

Abstract
Alterations in dopamine (DA) signaling underlie the most widely held theories of molecular and circuit level perturbations that lead to risk for attention-deficit hyperactivity disorder (ADHD). The DA transporter (DAT), a presynaptic reuptake protein whose activity provides critical support for DA signaling by limiting DA action at pre- and postsynaptic receptors, has been consistently associated with ADHD through pharmacological, behavioral, brain imaging and genetic studies. Currently, the animal models of ADHD exhibit significant limitations, stemming in large part from their lack of construct validity. To remedy this situation, we have pursued the creation of a mouse model derived from a functional nonsynonymous variant in the DAT gene (SLC6A3) of ADHD probands. We trace our path from the identification of these variants to in vitro biochemical and physiological studies to the production of the DAT Val559 mouse model. We discuss our initial findings with these animals and their promise in the context of existing rodent models of ADHD.
Keywords: ADHD, Dopamine, Transporter, Transgenic, Mouse, Animal model
1. Introduction
Attention-deficit/hyperactivity disorder (ADHD) is the most commonly diagnosed neuropsychiatric disorder of childhood, affecting an estimated 4–12% of school-age children (Biederman and Faraone, 2005; Polanczyk et al., 2007; Willcutt, 2012). Adult ADHD is also fairly common, estimated at 4–5% of adults (Fayyad et al., 2007; de Graaf et al., 2008; Kessler et al., 2006). More recent studies suggest that the rates of adult ADHD may actually be greater than 10% (Cahill et al., 2012; Garnier-Dykstra et al., 2010). Indeed, though ADHD is often considered a disorder of childhood and adolescence, studies suggest that ADHD symptoms persist into adulthood in 60–70% of cases (Biederman et al., 2000; Kessler et al., 2005). Like all neuropsychiatric disorders, ADHD presents with a spectrum of behavioral alterations, with features of motor hyperactivity, impulsivity, and/or inattention providing the diagnostic criteria used for diagnosis (American Psychiatric Association, 1994). There are no biomarkers for ADHD, and therefore diagnoses are based on clinical observation, as well as parent and teacher reports (Visser et al., 2013; Wolraich et al., 2003, 2013). ADHD diagnoses exhibit an ~3:1 male:female bias (Gaub and Carlson, 1997; Getahun et al., 2013). Whether this sex bias arises from cultural or biological factors (or both) that impact ADHD risk is unknown. Interestingly, rates of ADHD diagnosis are consistent among different cultural groups, as studies of populations in Africa (Bakare, 2012), Asia (Chien et al., 2012), and Europe (Bianchini et al., 2013; Ezpeleta et al., 2013) report similar prevalence and sex bias. These findings suggest that although environmental factors may be shared across communities, strong biological risk factors likely drive features and risk of the disorder and ultimately, diagnosis.
1.1. Support for a DA Connection to ADHD
A large body of research demonstrates that the dopamine (DA) system underlies the hallmark symptoms of ADHD. For example, locomotor hyperactivity, a main feature of ADHD as well as ADHD animal models, can be induced by treatment with a DA D1 receptor agonist (Brent, 1991; Dreher and Jackson, 1989; Tirelli and Terry, 1993) or psychostimulants that block DAT and increase synaptic DA concentrations (van Rossum and Hurkmans, 1964; Smith, 1964; Zubrycki et al., 1990). Conversely, depletion of DA with reserpine (Johnels, 1982; Sugita et al., 1989) or lesion of DA neurons with 6-hydroxydopamine (Erinoff et al., 1979; Joyce and Koob, 1981) or MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) (Colotla et al., 1990; Gnanalingham et al., 1995; Sahgal et al., 1984; Tsai et al., 1991) leads to a hypokinetic state. Similar hypokinetic characteristics are observed in Parkinson’s disease (Morris et al., 1994; Sian et al., 1999), a brain disorder characterized by death of DA neurons in the substantia nigra.
In addition to locomotor hyperactivity, differences in the DA system have been reported to underlie impulsivity. Work in rodents (Puumala and Sirviö, 1998; Winstanley et al., 2005) and humans (Buckholtz et al., 2010) have demonstrated that differences in DA and DA receptor levels correlate with impulsive traits. Consistent with these observations, animal studies indicate that DA receptor antagonists can reduce impulsivity, whereas AMPH can increase impulsivity (Burton and Fletcher, 2012; Wade et al., 2000). With respect to human studies, striatal DAT (Costa et al., 2013a) and D2-like receptor (Ghahremani et al., 2012; Trifilieff and Martinez, 2014) availability correlate with impulsive traits in healthy human subjects, whereas treatment with DA receptor agonists increases impulsivity (Barake et al., 2013; Ondo and Lai, 2008; Voon et al., 2010). Consistent with these findings, genetic variation in a number of components of DA signaling have been associated with impulsivity (Dalley and Roiser, 2012; Forbes et al., 2009).
1.2. Support for a DAT connection to ADHD: pharmacological considerations
A major connection between DAT and ADHD is the utility of DAT-targeted agents for treatment of the disorder (Bitter et al., 2012; Vaughan and Kratochvil, 2012). Thus, both methylphenidate (MPH) (e.g. Ritalin®) and amphetamine (AMPH) formulations (e.g. Adderall®) are effective in the treatment of subjects with ADHD (Faraone et al., 2002; Janols et al., 2009; Minzenberg, 2012; Treuer et al., 2013). Psychostimulant treatment in ADHD is often labeled as “paradoxical”, since, as indicated by their categorical definition, ADHD medications, particularly d-amphetamine, can produce motor activation (Glick and Milloy, 1973; Ralph et al., 2001) and disruption of cognitive performance (Ornstein et al., 2000; Sanday et al., 2013; Stefani and Moghaddam, 2002), even psychosis (Bramness et al., 2012; Grant et al., 2012; Segal and Kuczenski, 1997; Wallis et al., 1949). Some researchers discount differences in response between ADHD and normal adolescents to psychostimulants, reporting that pre-pubertal adolescents respond to psychostimulants oppositely to that seen in adults (Rapoport et al., 1978; Zahn et al., 1980), and thus the “paradoxical” effect of psychostimulants in ADHD may be more a bias based on expectations from adult actions of the drugs. However, this position seems to be at odds with continued medication benefits seen by adults with the disorder. With respect to animal models, psychostimulant drugs increase locomotor activity in both adult and adolescent rodents (Cirulli and Laviola, 2000; Gainetdinov et al., 1999; Good and Radcliffe, 2011; Kameda et al., 2011). Whether a “normal animal” is an appropriate model for the actions of these drugs in ADHD is certainly debatable. As we discuss later in this report, psychostimulants decrease locomotor hyperactivity in a number of current animal models of ADHD (Gainetdinov, 2010; Russell, 2011). But such observations may be misleading if the underlying causes of ADHD are not mirrored in the model, leading to convergent behavioral characteristics that appear related to the human disorder but that may diverge when it with respect to utility in understanding ADHD mechanisms.
Support for a role of altered DA signaling also comes from the actions of ADHD medications. MPH is a competitive DAT antagonist like cocaine, though more slowly acting, whereas AMPH perturbs DA signaling through multiple mechanisms (Sulzer et al., 2005). First, AMPH is a competitive substrate for DAT, precluding normal DA clearance. Second, the drug acts to trigger depletion of vesicular DA stores by a weak-base action on the intravesicular pH gradient that is required to concentrate DA. Recently, this action has been reported to differentially impact readily releasable versus reserve pool vesicles (Covey et al., 2013), where depletion was observed to occur with a loss of DA from reserve pool vesicles and an enhancement of vesicular release involving the readily releasable vesicle pool. The latter observation may involve the ability of AMPH to elevate intracellular Ca2+, known also to support the phosphorylation of DAT on the transporter’s N-terminus (Fog et al., 2006; Gnegy et al., 2004; Khoshbouei et al., 2004; Wei et al., 2007). Phosphorylation (evidence implicates CaMKII, and possibly PKCβ) then leads to an increased probability for DAT-dependent efflux of cytoplasmic DA. Fourth, AMPH is an MAO antagonist, and through a lack of DA catabolism leads to further elevation of DA cytoplasmic levels.
1.3. Support for a DAT connection to ADHD: brain imaging studies
Positron emission tomography (PET) methods have afforded a direct inspection of DAT levels in the brain of human ADHD subjects (Varrone and Halldin, 2010; Zimmer, 2009). However, the findings with this approach have been mixed, possibly due to prior drug exposure in some studies (Fusar-Poli et al., 2012). Thus, whereas DAT binding in the basal ganglia of both children (Cheon et al., 2005) and adults (Dougherty et al., 1999; Dresel et al., 2000; Krause et al., 2000) has been reported to be increased (Spencer et al., 2007) in ADHD, others have seen no change (van Dyck et al., 2002) or decreased DAT density in ADHD (Volkow et al., 2007).
Subsequent studies have focused on brain imaging abnormalities within specific domains of ADHD. Volkow and colleagues correlate reduced DAT and D2-like receptor availability in ADHD subjects with motivational deficits stemming from dysfunction in dopamine reward pathways (Volkow et al., 2011). However, other groups report that increased DAT availability contributes to impulsivity (Costa et al., 2013a; Forbes et al., 2009). Studies examining DAT variability (namely the DAT 3′ VNTR, discussed in Section 2.4) have similarly disparate findings, with a study suggesting an association between the DAT VNTR 10-repeat allele and frontal, medial, and parietal activation during a response inhibition task (Braet et al., 2011), and meta-analysis of SPECT studies reporting no effect of the DAT VNTR on striatal DAT availability (Costa et al., 2011).
Some of the conflicting reports of DAT availability in ADHD may reflect a differential role for DAT in specific ADHD traits. However, methodological limitations, including inadequate sensitivity and imaging ligands affected by endogenous DA (Weyandt et al., 2013), may restrict our ability to interpret imaging studies. Further research is required to improve DAT imaging approaches and clarify our understanding of DAT in ADHD.
1.4. Support for a DAT connection to ADHD: genetic studies
Many lines of evidence implicate DAT and DA receptors in ADHD. For example, genetic studies have repeatedly demonstrated an association between ADHD and D1 (Bobb et al., 2005; Ribases et al., 2012), D2 (Nyman et al., 2007), D4 (Roman et al., 2001; Bidwell et al., 2011) and D5 (Manor et al., 2004) receptors, though how the genetic variants perturb receptor function in ADHD is unclear. Several studies have also observed association between core components of ADHD and variation in the DAT gene, including spatial working memory function (Brehmer et al., 2009; Li et al., 2012; Shang and Gau, 2013), attentional asymmetry (Bellgrove et al., 2005; Newman et al., 2012), impulsivity (Costa et al., 2013a; Forbes et al., 2009; Paloyelis et al., 2010), response inhibition (Cornish et al., 2005; Cummins et al., 2012), and reward-related striatal responsivity (Hahn et al., 2011; Hoogmann et al., 2013; Wittmann et al., 2013). In addition, DAT genotype is linked to how well a patient responds to psychostimulant treatment (Costa et al., 2013b; Gilbert et al., 2006; Kambeitz et al., 2013; Pasini et al., 2013).
The DAT messenger RNA (mRNA) possesses a large, 3′ untranslated region (UTR) that contains a variable number tandem repeat (VNTR) polymorphism (Vandenbergh et al., 1992a, 1992b) (Fig. 1). The DAT VNTR is a 40-base pair sequence that repeats 3 to 13 times, with 9- and 10-repeats being the most common in human populations (Doucette-Stamm et al., 1995; Gerlernter et al., 1998; Kang et al., 1999). Multiple studies have found a link between the 10-repeat VNTR allele and ADHD (Faraone et al., 2013; Kambeitz et al., 2013), however this finding has not been replicated in all studies (Holmes et al., 2000; Palmer et al., 1999; Todd et al., 2001). Interestingly, recent studies have linked the 10-repeat VNTR allele to risk for childhood ADHD and the 9-repeat VNTR allele with persistent adult ADHD symptoms (Dresler et al., 2010; Franke et al., 2010).
Fig. 1.
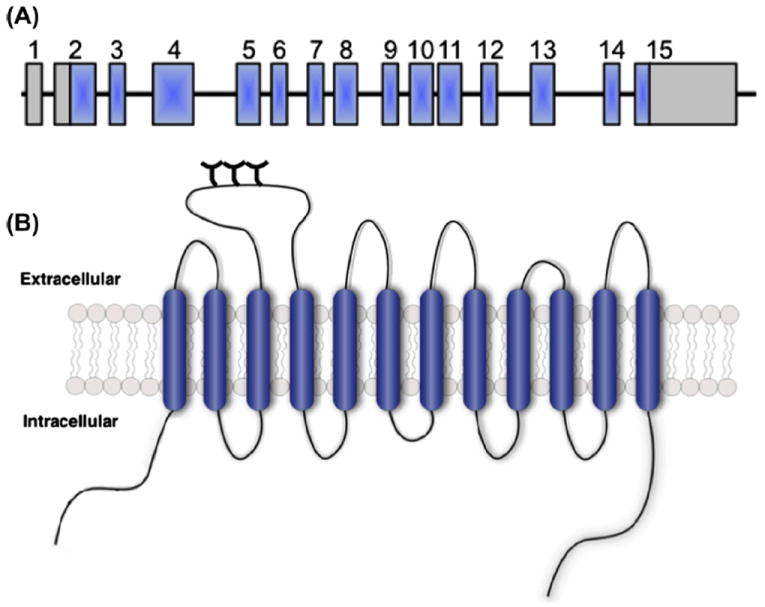
Gene and protein structure of the SLC6A3 gene encoding DAT. (A) The SLC6A3 gene consists of 15 exons (blue boxes), spanning 60 kb of genomic DNA sequence, with both 5′ and 3′ untranslated regions (gray boxes). Please note, gene structure is not depicted to scale. (B) Schematic representation of the DAT protein, a 620 amino acid protein with 12 transmembrane domains and intracellular N- and C-termini. Figure reproduced from Mazei-Robison and Blakely, 2006.
1.5. Support for a DAT Connection to ADHD: search for functional coding variation
Although DAT plays a significant role in normal DA signaling, is a prominent drug target, and has been repeatedly implicated by genetic association studies in disorders such as ADHD, only limited efforts have sought evidence for functional coding variation associated with the DAT gene (SLC6A3) that could contribute to ADHD risk. Thus far, several studies have sought evidence for SLC6A3 coding polymorphisms, identifying multiple subjects both heterozygous (Cargill et al., 1999; Grünhage et al., 2000; Hamilton et al., 2013; Mazei-Robison et al., 2005; Mergy MA and Blakely RD, unpublished data; Puffenberger et al., 2012; Sakrikar et al., 2012; Vandenbergh et al., 2000), and homozygous (Kurian et al., 2009, 2011) for nonsynonymous variants. Screening of ADHD subjects for new DAT coding variants is also ongoing in various diseases including bipolar disorder and autism with comorbid ADHD (Davis and Kollins, 2012; Mahajan et al., 2012; Rommelse et al., 2011). Disease-associated DAT coding variants are listed in Table 1 and shown on the DAT structure in Fig. 2. Other heterozygous coding variants that have been characterized for functional deficits (V24M, V55A, R237Q, and E602G) have yet to demonstrate changes in transporter protein expression or DA transport function in vitro. V382A DAT, which has yet to be mapped to a specific clinical phenotype, demonstrated upon in vitro expression a reduction in transporter protein levels, reduced capacity for both DA transport, and a reduced capacity for phorbol ester (PMA)-induced trafficking (Mazei-Robison and Blakely, 2005). The authors suggest that DAT Ala382 stabilizes an inactive conformation in the plasma membrane such that the loss of uptake exceeds that expected by PKC-induced internalization. Sakrikar and colleagues identified the variant R615C in an ADHD subject and found the Cys615 variant to display a significant reduction in cell surface DAT levels (approximately 50% of WT DAT) (Sakrikar et al., 2012), a commensurate reduction in DA transport velocity, and a shift in surface distribution from regulated to constitutive recycling. Accompany these changes was an insensitivity to the endocytic acceleration of DAT endocytosis by AMPH or protein kinase C (PKC) activation. The constitutive endocytic phenotype of DAT Cys615 was accompanied by hyperphosphorylation, increased association with calcium/calmodulin-dependent protein kinase II (CaMKII), decreased flotillin-1 association, and a disrupted localization to GM1 ganglioside-enriched membrane microdomains. These findings led Sakrikar and colleagues to propose a model whereby the distal DAT C-terminus dictates targeting to surface membrane microdomains where the transporter acquires its post-translational regulatory influences.
Table 1.
Disease-associated DAT coding variants. ADHD = attention deficit hyperactivity disorder; DTDS = dopamine transporter deficiency syndrome (juvenile dystonia/Parkinsonism).
| Variant | Disease Association | References |
|---|---|---|
| Val24Met | ADHD | M. Mergy, unpublished findings |
| Val55Ala | ? | Vandenbergh et al. (2000) |
| Val158Phe | DTDS | Kurian et al., 2011 |
| Leu167Phe | ADHD | M. Mazei-Robison, unpublished findings |
| Leu224Pro | DTDS | Kurian et al. (2011) |
| Arg237Gln | ? | Cargill et al., 1999 |
| Gly327Arg | DTDS | Kurian et al. (2011) |
| Leu368Gln | DTDS | Kurian et al. (2009, 2011) |
| Val382Ala | ? | Vandenbergh et al. (2000) |
| Pro395Leu | DTDS | Kurian et al. (2009, 2011) |
| Arg521Trp | DTDS | Kurian et al. (2011) |
| Pro529Leu | DTDS | Kurian et al. (2011) |
| Pro554Leu | DTDS | Kurian et al. (2011) |
| Ala559Val | Bipolar, ADHD | Grünhage et al. (2000); Mazei-Robison et al. (2005) |
| Glu602Gly | Bipolar | Grünhage et al. (2000) |
| Arg615Cys | ADHD | Sakrikar et al. (2012) |
Fig. 2.
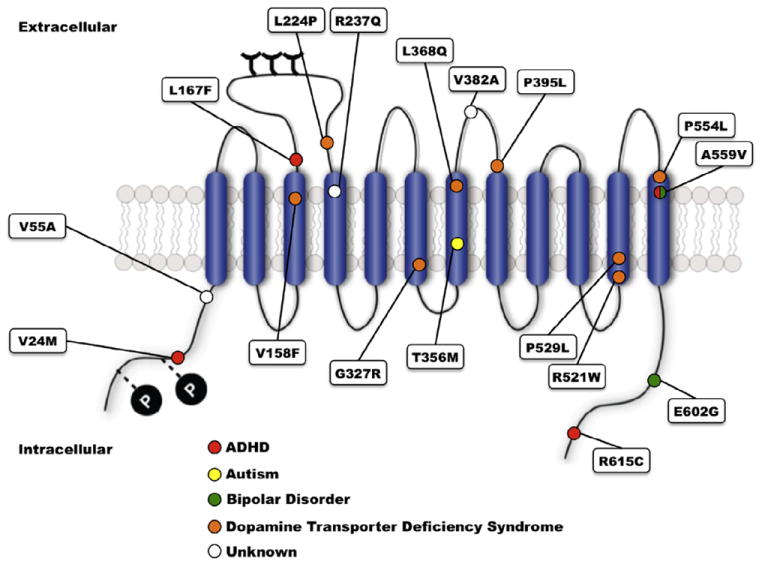
Location of disease-associated hDAT variants. ADHD-associated variants are marked with red circles, ASD-associated variants with yellow circles, bipolar disorder-associated variants with green circles, DAT deficiency syndrome-associated variants with orange circles, and variants without disease association with white circles.
Recent advances in whole-exome and whole-genome sequencing (1000 Genomes Project Consortium, 2010) have resulted in a plethora of new SLC6A3 nonsynonymous variants. Since these exome screening projects target either subjects without a clinical diagnosis or community samples with no available clinical history, variants identified are generally not associated with a particular disorder. Reported DAT coding variants (D = found in dbSNP database, G = found by 1000 Genomes, N = found in National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project) without known disease association are as follows: K3N (D, G, N), M11I (D, G, N), M11V (D, G), S12P (D, G, N), V14M (D, G), A16T (G), P17L (G,N), E20(stop) (D), E20V (G, N), I32M (G, N), V24A (G), G39R (G), L42F (D, G), P50L (D, G, N), S53R (D, G, N), V73I (D, G, N), L104I (D, G, N), G121S (G), V131I (D, G), L138P (G), L138R (G), A161T (G), A163V (G), A192T (D, G), S198T (G), S202L (D, G), S202W (D, G, N), G209R (G, N), V221M (D, G, N), R237W (G, N), V245A (D, G), I268V (G, N), T271N (G), V275L (D, G, N), L281P (G), G289R (G), G293S (G), V300I (D, G, N), E307K (D, G), A308V (G), A314V (G), D345G (G, N), A346T (G, N), A346V (D, G, N), F362L (G), L368Q (G), Q373R (D, G, N), G380R (G, N), P395L (G) G433R (G), D436N (G), R445G (G), A455V (G, N), V464I (D, G, N), V471I (D, G), I490V (D, G, N), V501A (G, N), Q509H (D, G), R515W (G), S517T (G), G538A (D), V538I (G, N), R544S (G), P545T (G, N), H547Q (D, N), A559T (D, G), A576E (G), K579R (G, N), R588Q (G, N), G607W (G), R610H (D, G), T613M (G, N), K619N (D, G, N). The reader is referred to the 1000 Genomes website (http://www.1000genomes.org) for information regarding the estimated frequencies of SLC6A3 variants, which generally are quite low, consistent with the existence of only rare nonsynonymous variation in the genes of the SLC6 transporter family. Interestingly, of the variants in DAT identified in disease-targeted studies, V24M, L167F, R237Q, A346V, V464I, A559V, and E602G are present in these whole genome efforts, though with no clinical information. Given the frequency of mental illness, both diagnosed and undiagnosed, it is likely that a significant fraction of the carriers of these variants meet criteria for a DSM-IV disorder, including ADHD.
1.6. Support for a DAT connection to ADHD: DAT Val559 and anomalous DA efflux
Our efforts to identify functional variation in DAT that impact risk for brain disorders originated with a screening of subjects diagnosed with ADHD for variations in DAT coding sequences and SLC6A3 exon splice junctions (Mazei-Robison et al., 2005). These efforts identified A559V in a pair of brothers, both diagnosed with ADHD. Interestingly, the DAT Val559 variant had been identified previously in a female diagnosed with bipolar disorder, though information related to the mode of transmission or shared traits could not be pursued in the parents (Grünhage et al., 2000). Given the association of DAT with both ADHD and bipolar disorder (Greenwood et al., 2013), it is possible that DAT Val559 variant may have contributed to this subject’s disorder.
Heterologous expression studies revealed that DAT Val559 supports comparable total and cell surface DAT protein expression, as well as DA transport kinetics, as DAT Ala559. However, the variant transporter induces an anomalous, DAT-mediated, outward “leak” of cytoplasmic DA when cells are pre-loaded with DA, which we have termed anomalous DA efflux (ADE) (Fig. 3A). The ADE of DAT Val559 was shown to be voltage-dependent, with DA efflux more prominent at depolarized membrane potentials (Fig. 3B). AMPH has been shown to trigger a rise in intracellular Na+ that contributes to the mechanism supporting DA efflux (Khoshbouei et al., 2003). In DAT Val559 transfected cells, the transporter’s sensitivity to intracellular Na+ increases, providing one mechanism by which ADE may be facilitated. Surprisingly, we found that AMPH lacked an ability to induce DA efflux in DAT Val559-transfected cells, instead acting only as a DAT antagonist, blocking ADE (Fig. 3C). These findings led to the development of a synaptic model for the ADHD of Val559 carriers, described in Figs. 4A-C. As illustrated, at wild-type (WT) DAT Ala559 containing synapses, DA is released by fusion of synaptic vesicles, triggered by afferent electrical stimulation (Fig. 4A) followed by efficient DAT-mediated DA uptake that inactivates DA signaling. In contrast, at DAT Val559 expressing synapses, ADE exists in parallel with vesicular DA release, mimicking the affects of AMPH on WT synapses, and leading to a loss of a tight coupling of DA release to afferent input (Fig. 4B). Although psychostimulant treatment reduces DA clearance by blocking DAT, this interaction also prevents ADE, restoring a more normal relationship between DA neuron activity and extracellular DA levels (Fig. 4C). This mechanism also provides an explanation for why only a single Val559-encoding transporter could lead to ADE and elevate risk for ADHD.
Fig. 3.
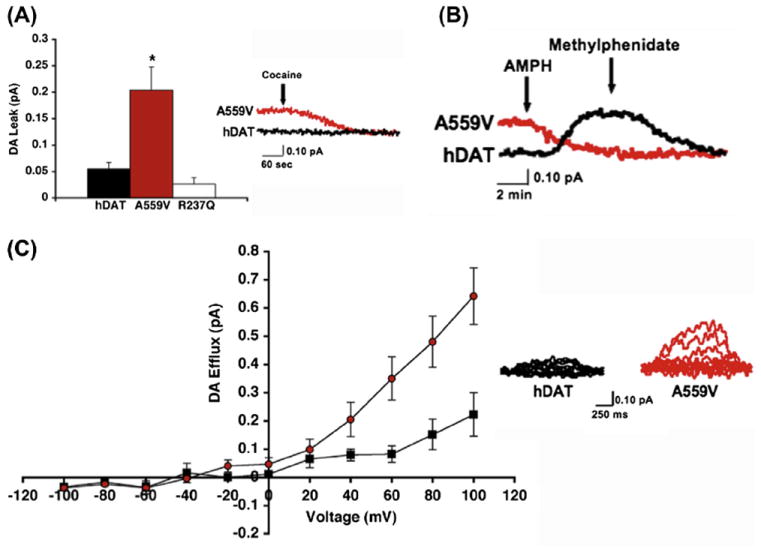
The hDAT A559V mutation supports anomalous DA efflux. (A) DAT Val559 mediates enhanced outward DA leak, reported as mean amperometric current ± SEM. (B) DAT Val559-transfected cells display enhanced voltage-dependent DA amperometric currents; left: mean amperometric current ± SEM, right: representative amperometric traces from WT DAT and DAT Val559 cells. (C) Representative amperometric trace showing that AMPH-evoked DA release can be blocked by methylphenidate in WT DAT-transfected cells (black trace), whereas AMPH blocks basal DA efflux in DAT Val559-transfected cells (red trace). No further reduction in amperometric current is observed upon application of MPH with AMPH. Figure reproduced from Mazei-Robison et al., 2008.
Fig. 4.
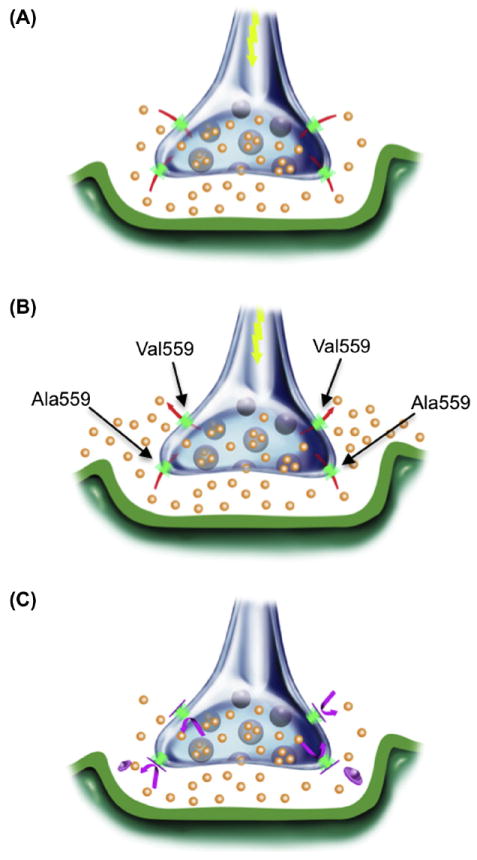
Illustration of disrupted synaptic transmission as a result of DAT-mediated ADE in ADHD subjects (A) Under normal circumstances, presynaptic DAT (green) inactivates and recycles DA (orange) released via vesicular fusion. (B) AMPH (red) acts both as a DAT substrate and as a reuptake inhibitor, eliciting reverse transport and blocking normal DA reuptake, thereby increasing synaptic DA levels. (C) At heterozygous DAT Val559 synapses, basal DAT-mediated DA efflux is a second, unexpected and temporally distinct mechanism for DA release, although normal DA reuptake can still occur. (D) Drugs such as MPH or cocaine (magenta) that block DAT-mediated ADE limit DA released only to that arising from vesicular fusion. Figure reproduced from Mazei-Robison et al., 2008.
Subsequent studies of the DAT Val559 variant (Bowton et al., 2010) revealed that ADE is sustained by tonic D2 receptor (D2R) signaling, as well as by CaMKII action. CaMKII-mediated phosphorylation of the WT DAT N-terminus is known to support reverse transport of DA in response to AMPH treatment (Fog et al., 2006). In keeping with a role for CaMKII in supporting ADE, Bowton and colleagues found that the N-terminus of DAT Val559 to be hyperphosphorylated (Fig. 5) (Bowton et al., 2010), suggesting that the DAT Val559N-terminus is more accessible to CamKII, consistent with elevated Val559 association with the kinase, and the ability of CamKII inhibitors to block ADE. Finally, the DAT Val559 variant induces an increase in outward channel states, previously associated with AMPH action (Kahlig et al., 2005). These studies support a model whereby DAT Val559 exhibits an abnormal residence of the transporter in an “efflux-willing” state as if being constantly stimulated with AMPH.
Fig. 5.
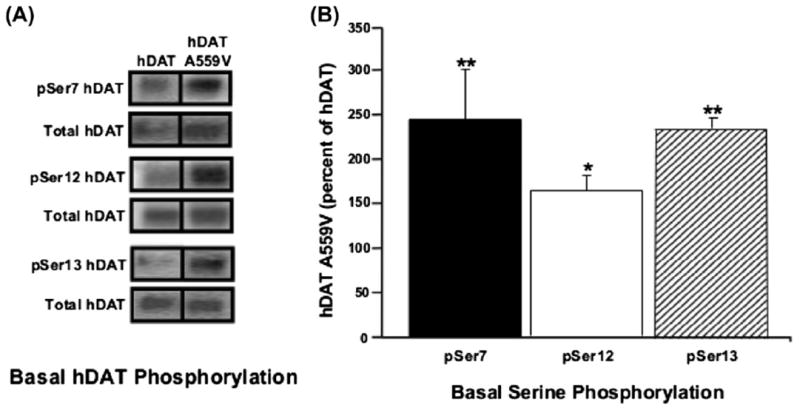
CaMKII-mediated hyperphosphorylation of the DAT Val559N-terminus. (A) Representative immunoblots of WT DAT and DAT Val559-transfected cells for phosphorylated serines at amino acids 7, 12, and 13 (pSer7, pSer12, and pSer13, respectively), and for total DAT. Inset: immunoblot for dopamine D2 receptor (top) and CaMKII (bottom), confirming their presence in the heterologous cell system. (B) Quantification of pSer DAT band intensities, normalized to total DAT. Figure reproduced from Bowton et al., 2010.
Recently, Hamilton and colleagues characterized a functional SLC6A3 variant, T356M, identified in a subject with autism spectrum disorder (ASD) (Hamilton et al., 2013). Key to the findings is the de novo status of the variant. ASD has been found to exhibit an increased number of de novo variants (Kenny et al., 2013; Jiang et al., 2013), including copy number variants (CNVs) (Griswold et al., 2012; Holt et al., 2012; Krumm et al., 2013; Menashe et al., 2013; Poultney et al., 2013; Vaishnavi et al., 2013) as well as single nucleotide polymorphisms (SNPs). Because de novo coding mutations are low in probability, particularly in the coding regions of genes, geneticists consider de novo variations to be of particular significance in establishing genetic risk. Approximately one third of subjects with ASD meet DSM-IV criteria for ADHD and subjects with ADHD are more likely to have ASD traits than found in the general population (Kotte et al., 2013; Leyfer et al., 2006). Functional studies indicate that the DAT Met356 variant exhibits significantly reduced DA uptake, as well as ADE. Structural evidence suggests that the variant may induce a more prominent outward-facing conformation which may be more conducive to ADE. Finally, in keeping with the reduced activity of DAT Met356, the mutant fails to reverse the hyperactivity of flies that lack DAT.
Our discovery of two cases of ADHD-associated, functional variation in DAT (Val559 and Cys615), along with several other variants being characterized, as well as the recent discovery of DAT Met356, provides both rationale and opportunity for the generation of construct-valid, transgenic mouse models of ADHD. Below, we describe our initial efforts to capture this opportunity using the DAT Val559 variant, followed by a discussion of our efforts in the context of existing rodent models of ADHD.
2. Materials and methods
2.1. Generation of DAT Val559 knock-in mouse
All procedures with animals were performed according to a protocol approved by the Vanderbilt Institutional Animal Care and Use Committee (IACUC). Transgenic mice were produced using a linearized construct, where 5′ and 3′ arms were derived from 129S6 genomic DNA that was mutated in the 5′ arm to encode Val at amino acid 559 by oligonucleotide mediated site-directed mutagenesis (Stratagene Quik-Change Mutagenesis Kit, Agilent Technologies, Santa Clara, CA). Our construct includes self-excising Cre recombinase and neomycin-resistance cassettes for positive selection and a thymidine kinase cassette for negative selection. The construct was electroporated into 129S6/SvEvTac-derived embryonic stem cells (TL-1) and successful homologous recombination was confirmed by Southern blotting, with the presence of the Val559 substitution confirmed directly by Sanger sequencing. Targeted stem cells were then injected into C57BL/6J blastocysts and implanted in pseudo-pregnant females. Chimeric offspring were mated with WT C57BL/ 6J mice to test for germline transmission of the DAT Val559 allele. Thereafter, genotypes of all mice were determined by PCR of genomic DNA isolated from tail snips using the REDExtract-N-Amp Tissue PCR Kit (Sigma, St. Louis, MO) using as forward primer, CAG CAT GGA AAA AAT CCA TGA A and as reverse primer, AGC TAT ATT CAC CAT CAA AAG G. Products generated in this analysis are WT, 490 base pair (bp), homozygous Val559, 561 bp product and both 490 and 561 bp products for heterozygous animals. A male founder was then crossed with a 129S6 female to generate the initial colony for breeding. Mice were maintained under standard housing conditions on a 12-h light/dark cycle (lights on 0600–1800 for breeding colony; lights off 0200–1400 for all animals used in experiments) with food (irradiated LabDiet 5L0D chow, PMI Nutrition International, St. Louis, MO) and water provided ad libitum.
2.2. Assessment of growth and reproduction of DAT Val559 mice
To assess growth, male mice of the three genotypes were weighed weekly from 3 to 12 weeks of age. Data were plotted and analyzed statistically by a two-way analysis of variance (ANOVA) using Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA), with P < .05 taken as indication of statistical significance. To assess reproduction, the number and sex of pups generated were tabulated over a course of 33 months. All animals studied derived from heterozygous breedings (75% 129S6, 25% C57BL/6J). Assessment of genotype and gender distributions were compared to expectations derived from Hardy–Weinberg equilibrium using χ2 tests, implemented in Prism 5.0 software, with P < .05 taken as indication of statistical significance.
2.3. Analysis of sensorimotor function in DAT Val559 mice
To assess sensorimotor function in the DAT Val559 mice, we implemented a modified version of the Irwin test battery (Irwin, 1968). Mice were observed and recorded over 2 consecutive days during the light phase (1600–2000).
2.3.1. General physical condition
The presence of whiskers, quality of fur, general coat condition, bald patches, piloerection, limb tone, and body tone were observed in a clean cage containing corncob bedding.
2.3.2. Motor abilities and reflexes
Trunk curl: Each mouse was lifted up 12–18 inches by the base of the tail followed by assessment of the presence of nose-to-tail curling. Forepaw reaching: Each mouse was lifted by the base of the tail and moved horizontally towards a metal wire food bin/cage insert. The degree to which the mouse extended its forepaws as it approached the wire food bin was scored (no reaching = 0, reaching upon nose contact = 1, reaching upon whisker contact = 2, reaching before whisker contact (18–20 mm) = 3, early, vigorous reaching (>25 mm) = 4). Wire hang: Two to four mice were placed on a metal grid screen (10 × 14 cm) into individual compartments. After placement, mice were given time to establish a grip on a wire screen before it was inverted 60 cm over a clean plastic cage containing fresh corncob bedding. The latency to fall was recorded up to 60 s, at which point the mice were removed from the apparatus and returned to the home cage. Positional passivity: Each mouse was subjected to sequential handling and the reaction to handling assessed. Mice were first restrained by the tail (score = 0), then gently restrained by the neck (score = 1), restrained by the nape of the neck (scruffed) and held supine (score = 2), and finally restrained by the hind legs (score = 3). Data are presented as the number of mice showing any positional passivity. Rotarod: Motor coordination and balance were assessed using an accelerating Rotarod apparatus (Ugo Basile, Comerio VA, Italy). Mice were placed on the rotating cylinder (3 cm in diameter) and confined to a segment of the cylinder approximately 6 cm wide by gray plastic dividers. The rotational speed of the cylinder was increased from 5 to 40 rpm over the 5-min testing period. The latency at which mice fell off of the rotating cylinder was measured. Each mouse was given a trial on the Rotarod before performance was assessed. Grip strength: Grip strength was measured using a force gauge attached to a small metal grate (8 × 8 cm). Each mouse was allowed to grip the metal grate with its forepaws, then gently pulled backwards by the base of the tail until it released the grate. Grip strength was recorded with a digital gauge that returned the maximum force during each trial. The average grip strength, given in Newtons, is the average of three trials. Righting reflex: Mice were inverted to a supine position in the researcher’s hand. The mouse was released and the ability of the mouse to right itself assessed. Air righting reflex: Mice were inverted to a supine position in the researcher’s hand while being held approximately 30 cm above a cage containing 8–10 cm of clean corncob bedding. Mice were then released and allowed to drop into the cage and bedding below. The ability of the mouse to right itself while falling was assessed. Data for all measures of motor function were analyzed using a one-way ANOVA and Bonferroni’s post hoc test for multiple comparisons, with P < .05 taken as indication of statistical significance.
2.3.3. Sensory testing
Ear twitch: Mice were gently restrained and the auditory meatus gently touched with the tip of a size 6.1 Touch Test (Von Frey) Sensory Evaluator (Stoelting, Wood Dale, IL). The presence of a reaction (active retraction and/or flick of the ear) was assessed. Petting escape: Mice were stroked down the length of the body starting with a light touch and gradually increased pressure as the researcher approached the mouse’s tail. The escape reaction was assessed as follows: no response (score = 0), mild (escape from firm pressure; score = 1), moderate (escape from light pressure; score = 2), or vigorous (escape from approach; score = 3). Data are presented as the number of mice showing an escape response. Data for all measures of sensory function were analyzed using a one-way ANOVA and Bonferroni’s post hoc test for multiple comparisons, with P < .05 taken as indication of statistical significance.
3. Results
3.1. Generation of DAT Val559 mice
In order to express the DAT Val559 variant from the endogenous mouse Slc6a3 locus, we pursued a homologous recombination, knock-in strategy with a construct bearing an altered codon encoding the Val substitution in exon 13 as described in Materials and Methods, along with a self-excising loxP-flanked Cre recombinase and neomycin resistance (NeoR) cassettes in the 3′ intron (Fig. 6A). Following the screening of embryonic stem cell clones that passed both positive and negative selection by Southern blots, we identified two clones that appeared to be successfully targeted. Subsequent sequencing verified that one of the two stem cell clones possessed the sequence encoding DAT Val559 and no evidence of other coding mutations.
Fig. 6.
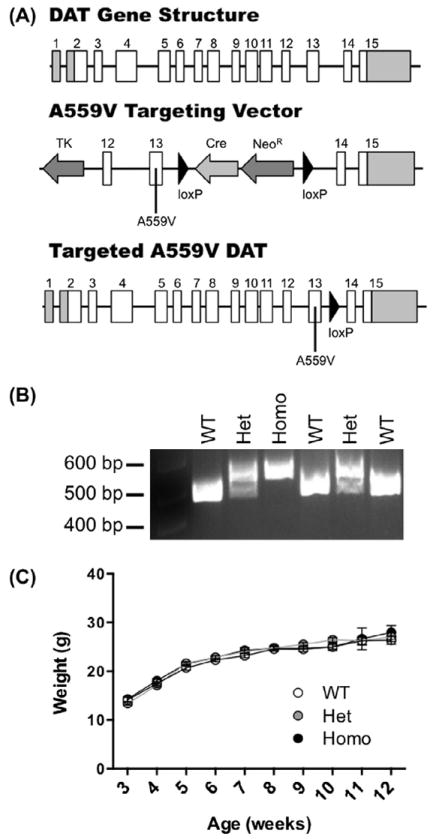
Generation of DAT Val559 mice by homologous recombination. (A) Structure of the Slc6a3 gene (top), DAT Val559 targeting construct (middle), and successfully recombined DAT gene (bottom). Coding exons are displayed as white boxes and non-coding exonic sequence as gray boxes. Following successful recombination and excision of Cre recombinase and neomycin resistance (NeoR) cassettes, (B) genotypes of all mice are determined by PCR (490 base pair (bp) band = WT, 561 bp band = DAT Val559 homozygote, 490 and 561 bp bands = DAT Val559 heterozygote). (C) Growth and development of mice post-weaning does not differ among DAT genotypes.
DAT Val559-positive stem cells were injected into blastocysts, producing four chimeric males. Three of the chimeras achieved germline transmission, as assessed by genotyping of pups (Fig. 6B). One of the chimeras positive for germline transmission of both the Val559 variant as well as Neor excision was selected to establish a breeding colony. We also backcrossed to C57BL/6 and 129S6 strains to generate congenic lines. (data not shown). All experiments to date have been performed with animals maintained on a 75% 129S6/25% C57BL/6 hybrid background.
3.2. Breeding of DAT Val559 mice
Considering 514 separate breeding events of heterozygous DAT Val559 parents, we observed 5.2 ± 0.1 pups per litter, assessed at 3 weeks of age when animals were weaned and genotyped. This value is in line with the categorization of 129 strains as good breeders with three to seven pups per litter (http://jax-mice.jax.org/support/husbandry/breeding-considerations.html). When the genotypes of male and female offspring within these litters were pooled, a significant deviation from Hardy–Weinberg distribution was detected (χ2 test: P < .05). This effect did not arise from an alteration in the genotype distribution of females (P > .05). Rather, males drove the effect, demonstrating a significant overall deviation (P < .05) arising from a significant reduction in the presence of homozygous Val559 males (Table 2).
Table 2.
Expected and observed allele frequencies of DAT Val559. The DAT Val559 allele is significantly under-represented in the total population of post-weaning mice (P < 0.01, χ2 test), driven primarily by a reduction in male mice possessing the DAT Val559 allele (P = 0.001, χ2 test). Males were obtained from 446 litters, females were obtained from 367 litters.
| Male | Female | Total | |
|---|---|---|---|
| Expected | |||
| WT | 332 (25%) | 286.5 (25%) | 618.5 (25%) |
| Het | 664 (50%) | 573 (50%) | 1237 (50%) |
| Homo | 332 (25%) | 286.5 (25%) | 618.5 (25%) |
| Male*** | Female | Total** | |
|
| |||
| Observed | |||
| WT | 386 (29.07%) | 299 (26.09%) | 685 (27.69%) |
| Het | 650 (48.95%) | 561 (48.95%) | 1211 (48.95%) |
| Homo | 292 (21.99%) | 286 (24.96%) | 578 (23.36%) |
P = 0.0057
P = 0.0010,
3.3. Growth of DAT Val559 mice
After weighing male mice weekly from 3 to 12 weeks of age, we assessed the growth relationships between WT, heterozygous and homozygous DAT Al559 mice. No significant genotype, time, or genotype × time interactions were observed (P > .05, two-way ANOVA) (Fig. 6C). Similarly, no genotype effects were evident in comparing body length (rump to nose, cm): WT, 8.10 ± 0.39 (n = 10); het, 8.36 ± 0.23 (n = 11); homo, 8.10 ± 0.32 (n = 10).
3.4. Sensorimotor characteristics of DAT Val559 mice
General sensorimotor assessment of mice at 7 weeks revealed no significant genotype differences in health, reflexes or basic motor functions (Table 3). Although the overall ANOVA did not detect a genotype difference in time to fall in the wire hang test, this measure displayed a trend for a progressive genotype decrease in latency, with an ~50% reduction occurring in homozygous DAT Val559 vs. WT animals (P = .06, unpaired Student’s t-test).
Table 3.
Sensorimotor evaluation of DAT Val559 mice. DAT Val559 hetero- and homozygous mice display no deficiencies in measures of general health, motor capability, or reflexes (each test analyzed by one-way ANOVA, P > .05 for all measures).
| WT | Het | Homo | |
|---|---|---|---|
| General | |||
| Missing whiskers | 1/8 | 0/8 | 0/8 |
| Poor coat condition | 0/8 | 0/8 | 0/8 |
| Piloerection | 0/8 | 0/8 | 0/8 |
| Bald patches | 0/8 | 0/8 | 1/8 |
| Body tone (# with normal tone) | 8/8 | 8/8 | 8/8 |
| Limb tone (# with normal tone) | 8/8 | 8/8 | 8/8 |
| Respiration rate (# abnormal) | 0/8 | 0/8 | 0/8 |
| Heart rate (# abnormal) | 0/8 | 0/8 | 0/8 |
| Tremor | 0/8 | 0/8 | 0/8 |
| Gait (# abnormal) | 0/8 | 0/8 | 0/8 |
| Motor ability | |||
| Trunk curl | 1/8 | 0/8 | 3/8 |
| Forepaw reaching | 8/8 | 8/8 | 8/8 |
| Horizontal wire hang | 8/8 | 8/8 | 8/8 |
| Vertical wire hang | 8/8 | 8/8 | 8/8 |
| Pole climb | 8/8 | 8/8 | 8/8 |
| Inverted screen (latency to fall, sec) | 34.9 ± 8.1 | 22.9 ± 8.3 | 16.7 ± 3.7 |
| Positional passivity | 0/8 | 0/8 | 0/8 |
| Rotorod (latency to fall, sec) | 171.9 ± 25.8 | 204.0 ± 10.1 | 166.9 ± 23.5 |
| Grip strength (N) | 1.71 ± 0.17 | 1.43 ± 0.04 | 1.71 ± 0.13 |
| Reflexes | |||
| Righting reflex | 8/8 | 8/8 | 8/8 |
| Air righting reflex | 8/8 | 8/8 | 8/8 |
| Ear twitch | 5/8 | 7/8 | 8/8 |
| Petting escape | 7/8 | 6/8 | 8/8 |
4. Discussion
Our successful generation and breeding of the DAT Val559 mice indicates that the introduction of the variant results in viable offspring that grow normally. In contrast, DAT KO mice demonstrate reduced weight gain and survival (Giros et al., 1996). These findings likely speak to the relatively conservative nature of the Val for Ala substitution, as compared to full elimination of DAT. DAT is expressed in the mouse during embryonic development (Chung et al., 2012) and full loss of the transporter leads to profound changes in a number of biochemical and physiological processes including more than a 90% reduction in tyrosine hydroxylase (TH) expression (Jones et al., 1998), as well as significant reductions in D2R mRNA levels (~50%) (Giros et al., 1996) and protein expression (~55%) (Jones et al., 1999). DAT KO mice also demonstrate a reduction in striatal volume that includes a fewer number of neurons (Cyr et al., 2005), as well as a loss of dendritic spines in proximal portions of dendrites (Berlanga et al., 2011). Although the profound hyperactivity evident in the DAT KO model (Giros et al., 1996) initially suggested that these animals might prove a relevant ADHD model, recent findings indicate that humans homozygous for DAT loss-of-function alleles display a complex motor phenotype characterized by infantile dystonia and Parkinsonian symptoms (Kurian et al., 2011). Interestingly, during early postnatal life, these subjects display hyperkinetic features characterized by twitching and frequent limb movements. These observations suggest that the DAT KO mice may be a useful model for a movement disorder rather than ADHD. Additionally, these studies indicate that hyperactivity may be a misleading expectation for animal models of the latter disorder.
Although we did not note differences in the growth profiles of the DAT Val559 mice, our measures were confined to ages post-weaning, after our animals were genotyped. Thus, an impact of the variant on embryonic growth or early neonatal survival could have been missed. This point may relate to our finding of a small, but significant reduction in the number of males homozygous for the Val559 variant. How this reduction could arise is a matter of speculation, though we note that sex-specific differences in the expression of genes that determine a dopaminergic phenotype, including DAT, have been detected in the mouse (Tao et al., 2012). A further investigation of this issue may be important given the reported male-bias in ADHD diagnoses (Gaub and Carlson, 1997; Getahun et al., 2013). We also note that this change is only evident in homozygous DAT Val559 mice whereas the two boys from whom we identified the allele are heterozygous carriers of the Val559 allele (Mazei-Robison et al., 2005). Genetic background can have a profound impact on the penetrance of modifications introduced into the mouse genome (see Kerr et al., 2013, for a recent example), and are expected for modest alterations that modulate risk versus determinant genetic changes. In relation to our efforts, we also note that a number of the phenotypes of the DAT KO mice vary by genetic background (Morice et al., 2004). As such, one cannot always predict whether phenotypes will emerge from heterozygous or homozygous animals, nor whether all traits will be exhibited on all genetic backgrounds.
In keeping with the relatively benign influence of the DAT Val559 variant, no significant changes were noted in initial sensorimotor tests. Certainly, future studies should examine these aspects in greater depth, particularly given the role of DA in basal ganglia control of motor function and in specific sensory modalities, such as the initial stages of visual perception (Feldkaemper and Schaeffel, 2013; Reis et al., 2007; Witkovsky, 2004; Xi et al., 2002). We did detect a trend toward a genotype-dependent reduction in latency to fall in the wire hang test. A reduction in latency is generally taken as a sign of motor weakness, though we saw no reductions in performance on the grip strength test. The wire-hang test however is likely more taxing than the grip strength test given that the animal is asked to support the full weight of the body. Alternatively, DAT Val559 animals may simply lack motivation for performance in this test, something that obviously the basic measures implemented to date do not assess. Other tests that have been reported to measure “behavioral despair” such as the Forced Swim Test or the Tail Suspension Test, may provide further insights.
The absence of gross changes in growth or sensorimotor function lays an important foundation for future tests of these animals which involve cue detection and motor engagement. These tests are often utilized to assess face and predictive validity of rodent disease models. Many such ADHD models with face and/or predictive validity have been advanced (Table 4), though none to date derive from genetic variation present in ADHD subjects, as introduced with the creation of the DAT Val559 model. Our in vitro studies with the DAT Val559 variant predict a reduction in sensitivity to AMPH given that the psychostimulant loses the ability to produce DAT-dependent, DA efflux. A number of the prior animal models display a reduced sensitivity to AMPH as well as MPH. We do not expect to lose sensitivity to AMPH completely, as the ability of AMPH to antagonize DA uptake is retained (Mazei-Robison et al., 2008), as well as the compound’s MAO inhibitory actions, should be intact. MPH sensitivity relies on vesicular DA release. The degree to which vesicular DA release or postsynaptic DA responses are impacted in vivo cannot be inferred from in the in vitro models we have used to date to characterize DAT Val559. It would not be surprising therefore to find that in vivo DA ADE could lead to compensations in vesicular DA release, such that the psychostimulant properties of MPH are also altered.
Table 4.
Rodent models of ADHD. + = phenotype observed in model; − = phenotype absent from model; ND = phenotype not determined for model.
| Hyperactivity | Inattention | AMPH/MPH response | |
|---|---|---|---|
| Lesion/insult models | |||
| 6-OHDA (rat) | + | ND | + |
| Prenatal nicotine exposure (mouse) | + | ND | + |
| Prenatal BrdU exposure (rat) | + | ND | − |
| Prenatal ethanol exposure (rat) | ND | + | ND |
| Neonatal hypoxia (rat) | + | − | ND |
| Neonatal PCB exposure (rat) | + | ND | ND |
| Neonatal BPA exposure (rat) | + | ND | − |
| Neonatal X-ray exposure (rat) | ND | + | + |
| Neonatal chronic GBR 12909 (rat) | + | + | − |
| Genetic KOs | |||
| DAT (mouse) | + | + | − |
| Steroid sulfatase (mouse) | + | − | ND |
| GIT1 (mouse) | + | + | + |
| NK1 receptor (mouse) | + | + | + |
| SNAP-25/coloboma (mouse) | + | ND | + |
| p35 (mouse) | + | ND | + |
| Guanylyl cyclase C (mouse) | + | ND | + |
| Other genetic manipulations | |||
| Grin1 (missense mutant mouse) | + | ND | + |
| SynCAM1 dominant negative | + | ND | + |
| Cocaine-insensitive DAT | + | ND | + |
| DAT knock-down | + | − | + |
| CK1-delta overexpression | + | ND | + |
| Thyroid hormone receptor beta-PV | + | + | + |
| Selective breeding models | |||
| SHR | + | + | + |
| Genetically Hypertensive (GH) rat | + | ND | ND |
| NHE rat | + | + | ND |
| WKHA rat | + | ND | − |
| Wig rat | + | ND | − |
| I/LnJ acallosal mouse | + | + | ND |
| Developmental models | |||
| Social Isolation | + | + | + |
| Dirupted habenula development | + | + | + |
| Maternal separation | + | + | ND |
One of the most widely studied rodent models of ADHD is the spontaneously hypertensive rat (SHR). Originally derived from selective breeding of Wistar rats (Okamoto and Aoki, 1963) and used to study essential hypertension (Yamori, 1977), researchers soon recognized the spontaneous hyperactivity of SHRs (McCarty and Kopin, 1979). Investigators then demonstrated that AMPH (Myers et al., 1982) and MPH (Wultz et al., 1990) reduce the motor hyperactivity of the SHRs, versus triggering motor activation, often denoted as a central feature of the psychostimulant response of subjects with ADHD. Further studies have demonstrated increased impulsivity (Fox et al., 2008; Sagvolden et al., 1992; Sagvolden, 2011), impaired attention (Sagvolden and Xu, 2008; Sagvolden, 2011), and cognitive deficits (Brackney et al., 2012; Clements and Wainwright, 2006; Kantak et al., 2008). Accordingly, the SHR has been lauded as the best validated model of ADHD (Sagvolden, 2011). However, as no genetic alterations have been identified that account for ADHD-like traits in SHR animals, the model cannot be said to possess construct validity. Additionally, although the SHR model displays a number of ADHD-like characteristics, these traits are not all improved by psychostimulant treatment. Moreover, although psychostimulants improve SHR performance on attention tasks in some studies (Kantak et al., 2008; Sagvolden, 2011) results of other studies are discordant (van den Bergh et al., 2006; Ferguson et al., 2007). Similarly, although AMPH typically reduces locomotor hyperactivity in the model (Sagvolden and Xu, 2008), the drug has been shown to potentiate locomotor behavior in some studies (Calzavara et al., 2011). Finally, SHRs display deficits in sensorimotor gating, as measured by pre-pulse inihibiton tests (Levin et al., 2011; Li et al., 2007) Humans with ADHD, however do not typically display this phenotype (Feifel et al., 2009; Hanlon et al., 2009; Holstein et al., 2011), unless the task requires sustained attention (Hawk et al., 2003). The PPI deficits observed in SHRs suggest that these animals could be a better model for schizophrenia than ADHD (Levin et al., 2011).
Another concerning aspect of the SHR model of ADHD is the lack of evidence that subjects with ADHD demonstrate a propensity for hypertension. One study did identify such an association, but when data were adjusted for body mass index (BMI), this effect was eliminated, indicating that the main relationship in the study was one between ADHD and obesity (Fuemmeler et al., 2011). Furthermore, neither hypertension nor hypotension arise with chronic psychostimulant treatment of ADHD subjects (Vitiello et al., 2012), adding further concern that a model with both cardiovascular and cognitive features fails to pass a test of face validity.
Having established the DAT Val559 model, what in vivo phenotypes may be predicted from in vitro studies? As noted above in our discussion of DAT KO animals, overt hyperactivity in a mouse model likely signals a profound disruption of DA signaling such as occurs with complete loss of the transporter. In vitro studies indicate that although DAT Val559 exhibits ADE, the variant supports normal total and surface expression, as well as DA recognition and uptake (Mazei-Robison et al., 2008). How, in a rodent, this combination of functional properties plays out in terms of biochemical, physiological and behavioral changes can only be determined empirically. Even ADE, the most conspicuous phenotype of the Val559 variant in transfected cells is capable of being suppressed by alterations in D2 DA receptor and CamKII signaling (Bowton et al., 2010), which could well occur as a compensation to constitutive DA efflux. If such compensations do not occur, reasonable in vivo phenotypes include an elevation in basal extracellular DA levels and a lack of AMPH-induced DA efflux in the context of normal DAT protein expression. Elevations in extracellular DA may be derived from both ADE and elevated vesicular DA release if presynaptic D2 DA receptors that normally limit vesicular release become densensitized by maintained, high levels of synaptic DA. Our preliminary in vivo microdialysis and brain slice release studies are consistent with these ideas (Mergy et al., 2012).
Beyond constitutive elevations in extracellular DA and a reduction in AMPH-evoked DA efflux, predictions of circuit and behavioral level phenotypes in the DAT Val559 model are more conjectural and await experimental evaluation. No model generated to our knowledge provides for elevations in extracellular DA throughout development in the context of normal DA uptake capacity. Thus, whether these animals should exhibit overt hyperactivity is unclear, though it seems likely that such behavior will be milder than that seen in the DAT KO, if evident at all. Similarly, the degree of impulsivity and inattention evident in these mice cannot be predicted. However, it seems reasonable to consider that a constitutive elevation in extracellular DA will reduce the dynamic range across which DA release can signal changes in presynaptic excitation and may therefore reduce cue saliency (Gowrishankar et al., 2013). At a behavioral level, a diminished dynamic range of DA signaling may lead to inattention, poor reward recognition, and diminished motivation. In such a context, restlessness and impulsivity may arise as subjects fail to constrain behavior as a result of learning the connections between appropriate response patterns and reward. Finally, we anticipate that constitutive changes in DA signaling will perturb molecular networks that support synaptic structure, strength and plasticity. Although a demonstration of behavioral alterations that mimic changes seen in ADHD subjects appears, on the surface, a test of the relevance of an ADHD animal model, we suspect that we will more often than not find difficulty with aspects of face validity, owing to differences in the mouse and human brain. Perhaps a better test of the value of the DAT Val559 model is the degree to which these animals afford insights into pathophysiogical mechanisms, subsequently subjected to testing in human subjects. Although the DAT Val559 variant is rare, we propose that some ADHD subjects exhibit ADE derived from changes not in DAT, but in the cellular signaling pathways that insure a biased inward flow of DA across the transporter at the presynaptic membrane.
Acknowledgments
This work was supported by NIH Awards MH090738 (M.A.M.), DA07390 (R.D.B.) and MH073159 (R.D.B.). We thank the outstanding laboratory support of Chris Svitek, Qiao Han, Sarah Whitaker, Tracy Moore-Jarrett and Angela Steele.
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM IV. 4. Am Psychiatric Assoc; Washington, DC: 1994. Attention-deficit and disruptive behavior disorders. [Google Scholar]
- Bakare MO. Attention deficit hyperactivity symptoms and disorder (ADHD) among African children: a review of epidemiology and co-morbidities. Afr J Psychiatry. 2012;15:358–361. doi: 10.4314/ajpsy.v15i5.45. [DOI] [PubMed] [Google Scholar]
- Barake M, Evins AE, Stoeckel L, Pachas GN, Nachtigall LB, Miller KK, Biller BMK, Tritos NA, Klibanski A. Pituitary. 2013 doi: 10.1007/s11102-013-0480-6. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellgrove MA, Hawi Z, Kirley A, Gill M, Robertson IH. Dissecting the attention deficit hyperactivity disorder (ADHD) phenotype: sustained attention, response variability and spatial attentional asymmetries in relation to dopamine transporter (DAT1) genotype. Neuropsychologia. 2005;43:1847–1857. doi: 10.1016/j.neuropsychologia.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Berlanga ML, Price DL, Phung BS, Giuly R, Terada M, Yamada N, Cyr M, Caron MG, Laasko A, Martone ME, Ellisman MH. Multiscale imaging characterization of dopamine transporter knockout mice reveals regional alterations in spine density of medium spiny neurons. Brain Res. 2011;1390:41–49. doi: 10.1016/j.brainres.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchini R, Postorino V, Grasso R, Santoro B, Migliore S, Burlò C, Tata C, Mazzone L. Prevalence of ADHD in a sample of Italian students: a population-based study. Res Dev Disabil. 2013;34:2543–2550. doi: 10.1016/j.ridd.2013.05.027. [DOI] [PubMed] [Google Scholar]
- Bidwell LC, Willcutt EG, McQueen MB, DeFries JC, Olson RK, Smith SD, Pennington BF. A family based association study of DRD4, DAT1, and 5HTT and continuous traits of attention-deficit hyperactivity disorder. Behav Genet. 2011;41:165–174. doi: 10.1007/s10519-010-9437-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederman J, Faraone SV. Attention-deficit hyperactivity disorder. Lancet. 2005;366:237–248. doi: 10.1016/S0140-6736(05)66915-2. [DOI] [PubMed] [Google Scholar]
- Biederman J, Mick E, Faraone SV. Age-dependent decline of symptoms of attention deficit hyperactivity disorder: impact of remission definition and symptom type. Am J Psychiatry. 2000;157:816–818. doi: 10.1176/appi.ajp.157.5.816. [DOI] [PubMed] [Google Scholar]
- Bitter I, Angyalosi A, Czobar P. Pharmacological treatment of adult ADHD. Curr Opin Psychiatry. 2012;25:529–534. doi: 10.1097/YCO.0b013e328356f87f. [DOI] [PubMed] [Google Scholar]
- Bobb AJ, Addington AM, Sidransky E, Gornick MC, Lerch JP, Greenstein DK, Clasen LS, Sharp WS, Inoff-Germain G, Wavrant-De Vrièze F, Arcos-Burgos M, Straub RE, Hardy JA, Castellanos FX, Rapoport JL. Support for association between ADHD and two candidate genes: NET1 and DRD1. Am J Med Genet Part B. 2005;134B:67–72. doi: 10.1002/ajmg.b.30142. [DOI] [PubMed] [Google Scholar]
- Bowton E, Saunders C, Erreger K, Sakrikar D, Matthies HJ, Sen N, Jessen T, Colbran RJ, Caron MG, Javitch JA, Blakely RD, Galli A. Dysregulation of dopamine transporters via dopamine D2 autoreceptors triggers anomalous dopamine efflux associated with attention-deficit hyperactivity disorder. J Neurosci. 2010;30(17):6048–6057. doi: 10.1523/JNEUROSCI.5094-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackney RJ, Cheung THC, Herbst K, Hill JC, Sanabria F. Extinction learning deficit in a rodent model of attention-deficit hyperactivity disorder. Behav Brain Funct. 2012;8:59. doi: 10.1186/1744-9081-8-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braet W, Johnson KA, Tobin CT, Acheson R, McDonnell C, Hawi Z, Barry E, Mulligan A, Gill M, Bellgrove MA, Robertson IH, Garavan H. FMRI activation during response inhibition and error processing: the role of the DAT1 gene in typically developing adolescents and those diagnosed with ADHD. Neuropsychologia. 2011;49:1641–1650. doi: 10.1016/j.neuropsychologia.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Bramness JG, Gundersen ØH, Guterstam J, Rognli EB, Konstenius M, Løberg EM, Medhus S, Tanum L, Franck J. Amphetamine-induced psychosis–a separate diagnostic entity or primary psychosis triggered in the vulnerable? BMC Psychiatry. 2012;12:221. doi: 10.1186/1471-244X-12-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehmer Y, Westerberg H, Bellander M, Fürth D, Karlsson S, Bäckman Working memory plasticity modulated by dopamine transporter genotype. Neurosci Lett. 2009;467L:117–120. doi: 10.1016/j.neulet.2009.10.018. [DOI] [PubMed] [Google Scholar]
- Brent PJ. Behavioural effects of selective dopamine D-1 and D-2 agonists and antagonists in guinea pigs. Psychopharmacology. 1991;104:201–207. doi: 10.1007/BF02244179. [DOI] [PubMed] [Google Scholar]
- Buckholtz JW, Treadway MT, Cowan RL, Woodward ND, Li R, Ansari S, Baldwin RM, Schwartzman AN, Shelby ES, Smith CE, Kessler RM, Zald DH. Dopaminergic network differences in human impulsivity. Science. 2010;329:532. doi: 10.1126/science.1185778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton CL, Fletcher PJ. Age and sex differences in impulsive action in rats: the role of dopamine and glutamate. Behav Brain Res. 2012;230:21–33. doi: 10.1016/j.bbr.2012.01.046. [DOI] [PubMed] [Google Scholar]
- Cahill BS, Coolidge FL, Segal DL, Klebe KJ, Marle PD, Overmann KA. Prevalence of ADHD and its subtypes in male and female adult prison inmates. Behav Sci Law. 2012;30:154–166. doi: 10.1002/bsl.2004. [DOI] [PubMed] [Google Scholar]
- Calzavara MB, Levin R, Medrano WA, Almeida V, Sampaio APF, Barone LC, Frussa-Filho R, Abílio VC. Effects of antipsychotics and amphetamine on social behaviors in spontaneously hypertensive rats. Behav Brain Res. 2011;225:15–22. doi: 10.1016/j.bbr.2011.06.026. [DOI] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardile K, Patil N, Lane CR, Lim EP, Palyanaraman N, Nemesh J, Ziaugra L, Friedland L, Rolfe A, Warrington J, Lipshutz R, Daley GQ, Lander ES. Characterization of single nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Cheon K-A, Ryu Y-H, Kim J-W, Cho D-Y. The homozygosity for 10-repeat allele at dopamine transporter gene and dopamine transporter density in Korean children with attention deficit hyperactivity disorder: relating to treatment response to methylphenidate. Eur Neuropsychopharmacol. 2005;15:95–101. doi: 10.1016/j.euroneuro.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Chien I-C, Lin C-H, Chou Y-J, Chou P. Prevalence, incidence, and stimulant use of attention-deficit hyperactivity disorder in Taiwan, 1996–2005: a national population-based study. Soc Psychiatry Psychiatr Epidemiol. 2012;47:1885–1890. doi: 10.1007/s00127-012-0501-1. [DOI] [PubMed] [Google Scholar]
- Chung S, Kim C-H, Kim K-S. Lmx1a regulates dopamine transporter gene expression during ES cell differentiation and mouse embryonic development. J Neurochem. 2012;122:244–250. doi: 10.1111/j.1471-4159.2012.07779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli F, Laviola G. Paradoxical effects of d-amphetamine in infant and adolescent mice. role of gender and environmental risk factors. Neurosci Biobehav Rev. 2000;24:73–84. doi: 10.1016/s0149-7634(99)00047-0. [DOI] [PubMed] [Google Scholar]
- Clements KM, Wainwright PE. Spontaneously hypertensive, Wistar-Kyoto and Sprague–Dawley rats differ in performance on a win-shift task in the water radial arm maze. Behav Brain Res. 2006;167:295–304. doi: 10.1016/j.bbr.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Colotla VA, Flores E, Oscos A, Meneses A, Tapia R. Effects of MPTP on locomotor activity in mice. Neurotoxicol Teratol. 1990;12:405–407. doi: 10.1016/0892-0362(90)90061-g. [DOI] [PubMed] [Google Scholar]
- Cornish KM, Manly T, Savage R, Swanson J, Morisano D, Butler N, Grant C, Cross G, Bentley L, Hollis CP. Association of the dopamine transporter (DAT1) 10/10-repeat genotype with ADHD symptoms and response inhibition in a general population sample. Mol Psychiatry. 2005;10:686–698. doi: 10.1038/sj.mp.4001641. [DOI] [PubMed] [Google Scholar]
- Costa A, Riedel M, Müller U, Möller H-J, Ettinger U. Relationship between SLC6A3 genotype and striatal dopamine transporter availability: a meta-analysis of human single photon emission computed tomography studies. Synapse. 2011;65:998–1005. doi: 10.1002/syn.20927. [DOI] [PubMed] [Google Scholar]
- Costa A, la Fougère C, Pogarell O, Möller H-J, Riedel M, Ettinger U. Impulsivity is related to striatal dopamine transporter availability in healthy males. Psychiatry Res. 2013a;211:251–256. doi: 10.1016/j.pscychresns.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Costa A, Riedel M, Pogarell O, Menzel-Zelnitschek F, Schwarz M, Reiser M, Möller H-J, Rubia K, Meindl T, Ettinger U. Methylphenidate effects on neural activity during response inhibition in healthy humans. Cereb Cortex. 2013b;23:1179–1189. doi: 10.1093/cercor/bhs107. [DOI] [PubMed] [Google Scholar]
- Covey DP, Juliano SA, Garris PA. Amphetamine elicits opposing actions on readily releasable and reserve pools for dopamine. PLoS ONE. 2013;8:e60763. doi: 10.1371/journal.pone.0060763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TDR, Hawi Z, Hocking J, Strudwick M, Hester R, Garavan H, Wagner J, Chambers CD, Bellgrove MA. Dopamine transporter genotype predicts behavioural and neural measures of response inhibition. Mol Psychiatry. 2012;17:1086–1092. doi: 10.1038/mp.2011.104. [DOI] [PubMed] [Google Scholar]
- Cyr M, Caron MG, Johnson GA, Laasko A. Magnetic resonance imaging at microscopic resolution reveals subtle morphological changes in a mouse model of dopaminergic hyperfunction. Neuroimage. 2005;26:83–90. doi: 10.1016/j.neuroimage.2005.01.039. [DOI] [PubMed] [Google Scholar]
- Dalley JW, Roiser JP. Dopamine, serotonin and impulsivity. Neuroscience. 2012;215:42–58. doi: 10.1016/j.neuroscience.2012.03.065. [DOI] [PubMed] [Google Scholar]
- Davis NO, Kollins SH. Treatment for co-occurring attention deficit/ hyperactivity disorder and autism spectrum disorder. Neurotherapeutics. 2012;9:518–530. doi: 10.1007/s13311-012-0126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf R, Kessler RC, Fayyad J, ten Have M, Alonso J, Angermeyer M, Borges G, Demyttenaere K, Gasquet I, de Girolamo G, Haro JM, Jin R, Karam EG, Ormel J, Posada-Villa J. The prevalence and effects of adult attention-deficit/hyperactivity disorder (ADHD) on the performance of workers: results from the WHO World Mental Health Survey Initiative. Occup Environ Med. 2008;65:835–842. doi: 10.1136/oem.2007.038448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucette-Stamm LA, Blakely DJ, Tian J, Mockus S, Mao J. Population genetic study of the human dopamine transporter gene (DAT1) Genet Epidemiol. 1995;12:303–308. doi: 10.1002/gepi.1370120307. [DOI] [PubMed] [Google Scholar]
- Dougherty DD, Bonab AA, Spencer TJ, Rauch SL, Madras BK, Fischman AJ. Dopamine transporter density in patients with attention deficit hyperactivity disorder. Lancet. 1999;354:2132–2133. doi: 10.1016/S0140-6736(99)04030-1. [DOI] [PubMed] [Google Scholar]
- Dreher JK, Jackson DM. Role of D1 and D2 dopamine receptors in mediating locomotor activity elicited from the nucleus accumbens of rats. Brain Res. 1989;487:267–277. doi: 10.1016/0006-8993(89)90831-7. [DOI] [PubMed] [Google Scholar]
- Dresel S, Krause J, Krause H-H, LaFougere C, Brinkbäumer K, Kung HF, Hahn K, Tatsch K. Attention deficit hyperactivity disorder: binding of [99mTc]TRODAT-1 to the dopamine transporter before and after methylphenidate treatment. Eur J Nucl Med. 2000;27:1518–1524. doi: 10.1007/s002590000330. [DOI] [PubMed] [Google Scholar]
- Dresler T, Ehlis A-C, Heinzel S, Renner TJ, Reif A, Baehne CG, Meine M, Boreatti-Hümmer A, Jacob CP, Lesch K-P, Falgatter AJ. Dopamine transporter (SLC6A3) genotype impacts neurophysiological correlates of cognitive response control in an adult sample of patients with ADHD. Neuropsychopharmacology. 2010;35:2193–2202. doi: 10.1038/npp.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erinoff L, Macphail RC, Heller A, Seiden LS. Age-dependent effects of 6-hydroxydopamine on locomotor activity in the rat. Brain Res. 1979;164:195–205. doi: 10.1016/0006-8993(79)90015-5. [DOI] [PubMed] [Google Scholar]
- Ezpeleta L, de la Osa N, Doménech JM. Prevalence of DSM-IV disorders, comorbidity and impairment in 3-year-old Spanish preschoolers. Soc Psychiatry Psychiatr Epidemiol. 2013 doi: 10.1007/s00127-013-0683-1. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Biederman J, Roe C. Comparative efficacy of Adderall and methylphenidate in attention-deficit/hyperactivity disorder: a meta-analysis. J Clin Psychopharmacol. 2002;22:468–473. doi: 10.1097/00004714-200210000-00005. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Spencer TJ, Madras BK, Zhang-James Y, Biederman J. Functional effects of dopamine transporter gene genotypes on in vivo dopamine transporter functioning: a meta-analysis. Mol Psychiatry. 2013:1–10. doi: 10.1038/mp.2013.126. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Fayyad J, de Graaf R, Kessler R, Alonso J, Angermeyer M, Demyttenaere K, de Girolamo G, Haro JM, Karam EG, Lara C, Lépine J-P, Ormel J, Posada-Villa J, Zaslavsky AM, Jin R. Cross-national prevalence and correlates of adult attention-deficit hyperactivity disorder. Br J Psychiatry. 2007;190:402–409. doi: 10.1192/bjp.bp.106.034389. [DOI] [PubMed] [Google Scholar]
- Feifel D, Minassian A, Perry W. Prepulse inhibition of startle in adults with ADHD. J Psychiatr Res. 2009;43:484–489. doi: 10.1016/j.jpsychires.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldkaemper M, Schaeffel F. An updated view on the role of dopamine in myopia. Exp Eye Res. 2013;114:106–119. doi: 10.1016/j.exer.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Ferguson SA, Paule MG, Cada A, Fogle CM, Gray EP, Berry KJ. Baseline behavior, but not sensitivity to stimulant drugs, differs among spontaneously hypertensive, Wistar-Kyoto, and Sprague–Dawley rat strains. Neurotoxicol Teratol. 2007;29:547–561. doi: 10.1016/j.ntt.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Fog JU, Khoshbouei H, Holy M, Owens WA, Vaegter CB, Sen N, Nikandrova Y, Bowton E, McMahon DG, Colbran RJ, Daws LC, Sitte HH, Javitch JA, Galli A, Gether U. Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport. Neuron. 2006;51:417–429. doi: 10.1016/j.neuron.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Forbes EE, Brown SM, Kimak M, Ferrell RE, Manuck SB, Hariri AR. Genetic variation in components of dopamine neurotransmission impacts ventral striatal reactivity associated with impulsivity. Mol Psychiatry. 2009;14:60–70. doi: 10.1038/sj.mp.4002086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AT, Hand DJ, Reilly MP. Impulsive choice in a rodent model of attention-deficit/hyperactivity disorder. Behav Brain Res. 2008;187:146–152. doi: 10.1016/j.bbr.2007.09.008. [DOI] [PubMed] [Google Scholar]
- Franke B, Vasquez AA, Johansson S, Moogman M, Romanos J, Boreatti-Hümmer A, Heine M, Jacob CP, Lesch K-P, Casas M, Ribasés M, Bosch R, Sánchez-Mora C, Gómez-Barros N, Fernàndez-Castillo N, Bayés M, Halmøy A, Halleland H, Landaas ET, Fasmer OB, Knappskog PM, Heister AJGAM, Kiemeney LA, Kooij JJS, Boonstra AM, Kan CC, Asherson P, Faraone SV, Buitelaar JK, Haavik J, Cormand B, Ramos-Quiroga JA, Reif A. Multicenter analysis of the SLC6A3/DAT1 VNTR haplotype in persistent ADHD suggests differential involvement of the gene in childhood and persistent ADHD. Neuropsychopharmacology. 2010;35:656–664. doi: 10.1038/npp.2009.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuemmeler BF, Østbye T, Yang C, McClernon FJ, Kollins SH. Association between attention-deficit/hyperactivity disorder symptoms and obesity and hypertension in early adulthood: a population-based study. Int J Obes (Lond) 2011;35:852–862. doi: 10.1038/ijo.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusar-Poli P, Rubia K, Rossi G, Sartori G, Malottin U. Striatal dopamine transporter alterations in ADHD: pathophysiology or adaptation to psychostimulants? A meta-analysis. Am J Psychiatry. 2012;169:264–272. doi: 10.1176/appi.ajp.2011.11060940. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR. Strengths and limitations of genetic models of ADHD. Atten Defic Hyperact Disord. 2010;2:21–30. doi: 10.1007/s12402-010-0021-3. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Wetsel WC, Jones SR, Levin ED, Jaber M, Caron MG. Role of serotonin in the paradoxical calming effect of psychostimulants on hyperactivity. Science. 1999;283:397–401. doi: 10.1126/science.283.5400.397. [DOI] [PubMed] [Google Scholar]
- Garnier-Dykstra LM, Pinchevsky GM, Caldeira KM, Vincent KB, Arria AM. Self-reported adult attention deficit hyperactivity disorder symptoms among college students. J Am Coll Health. 2010;59(2):133–136. doi: 10.1080/07448481.2010.483718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaub M, Carlson CL. Gender differences in ADHD: a meta-analysis and critical review. J Am Acad Child Adolesc Psychiatry. 1997;36(8):1036–1045. doi: 10.1097/00004583-199708000-00011. [DOI] [PubMed] [Google Scholar]
- Gerlernter J, Kranzler H, Lacobelle J. Population studies of polymorphisms at loci of neuropsychiatric interest (tryptophan hydroxylase (TPH), dopamine transporter protein (SLC6A3), D2 dopamine receptor (DRD3), apolipoprotein E (APOE), μ opioid receptor (OPRM1), and ciliary neurotrophic factor (CNTF)) Genomics. 1998;52:289–297. doi: 10.1006/geno.1998.5454. [DOI] [PubMed] [Google Scholar]
- Getahun D, Jacobsen SJ, Fassett MJ, Chen W, Demissie K, Rhoads GG. Recent trends in childhood attention-deficit/hyperactivity disorder. JAMA Pediatr. 2013;167(3):282–288. doi: 10.1001/2013.jamapediatrics.401. [DOI] [PubMed] [Google Scholar]
- Ghahremani DG, Lee B, Robertson CL, Tabibnia G, Morgan AT, Shelter ND, Brown AK, Monterosso JR, Aron AR, Mandelkern MA, Poldrack RA, London ED. Striatal dopamine D2/D3 receptors mediate response inhibition and related activity in frontostriatal neural circuitry in humans. J Neurosci. 2012;32:7316–7324. doi: 10.1523/JNEUROSCI.4284-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert DL, Wang Z, Sallee FR, Ridel KR, Merhar S, Zhang J, Lipps TD, White C, Badreldin N, Wassermann EM. Dopamine transporter genotype influences the physiological response to medication in ADHD. Brain. 2006;129:2038–2046. doi: 10.1093/brain/awl147. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Glick SD, Milloy S. Rate-dependent effects of d-amphetamine on locomotor activity in mice. possible relationship to paradoxical amphetamine sedation in minimal brain dysfunction. Eur J Pharmacol. 1973;24:266–268. doi: 10.1016/0014-2999(73)90082-4. [DOI] [PubMed] [Google Scholar]
- Gnanalingham KK, Milkowski NA, Smith LA, Hunter AJ, Jenner P, Marsden CD. Short and long-term changes in cerebral [14C]-2-deoxyglucose uptake in the MPTP-treated marmoset: relationship to locomotor activity. J Neural Transm. 1995;101:65–82. doi: 10.1007/BF01271546. [Gen Sect] [DOI] [PubMed] [Google Scholar]
- Gnegy ME, Khoshbouei H, Berg KA, Javitch JA, Clarke WP, Zhang M, Galli A. Intracellular Ca2+ regulates amphetamine-induced dopamine efflux and currents mediated by the human dopamine transporter. Mol Pharmacol. 2004;66:137–143. doi: 10.1124/mol.66.1.137. [DOI] [PubMed] [Google Scholar]
- Good RL, Radcliffe RA. Methamphetamine-induced locomotor changes are dependent on age, dose and genotype. Pharmacol Biochem Behav. 2011;98:101–111. doi: 10.1016/j.pbb.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar R, Hahn MK, Blakely RD. Good Riddance to Dopamine: Roles for the Dopamine Transporter in Synaptic Function and Dopamine-associated Brain Disorders. 2013 doi: 10.1016/j.neuint.2013.10.016. Submitted for publication. [DOI] [PubMed] [Google Scholar]
- Grant KM, LeVan TD, Wells SM, Li M, Stoltenberg SF, Gendelman HE, Carlo G, Bevins RA. Methamphetamine-associated psychosis. J Neuroimmune Pharmacol. 2012;7:113–139. doi: 10.1007/s11481-011-9288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood TA, Joo E-J, Shekhtman T, Sadovnick AD, Remick RA, Keck PE, McElroy SL, Kelsoe JR. Association of dopamine transporter gene variants with childhood ADHD features in bipolar disorder. Am J Med Genet Part B. 2013;162B:137–145. doi: 10.1002/ajmg.b.32108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold AJ, Ma D, Cukier HN, Nations LD, Schmidt MA, Chung R-H, Jaworski JM, Salyakina D, Konidari I, Whitehead PL, Wright HH, Abramson RK, Williams SM, Menon R, Martin ER, Haines JL, Gilbert JR, Cuccaro ML, Pericak-Vance MA. Evaluation of copy number variations reveals novel candidate genes in autism spectrum disorder-associated pathways. Hum Mol Genet. 2012;21:3513–3523. doi: 10.1093/hmg/dds164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünhage F, Schulze TG, Müller DJ, Lanczik M, Franzek E, Albus M, Borrmann-Hassenback M, Knapp M, Cichon S, Maier W, Rietschel M, Propping P, Nöthen MM. Systematic screening for DNA sequence variation in the coding region of the human dopamine transporter gene (DAT1) Mol Psychiatry. 2000;5:275–282. doi: 10.1038/sj.mp.4000711. [DOI] [PubMed] [Google Scholar]
- Hahn T, Heinzel S, Dresler T, Plichta MM, Renner TJ, Markulin F, Jakob PM, Lesch K-P, Fallgatter AJ. Association between reward-related activation in the ventral striatum and trait reward sensitivity is moderated by dopamine transporter genotype. Hum Brain Mapp. 2011;32:1557–1565. doi: 10.1002/hbm.21127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton PJ, Campbell NG, Sharma S, Erreger K, Herborg Hansen F, Saunders C, Belovich AN, Sahai MA, Cook EH, Gether U, Mchaourab HS, Matthies HJG, Sutcliffe JS, Galli A NIH ARRA Autism Sequencing Consortium. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol Psychiatry. 2013:1–9. doi: 10.1038/mp.2013.102. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon MC, Karayanidis F, Schall U. Intact sensorimotor gating in adult attention deficit hyperactivity disorder. Int J Neuropsychopharmacol. 2009;12(5):701–707. doi: 10.1017/S1461145708009711. http://dx.doi.org/10.1017/S1461145708009711, Epub 2008 Dec 2. [DOI] [PubMed] [Google Scholar]
- Hawk LW, Jr, Yartz AR, Pelham WE, Jr, Lock TM. The effects of methylphenidate on prepulse inhibition during attended and ignored prestimuli among boys with attention-deficit hyperactivity disorder. Psychopharmacology. 2003;165:118–127. doi: 10.1007/s00213-002-1235-7. [DOI] [PubMed] [Google Scholar]
- Holmes J, Payton A, Barrett JH, Hever T, Fitzpatrick H, Trumper AL, Harrington R, McGuffin P, Owen M, Ollier W, Worthington J, Thapar A. A family-based and case-control association study of the dopamine D4 receptor gene and dopamine transporter gene in attention deficit hyperactivity disorder. Mol Psychiatry. 2000;5:523–530. doi: 10.1038/sj.mp.4000751. [DOI] [PubMed] [Google Scholar]
- Holstein DH, Csomor PA, Geyer MA, Huber T, Brugger N, Studerus E, Vollenwieder FX. The effects of sertindole on sensory gating, sensorimotor gating, and cognition in healthy volunteers. J Psychopharmacol. 2011;25(12):1600–1613. doi: 10.1177/0269881111415734. [DOI] [PubMed] [Google Scholar]
- Holt R, Sykes NH, Conceição IC, Cazier J-B, Anney RJL, Oliveira G, Gallagher L, Vicente A, Monaco AP, Pagnamenta AT. CNVs leading to fusion transcripts in individuals with autism spectrum disorder. Eur J Hum Genet. 2012;20:1141–1147. doi: 10.1038/ejhg.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogmann M, Onnink M, Cools R, Aarts E, Kan C, Vasquez AA, Buitelaar J, Franke B. The dopamine transporter haplotype and reward-related striatal responses in adult ADHD. Eur Neuropsychopharmacol. 2013;23:469–478. doi: 10.1016/j.euroneuro.2012.05.011. [DOI] [PubMed] [Google Scholar]
- Irwin S. Comprehensive observational assessment: Ia. A systematic, quantitative procedure for assessing the behavior and physiologic state of the mouse. Psychopharmacologia. 1968;13(3):222–257. doi: 10.1007/BF00401402. [DOI] [PubMed] [Google Scholar]
- Janols L-O, Liliemark J, Klintberg K, von Knorring A-L. Central stimulants in the treatment of attention-deficit hyperactivity disorder (ADHD) in children and adolescents. A naturalistic study of the prescription in Sweden, 1977–2007. Nord J Psychiatry. 2009;63:508–516. doi: 10.3109/08039480903154534. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Yuen RKC, Jin X, Wang M, Chen N, Wu X, Ju J, Mei J, Shi Y, He M, Wang G, Liang J, Wang Z, Cao D, Carter MT, Chrysler C, Drmic IE, Howe JL, Lau L, Marshall CR, Merico D, Nalpathamkalam T, Thiruvahindrapuram B, Thompson A, Uddin M, Walker S, Luo J, Anagnostou E, Zwaigenbaum L, Ring RH, Wang J, Lajonchere C, Wang J, Shih A, Szatmari P, Yang H, Dawson G, Li Y, Scherer SW. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am J Hum Genet. 2013;93:249–263. doi: 10.1016/j.ajhg.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnels B. Locomotor hypokinesia in the reserpine-treated rat: drug effects from the corpus striatum and nucleus accumbens. Pharmacol Biochem Behav. 1982;17:283–289. doi: 10.1016/0091-3057(82)90082-x. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci USA. 1998;95:4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Hu X-T, Cooper DC, Wightman RM, White FJ, Caron MG. Loss of autoreceptor functions in mice lacking the dopamine transporter. Nat Neurosci. 1999;2(7):649–655. doi: 10.1038/10204. [DOI] [PubMed] [Google Scholar]
- Joyce EM, Koob GF. Amphetamine-, scopolamine- and caffeine-induced locomotor activity following 6-hydroxydopamine lesions of the mesolimbic dopamine system. Psychopharmacology. 1981;73:311–313. doi: 10.1007/BF00426456. [DOI] [PubMed] [Google Scholar]
- Kahlig KM, Binda F, Khoshbouei H, Blakely RD, McMahon DG, Javitch JA, Galli A. Amphetamine induces dopamine efflux through a dopamine transporter channel. Proc Natl Acad Sci USA. 2005;102:3495–3500. doi: 10.1073/pnas.0407737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambeitz J, Romanos M, Ettinger U. Meta-analysis of the assocication between dopamine transporter genotype and response to methylphenidate treatment in ADHD. Pharmacogenomics J. 2013:1–8. doi: 10.1038/tpj.2013.9. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Kameda SR, Fukushiro DF, Trombin TF, Procópio-Souza R, Patti CL, Hollais AW, Calzavara MB, Abílio VC, Ribeiro RA, Tufik S, D’Almeida V, Frussa-Filho R. Adolescent mice are more vulnerable than adults to single injection-induced behavioral sensitization to amphetamine. Pharmacol Biochem Behav. 2011;98:320–324. doi: 10.1016/j.pbb.2011.01.013. [DOI] [PubMed] [Google Scholar]
- Kang AM, Palmatier MA, Kidd KK. Global variation of a 40-bp VNTR in the 3′-untranslated region of the dopamine transporter gene (SLC6A3) Biol Psychiatry. 1999;46:151–160. doi: 10.1016/s0006-3223(99)00101-8. [DOI] [PubMed] [Google Scholar]
- Kantak KM, Singh T, Kerstetter KA, Dembro KA, Mutebi MM, Harvey RC, Deschepper CF. Advancing the spontaneous hypertensive rat model of attention deficit/hyperactivity disorder. Behav Neurosci. 2008;122(2):340–357. doi: 10.1037/0735-7044.122.2.340. [DOI] [PubMed] [Google Scholar]
- Kenny EM, Cormican P, Furlong S, Heron E, Kenny G, Fahey C, Kelleher E, Ennis S, Tropea D, Anney R, Corvin AP, Donohoe G, Gallagher L, Gill M, Morris Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol Psychiatry. 2013:1–8. doi: 10.1038/mp.2013.127. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Kerr TM, Muller CL, Miah M, Jetter CS, Pfeiffer R, Shah C, Baganz N, Anderson GM, Crawley JN, Sutcliffe JS, Blakely RD, Veenstra-VanderWeele J. Genetic background modulated phenotypes of serotonin transporter Ala56 knock-in mice. Mol Autism. 2013;4:35. doi: 10.1186/2040-2392-4-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Adler LA, Barkley R, Biederman J, Conners CK, Faraone SV, Greenhill LL, Jaeger S, Secnik K, Spencer T, Üstün TB, Zaslavsky AM. Patterns and predictors of attention-deficit/hyperactivity disorder persistence into adulthood: results from the National Comorbidity Survey Replication. Biol Psychiatry. 2005;57:1442–1451. doi: 10.1016/j.biopsych.2005.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Adler L, Barkley R, Biederman J, Conners CK, Demler O, Faraone SV, Greenhill LL, Howes MJ, Secnik K, Spencer T, Ustun TB, Walters EE, Zaslavsky AM. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry. 2006;163:716–723. doi: 10.1176/appi.ajp.163.4.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME, Galli A, Javitch JA. N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol. 2004;2:0387–0393. doi: 10.1371/journal.pbio.0020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshbouei H, Wang H, Lechleiter JD, Javitch JA, Galli A. Amphetamine-induced dopamine efflux. A voltage-sensitive and intracellular Na+-dependent mechanism. J Biol Chem. 2003;278(14):12070–12077. doi: 10.1074/jbc.M212815200. [DOI] [PubMed] [Google Scholar]
- Kotte A, Joshi G, Fried R, Uchida M, Spencer A, Woodworth KY, Kenworthy T, Faraone SV, Biederman J. Pediatrics. 2013;132:e612–e622. doi: 10.1542/peds.2012-3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause K-H, Dresel SH, Krause J, Kung HF, Tatsch K. Increase striatal dopamine transporter in adult patients with attention deficit hyperactivity disorder: effects of methylphenidate as measured by single photon emission computed tomography. Neurosci Lett. 2000;285:107–110. doi: 10.1016/s0304-3940(00)01040-5. [DOI] [PubMed] [Google Scholar]
- Krumm N, O’Roak BJ, Karakoc E, Mohajeri K, Nelson B, Vives L, Jacquemont S, Munson J, Bernier R, Eichler EE. Transmission disequilibrium of small CNVs in simplex autism. Am J Hum Genet. 2013;93:595–606. doi: 10.1016/j.ajhg.2013.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurian MA, Zhen J, Cheng S-Y, Li Y, Mordekar SR, Jardine P, Morgan NV, Meyer E, Tee L, Pasha S, Wassmer E, Heales SJR, Gissen P, Reith MEA, Maher ER. Homozygous loss-of-function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism-dystonia. J Clin Invest. 2009;119:1595–1603. doi: 10.1172/JCI39060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurian MA, Li Y, Zhen J, Meyer E, Hai N, Christen H-J, Hoffmann GF, Jardine P, von Moers A, Mordekar SR, O’Callaghan F, Wassmer E, Wraige E, Dietrich C, Lewis T, Hyland K, Heales SJR, Sanger T, Gissen P, Assmann BE, Reith MEA, Maher ER. Clinical and molecular characterization of hereditary dopamine transporter deficiency syndrome: an observational cohort and experimental study. Lancet Neurol. 2011;10:54–62. doi: 10.1016/S1474-4422(10)70269-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin R, Calzavara MB, Santos CM, Medrano WA, Niigaki ST, Abílio VC. Spontaneously hypertensive rats (SHR) present deficits in prepulse inhibition of startle specifically reverted by clozapine. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:1748–1752. doi: 10.1016/j.pnpbp.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Leyfer OT, Folstein SE, Bacalman S, Davis NO, Dinh E, Morgan J, Tager-Flusberg H, Lainhart JE. Comorbid psychiatric disorders in children with autism: interview development and rates of disorders. J Autism Dev Disord. 2006;36:849–461. doi: 10.1007/s10803-006-0123-0. [DOI] [PubMed] [Google Scholar]
- Li Q, Lu G, Antonio GE, Mak YT, Rudd JA, Fan M, Yew DT. The usefulness of the spontaneously hypertensive rat to model attention-deficit/hyperactivity disorder (ADHD) may be explained by the differential expression of dopamine-related genes in the brain. Neurochem Int. 2007;50:848–857. doi: 10.1016/j.neuint.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Li X, Sroubek A, Kelly MS, Lesser I, Sussman E, He Y, Branch C, Foxe JJ. Atypical pulvinar-cortical pathways during sustained attention performance in children with attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry. 2012;51:1197–1207. doi: 10.1016/j.jaac.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan R, Bernal MP, Panzer R, Whitaker A, Roberts W, Handen B, Hardan A, Anagnostou E, Veenstra-VanderWeele J. Clinical practice pathways for evaluation and medication choice for attention-deficit/hyperactivity disorder symptoms in autism spectrum disorders. Pediatrics. 2012;130:S125–S138. doi: 10.1542/peds.2012-0900J. [DOI] [PubMed] [Google Scholar]
- Manor I, Corbex M, Eisenberg J, Gritsenkso I, Bachner-Melman R, Tyano S, Ebstein RP. Association of the dopamine D5 receptor with attention deficit hyperactivity disorder (ADHD) and scores on a continuous performance test (TOVA) Am J Med Genet Part B. 2004;127B:73–77. doi: 10.1002/ajmg.b.30020. [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Blakely RD. Expression studies of naturally occurring human dopamine transporter variants identifies a novel state of transporter inactivation associated with Val382Ala. Neuropharmacology. 2005;49:737–749. doi: 10.1016/j.neuropharm.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Blakely RD. ADHD and the dopamine transporter: are there reasons to pay attention? Handb Exp Pharmacol. 2006;175:373–415. doi: 10.1007/3-540-29784-7_17. [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Couch RS, Shelton RC, Stein MA, Blakely RD. Sequence variation in the human dopamine transporter gene in children with attention deficit hyperactivity disorder. Neuropharmacology. 2005;49:724–736. doi: 10.1016/j.neuropharm.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Bowton E, Holy M, Schmudermaier M, Freissmuth M, Sitte HH, Galli A, Blakely RD. Anomalous dopamine release associated with a human dopamine transporter coding variant. J Neurosci. 2008;28(28):7040–7046. doi: 10.1523/JNEUROSCI.0473-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty R, Kopin IJ. Patterns of behavioral development in spontaneously hypertensive rats and Wistar-Kyoto normotensive controls. Dev Psychobiol. 1979;12(3):239–243. doi: 10.1002/dev.420120307. [DOI] [PubMed] [Google Scholar]
- Menashe I, Larsen EC, Banerjee-Basu S. Prioritization of copy number variation loci associated with autism from AutDB-an integrative multi-study genetic database. PLoS ONE. 2013;8:e66707. doi: 10.1371/journal.pone.0066707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergy MA, Jessen TN, Gowrishankar R, Wheeler CA, Davis GL, Waters WE, Ochoa-Vargas FJ, Belova Y, Gresch PJ, Wright J, Stanwood GD, Galli A, Blakely RD. Rare opportunities for progress in ADHD: behavioral and pharmacological analysis of knock-in mice expressing an ADHD-associated dopamine transporter gene variant. Poster Session Presented at: Tenth International Catecholamine Symposium; Pacific Grove, CA. September 9–13, 2012.2012. [Google Scholar]
- Minzenberg MJ. Pharmacotherapy for attention-deficit/hyperactivity disorder: from cells to circuits. Neurotherapeutics. 2012;9:610–621. doi: 10.1007/s13311-012-0128-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morice E, Denis C, Giros B, Nosten-Bertrand M. Phenotypic expression of the targeted null-mutation in the dopamine transporter gene varies as a function of the genetic background. Eur J Neurosci. 2004;20:120–126. doi: 10.1111/j.1460-9568.2004.03465.x. [DOI] [PubMed] [Google Scholar]
- Morris ME, Iansek R, Matyas TA, Summers JJ. The pathogenesis of gait hypokinesia in Parkinson’s disease. Brain. 1994;117:1169–1181. doi: 10.1093/brain/117.5.1169. [DOI] [PubMed] [Google Scholar]
- Myers MM, Musty RE, Hendley ED. Attenuation of hyperactivity in the spontaneously hypertensive rat by amphetamine. Behav Neural Biol. 1982;34:42–54. doi: 10.1016/s0163-1047(82)91397-8. [DOI] [PubMed] [Google Scholar]
- Newman DP, O’Connell RG, Nathan PJ, Bellgrove MA. Dopamine transporter genotype predicts attentional asymmetry in healthy adults. Neuropsychologia. 2012;50:2823–2829. doi: 10.1016/j.neuropsychologia.2012.08.012. [DOI] [PubMed] [Google Scholar]
- Nyman ES, Ogdie MN, Loukola A, Varilo T, Taanila A, Hurtig T, Moilanen IK, Loo SK, McGough JJ, Järvelin M-R, Smalley SL, Nelson SF, Peltonen L. ADHD candidate gene study in a population-based birth cohort: association with DBH and DRD2. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1614–1621. doi: 10.1097/chi.0b013e3181579682. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Aoki K. Development of a strain of spontaneously hypertensive rats. Jpn Circ J. 1963;27:282–293. doi: 10.1253/jcj.27.282. [DOI] [PubMed] [Google Scholar]
- Ondo WG, Lai D. Predictors of impulsivity and reward seeking behavior with dopamine agonists. Parkinsonism Relat Disord. 2008;14:28–32. doi: 10.1016/j.parkreldis.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Ornstein TJ, Iddon JL, Baldacchino AM, Sahakian BJ, London M, Everitt BJ, Robbins TW. Profiles of cognitive dysfunction in chronic amphetamine and heroin abusers. Neuropsychopharmacology. 2000;23:113–126. doi: 10.1016/S0893-133X(00)00097-X. [DOI] [PubMed] [Google Scholar]
- Palmer CGS, Bailey JN, Ramsey C, Cantwell D, Sinsheimer JS, Del’Homme M, McGough J, Woodward JA, Asarnow R, Asarnow J, Nelson S, Smalley SL. No evidence of linkage or linkage disequilibrium between DAT1 and attention deficit hyperactivity disorder in a large sample. Psychiatr Genet. 1999;9:157–160. doi: 10.1097/00041444-199909000-00009. [DOI] [PubMed] [Google Scholar]
- Paloyelis Y, Asherson P, Mehta MA, Faraone SV, Kuntsi J. DAT1 and COMT effects on delay discounting and trait impulsivity in male adolescents with attention deficit/hyperactivity disorder and healthy controls. Neuropsychopharmacology. 2010;35:2414–2426. doi: 10.1038/npp.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini A, Sinibaldi L, Paloscia C, Douzgou S, Pitzianti MB, Romeo E, Curatolo P, Pizzuto A. Neurocognitive effects of methylphenidate on ADHD children with different DAT genotypes: a longitudinal open label trial. Eur J Paediatr Neurol. 2013;17:407–414. doi: 10.1016/j.ejpn.2013.02.002. [DOI] [PubMed] [Google Scholar]
- Polanczyk G, de Lima MS, Horta BL, Biederman J, Rohde LA. The worldwide prevalence of ADHD: a systematic review and metaregression analysis. Am J Psychiatry. 2007;164:942–948. doi: 10.1176/ajp.2007.164.6.942. [DOI] [PubMed] [Google Scholar]
- Poultney CS, Goldberg AP, Drapeau E, Kou Y, Harony-Nicolas H, Kajiwara Y, De Rubeis S, Durand S, Stevens C, Rehnström K, Palotie A, Daly MJ, Ma’ayan A, Fromer M, Buxbaum JD. Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. Am J Hum Genet. 2013;93:607–619. doi: 10.1016/j.ajhg.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puffenberger EG, Junks RN, Sougnez C, Cibulskis K, Willert RA, Achilly NP, Cassidy RP, Fiorentini CJ, Heiken KF, Lawrence JJ, Mahoney MH, Miller CJ, Nair DT, Politi KA, Worcester KN, Setton RA, DiPiazza R, Sherman EA, Eastman JT, Francklyn C, Robey-Bond S, Rider NL, Gabriel S, Morton DH, Strauss KA. Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS ONE. 2012;7:e28396. doi: 10.1371/journal.pone.0028936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puumala T, Sirviö J. Changes in activities of dopamine and serotonin systems in the frontal cortex underlie poor choice accuracy and impulsivity of rats in an attention task. Neuroscience. 1998;83:489–499. doi: 10.1016/s0306-4522(97)00392-8. [DOI] [PubMed] [Google Scholar]
- Ralph RJ, Paulus MP, Geyer MA. Strain-specific effects of amphetamine on prepulse inhibition and patterns of locomotor behavior in mice. J Pharmacol Exp Ther. 2001;298:148–155. [PubMed] [Google Scholar]
- Rapoport JL, Buchsbaum MS, Zahn TP, Weingartner H, Ludlow C, Mikkelsen EJ. Dextroamphetamine: cognitive and behavioral effects in normal prepubertal boys. Science. 1978;199:560–563. doi: 10.1126/science.341313. [DOI] [PubMed] [Google Scholar]
- Reis RAM, Ventura ALM, Kubrusly RCC, de Mello MCF, de Mello FG. Dopaminergic signaling in the developing retina. Brain Res Rev. 2007;54:181–188. doi: 10.1016/j.brainresrev.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Ribases M, Ramos-Quiroga JA, Hervás A, Sánchez-Mora C, Bosch R, Bielsa A, Gastaminza X, Lesch K-P, Reif A, Renner TJ, Romanos M, Warnke A, Walitza S, Freitag C, Meyer J, Palmason H, Casas M, Bayés M, Cormand B. Candidate system analysis in ADHD: evaluation of nine genes involved in dopaminergic neurotransmission identifies association with DRD1. World J Biol Psychiatry. 2012;13:281–292. doi: 10.3109/15622975.2011.584905. [DOI] [PubMed] [Google Scholar]
- Roman T, Schmitz M, Polanczyk G, Eizirik M, Rohde LA, Hutz MH. Attention-deficit hyperactivity disorder: a study of association with both the dopamine transporter gene and the dopamine D4 receptor gene. Am J Med Genet Part B. 2001;105B:471–478. doi: 10.1002/ajmg.1408. [DOI] [PubMed] [Google Scholar]
- Rommelse NNJ, Geurts HM, Franke B, Buitelaar JK, Hartman CA. A review on cognitive and brain endophenotypes that may be common in autism spectrum disorder and attention-deficit/hyperactivity disorder and facilitate the search for pleiotropic genes. Neurosci Biobehav Rev. 2011;35:1363–1396. doi: 10.1016/j.neubiorev.2011.02.015. [DOI] [PubMed] [Google Scholar]
- Russell VA. Overview of animal models of attention deficit hyperactivity disorder (ADHD) Curr Protoc Neurosci. 2011;54:9.35.1–9.35.25. doi: 10.1002/0471142301.ns0935s54. [DOI] [PubMed] [Google Scholar]
- Sagvolden T. Impulsiveness, overactivity, and poorer sustained attention improve by chronic treatment with low doses of l-amphetamine in an animal model of attention-deficit/hyperactivity disorder (ADHD) Behav Brain Funct. 2011;7:6. doi: 10.1186/1744-9081-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagvolden T, Xu T. l-Amphetamine improves poor sustained attention while D-amphetamine reduces overactivity and impulsiveness as well as improves sustained attention in an animal model of attention-deficit/hyperactivity disorder (ADHD) Behav Brain Funct. 2008;4:3. doi: 10.1186/1744-9081-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagvolden T, Metzger MA, Schiørbeck HK, Rugland A-L, Spinnangr I, Sagvolden G. The spontaneously hypertensive rat (SHR) as an animal model of childhood hyperactivity (ADHD): changed reactivity to reinforcers and to psychomotor stimulants. Behav Neural Biol. 1992;58:103–112. doi: 10.1016/0163-1047(92)90315-u. [DOI] [PubMed] [Google Scholar]
- Sahgal A, Andrews JS, Biggins JA, Candy JM, Edwardson JA, Keith AB, Turner JD, Wright C. N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) affects locomotor activity without producing a nigrostriatal lesion in the rat. Neurosci Lett. 1984;48:179–184. doi: 10.1016/0304-3940(84)90016-8. [DOI] [PubMed] [Google Scholar]
- Sakrikar D, Mazei-Robison MS, Mergy MA, Richtand NW, Han Q, Hamilton PJ, Bowton E, Galli A, Veenstra-VanderWeele J, Gill M, Blakely RD. Attention deficit/hyperactivity disorder-derived coding variation in the dopamine transporter disrupts microdomain targeting and trafficking regulation. J Neurosci. 2012;32(16):5385–5397. doi: 10.1523/JNEUROSCI.6033-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanday L, Patti CL, Zanin KA, Tufik S, Frussa-Filho R. Amphetamine-induced memory impairment in a discriminative avoidance task is state-dependent in mice. Int J Neuropsychopharmacol. 2013;16:583–592. doi: 10.1017/S1461145712000296. [DOI] [PubMed] [Google Scholar]
- Segal DS, Kuczenski R. An escalating dose “binge” model of amphetamine psychosis: behavioral and neurochemical characteristics. J Neurosci. 1997;17:2551–2566. doi: 10.1523/JNEUROSCI.17-07-02551.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang C-Y, Gau SS-F. Association between the DAT1 gene and spatial working memory in attention deficit hyperactivity disorder. Int J Neuropsychopharmacol. 2013 Sep 6;:1–13. doi: 10.1017/S1461145713000783. [DOI] [PubMed] [Google Scholar]
- Sian J, Gerlach M, Youdim MBH, Riederer P. Parkinson’s disease: a major hypokinetic basal ganglia disorder. J Neural Transm. 1999;106:443–476. doi: 10.1007/s007020050171. [DOI] [PubMed] [Google Scholar]
- Smith CB. Effects of d-amphetamine upon brain amine content and locomotor activity of mice. J Pharmacol Exp Ther. 1964;147:96–102. [PubMed] [Google Scholar]
- Spencer TJ, Biederman J, Madras BK, Dougherty DD, Bonab AA, Livni E, Meltzer PC, Martin J, Rauch S, Fischman AJ. Further evidence of dopamine transporter dysregulation in ADHD: a controlled PET imaging study using altropane. Biol Psychiatry. 2007;62:1059–1061. doi: 10.1016/j.biopsych.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani MR, Moghaddam B. Effects of repeated treatment with amphetamine or phencyclidine on working memory in the rat. Behav Brain Res. 2002;134:267–274. doi: 10.1016/s0166-4328(02)00040-2. [DOI] [PubMed] [Google Scholar]
- Sugita R, Sawa Y, Nomura S, Zorn SH, Tamauchi T. Effects of reserpine on dopamine metabolite in the nucleus accumbens and locomotor activity in freely moving rats. Neurochem Res. 1989;14:267–270. doi: 10.1007/BF00971322. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Tao Q, Fan X, Li T, Tang Y, Yang D, Le W. Gender segregation in gene expression and vulnerability to oxidative stress induced injury in ventral mesencephalic cultures of dopamine neurons. J Neurosci Res. 2012;90:167–178. doi: 10.1002/jnr.22729. [DOI] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirelli E, Terry P. Biphasic locomotor effects of the dopamine D1 agonist SKF 38393 and their attenuation in non-habituated mice. Psychopharmacology. 1993;110:69–75. doi: 10.1007/BF02246952. [DOI] [PubMed] [Google Scholar]
- Todd RD, Jong Y-JI, Lobos EA, Reich W, Heath AC, Neuman RJ. No association of the dopamine transporter gene 3′ VNTR polymorphism with ADHD subtypes in a population sample of twins. Am J Med Genet Part B. 2001;105B:745–748. doi: 10.1002/ajmg.1611. [DOI] [PubMed] [Google Scholar]
- Treuer T, Gau SS-F, Méndez L, Montgomery W, Monk JA, Altin M, Wu S, Lin CCH, Dueñas HJ. A systematic review of combination therapy with stimulants and atomoxetine for attention-deficit/hyperactivity disorder, including patient characteristics, treatment strategies, effectiveness, and tolerability. J Child Adolesc Psychopharmacol. 2013;23:179–193. doi: 10.1089/cap.2012.0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifilieff P, Martinez D. Imaging addiction D2 receptors and dopamine signaling in the striatum as biomarkers for impulsivity. Neuropharmacology. 2014;76:498–509. doi: 10.1016/j.neuropharm.2013.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai Y-F, Tsai H-W, Tai M-Y, Lu K-S. Age-related changes in locomotor behavior induced by MPTP in rats. Neurosci Lett. 1991;129:153–155. doi: 10.1016/0304-3940(91)90743-d. [DOI] [PubMed] [Google Scholar]
- Vaishnavi V, Manikandan M, Tiwary BK, Munirajan AK. Insights on the functional impact of microRNAs present in autism-associated copy number variants. PLoS ONE. 2013;8:e56781. doi: 10.1371/journal.pone.0056781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bergh FS, Bloemarts E, Chan JSW, Groenink L, Olivier B, Oosting RS. Spontaneously hypertensive rats do not predict symptoms of attention-deficit hyperactivity disorder. Pharmacol Biochem Behav. 2006;83:380–390. doi: 10.1016/j.pbb.2006.02.018. [DOI] [PubMed] [Google Scholar]
- van Dyck CH, Quinlan DM, Cretella LM, Staley JK, Malison RT, Baldwin RM, Seibyl JP, Innis RB. Unaltered dopamine transporter availability in adult attention deficit hyperactivity disorder. Am J Psychiatry. 2002;159:309–312. doi: 10.1176/appi.ajp.159.2.309. [DOI] [PubMed] [Google Scholar]
- van Rossum JM, Hurkmans JATM. Mechanism of action of psychomotor stimulant drugs: significance of dopamine in locomotor stimulant action. Int J Neuropharmacol. 1964;3:227–239. doi: 10.1016/0028-3908(64)90012-7. [DOI] [PubMed] [Google Scholar]
- Vandenbergh DJ, Persico AM, Hawkins AL, Griffin CA, Li X, Jabs EW, Uhl GR. Human dopamine transporter gene (DAT1) maps to chromosome 5p15.3 and displays a VNTR. Genomics. 1992a;14:1104–1106. doi: 10.1016/s0888-7543(05)80138-7. [DOI] [PubMed] [Google Scholar]
- Vandenbergh DJ, Persico AM, Uhl GR. A human dopamine transporter cDNA predicts reduced glycosylation, displays a novel repetitive element and provides racially-dimorphic TaqI RFLPs. Brain Res Mol Brain Res. 1992b;15:161–166. doi: 10.1016/0169-328x(92)90165-8. [DOI] [PubMed] [Google Scholar]
- Vandenbergh DJ, Thompson MD, Cook EH, Bendahhou E, Nguyen T, Krasowski MD, Zarrabian D, Comings D, Sellers EM, Tyndale RF, George SR, O’Dowd BF, Uhl GR. Human dopamine transporter gene: coding region conservation among normal, Tourette’s disorder, alcohol dependence and attention-deficit hyperactivity disorder populations. Mol Psychiatry. 2000;5:283–292. doi: 10.1038/sj.mp.4000701. [DOI] [PubMed] [Google Scholar]
- Varrone A, Halldin C. Molecular imaging of the dopamine transporter. J Nucl Med. 2010;51:1331–1334. doi: 10.2967/jnumed.109.065656. [DOI] [PubMed] [Google Scholar]
- Vaughan B, Kratochvil CJ. Pharmacotherapy of pediatric attention-deficit/ hyperactivity disorder. Child Adolesc Psychiatr Clin N Am. 2012;21:941–955. doi: 10.1016/j.chc.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Visser SN, Danielson ML, Bitsko RH, Perou R, Blumberg SJ. Convergent validity of parent-reported attention-deficit/hyperactivity disorder diagnosis: a cross-study comparison. JAMA Pediatr. 2013;167(7):674–675. doi: 10.1001/jamapediatrics.2013.2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitiello B, Elliott GR, Swanson JM, Arnold LE, Hechtman L, Abikoff H, Molina BS, Wells K, Wigal T, Jensen PS, Greenhill LL, Kaltman JR, Severe JB, Odbert C, Hur K, Gibbons R. Blood pressure and heart rate over 10 years in the multimodal treatment study of children with ADHD. Am J Psychiatry. 2012;169:167–177. doi: 10.1176/appi.ajp.2011.10111705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang G-J, Newcorn J, Fowler JS, Telang F, Solanto MV, Logan J, Wong C, Ma Y, Swanson JM, Schulz K, Pradhan K. Brain dopamine transporter levels in treatment and drug naïve adults with ADHD. Neuroimage. 2007;34:1182–1190. doi: 10.1016/j.neuroimage.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang G-J, Newcorn JH, Kollins SH, Wigal TL, Telang F, Fowler JS, Goldstein RZ, Klein N, Logan J, Wong C, Swanson JM. Motivation deficit in ADHD is associated with dysfunction of the dopamine reward pathway. Mol Psychiatry. 2011;16:1147–1154. doi: 10.1038/mp.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voon V, Reynolds B, Brezing C, Gallea C, Skaljic M, Ekanayake V, Fernandez H, Potenza MN, Dolan RJ, Hallett M. Impulsive choice and response in dopamine agonist-related impulse control behaviors. Psychopharmacology. 2010;207:645–659. doi: 10.1007/s00213-009-1697-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade TR, de Wit H, Richards JB. Effects of dopaminergic drugs on delayed reqard as a measure of impulsive behavior in rats. Psychopharmacology. 2000;150:90–101. doi: 10.1007/s002130000402. [DOI] [PubMed] [Google Scholar]
- Wallis GG, McHarg JF, Scott OCA. Acute psychosis caused by dextro-amphetamine. BMJ. 1949;2:1394. doi: 10.1136/bmj.2.4641.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Williams JM, Dipace C, Sung U, Javitch JA, Galli A, Saunders C. Dopamine transporter activity mediates amphetamine-induced inhibition of Akt through a Ca2+/calmodulin-dependent kinase II-dependent mechanism. Mol Pharmacol. 2007;71:835–842. doi: 10.1124/mol.106.026351. [DOI] [PubMed] [Google Scholar]
- Weyandt L, Swentosky A, Gudmundsdottir BG. Neuroimaging and ADHD: fMRI, PET, DTI findings, and methodological limitations. Dev Neuropsychol. 2013;38:211–225. doi: 10.1080/87565641.2013.783833. [DOI] [PubMed] [Google Scholar]
- Willcutt EG. The prevalence of DSM-IV attention-deficit/hyperactivity disorder: a meta-analytic review. Neurotherapeutics. 2012;9:490–499. doi: 10.1007/s13311-012-0135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winstanley CA, Theobald DEH, Dalley JW, Robbins TW. Interactions between serotonin and dopamine in the control of impulsive choice in rats: therapeutic implications for impulse control disorders. Neuropsychopharmacology. 2005;30:669–682. doi: 10.1038/sj.npp.1300610. [DOI] [PubMed] [Google Scholar]
- Witkovsky P. Dopamine and retinal function. Doc Ophthalmol. 2004;108:17–40. doi: 10.1023/b:doop.0000019487.88486.0a. [DOI] [PubMed] [Google Scholar]
- Wittmann BC, Tan GC, Lisman JE, Dolan RJ, Düzel E. DAT genotype modulates striatal processing and long-term memory for items associated with reward and punishment. Neuropsychologia. 2013;51:2184–2193. doi: 10.1016/j.neuropsychologia.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolraich ML, Lambert W, Doffing MA, Bickman L, Simmons T, Worley K. Psychometric properties of the Vanderbilt ADHD Diagnostic Parent Rating Scale in a referred population. J Pediatr Psychol. 2003;28(8):559–568. doi: 10.1093/jpepsy/jsg046. [DOI] [PubMed] [Google Scholar]
- Wolraich ML, Bard DE, Neas B, Doffing M, Beck L. The psychometric properties of the Vanderbilt Attention-Deficit Hyperactivity Disorder Diagnostic Teacher Rating Scale in a community population. J Dev Behav Pediatr. 2013;34:83–93. doi: 10.1097/DBP.0b013e31827d55c3. [DOI] [PubMed] [Google Scholar]
- Wultz B, Sagvolden T, Moser EI, Moser M-B. The spontaneously hypertensive rat as an animal model of attention-deficit hyperactivity disorder: effects of methylphenidate on exploratory behavior. Behav Neural Biol. 1990;53:88–102. doi: 10.1016/0163-1047(90)90848-z. [DOI] [PubMed] [Google Scholar]
- Xi X, Chu R, Zhou X, Lu Y, Liu X. Retinal dopamine transporter in experimental myopia. Chin Med J (Engl) 2002;115:1027–1030. [PubMed] [Google Scholar]
- Yamori Y. Pathogenesis of spontaneous hypertension as a model for essential hypertension. Jpn Circ J. 1977;41:259–266. doi: 10.1253/jcj.41.259. [DOI] [PubMed] [Google Scholar]
- Zahn TP, Rapoport JL, Thompson CL. Autonomic and behavioral effects of dextroamphetamin and placebo in normal and hyperactive prepubertal boys. J Abnorm Child Psychol. 1980;8:145–160. doi: 10.1007/BF00919060. [DOI] [PubMed] [Google Scholar]
- Zimmer L. Positron emission tomography neuroimaging for a better understanding of the biology of ADHD. Neuropharmacology. 2009;57:601–607. doi: 10.1016/j.neuropharm.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Zubrycki EM, Giordano M, Sanberg PR. The effects of cocaine on multivariate locomotor behavior and defecation. Behav Brain Res. 1990;36:155–159. doi: 10.1016/0166-4328(90)90169-f. [DOI] [PubMed] [Google Scholar]