

Abstract
Angiogenesis is a complex process finely regulated by the balance between angiogenesis stimulators and inhibitors. As a result of proangiogenic factors overexpression, it plays a crucial role in cancer development. Although initially mast cells (MCs) role has been defined in hypersensitivity reactions and in immunity, it has been discovered that MCs have a crucial interplay on the regulatory function between inflammatory and tumor cells through the release of classical proangiogenic factors (e.g., vascular endothelial growth factor) and nonclassical proangiogenic mediators granule-associated (mainly tryptase). In fact, in several animal and human malignancies, MCs density is highly correlated with tumor angiogenesis. In particular, tryptase, an agonist of the proteinase-activated receptor-2 (PAR-2), represents one of the most powerful angiogenic mediators released by human MCs after c-Kit receptor activation. This protease, acting on PAR-2 by its proteolytic activity, has angiogenic activity stimulating both human vascular endothelial and tumor cell proliferation in paracrine manner, helping tumor cell invasion and metastasis. Based on literature data it is shown that tryptase may represent a promising target in cancer treatment due to its proangiogenic activity. Here we focused on molecular mechanisms of three tryptase inhibitors (gabexate mesylate, nafamostat mesylate, and tranilast) in order to consider their prospective role in cancer therapy.
1. Introduction
Angiogenesis is a complex process, mainly mediated by endothelial cells, consisting in the formation of new blood capillaries from existing vessels [1–4]. It is finely regulated by the balance between several angiogenesis stimulators, such as vascular endothelial growth factor (VEGF), fibroblast growth factor-2 (FGF-2), platelet derived growth factor (PDGF), angiopoietins, tryptase, and some angiogenesis inhibitors, including thrombospondin, angiostatin, and endostatin [5–11]. Angiogenesis, further than being involved in normal physiological processes, has been demonstrated to play a crucial role in cancer development inducing tumor growth, invasion, and metastasis [12, 13].
Mast cells (MCs) intervene in tissue angiogenesis through several classical proangiogenic factors such as VEGF, FGF-2, PDGF, interleukin-6 (IL-6), and nonclassical proangiogenic factors, such as tryptase and chymase, stored in their secretory granules [14–18]. In fact, MCs density is highly correlated with the extent of tumor angiogenesis both in benign tumors (e.g., in keloids) and in animal and human malignancies (systemic mastocytosis, head and neck, colorectal, lung, and cutaneous cancer) [19–24]. Tryptase and chymase stimulate angiogenesis and the response is similar to that obtained with VEGF [16]. This evidence confirms even more the angiogenic activity of these two proteases stored in MCs granules [16].
2. Role of Mast Cell Tryptase in Angiogenesis and Tumor Growth
MCs are tissue leukocytes originating from hematopoietic stem cells in bone marrow. Generally, these precursor cells circulate in blood as agranular cells; then, MCs migrate into different tissues completing their maturation into granulated cells under the influence of several microenvironmental growth factors. One of these crucial factors is the stem cell factor (SCF), the ligand of c-Kit receptor (c-KitR) secreted by fibroblasts and stromal and endothelial cells. SCF is critically involved in MCs activation [25, 26]. MCs can be naturally found in association with connective tissue structures (i.e., blood vessels, lymphatic vessels, and nerves) and in the proximity of skin and mucosa of the gastrointestinal, respiratory, and genitourinary tracts [27], which represent common portals of infections [26, 28]. Accordingly, for many years, MCs have been implicated in the pathogenesis of IgE-associated allergic reactions and certain protective responses to parasites, bacteria, viruses, and fungi [29–31]. However, increasing evidence suggests the involvement of these cells in several biological settings, such as inflammation, immunomodulation, angiogenesis, wound healing, tissue remodeling, and cancer [17, 32–41]. Specifically, the multiple functions of MCs depend on their capability to release panoply of biologically active products upon suitable immunological and nonimmunological stimulation [42]. These mediators are either preformed in their secretory granules (biogenic amines, neutral serine proteases) or synthesized de novo (metabolites of arachidonic acid, cytokines) [43, 44]. MCs granules represent key functional elements, whose content can be released by two distinct secretory mechanisms: exocytosis (anaphylactic degranulation) or piecemeal degranulation [25]. Interestingly, the latter process is the most frequent secretory mechanism observed in chronic inflammatory settings, such as cancer [31, 45].
A possible causal relationship between MCs, chronic inflammation, and cancer has long been suggested. Accordingly, as most tumors contain inflammatory cell infiltrates, often including abundant MCs, the question about the possible contribution of MCs to tumor development has progressively been emerging [31, 39]. MCs have been recognized as one of the earliest cell types to infiltrate many developing tumors, particularly malignant melanoma and breast and colorectal cancer (CRC) [8, 17, 21, 23, 40, 70, 71]. Ample evidence highlights that MCs accumulate predominantly around several types of tumors, at the boundary between malignant and healthy tissues [8, 17]. In particular, these cells are often strategically located in proximity of blood vessels within the tumor microenvironment, suggesting an early role of MCs in angiogenesis and tumor growth; in fact angiogenesis generates a new vascular supply that delivers oxygen and nutrients to the rapidly proliferating malignant tissue [25, 39, 72]. In agreement with this role, MCs are an abundant source of potent proangiogenic factors, which represent a major issue linking these cells to cancer [26, 73]. In many experimental tumor settings, MCs promote angiogenesis by releasing preformed mediators or by activating proteolytic release of extracellular matrix-bound angiogenic molecules [25, 32, 72]. In vitro studies have demonstrated that MC granular components can induce vascularization [25]. Indeed, the addition of either human recombinant tryptase or chymase is able to stimulate neovascularization in the chick embryo chorioallantoic membrane assay (CAM) [32, 72]. Based on these results, treatment with cromolyn, an inhibitor of MCs degranulation, has been shown to restrain expansion and survival of pancreatic cancer and endothelial cells [15].
Tryptase and chymase are preformed active serine proteases and are stored in large amounts in MCs secretory granules [74], whose angiogenic role has been established [16, 75]. In particular, tryptase represents one of the most powerful angiogenic mediators released by human MCs upon c-KitR activation, and it may be angiogenic via several mechanisms [24]. This protease directly stimulates human vascular endothelial cell proliferation acting on protease-activated receptor-2 (PAR-2) by its proteolytic activity [24, 75, 76], leading to direct angiogenic effect (Figure 1). This particular proliferative pathway has been showed by Yoshii et al. [50] who have demonstrated that tryptase induces PAR-2-mediated proliferative effects on a human colon carcinoma cell line (DLD-1 cells) in a mitogen-activated protein kinase (MAP) kinase- and cyclooxygenase- (COX-) dependent manner. PAR-2 activation also leads to the release of IL-6 and granulocyte-macrophage colony stimulating factor (GM-CSF), which, in turn, act as angiogenic factors [77]. The important role of tryptase in neovascularization is also shown by its ability to degrade connective tissue matrix in order to provide rooms for neovascular growth. Tryptase may also contribute indirectly to tissue neovascularization by activating latent matrix-metalloproteinases (MMPs) and plasminogen activator, which, in turn, degrade extracellular matrix (ECM) with consequent release of ECM-bound angiogenic factors, such as VEGF and FGF-2 [25, 26, 52]. The disruption of local ECM leads also to release of SCF. Interestingly, tumor-derived SCF has been recently implicated both in MCs recruitment into the tumor environment as well as in increased MCs release and production of VEGF and FGF-2 [78, 79].
Figure 1.
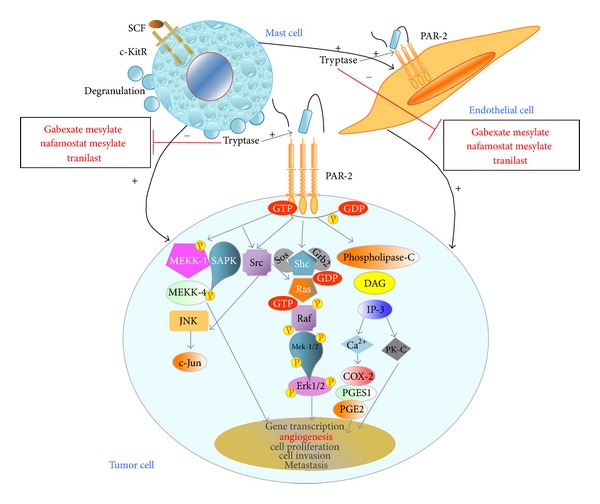
Tryptase, released after MCs activation of c-KitR/SCF-mediated, acting on PAR-2 by its proteolytic activity, has angiogenic activity stimulating both human vascular endothelial and tumor cell proliferation in paracrine manner, helping tumor cell invasion and metastasis. In cancer treatment, tryptase may represent a promising target by tryptase inhibitors (gabexate mesylate, nafamostat mesylate, tranilast) due to their potential antiangiogenic activity. c-KitR, c-Kit receptor; PAR-2, proteinase-activated receptor-2; VEGFR, vascular endothelial growth factor receptor; SCF, stem cell factor, VEGF, vascular endothelial growth factor; NHERF-1, Na+/H+ exchanger regulatory factor-1; MEKK-1, mitogen-activated protein kinase/extracellular signal-related kinase-1; MEKK-4, mitogen-activated protein kinase/extracellular signal-related kinase-4; JNK, c-Jun N-terminal kinase; c-Jun, Jun protooncogene; SAPK, mitogen-activated protein kinase-9; GEF, rho/rac guanine nucleotide exchange factor; Rho, rhodopsin transcription termination factor; SOS, SOn of sevenless protein; Grb2, growth factor receptor-bound protein 2; Shc, Shc transforming protein kinase; Ras, Ras protein kinase; Raf, Raf protein kinase; mitogen-activated protein kinase/extracellular signal-related kinase-1/2; Erk, Elk-related tyrosine kinase; DAG, Diacylglycerol; IP-3, inositol triphosphate; PK-C, protein kinase-C; COX-2, cyclooxygenase-2; PGE2, prostaglandin E2; PGES-1, prostaglandin E synthase-1; PK-A, protein kinase-A.
With reference to the above-described mechanisms that link tryptase to tumor angiogenesis and cancer progression, several studies have reported a linear correlation between mast cells density positive to tryptase (MCDPT) and angiogenesis in solid tumors, such as human malignant melanoma [80, 81], endometrial carcinoma [41], breast cancer [8, 82], uterine leiomyomas [83], gastric cancer [23, 24, 40], and CRC [21, 84]. Regarding hematological tumors, angiogenesis has been shown to increase with the MCDPT in B cell non-Hodgkin's lymphomas [85] as well as in the bone marrow of patients with multiple myeloma, monoclonal gammopathies of undetermined significance [86], myelodysplastic syndrome [87], and B-cell chronic lymphocytic leukemia [85]. In the majority of studies, MCDPT correlates with angiogenesis, tumor aggressiveness, and poor prognosis [25], even if some human studies have demonstrated a correlation between high mast cells density (MCD) and improved overall survival [88–91], suggesting that MCs effects on tumor fate may depend on some bias related to cancer (e.g., type of surgical treatment with relative lymph node collection, histology, stage tumor, small sample size) and different methods of MCs evaluation (e.g., histochemistry with toluidine blue, Giemsa stain, primary antibody antitryptase or antichymase for immunohistochemistry, standardization of MCs count with reference to magnification, MCs location, and microscopic field of evaluation).
Overall, despite conflicting reports on the role of MCD- and MCDPT-mediated angiogenesis in tumor development, literature data indicate that tryptase may represent a promising target in adjuvant cancer treatment [25, 26], leading to considering the therapeutic use of drugs which specifically inhibit its angiogenic activity. Therefore, tryptase inhibitors, such as gabexate mesylate and nafamostat mesylate [92–94], might be evaluated in clinical trials as new antiangiogenic agents in combination with chemotherapy in the treatment of cancer.
3. Potential Role of Mast Cells Tryptase Inhibitors in Cancer
In the light of the aforementioned complex relationship between MCs tryptase and angiogenesis in tumor development, we have described the possible molecular mechanisms of three drugs targeting tryptase functions, such as gabexate mesylate, nafamostat mesylate, and tranilast, in order to discuss their prospective role in cancer therapy.
3.1. Gabexate Mesylate
Gabexate mesylate (GM) is a synthetic inhibitor of trypsin-like serine proteases [95–97] that shows an antiproteinase activity on various kinds of plasma proteases, such as thrombin, plasmin, trypsin, kallikrein, C1 esterase in the complement system, and factor Xa in the coagulation cascade. Accordingly, GM has been therapeutically used for disseminated intravascular coagulation (DIC) and acute pancreatitis [95] in Japan, Italy, Korea, and Taiwan. In addition, several recent studies have reported that this protease inhibitor exerts a significant antitumorigenic effect, both in vitro and in vivo [46, 47, 94].
Proteolytic degradation of ECM components is a crucial step for tumor cell invasion and metastasis. Among several classes of degrading ECM proteinases, MMPs (MMP-2 and MMP-9) and urokinase-type plasminogen activator (uPA) have been closely associated with the metastatic phenotype of cancer cells [98–102]. These enzymes are also implicated in tumor angiogenesis [103]. Therefore, inhibitors of MMPs and uPA are able to inhibit invasion and metastasis [104, 105] by reducing angiogenesis in vitro and in vivo [106–109]. Furthermore, serine plasma proteases, such as thrombin and plasmin, are closely associated with activation pathways of certain MMPs (MMP-2, MMP-3, and MMP-9) [110, 111], indicating that multispecific protease inhibitors could be useful tools for an antimetastatic and antiangiogenic strategy. Based on these findings, GM has been shown to inhibit proliferation, invasion, and metastasis of human colon cancer cell lines through the inhibition of both MMPs and uPA-plasmin system, consequentially limiting angiogenesis [46]. Although the inhibition of the uPA system may be involved in downregulation of MMP activity, the results of this study have suggested that GM has a direct inhibitory effect on MMPs, whose related-mechanism is unknown [46].
Interestingly, the inhibition of MMPs by GM and, in general, its anti-invasive, antimetastatic, and antiangiogenic properties could also be explained through its potent and selective inhibition of human tryptase [92]. Indeed, as above described, in the early stages of tumor development several tumor-derived factors (i.e., SCF, adrenomedullin) recruit and activate MCs in tumor microenvironment, leading to the release of tryptase [25, 26], which, in turn, can indirectly stimulate tumor angiogenesis by activating latent MMPs and uPA [23, 24, 75]. Accordingly, Yoshii et al. [50] demonstrated the specific localization of MCDPT in the invasive front of tumor tissues by examining 30 cases of human colon adenocarcinoma. A previous study [49] has found the proliferation of DLD-1 colon cancer cells expressing PAR-2 in response to PAR-2 activating peptide (AP). Moreover, tryptase also enhanced DLD-1 cell proliferation by means of a specific stimulation of PAR-2 via MAPK- and COX-dependent manners. Furthermore, these proliferative effects were concentration-dependently inhibited by nafamostat mesylate, a very potent inhibitor of human tryptase [93, 112], suggesting that PAR-2 activation was dependent on tryptase proteolytic activity. In the same study, PAR-2 density in tumor tissues was higher than that in the normal tissues, as revealed by the immunohistochemical analysis. This suggests that tryptase released by MCs surrounding tumor tissues may induce the PAR-2-mediated proliferation of colon cancer cells in a paracrine way [49]. Similarly, tryptase has been reported to stimulate angiogenesis directly [75] via PAR-2 activation on vascular endothelial cells [24, 76]. Moreover, increasing evidences support that MCs tryptase is involved in angiogenesis through the direct degradation of connective tissue matrix [25, 26, 75], with consequent release of matrix-associated angiogenic substances, such as VEGF or FGF-2 [40, 72, 113–116]. These findings as a whole suggest that MCs tryptase may sustain colon cancer cell growth in two ways: direct proliferative effect via PAR-2 stimulation and indirect support through angiogenesis stimulation. Thus, tryptase may be considered a novel target of colon cancer therapy. Taken together, all the reported evidences suggest that the inhibition of colon cancer growth, invasion, and metastasis by GM may be also due to its selective inhibition of MC tryptase. Therefore, GM could be potentially useful for antimetastatic and antiangiogenic treatment of colon cancers. Noteworthy, this assumption is corroborated by a recent study by Brandi et al. [47] aimed to investigate the antitumor efficacy of GM, alone, and in combination with the antiepidermal growth factor receptor (EGFR) monoclonal antibody cetuximab, in a group of human CRC cell lines with a different expression pattern of wild-type/mutated V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (K-RAS), protooncogene B-Raf murine sarcoma viral oncogene homolog B1 (BRAF), and phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha oncogene (PIK3CA). Besides confirming the lack of response to cetuximab in CRC cells bearing such mutations [117–119], results demonstrated that GM significantly inhibited the growth, invasiveness, and tumor-induced angiogenesis in all CRC cells tested in this study [47]. In particular, the antiangiogenic effect of GM in combination with the anti-EGFR antibody was found to be not superior than that observed with GM as single agent, suggesting that the inhibition of tumor angiogenesis may be largely related to GM mechanism of action, most notably the inhibition of MCs tryptase. Therefore, also considering its good toxicological profile, these findings indicate that GM could represent a valuable therapeutic option for patients with EGFR-expressing metastatic CRC (mCRC), particularly for those ones bearing KRAS, BRAF, and PIK3CA mutations, either as monotherapy or in combination with standard chemotherapy [47].
The antimetastatic and antiangiogenic mechanisms of GM have also been investigated in pancreatic cancer cell lines. As in colon cancer, MMPs and uPA play a crucial role also in the progression of pancreatic cancer [48]. In addition, a previous study by Uchima et al. [48] reported the involvement of tumor-associated trypsinogen (TAT) and pancreatic acinar trypsinogen (PAT) in pancreatic cancer invasion and metastasis. Both these serine proteases can be activated by uPA, which is produced by pancreatic cancer. Following, they are able to degrade ECM components and can also directly activate TAT, PAT, pro-MMPs, and pro-uPA, leading to further ECM breakdown. The resulting vicious cycle would activate latent ECM-degrading proteases, thereby promoting tumor cell invasion and metastasis. In particular, PAT and TAT had been shown to continuously stimulate pancreatic cancer cell proliferation by activating PAR-2 [120]. Furthermore, several investigations reported that transforming growth factor-beta 1 (TGF-β1), produced in the tumor microenvironment, could be a strong mediator of pancreatic cancer cell invasion, metastasis, and angiogenesis by upregulating VEGF, MMP-2, and uPA secretion [121–123]. High uPA levels, in turn, could activate latent TGF-beta1, resulting in a positive feedback loop on tumor progression [123]. Starting from these data, Uchima et al. [94] suggested that GM inhibited the invasiveness, proliferation, and potential liver metastatic of pancreatic cancer cell lines by downregulating TAT and uPA activities, reducing PAR-2 activation, and inhibiting the production of TGF-β1 and VEGF. Moreover, in pancreatic cancer the inhibitory effects of GM may be, in part, associated with tryptase inhibition. In fact, similarly to TAT, MCs tryptase may be responsible for PAR-2-mediated pancreatic cell proliferation, since tryptase is a natural agonist of this receptor [124]. Moreover, tryptase-mediated activation of latent MMPs and uPA [25, 26, 75] may induce further TAT, MMPs, and uPA activation and ECM degradation, thus triggering an ECM-protease network responsible for tumor cell invasion and metastasis [48, 94]. In this context, tryptase inhibition by GM may downregulate TAT and uPA enzymatic activities. The resulting downregulation of uPA levels may decrease the activation of latent TGF-β1, thereby impairing the abovementioned cycle vicious of uPA and TGF-β1 and downregulating VEGF production. On the other hand, tryptase inhibition may also directly suppress the production of TGF-β1 and VEGF involved in tumor growth and angiogenesis. In agreement with this proposed mechanism, tryptase has been reported to increase the production of TGF-β1 in other pathophysiological settings [125, 126]. The findings about the GM mechanism of action in pancreatic cancer cells, together with our considerations, indicate that this protease inhibitor could be a useful therapeutic option for antimetastatic and antiangiogenic treatment of pancreatic cancer.
The above studies are summarized in Table 1.
Table 1.
All preclinical studies mentioned above that have considered gabexate mesylate.
| Author, reference, year | Drug/s | Tumor target | Molecular mechanisms of action | Results |
|---|---|---|---|---|
| Yoon et al. [46] 2004 | gabexate mesylate | several human colon cancer cell lines | (1) down-regulation of MMPs (2) inhibition of uPA-plasmin system |
inhibition of angiogenesis, tumor cell growth, invasion, metastasis |
|
| ||||
| Brandi et al. [47] 2012 | (1) gabexate mesylate
(2) gabexate mesylate plus cetuximab |
several human colorectal cancer cell lines (wt/mut KRAS, BRAF, PIK3CA) | not analyzed | (1) inhibition of tumor cell growth, angiogenesis, invasion, metastasis (2) antitumoral efficacy of the combination therapy was not superior than gabexate mesylate alone |
|
| ||||
| Uchima et al. [48] 2003 | gabexate mesylate | several human pancreatic cancer cell lines | down-regulation of uPA, TAT, PAT, MMPs, TGF-β1, VEGF | inhibition of angiogenesis, cell growth, invasion, metastasis |
MMPs, Metalloproteinases; uPA, urokinase-type plasminogen activator; wt, wild-type; mut, mutated; TAT, Tumor-associated trypsinogen; PAT, Pancreatic acinar trypsinogen; TGF-β1, Tumor growth factor-beta1; VEGF, Vascular endothelial growth factor.
3.2. Nafamostat Mesylate
Similarly to GM, nafamostat mesylate (NM) is able to inhibit a variety of trypsin-like serine proteases and some proteases implicated in the coagulation cascade [127, 128]. Interestingly, Mori et al. [93] have demonstrated that NM inhibits human tryptase with potency 1000 times higher than that of GM, concluding that NM is an extremely potent and selective inhibitor when employed at relatively low concentration. They have also suggested that such inhibitory action on tryptase activity can account for some therapeutic effects of NM in specific clinical conditions. Indeed, human tryptase may be involved in the pathogenesis of several MCs-mediated allergic and inflammatory diseases, such as rhinitis and asthma. It is also implicated in specific gastrointestinal, dermatological, and cardiovascular disorders [129–131]. Therefore, NM has been widely used for the treatment of acute pancreatitis and DIC in Japan [132, 133].
The antitumor potential of NM is suggested by Yoshii et al.'s study previously described [50]. In fact, the in vitro analysis showed that NM concentration-dependently inhibited the tryptase-induced enhancement of proliferation of DLD-1 cells, thus suggesting that tryptase inhibition may mediate the anticancer effect of NM. It has also been reported that NM inhibits liver metastases of colon cancer cells in mice [134]. Moreover, previous studies showed that NM inhibited the proliferation and invasion of pancreatic cancer cells by antagonizing TAT-induced activation of PAR-2 in vitro, in the same fashion of GM [51, 52]. Indeed, several studies have recently revealed that NM exerts antiproliferative, antiangiogenic, and antimetastatic effects also in pancreatic cancer, proposing the use of this serine protease inhibitor in combination with standard chemotherapy regimens for pancreatic cancer management [53–56]. In particular, the blockade of nuclear factor kappa-B (NF-κB) activation has been reported to underlie antitumor effects of NM [55]. In this regard, Karin and Lin have demonstrated that NF-κB plays an important role in the modulation of inflammatory responses, cell proliferation, apoptosis, and oncogenesis including invasion and angiogenesis [55]. Typically, inactive NF-κB is sequestered in the cytoplasm by nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha (IkBα); however, a specific activation signaling leads to IkBα phosphorylation and consequent release of NF-κB protein, which translocates into the nucleus, where it induces the transcription of target genes [135]. Most notably, constitutive activation of NF-κB has been identified in a variety of tumors including pancreatic cancer [135] and it is known to contribute to the aggressive phenotype [136] and chemoresistance [137]. The resulting overexpression of downstream target genes of NF-κB, such as intercellular adhesion molecule-1 (ICAM-1) [138], IL-8 [136, 139], VEGF [136, 139, 140], MMP-9 [140], and uPA [141], promotes cell adhesion, angiogenesis, invasion, and metastasis. Interestingly, some cancer chemotherapy drugs, such as oxaliplatin and gemcitabine, have been shown to activate NF-κB by themselves, thereby reducing their antitumor efficacy [142–144]. Based on these findings, a NF-κB inhibitor like NM may be able to suppress proliferation, angiogenesis, and metastasis both in pancreatic cancer and in other malignancies, opening an avenue for novel therapeutic approaches. In the study by Fujiwara et al. [55], NM has been shown to downregulate activities of phosphorylated IkBα, NF-κB, and its target genes, resulting in inhibition of cell adhesion, invasion, and increase of a particular programmed cell death (anoikis) in human pancreatic tumor cell lines. In vivo, intraperitoneal administration of pancreatic cancer cells, pretreated with NM, in nude mice revealed reduced peritoneal metastasis and neovascularization and increased survival compared with controls. This suggests that NM may potentially reduce the incidence of postoperative recurrences due to peritoneal dissemination in pancreatic cancer patients [145]. In accordance with these findings, the authors have already reported the ability of NM to inhibit NF-κB activation and induce caspase-8-mediated apoptosis when this serine protease inhibitor was used as monotherapy or with gemcitabine, in vitro and in vivo [53, 146, 147]. Most notably, they reported a better clinical outcome of combination therapy of gemcitabine or paclitaxel with NM in comparison with gemcitabine or paclitaxel alone in pancreatic cancer-bearing mice through the inhibition of chemotherapeutic drug-induced NF-κB activation [53, 55]. It was also demonstrated the clinical usefulness of intra-arterial NM administration combined with gemcitabine in patients with unresectable pancreatic cancer [54, 148]. Accordingly, Gocho and colleagues [56] have recently proven that NM enhances the antitumor effect of oxaliplatin by inhibiting oxaliplatin-induced NF-κB activation. This leads to downregulation of the cellular inhibitor of apoptosis proteins, c-IAP1 and c-IAP2, resulting in cleavage of poly ADP-ribose polymerase (PARP) and caspase-8-mediated apoptosis in vitro and in vivo: the inhibition of NF-κB activity results in chemosensitization of pancreatic cancer. Therefore, combination chemotherapy with NM and oxaliplatin exerts a synergistic cytotoxic effect in pancreatic cancer both in vitro and in vivo.
Taking into account the above-illustrated pathophysiological pathways, we propose that the potent inhibition of MCs tryptase may also be involved in the antitumor activities of NM. Firstly, this hypothesis is supported by the ability of tryptase to stimulate cell proliferation and invasion of cancer cells in vitro through the activation of PAR-2. We herein report the evidence of these tryptase-mediated proliferative effects only in colon cancer cells [50]; however, tryptase, being a natural agonist of PAR-2 [124], may be potentially able to activate this receptor class expressed also in the gastrointestinal tract, pancreas, liver, kidney, and sensory neurons [149–151], triggering a proliferative response. Moreover, the above mentioned antiproliferative effect of NM in pancreatic cancer cells by blocking TAT-induced PAR-2 stimulation [51, 52] may be indirectly related to tryptase inhibition. In fact, we have previously reported that tryptase can activate the uPA system [25, 26, 75], which, in turn, activates TAT leading to stimulation of PAR-2 on the surface of pancreatic cancer cells [94]. On the other hand, the inhibition of tryptase-mediated activation of PAR-2 on vascular endothelial cells could contribute to antiangiogenic effects of NM.
MCs tryptase may contribute to cancer pathways triggered by the constitutive activation of NF-κB. In particular, tryptase may upregulate the levels of several target genes overexpressed owing to the pathological NF-κB activation, such as VEGF, IL-8, MMP-9, and uPA, thereby contributing to promote angiogenesis, invasion, and metastasis in a variety of tumors. Interestingly, several studies have reported that PAR-2 is able to mediate some important tryptase-induced inflammatory processes, such as microglia activation and skin inflammation [152, 153]. In particular, it has been shown that MC tryptase, via PAR-2, may induce the upregulation/release of proinflammatory cytokines (i.e., IL-6, IL-8, TNF-α) and activate important inflammatory signaling cascades such as NF-κB pathway in human dermal microvascular endothelial cells and microglia: MAPK signaling pathways are involved in NF-κB activation and consequent production/release of proinflammatory cytokines by tryptase [152, 153]. Furthermore, according to Ma et al. [154] tryptase could phosphorylate protein-kinase B (PKB, also known as AKT) through PAR-2, activate phosphoinositol-3-kinase (PI3K)/PKB pathway, and upregulate the expression of NF-κB in inflammatory settings. Most notably, PKB/AKT is involved in cellular survival pathways by inhibiting apoptotic processes [155]; hence, it has been implicated as a major factor in many types of cancers [156].
In the light of these last findings, MCs tryptase may probably contribute to the aggressive behavior and chemoresistance of pancreatic cancer cells, by activating NF-κB. Therefore, the inhibitory effect of NM on NF-κB activities may also indirectly depend on the selective tryptase inhibition. On the other hand, tryptase inhibition could also justify the apoptotic effect of NM through the downregulation of PI3K/protein kinase B (PKB) signaling pathway. As a whole, the above detailed findings and mechanisms suggest a potential usefulness of NM in preoperative management of pancreatic cancer patients, because its use may reduce postoperative recurrences and improve survival by inhibition of metastasis induced by surgical resection [157]. Moreover, taking into account the improved outcomes and relatively low toxicity of preclinical and clinical studies of the combination therapy with traditional chemotherapeutic agents and NM, these combination chemotherapy regimens could represent a novel promising strategy for pancreatic cancer treatment.
The above studies are summarized in Table 2.
Table 2.
All studies mentioned above that have considered nafamostat mesylate.
| Author, reference, year | Drug/s | Tumor target | Molecular mechanisms of action | Results |
|---|---|---|---|---|
| Jikuhara et al. [49] 2003 | nafamostat mesylate | human colon cancer cell line (DLD-1) | (1) inhibition of PAR-2 stimulation via MAPK- and COX-dependent manner (2) inhibition of VEGF and FGF-2 levels |
inhibition of tumor cell growth, angiogenesis, invasion, metastasis |
|
| ||||
| Yoshii et al. [50] 2005 | nafamostat mesylate | human colon cancer cell line (DLD-1) | (1) Inhibition of PAR-2 stimulation via MAPK- and COX-dependent manner (2) Inhibition of the release of IL-6 and GM-CSF |
inhibition of angiogenesis, cell growth, invasion, metastasis |
|
| ||||
| Tajima et al. [51] 2001 | nafamostat mesylate | several human pancreatic cancer cell lines | antagonizing TAT-induced activation of PAR-2 | inhibition of tumor cell growth and invasion |
|
| ||||
| Ohta et al. [52] 2003 | nafamostat mesylate | several human pancreatic cancer cell lines | antagonizing TAT-induced activation of PAR-2 | inhibition of tumor cell growth and invasion |
|
| ||||
| Uwagawa et al. [53] 2009 | (1) nafamostat mesylate
(2) nafamostat mesylate plus gemcitabine |
human pancreatic cancer cell line (Panc-1) | Down-regulation of NF-κB with reduction of ICAM-1, IL-8, VEGF, MMP-9, uPA, RRM1 | (1) inhibition of tumor cell adhesion and growth, angiogenesis, invasion metastasis (2) increase of apoptosis (3) increase of body weight loss of mice |
|
| ||||
| Uwagawa et al. [54] 2009 | nafamostat mesylate plus intra-arterial gemcitabine | unresectable locally advanced or metastatic pancreatic cancer (20 pts) |
not analyzed | (1) CBR of 60% (2) reduction of CA19-9 serum level in 90% of pts (3) improvement in health-related quality of life |
|
| ||||
| Fujiwara et al. [55] 2011 | nafamostat mesylate | human pancreatic cancer cell lines (AsPC-1, BxPC-3, PANC-1) |
down-regulation of IkBα, NF-κB with reduction of ICAM-1, IL-8, VEGF, MMP-9, uPA | (1) increase of cell adhesion, programmed cell death (2) inhibition of angiogenesis, invasion, metastasis in peritoneal dissemination |
|
| ||||
| Gocho et al. [56] 2013 | (1) nafamostat mesylate
(2) nafamostat mesylate plus oxaliplatin |
human pancreatic cancer cell line (Panc-1) and pancreatic cancer mouse model |
down-regulation of NF-κB with reduction of ICAM-1, IL-8, VEGF, MMP-9, uPA, c-IAP1, c-IAP2 | (1) increase of cell adhesion, caspase-8-mediated apoptosis (2) inhibition of PARP, angiogenesis, invasion and metastasis, (3) synergistic cytotoxic effect |
PAR-2, Protease-activated receptor-2; MAPK, mitogen-activated protein kinase; COX, cyclooxygenase; IL, Interleukin; GM-CSF, Granulocyte-macrophage colony stimulating factor; TAT, Tumor-associated trypsinogen; IkB, Inhibitor of NF-κB; NF-κB, Nuclear factor-kappaB; MMPs, metalloproteinases; uPA, urokinase-type plasminogen activator; ICAM-1, Intercellular Adhesion Molecule-1, VEGF, Vascular endothelial growth factor, IAP, Inhibitors of apoptosis.
3.3. Tranilast
Among pharmacological agents that affect several inflammatory and allergic pathways mediated by MCs tryptase, also tranilast (TN) has progressively attracted considerable attention because of its antitumor potential. Since 1982, this drug has been approved in Japan and Korea for the systemic and topical treatment of bronchial asthma, atopic dermatitis, and allergic conjunctivitis, with indications for keloids and hypertrophic scar added in 1993 [158]. Follow-up studies have revealed that clinical effectiveness of TN in such applications depends on inhibition of the release of biologically active mediators from MCs [158, 159]. Moreover, tranilast was reported to inhibit the VEGF-induced angiogenesis both in vitro and in vivo, and most notably, these antiangiogenic activities have been shown to be concomitant with inhibitory effects on MCs degranulation [160].
TN was also reported to inhibit the release of TGF-beta, IL-1beta, prostaglandin (PG) E2, and IL-2 from human monocytes and macrophages [161, 162]. In the late 1980s, Isaji et al. [160] discovered the antiproliferative properties of TN. In particular, it was found that this agent inhibited fibroblast proliferation in vitro, resulting in suppression of proliferative inflammation in vivo. Subsequent studies confirmed the ability of TN in inhibiting tumor cell growth and proliferation in various models of cancer [59, 163, 164]. Overall, data from in vitro and in vivo models for proliferative disorders, clinical studies, and case reports have corroborated the antiproliferative and antitumor potential of TN [165], providing important insights into its mechanisms of action. Two studies, addressing antiproliferative activity of TN in several breast cancer cell lines, revealed that TN inhibits cell proliferation, by arresting cell cycle progression, and downregulates TGF-β signaling pathway [57, 58]. Moreover, Chakrabarti et al. [57] demonstrated that TN is able to inhibit MAPK signaling pathway.
TN was also reported to suppress the proliferation of cultured human leiomyoma cells by inhibiting cell cycle modulators, such as cyclin-dependent kinase 2 (CDK-2) [164].
As concerns pancreatic cancer, Hiroi et al. [59] reported that TN significantly inhibited proliferation of PGHAM-1, a hamster pancreatic cancer cell line. Moreover, TN was able to inhibit tumor angiogenesis in response to VEGF. Interestingly, in another study by Mitsuno et al. [60] TN was found to enhance chemotherapeutic effect of gemcitabine, as above reported for NM [53, 54]. However, unlike NM-induced effect, this chemosensitization was associated with the downregulation of ribonucleotide reductase M1 (RRM1) [53, 54].
Further experiments revealed that TN treatment inhibited prostate cancer cell proliferation in vitro by promoting apoptosis. In addition, it was reported the ability of TN to downregulate TGF-beta production from bone stromal cells and other different cell types, thereby suppressing TGF-β-stimulated osteoclast differentiation which underlies, in part, osteoblastic bone metastasis [61, 166]. Noguchi et al. [62] demonstrated that three weeks of TN treatment significantly reduced the tumor growth and metastasis, when administered daily by intraperitoneal injection (4 mg/animal), in a mouse model of oral squamous cell carcinoma. TN has also been reported to exert antitumor effects in gastric cancer [63] and malignant glioma [64] through different mechanisms. Izumi et al. [61] have reported that the treatment with oral TN (300 mg/day) promoted a reduction of prostate-specific antigen (PSA) levels in 4 out of 16 patients with advanced castration-resistant prostate cancer (CRPC). Accordingly, in the subsequent follow-up pilot study, oral treatment with TN (300 mg/day) for a median period of five months documented a continuous PSA inhibition in 3 out of 21 patients with advanced CRPC. Overall survival rates at 12 and 24 months were 74.5% and 61.5%, respectively [167]. As a whole, these results suggest that TN could be used to improve the prognosis of patients with advanced CRPC. However, the two clinical investigations had some limitations: (1) open-label studies with one arm; (2) short follow-up period; (3) small sample size; (4) all patients were Japanese. Therefore, the reported findings need further confirmation. Finally, several case studies have reported that transdermal application of TN was able to relieve both itching and pain associated with hypertrophic, keloid scars [168].
Several important pathways have been recognized as potential targets of TN antitumor activity. In particular, the TN inhibitory effects on cell proliferation depend mainly on its ability to interfere with TGF-β signaling and also reduce TGF-β secretion [57, 61, 63, 64]. Also, TN-mediated inhibition of cell proliferation has been markedly associated with blockade of cell cycle progression and consequent cell arrest in the G 0/G 1 transition [58, 59, 164, 169]. Probably, TN can induce cell cycle arrest also through the inhibition of calcium influx, which is crucial for G 1/S transition, as demonstrated by Nie et al. [65]. After TN treatment, the induction of apoptosis has been reported in several breast and prostate cancer cell lines [61, 66]. In particular, Subramaniam et al. [58] showed that TN induced p53 upregulation, enhanced RAC-alpha serine/threonine-protein kinase (AKT1) phosphorylation, and reduced phosphorylation of extracellular regulated kinase 2 (ERK2). Another work by Subramaniam et al. [66] detected an increased level of a PARP-cleavage product in human cancer cell lines treated with TN.
TN also acts as a nontoxic agonist of the aryl hydrocarbon receptor (ARH) [58, 170], whose function is involved in anticancer effects [67, 68]: ARH presence in the cell is critical for TN-mediated cell cycle arrest. Interestingly, the AHR also antagonizes TGF-β activity [171] and exerts ligand-dependent inhibitory effects on NF-κB signaling [172]. These AHR-mediated activities may contribute to the antiproliferative, antiangiogenic, and antimetastatic effects of TN.
The ability of TN to inhibit MAPK signaling pathway could also explain its antimetastatic potential, because this pathway is known to be implicated during the epithelial to mesenchymal transition (EMT), which is important for tumor cell invasion [57]. Moreover, the downregulation of certain MMPs, such as MMP-9, contributes to TN-mediated inhibition of tumor cell invasion during metastasis: such reduction of MMP-9 levels has been also linked to inhibition of TGF-β signaling [58].
In addition to the above detailed potential targets, the antitumor action of TN relies on the blocking of the release of chemical mediators from MCs [57, 61, 64, 159], which is also the mechanism responsible for its antiallergic and anti-inflammatory efficacy [158]. In agreement with this correlation, Yamamoto et al. [158] have recently documented that TN downregulated neurofibroma cell (NF1 cells) proliferation through not only suppression of cell-growth promoting pathways but also the inhibition of biologically active mediators by MCs. Interestingly, this study supports the involvement of tryptase in the antitumor activities of TN. Indeed, following its addition to NF1 cells cocultured with MCs, this agent was reported to significantly inhibit NF1 cell proliferation and lower the levels of TGF-β, SCF, and tryptase. These findings suggest that TN inhibits tumor proliferation also through the downregulation of MC tryptase, whose PAR-2-mediated proliferative and angiogenic effects have been previously described [50, 76]. Furthermore, tryptase has been reported to activate PI3K/PKB pathway via PAR-2 cleavage/activation and subsequently upregulate NF-κB expression [154], promoting tumor cell survival and chemoresistance [56, 155, 156]. Thus, the inhibition of tryptase release may represent a further molecular mechanism involved in the induction of apoptosis and cell cycle arrest upon TN treatment. Because tryptase-mediated PAR-2 activation triggers the MAPK signaling pathway, which is involved in the EMT process [57], tryptase inhibition by TN may also mediate its anti-invasion and antimetastatic properties.
The inhibitory effect on tryptase release could contribute to the ability of TN treatment to target TGF-beta-regulated signaling cascade and reduce TGF-β production. As above described, indeed, in the tumor microenvironment, tryptase may upregulate uPA levels, [25, 26, 75] thereby activating latent TGF-β which, in turn, upregulates the production of uPA, MMP-2, and VEGF. This vicious cycle has been implicated in angiogenesis, tumor cell invasion, and metastasis [94]. By the way, tryptase can also participate to the neovascular growth by activating latent MMPs, which, in turn, promote tumor invasiveness and release of angiogenic factors (VEGF or FGF-2) from their matrix-bound state [25, 26, 75]. Therefore, also taking into account the previously reported study by Isaji et al. [69], the downregulation of tryptase release may probably contribute to the TN-induced inhibition of tumor angiogenesis in response to VEGF, as observed in experimental pancreatic cancer [59].
In the light of the exposed considerations, we suggest that the inhibition of tryptase functions may underlie the anti-invasion, antimetastatic, and antiangiogenic effects of TN treatment. As concerns safety, TN shows relatively low toxicity in [61, 69, 173], making it a promising candidate for further clinical investigations. Based on the encouraging in vitro and in vivo research data, TN seems to be a safe and effective agent for the treatment of several proliferative and angiogenic diseases.
The above studies are summarized in Table 3.
Table 3.
All studies mentioned above that have considered tranilast.
| Author, reference, year | Drug/s | Tumor target | Molecular mechanisms of action | Results |
|---|---|---|---|---|
| Chakrabarti et al. [57] 2009 | tranilast | several mouse, rat and human breast cancer cell lines | (1) down-regulation of TGF-β pathway (2) inhibition of MAPK pathway |
inhibition of tumor cell proliferation, angiogenesis, apoptosis, migration |
|
| ||||
| Subramaniam et al. [58] 2010 | tranilast | mouse breast cancer cell line (4T1) | (1) down-regulation of TGF-β pathway (2) induction cell arrest in the G0/G1 transition, PARP cleavage, AKT1 phosphorylation (3) up-regulation of p53 (4) reduction of ERK1/2 phosphorylation |
inhibition of tumor cell proliferation, angiogenesis, apoptosis, migration |
|
| ||||
| Hiroi et al. [59] 2002 | tranilast | hamster pancreatic cancer cell line (PGHAM-1) | (1) down-regulation of TGF-β pathway with reduction of MMP-9 and VEGF levels (2) induction cell arrest in the G0/G1 transition |
inhibition of tumor cell proliferation, angiogenesis |
|
| ||||
| Mitsuno et al. [60] 2010 | (1) tranilast plus gemcitabine
(2) gemcitabine |
human pancreatic cancer cell line (KP4) | decrease of RRM1 expression | (1) inhibition of tumor cell proliferation, angiogenesis, apoptosis (2) synergistic cytotoxic effect of combination therapy |
|
| ||||
| Izumi et al. [61] 2009 | tranilast | (1) prostate cancer cell lines and bone-derived stromal cells (2) SCID mice (3) advanced hormone-refractory prostaste cancer (21 pts) |
down-regulation of TGF-β1 pathway | (1) induction of apoptosis (2) reduction of invasion and bone metastasis, PSA levels, improve prognosis |
|
| ||||
| Noguchi et al. [62] 2003 | tranilast | mouse model of oral squamous cell carcinoma | not analyzed | decrease of tumor growth, angiogenesis, cervical lymph node metastases |
|
| ||||
| Yashiro et al. [63] 2003 | tranilast | human gastric carcinoma cell line (OCUM-2D) and gastric fibroblast cell line (NF-10) | down-regulation of TGF-β pathway | decrease of tumor growth, angiogenesis, invasion |
|
| ||||
| Platten et al. [64] 2001 | tranilast | human malignant glioma cell line | down-regulation of TGF-β1-2 pathway | decrease of tumor growth, angiogenesis, migration, invasion |
|
| ||||
| Nie et al. [65] 1997 | tranilast | breast cancer cell lines (MCF-7) | induction cell arrest in the G0/G1 transition | decrease of tumor growth |
|
| ||||
| Subramaniam et al. [66] 2011 | tranilast | human breast cancer cell lines (triple positive-BT-474, triple negative-MDA-MB-231) | (1) up-regulation of p53 (2) induction cell arrest in the G0/G1 transition, AKT1 and ERK2 phosphorylation, PARP-cleavage product |
induction of apoptosis, tumor growth, migration |
|
| ||||
| Zhang et al. [67] 2009 | tranilast | several ER negative human breast cancer cell lines | agonizing ARH with down-regulation of TGF-β and NF-κB pathways | (1) induction of apoptosis (2) inhibition of angiogenesis, cell growth, invasion and metastasis |
|
| ||||
| Hall et al. [68] 2010 | tranilast | several human breast cancer cell lines | agonizing ARH with down-regulation of TGF-β and NF-κB pathways | (1) induction of apoptosis (2) inhibition of angiogenesis, cell growth, invasion and metastasis |
|
| ||||
| Isaji et al. [69] 1997 | tranilast | human pancreatic cancer cell lines | decrease of VEGF and MMPs levels | inhibition of angiogenesis, cell growth, migration |
TGF-β1, Tumor growth factor-beta1, MMPs, metalloproteinases; MAPK, mitogen-activated protein kinase uPA, PARP, poly ADP-ribose polymerase; urokinase-type plasminogen activator; AKT1, RAC-alpha serine/threonine-protein kinase; ERK, Extracellular regulated kinase 2; VEGF, Vascular endothelial growth factor; RRM1, Ribonucleotide reductase M1.
4. Concluding Remarks
Several literature data support a potential implication of MCs tryptase in three pivotal processes involved in cancer development and metastasization: cell growth, tumor-induced angiogenesis, and invasion [174, 175]. Therefore, this serine protease may be considered a novel promising target for the adjuvant treatment of tumors through the selective inhibition of angiogenesis, proliferation, and tissue remodelling. In agreement with these considerations, compounds targeting tryptase functions, although designed as antiallergic drugs, could exert a useful antitumor activity as well. In this regard, it is of interest to underline that many new anticancer drugs used in clinical field, such as sorafenib [18], sunitinib [176], pazopanib [177] axitinib [178], and masitinib [179] are all targeted against c-KitR, whose activation leads to the release of tryptase by MCs [24].
In particular, we herein discuss the antitumor and antiangiogenic potential of three agents which are able to inhibit the functions of MCs tryptase: gabexate mesylate, nafamostat mesylate, and tranilast. Although no definitive experimental data are available to confirm the role that tryptase released from mast cells stimulate tumor angiogenesis, the above hypothesis is supported by a pilot study in the in vivo chorioallantoic membrane assay [16]. In this study an angiogenic activity of human recombinant tryptase comparable to the angiogenic activity induced by the VEGF has been demonstrated. Data from this study suggest that the inhibition of tryptase is intriguing hypothesis worthy to further investigation.
The new antiangiogenic approach here reviewed should be substantially strengthened by future awaited clinical studies having the aim to evaluate the truly efficacy of the tryptase inhibitors as a novel tumor antiangiogenic therapy.
Disclosure
All authors have no financial or personal relationships with other people or organizations that could inappropriately influence their work.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Folkman J, Shing Y. Angiogenesis. Journal of Biological Chemistry. 1992;267(16):10931–10934. [PubMed] [Google Scholar]
- 2.Breier G. Angiogenesis in embryonic development—a review. Placenta. 2000;21(supplement A):S11–S15. doi: 10.1053/plac.1999.0525. [DOI] [PubMed] [Google Scholar]
- 3.Iruela-Arispe ML, Dvorak HF. Angiogenesis: a dynamic balance of stimulators and inhibitors. Thrombosis and Haemostasis. 1997;78(1):672–677. [PubMed] [Google Scholar]
- 4.Daniel TO, Abrahamson D. Endothelial signal integration in vascular assembly. Annual Review of Physiology. 2000;62:649–671. doi: 10.1146/annurev.physiol.62.1.649. [DOI] [PubMed] [Google Scholar]
- 5.Petrova TV, Makinen T, Alitalo K. Signaling via vascular endothelial growth factor receptors. Experimental Cell Research. 1999;253(1):117–130. doi: 10.1006/excr.1999.4707. [DOI] [PubMed] [Google Scholar]
- 6.Nugent MA, Iozzo RV. Fibroblast growth factor-2. The International Journal of Biochemistry and Cell Biology. 2000;32(2):115–120. doi: 10.1016/s1357-2725(99)00123-5. [DOI] [PubMed] [Google Scholar]
- 7.Gadaleta CD, Ranieri G. Trans-arterial chemoembolization as a therapy for liver tumours: new clinical developments and suggestions for combination with angiogenesis inhibitors. Critical Reviews in Oncology/Hematology. 2011;80(1):40–53. doi: 10.1016/j.critrevonc.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Ranieri G, Ammendola M, Patruno R, et al. Tryptase-positive mast cells correlate with angiogenesis in early breast cancer patients. International Journal of Oncology. 2009;35(1):115–120. doi: 10.3892/ijo_00000319. [DOI] [PubMed] [Google Scholar]
- 9.Roberts DD. Regulation of tumor growth and metastasis by thrombospondin-1. FASEB Journal. 1996;10(10):1183–1191. [PubMed] [Google Scholar]
- 10.O'Reilly MS, Holmgren L, Shing Y, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79(2):315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 11.O'Reilly MS, Boehm T, Shing Y, et al. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88(2):277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 12.Ranieri G, Pantaleo M, Piccinno M, et al. Tyrosine kinase inhibitors (TKIs) in human and pet tumours with special reference to breast cancer: a comparative review. Critical Reviews in Oncology/Hematology. 2013;88(2):293–308. doi: 10.1016/j.critrevonc.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 13.Ranieri G, Gadaleta CD, Patruno R, et al. A model of study for human cancer: spontaneous occurring tumors in dogs: biological features and translation for new anticancer therapies. Critical Reviews in Oncology/Hematology. 2013;88(1):187–197. doi: 10.1016/j.critrevonc.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 14.Kankkunen JP, Harvima IT, Naukkarinen A. Quantitative analysis of tryptase and chymase containing mast cells in benign and malignant breast lesions. International Journal of Cancer. 1997;72(3):385–338. doi: 10.1002/(sici)1097-0215(19970729)72:3<385::aid-ijc1>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 15.Soucek L, Lawlor ER, Soto D, Shchors K, Swigart LB, Evan GI. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nature Medicine. 2007;13(10):1211–1218. doi: 10.1038/nm1649. [DOI] [PubMed] [Google Scholar]
- 16.Ribatti D, Ranieri G, Nico B, Benagiano V, Crivellato E. Tryptase and chymase are angiogenic in vivo in the chorioallantoic membrane assay. International Journal of Developmental Biology. 2011;55(1):99–102. doi: 10.1387/ijdb.103138dr. [DOI] [PubMed] [Google Scholar]
- 17.Mangia A, Malfettone A, Rossi R, et al. Tissue remodelling in breast cancer: human mast cell tryptase as an initiator of myofibroblast differentiation. Histopathology. 2011;58(7):1096–1106. doi: 10.1111/j.1365-2559.2011.03842.x. [DOI] [PubMed] [Google Scholar]
- 18.Ranieri G, Gadaleta-Caldarola G, Goffredo V, et al. Sorafenib (BAY 43-9006) in hepatocellular carcinoma patients: from discovery to clinical development. Current Medicinal Chemistry. 2012;19(7):938–944. doi: 10.2174/092986712799320736. [DOI] [PubMed] [Google Scholar]
- 19.Ranieri G, Passantino L, Patruno R, et al. The dog mast cell tumour as a model to study the relationship between angiogenesis, mast cell density and tumour malignancy. Oncology Reports. 2003;10(5):1189–1193. [PubMed] [Google Scholar]
- 20.Ranieri G, Labriola A, Achille G, et al. Microvessel density, mast cell density and thymidine phosphorylase expression in oral squamous carcinoma. International Journal of Oncology. 2002;21(6):1317–1323. [PubMed] [Google Scholar]
- 21.Gulubova M, Vlaykova T. Prognostic significance of mast cell number and microvascular density for the survival of patients with primary colorectal cancer. Journal of Gastroenterology and Hepatology. 2009;24(7):1265–1275. doi: 10.1111/j.1440-1746.2007.05009.x. [DOI] [PubMed] [Google Scholar]
- 22.Yano H, Kinuta M, Tateishi H, et al. Mast cell infiltration around gastric cancer cells correlates with tumour angiogenesis and metastasis. Gastric Cancer. 1999;2(1):26–32. doi: 10.1007/s101200050017. [DOI] [PubMed] [Google Scholar]
- 23.Ammendola M, Sacco R, Donato G, et al. Mast cell positivity to tryptase correlates with metastatic lymph nodes in gastrointestinal cancer patients treated surgically. Oncology. 2013;85(2):111–116. doi: 10.1159/000351145. [DOI] [PubMed] [Google Scholar]
- 24.Moon TC, Lee E, Baek S-H, et al. Degranulation and cytokine expression in human cord blood-derived mast cells cultured in serum-free medium with recombinant human stem cell factor. Molecules and Cells. 2003;16(2):154–160. [PubMed] [Google Scholar]
- 25.Sperr WR, Czerwenka K, Mundigler G, et al. Specific activation of human mast cells by the ligand for c-Kit: comparison between lung, uterus and heart mast cells. International Archives of Allergy and Immunology. 1993;102(2):170–175. doi: 10.1159/000236568. [DOI] [PubMed] [Google Scholar]
- 26.Iemura A, Tsai M, Ando A, Wershil BK, Galli SJ. The c-kit ligand, stem cell factor, promotes mast cell survival by suppressing apoptosis. The American Journal of Pathology. 1994;144(2):321–328. [PMC free article] [PubMed] [Google Scholar]
- 27.Gurish MF, Austen KF. The diverse roles of mast cells. The Journal of Experimental Medicine. 2001;194(1):1–5. doi: 10.1084/jem.194.1.f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaik-Dasthagirisaheb YB, Varvara G, Murmura G, et al. Vascular endothelial growth factor (VEGF), mast cells and inflammation. International Journal of Immunopathology and Pharmacology. 2013;26(2):327–335. doi: 10.1177/039463201302600206. [DOI] [PubMed] [Google Scholar]
- 29.Puxeddu I, Ribatti D, Crivellato E, Levi-Schaffer F. Mast cells and eosinophils: a novel link between inflammation and angiogenesis in allergic diseases. Journal of Allergy and Clinical Immunology. 2005;116(3):531–536. doi: 10.1016/j.jaci.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Marshall JS. Mast-cell responses to pathogens. Nature Reviews Immunology. 2004;4(10):787–799. doi: 10.1038/nri1460. [DOI] [PubMed] [Google Scholar]
- 31.Crivellato E, Ribatti D. The mast cell: an evolutionary perspective. Biological Reviews of the Cambridge Philosophical Society. 2010;85(2):347–360. doi: 10.1111/j.1469-185X.2009.00105.x. [DOI] [PubMed] [Google Scholar]
- 32.Ribatti D, Crivellato E, Candussio L, et al. Mast cells and their secretory granules are angiogenic in the chick embryo chorioallantoic membrane. Clinical and Experimental Allergy. 2001;31(4):602–608. doi: 10.1046/j.1365-2222.2001.00986.x. [DOI] [PubMed] [Google Scholar]
- 33.Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CMM, Tsai M. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annual Review of Immunology. 2005;23:749–786. doi: 10.1146/annurev.immunol.21.120601.141025. [DOI] [PubMed] [Google Scholar]
- 34.Beaven MA. Our perception of the mast cell from Paul Ehrlich to now. European Journal of Immunology. 2009;39(1):11–25. doi: 10.1002/eji.200838899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oda K, Kitano H. A comprehensive map of the toll-like receptor signaling network. Molecular Systems Biology. 2006;2 doi: 10.1038/msb4100057.2006.0015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coussens LM, Raymond WW, Bergers G, et al. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes and Development. 1999;13(11):1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakayama T, Yao L, Tosato G. Mast cell-derived angiopoietin-1 plays a critical role in the growth of plasma cell tumors. The Journal of Clinical Investigation. 2004;114(9):1317–1325. doi: 10.1172/JCI22089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Theoharides TC, Conti P. Mast cells: the JEKYLL and HYDE of tumor growth. Trends in Immunology. 2004;25(5):235–241. doi: 10.1016/j.it.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 39.Ribatti D, Crivellato E. Mast cells, angiogenesis and cancer. Advances in Experimental Medicine and Biology. 2011;716:270–288. doi: 10.1007/978-1-4419-9533-9_14. [DOI] [PubMed] [Google Scholar]
- 40.Ribatti D, Guidolin D, Marzullo A, et al. Mast cells and angiogenesis in gastric carcinoma. International Journal of Experimental Pathology. 2010;91(4):350–356. doi: 10.1111/j.1365-2613.2010.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ribatti D, Finato N, Crivellato E, et al. Neovascularization and mast cells with tryptase activity increase simultaneously with pathologic progression in human endometrial cancer. The American Journal of Obstetrics and Gynecology. 2005;193(6):1961–1965. doi: 10.1016/j.ajog.2005.04.055. [DOI] [PubMed] [Google Scholar]
- 42.Galli SJ, Tsai M. Mast cells: versatile regulators of inflammation, tissue remodeling, host defense and homeostasis. Journal of Dermatological Science. 2008;49(1):7–19. doi: 10.1016/j.jdermsci.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilfillan AM, Tkaczyk C. Integrated signalling pathways for mast-cell activation. Nature Reviews Immunology. 2006;6(3):218–230. doi: 10.1038/nri1782. [DOI] [PubMed] [Google Scholar]
- 44.Bischoff SC. Physiological and pathophysiological functions of intestinal mast cells. Seminars in Immunopathology. 2009;31(2):185–205. doi: 10.1007/s00281-009-0165-4. [DOI] [PubMed] [Google Scholar]
- 45.Theoharides TC, Kempuraj D, Tagen M, Conti P, Kalogeromitros D. Differential release of mast cell mediators and the pathogenesis of inflammation. Immunological Reviews. 2007;217(1):65–78. doi: 10.1111/j.1600-065X.2007.00519.x. [DOI] [PubMed] [Google Scholar]
- 46.Yoon W-H, Jung Y-J, Kim T-D, et al. Gabexate mesilate inhibits colon cancer growth, invasion, and metastasis by reducing matrix metalloproteinases and angiogenesis. Clinical Cancer Research. 2004;10(13):4517–4526. doi: 10.1158/1078-0432.CCR-04-0084. [DOI] [PubMed] [Google Scholar]
- 47.Brandi G, Tavolari S, de Rosa F, et al. Antitumoral efficacy of the protease inhibitor gabexate mesilate in colon cancer cells harbouring KRAS, BRAF and PIK3CA mutations. PLoS ONE. 2012;7(7) doi: 10.1371/journal.pone.0041347.e41347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uchima Y, Sawada T, Nishihara T, et al. Identification of a trypsinogen activity stimulating factor produced by pancreatic cancer cells: its role in tumor invasion and metastasis. International Journal of Molecular Medicine. 2003;12(6):871–878. [PubMed] [Google Scholar]
- 49.Jikuhara A, Yoshii M, Iwagaki H, Mori S, Nishibori M, Tanaka N. MAP kinase-mediated proliferation of DLD-1 carcinoma by the stimulation of protease-activated receptor 2. Life Sciences. 2003;73(22):2817–2829. doi: 10.1016/s0024-3205(03)00702-1. [DOI] [PubMed] [Google Scholar]
- 50.Yoshii M, Jikuhara A, Mori S, et al. Mast cell tryptase stimulates DLD-1 carcinoma through prostaglandin-and MAP kinase-dependent manners. Journal of Pharmacological Sciences. 2005;98(4):450–458. doi: 10.1254/jphs.fpj05002x. [DOI] [PubMed] [Google Scholar]
- 51.Tajima H, Ohta T, Elnemr A, et al. Enhanced invasiveness of pancreatic adenocarcinoma cells stably transfected with cationic trypsinogen cDNA. International Journal of Cancer. 2001;94(5):699–704. doi: 10.1002/ijc.1531. [DOI] [PubMed] [Google Scholar]
- 52.Ohta T, Shimizu K, Yi S, et al. Protease-activated receptor-2 expression and the role of trypsin in cell proliferation in human pancreatic cancers. International Journal of Oncology. 2003;23(1):61–66. [PubMed] [Google Scholar]
- 53.Uwagawa T, Chiao PJ, Gocho T, Hirohara S, Misawa T, Yanaga K. Combination chemotherapy of nafamostat mesilate with gemcitabine for pancreatic cancer targeting NF-κB activation. Anticancer Research. 2009;29(8):3173–3178. [PubMed] [Google Scholar]
- 54.Uwagawa T, Misawa T, Sakamoto T, et al. A phase I study of full-dose gemcitabine and regional arterial infusion of nafamostat mesilate for advanced pancreatic cancer. Annals of Oncology. 2009;20(2):239–243. doi: 10.1093/annonc/mdn640. [DOI] [PubMed] [Google Scholar]
- 55.Fujiwara Y, Furukawa K, Haruki K, et al. Nafamostat mesilate can prevent adhesion, invasion and peritoneal dissemination of pancreatic cancer thorough nuclear factor kappa-B inhibition. Journal of Hepato-Biliary-Pancreatic Sciences. 2011;18(5):731–739. doi: 10.1007/s00534-011-0390-9. [DOI] [PubMed] [Google Scholar]
- 56.Gocho T, Uwagawa T, Furukawa K, et al. Combination chemotherapy of serine protease inhibitor nafamostat mesilate with oxaliplatin targeting NF-κB activation for pancreatic cancer. Cancer Letters. 2013;333(1):89–95. doi: 10.1016/j.canlet.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 57.Chakrabarti R, Subramaniam V, Abdalla S, Jothy S, Prud’homme GJ. Tranilast inhibits the growth and metastasis of mammary carcinoma. Anti-Cancer Drugs. 2009;20(5):334–345. doi: 10.1097/CAD.0b013e328327994e. [DOI] [PubMed] [Google Scholar]
- 58.Subramaniam V, Chakrabarti R, Prud'Homme GJ, Jothy S. Tranilast inhibits cell proliferation and migration and promotes apoptosis in murine breast cancer. Anti-Cancer Drugs. 2010;21(4):351–361. doi: 10.1097/CAD.0b013e328334992c. [DOI] [PubMed] [Google Scholar]
- 59.Hiroi M, Onda M, Uchida E, Aimoto T. Anti-tumor effect of N-[3,4-dimethoxycinnamoyl]-anthranilic acid (tranilast) on experimental pancreatic cancer. Journal of Nippon Medical School. 2002;69(3):224–234. doi: 10.1272/jnms.69.224. [DOI] [PubMed] [Google Scholar]
- 60.Mitsuno M, Kitajima Y, Ohtaka K, et al. Tranilast strongly sensitizes pancreatic cancer cells to gemcitabine via decreasing protein expression of ribonucleotide reductase 1. International Journal of Oncology. 2010;21(4):351–361. [PubMed] [Google Scholar]
- 61.Izumi K, Mizokami A, You QL, et al. Tranilast inhibits hormone refractory prostate cancer cell proliferation and suppresses transforming growth factor β1-associated osteoblastic changes. Prostate. 2009;69(11):1222–1234. doi: 10.1002/pros.20975. [DOI] [PubMed] [Google Scholar]
- 62.Noguchi N, Kawashiri S, Tanaka A, Kato K, Nakaya H. Effects of fibroblast growth inhibitor on proliferation and metastasis of oral squamous cell carcinoma. Oral Oncology. 2003;39(3):240–247. doi: 10.1016/s1368-8375(02)00092-1. [DOI] [PubMed] [Google Scholar]
- 63.Yashiro M, Murahashi K, Matsuoka T, et al. Tranilast (N-3 ,4 -dimethoxycinamoyl anthranilic acid): a novel inhibitor of invasion-stimulating interaction between gastric cancer cells and orthotopic fibroblasts. Anticancer Research. 2003;23(5A):3899–3904. [PubMed] [Google Scholar]
- 64.Platten M, Wild-Bode C, Wick W, Leitlein J, Dichgans J, Weller M. N-[3,4-dimethoxycinnamoyl]-anthranilic acid (tranilast) inhibits transforming growth factor-β release and reduces migration and invasiveness of human malignant glioma cells. International Journal of Cancer. 2001;93(1):53–61. doi: 10.1002/ijc.1289. [DOI] [PubMed] [Google Scholar]
- 65.Nie L, Oishi Y, Doi I, Shibata H, Kojima I. Inhibition of proliferation of MCF-7 breast cancer cells by a blocker of Ca2+-permeable channel. Cell Calcium. 1997;22(2):75–82. doi: 10.1016/s0143-4160(97)90107-x. [DOI] [PubMed] [Google Scholar]
- 66.Subramaniam V, Ace O, Prud’homme GJ, Jothy S. Tranilast treatment decreases cell growth, migration and inhibits colony formation of human breast cancer cells. Experimental and Molecular Pathology. 2011;90(1):116–122. doi: 10.1016/j.yexmp.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 67.Zhang S, Lei P, Liu X, et al. The aryl hydrocarbon receptor as a target for estrogen receptor-negative breast cancer chemotherapy. Endocrine-Related Cancer. 2009;16(3):835–844. doi: 10.1677/ERC-09-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hall JM, Barhoover MA, Kazmin D, McDonnell DP, Greenlee WF, Thomas RS. Activation of the aryl-hydrocarbon receptor inhibits invasive and metastatic features of human breast cancer cells and promotes breast cancer cell differentiation. Molecular Endocrinology. 2010;24(2):359–369. doi: 10.1210/me.2009-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Isaji M, Miyata H, Ajisawa Y, Takehana Y, Yoshimura N. Tranilast inhibits the proliferation, chemotaxis and tube formation of human microvascular endothelial cells in vitro and angiogenesis in vivo. British Journal of Pharmacology. 1997;122(6):1061–1066. doi: 10.1038/sj.bjp.0701493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ch'ng S, Wallis RA, Yuan L, Davis PF, Tan ST. Mast cells and cutaneous malignancies. Modern Pathology. 2006;19(1):149–159. doi: 10.1038/modpathol.3800474. [DOI] [PubMed] [Google Scholar]
- 71.Amini R-M, Aaltonen K, Nevanlinna H, et al. Mast cells and eosinophils in invasive breast carcinoma. BMC Cancer. 2007;7, article 165 doi: 10.1186/1471-2407-7-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maltby S, Khazaie K, McNagny KM. Mast cells in tumor growth: angiogenesis, tissue remodelling and immune-modulation. Biochimica et Biophysica Acta—Reviews on Cancer. 2009;1796(1):19–26. doi: 10.1016/j.bbcan.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Norrby K. Mast cells and angiogenesis. APMIS. 2002;110(5):355–371. doi: 10.1034/j.1600-0463.2002.100501.x. [DOI] [PubMed] [Google Scholar]
- 74.Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiological Reviews. 1997;77(4):1033–1079. doi: 10.1152/physrev.1997.77.4.1033. [DOI] [PubMed] [Google Scholar]
- 75.Blair RJ, Meng H, Marchese MJ, et al. Human mast cells stimulate vascular tube formation. Tryptase is a novel, potent angiogenic factor. The Journal of Clinical Investigation. 1997;99(11):2691–2700. doi: 10.1172/JCI119458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Milia AF, Salis MB, Stacca T, et al. Protease-activated receptor-2 stimulates angiogenesis and accelerates hemodynamic recovery in a mouse model of hindlimb ischemia. Circulation Research. 2002;91(4):346–352. doi: 10.1161/01.res.0000031958.92781.9e. [DOI] [PubMed] [Google Scholar]
- 77.Liu Y, Mueller BM. Protease-activated receptor-2 regulates vascular endothelial growth factor expression in MDA-MB-231 cells via MAPK pathways. Biochemical and Biophysical Research Communications. 2006;344(4):1263–1270. doi: 10.1016/j.bbrc.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 78.Zhang W, Stoica G, Tasca SI, Kelly KA, Meininger CJ. Modulation of tumor angiogenesis by stem cell factor. Cancer Research. 2000;60(23):6757–6762. [PubMed] [Google Scholar]
- 79.Huang B, Lei Z, Zhang G-M, et al. SCF-mediated mast cell infiltration and activation exacerbate the inflammation and immunosuppression in tumor microenvironment. Blood. 2008;112(4):1269–1279. doi: 10.1182/blood-2008-03-147033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ribatti D, Vacca A, Ria R, et al. Neovascularisation, expression of fibroblast growth factor-2, and mast cells with tryptase activity increase simultaneously with pathological progression in human malignant melanoma. European Journal of Cancer. 2003;39(5):666–674. doi: 10.1016/s0959-8049(02)00150-8. [DOI] [PubMed] [Google Scholar]
- 81.Ribatti D, Ennas MG, Vacca A, et al. Tumor vascularity and tryptase-positive mast cells correlate with a poor prognosis in melanoma. European Journal of Clinical Investigation. 2003;33(5):420–425. doi: 10.1046/j.1365-2362.2003.01152.x. [DOI] [PubMed] [Google Scholar]
- 82.Ribatti D, Finato N, Crivellato E, et al. Angiogenesis and mast cells in human breast cancer sentinel lymph nodes with and without micrometastases. Histopathology. 2007;51(6):837–842. doi: 10.1111/j.1365-2559.2007.02869.x. [DOI] [PubMed] [Google Scholar]
- 83.Ribatti D, Belloni AS, Nico B, et al. Tryptase- and leptin-positive mast cells correlate with vascular density in uterine leiomyomas. The American Journal of Obstetrics and Gynecology. 2007;196(5):470.e1–470.e7. doi: 10.1016/j.ajog.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 84.Yodavudh S, Tangjitgamol S, Puangsa-art S. Prognostic significance of microvessel density and mast cell density for the survival of Thai patients with primary colorectal cancer. Journal of the Medical Association of Thailand. 2008;91(5):723–732. [PubMed] [Google Scholar]
- 85.Ribatti D, Molica S, Vacca A, et al. Tryptase-positive mast cells correlate positively with bone marrow angiogenesis in B-cell chronic lymphocytic leukemia. Leukemia. 2003;17(7):1428–1430. doi: 10.1038/sj.leu.2402970. [DOI] [PubMed] [Google Scholar]
- 86.Ribatti D, Vacca A, Nico B, et al. Bone marrow angiogenesis and mast cell density increase simultaneously with progression of human multiple myeloma. British Journal of Cancer. 1999;79(3-4):451–455. doi: 10.1038/sj.bjc.6690070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ribatti D, Polimeno G, Vacca A, et al. Correlation of bone marrow angiogenesis and mast cells with tryptase activity in myelodysplastic syndromes. Leukemia. 2002;16(9):1680–1684. doi: 10.1038/sj.leu.2402586. [DOI] [PubMed] [Google Scholar]
- 88.Nielsen HJ, Hansen U, Christensen IJ, Reimert CM, Brünner N, Moesgaard F. Independent prognostic value of eosinophil and mast cell infiltration in colorectal cancer tissue. The Journal of Pathology. 1999;189(4):487–495. doi: 10.1002/(SICI)1096-9896(199912)189:4<487::AID-PATH484>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 89.Aaltomaa S, Lipponen P, Papinaho S, Kosma V-M. Mast cells in breast cancer. Anticancer Research. 1993;13(3):785–788. [PubMed] [Google Scholar]
- 90.Dabiri S, Huntsman D, Makretsov N, et al. The presence of stromal mast cells identifies a subset of invasive breast cancers with a favorable prognosis. Modern Pathology. 2004;17(6):690–695. doi: 10.1038/modpathol.3800094. [DOI] [PubMed] [Google Scholar]
- 91.Welsh TJ, Green RH, Richardson D, Waller DA, O'Byrne KJ, Bradding P. Macrophage and mast-cell invasion of tumor cell islets confers a marked survival advantage in non-small-cell lung cancer. Journal of Clinical Oncology. 2005;23(35):8959–8967. doi: 10.1200/JCO.2005.01.4910. [DOI] [PubMed] [Google Scholar]
- 92.Erba F, Fiorucci L, Pascarella S, Menegatti E, Ascenzi P, Ascoli F. Selective inhibition of human mast cell tryptase by gabexate mesylate, an antiproteinase drug. Biochemical Pharmacology. 2001;61(3):271–276. doi: 10.1016/s0006-2952(00)00550-5. [DOI] [PubMed] [Google Scholar]
- 93.Mori S, Itoh Y, Shinohata R, Sendo T, Oishi R, Nishibiro M. Nafamostat mesilate is an extremely potent inhibitor of human tryptase. Journal of Pharmacological Sciences. 2003;92(4):420–423. doi: 10.1254/jphs.92.420. [DOI] [PubMed] [Google Scholar]
- 94.Uchima Y, Sawada T, Hirakawa K. Action of antiproteases on pancreatic cancer cells. Journal of the Pancreas. 2007;8(supplement 4):479–487. [PubMed] [Google Scholar]
- 95.Martindale JJ. The Extra Pharmacopeia. 31st edition. London, UK: The Royal Pharmaceutical Society; 1996. [Google Scholar]
- 96.Menegatti E, Bolognesi M, Scalia S, Bortolotti F, Guarneri M, Ascenzi P. Gabexate mesylate inhibition of serine proteases: thermodynamic and computer-graphics analysis. Journal of Pharmaceutical Sciences. 1986;75(12):1171–1174. doi: 10.1002/jps.2600751211. [DOI] [PubMed] [Google Scholar]
- 97.Cortes R, Ascenzi P, Colasanti M, et al. Cross-enzyme inhibition by gabexate mesylate: formulation and reactivity study. Journal of Pharmaceutical Sciences. 1998;87(11):1335–1340. doi: 10.1021/js980079u. [DOI] [PubMed] [Google Scholar]
- 98.Sappino A-P, Busso N, Belin D, Vassalli J-D. Increase of urokinase-type plasminogen activator gene expression in human lung and breast carcinomas. Cancer Research. 1987;47(15):4043–4046. [PubMed] [Google Scholar]
- 99.Gottesman M. The role of proteases in cancer. Seminars in Cancer Biology. 1990;1:97–160. [PubMed] [Google Scholar]
- 100.Liotta LA, Stetler-Stevenson WG. Tumor invasion and metastasis: an imbalance of positive and negative regulation. Cancer Research. 1991;51(supplement 18):5045–5059. [PubMed] [Google Scholar]
- 101.Dahiya R, Yoon W-H, Boyle R, Schoenberg T, Yen T-SB, Narayan P. Biochemical,cytogenetic, and morphological characteristics of human primary and metastatic prostate cancer cell lines. Biochemistry International. 1992;27(4):567–577. [PubMed] [Google Scholar]
- 102.Stetler-Stevenson WG. Progelatinase A activation during tumor cell invasion. Invasion and Metastasis. 1994;95(14):259–268. [PubMed] [Google Scholar]
- 103.Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21(7):1104–1117. doi: 10.1161/hq0701.093685. [DOI] [PubMed] [Google Scholar]
- 104.Naito K, Kanbayashi N, Nakajima S, et al. Inhibition of growth of human tumor cells in nude mice by a metalloproteinase inhibitor. International Journal of Cancer. 1994;58(5):730–735. doi: 10.1002/ijc.2910580518. [DOI] [PubMed] [Google Scholar]
- 105.Watson SA, Morris TM, Robinson G, Crimmin MJ, Brown PD, Hardcastle JD. Inhibition of organ invasion by the matrix metalloproteinase inhibitor batimastat (BB-94) in two human colon carcinoma metastasis models. Cancer Research. 1995;55(16):3629–3633. [PubMed] [Google Scholar]
- 106.Reich R, Thompson EW, Iwamoto Y, et al. Effects of inhibitors of plasminogen activator, serine proteinases, and collagenase IV on the invasion of basement membranes by metastatic cells. Cancer Research. 1988;48(12):3307–3312. [PubMed] [Google Scholar]
- 107.Benelli R, Adatia R, Ensoli B, Stetler-Stevenson WG, Santi L, Albini A. Inhibition of AIDS-Kaposi's sarcoma cell induced endothelial cell invasion by TIMP-2 and a synthetic peptide from the metalloproteinase propeptide: Implications for an anti-angiogenic therapy. Oncology Research. 1994;6(6):251–257. [PubMed] [Google Scholar]
- 108.Anand-Apte B, Pepper MS, Voest E, et al. Inhibition of angiogenesis by tissue inhibitor of metalloproteinase-3. Investigative Ophthalmology and Visual Science. 1997;38(5):817–823. [PubMed] [Google Scholar]
- 109.Hiraoka N, Allen E, Apel IJ, Gyetko MR, Weiss SJ. Matrix metalloproteinases regulate neovascularization by acting as pericellular fibrinolysins. Cell. 1998;95(3):365–377. doi: 10.1016/s0092-8674(00)81768-7. [DOI] [PubMed] [Google Scholar]
- 110.Nguyen M, Arkell J, Jackson CJ. Thrombin rapidly and efficiently activates gelatinase A in human microvascular endothelial cells via a mechanism independent of active MT1 matrix metalloproteinase. Laboratory Investigation. 1999;79(4):467–475. [PubMed] [Google Scholar]
- 111.Ramos-DeSimone N, Hahn-Dantona E, Sipley J, Nagase H, French DL, Quigley JP. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. The Journal of Biological Chemistry. 1999;274(19):13066–13076. doi: 10.1074/jbc.274.19.13066. [DOI] [PubMed] [Google Scholar]
- 112.Sendo T, Sumimura T, Itoh Y, et al. Involvement of proteinase-activated receptor-2 in mast cell tryptase-induced barrier dysfunction in bovine aortic endothelial cells. Cellular Signalling. 2003;15(8):773–781. doi: 10.1016/s0898-6568(03)00014-7. [DOI] [PubMed] [Google Scholar]
- 113.Meininger CJ. Mast cells and tumor-associated angiogenesis. Chemical Immunology. 1995;61(1):239–257. [PubMed] [Google Scholar]
- 114.Brown LF, Detmar M, Claffey K, et al. Vascular permeability factor/vascular endothelial growth factor: a multifunctional angiogenic cytokine. EXS. 1997;79:233–269. doi: 10.1007/978-3-0348-9006-9_10. [DOI] [PubMed] [Google Scholar]
- 115.Friedl A, Chang Z, Tierney A, Rapraeger AC. Differential binding of fibroblast growth factor-2 and -7 to basement membrane heparan sulfate: comparison of normal and abnormal human tissues. The American Journal of Pathology. 1997;150(4):1443–1455. [PMC free article] [PubMed] [Google Scholar]
- 116.Poltorak Z, Cohen T, Sivan R, et al. VEGF145, a secreted vascular endothelial growth factor isoform that binds to extracellular matrix. The Journal of Biological Chemistry. 1997;272(11):7151–7158. doi: 10.1074/jbc.272.11.7151. [DOI] [PubMed] [Google Scholar]
- 117.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. The New England Journal of Medicine. 2008;359(17):1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 118.Normanno N, Tejpar S, Morgillo F, de Luca A, van Cutsem E, Ciardiello F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nature Reviews Clinical Oncology. 2009;6(9):519–527. doi: 10.1038/nrclinonc.2009.111. [DOI] [PubMed] [Google Scholar]
- 119.Leporini C, Saullo F, Filippelli G, et al. Management of dermatologic toxicities associated with monoclonal antibody epidermal growth factor receptor inhibitors: a case review. Journal of Pharmacology & Pharmacotherapeutics. 2013;4(supplement 1):S78–S85. doi: 10.4103/0976-500X.120966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shimamoto R, Sawada T, Uchima Y, et al. A role for protease-activated receptor-2 in pancreatic cancer cell proliferation. International Journal of Oncology. 2004;24(6):1401–1406. [PubMed] [Google Scholar]
- 121.Ellenrieder V, Hendler SF, Ruhland C, Boeck W, Adler G, Gress TM. TGF-β-induced invasiveness of pancreatic cancer cells is mediated by matrix metalloproteinase-2 and the urokinase plasminogen activator system. International Journal of Cancer. 2001;93(2):204–211. doi: 10.1002/ijc.1330. [DOI] [PubMed] [Google Scholar]
- 122.Teraoka H, Sawada T, Yamashita Y, et al. TGF-beta1 promotes liver metastasis of pancreatic cancer by modulating the capacity of cellular invasion. International Journal of Oncology. 2001;19(4):709–715. doi: 10.3892/ijo.19.4.709. [DOI] [PubMed] [Google Scholar]
- 123.Teraoka H, Sawada T, Nishihara T, et al. Enhanced VEGF production and decreased immunogenicity induced by TGF-β 1 promote liver metastasis of pancreatic cancer. British Journal of Cancer. 2001;85(4):612–617. doi: 10.1054/bjoc.2001.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Malamud V, Vaaknin A, Abramsky O, et al. Tryptase activates peripheral blood mononuclear cells causing the synthesis and release of TNF-α, IL-6 and IL-1β: possible relevance to multiple sclerosis. Journal of Neuroimmunology. 2003;138(1-2):115–122. doi: 10.1016/s0165-5728(03)00090-0. [DOI] [PubMed] [Google Scholar]
- 125.Woodman L, Siddiqui S, Cruse G, et al. Mast cells promote airway smooth muscle cell differentiation via autocrine up-regulation of TGF-β1. The Journal of Immunology. 2008;181(7):5001–5007. doi: 10.4049/jimmunol.181.7.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zeng X, Zhang S, Xu L, Yang H, He S. Activation of protease-activated receptor 2-mediated signaling by mast cell tryptase modulates cytokine production in primary cultured astrocytes. Mediators of Inflammation. 2013;2013:10 pages. doi: 10.1155/2013/140812.140812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Aoyama T, Ino Y, Ozeki M, et al. Pharmacological studies of FUT-175, nafamstat mesilate. I. Inhibition of protease activity in in vitro and in vivo experiments. Japanese Journal of Pharmacology. 1984;35(3):203–227. doi: 10.1254/jjp.35.203. [DOI] [PubMed] [Google Scholar]
- 128.Uchiba M, Okajima K, Abe H, Okabe H, Takatsuki K. Effect of nafamostat mesilate, a synthetic protease inhibitor, on tissue factor-factor VIIa complex activity. Thrombosis Research. 1994;74(2):155–161. doi: 10.1016/0049-3848(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 129.Schwartz LB. Mast cell tryptase: properties and roles in human allergic responses. In: Caughey GM, editor. Mast Cell Proteases in Immunology and Biology. New York, NY, USA: Marcel Dekker; 1995. pp. 9–23. [Google Scholar]
- 130.Clark JM, Moore WR, Tanaka RD. Tryptase inhibitors: a new class of antiinflammatory drugs. Drugs Future. 1996;21(8):811–816. [Google Scholar]
- 131.Sommerhoff CP, Bode W, Matschiner G, Bergner A, Fritz H. The human mast cell tryptase tetramer: a fascinating riddle solved by structure. Biochimica et Biophysica Acta. 2000;1477(1-2):75–89. doi: 10.1016/s0167-4838(99)00265-4. [DOI] [PubMed] [Google Scholar]
- 132.Yoshikawa T, Murakami M, Furukawa Y, Kato H, Takemura S, Kondo M. Effects of FUT-175, a new synthetic protease inhibitor on endotoxin-induced disseminated intravascular coagulation in rats. Haemostasis. 1983;13(6):374–378. doi: 10.1159/000214825. [DOI] [PubMed] [Google Scholar]
- 133.Takahashi H, Takizawa S, Tatewaki W, et al. Nafamostat mesilate (FUT-175) in the treatment of patients with disseminated intravascular coagulation. Thrombosis and Haemostasis. 1989;62:p. 372. [Google Scholar]
- 134.Kimura T, Fuchimoto S, Iwagaki H, Hizuta A, Orita K. Inhibitory effect of nafamostat mesilate on metastasis into the livers of mice and on invasion of the extracellular matrix by cancer cells. Journal of International Medical Research. 1992;20(4):343–352. doi: 10.1177/030006059202000405. [DOI] [PubMed] [Google Scholar]
- 135.Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-κB RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clinical Cancer Research. 1999;5(1):119–127. [PubMed] [Google Scholar]
- 136.Fujioka S, Sclabas GM, Schmidt C, et al. Function of nuclear factor kappaB in pancreatic cancer metastasis. Clinical Cancer Research. 2003;9(1):346–354. [PubMed] [Google Scholar]
- 137.Arlt A, Schäfer H. NFκB-dependent chemoresistance in solid tumors. International Journal of Clinical Pharmacology and Therapeutics. 2002;40(8):336–347. doi: 10.5414/cpp40336. [DOI] [PubMed] [Google Scholar]
- 138.Rosette C, Roth RB, Oeth P, et al. Role of ICAM1 in invasion of human breast cancer cells. Carcinogenesis. 2005;26(5):943–950. doi: 10.1093/carcin/bgi070. [DOI] [PubMed] [Google Scholar]
- 139.Huang S, Robinson JB, DeGuzman A, Bucana CD, Fidler IJ. Blockade of nuclear factor-κb signaling inhibits angiogenesis and tumorigenicity of human ovarian cancer cells by suppressing expression of vascular endothelial growth factor and interleukin 8. Cancer Research. 2000;60(19):5334–5339. [PubMed] [Google Scholar]
- 140.Nagakawa Y, Aoki T, Kasuya K, Tsuchida A, Koyanagi Y. Histologic features of venous invasion, expression of vascular endothelial growth factor and matrix metalloproteinase-2 and matrix metalloproteinase-9, and the relation with liver metastasis in pancreatic cancer. Pancreas. 2002;24(2):169–178. doi: 10.1097/00006676-200203000-00008. [DOI] [PubMed] [Google Scholar]
- 141.Wang W, Abbruzzese JL, Evans DB, Chiao PJ. Overexpression of urokinase-type plasminogen activator in pancreatic adenocarcinoma is regulated by constitutively activated RelA. Oncogene. 1999;18(32):4554–4563. doi: 10.1038/sj.onc.1202833. [DOI] [PubMed] [Google Scholar]
- 142.Rakitina TV, Vasilevskaya IA, O'Dwyer PJ. Additive interaction of oxaliplatin and 17-allylamino-17-demethoxygeldanamycin in colon cancer cell lines results from inhibition of nuclear factor κB signaling. Cancer Research. 2003;63(24):8600–8605. [PubMed] [Google Scholar]
- 143.Wilson C, Purcell C, Seaton A, et al. Chemotherapy-induced CXC-chemokine/CXC-chemokine receptor signaling in metastatic prostate cancer cells confers resistance to oxaliplatin through potentiation of nuclear factor-κB transcription and evasion of apoptosis. The Journal of Pharmacology and Experimental Therapeutics. 2008;327(3):746–759. doi: 10.1124/jpet.108.143826. [DOI] [PubMed] [Google Scholar]
- 144.Banerjee S, Kaseb AO, Wang Z, et al. Antitumor activity of gemcitabine and oxaliplatin is augmented by thymoquinone in pancreatic cancer. Cancer Research. 2009;69(13):5575–5583. doi: 10.1158/0008-5472.CAN-08-4235. [DOI] [PubMed] [Google Scholar]
- 145.Sperti C, Pasquali C, Piccoli A, Pedrazzoli S. Recurrence after resection for ductal adenocarcinoma of the pancreas. World Journal of Surgery. 1997;21(2):195–200. doi: 10.1007/s002689900215. [DOI] [PubMed] [Google Scholar]
- 146.Uwagawa T, Li Z, Chang Z, et al. Mechanisms of synthetic serine protease inhibitor (FUT-175)-mediated cell death. Cancer. 2007;109(10):2142–2153. doi: 10.1002/cncr.22658. [DOI] [PubMed] [Google Scholar]
- 147.Furukawa K, Iida T, Shiba H, et al. Anti-tumor effect by inhibition of NF-κB activation using nafamostat mesilate for pancreatic cancer in a mouse model. Oncology Reports. 2010;24(4):843–850. doi: 10.3892/or.2010.843. [DOI] [PubMed] [Google Scholar]
- 148.Uwagawa T, Misawa T, Tsutsui N, et al. Phase II study of gemcitabine in combination with regional arterial infusion of nafamostat mesilate for advanced pancreatic cancer. American Journal of Clinical Oncology: Cancer Clinical Trials. 2013;36(1):44–48. doi: 10.1097/COC.0b013e31823a53b2. [DOI] [PubMed] [Google Scholar]
- 149.Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(20):9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Böhm SK, Kong W, Brömme D, et al. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochemical Journal. 1996;314(3, part 3):1009–1016. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kawabata A, Kuroda R, Nishida M, et al. Protease-activated receptor-2 (PAR-2) in the pancreas and parotid gland: immunolocalization and involvement of nitric oxide in the evoked amylase secretion. Life Sciences. 2002;71(20):2435–2446. doi: 10.1016/s0024-3205(02)02044-1. [DOI] [PubMed] [Google Scholar]
- 152.Shpacovitch VM, Brzoska T, Buddenkotte J, et al. Agonists of proteinase-activated receptor 2 induce cytokine release and activation of nuclear transcription factor κB in human dermal microvascular endothelial cells. Journal of Investigative Dermatology. 2002;118(2):380–385. doi: 10.1046/j.0022-202x.2001.01658.x. [DOI] [PubMed] [Google Scholar]
- 153.Zhang S, Zeng X, Yang H, Hu G, He S. Mast cell tryptase induces microglia activation via protease-activated receptor 2 signaling. Cellular Physiology and Biochemistry. 2012;29(5-6):931–940. doi: 10.1159/000171029. [DOI] [PubMed] [Google Scholar]
- 154.Ma Y, Zhang B, Qian R, Lu C, Zhao F, Yin L. Tryptase activates PKB in inflammatory reaction in ECV304 cells. Biochimica et Biophysica Acta—Molecular Cell Research. 2006;1763(3):313–321. doi: 10.1016/j.bbamcr.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 155.Chen WS, Xu P-Z, Gottlob K, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the akt1 gene. Genes & Development. 2001;15(17):2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cheung M, Testa JR. Diverse mechanisms of AKT pathway activation in human malignancy. Current Cancer Drug Targets. 2013;13(3):234–244. doi: 10.2174/1568009611313030002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kobayashi S, Asano T, Ochiai T. A proposal of no-touch isolation technique in pancreatoduodenectomy for periampullary carcinomas. Hepato-Gastroenterology. 2001;48(38):372–374. [PubMed] [Google Scholar]
- 158.Azuma H, Banno K, Yoshimura T. Pharmacological properties of N-(3′,4′ dimethoxycinnamoyl) anthranilic acid (N-5′), a new anti atopic agent. British Journal of Pharmacology. 1976;58(4):483–488. doi: 10.1111/j.1476-5381.1976.tb08614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Komatsu H, Kojima M, Tsutsumi N, et al. Study of the mechanism of inhibitory action of tranilast on chemical mediator release. Japanese Journal of Pharmacology. 1988;46(1):43–51. doi: 10.1254/jjp.46.43. [DOI] [PubMed] [Google Scholar]
- 160.Isaji M, Nakajoh M, Naito J. Selective inhibition of collagen accumulation by N-(3,4-dimethoxycinnamoyl)anthranilic acid (N-5') in granulation tissue. Biochemical Pharmacology. 1987;36(4):469–474. doi: 10.1016/0006-2952(87)90353-4. [DOI] [PubMed] [Google Scholar]
- 161.Yanagi T, Watanabe M, Fukuda S, Tsuji Y. Suppressive effects of tranilast (TN) on human mononuclear cells. Japanese Journal of Inflammation. 1987;7:169–173. [Google Scholar]
- 162.Suzawa H, Kikuchi S, Arai N, Koda A. The mechanism involved in the inhibitory action of tranilast on collagen biosynthesis of keloid fibroblasts. Japanese Journal of Pharmacology. 1992;60(2):91–96. doi: 10.1254/jjp.60.91. [DOI] [PubMed] [Google Scholar]
- 163.Yashiro M, Chung Y-S, Sowa M. Tranilast (N-(3,4-dimethoxycinnamoyl) anthranilic acid) down-regulates the growth of scirrhous gastric cancer. Anticancer Research A. 1997;17(2):895–900. [PubMed] [Google Scholar]
- 164.Shime H, Kariya M, Orii A, et al. Tranilast inhibits the proliferation of uterine leiomyoma cells in vitro through G1 arrest associated with the induction of p21waf1 and p53. Journal of Clinical Endocrinology and Metabolism. 2002;87(12):5610–5617. doi: 10.1210/jc.2002-020444. [DOI] [PubMed] [Google Scholar]
- 165.Rogosnitzky M, Danks R, Kardash E. Therapeutic potential of tranilast, an anti-allergy drug, in proliferative disorders. Anticancer Research. 2012;32(7):2471–2478. [PubMed] [Google Scholar]
- 166.Padua D, Massagué J. Roles of TGFbeta in metastasis. Cell Research. 2009;19(1):89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 167.Izumi K, Mizokami A, Shima T, et al. Preliminary results of tranilast treatment for patients with advanced castration-resistant prostate cancer. Anticancer Research. 2010;30(7):3077–3081. [PubMed] [Google Scholar]
- 168.Shigeki S, Murakami T, Yata N, Ikuta Y. Treatment of keloid and hypertrophic scars by iontophoretic transdermal delivery of tranilast. Scandinavian Journal of Plastic and Reconstructive Surgery and Hand Surgery. 1997;31(2):151–158. doi: 10.3109/02844319709085482. [DOI] [PubMed] [Google Scholar]
- 169.Isaji M, Miyata H, Ajisawa Y. Tranilast: a new application in the cardiovascular field as an antiproliferative drug. Cardiovascular Drug Reviews. 1998;16(3):288–299. [Google Scholar]
- 170.Kerkvliet NI. AHR-mediated immunomodulation: the role of altered gene transcription. Biochemical Pharmacology. 2009;77(4):746–760. doi: 10.1016/j.bcp.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gomez-Duran A, Carvajal-Gonzalez JM, Mulero-Navarro S, Santiago-Josefat B, Puga A, Fernandez-Salguero PM. Fitting a xenobiotic receptor into cell homeostasis: How the dioxin receptor interacts with TGFβ signaling. Biochemical Pharmacology. 2009;77(4):700–712. doi: 10.1016/j.bcp.2008.08.032. [DOI] [PubMed] [Google Scholar]
- 172.Ruby CE, Leid M, Kerkvliet NI. 2,3,7,8-Tetrachlorodibenzo-p-dioxin suppresses tumor necrosis factor-α and anti-CD40-induced activation of NF-κB/Rel in dendritic cells: p50 homodimer activation is not affected. Molecular Pharmacology. 2002;62(3):722–728. doi: 10.1124/mol.62.3.722. [DOI] [PubMed] [Google Scholar]
- 173.Ammendola M, Sacco R, Sammarco G, et al. Correlation between serum tryptase, mast cells positive to tryptase and microvascular density in colo-rectal cancer patients: possible biological-clinical significance. Plos ONE. 2014;9(6) doi: 10.1371/journal.pone.0099512.e99512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Marech I, Ammendola M, Sacco R, et al. Serum tryptase, mast cells positive to tryptase and microvascular density evaluation in early breast cancer patients: possible translational significance. BMC Cancer. 2014;14(article 534) doi: 10.1186/1471-2407-14-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nanba K, Oura T, Soeda S, Sioya N, Tsukada S, Hanaoka K. Clinical evaluation of tranilast for keloid and hypertrophic scarring. Optimal dose finding study in a double blind study. Nessho. 1992;18(30–45) [Google Scholar]
- 176.Ashman LK, Griffith R. Therapeutic targeting of c-KIT in cancer. Expert Opinion on Investigational Drugs. 2013;22(1):103–115. doi: 10.1517/13543784.2013.740010. [DOI] [PubMed] [Google Scholar]
- 177.Ranieri G, Mammì M, Donato Di Paola E, et al. Pazopanib a tyrosine kinase inhibitor with strong anti-angiogenetic activity: a new treatment for metastatic soft tissue sarcoma. Critical Review in Oncology/Hematology. 2014;89(2):322–329. doi: 10.1016/j.critrevonc.2013.08.012. [DOI] [PubMed] [Google Scholar]
- 178.Kessler ER, Bowles DW, Flaig TW, Lam ET, Jimeno A. Axitinib, a new therapeutic option in renal cell carcinoma. Drugs of Today. 2012;48(10):633–644. doi: 10.1358/dot.2012.48.10.1860768. [DOI] [PubMed] [Google Scholar]
- 179.Marech I, Patruno R, Zizzo N, et al. Masitinib (AB1010), from canine tumor model to human clinical development: where we are? Critical Reviews in Oncology/Hematology. 2014;91(1):98–111. doi: 10.1016/j.critrevonc.2013.12.011. [DOI] [PubMed] [Google Scholar]