

Abstract
Haploinsufficiency for Nipbl, a cohesin loading protein, causes Cornelia de Lange Syndrome (CdLS), the most common “cohesinopathy”. It has been proposed that the effects of Nipbl-haploinsufficiency result from disruption of long-range communication between DNA elements. Here we use zebrafish and mouse models of CdLS to examine how transcriptional changes caused by Nipbl deficiency give rise to limb defects, a common condition in individuals with CdLS. In the zebrafish pectoral fin (forelimb), knockdown of Nipbl expression led to size reductions and patterning defects that were preceded by dysregulated expression of key early limb development genes, including fgfs, shha, hand2 and multiple hox genes. In limb buds of Nipbl-haploinsufficient mice, transcriptome analysis revealed many similar gene expression changes, as well as altered expression of additional classes of genes that play roles in limb development. In both species, the pattern of dysregulation of hox-gene expression depended on genomic location within the Hox clusters. In view of studies suggesting that Nipbl colocalizes with the mediator complex, which facilitates enhancer-promoter communication, we also examined zebrafish deficient for the Med12 Mediator subunit, and found they resembled Nipbl-deficient fish in both morphology and gene expression. Moreover, combined partial reduction of both Nipbl and Med12 had a strongly synergistic effect, consistent with both molecules acting in a common pathway. In addition, three-dimensional fluorescent in situ hybridization revealed that Nipbl and Med12 are required to bring regions containing long-range enhancers into close proximity with the zebrafish hoxda cluster. These data demonstrate a crucial role for Nipbl in limb development, and support the view that its actions on multiple gene pathways result from its influence, together with Mediator, on regulation of long-range chromosomal interactions.
Author Summary
Limb malformations are a striking feature of Cornelia de Lange Syndrome (CdLS), a multi-system birth defects disorder most commonly caused by haploinsufficiency for NIPBL. In addition to its role as a cohesin-loading factor, Nipbl also regulates gene expression, but how partial Nipbl deficiency causes limb defects is unknown. Using zebrafish and mouse models, we show that expression of multiple key regulators of early limb development, including shha, hand2 and hox genes, are sensitive to Nipbl deficiency. Furthermore, we find morphological and gene expression abnormalities similar to those of Nipbl-deficient zebrafish in the limb buds of zebrafish deficient for the Med12 subunit of Mediator—a protein complex that mediates physical interactions between enhancers and promoters—and genetic interaction studies support the view that Mediator and Nipbl act together. Strikingly, depletion of either Nipbl or Med12 leads to characteristic changes in hox gene expression that reflect the locations of genes within their chromosomal clusters, as well as to disruption of large-scale chromosome organization around the hoxda cluster, consistent with impairment of long-range enhancer-promoter interaction. Together, these findings provide insights into both the etiology of limb defects in CdLS, and the mechanisms by which Nipbl and Mediator influence gene expression.
Introduction
Cohesin, a ring-shaped, DNA-associated protein complex, is best known for its role in tethering sister chromatids together until mitosis [1], [2]. However, growing evidence indicates that cohesin, and proteins such as Nipped-B-like (Nipbl) that regulate cohesin loading onto DNA, also play critical roles in gene regulation [3]–[13]. In particular, it has been suggested that Nipbl and cohesin mediate interactions between promoters and distant enhancers, a process thought to involve the physical looping out of intervening DNA sequences [14]–[16]. For example, in Drosophila, Nipped-B (the orthologue of Nipbl) and cohesin regulate cut gene expression by controlling long-range interactions between the cut promoter and a wing-specific remote enhancer [5]. In mice, haploinsufficiency for Nipbl impairs looping that controls the selective expression of beta-globin isoforms by erythroid cells [13].
Recently, it was found that Nipbl co-localizes with the Mediator complex at promoters/enhancers of actively transcribed genes in mouse embryonic stem cells [17]. Thought to play a pivotal role in transmitting regulatory signals from gene-specific activators/repressors to RNA polymerase II [18], [19], Mediator is a large complex composed of a core that interacts with RNA polymerase II and gene-specific transcriptional regulators, and a Cdk8 submodule (containing Cdk8, CyclinC, Med12 and Med13) and can either negatively [20]–[22] or positively [23], [24] regulate transcription. The reported physical interaction between Mediator and Nipbl at active genes suggests that they function together in promoter-enhancer communication, but exactly how this occurs is unknown.
Much insight into the physiological significance of cohesin's influence on transcription has come from the study of Cornelia de Lange Syndrome (CdLS) and other “cohesinopathies”. CdLS is a congenital syndrome characterized by growth retardation, neurological dysfunction, and structural defects in multiple organs [25]–[30], and is caused, in most cases, by haploinsufficiency for NIPBL [31], [32]. More recently it has been shown that mutations in cohesin subunits SMC1A or SMC3 [33], [34] or the SMC3 deacetylase, HDAC8 [35], are less common causes of CdLS. Analysis of both patient samples and animal models indicate that Nipbl haploinsufficiency causes small changes (usually less than 1.5-fold) in the expression of many hundreds of genes [3], [4], [11]. Analysis of both mouse and fish models of Nipbl deficiency suggests that pervasive phenotypic abnormalities result from the collective, and sometimes synergistic, effects of such small changes in gene expression [3], [11].
Among the most striking abnormalities in CdLS are limb defects, which range from mild brachydactyly and clinodactyly to severe digit and limb truncations, the latter in about 1/3 of cases [26], [28], [36]. Limb reduction is one of the few structural defects in CdLS that is not replicated in the Nipbl-haploinsufficient mouse model, as these mice exhibit only minor changes in the shape of the olecranon process, and delays in the ossification of limb bones [3]. Hypothesizing that this difference might reflect slight differences in the threshold for triggering such defects in mouse versus man, we decided to look at development of the pectoral fin (the homologue of the mammalian forelimb) in a zebrafish model of Nipbl-deficiency, produced by injection of morpholino oligonucleotides (MO) directed against the two zebrafish nipbls [11]. Here we show that Nipbl-deficient fish display a marked reduction in pectoral fin size, which is already apparent early in fin bud development. We demonstrate that Nipbl is required for normal expression of conserved regulators of vertebrate limb growth and patterning, including fgfs in the apical ectodermal ridge (AER), shh in the zone of polarizing activity (ZPA), and several hox genes of the hoxab, hoxca and hoxda clusters. We also show that that Nipbl-haploinsufficient mouse limb buds display a pattern of gene expression changes strikingly similar to those observed in Nipbl-deficient pectoral fin buds.
Pectoral fin defects have also been reported in med12-mutant zebrafish, in which Mediator function is disrupted [37]. Interestingly, we find that both the morphological and gene expression changes that occur in Nipbl-deficient fin buds are mimicked when med12 is knocked down. In particular, expression of multiple hox genes in different clusters is affected in a similar position-specific manner in both Nipbl- and Med12-deficient fish embryos, and results of experiments in which we simultaneously knock down both Nipbl and Med12 suggest that they interact genetically. Using 3-dimensional fluorescent in situ hybridization (3D-FISH) in zebrafish fin buds, we further show that Nipbls and Med12 are required for higher-order chromatin organization near the hoxda cluster. Overall, the data point to a shared, conserved role for Nipbl and the Mediator complex in the regulation of long-range enhancer-promoter interactions underlying growth and patterning of the vertebrate limb.
Results
Impaired pectoral fin development in Nipbl-deficient zebrafish
Both nipbl genes in zebrafish, nipbla and nipblb [11], are expressed in developing pectoral fin bud mesenchyme (Fig. S1A, B). To investigate their requirements in forelimb development, we generated “Nipbl-deficient” embryos in which both nipbla and nipblb were depleted by injecting either of two different sets of antisense morpholinos (MOs) designed against distinct MO target sites, as described previously [11]. Pectoral fins of Nipbl-deficient larvae were 40% shorter in length at 72 hours post fertilization (hpf) than those of control embryos (Figure 1A, B, E). This reduced size was not simply due to developmental delay, since it was much more severe than expected given the delay in development of the eye and lower jaw, which we estimated at approximately 16 hrs (Figure S2, see below). Pectoral fin defects were more severe when MOs to both nipbl mRNAs were injected, compared with either one alone (Figure S3) [11]; thus, injections of both MOs were used in subsequent experiments. Pectoral fin defects were partially rescued by co-injection of exogenous nipbla mRNA, confirming MO specificity (Figure 1C–E).
Figure 1. Nipbl knockdown disrupts pectoral fin development.

(A–D) Reduced pectoral fins in live Nipbl-deficient embryos at 76 hpf. Dorsal views, anterior to the left. Uninjected control (A), Nipbl-deficient (nipbla/b-MO) (B), injected with 400 pg of nipbla mRNA alone (C) and co-injected with nipbla/b-MO+nipbla mRNA (D). (E) Whisker plots of fin length at 76 hpf; medians: 431.8 µm, n = 50 (control), 258.5 µm, n = 88 (nipbla/b-MOs), 423.0 µm, n = 22 (nipbla mRNA alone), and 319.5 µm, n = 70 (nipbla/b-MOs+nipbla mRNA). p-values are indicated in the graph. (F, F′, G, G′) Alcian blue staining of cartilages in pectoral fins in control (F, F′) and Nipbl-deficient embryos (G, G′) at 120 hpf. F′ and G′ are higher magnification pictures of boxed areas of endoskeletal discs in F and G, respectively. ac, actinotrichs; cl, cleithrum; ed, endoskeletal disc; sco, scapulocoracoid. (H) Numbers of endoskeletal cells in pectoral fins along proximodistal (PD) and anteroposterior (AP) axes (control; n = 13, Nipbl-deficient embryos; n = 16). PD (Ave ± S.D.): 33.2±1.5 (control) and 22.4±4.2 (Nipbl-deficient embryos), p<10−8. AP (Ave ± S.D.): 27.9±1.5 (control) and 17.7±2.2 (Nipbl-deficient embryos), p<10−13. (I-X) Morphology of developing pectoral fin buds in live embryos. Lateral views, anterior and dorsal to the left and top, respectively.
Alcian Blue staining at 5 dpf revealed that pectoral fin cartilages of the endoskeletal discs form in Nipbl-deficient larvae but are smaller (Figure 1F, G). Cell numbers in these discs were reduced by 37% and 33% along anterior-posterior (A-P) and proximo-distal (P-D) axes, respectively (Figure 1H), whereas cell size resembled controls (Figure 1F′, G′) and we found no change in cell death (Figure S4A, B), suggesting impaired growth of cartilage progenitors at earlier stages. In addition, the orderly arrangement and spacing of chondrocytes in the endoskeletal disc cells was noticeably disrupted in Nipbl-deficient fins (Figure 1F′, G′).
In zebrafish embryos, pectoral fin buds first appear at 30 hpf as shallow domes along the A-P axis, and grow and begin to fold posteriorly by 42 hpf. In Nipbl-deficient embryos, pectoral fin buds also initiate at 30 hpf but grow more slowly than controls (Figure 1I–R). TUNEL assays (Figure S4C–E) showed no increase in cell death in Nipbl-deficient fin buds (Figure S4A–E). In contrast, numbers of BrdU+ cells decreased significantly in the mesenchyme of Nipbl-deficient pectoral fin buds (Figure S4F–I). These data suggest that endoskeletal disc size reduction in Nipbl-deficient limb buds reflects cumulative effects of slower rates of cell division.
Since Nipbl is required for embryonic growth in both fish and mice [3], [11], we stage-matched embryos using an independent criterion – i.e. the A-P position of the migrating posterior lateral line (pLL) primordium labeled by in situ hybridization (ISH) for fgf10a; Figure S5A, red arrows). In controls, pLL primordia lie just posterior to the pectoral fin buds at 22 hpf, and continue to migrate posteriorly. Based on this staging criterion the developmental delay in Nipbl-deficient embryos (summarized in Figure S5B) cannot account for the severe limb reductions in Nipbl-deficient larvae (Figure 1S–X).
Nipbls are required for fgf expression in the AER but not in fin bud mesenchyme
Early limb development is highly conserved from fish to mammals [38]–[40]. Each fin/limb bud possesses an apical ectodermal ridge (AER) and zone of polarizing activity (ZPA) [40], [41], which play important roles in growth and patterning [42]–[44]. The AER, a thickened epithelium that rims the distal ends of the buds, is the source of Fgf signals required for P-D limb outgrowth [45]–[48]. The zebrafish AER expresses 4 fgf genes: fgf4, fgf8a, fgf16 and fgf24 [49], [50]. Of these, expression of fgf4, fgf8a and fgf16 was dramatically reduced in pectoral fin buds of Nipbl-deficient embryos (Figure 2A–C), which was rescued by over-expression of full-length nipbla mRNA (Figure S6). This was not simply due to loss of AER cells since fgf24 expression was not downregulated in the Nipbl-deficient AER (Figure 2D).
Figure 2. Reduced expression of fgfs in the AER of Nipbl-deficient embryos.

Expression of fgf4 (A), fgf8a (B), fgf16 (C) and fgf24 (D) in the AER (arrows) at indicated stages in control and Nipbl-deficient embryos examined by ISH. Dorsal views, anterior to the top.
Limb bud development also requires expression of fgf10a and fgf24 in the mesenchyme [49], [51], [52]. However, we found no differences in fgf10a expression between wild type and Nipbl-deficient limb buds between 22–48 hpf (Figure S7A), as well as no differences in expression of tbx5a and fgf24, which control the expression of fgf10a (Figure S7B–C).
Nipbls are required for shh expression in the ZPA and its regulation in fin bud mesenchyme
The ZPA acts as an organizing center in the posterior limb/fin bud mesenchyme in part because it produces Shh [38], [40], [45], [50], [53]–[55]. Shh is required for limb A-P polarity, outgrowth and Fgf expression in the AER [55]. Zebrafish Shh (shha) and its receptor (and transcriptional target) ptch2 are first expressed in the ZPA at 24 hpf and expression progressively increases until 36 hpf (Figure 3A–B) [56]. In Nipbl-deficient limb buds, shha and ptch2 expression was reduced at these stages (Figure 3A, B). shha and ptch2 expression levels were also reduced in the intestine (where Nipbl is also required for development [11]; Figure 3A, B, asterisks), but unaffected in the notochord and neural tube (Figure 3A, B and unpublished data), suggesting a tissue-specific requirement for Nipbl in the expression of Shh and its receptor.
Figure 3. Reduction of genes involved in the shh-related gene regulatory cassette in developing pectoral fin mesenchyme of Nipbl-deficient embryos.

Expression of fin mesenchymal genes at indicated stages in control and Nipbl-deficient embryos examined by ISH. Dorsal views, anterior to the top. (A, B) Expression of shha and ptch2 in pectoral fin buds (arrows) was significantly reduced in Nipbl-deficient embryos, while midline, neural tube expression was unaffected. Expression in endoderm derived-tissues (asterisks) is also reduced. (C) Expression of hand2 was also significantly reduced in stage-matched pectoral fin buds. hand2 expression in the fin buds and posterior lateral plate mesoderm is marked by brackets and arrowheads, respectively. (D) gli3 expression was not significantly affected in a stage-matched comparison.
Hand2 regulates Shh expression in fin/limb buds [56]–[58], and we found that hand2 expression was also reduced in Nipbl-deficient fin buds (36 and 40 hpf) compared with stage-matched controls (32 and 36 hpf) (Figure 3C). In mouse limb buds, anterior expression of the transcriptional repressor, Gli3, restricts expression of Hand2 posteriorly [59]. Zebrafish pectoral fin buds also express gli3 [60] but its expression was not affected by reduction of Nipbl (Figure 3D).
Mammalian Hand2 acts together with the products of 5′-Hoxd genes [58] in the regulation of Shh expression. In pectoral fin buds of Nipbl-deficient embryos, we found that 5′-hoxd genes, including hoxd9a-d13a (Figure 4A), were significantly downregulated (Figure 4B). Importantly, fin bud expression of hand2, hoxd10a, shha and ptch2 could all be partially rescued by exogenous nipbla mRNA (Figure S8).
Figure 4. Nipbls are required for spatial patterns of hoxd expression in pectoral fin buds.

(A) Diagram of zebrafish hoxda cluster. (B, C) Expression of 3′-hoxd genes including hoxd3a and d4a (B) and 5′-hoxd genes including hoxd9a-d13a (C) was examined by ISH at 32 and 36 hpf to show both time-matched and stage-matched (nipbla/b-MO at 36 hpf and control at 32 hpf) comparisons. Dorsal views, anterior to the top.
Retinoic acid (RA) produced in anterior somites also regulates shha expression in pectoral fin buds (12–22 hpf [42]–[44], [61]), as well as fin bud expression of fgf10a. However, we found no differences in expression of either the RA synthesizing enzyme aldh1a2 or the RA degradation enzyme and target gene, cyp26a1, at 13 and 19 hpf in Nipbl-deficient embryos (Figure S9).
Together, these findings indicate that Nipbls regulate the 5′-hoxd/hand2/shha signaling cascade, but do not affect the tbx5a/fgf24/fgf10a pathway that lies downstream of RA signaling, during vertebrate limb development.
Nipbls regulate expression of hox genes according to their genomic location
Hox genes belong to 13 paralog groups organized in four (mammals) or seven (zebrafish) clusters; the HoxA and D clusters are crucial for limb/fin development [56], [62], [63]. The most 3′-located genes (3′-Hox), such as Hoxd1, are expressed earliest in mouse limb buds, whereas expression of 5′-located genes (5′-Hox, d10-d13) begins later [64], [65]. 5′-Hoxd gene expression occurs first in proximal limb buds, where it is required for Shh expression in the ZPA to establish A-P patterning [55], [66], and is later restricted distally in limb buds, where it is required for proper digit formation [64], [65]. Expression of hoxd genes in zebrafish fin buds is reminiscent of that in proximal mouse limb buds but appears to lack the second wave of distal expression, consistent with the lack of digits in ray-finned fish [64].
Examination of expression of multiple hox genes from the Hoxa (hoxab), Hoxc (hoxca), and Hoxd (hoxda) clusters in the fin buds of Nipbl-deficient embryos revealed that changes in expression correlated strongly with positions of genes within clusters (Figures 4–5). Expression of five hoxd genes located at the 5′ ends of the hoxda cluster (hoxd9a-d13a) was severely reduced (Figure 4B), while expression of two hoxd genes located more 3′ in the cluster, hoxd3a and hoxd4a, expanded to encompass the entire bud (Figure 4C). Similarly, expression of 5′-genes in the hoxab cluster—such as hoxa9b, a10b, and a13b—was significantly reduced in Nipbl-deficient fin buds, while a 3′ gene, hoxa2b, was upregulated (Figure 5A, B). Likewise, expression of hoxc8a and hoxc9a was reduced in Nipbl-deficient fin buds while expression of hoxc1a, hoxc4a, and hoxc6a expanded posteriorly (Figure 5C, D). Thus, in all three hox clusters expressed in the pectoral fin buds, expression of genes near the 3′ end of the cluster expands, whereas expression of those closer to the 5′ end is reduced (Figure 5E). Interestingly, this position-specific regulation of hox gene expression is specific to pectoral fin buds, since hox expression patterns in the neural tube were unaffected in Nipbl-deficient embryos (Figure S10).
Figure 5. Nipbls regulate hox gene expression according to genomic location.

(A–D) Expression of genes in hoxab (A,B) and hoxca (C,D) clusters was examined by ISH. (A) 5′-hoxa, (B) 3′-hoxa, (C) 5′-hoxc, and (D) 3′-hoxc genes. Dorsal views, anterior to the top. (E) Diagram summarizing effects of Nipbl reduction on hox genes. Genes located closer to 5′-ends show reduced expression (red boxes) whereas those closer to 3′-ends become expressed across entire fin buds (green boxes).
Shh signaling from the ZPA regulates expression of several hox genes along the A-P axis of limb buds, and reduced expression of 5′-hoxa/hoxd genes as well as posterior expansion of hoxc6a expression, similar to that described above, occurs in Shh-deficient zebrafish [55]. To test if the Shh reductions resulting from Nipbl deficiency might cause the defects in hox gene expression, we treated wild-type embryos with the Shh signaling inhibitor, cyclopamine (CyA). Although CyA treatment caused some developmental delay, (∼4–5 hr, based on the A-P positions of pLL primordia [compare Fig. S11A with Fig. S5A], and no more than 12 hr based on pectoral fin development), it strongly reduced expression of ptch2 as well as hoxa13b, hoxd10a and hoxd13a, while expression of hoxc4a and hoxc6a expanded posteriorly (compared with stage-matched controls, Figure S11B). These effects of CyA treatment resembled those of Nipbl depletion, but others did not - e.g. hoxd4a expression was severely reduced, and hoxc8a expression expanded posteriorly in CyA-treated embryos (Figure S11B), in contrast to Nipbl-deficient embryos (Fig. 4C, 5C). Thus, loss of Shh signaling cannot explain all of the changes in hox gene expression in Nipbl-deficient embryos, suggesting that either Nipbls regulate the expression of hox genes directly, or they do so via regulators other than (or in addition to) Shh.
Gene expression changes in limb buds of Nipbl-haploinsufficient mice mirror those in Nipbl-deficient fish
Nipbl +/− mutant mice fail to display obvious limb reductions, but do show some limb patterning and bone calcification defects [3]. Given the gene expression changes we found in pectoral fin buds of Nipbl-deficient fish, we decided to investigate if Nipbl-deficient mouse limb buds show some of the same changes. ISH for Shh in E10.5 limb buds of Nipbl +/− mice revealed a marked reduction in Shh expression in the ZPA, similar to Nipbl-deficient fin buds (compare Figure 3A and Figure 6). This was confirmed by both Q-RT-PCR and expression microarray analysis, using RNA extracted from E10.5 limb buds harvested from stage-matched Nipbl+/− (n = 12) and wildtype (n = 12) littermate embryos (Table 1; also see Methods). Microarray analysis identified approximately 1000 genes as significantly over- or under-expressed in Nipbl +/− limb buds (Table 1 and data publically deposited) and, similar to tissues and cells of Nipbl +/− mice and individuals with CdLS, most gene expression changes were typically less than 1.5-fold [3], [4]. Nonetheless, statistically-significant changes in expression (mostly decreases) were observed for multiple genes in the Fgf, Bmp and Shh pathways, as well as numerous genes in the Wnt/planar cell polarity signaling pathway. In addition, multiple genes at the 5′ and 3′ ends of the Protocadherin B cluster were downregulated (not shown), while Stag1 (which encodes a cohesin subunit) was upregulated; both of these changes are hallmarks of Nipbl deficiency in other tissues [3].
Figure 6. Reduced ZPA expression of Shh in Nipbl +/− mouse limb buds.
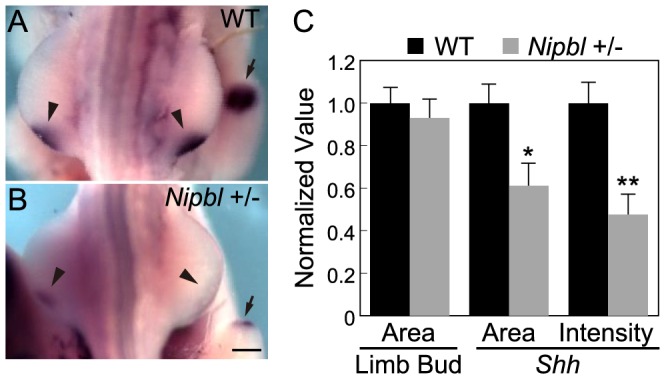
(A–B) Whole mount ISH for Shh in the hindlimb buds of E10.5 wild type (A) and Nipbl +/− (B) mice. In these dorsal views, anterior to the top, the left and right ZPA are seen as localized patches of staining on the posteriolateral edge of each bud (arrowheads). The ZPA of the right forelimb bud is also visible in the background (arrows). Scale bar = 0.5 mm. (C) Quantification of ISH patterns from 5 wild type and 5 mutant embryos. Limb bud and ZPA size were estimated from image areas. Hybridization intensity was measured as mean pixel intensity in the ZPA multiplied by ZPA area. Data are normalized to wild type values. * = p<0.05, ** = p<0.01.
Table 1. Gene expression changes in Nipbl+/− mouse limb buds.
| Expression by microarray | Expression by Q-RT-PCR | Expression by microarray | Expression by Q-RT-PCR | ||||||
| Gene | FDR | mut/wt | mut/wt | Notes | Gene | FDR | mut/wt | mut/wt | Notes |
| Shh pathway | |||||||||
| Shh | 1.0% | 0.82 | 0.48 (p<0.01) | Hmgb2 | 8.6% | 0.71 | i | ||
| Gli3 | 9.5% | 1.09 | Pbx2 | 18.3% | 1.03 | ii | |||
| Hhip | 2.4% | 0.76 | Disp1 | 1.0% | 1.08 | ||||
| Hand2 | 2.4% | 0.92 | 0.85 (p = 0.09) | iii , iv | Ptch1 | 1.0% | 0.90 | 0.82 (p = 0.03) | |
| FGF signaling | |||||||||
| Fgf15 | 20.2% | 0.91 | Ets1 | 3.0% | 0.93 | v | |||
| Fgf18 | 1.8% | 0.83 | vi | Ets2 | 1.0% | 0.92 | |||
| Fgf9 | 18.8% | 0.88 | vii | Spred2 | 1.0% | 1.16 | |||
| Spry2 | 4.1% | 0.89 | Gpc1 | 2.4% | 0.92 | ||||
| Hox Gene Expression and Function | |||||||||
| Hoxa13 | 22.1% | 0.73 | 0.46 (p = 0.03) | Hoxd12 | 1.0% | 0.74 | 0.53 (p<0.01) | ||
| Hoxc9 | 7.5% | 0.89 | Hoxd13 | 1.0% | 0.65 | 0.51 (p<0.01) | |||
| Hoxc13 | 2.4% | 0.85 | 0.57 (p<0.01) | Enpp2 | 1.0% | 0.75 | viii | ||
| Hoxd10 | 1.0% | 1.06 | 1.27 (p<0.01) | Epha7 | 1.0% | 0.82 | ix | ||
| Hoxd11 | 23.6% | 0.93 | 0.75 (p = 0.04) | Pbx1 | 1.0% | 1.12 | x | ||
| BMP signaling | |||||||||
| Bmp2 | 1.0% | 0.79 | iv | Bambi | 1.0% | 0.89 | |||
| Bmp4 | 1.0% | 0.90 | iv | Rgmb | 3.5% | 0.90 | |||
| Bmp7 | 6.4% | 1.10 | Msx2 | 9.9% | 1.10 | ||||
| Bmpr1a | 1.0% | 1.10 | |||||||
| Cohesin Function | |||||||||
| Nipbl | 1.0% | 0.63 | 0.62 (p<0.01) | Esco1 | 22.3% | 1.13 | |||
| Stag1 | 1.0% | 1.16 | |||||||
| Mediator Complex | |||||||||
| Med12l | 1.8% | 1.11 | Med19 | 1.0% | 1.09 | ||||
| Med14 | 4.1% | 1.05 | |||||||
| Wnt/Planar Cell Polarity Pathway | |||||||||
| Wnt11 | 1.0% | 0.87 | xi | Csnk2a1 | 1.0% | 1.09 | |||
| Wnt8a | 9.9% | 1.12 | xii | Csnk2a2 | 7.5% | 1.05 | |||
| Fzd2 | 2.4% | 0.92 | Cthrc1 | 1.0% | 0.72 | xiii | |||
| Fzd8 | 1.0% | 0.86 | Gpc4 | 1.0% | 1.14 | ||||
| Dkk2 | 1.0% | 0.82 | Gpc6 | 1.0% | 1.16 | ||||
| Rspo2 | 1.8% | 0.84 | xiv | Daam1 | 8.6% | 0.91 | |||
| Rspo3 | 1.0% | 0.74 | Daam2 | 1.0% | 0.75 | ||||
| Sfrp1 | 4.6% | 0.81 | iv | Nlk | 1.0% | 1.15 | |||
| Sulf1 | 1.0% | 0.81 | Ppap2b | 1.0% | 0.85 | ||||
| Fat1 | 1.0% | 0.89 | Prickle1 | 1.0% | 0.91 | ||||
| Fat3 | 1.0% | 0.78 | Ror1 | 2.4% | 0.88 | ||||
| Fat4 | 1.0% | 0.65 | |||||||
Relative gene expression levels in limb buds of stage-matched, E10.5 wildtype and Nipbl+/− mice were determined as described (see Materials and Methods), and in certain cases confirmed by Q-RT-PCR. Selected transcripts are shown.
Involved in control of Shh expression in limb bud [101].
Also controls Hox gene expression in the limb bud [102].
Also controls Hox gene expression in the limb bud [69].
Direct target of Hoxd13 in limb buds [103].
Directs the position of the Shh expression boundary delineating the experimentally defined ZPA [104].
Mesenchymal, involved in chondrocyte proliferation [105].
AER-Fgf [106].
Strongly activated by HOXA13 [67].
HOXA13 target in limb buds [68].
Functions as a HOX cofactor during development; complexes with HOXA9; also controls Hox and Shh expression [102].
Non-canonical Wnt [107].
Canonical Wnt [108].
Interacts with some Wnts and Frizzleds and supports Wnt-Fz-Ror2 complex formation, and at the same reduces Wnt-Fz-LRP complex formation, thus favoring non-canonical Wnt signaling [109].
Wnt regulator; required for maintenance of AER and Shh signaling [110].
Similar to Nipbl-deficient fin buds, Nipbl+/− limb buds displayed reductions in the expression of 5′-Hox genes (Table 1 and Figure S12). This was particularly obvious for genes at the extreme 5′ end of Hox clusters, such as Hoxa13, Hoxc13, Hoxd12, and Hoxd13, expression of which was reduced between 15% and 35% by microarray, although Q-RT-PCR measurements (Table 1) and ISH (Figure S12) suggested that the true decrease is probably closer to 50%. Also downregulated were Enpp2 and Epha7, which are known targets of 5′-Hox genes (Table 1 and [67], [68]). Hand2, which lies upstream of both Shh and Hox gene expression [58], [69], was also modestly downregulated (Table 1) similar to Nipbl-deficient fish fin buds (Figure 3C).
Overall, reductions in 5′-Hox gene expression in Nipbl-deficient mouse limb buds were not as large as those observed in Nipbl-deficient zebrafish, most likely reflecting the fact that Nipbl expression is more severely reduced in MO-injected fish embryos than in haploinsufficient mice (which, due to compensatory mechanisms, only show a 37% reduction in Nipbl transcript levels; cf. Table 1). Nonetheless, at least for the HoxD cluster, the downregulation of the most 5′- genes in Nipbl+/− mouse limbs was accompanied by upregulation of at least some genes lying more 3′ in the same cluster (Figure S12A–D).
Med12 and Nipbl regulate spatial expression of hox gene expression and act together in pectoral fin development
Recent studies indicate that Nipbl and cohesin can co-localize at enhancer and promoter regions with the Mediator complex, suggesting that Nipbl participates with Mediator in linking distant transcriptional regulators to basal transcriptional machinery [17]. Interestingly, in zebrafish, a loss-of-function mutation in med12, which encodes a subunit of Mediator, disrupts pectoral fin development [37]. We injected embryos with varying amounts of med12-MO ([70]; up to 6 ng/embryo), and observed severe reductions in pectoral fins at 52–120 hpf (Figure 7A–E) that resembled Nipbl-deficient embryos. Moreover, Med12 depletion caused changes in gene expression in pectoral fin buds strikingly similar to those observed in Nipbl-deficient embryos (Figures 7F and S13), particularly changes in expression of hox genes. Notably, the same 3′ genes of the hoxab, hoxca and hoxda clusters were expanded posteriorly following knockdown of med12, while expression of the same 5′ genes was reduced (Figure 8A).
Figure 7. Med12 depletion disrupts pectoral fin morphology and gene expression similar to Nipbl depletion.

(A, B) Morphology of live embryos at 52 hpf (A, lateral views) and 76 hpf (B, dorsal views). (A) Anterior halves of control and med12-MO-injected embryos (left column) and higher magnification pictures of their pectoral fin buds (right column). (B) Dorsal views of embryos at 76 hpf. (C) Whisker plots of fin length. Fin lengths (medians) are 430.0 µm, n = 18 (control), 275.6 µm, n = 20 (med12-MO, 2 ng), and 183.8 µm, n = 20 (med12-MO, 4 ng). *: p<10−4. (D) Alcian blue staining of pectoral fin cartilage of control (upper) and Med12-deficient (med12-MO, 4 ng; lower) embryos at 120 hpf. Dorsal view, anterior to the top. Right column, higher magnification pictures of boxed areas of endoskeletal discs. ac, actinotrichs; ed, endoskeletal disc; sco, scapulocoracoid. (E) Controls for med12-MO efficiency. RT-PCR, 36 hpf. Both pairs of med12 primers (Primer #1 and #2) show that splicing of med12 mRNA is significantly suppressed by med12-MO, with a slightly higher efficiency at 6 ng. Primer pair #1 detects both precursor and mature mRNA, whereas primer pair #2 only detects mature mRNA (see Materials and Methods). nipbla and nipblb expression was not affected by Med12 depletion. ef1a was used as a control. (F) Expression of genes involved in the 5′-hox/hand2/shha gene cassette and AER fgf genes in pectoral fin buds examined by ISH at 36 hpf. Dorsal views, anterior to the top. Similar to Nipbl-deficient embryos, shha, hand2 and 5′-hoxd genes in mesenchyme as well as fgf16 and fgf8a in the AER are reduced in Med12-deficient embryos (4 ng/embryo med12-MO).
Figure 8. Functional interactions between Nipbl and Med12 in pectoral fin development.

(A) hox gene expression in pectoral fin buds of Med12-deficient embryos examined by ISH at 36 hpf. Dorsal views with anterior to the top. (B) Lateral views of pectoral fins in living larvae at 76 hpf in controls or injected with 0.5 ng med12-MO alone (low-med12-MO). (C) Pectoral fin lengths in larvae injected with low-med12-MO alone or combined with low amounts of nipbl-MOs (0.05 ng nipbla-MO+0.75 ng of nipblb-MO; low-nipbla/b-MO). Medians: 410.1 µm, n = 16 (control), 382.2 µm, n = 24 (low-nipbla/b-MOs), 385.4 µm, n = 16 (low-med12-MO alone), and 341.4 µm, n = 16 (low-med12-MO+low-nipbla/b-MOs). Asterisks indicate statistical significance (p-values<0.001). (D) hox expression in larvae injected with low-med12-MO alone or combined with low nipbla/b-MO. Dorsal views, anterior to the top.
The possibility that these similarities reflect a transcriptional relationship between Nipbl and Med12—e.g. Nipbl positively regulates Med12 expression (or vice versa)—was ruled out by direct measurements of transcript levels in the fin buds of MO-injected embryos (Figures 7E and S14). This conclusion also agrees with the mouse microarray data, which show no decrease in expression of any Mediator subunit in Nipbl +/− limb buds. Indeed, some Mediator genes (Med14, Med19, and Med12l) exhibit modest increases in expression, suggesting, if anything, a negative role for Nipbl in Mediator expression (Table 1).
To test for a genetic interaction between Nipbl and Mediator, nipbla/b-MOs and med12-MO were co-injected at subthreshold doses, and assayed for changes in pectoral fin development and gene expression. Small amounts of med12-MO (0.5 ng/embryo; low-med12-MO) caused only slight reductions in pectoral fin size and 5′-hoxa/hoxd gene expression in fin buds (Figure 8B–D). However, when combined with low doses of nipbla/b-MOs (a combination of 0.05 ng/embryo of nipbla-MO and 0.75 ng/embryo of nipblb-MO; low-nipbla/b-MO), low-med12-MO caused reductions in 5′-hox gene expression and expansion of 3′-hox gene expression similar to those observed with higher doses of either nipbla/b- or med12-MOs alone (Figure 8D). These results suggest that Nipbl and Mediator interact functionally to regulate spatial patterning of hox gene expression in the developing limb.
Interestingly, depletion of the cohesin subunit Rad21 caused very different defects in pectoral fin development and gene expression than deficiencies for Nipbl or Med12. Rad21 depletion delayed development (by approximately 10 hrs, based on the A-P positions of pLL primordia; Figure S15), consistent with a previous report [71], but when compared with stage-matched controls all fin mesenchymal genes (including 3′-hox genes, hoxc6a and hoxd4a) were downregulated (Figure S16). Reductions in hox gene expression became more severe at later stages, although, interestingly, only in fin buds, and not in the neural tube (Figure S16).
Nipbl and Med12 regulate chromatin conformation around the hoxda cluster
Spatial- and temporal patterns of Hox gene expression are achieved through regulation of chromatin organization around Hox clusters. In mouse limb buds, for example, remote enhancers located in flanking “gene deserts” found at the telomeric (3′) and centromeric (5′) sides of the clusters regulate the proximal versus distal expression of 5′-Hox genes [15], [72] ; these enhancers are distinct from cis-regulatory elements within the clusters that regulate co-linear expression along the body axis [73], [74]. Although these remote enhancers have been most extensively studied in mammals, some are clearly conserved and functional in teleosts [75]–[77]. For example, of two distinct regions in the gene desert telomeric to the mouse HoxD cluster recently shown to have proximal limb-specific enhancer activity [72], we located sequences homologous to one, CNS65, about 200 kb telomeric to the hoxda cluster in the zebrafish genome (Figure 9A).
Figure 9. Nipbls and Med12 play roles in regulation of higher-order chromosome conformation at the Hoxd locus in pectoral fin buds.

(A) Diagram of the genomic organization at the zebrafish hoxda locus. Genes in the hoxda cluster and flanking genes are shown as black boxes. Putative regulatory elements conserved between zebrafish and mouse and probes used for FISH are shown as colored ovals and lines, respectively. (B–D) Typical images of FISH. (B) Low magnification picture of a sagittal section of pectoral fin bud. Scale bar = 10 µm. (C,D) Higher magnification images of nuclei with colocalized (C) and separate signals (D). Hybridized probes are detected as green and red fluorescent dots in DAPI-stained nucleus. Scale bar = 2 µm. (E,F) Whisker plots of interprobe distances between hoxd and 3′ probes (E) or hoxd and 5′ probes (F) at 38 hpf. Medians, numbers of nuclei and embryos, and p-values calculated by the non-parametric Mann-Whitney U-test are shown in Table 2. Dotted lines indicate thresholds for separated (upper) and closed (lower) signals in Table 2. (G) Sizes of nuclei in pectoral fin buds (n = 30 each) were estimated at 38 hpf by measuring major and minor axes. Major axis (Ave ± S.D.): 8.58±1.63 µm (control), 8.22±1.76 µm (nipbla/b-MOs, p = 0.412), and 8.14±1.43 µm (med12-MO, p = 0.280). Minor axis (Ave ± S.D.): 4.41±1.28 µm (control), 4.70±0.92 µm (nipbla/b-MOs, p = 0.314), and 4.56±0.73 µm (med12-MO, p = 0.577). p-values were calculated by Student's t-test.
Such results suggest that, in both fish and mice, limb bud hox gene expression depends on long-range chromosomal interactions the formation of which may be regulated by Nipbl and Mediator [17]. We tested this hypothesis by looking for changes in chromatin architecture around the hoxda cluster following Nipbl or Mediator depletion, using probes for 3D-FISH with which we can measure physical distances between the hoxda cluster and distant flanking regions on both centromeric and telomeric sides (Figure 9). When 3D-FISH was performed on cryosections of 38 hpf pectoral fin buds, using a hoxd probe and a distant flanking probe (either 3′ or 5′), each nucleus typically contained one or two pairs of closely-spaced fluorescent spots (Figure 9B–D). The separation between spots varied among nuclei (Figure 9E, F), even within a single fin bud, consistent with the dynamic nature of chromatin interactions [78], [79]. However, when measured in large numbers (≥180 nuclei per condition) we observed a significant increase in inter-probe distances in both Nipbl-deficient and Med12-deficient fin buds, both when centromeric and telomeric flanking probes were used (Figure 9E, F, Table 2). Indeed, a significant percentage of Nipbl- and Med12-deficient nuclei showed inter-probe distances more than double those of controls, and the number of nuclei with probes in close proximity was significantly reduced (Table 2). These effects were not due to changes in nuclear size (Figure 9G). Overall, the results strongly suggest that Nipbl and Mediator regulate expression of hoxd genes in developing limbs by modulating the interaction of promoters with remote enhancers.
Table 2. Results from 3D-FISH around the zebrafish hoxda locus.
| median (µm) | nuclei (embryos) | p* | % of nuclei** | ||
| closed | separated | ||||
| 5′-hoxd (centromeric) | |||||
| pectoral fin buds | |||||
| control | 0.278 | 240 (4) | 9.58 | 5.00 | |
| nipbla/b-MO | 0.357 | 240 (4) | 4.8×10−7 | 6.25 | 15.9 |
| med12-MO | 0.362 | 180 (3) | 7.9×10−9 | 5.56 | 17.2 |
| hindbrain | |||||
| control | 0.295 | 165 (4) | 8.48 | 3.64 | |
| nipbla/b-MO | 0.312 | 160 (4) | 0.092 | 8.12 | 8.12 |
| med12-MO | 0.306 | 125 (3) | 0.716 | 9.60 | 2.40 |
| 3′-hoxd (telomeric) | |||||
| pectoral fin buds | |||||
| control | 0.220 | 240 (4) | 9.58 | 1.67 | |
| nipbla/b-MO | 0.328 | 420 (7) | 5.5×10−19 | 4.76 | 25.0 |
| med12-MO | 0.317 | 180 (3) | 2.2×10−9 | 6.11 | 28.3 |
| hindbrain | |||||
| control | 0.237 | 160 (4) | 15.0 | 6.25 | |
| nipbla/b-MO | 0.349 | 160 (4) | 2.2×10−11 | 4.38 | 26.9 |
| med12-MO | 0.337 | 160 (4) | 1.3×10−8 | 3.13 | 21.9 |
* Evaluated by the Mann-Whitney test.
** Proportions of nuclei exhibiting interprobe distances less than half of (closed) and longer than double (separated) the control medians.
We also used 3D-FISH to examine chromosome conformation at the hoxda cluster in cells of the hindbrain, where Nipbl deficiency does not alter hox gene expression (Fig. S10). Similar to pectoral fin buds, we observed close apposition between the hox cluster and its 3′- and 5′- flanking regions (Figure S17, Table 2), which agrees with data showing that long-range HoxD interactions in the mouse occur in both the limbs and the CNS [15], [77], [80]. Interestingly, while Nipbl- or Med12-depletion both increased the separation between the hox cluster and 3′ flanking sequences in the CNS (similar to the fin buds), they did not alter separation between the hox cluster and 5′ flanking sequences in the hindbrain. The potential significance of these results is discussed below.
Discussion
Multiple genes are dysregulated in fin/limb buds of Nipbl-deficient embryos
Limb reductions are among the most striking structural birth defects in CdLS [26], [28], [36]. Previous studies of both fish and mouse models of Nipbl deficiency, as well as of cell lines derived from human patients with CdLS, strongly suggest that such defects result from the collective and sometimes synergistic effects of numerous small changes in gene expression during development [3], [4], [11]. Distinct sets of gene expression changes have been found in every tissue studied thus far, providing insights into genetic pathways that underlie defects in different tissues and organs [3], [4], [11]. Until now, identifying gene expression changes underlying limb reductions in CdLS has not been possible, since limb reduction is one of the few structural defects in CdLS that is not obviously replicated in the Nipbl-haploinsufficient mouse model [3]. However, by combining studies of zebrafish and mice in the present study, we show that Nipbl levels are critical for limb development (Figure 1), and that Nipbl regulates expression of specific sets of genes in the embryonic limb, including many key developmental regulators that are conserved between fish and mice. Among these Fgfs, Shh, and 5′-Hox genes (Figures 2, 3, 5, and Table 1) are of particular note because of the central and conserved roles these genes play in early limb bud growth and patterning.
In the E10.5 mouse embryo, where the larger size of the limb bud (compared with zebrafish) made genome-wide transcriptional profiling feasible, levels of more than 1000 transcripts were significantly altered (Table 1 and data publically deposited). Both the large number of affected genes and the relatively small sizes of the effects were similar to what has been observed in other tissues of Nipbl +/− mice and in cells from individuals with CdLS [3], [4]. It may be noteworthy that in the mouse limb a large number of Nipbl-sensitive genes are involved in Wnt/planar cell polarity signaling. Although this finding was not further investigated here, it is possible that disruption of this pathway is related to the disorderly arrangement of endoskeletal cells that we consistently observe in developing, Nipbl-deficient fins (Figure 1F′, G′). It may also be noteworthy that, in Nipbl-deficient mouse limbs, several Mediator subunits are (slightly) upregulated (Table 1). As described below, upregulated Mediator function might potentially provide some compensation for Nipbl deficiency.
Interactions between Nipbl and Mediator in gene regulation
Chromatin binding studies have shown that Nipbl co-localizes with cohesin and the Mediator complex at putative regulatory elements of actively transcribed genes, suggesting that Nipbl and Mediator act together to regulate gene expression [13], [17], [81]. Here we provide the first in vivo evidence in support of this hypothesis: 1) Med12- and Nipbl-deficient pectoral fin buds display similar size reductions and gene expression changes—particularly within hox gene clusters; 2) subthreshold doses of nipbl- and med12-MOs synergize to reduce limb size and disrupt gene expression; and 3) both nipbl- and med12-MOs cause similar changes in chromatin conformation at the hoxda locus.
These results support the view that Nipbl and Mediator play roles in the long-range coordination of gene expression. Moreover, the observed differential effects on expression of 3′- versus 5′-hox genes suggest an important role for Nipbl and mediator in transcriptional coordination at multi-gene loci, a result also supported by position-specific effects seen at the protocadherin beta locus in Nipbl-haploinsufficient mice [3], and by studies on the role of Nipbl in long-range control of the beta-globin locus [13].
Interestingly, instead of having position-specific effects, depletion of the cohesin subunit Rad21 led to downregulation of all 3′- and 5′-hox genes that we tested, suggesting that the gene regulatory effects of Nipbl/Mediator are not equivalent to those of cohesin. Indeed, although cohesin has been implicated in long-range chromatin interactions [82]–[84], and Rad21 co-localizes at promoters and enhancers with Nipbl and Mediator [17], this co-localization only occurs at a subset of cohesin binding sites. Moreover, recent work suggests that Nipbl, but not cohesin, co-localizes with certain transcription factors [85]. Such differences may explain the markedly different results that have been observed, in both cell lines and embryos, in the changes in gene expression and chromatin organization that occur in response to depletion of cohesin versus Nipbl [11], [85], [86].
Direct versus indirect effects of Nipbl and Mediator in limb development
Previous studies have proposed that limb development is controlled by a positive feedback loop in which Shh from the ZPA and Fgfs from the AER maintain one another's expression [38], [40], [53]. Consistent with this, we found that expression of both Shh and Fgf genes were reduced in Nipbl-deficient limb and fin buds (Figures 2, 3 and Table 1). As nipbla and nipblb are expressed most highly in fin bud mesenchyme (Figure S1), it is possible that Nipbls regulate the expression of mesenchymal genes such as shha directly, whereas regulation of fgf expression in the AER may be indirect.
On the other hand, hox genes could be the major direct targets of Nipbl deficiency, with effects on shha expression being secondary. Both HoxD and Hand2 regulate Shh expression in early limb/fin buds [57], [58], [87], and Hox proteins also regulate Hand2 expression [88]. In Drosophila, Nipped-B and cohesin bind to genes in the bithorax (Hox) complex (BX-C), specifically in cells that express BX-C genes [81]. More recently, it has been shown that human cohesin binds to the HOXA and HOXB clusters, and disruption of its function reduces expression of multiple HOX genes [83]. Our finding that three distinct hox clusters (A, C, and D) are all affected similarly in Nipbl- and Med12-deficient zebrafish suggests that Nipbl and Mediator play a common role in hox locus control. Results of 3D-FISH experiments at the hoxda cluster further suggest that Nipbl/Mediator-dependent regulation of long-range chromatin interactions is an important part of this role, as discussed below.
Regulation of chromatin conformation by Nipbl and Mediator
The position-specific effects of depleting Nipbl or Med12 on hox gene expression in the zebrafish pectoral fin bud—with 5′-genes down-regulated and 3′ genes up-regulated—suggest a coupling of transcriptional regulation between the two ends of hox clusters. Our 3D-FISH results, which show that Nipbl and Med12 are required in fin buds for long-range interactions on both sides of the hoxda cluster, raise two possibilities for explaining the effects of depleting Nipbl and Med12 on hox gene transcription (Figure 10).
Figure 10. Model of hox gene regulation by Nipbls and mediator.

Along topological domains, 3′- and 5′-hox genes tend to interact with limb-specific regulatory elements in telomeric and centromeric landscapes, respectively. These interactions are required to establish proper patterns of hox gene expression in limb/fin buds and depend on Nipbl/Mediator. The long-range enhancer-promoter interactions are disrupted in the absence of Nipbl and Med12, leading to dysregulation of hox genes. (A) Expanded expression of 3′ -hox genes might be allowed when released from putative remote repressors in Nipbl/Med12-deficient fin buds. (B) Alternatively, disruption of chromosomal conformation may lead to replacement of 3′ remote enhancers with (more closely located) putative ectopic enhancers that can activate 3′-hox genes strongly through long-range interactions.
According to one model, disruption of long-range chromosomal interactions leads to a loss of long-range activation at the 5′ ends and long-range repression at the 3′ ends of hox clusters (Figure 10A). Alternatively, disruption of chromosomal conformation may allow the 3′ remote enhancers to be replaced with other (probably more closely located) regulatory elements, leading to their ectopic activation (Figure 10B). These putative regulatory elements might be fish-specific since, the orthologous 3′-Hox genes are not upregulated in Nipbl-deficient mouse limb buds.
On the other hand, direct comparisons between mice and fish could be misleading, due to the dynamics of hox gene expression. In both tetrapod and zebrafish limb buds, hox gene expression progresses through distinct stages, first being biased toward 3′ genes and later toward 5′ ones [64], as the balance of long-range interactions shifts from telomeric to centromeric [72], [77]. If E10.5 mouse hindlimb buds are not at exactly the same stage as the pectoral fin (forelimb) buds examined here, they may not possess the same potential to express 3′-Hox genes.
A third possibility is that some upregulation of Hox genes does take place in the Nipbl-deficient mouse limb, similar to fish, but the genes affected are not as close to the 3′-end of the cluster. For example, among Hoxd genes, we observed that significant up-regulation of Hoxd10, and possibly also Hoxd8, accompanies the down-regulation Hoxd11, 12 and 13 in Nipbl +/− limbs.
Interestingly, in the zebrafish hindbrain, the effects of depletion of Nipbl or Med12 on hox gene expression and chromosomal interactions differ from those observed in fin buds. 3D-FISH in wild-type hindbrain cells reveals chromosomal interactions of hoxda with both 3′- and 5′-territories—despite the fact that the hindbrain expresses only 3′-hox genes. Moreover, the expression of hindbrain hox genes is unaffected in Nipbl- or Med12-deficient embryos, even though long-range interactions on the 3′ side of the hoxda cluster are markedly diminished. These results suggest: 1) that hindbrain hox gene expression is not primarily controlled by long-range enhancers (at least not on the 3′ side), and 2) that long-range interactions of hox genes are not necessarily associated with active transcription (i.e. they may sometimes represent a poised, or latent state). Consistent with the latter idea, in mouse forebrain, where Hox genes are not expressed, the Hoxd locus still interacts with many of the same long-range elements as it does in limb bud cells [15]. Similar examples of long-range promoter-enhancer associations that do not necessarily correlate with gene expression have also been described for Shh in the mouse limb [14].
Whereas Nipbl and Med12 depletion inhibits both 3′- and 5′- chromosomal interactions of the hoxda cluster in the pectoral fin buds, in the hindbrain such depletion fails to affect 5′ interactions, suggesting a distinct underlying mechanism. In the trunk, the activation of hox gene expression is thought to reflect an anterior-to-posterior wave of chromatin decompaction, from 3′ to 5′, such that in anterior structures (such as the hindbrain) 5′-hox genes and adjacent sequences remain in a highly condensed state [89], associated with high levels of H3K27me3 modification [80]. One possible interpretation of our results is that Nipbl and Med12 play essential roles in long-range interactions, but are not required for the maintenance of the condensed state. The full explanation, however, is likely not this simple, in view of a recent study involving siRNA-treated cell lines and cells from CdLS patients, which shows that chromatin compaction at some loci is highly sensitive to reductions in Nipbl function [86]. Interestingly, the effects on compaction reported in that study were not reproduced by knockdown of the Smc3 cohesin subunit, underscoring the idea, discussed earlier, that the transcriptional function of Nipbl is distinct from that of cohesin.
Understanding the variability of limb defects caused by Nipbl deficiency
Many of the mesenchymal genes (e.g. shh, hand2, 5′-hox) we find downregulated in Nipbl-deficient fin buds are essential for growth and patterning of mouse limbs. Shh −/− mice, for example, have limb truncations [90], [91], and Hand2 is required for Shh expression in the ZPA [87]. Mice lacking certain genes within the HoxA or HoxD clusters have mild digit defects, while a simultaneous deletion of both HoxA and HoxD clusters causes dramatic forelimb truncations [62], [63]. Our finding that expression of Shh and multiple Hox genes is reduced in the limb buds of Nipbl +/− mutant mice indicates that these genes are common targets of Nipbl in the vertebrate limb, and the dysregulation of their expression is likely to be central to the etiology of limb defects in CdLS.
Nonetheless, Nipbl+/− mice display very mild limb abnormalities [3]. One likely explanation for this difference is that haploinsufficiency does not lower Nipbl levels as much as is achieved in MO-injected zebrafish embryos. Indeed, it has been observed that, due to unknown compensatory mechanisms, Nipbl+/− mice display only a 35–40% reduction in Nipbl transcripts (cf. [3], and Table 1), whereas nipbl MOs can lower nipbla and nipblb transcript levels to a much larger degree [11].
The idea that the strength of limb phenotypes is related to the degree of nipbl depletion is further supported by the observation, in zebrafish, that fin reductions are more severe when larger amounts of nipbla-MO are injected, or when both nipbla and nipblb are knocked-down, as opposed to either one alone (Figure S3). In light of this observation, it is noteworthy that only about a third of individuals with CdLS display limb abnormalities at the severe end of the spectrum [26]. A subset of this phenotypic variability likely relates to the strengths of different mutations on Nipbl protein expression (severe forelimb defects tend to correlate with nonsense or frame shift mutations [92], [93]). However, it likely also reflects inter-individual variability in the functions of genes that control Nipbl expression or, like components of the Mediator complex, work together with Nipbl in the control of gene expression.
Materials and Methods
Ethics statement
All animals were handled in strict accordance with good animal practice as defined by the relevant national and/or local animal welfare bodies, and all animal work was approved by the University of California, Irvine, Institutional Animal Care and Use Committee.
Fish and mouse maintenance, embryo raising and staging
Zebrafish (AB strain) were maintained and staged as described [94], [95]. Embryos were stage-matched based on relative positions of posterior lateral line primordial along the A-P axis, detected by ISH with a fgf10a probe. Pectoral fin buds and the posterior end of the yolk sac extension were used as landmarks (Figure S5). Nipbl+/− (RRS strain) mice were housed, mated, and staged as described previously [3].
Microinjection of morpholino antisense oligonucleotides (MOs) and mRNA
MOs were designed to block translation (Gene Tools, Inc.), prepared at 20 mg/ml and diluted in 1× Danieau buffer [58 mM NaCl, 0.7 mM KCl, 0.4 mM MgSO4, 0.6 mM Ca (NCO3)2, 5 mM HEPES (pH 7.6)] and stored at −20°C. MO sequences are shown elsewhere (all nipbl-MOs and rad21-MO [11], and med12-MO [70]).
Full-length cDNA of nipbla was prepared by fusing partial cDNA fragments amplified by RT-PCR in pCRII-TOPO, fused with SV40 polyA sequence derived from pCS2+ and subcloned into pBS-KS+ for in vitro mRNA synthesis. Full-length capped nipbla mRNA was synthesized using mMESSAGE mMACHINE (T3) kit (Ambion) in the presence of rGTP according to the manufacturer's instructions. Synthesized mRNA was electrophoretically separated and a full-length mRNA was gel-isolated using RECOCHIP (TAKARA). MOs and full-length nipbla mRNA were injected into embryos at the 1–4-cell stage. A combination of nipbla-MO and nipblb-MO were injected to generate Nipbl-deficient embryos, at 0.75 ng/embryo each or otherwise as indicated in figure legends.
Whole mount in situ hybridization (ISH)
Whole mount ISH of zebrafish embryos was performed using digoxigenin (DIG)-labeled antisense RNA probes as previously indicated [11]. Whole mount ISH of E10.5 mouse embryos was performed according to published protocols [96]. The 642 bp mouse Shh probe has been previously described [97]. The Hoxd12 and Hoxd13 probes were a kind gift from Denis Duboule.
Measurement of fin length
Pectoral fin lengths were measured using ImageJ from the proximal base to the distal tip in dorsal views (Whisker plots). The interquartile ranges (IQR) are shown as boxes, with the median as the horizontal lines within the boxes. The upper and lower whiskers are the highest and lowest data points within 1.5× the IQR from the top and bottom of the box, respectively. Individual data including outliers are shown as dots. p-values are calculated by the non-parametric Mann-Whitney U test with the Bonferroni adjustment.
RNA preparation and RT-PCR
Total RNA was extracted from 20 whole zebrafish embryos for each sample, and subjected to cDNA synthesis using ProtoScript M-MuLV First Strand cDNA Synthesis Kit (New England BioLabs). mRNA levels were examined by RT-PCR using ef1a as a control. Primers used in RT-PCR are:
med12-primer #1, sense, 5′-GCTCTGGTCTGGCACTACTC-3′, antisense, 5′-CTGTTGTCTCCTGACACTTG-3′; med12-primer #1, sense, 5′-CTAAGCTGCATGCTACAGAGTAT-3′, antisense, 5′-CCTTTGCCCG AACCTGTTG-3′; nipbla, sense, 5′-GGCTACATGCAGTACAGCCA-3′, antisense, 5′-CATCGTACGGGGTTCCACTA-3′; nipblb, sense, 5′-CAGACCCAGAAGGAGAGCT-3′, antisense, 5′-CTTGGTCCGAGTCGTCGTAT-3′; ef1a, sense, 5′-TCAGCGCATACATCAAGAAGA-3′, antisense, 5′-CTGTGCAGACTTTGTGACCT-3′.
The med12-primer #1 was designed to detect both precursor (including an intron of about 600 bases) and mature mRNA, whereas med12-primer #2 was designed at junctions of exons to detect only mature mRNA [98].
For Q-RT-PCR of mouse tissue, total RNA was isolated from somite-staged mouse hindlimbs from E10.5 embryos (WT n = 6, mutant n = 7) using the RNeasy minikit (QIAGEN). cDNA was synthesized from RNA using the iScript Reverse Transcription Supermix for RT-qPCR (BioRad). cDNA was PCR amplified using the iQ SYBR green Supermix (BioRad) with a CFX96 Real-Time System (Bio-Rad). Expression changes were normalized to beta-2 microglobulin, and the expression of each gene was calculated using the 2−ΔΔCt method. A Student's t test was used for statistical analysis.
Primers:
B2m atgggaagccgaacatactg cagtctcagtgggggtgaat
Nipbl agtccatatgccccacagag accggcaacaataggacttg
Shh ggaactcacccccaattaca tcatcacagagatggccaag
Ptch1 gccacagcccctaacaaaaat acccacaatcaactcctcctg
Hand2 ccgacaccaaactctccaag tcttgtcgttgctgctcact
Hoxa13 ctggaacggccaaatgtact cctataggagctggcgtctg
Hoxc4 ccagcaagcaacccatagtc ctcagagaggcacagcgagt
Hoxc6 ccaggaccagaaagccagta ccgagttaggtagcggttga
Hoxc13 taccagcactgggctctttc gaatttgctggctgcgtact
Hoxd4 ccctgggaaccactgttct ctccctgggctgagactgt
Hoxd8 gaggccgagctggtacaata ctagggtttggaagcgactg
Hoxd9 gctgaaggaggaggagaagc gcgtctggtatttggtgtagg
Hoxd10 ggagcccactaaagtctccc tttccttctcctgcacttcg
Hoxd11 aaagagcggcggcacagt aaagaaaaactcgcgttcca
Hoxd12 aaggcaccaagtatgactacgc atctgctgctttgtgtagggt
Hoxd13 tggaacagccaggtgtactg tggtgtaaggcacccttttc
Cyclopamine (CyA) treatment
CyA was prepared at 10 mM in ethanol and stored at −20°C. Zebrafish embryos were incubated in CyA at 50 µM in embryo medium starting at 8 hpf in the dark and fixed with 4% paraformaldehyde (PFA) at indicated stages for ISH.
Proliferation and cell death
Cell proliferation was examined by bromodeoxy uridine (BrdU) incorporation assay as previously reported [98]. Incorporated BrdU was detected by staining with a rat monoclonal anti-BrdU antibody (Abcam, 1∶100) and an anti-rat Alexa488-conjugated secondary antibody (Invitrogen, 1∶200). Nuclei of acid-treated samples were stained with DAPI (0.5 µg/mL). Levels of proliferation were quantified by calculating a ratio of BrdU-positive cells to DAPI-stained total cells. P-values were determined by student t-test
Cell death was examined by the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay and acridine orange staining. For TUNEL assays, embryos were fixed at indicated stages with 2% PFA for 2 hr at room temperature and then washed in PBS containing 0.1% Triton-X-100. The fixed embryos were dehydrated in a graded series of methanol, permeabilized in cold-acetone for 10 min at −20°C, and then treated with proteinase K (10 µg/ml for 10 min at room temperature). Fragmented genomic DNA in dying cells was detected by using In Situ Cell Death Detection Kit (Roche). Dying cells were also detected by staining whole live embryos with acridine orange (5 µg/mL) for 5 min.
Microarray analysis
Total RNA was extracted from hindlimbs (left and right) from each of 12 Nipbl+/+ and 12 Nipbl+/− mouse embryos (E10.5, somite stages 35–38) [3]. The RNA was further processed by the UCI Genomics High-Throughput Facility for microarray analysis using Affymetrix Mouse Gene 1.0 ST arrays. The 24 probe cell intensity files (.Cel) were pre-processed using the Expression File Creator program of GenePattern (Broad Institute) and statistical analysis was performed using the Comparative Markers Selection module. Raw data will be made freely available to the public through Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60932; accession number GSE60932).
Three dimensional-fluorescence in situ hybridization (3D-FISH)
Zebrafish embryos were fixed at indicated stages in 4% PFA and sagittal cryosections cut at a thickness of 20 µm. FISH was performed on sections as described elsewhere [99]. Briefly, sections were permeabilized in 0.5% Triton X-100 in PBS and then genomic DNA was unmasked by 9 cycles of incubation at 90°C using a microwave and cooling for 2 min in 10 mM sodium citrate buffer (pH 6.0). Sections were then permeabilized in acetone for 5 min at −20°C and incubated in 50% formamide in 1× SSC for at least 4 hours. Pretreated sections were loaded with probe solution prepared in hybridization buffer (50% formamide, 10% dextran sulfate in 1× SSC), covered with a cover slip, sealed with rubber cement and prehybridized for at least 2 hr at 37°C. Probes were heat-denatured by incubating the slides at 80°C for 5 min, and hybridized at 37°C for 2–3 days. After washing in 0.1× SSC at 60°C, nuclei were stained with DAPI (0.05 µg/mL) and slides were mounted for fluorescence microscopy.
Fluorescent probes for FISH were labeled with dUTP conjugated with Alexa 488 or Alexa 568 (Invitrogen) using a Nick Translation Mix (Roche) according to the manufacturer's instructions and purified as described elsewhere [100]. For labeling, 1 µg of BAC DNA purchased from the BACPAC resource center was used as a template, and 250 ng each of the labeled probes per slide were used for hybridization. Zebrafish BAC clones used for hoxd, 3′, and 5′ probes are CH73-86I10, CH73-267A7, and CH73-381A1, respectively.
Image analysis
Slides were examined using an Olympus confocal microscope (FV1000) and multiple optical sections along the z-axis were taken in 0.1 µm intervals. Captured images were analyzed using ImageJ. Outlines, areas, and central coordinates along x, y and z axes were measured for each fluorescent signal using the Wand tool in Image J in combination with ROI manager, and spatial distances between two closely located and differently colored signals were calculated. 60 nuclei from pectoral fin buds and 40–45 nuclei from hindbrain were analyzed for each embryo, and 3–7 embryos were used for each condition/probe set tested. Normalized inter-probe distances were plotted in probability histograms showing the mean percentage (± SD) of total nuclei from each sample displaying a given separation between fluorescent dots. Statistical significance was determined by the Mann-Whitney U test.
Supporting Information
Expression of nipbla and nipblb in developing pectoral fin buds. (A) Expression of nipbla and nipblb in pectoral fin buds examined by ISH. Dorsal views, anterior to the top. (B) Transverse sections of pectoral fin buds at 36 hpf showing nipbla and nipblb expression in fin mesenchyme (me) rather than apical ectoderm (ec). Expression is also detected in endoderm (en) and neural tube (nt) but not somites (so).
(EPS)
Reduced pectoral fins in Nipbl-deficient embryos. Morphologies of live control (A, B, E, F) and Nipbl-deficient embryos (C, D, G, H) at 60 hpf (A–D) and 76 hpf (E–H). Dorsal (A, C, E, G) and lateral (B, D, F, H) views, anterior to the left. Pectoral fins are reduced in Nipbl-deficient embryos (arrows) and anterior ends of lower jaws are indicated (arrowheads).
(EPS)
Nipbla and Nipblb act cooperatively in pectoral fin development. Morphologies of live control (A), Nipbl-deficient (B) and embryos injected with either nipbla-MO (C, D) or nipblb-MO (E, F) at indicated amounts. Dorsal views, anterior to the left. (G) Whisker plots of fin length (medians): 431.8 µm, n = 20 (control), 259.1 µm, n = 40 (nipbla/b-MO), 385.3 µm, n = 40 (nipbla-MO, 0.75 ng), 351.0 µm, n = 40 (nipbla-MO, 1.5 ng), 417.6 µm, n = 40 (nipblb-MO, 0.75 ng), and 399.7 µm, n = 20 (nipblb-MO, 1.5 ng). *: p<10−6.
(EPS)
Small fin phenotypes could be caused by reduced proliferation rather than cell death of fin mesenchymal cells. (A, B) Cell death in pectoral fins of control (A) and Nipbl-deficient larvae (B) at 120 hpf was examined by TUNEL assay. (C–E) Cell death in pectoral fin buds of Nipbl-deficient at 40 hpf was examined by TUNEL assay in comparison with stage-matched (C, 36 hpf) and time-matched (D, 40 hpf) comparisons. TUNEL positive cells are shown in red. Lateral views with anterior to the left. (F–I) Proliferation of pectoral fin buds was examined by BrdU incorporation assay. Stage-matched (F, 36 hpf) and time-matched (G, 40 hpf) controls were compared with Nipbl-deficient embryos (H, 40 hpf). BrdU-positive cells (green) and DAPI stained cells (blue). Lateral views, anterior to the left. (I) Cell proliferation was quantitatively analyzed by determining proportions of BrdU-positive cells in fin mesenchymal cells stained with DAPI. Ave ± S.D.: 73.9±5.29% (n = 8, control at 36 hpf), 73.0±3.78% (n = 9, control at 40 hpf), 48.7±12.0% (n = 12, nipbla/b-MOs-injected embryos at 40 hpf). p-values compared with control at 36 hpf were determined by student t-test. *: p<0.001.
(EPS)
Staging embryos by posterior lateral line primordium position. (A) Posterior lateral line (pLL) primordia (red arrows) detected by ISH for fgf10a at indicated stages move progressively posterior relative to pectoral fin buds (yellow arrows) from 22–48 hpf, indicating a 3–4 hour and 6 hour developmental delay in Nipbl-deficient embryos at 36 hpf and 48 hpf, respectively. Duplicated signals at the tail tips in some panels reflect dorsoventral bifurcation of the tail, rather than ectopic expression of fgf10a, which is a typical phenotype observed in Nipbl-deficient embryos [11]. Lateral views. (B) Summary of stage-match comparisons between control and Nipbl-deficient embryos.
(EPS)
Rescue of AER-fgf gene expression in pectoral fin buds of Nipbl-deficient embryos by exogenous nipbla mRNA. Expression of the AER fgf genes, fgf16 and fgf8a, in pectoral fin buds (arrows) of controls, embryos injected with nipbla/b-MOs, and those co-injected with nipbla/b-MO and 400 pg of nipbla mRNA was examined by ISH at 48 hpf. Dorsal views, anterior to the top.
(EPS)
Expression of genes in the fgf10a signaling pathways is unaffected in Nipbl-deficient embryos. Expression of fgf10a (A; arrows), tbx5a (B) and fgf24 (C) in control and Nipbl-deficient embryos was examined by ISH at indicated stages. fgf10a-expressing pLL primordia are marked by asterisks. Dorsal views, anterior to the top.
(EPS)
Rescue of mesenchymal gene expression in pectoral fin buds of Nipbl-deficient embryos by exogenous nipbla mRNA. Expression of genes in the shha/hand2/5′-hox gene cassette at 36 hpf in pectoral fin buds of controls, embryos injected with nipbla/b-MOs, and those co-injected with nipbla/b-MO and 400 pg of nipbla mRNA was examined by ISH. Dorsal views, anterior to the top.
(EPS)
Expression of genes in the RA signaling pathways is unaffected in Nipbl-deficient embryos. Expression of the RA synthesizing enzyme aldh1a2 and the RA-degrading enzyme cyp26a1, a target of the RA signaling, at 13 (upper) and 19 (lower) hpf in Nipbl-deficient embryos. Anterior somites are indicated by brackets. Dorsal views, anterior to the top.
(EPS)
Expression of hox genes along the anterior-posterior axis of the neural tube is unaffected in Nipbl-deficient embryos. hox gene expression at 36 hpf examined by ISH. Lateral views, anterior to the left. Pectoral fin buds are indicated by black arrows. Anterior and posterior limits of hox expression in the neural tube are indicated by red arrowheads.
(EPS)
Gene expression in pectoral fin buds of CyA-treated embryos. (A) Effects of CyA treatment on development was examined by A-P positioning of pLL primordial expressing fgf10a. Lateral views with anterior to the left. Pectoral fin buds (yellow arrows) and pLL primordial (red arrows) are pointed. Expression of fgf10a in pLL primordial and pectoral fin buds is reduced by CyA treatment and becomes undetectable in pLL primordia by 36 hpf. (B) Expression of mesenchymal genes in pectoral fin buds was examined by ISH in possible stage-match comparisons. Dorsal views, anterior to the top.
(EPS)
Analysis of expression of all mouse Hox genes in wildtype and Nipbl +/− hindlimb buds. (A–D) Expression values show hybridization intensity for probe sets representing all of genes in the four Hox clusters (although the relationship between hybridization intensity and transcript abundance is not necessarily the same for different probesets, intensity gives a rough sense of abundance). Data are graphed as mean ± SEM for the mutant (red) and wild type (blue) samples (see Experimental Procedures). Asterisks indicate genes also shown in Table 1, for which expression changes were observed with strong or moderate statistical significance (1%<FDR<7.5%; double asterisk) or weak statistical significance (FDR<25%; single asterisk); filled arrows show the directions in which expression changes were observed. Open arrows show directional changes that were also tested and confirmed by Q-RT-PCR (see Table 1), whereas “n.c.” marks genes that were tested by Q-RT-PCR and showed no detectable change in expression. The open arrow by Hoxd8 is marked with a question mark because Q-RT-PCR showed a 27% elevation in expression in Nipbl+/− limb buds, but the result was not statistically significant (P = 0.14). The tight error bars on the microarray data for most transcripts, and the confirmation of all significant results by PCR, justify the ability to attach statistical significance even to these modest effect sizes. Overall, the data illustrate that expression changes in Nipbl +/− limb buds are biased toward the 5′ ends of the Hox clusters, and suggest that, as in zebrafish, effects occur at all of the expressed clusters (HoxA, C and D). In the HoxD cluster, which exhibits the highest overall hybridization intensities, the largest fold decreases are seen at the extreme 5′ end (Hoxd12, Hoxd13), and give way, as one moves in the 3′ direction, to a weak decrease (Hoxd11) and then a statistically significant increase (Hoxd10, and possibly Hoxd8). Thus, even though these effects are much smaller in magnitude than in zebrafish (where nipbl knockdown is likely much greater than in Nipbl +/− mice), they appear to show similar positional trends. (E–F) Whole mount in situ hybridization for Hoxd12 (E) and Hoxd13 (F) in E10.5 limb buds of Nipbl+/− and wildtype mice. For both Hox genes, expression is reduced significantly in both forelimbs and hindlimbs, consistent with the results of microarray analysis and Q-RT-PCR (panels A–D, and Table 1).
(EPS)
Expression of genes in the fgf10a pathways is unaffected in Med12-deficient embryos. Expression of fgf24 (30, 48 hpf) and fgf10a (36 hpf) in Med12-deficient embryos examined by ISH. Dorsal views, anterior to the top.
(EPS)
Expression of med12 in Nipbl-deficient embryos. Expression of med12 in embryos injected with nipbla/b-MOs was examined by RT-PCR at 8 and 36 hpf. ef1a was used as a control.
(EPS)
Developmental delay of Rad21-deficient embryos. Effects of Rad21-deficiency (rad21-MO at 2.5 ng/embryo) on embryonic development were examined by A-P positions of pLL primordia by ISH for fgf10a. Lateral views, anterior to the left. Pectoral fin buds (yellow arrows) and pLL primordial (red arrows) are pointed. Expression of fgf10a in pLL primordial and pectoral fin buds of Rad21-deficient embryos were detected at levels significantly lower than control and lost by 54 hpf.
(EPS)
Expression of genes in pectoral fin buds of Rad21-deficient embryos. Effects of Rad21 reduction of expression of mesenchymal genes in pectoral fin buds were examined by ISH in possible stage-match comparisons. Dorsal views, anterior to the top.
(EPS)
Chromosome conformation around the hoxda locus in hindbrain. (A, B) Effects of Nipbl- and Med12-reduction on a higher-order chromatin conformation in hindbrain was examined by 3D-FISH at 38 hpf. Interprobe distances between (A) hoxd and 3′ probes and (B) hoxd and 5′ probes shown by Whisker plots. Details of medians, numbers of nuclei and embryos, and p-values are shown in Table 2. Dotted lines indicate thresholds for separated (upper) and closed (lower) signals in Table 2.
(EPS)
Acknowledgments
We are grateful to Shimako Kawauchi and Rosaysela Santos for assistance in obtaining, dissecting, and staging mouse embryo limb buds for expression microarray analysis, and Marissa Macchietto for help with some ISH experiments. We thank Tailin Zhang and Ines Gehring for fish care. Microarray analysis was performed at the UCI Genomics High-Throughput Facility.
Funding Statement
This work was supported by the US National Institute of Health (P01-HD052860 and P50-GM076516) and the Cornelia de Lange Syndrome Foundation. This project used the UCI Genomics High-Throughput Facility, which is partially supported by Award Number P30CA062203 from the National Cancer Institute to the Chao Comprehensive Cancer Center. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Peters JM, Tedeschi A, Schmitz J (2008) The cohesin complex and its roles in chromosome biology. Genes Dev 22: 3089–3114. [DOI] [PubMed] [Google Scholar]
- 2. Remeseiro S, Losada A (2013) Cohesin, a chromatin engagement ring. Curr Opin Cell Biol 25: 63–71. [DOI] [PubMed] [Google Scholar]
- 3. Kawauchi S, Calof AL, Santos R, Lopez-Burks ME, Young CM, et al. (2009) Multiple Organ System Defects and Transcriptional Dysregulation in the Nipbl +/2 Mouse, a Model of Cornelia de Lange Syndrome. PLoS Genet 5: e1000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu J, Zhang Z, Bando M, Itoh T, Deardorff MA, et al. (2009) Transcriptional Dysregulation in NIPBL and Cohesin Mutant Human Cells. PLoS Biol 7: e1000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rollins RA, Korom M, Aulner N, Martens A, Dorsett D (2004) Drosophila Nipped-B Protein Supports Sister Chromatid Cohesion and Opposes the Stromalin/Scc3 Cohesion Factor To Facilitate Long-Range Activation of the cut Gene. Mol Cell Biol 24: 3100–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dorsett D, Eissenberg JC, Misulovin Z, Martens A, Redding B, et al. (2005) Effects of sister chromatid cohesion proteins on cut gene expression during wing development in Drosophila. Development 132: 4743–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, et al. (2008) Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 451: 796–801. [DOI] [PubMed] [Google Scholar]
- 8. Parelho V, Hadjur S, Spivakov M, Leleu M, Sauer S, et al. (2008) Cohesins Functionally Associate with CTCF on Mammalian Chromosome Arms. Cell 132: 422–433. [DOI] [PubMed] [Google Scholar]
- 9. Rhodes JM, Bentley FK, Print CG, Dorsett D, Misulovin Z, et al. (2010) Positive regulation of c-Myc by cohesin is direct, and evolutionarily conserved. Dev Biol 344: 637–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dorsett D (2011) Cohesin: genomic insights into controlling gene transcription and development. Curr Opin Genet Dev 21: 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Muto A, Calof AL, Lander AD, Schilling TF (2011) Multifactorial Origins of Heart and Gut Defects in nipbl-Deficient Zebrafish, a Model of Cornelia de Lange Syndrome. PLoS Biol 9: e1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dorsett D, Strom L (2012) The ancient and evolving roles of cohesin in gene expression and DNA repair. Curr Biol 22: R240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chien R, Zeng W, Kawauchi S, Bender MA, Santos R, et al. (2011) Cohesin mediates chromatin interactions that regulate mammalian beta-globin expression. J Biol Chem 286: 17870–17878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Amano T, Sagai T, Tanabe H, Mizushina Y, Nakazawa H, et al. (2009) Chromosomal dynamics at the Shh locus: limb bud-specific differential regulation of competence and active transcription. Dev Cell 16: 47–57. [DOI] [PubMed] [Google Scholar]
- 15. Montavon T, Soshnikova N, Mascrez B, Joye E, Thevenet L, et al. (2011) A regulatory archipelago controls Hox genes transcription in digits. Cell 147: 1132–1145. [DOI] [PubMed] [Google Scholar]
- 16. Ferrai C, Pombo A (2009) 3D chromatin regulation of Sonic hedgehog in the limb buds. Dev Cell 16: 9–11. [DOI] [PubMed] [Google Scholar]
- 17. Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, et al. (2010) Mediator and cohesin connect gene expression and chromatin architecture. Nature 467: 430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Borggrefe T, Yue X (2011) Interactions between subunits of the Mediator complex with gene-specific transcription factors. Semin Cell Dev Biol 22: 759–768. [DOI] [PubMed] [Google Scholar]
- 19. Ries D, Meisterernst M (2011) Control of gene transcription by Mediator in chromatin. Semin Cell Dev Biol 22: 735–740. [DOI] [PubMed] [Google Scholar]
- 20. Knuesel MT, Meyer KD, Bernecky C, Taatjes DJ (2009) The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev 23: 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hengartner CJ, Myer VE, Liao SM, Wilson CJ, Koh SS, et al. (1998) Temporal regulation of RNA polymerase II by Srb10 and Kin28 cyclin-dependent kinases. Mol Cell 2: 43–53. [DOI] [PubMed] [Google Scholar]
- 22. Akoulitchev S, Chuikov S, Reinberg D (2000) TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature 407: 102–106. [DOI] [PubMed] [Google Scholar]
- 23. Furumoto T, Tanaka A, Ito M, Malik S, Hirose Y, et al. (2007) A kinase subunit of the human mediator complex, CDK8, positively regulates transcriptional activation. Genes Cells 12: 119–132. [DOI] [PubMed] [Google Scholar]
- 24. Belakavadi M, Fondell JD (2010) Cyclin-dependent kinase 8 positively cooperates with Mediator to promote thyroid hormone receptor-dependent transcriptional activation. Mol Cell Biol 30: 2437–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ireland M, Donnai D, Burn J (1993) Brachmann-de Lange Syndrome. Delineation of the Clinical Phenotype. Am J Med Genet 47: 959–964. [DOI] [PubMed] [Google Scholar]
- 26. Jackson L, Kline AD, Barr MA, Koch S (1993) de Lange Syndrome: A Clinical Review of 310 Individuals. Am J Med Genet 47: 940–946. [DOI] [PubMed] [Google Scholar]
- 27. Kline AD, Krantz ID, Sommer A, Kliewer M, Jackson LG, et al. (2007) Cornelia de Lange Syndrome: Clinical Review, Diagnostic and Scoring Systems, and Anticipatory Guidance. Am J Med Genet A 143A: 1287–1296. [DOI] [PubMed] [Google Scholar]
- 28. Liu J, Krantz ID (2008) Cohesin and Human Disease. Annu Rev Genomics Hum Genet 9: 303–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Strachan T (2005) Cornelia de Lange Syndrome and the link between chromosomal function, DNA repair and developmental gene regulation. Curr Opin Genet Dev 15: 258–264. [DOI] [PubMed] [Google Scholar]
- 30. Bose T, Gerton JL (2010) Cohesinopathies, gene expression, and chromatin organization. J Cell Biol 189: 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tonkin ET, Wang T-J, Lisgo S, Bamshad MJ, Strachan T (2004) NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet 36: 636–641. [DOI] [PubMed] [Google Scholar]
- 32. Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, et al. (2004) Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet 36: 631–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Musio A, Selicorni A, Focarelli ML, Gervasini C, Milani D, et al. (2006) X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet 38: 528–530. [DOI] [PubMed] [Google Scholar]
- 34. Deardorff MA, Kaur M, Yaeger D, Rampuria A, Korolev S, et al. (2007) Mutations in Cohesin Complex Members SMC3 and SMC1A Cause a Mild Variant of Cornelia de Lange Syndrome with Predominant Mental Retardation. Am J Hum Genet 80: 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, et al. (2012) HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 489: 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dorsett D, Krantz ID (2009) On the Molecular Etiology of Cornelia de Lange Syndrome. Ann N Y Acad Sci 1151: 21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rau MJ, Fischer S, Neumann CJ (2006) Zebrafish Trap230/Med12 is required as a coactivator for Sox9-dependent neural crest, cartilage and ear development. Dev Biol 296: 83–93. [DOI] [PubMed] [Google Scholar]
- 38. Mercader N (2007) Early steps of paired fin development in zebrafish compared with tetrapod limb development. Dev Growth Differ 49: 421–437. [DOI] [PubMed] [Google Scholar]
- 39. Abbasi AA (2011) Evolution of vertebrate appendicular structures: Insight from genetic and palaeontological data. Dev Dyn 240: 1005–1016. [DOI] [PubMed] [Google Scholar]
- 40. Benazet JD, Zeller R (2009) Vertebrate limb development: moving from classical morphogen gradients to an integrated 4-dimensional patterning system. Cold Spring Harb Perspect Biol 1: a001339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mari-Beffa M, Murciano C (2010) Dermoskeleton morphogenesis in zebrafish fins. Dev Dyn 239: 2779–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gibert Y, Gajewski A, Meyer A, Begemann G (2006) Induction and prepatterning of the zebrafish pectoral fin bud requires axial retinoic acid signaling. Development 133: 2649–2659. [DOI] [PubMed] [Google Scholar]
- 43. Mic FA, Sirbu IO, Duester G (2004) Retinoic acid synthesis controlled by Raldh2 is required early for limb bud initiation and then later as a proximodistal signal during apical ectodermal ridge formation. J Biol Chem 279: 26698–26706. [DOI] [PubMed] [Google Scholar]
- 44. Niederreither K, Vermot J, Schuhbaur B, Chambon P, Dolle P (2002) Embryonic retinoic acid synthesis is required for forelimb growth and anteroposterior patterning in the mouse. Development 129: 3563–3574. [DOI] [PubMed] [Google Scholar]
- 45. Prykhozhij SV, Neumann CJ (2008) Distinct roles of Shh and Fgf signaling in regulating cell proliferation during zebrafish pectoral fin development. BMC Dev Biol 8: 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Grandel H, Draper BW, Schulte-Merker S (2000) dackel acts in the ectoderm of the zebrafish pectoral fin bud to maintain AER signaling. Development 127: 4169–4178. [DOI] [PubMed] [Google Scholar]
- 47. Grandel H, Schulte-Merker S (1998) The development of the paired fins in the zebrafish (Danio rerio). Mech Dev 79: 99–120. [DOI] [PubMed] [Google Scholar]
- 48. Yano T, Abe G, Yokoyama H, Kawakami K, Tamura K (2012) Mechanism of pectoral fin outgrowth in zebrafish development. Development 139: 2916–2925. [DOI] [PubMed] [Google Scholar]
- 49. Fischer S, Draper BW, Neumann CJ (2003) The zebrafish fgf24 mutant identifies an additional level of Fgf signaling involved in vertebrate forelimb initiation. Development 130: 3515–3524. [DOI] [PubMed] [Google Scholar]
- 50. Nomura R, Kamei E, Hotta Y, Konishi M, Miyake A, et al. (2006) Fgf16 is essential for pectoral fin bud formation in zebrafish. Biochem Biophys Res Commun 347: 340–346. [DOI] [PubMed] [Google Scholar]
- 51. Norton WH, Ledin J, Grandel H, Neumann CJ (2005) HSPG synthesis by zebrafish Ext2 and Extl3 is required for Fgf10 signalling during limb development. Development 132: 4963–4973. [DOI] [PubMed] [Google Scholar]
- 52. Ohuchi H, Nakagawa T, Yamamoto A, Araga A, Ohata T, et al. (1997) The mesenchymal factor, FGF10, initiates and maintains the outgrowth of the chick limb bud through interaction with FGF8, an apical ectodermal factor. Development 124: 2235–2244. [DOI] [PubMed] [Google Scholar]
- 53. Camarata T, Snyder D, Schwend T, Klosowiak J, Holtrup B, et al. (2010) Pdlim7 is required for maintenance of the mesenchymal/epidermal Fgf signaling feedback loop during zebrafish pectoral fin development. BMC Dev Biol 10: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hill RE (2007) How to make a zone of polarizing activity: insights into limb development via the abnormality preaxial polydactyly. Dev Growth Differ 49: 439–448. [DOI] [PubMed] [Google Scholar]
- 55. Neumann CJ, Grandel H, Gaffield W, Schulte-Merker S, Nusslein-Volhard C (1999) Transient establishment of anteroposterior polarity in the zebrafish pectoral fin bud in the absence of sonic hedgehog activity. Development 126: 4817–4826. [DOI] [PubMed] [Google Scholar]
- 56. Sakamoto K, Onimaru K, Munakata K, Suda N, Tamura M, et al. (2009) Heterochronic shift in Hox-mediated activation of sonic hedgehog leads to morphological changes during fin development. PLoS One 4: e5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yelon D, Ticho B, Halpern ME, Ruvinsky I, Ho RK, et al. (2000) The bHLH transcription factor hand2 plays parallel roles in zebrafish heart and pectoral fin development. Development 127: 2573–2582. [DOI] [PubMed] [Google Scholar]
- 58. Galli A, Robay D, Osterwalder M, Bao X, Benazet JD, et al. (2010) Distinct roles of Hand2 in initiating polarity and posterior Shh expression during the onset of mouse limb bud development. PLoS Genet 6: e1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Buscher D, Bosse B, Heymer J, Ruther U (1997) Evidence for genetic control of Sonic hedgehog by Gli3 in mouse limb development. Mech Dev 62: 175–182. [DOI] [PubMed] [Google Scholar]
- 60. Tyurina OV, Guner B, Popova E, Feng J, Schier AF, et al. (2005) Zebrafish Gli3 functions as both an activator and a repressor in Hedgehog signaling. Dev Biol 277: 537–556. [DOI] [PubMed] [Google Scholar]
- 61. Mercader N, Fischer S, Neumann CJ (2006) Prdm1 acts downstream of a sequential RA, Wnt and Fgf signaling cascade during zebrafish forelimb induction. Development 133: 2805–2815. [DOI] [PubMed] [Google Scholar]
- 62. Zakany J, Duboule D (2007) The role of Hox genes during vertebrate limb development. Curr Opin Genet Dev 17: 359–366. [DOI] [PubMed] [Google Scholar]
- 63. Kmita M, Tarchini B, Zakany J, Logan M, Tabin CJ, et al. (2005) Early developmental arrest of mammalian limbs lacking HoxA/HoxD gene function. Nature 435: 1113–1116. [DOI] [PubMed] [Google Scholar]
- 64. Ahn D, Ho RK (2008) Tri-phasic expression of posterior Hox genes during development of pectoral fins in zebrafish: implications for the evolution of vertebrate paired appendages. Dev Biol 322: 220–233. [DOI] [PubMed] [Google Scholar]
- 65. Tarchini B, Duboule D (2006) Control of Hoxd genes' collinearity during early limb development. Dev Cell 10: 93–103. [DOI] [PubMed] [Google Scholar]
- 66. Anderson E, Peluso S, Lettice LA, Hill RE (2012) Human limb abnormalities caused by disruption of hedgehog signaling. Trends Genet 28: 364–373. [DOI] [PubMed] [Google Scholar]
- 67. Williams TM, Williams ME, Kuick R, Misek D, McDonagh K, et al. (2005) Candidate downstream regulated genes of HOX group 13 transcription factors with and without monomeric DNA binding capability. Dev Biol 279: 462–480. [DOI] [PubMed] [Google Scholar]
- 68. Salsi V, Zappavigna V (2006) Hoxd13 and Hoxa13 directly control the expression of the EphA7 Ephrin tyrosine kinase receptor in developing limbs. J Biol Chem 281: 1992–1999. [DOI] [PubMed] [Google Scholar]
- 69. Zakany J, Kmita M, Duboule D (2004) A dual role for Hox genes in limb anterior-posterior asymmetry. Science 304: 1669–1672. [DOI] [PubMed] [Google Scholar]
- 70. Shin CH, Chung WS, Hong SK, Ober EA, Verkade H, et al. (2008) Multiple roles for Med12 in vertebrate endoderm development. Dev Biol 317: 467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Horsfield JA, Anagnostou SH, Hu JK-H, Cho KHY, Geisler R, et al. (2007) Cohesin-dependent regulation of Runx genes. Development 134: 2639–2649. [DOI] [PubMed] [Google Scholar]
- 72. Andrey G, Montavon T, Mascrez B, Gonzalez F, Noordermeer D, et al. (2013) A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science 340: 1234167. [DOI] [PubMed] [Google Scholar]
- 73. Tschopp P, Duboule D (2011) A genetic approach to the transcriptional regulation of Hox gene clusters. Annu Rev Genet 45: 145–166. [DOI] [PubMed] [Google Scholar]
- 74. Spitz F (2010) Control of vertebrate Hox clusters by remote and global cis-acting regulatory sequences. Adv Exp Med Biol 689: 63–78. [DOI] [PubMed] [Google Scholar]
- 75. Schneider I, Aneas I, Gehrke AR, Dahn RD, Nobrega MA, et al. (2011) Appendage expression driven by the Hoxd Global Control Region is an ancient gnathostome feature. Proc Natl Acad Sci U S A 108: 12782–12786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Amemiya CT, Alfoldi J, Lee AP, Fan S, Philippe H, et al. (2013) The African coelacanth genome provides insights into tetrapod evolution. Nature 496: 311–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Woltering JM, Noordermeer D, Leleu M, Duboule D (2014) Conservation and divergence of regulatory strategies at hox Loci and the origin of tetrapod digits. PLoS Biol 12: e1001773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mateos-Langerak J, Bohn M, de Leeuw W, Giromus O, Manders EM, et al. (2009) Spatially confined folding of chromatin in the interphase nucleus. Proc Natl Acad Sci U S A 106: 3812–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bohn M, Heermann DW (2010) Diffusion-driven looping provides a consistent framework for chromatin organization. PLoS One 5: e12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Noordermeer D, Leleu M, Splinter E, Rougemont J, De Laat W, et al. (2011) The dynamic architecture of Hox gene clusters. Science 334: 222–225. [DOI] [PubMed] [Google Scholar]
- 81. Misulovin Z, Schwartz YB, Li X-Y, Kahn TG, Gause M, et al. (2008) Association of cohesin and Nipped-B with transcriptionally active regions of the Drosophila melanogaster genome. Chromosoma 117: 89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. DeMare LE, Leng J, Cotney J, Reilly SK, Yin J, et al. (2013) The genomic landscape of cohesin-associated chromatin interactions. Genome Res 23: 1224–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zuin J, Dixon JR, van der Reijden MI, Ye Z, Kolovos P, et al. (2014) Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc Natl Acad Sci U S A 111: 996–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ong CT, Corces VG (2011) Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet 12: 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zuin J, Franke V, van Ijcken WF, van der Sloot A, Krantz ID, et al. (2014) A cohesin-independent role for NIPBL at promoters provides insights in CdLS. PLoS Genet 10: e1004153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Nolen LD, Boyle S, Ansari M, Pritchard E, Bickmore WA (2013) Regional chromatin decompaction in Cornelia de Lange syndrome associated with NIPBL disruption can be uncoupled from cohesin and CTCF. Hum Mol Genet 22: 4180–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Charite J, McFadden DG, Olson EN (2000) The bHLH transcription factor dHAND controls Sonic hedgehog expression and establishment of the zone of polarizing activity during limb development. Development 127: 2461–2470. [DOI] [PubMed] [Google Scholar]
- 88. Xu B, Wellik DM (2011) Axial Hox9 activity establishes the posterior field in the developing forelimb. Proc Natl Acad Sci U S A 108: 4888–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chambeyron S, Da Silva NR, Lawson KA, Bickmore WA (2005) Nuclear re-organisation of the Hoxb complex during mouse embryonic development. Development 132: 2215–2223. [DOI] [PubMed] [Google Scholar]
- 90. Kraus P, Fraidenraich D, Loomis CA (2001) Some distal limb structures develop in mice lacking Sonic hedgehog signaling. Mech Dev 100: 45–58. [DOI] [PubMed] [Google Scholar]
- 91. Chiang C, Litingtung Y, Harris MP, Simandl BK, Li Y, et al. (2001) Manifestation of the limb prepattern: limb development in the absence of sonic hedgehog function. Dev Biol 236: 421–435. [DOI] [PubMed] [Google Scholar]
- 92. Yan J, Saifi GM, Wierzba TH, Withers M, Bien-Willner GA, et al. (2006) Mutational and Genotype–Phenotype Correlation Analyses in 28 Polish Patients With Cornelia de Lange Syndrome. Am J Med Genet A 140A: 1531–1541. [DOI] [PubMed] [Google Scholar]
- 93. Oliveira J, Dias C, Redeker E, Costa E, Silva J, et al. (2010) Development of NIPBL locus-specific database using LOVD: from novel mutations to further genotype-phenotype correlations in Cornelia de Lange Syndrome. Hum Mutat 31: 1216–1222. [DOI] [PubMed] [Google Scholar]
- 94.Westerfield M (1995) The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio). Eugene, OR: University of Oregon Press. [Google Scholar]
- 95. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF (1995) Stages of embryonic development of the zebrafish. Dev Dyn 203: 253–310. [DOI] [PubMed] [Google Scholar]
- 96. Kawauchi S, Takahashi S, Nakajima O, Ogino H, Morita M, et al. (1999) Regulation of lens fiber cell differentiation by transcription factor c-Maf. J Biol Chem 274: 19254–19260. [DOI] [PubMed] [Google Scholar]
- 97. Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, et al. (1993) Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 75: 1417–1430. [DOI] [PubMed] [Google Scholar]
- 98. Shepard JL, Stern HM, Pfaff KL, Amatruda JF (2004) Analysis of the cell cycle in zebrafish embryos. Methods Cell Biol 76: 109–125. [DOI] [PubMed] [Google Scholar]
- 99. Solovei I, Grasser F, Lanctot C (2007) FISH on Histological Sections. CSH Protoc 2007: pdb prot4729. [DOI] [PubMed] [Google Scholar]
- 100. Muller S, Neusser M, Kohler D, Cremer M (2007) Preparation of Complex DNA Probe Sets for 3D FISH with up to Six Different Fluorochromes. CSH Protoc 2007: pdb prot4730. [DOI] [PubMed] [Google Scholar]
- 101. Itou J, Taniguchi N, Oishi I, Kawakami H, Lotz M, et al. (2011) HMGB factors are required for posterior digit development through integrating signaling pathway activities. Dev Dyn 240: 1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Capellini TD, Di Giacomo G, Salsi V, Brendolan A, Ferretti E, et al. (2006) Pbx1/Pbx2 requirement for distal limb patterning is mediated by the hierarchical control of Hox gene spatial distribution and Shh expression. Development 133: 2263–2273. [DOI] [PubMed] [Google Scholar]
- 103. Salsi V, Vigano MA, Cocchiarella F, Mantovani R, Zappavigna V (2008) Hoxd13 binds in vivo and regulates the expression of genes acting in key pathways for early limb and skeletal patterning. Dev Biol 317: 497–507. [DOI] [PubMed] [Google Scholar]
- 104. Lettice LA, Williamson I, Wiltshire JH, Peluso S, Devenney PS, et al. (2012) Opposing functions of the ETS factor family define Shh spatial expression in limb buds and underlie polydactyly. Dev Cell 22: 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Liu Z, Lavine KJ, Hung IH, Ornitz DM (2007) FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev Biol 302: 80–91. [DOI] [PubMed] [Google Scholar]
- 106. Hung IH, Yu K, Lavine KJ, Ornitz DM (2007) FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev Biol 307: 300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Pandur P, Lasche M, Eisenberg LM, Kuhl M (2002) Wnt-11 activation of a non-canonical Wnt signalling pathway is required for cardiogenesis. Nature 418: 636–641. [DOI] [PubMed] [Google Scholar]
- 108. Darken RS, Wilson PA (2001) Axis induction by wnt signaling: Target promoter responsiveness regulates competence. Dev Biol 234: 42–54. [DOI] [PubMed] [Google Scholar]
- 109. Yamamoto S, Nishimura O, Misaki K, Nishita M, Minami Y, et al. (2008) Cthrc1 selectively activates the planar cell polarity pathway of Wnt signaling by stabilizing the Wnt-receptor complex. Dev Cell 15: 23–36. [DOI] [PubMed] [Google Scholar]
- 110. Nam JS, Park E, Turcotte TJ, Palencia S, Zhan X, et al. (2007) Mouse R-spondin2 is required for apical ectodermal ridge maintenance in the hindlimb. Dev Biol 311: 124–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expression of nipbla and nipblb in developing pectoral fin buds. (A) Expression of nipbla and nipblb in pectoral fin buds examined by ISH. Dorsal views, anterior to the top. (B) Transverse sections of pectoral fin buds at 36 hpf showing nipbla and nipblb expression in fin mesenchyme (me) rather than apical ectoderm (ec). Expression is also detected in endoderm (en) and neural tube (nt) but not somites (so).
(EPS)
Reduced pectoral fins in Nipbl-deficient embryos. Morphologies of live control (A, B, E, F) and Nipbl-deficient embryos (C, D, G, H) at 60 hpf (A–D) and 76 hpf (E–H). Dorsal (A, C, E, G) and lateral (B, D, F, H) views, anterior to the left. Pectoral fins are reduced in Nipbl-deficient embryos (arrows) and anterior ends of lower jaws are indicated (arrowheads).
(EPS)
Nipbla and Nipblb act cooperatively in pectoral fin development. Morphologies of live control (A), Nipbl-deficient (B) and embryos injected with either nipbla-MO (C, D) or nipblb-MO (E, F) at indicated amounts. Dorsal views, anterior to the left. (G) Whisker plots of fin length (medians): 431.8 µm, n = 20 (control), 259.1 µm, n = 40 (nipbla/b-MO), 385.3 µm, n = 40 (nipbla-MO, 0.75 ng), 351.0 µm, n = 40 (nipbla-MO, 1.5 ng), 417.6 µm, n = 40 (nipblb-MO, 0.75 ng), and 399.7 µm, n = 20 (nipblb-MO, 1.5 ng). *: p<10−6.
(EPS)
Small fin phenotypes could be caused by reduced proliferation rather than cell death of fin mesenchymal cells. (A, B) Cell death in pectoral fins of control (A) and Nipbl-deficient larvae (B) at 120 hpf was examined by TUNEL assay. (C–E) Cell death in pectoral fin buds of Nipbl-deficient at 40 hpf was examined by TUNEL assay in comparison with stage-matched (C, 36 hpf) and time-matched (D, 40 hpf) comparisons. TUNEL positive cells are shown in red. Lateral views with anterior to the left. (F–I) Proliferation of pectoral fin buds was examined by BrdU incorporation assay. Stage-matched (F, 36 hpf) and time-matched (G, 40 hpf) controls were compared with Nipbl-deficient embryos (H, 40 hpf). BrdU-positive cells (green) and DAPI stained cells (blue). Lateral views, anterior to the left. (I) Cell proliferation was quantitatively analyzed by determining proportions of BrdU-positive cells in fin mesenchymal cells stained with DAPI. Ave ± S.D.: 73.9±5.29% (n = 8, control at 36 hpf), 73.0±3.78% (n = 9, control at 40 hpf), 48.7±12.0% (n = 12, nipbla/b-MOs-injected embryos at 40 hpf). p-values compared with control at 36 hpf were determined by student t-test. *: p<0.001.
(EPS)
Staging embryos by posterior lateral line primordium position. (A) Posterior lateral line (pLL) primordia (red arrows) detected by ISH for fgf10a at indicated stages move progressively posterior relative to pectoral fin buds (yellow arrows) from 22–48 hpf, indicating a 3–4 hour and 6 hour developmental delay in Nipbl-deficient embryos at 36 hpf and 48 hpf, respectively. Duplicated signals at the tail tips in some panels reflect dorsoventral bifurcation of the tail, rather than ectopic expression of fgf10a, which is a typical phenotype observed in Nipbl-deficient embryos [11]. Lateral views. (B) Summary of stage-match comparisons between control and Nipbl-deficient embryos.
(EPS)
Rescue of AER-fgf gene expression in pectoral fin buds of Nipbl-deficient embryos by exogenous nipbla mRNA. Expression of the AER fgf genes, fgf16 and fgf8a, in pectoral fin buds (arrows) of controls, embryos injected with nipbla/b-MOs, and those co-injected with nipbla/b-MO and 400 pg of nipbla mRNA was examined by ISH at 48 hpf. Dorsal views, anterior to the top.
(EPS)
Expression of genes in the fgf10a signaling pathways is unaffected in Nipbl-deficient embryos. Expression of fgf10a (A; arrows), tbx5a (B) and fgf24 (C) in control and Nipbl-deficient embryos was examined by ISH at indicated stages. fgf10a-expressing pLL primordia are marked by asterisks. Dorsal views, anterior to the top.
(EPS)
Rescue of mesenchymal gene expression in pectoral fin buds of Nipbl-deficient embryos by exogenous nipbla mRNA. Expression of genes in the shha/hand2/5′-hox gene cassette at 36 hpf in pectoral fin buds of controls, embryos injected with nipbla/b-MOs, and those co-injected with nipbla/b-MO and 400 pg of nipbla mRNA was examined by ISH. Dorsal views, anterior to the top.
(EPS)
Expression of genes in the RA signaling pathways is unaffected in Nipbl-deficient embryos. Expression of the RA synthesizing enzyme aldh1a2 and the RA-degrading enzyme cyp26a1, a target of the RA signaling, at 13 (upper) and 19 (lower) hpf in Nipbl-deficient embryos. Anterior somites are indicated by brackets. Dorsal views, anterior to the top.
(EPS)
Expression of hox genes along the anterior-posterior axis of the neural tube is unaffected in Nipbl-deficient embryos. hox gene expression at 36 hpf examined by ISH. Lateral views, anterior to the left. Pectoral fin buds are indicated by black arrows. Anterior and posterior limits of hox expression in the neural tube are indicated by red arrowheads.
(EPS)
Gene expression in pectoral fin buds of CyA-treated embryos. (A) Effects of CyA treatment on development was examined by A-P positioning of pLL primordial expressing fgf10a. Lateral views with anterior to the left. Pectoral fin buds (yellow arrows) and pLL primordial (red arrows) are pointed. Expression of fgf10a in pLL primordial and pectoral fin buds is reduced by CyA treatment and becomes undetectable in pLL primordia by 36 hpf. (B) Expression of mesenchymal genes in pectoral fin buds was examined by ISH in possible stage-match comparisons. Dorsal views, anterior to the top.
(EPS)
Analysis of expression of all mouse Hox genes in wildtype and Nipbl +/− hindlimb buds. (A–D) Expression values show hybridization intensity for probe sets representing all of genes in the four Hox clusters (although the relationship between hybridization intensity and transcript abundance is not necessarily the same for different probesets, intensity gives a rough sense of abundance). Data are graphed as mean ± SEM for the mutant (red) and wild type (blue) samples (see Experimental Procedures). Asterisks indicate genes also shown in Table 1, for which expression changes were observed with strong or moderate statistical significance (1%<FDR<7.5%; double asterisk) or weak statistical significance (FDR<25%; single asterisk); filled arrows show the directions in which expression changes were observed. Open arrows show directional changes that were also tested and confirmed by Q-RT-PCR (see Table 1), whereas “n.c.” marks genes that were tested by Q-RT-PCR and showed no detectable change in expression. The open arrow by Hoxd8 is marked with a question mark because Q-RT-PCR showed a 27% elevation in expression in Nipbl+/− limb buds, but the result was not statistically significant (P = 0.14). The tight error bars on the microarray data for most transcripts, and the confirmation of all significant results by PCR, justify the ability to attach statistical significance even to these modest effect sizes. Overall, the data illustrate that expression changes in Nipbl +/− limb buds are biased toward the 5′ ends of the Hox clusters, and suggest that, as in zebrafish, effects occur at all of the expressed clusters (HoxA, C and D). In the HoxD cluster, which exhibits the highest overall hybridization intensities, the largest fold decreases are seen at the extreme 5′ end (Hoxd12, Hoxd13), and give way, as one moves in the 3′ direction, to a weak decrease (Hoxd11) and then a statistically significant increase (Hoxd10, and possibly Hoxd8). Thus, even though these effects are much smaller in magnitude than in zebrafish (where nipbl knockdown is likely much greater than in Nipbl +/− mice), they appear to show similar positional trends. (E–F) Whole mount in situ hybridization for Hoxd12 (E) and Hoxd13 (F) in E10.5 limb buds of Nipbl+/− and wildtype mice. For both Hox genes, expression is reduced significantly in both forelimbs and hindlimbs, consistent with the results of microarray analysis and Q-RT-PCR (panels A–D, and Table 1).
(EPS)
Expression of genes in the fgf10a pathways is unaffected in Med12-deficient embryos. Expression of fgf24 (30, 48 hpf) and fgf10a (36 hpf) in Med12-deficient embryos examined by ISH. Dorsal views, anterior to the top.
(EPS)
Expression of med12 in Nipbl-deficient embryos. Expression of med12 in embryos injected with nipbla/b-MOs was examined by RT-PCR at 8 and 36 hpf. ef1a was used as a control.
(EPS)
Developmental delay of Rad21-deficient embryos. Effects of Rad21-deficiency (rad21-MO at 2.5 ng/embryo) on embryonic development were examined by A-P positions of pLL primordia by ISH for fgf10a. Lateral views, anterior to the left. Pectoral fin buds (yellow arrows) and pLL primordial (red arrows) are pointed. Expression of fgf10a in pLL primordial and pectoral fin buds of Rad21-deficient embryos were detected at levels significantly lower than control and lost by 54 hpf.
(EPS)
Expression of genes in pectoral fin buds of Rad21-deficient embryos. Effects of Rad21 reduction of expression of mesenchymal genes in pectoral fin buds were examined by ISH in possible stage-match comparisons. Dorsal views, anterior to the top.
(EPS)
Chromosome conformation around the hoxda locus in hindbrain. (A, B) Effects of Nipbl- and Med12-reduction on a higher-order chromatin conformation in hindbrain was examined by 3D-FISH at 38 hpf. Interprobe distances between (A) hoxd and 3′ probes and (B) hoxd and 5′ probes shown by Whisker plots. Details of medians, numbers of nuclei and embryos, and p-values are shown in Table 2. Dotted lines indicate thresholds for separated (upper) and closed (lower) signals in Table 2.
(EPS)