiannis A, Lambropoulos S, Elisaf M. Association of drinking pattern and alcohol beverage type with the prevalence of metabolic syndrome, diabetes, coronary heart disease, stroke, and peripheral arterial disease in a Mediterranean cohort. Angiology. 2007;58:689–697. doi: 10.1177/0003319707306146. [DOI] [PubMed] [Google Scholar]
- 314.Djoussé L, Arnett DK, Eckfeldt JH, Province MA, Singer MR, Ellison RC. Alcohol consumption and metabolic syndrome: does the type of beverage matter? Obes Res. 2004;12:1375–1385. doi: 10.1038/oby.2004.174. [DOI] [PubMed] [Google Scholar]
- 315.Freiberg MS, Cabral HJ, Heeren TC, Vasan RS, Curtis Ellison R. Alcohol consumption and the prevalence of the Metabolic Syndrome in the US.: a cross-sectional analysis of data from the Third National Health and Nutrition Examination Survey. Diabetes Care. 2004;27:2954–2959. doi: 10.2337/diacare.27.12.2954. [DOI] [PubMed] [Google Scholar]
- 316.Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decreased prevalence of suspected nonalcoholic fatty liver disease. Hepatology. 2008;47:1947–1954. doi: 10.1002/hep.22292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Dunn W, Sanyal AJ, Brunt EM, Unalp-Arida A, Donohue M, McCullough AJ, Schwimmer JB. Modest alcohol consumption is associated with decreased prevalence of steatohepatitis in patients with non-alcoholic fatty liver disease (NAFLD) J Hepatol. 2012;57:384–391. doi: 10.1016/j.jhep.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Gunji T, Matsuhashi N, Sato H, Fujibayashi K, Okumura M, Sasabe N, Urabe A. Light and moderate alcohol consumption significantly reduces the prevalence of fatty liver in the Japanese male population. Am J Gastroenterol. 2009;104:2189–2195. doi: 10.1038/ajg.2009.361. [DOI] [PubMed] [Google Scholar]
- 319.Moriya A, Iwasaki Y, Ohguchi S, Kayashima E, Mitsumune T, Taniguchi H, Ikeda F, Shiratori Y, Yamamoto K. Alcohol consumption appears to protect against non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2011;33:378–388. doi: 10.1111/j.1365-2036.2010.04520.x. [DOI] [PubMed] [Google Scholar]
- 320.Suzuki A, Angulo P, St Sauver J, Muto A, Okada T, Lindor K. Light to moderate alcohol consumption is associated with lower frequency of hypertransaminasemia. Am J Gastroenterol. 2007;102:1912–1919. doi: 10.1111/j.1572-0241.2007.01274.x. [DOI] [PubMed] [Google Scholar]
- 321.Liangpunsakul S, Chalasani N. What should we recommend to our patients with NAFLD regarding alcohol use? Am J Gastroenterol. 2012;107:976–978. doi: 10.1038/ajg.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Loomba R, Bettencourt R, Barrett-Connor E. Synergistic association between alcohol intake and body mass index with serum alanine and aspartate aminotransferase levels in older adults: the Rancho Bernardo Study. Aliment Pharmacol Ther. 2009;30:1137–1149. doi: 10.1111/j.1365-2036.2009.04141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Loomba R, Yang HI, Su J, Brenner D, Barrett-Connor E, Iloeje U, Chen CJ. Synergism between obesity and alcohol in increasing the risk of hepatocellular carcinoma: a prospective cohort study. Am J Epidemiol. 2013;177:333–342. doi: 10.1093/aje/kws252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Bellentani S, Saccoccio G, Masutti F, Crocè LS, Brandi G, Sasso F, Cristanini G, Tiribelli C. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann Intern Med. 2000;132:112–117. doi: 10.7326/0003-4819-132-2-200001180-00004. [DOI] [PubMed] [Google Scholar]
- 325.Ruhl CE, Everhart JE. Joint effects of body weight and alcohol on elevated serum alanine aminotransferase in the United States population. Clin Gastroenterol Hepatol. 2005;3:1260–1268. doi: 10.1016/s1542-3565(05)00743-3. [DOI] [PubMed] [Google Scholar]
- 326.Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology. 2010;51:1972–1978. doi: 10.1002/hep.23527. [DOI] [PubMed] [Google Scholar]
- 327.Fukushima Y, Kasuga M, Nakao K, Shimomura I, Matsuzawa Y. Effects of coffee on inflammatory cytokine gene expression in mice fed high-fat diets. J Agric Food Chem. 2009;57:11100–11105. doi: 10.1021/jf901278u. [DOI] [PubMed] [Google Scholar]
- 328.Matsuda Y, Kobayashi M, Yamauchi R, Ojika M, Hiramitsu M, Inoue T, Katagiri T, Murai A, Horio F. Coffee and caffeine improve insulin sensitivity and glucose tolerance in C57BL/6J mice fed a high-fat diet. Biosci Biotechnol Biochem. 2011;75:2309–2315. doi: 10.1271/bbb.110452. [DOI] [PubMed] [Google Scholar]
- 329.Murase T, Misawa K, Minegishi Y, Aoki M, Ominami H, Suzuki Y, Shibuya Y, Hase T. Coffee polyphenols suppress diet-induced body fat accumulation by downregulating SREBP-1c and related molecules in C57BL/6J mice. Am J Physiol Endocrinol Metab. 2011;300:E122–E133. doi: 10.1152/ajpendo.00441.2010. [DOI] [PubMed] [Google Scholar]
- 330.Choi EY, Cho YO. Interaction of physical trainings and coffee intakes in fuel utilization during exercise in rats. Nutr Res Pract. 2013;7:178–184. doi: 10.4162/nrp.2013.7.3.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Abrahão SA, Pereira RG, de Sousa RV, Lima AR, Crema GP, Barros BS. Influence of coffee brew in metabolic syndrome and type 2 diabetes. Plant Foods Hum Nutr. 2013;68:184–189. doi: 10.1007/s11130-013-0355-z. [DOI] [PubMed] [Google Scholar]
- 332.Hino A, Adachi H, Enomoto M, Furuki K, Shigetoh Y, Ohtsuka M, Kumagae S, Hirai Y, Jalaldin A, Satoh A, et al. Habitual coffee but not green tea consumption is inversely associated with metabolic syndrome: an epidemiological study in a general Japanese population. Diabetes Res Clin Pract. 2007;76:383–389. doi: 10.1016/j.diabres.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 333.Takami H, Nakamoto M, Uemura H, Katsuura S, Yamaguchi M, Hiyoshi M, Sawachika F, Juta T, Arisawa K. Inverse correlation between coffee consumption and prevalence of metabolic syndrome: baseline survey of the Japan Multi-Institutional Collaborative Cohort (J-MICC) Study in Tokushima, Japan. J Epidemiol. 2013;23:12–20. doi: 10.2188/jea.JE20120053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Gutiérrez-Grobe Y, Chávez-Tapia N, Sánchez-Valle V, Gavilanes-Espinar JG, Ponciano-Rodríguez G, Uribe M, Méndez-Sánchez N. High coffee intake is associated with lower grade nonalcoholic fatty liver disease: the role of peripheral antioxidant activity. Ann Hepatol. 2012;11:350–355. [PubMed] [Google Scholar]
- 335.Catalano D, Martines GF, Tonzuso A, Pirri C, Trovato FM, Trovato GM. Protective role of coffee in non-alcoholic fatty liver disease (NAFLD) Dig Dis Sci. 2010;55:3200–3206. doi: 10.1007/s10620-010-1143-3. [DOI] [PubMed] [Google Scholar]
- 336.Birerdinc A, Stepanova M, Pawloski L, Younossi ZM. Caffeine is protective in patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2012;35:76–82. doi: 10.1111/j.1365-2036.2011.04916.x. [DOI] [PubMed] [Google Scholar]
- 337.Anty R, Marjoux S, Iannelli A, Patouraux S, Schneck AS, Bonnafous S, Gire C, Amzolini A, Ben-Amor I, Saint-Paul MC, et al. Regular coffee but not espresso drinking is protective against fibrosis in a cohort mainly composed of morbidly obese European women with NAFLD undergoing bariatric surgery. J Hepatol. 2012;57:1090–1096. doi: 10.1016/j.jhep.2012.07.014. [DOI] [PubMed] [Google Scholar]
- 338.Bambha K, Wilson LA, Unalp A, Loomba R, Neuschwander-Tetri BA, Brunt EM, Bass NM. Coffee consumption in NAFLD patients with lower insulin resistance is associated with lower risk of severe fibrosis. Liver Int. 2014;34:1250–1258. doi: 10.1111/liv.12379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Molloy JW, Calcagno CJ, Williams CD, Jones FJ, Torres DM, Harrison SA. Association of coffee and caffeine consumption with fatty liver disease, nonalcoholic steatohepatitis, and degree of hepatic fibrosis. Hepatology. 2012;55:429–436. doi: 10.1002/hep.24731. [DOI] [PubMed] [Google Scholar]
- 340.Zelber-Sagi S, Kessler A, Brazowsky E, Webb M, Lurie Y, Santo M, Leshno M, Blendis L, Halpern Z, Oren R. A double-blind randomized placebo-controlled trial of orlistat for the treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2006;4:639–644. doi: 10.1016/j.cgh.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 341.Gloy VL, Briel M, Bhatt DL, Kashyap SR, Schauer PR, Mingrone G, Bucher HC, Nordmann AJ. Bariatric surgery versus non-surgical treatment for obesity: a systematic review and meta-analysis of randomised controlled trials. BMJ. 2013;347:f5934. doi: 10.1136/bmj.f5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Ikramuddin S, Korner J, Lee WJ, Connett JE, Inabnet WB, Billington CJ, Thomas AJ, Leslie DB, Chong K, Jeffery RW, et al. Roux-en-Y gastric bypass vs intensive medical management for the control of type 2 diabetes, hypertension, and hyperlipidemia: the Diabetes Surgery Study randomized clinical trial. JAMA. 2013;309:2240–2249. doi: 10.1001/jama.2013.5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Mummadi RR, Kasturi KS, Chennareddygari S, Sood GK. Effect of bariatric surgery on nonalcoholic fatty liver disease: systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2008;6:1396–1402. doi: 10.1016/j.cgh.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 344.Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–2307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 345.Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, Austin AS, Freeman JG, Morgan L, Webber J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135:1176–1184. doi: 10.1053/j.gastro.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 346.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Mahady SE, Webster AC, Walker S, Sanyal A, George J. The role of thiazolidinediones in non-alcoholic steatohepatitis - a systematic review and meta analysis. J Hepatol. 2011;55:1383–1390. doi: 10.1016/j.jhep.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 348.Musso G, Cassader M, Rosina F, Gambino R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of randomised trials. Diabetologia. 2012;55:885–904. doi: 10.1007/s00125-011-2446-4. [DOI] [PubMed] [Google Scholar]
- 349.Lutchman G, Modi A, Kleiner DE, Promrat K, Heller T, Ghany M, Borg B, Loomba R, Liang TJ, Premkumar A, et al. The effects of discontinuing pioglitazone in patients with nonalcoholic steatohepatitis. Hepatology. 2007;46:424–429. doi: 10.1002/hep.21661. [DOI] [PubMed] [Google Scholar]
- 350.Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA. 2007;298:1180–1188. doi: 10.1001/jama.298.10.1180. [DOI] [PubMed] [Google Scholar]
- 351.Colhoun HM, Livingstone SJ, Looker HC, Morris AD, Wild SH, Lindsay RS, Reed C, Donnan PT, Guthrie B, Leese GP, et al. Hospitalised hip fracture risk with rosiglitazone and pioglitazone use compared with other glucose-lowering drugs. Diabetologia. 2012;55:2929–2937. doi: 10.1007/s00125-012-2668-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Lewis JD, Ferrara A, Peng T, Hedderson M, Bilker WB, Quesenberry CP, Vaughn DJ, Nessel L, Selby J, Strom BL. Risk of bladder cancer among diabetic patients treated with pioglitazone: interim report of a longitudinal cohort study. Diabetes Care. 2011;34:916–922. doi: 10.2337/dc10-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Haukeland JW, Konopski Z, Eggesbø HB, von Volkmann HL, Raschpichler G, Bjøro K, Haaland T, Løberg EM, Birkeland K. Metformin in patients with non-alcoholic fatty liver disease: a randomized, controlled trial. Scand J Gastroenterol. 2009;44:853–860. doi: 10.1080/00365520902845268. [DOI] [PubMed] [Google Scholar]
- 354.Hyogo H, Tazuma S, Arihiro K, Iwamoto K, Nabeshima Y, Inoue M, Ishitobi T, Nonaka M, Chayama K. Efficacy of atorvastatin for the treatment of nonalcoholic steatohepatitis with dyslipidemia. Metabolism. 2008;57:1711–1718. doi: 10.1016/j.metabol.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 355.Fernández-Miranda C, Pérez-Carreras M, Colina F, López-Alonso G, Vargas C, Solís-Herruzo JA. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig Liver Dis. 2008;40:200–205. doi: 10.1016/j.dld.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 356.Athyros VG, Mikhailidis DP, Didangelos TP, Giouleme OI, Liberopoulos EN, Karagiannis A, Kakafika AI, Tziomalos K, Burroughs AK, Elisaf MS. Effect of multifactorial treatment on non-alcoholic fatty liver disease in metabolic syndrome: a randomised study. Curr Med Res Opin. 2006;22:873–883. doi: 10.1185/030079906X104696. [DOI] [PubMed] [Google Scholar]
- 357.Ishitobi T, Hyogo H, Tokumo H, Arihiro K, Chayama K. Efficacy of probucol for the treatment of non-alcoholic steatohepatitis with dyslipidemia: An open-label pilot study. Hepatol Res. 2013:Epub ahead of print. doi: 10.1111/hepr.12135. [DOI] [PubMed] [Google Scholar]
- 358.Merat S, Malekzadeh R, Sohrabi MR, Sotoudeh M, Rakhshani N, Sohrabpour AA, Naserimoghadam S. Probucol in the treatment of non-alcoholic steatohepatitis: a double-blind randomized controlled study. J Hepatol. 2003;38:414–418. doi: 10.1016/s0168-8278(02)00441-5. [DOI] [PubMed] [Google Scholar]
- 359.Park H, Shima T, Yamaguchi K, Mitsuyoshi H, Minami M, Yasui K, Itoh Y, Yoshikawa T, Fukui M, Hasegawa G, et al. Efficacy of long-term ezetimibe therapy in patients with nonalcoholic fatty liver disease. J Gastroenterol. 2011;46:101–107. doi: 10.1007/s00535-010-0291-8. [DOI] [PubMed] [Google Scholar]
- 360.Yoneda M, Fujita K, Nozaki Y, Endo H, Takahashi H, Hosono K, Suzuki K, Mawatari H, Kirikoshi H, Inamori M, et al. Efficacy of ezetimibe for the treatment of non-alcoholic steatohepatitis: An open-label, pilot study. Hepatol Res. 2010;40:566–573. doi: 10.1111/j.1872-034X.2010.00644.x. [DOI] [PubMed] [Google Scholar]
- 361.Athyros VG, Tziomalos K, Gossios TD, Griva T, Anagnostis P, Kargiotis K, Pagourelias ED, Theocharidou E, Karagiannis A, Mikhailidis DP. Safety and efficacy of long-term statin treatment for cardiovascular events in patients with coronary heart disease and abnormal liver tests in the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) Study: a post-hoc analysis. Lancet. 2010;376:1916–1922. doi: 10.1016/S0140-6736(10)61272-X. [DOI] [PubMed] [Google Scholar]
- 362.Athyros VG, Katsiki N, Karagiannis A, Mikhailidis DP. Are statins "IDEAL" for non-alcoholic fatty liver disease? Curr Med Res Opin. 2014;30:229–231. doi: 10.1185/03007995.2013.855192. [DOI] [PubMed] [Google Scholar]
- 363.Yokohama S, Tokusashi Y, Nakamura K, Tamaki Y, Okamoto S, Okada M, Aso K, Hasegawa T, Aoshima M, Miyokawa N, et al. Inhibitory effect of angiotensin II receptor antagonist on hepatic stellate cell activation in non-alcoholic steatohepatitis. World J Gastroenterol. 2006;12:322–326. doi: 10.3748/wjg.v12.i2.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Yokohama S, Yoneda M, Haneda M, Okamoto S, Okada M, Aso K, Hasegawa T, Tokusashi Y, Miyokawa N, Nakamura K. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology. 2004;40:1222–1225. doi: 10.1002/hep.20420. [DOI] [PubMed] [Google Scholar]
- 365.Hirata T, Tomita K, Kawai T, Yokoyama H, Shimada A, Kikuchi M, Hirose H, Ebinuma H, Irie J, Ojiro K, et al. Effect of Telmisartan or Losartan for Treatment of Nonalcoholic Fatty Liver Disease: Fatty Liver Protection Trial by Telmisartan or Losartan Study (FANTASY) Int J Endocrinol. 2013;2013:587140. doi: 10.1155/2013/587140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Benson SC, Pershadsingh HA, Ho CI, Chittiboyina A, Desai P, Pravenec M, Qi N, Wang J, Avery MA, Kurtz TW. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity. Hypertension. 2004;43:993–1002. doi: 10.1161/01.HYP.0000123072.34629.57. [DOI] [PubMed] [Google Scholar]
- 367.Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142:37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- 368.Klein EA, Thompson IM, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, Minasian LM, Ford LG, Parnes HL, Gaziano JM, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2011;306:1549–1556. doi: 10.1001/jama.2011.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Schürks M, Glynn RJ, Rist PM, Tzourio C, Kurth T. Effects of vitamin E on stroke subtypes: meta-analysis of randomised controlled trials. BMJ. 2010;341:c5702. doi: 10.1136/bmj.c5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Lindor KD, Kowdley KV, Heathcote EJ, Harrison ME, Jorgensen R, Angulo P, Lymp JF, Burgart L, Colin P. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: results of a randomized trial. Hepatology. 2004;39:770–778. doi: 10.1002/hep.20092. [DOI] [PubMed] [Google Scholar]
- 371.Dufour JF, Oneta CM, Gonvers JJ, Bihl F, Cerny A, Cereda JM, Zala JF, Helbling B, Steuerwald M, Zimmermann A. Randomized placebo-controlled trial of ursodeoxycholic acid with vitamin e in nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2006;4:1537–1543. doi: 10.1016/j.cgh.2006.09.025. [DOI] [PubMed] [Google Scholar]
- 372.Leuschner UF, Lindenthal B, Herrmann G, Arnold JC, Rössle M, Cordes HJ, Zeuzem S, Hein J, Berg T. High-dose ursodeoxycholic acid therapy for nonalcoholic steatohepatitis: a double-blind, randomized, placebo-controlled trial. Hepatology. 2010;52:472–479. doi: 10.1002/hep.23727. [DOI] [PubMed] [Google Scholar]
- 373.Ratziu V, de Ledinghen V, Oberti F, Mathurin P, Wartelle-Bladou C, Renou C, Sogni P, Maynard M, Larrey D, Serfaty L, et al. A randomized controlled trial of high-dose ursodesoxycholic acid for nonalcoholic steatohepatitis. J Hepatol. 2011;54:1011–1019. doi: 10.1016/j.jhep.2010.08.030. [DOI] [PubMed] [Google Scholar]
- 374.Balmer ML, Siegrist K, Zimmermann A, Dufour JF. Effects of ursodeoxycholic acid in combination with vitamin E on adipokines and apoptosis in patients with nonalcoholic steatohepatitis. Liver Int. 2009;29:1184–1188. doi: 10.1111/j.1478-3231.2009.02037.x. [DOI] [PubMed] [Google Scholar]
- 375.Adams LA, Zein CO, Angulo P, Lindor KD. A pilot trial of pentoxifylline in nonalcoholic steatohepatitis. Am J Gastroenterol. 2004;99:2365–2368. doi: 10.1111/j.1572-0241.2004.40064.x. [DOI] [PubMed] [Google Scholar]
- 376.Lee YM, Sutedja DS, Wai CT, Dan YY, Aung MO, Zhou L, Cheng CL, Wee A, Lim SG. A randomized controlled pilot study of Pentoxifylline in patients with non-alcoholic steatohepatitis (NASH) Hepatol Int. 2008;2:196–201. doi: 10.1007/s12072-008-9058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Van Wagner LB, Koppe SW, Brunt EM, Gottstein J, Gardikiotes K, Green RM, Rinella ME. Pentoxifylline for the treatment of non-alcoholic steatohepatitis: a randomized controlled trial. Ann Hepatol. 2011;10:277–286. [PubMed] [Google Scholar]
- 378.Zein CO, Yerian LM, Gogate P, Lopez R, Kirwan JP, Feldstein AE, McCullough AJ. Pentoxifylline improves nonalcoholic steatohepatitis: a randomized placebo-controlled trial. Hepatology. 2011;54:1610–1619. doi: 10.1002/hep.24544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Aller R, De Luis DA, Izaola O, Conde R, Gonzalez Sagrado M, Primo D, De La Fuente B, Gonzalez J. Effect of a probiotic on liver aminotransferases in nonalcoholic fatty liver disease patients: a double blind randomized clinical trial. Eur Rev Med Pharmacol Sci. 2011;15:1090–1095. [PubMed] [Google Scholar]
- 380.Loguercio C, De Simone T, Federico A, Terracciano F, Tuccillo C, Di Chicco M, Cartenì M. Gut-liver axis: a new point of attack to treat chronic liver damage? Am J Gastroenterol. 2002;97:2144–2146. doi: 10.1111/j.1572-0241.2002.05942.x. [DOI] [PubMed] [Google Scholar]
- 381.Loguercio C, Federico A, Tuccillo C, Terracciano F, D’Auria MV, De Simone C, Del Vecchio Blanco C. Beneficial effects of a probiotic VSL#3 on parameters of liver dysfunction in chronic liver diseases. J Clin Gastroenterol. 2005;39:540–543. doi: 10.1097/01.mcg.0000165671.25272.0f. [DOI] [PubMed] [Google Scholar]
- 382.Malaguarnera M, Vacante M, Antic T, Giordano M, Chisari G, Acquaviva R, Mastrojeni S, Malaguarnera G, Mistretta A, Li Volti G, et al. Bifidobacterium longum with fructo-oligosaccharides in patients with non alcoholic steatohepatitis. Dig Dis Sci. 2012;57:545–553. doi: 10.1007/s10620-011-1887-4. [DOI] [PubMed] [Google Scholar]
- 383.Wong VW, Won GL, Chim AM, Chu WC, Yeung DK, Li KC, Chan HL. Treatment of nonalcoholic steatohepatitis with probiotics. A proof-of-concept study. Ann Hepatol. 2013;12:256–262. [PubMed] [Google Scholar]