

Abstract
The inherited peroxisomal disorder X-linked adrenoleukodystrophy (X-ALD), associated with neurodegeneration and inflammatory cerebral demyelination, is caused by mutations in the ABCD1 gene encoding the peroxisomal ATP-binding cassette (ABC) transporter ABCD1 (ALDP). ABCD1 transports CoA-esters of very long-chain fatty acids (VLCFA) into peroxisomes for degradation by β-oxidation; thus, ABCD1 deficiency results in VLCFA accumulation. The closest homologue, ABCD2 (ALDRP), when overexpressed, compensates for ABCD1 deficiency in X-ALD fibroblasts and in Abcd1-deficient mice. Microglia/macrophages have emerged as important players in the progression of neuroinflammation. Human monocytes, lacking significant expression of ABCD2, display severely impaired VLCFA metabolism in X-ALD. Here, we used thioglycollate-elicited primary mouse peritoneal macrophages (MPMΦ) from Abcd1 and Abcd2 single- and double-deficient mice to establish how these mutations affect VLCFA metabolism. By quantitative RT-PCR, Abcd2 mRNA was about half as abundant as Abcd1 mRNA in wild-type and similarly abundant in Abcd1-deficient MPMΦ. VLCFA (C26∶0) accumulated about twofold in Abcd1-deficient MPMΦ compared with wild-type controls, as measured by gas chromatography-mass spectrometry. In Abcd2-deficient macrophages VLCFA levels were normal. However, upon Abcd1/Abcd2 double-deficiency, VLCFA accumulation was markedly increased (sixfold) compared with Abcd1-deficient MPMΦ. Elovl1 mRNA, encoding the rate-limiting enzyme for elongation of VLCFA, was equally abundant across all genotypes. Peroxisomal β-oxidation of C26∶0 amounted to 62% of wild-type activity in Abcd1-deficient MPMΦ and was significantly more impaired (29% residual activity) upon Abcd1/Abcd2 double-deficiency. Single Abcd2 deficiency did not significantly compromise β-oxidation of C26∶0. Thus, the striking accumulation of VLCFA in double-deficient MPMΦ compared with single Abcd1 deficiency was due to the loss of ABCD2-mediated, compensatory transport of VLCFA into peroxisomes. We propose that moderate endogenous expression of Abcd2 in Abcd1-deficient murine macrophages prevents the severe metabolic phenotype observed in human X-ALD monocytes, which lack appreciable expression of ABCD2. This supports upregulation of ABCD2 as a therapeutic concept in X-ALD.
Introduction
X-linked adrenoleukodystrophy (X-ALD, OMIM #300100) is an inherited, neurodegenerative disease with variable clinical presentation. Childhood cerebral ALD (CCALD) with rapidly progressive cerebral inflammation and demyelination and adulthood adrenomyeloneuropathy (AMN), characterized by slowly progressive spastic paraparesis, are the two most prominent clinical phenotypes of X-ALD in males [1]–[3]. In heterozygous female carriers X-ALD manifests as a milder variant with myelopathy and/or neuropathy [4], while homozygous females are extremely rare. All forms of X-ALD are caused by mutations in the ATP-binding cassette transporter sub-family D (ALD) member 1 (ABCD1) gene. The encoded protein, ABCD1 (formerly adrenoleukodystrophy protein, ALDP), constitutes a half-transporter in the peroxisomal membrane [5]. ABCD1 mediates the transport of CoA-activated very long-chain fatty acids (VLCFA; carbon chain ≥22 C) into peroxisomes, where they are degraded by β-oxidation [6], [7]. Accumulation of saturated VLCFA, in particular C26∶0, in the plasma and tissues of human X-ALD patients is a characteristic biochemical and diagnostic feature of the disease [8].
The endogenous biosynthesis of VLCFA by elongation of long-chain fatty acids (LCFA) accounts for the major pool of VLCFA in human X-ALD patients [9], [10] with only a minor contribution from dietary sources [11]. The enzyme family, elongation of very long-chain fatty acids (ELOVL), mediates the substrate-selective step of the elongation pathway [12]. ELOVL1 was identified to catalyze the synthesis of both saturated (C26∶0) and monounsaturated (C26∶1) VLCFA in human fibroblasts [13].
The capacity for peroxisomal β-oxidation of saturated VLCFA is reduced in patients with X-ALD [14] as well as in Abcd1-deficient mice [15]–[17]. In human fibroblasts, ABCD1 deficiency leads to impaired transport of CoA-esters of VLCFA across the peroxisomal membrane [7]. As a result, the majority of VLCFA escapes degradation via peroxisomal β-oxidation and accumulates in X-ALD conditions. Moreover, the accumulating VLCFA can serve as substrates for ELOVL1, which promotes further increase in chain length and amount of VLCFA culminating with peak levels of C26∶0 [13], [18]. The excess of VLCFA leads to disturbances of intracellular metabolism to varying extent depending on the cell type. Such perturbations may result in alterations of membrane structure, stability and function [19]–[21], oxidative stress [22], [23], energy metabolism [24], [25] and increased expression of inducible nitric oxide synthase [26], [27].
Another peroxisomal ATP-binding cassette (ABC) transporter, ABCD2 (formerly adrenoleukodystrophy-related protein, ALDRP), encoded by the ABCD2 gene, is the closest homologue of ABCD1 [28]. Overexpression of ABCD2 has been shown to functionally compensate for the impairment in peroxisomal β-oxidation and VLCFA accumulation resulting from ABCD1 deficiency in human X-ALD fibroblasts and in Abcd1-deficient mice [29]–[31]. Furthermore, expression of the human ABC transporters in a heterologous yeast system indicated different but overlapping substrate specificities for ABCD1 and ABCD2 [6], [32]. However, little is known about the endogenous capacity for cross-compensation between ABCD1 and ABCD2 under conditions of insufficiency in vivo. In Abcd2-deficient mice, saturated VLCFA do not generally accumulate; increased C26∶0 levels have been detected exclusively in the dorsal root ganglia of young Abcd2-deficient mice [33]. Instead, accumulation of some saturated LCFA and monounsaturated fatty acids; in particular C20∶0 and ω9-monounsaturated fatty acids was reported in several tissues of Abcd2-deficient mice including liver, adrenal gland and sciatic nerve. In other tissues, like spinal cord, the abundance of these fatty acids was normal; and in Abcd1/Abcd2-double deficient mice only the level of C22∶1ω9 was elevated compared with Abcd1 and Abcd2 single deficient mice [34].
Currently, there is no curative therapy available for most X-ALD patients [35]. Allogeneic bone marrow transplantation or hematopoietic stem cell transplantation [36], [37] and also gene therapy of autologous CD34+ hematopoietic stem cells, providing intact ABCD1, can arrest the inflammatory cerebral demyelination in patients at early stages of cerebral ALD [38]. The beneficial effects observed upon transplantation and gene therapy of CD34+ hematopoietic stem cells suggest an involvement of the bone marrow-derived macrophages and microglia in this process [39].
In human monocytes/macrophages, ABCD2 is virtually not expressed [40] and, therefore, cannot compensate for the functional loss of ABCD1 resulting in a severe metabolic phenotype in this cell lineage of X-ALD patients [41]. Interestingly, in the perilesional white matter surrounding demyelinating brain lesions in cerebral ALD, microglia, the resident macrophages of the brain, are particularly vulnerable to neurotoxicity and apoptosis [42], as reviewed in [43]. Furthermore, in X-ALD, VLCFA were found to be enriched in the lysophosphatidylcholine (LPC) fraction [44]; and C24∶0-containing LPC elicited microglial activation and apoptosis when injected into the cerebral cortex of mice [42]. In comparison to other sources of murine macrophages or microglia, large numbers of primary macrophages can be obtained from the peritoneal cavity after stimulation with thioglycollate [45], [46], permitting biochemical studies of VLCFA metabolism in primary macrophages of targeted mouse mutants. These preparations are highly enriched for macrophages and after adherence to plastic culture dishes, most of the cells are positive for the macrophage markers F4/80 or CD11b. In addition to macrophages, some eosinophils, which also express these surface markers, may be present [47]. The vast majority of thioglycollate-elicited primary mouse peritoneal macrophages (MPMФ) are derived from monocytes that are recruited into the peritoneum and partially activated by the inflammatory stimulus. These differ in size, surface markers and function from the residential peritoneal macrophages but show similar phagocytic activity [48]. Genome-wide transcriptome analysis indicates that thioglycollate-induced macrophages are closely related to microglial cells as well as to bone marrow derived macrophages [49].
Here, we used MPMΦ to study the contribution of endogenous Abcd2 expression to VLCFA metabolism in the macrophage-lineage under Abcd1 deficiency. We determined the mRNA levels for the peroxisomal fatty acid transporters ABCD1 and ABCD2, as well as for the fatty acyl chain-elongating enzyme ELOVL1, in primary macrophages from wild-type and mutant mice with single or combined Abcd1 and Abcd2 deficiencies. Next, we established how these expression patterns correlate with the extent of accumulation and the capacity for degradation of VLCFA. Our results indicate a strong compensatory effect of ABCD2 on the metabolic phenotype of Abcd1-deficient murine macrophages.
Materials and Methods
Ethics Statement
The care, handling and experiments involving mice were carried out in accordance with the national (Austrian) regulations (BGBl. II Nr. 522/2012) and the directive 2010/63/EU of the European Parliament and the council of the European Union. All procedures were reviewed and approved by the local Animal Care and Use Committee of the Medical University of Vienna and by the Austrian Ministry for Science and Research (BMWF-66.009/0100-II/3b/2013). Mice were euthanized by CO2 inhalation before isolation of peritoneal macrophages.
Animals
Mice with a targeted inactivation (knock-out) of the Abcd1 gene (B6.129-Abcd1tm1Kan) have been described previously [15]. Mice with a null mutation in the Abcd2 gene (B6.129-Abcd2tm1Sfp) were generated as described below. Both mutations had been backcrossed onto the C57BL/6J background for at least twelve generations, before cross-breeding to generate Abcd1 and Abcd2 single- and double-deficient mutants and wild-type littermates for this study. Mice of the different allelic combinations appeared at the expected frequencies, were viable and developed on schedule without any overt phenotypical abnormalities. Adult (3 to 6-months-old) male mice were used for isolation of primary peritoneal macrophages. The genotype was determined at weaning and confirmed at sacrifice of experimental animals by standard PCR analysis using the following primers: for Abcd1, forward 5′-TGTCGGGCGTAGACGCTGTCGT-3′ in combination with reverse 5′-CAGGACCACAGCTGTGCGCTTC-3′ for the wild-type allele (yielding a 597-bp PCR product) and reverse 5′-GCCTTCTATCGCCTTCTTGACGAG-3′ for the knock-out (neomycin resistance gene, neo) allele (yielding a 210-bp PCR product); and for Abcd2, forward 5′-TTCTAAGTGCCGCTGAGCATGC-3′ in combination with reverse 5′-CTGCTGCATTTAGCATGTGTATC-3′ for the wild-type allele (yielding a 466-bp PCR product) and reverse 5′-CCATCTTGTTCAATGGCCGATC-3′ for the knock-out (neo) allele (yielding a 322-bp PCR product). All mice for this study were bred and maintained at the local animal facility of the Medical University of Vienna. Mice were housed under standard conditions on a 12/12 h light/dark cycle in a temperature and humidity–controlled environment with standard mouse chow diet and water ad libitum.
Construction of the Abcd2 targeting vector
A mouse 129/SvJ genomic library (λFIXII; Stratagene) was screened with a probe covering nucleotides 16−1,078 of the murine Abcd2 cDNA (GenBank™ Accession No. NM 011994). From a phage λ-clone containing 17 kb of the Abcd2 gene, including exon 1 through intron 3 and 3.5 kb of 5′-flanking DNA, genomic DNA fragments were subcloned into plasmids to generate a gene-replacement vector for positive-negative selection [50]. The final targeting construct (Fig. 1A) contained in 5′ to 3′ order: a 2.2-kb XhoI−NdeI fragment carrying the thymidine kinase gene from pMC1TK (Stratagene); a 2.1-kb NdeI− SphI fragment (−2.5 kb to −422 relative to the translation start site of Abcd2) representing the 5′ (short) Abcd2 homology region, adapted with a 3′ HindIII site; a 1.15-kb neo gene cassette from pMCneopA (Stratagene) adapted with 3′ HindIII sites; and a 6-kb HindIII−NotI fragment accommodating the 3′ (long) Abcd2 homology region [from HindIII (+816) in exon 1 to XhoI in intron 3, cloned into a polylinker with a NotI site] inserted between SalI and NotI of the pBluescript KSII (Stratagene) vector backbone. Thus, the neo cassette replaces a 1,238-bp region of the Abcd2 gene, between SphI (−422) and HindIII (+816), which contains the promoter and most of exon 1. Both neo and thymidine kinase were inserted in sense orientation relative to Abcd2 (Fig. 1A).
Figure 1. Strategy and molecular evidence for targeted inactivation of the Abcd2 gene.

(A) Structure of the 5′ region of the Abcd2 gene, the targeting construct and the disrupted gene. Exons (Ex) 1–3 are depicted as boxes with the protein coding region in gray. After homologous recombination, 1.24 kb of the Abcd2 gene, including the transcription and translation start sites and most of exon 1, is replaced by the neomycin resistance gene cassette (Neo) of the targeting construct. Positions of the PCR primers (P1, P2) and the Southern blot probe (****) used in genotyping to detect homologous recombination are indicated. Restriction sites used for cloning or analysis are marked: H, HindIII; N, NdeI; Not, NotI; S, ScaI; Sp, SphI and X, XhoI. (B) Southern blot analysis of ScaI-digested DNA from Abcd2 −/−, Abcd2 +/+ and Abcd2 +/− mice. Chemiluminescent detection of a digoxygenin-labelled probe from the 5′ flanking DNA verified homologous recombination based on deletion of a ScaI-site in exon 1 of Abcd2 resulting in 3.9-kb and 4.4-kb fragments from the wild-type (WT) and knock-out (KO) alleles, respectively. (C) Northern blot analysis of mRNA from brain and skeletal muscle of adult Abcd2 −/− and Abcd2 +/+ mice. A 32P-labelled Abcd2 cDNA probe hybridized to the major 4.2 kb Abcd2 mRNA and minor (5.5 and 2.8 kb) variants in both tissues of wild-type mice, but not in Abcd2 −/− mice. As a loading control, the blot was re-probed with β-actin cDNA, which in skeletal muscle detects a shorter, more abundant mRNA than in brain. (D) Reverse transcription-coupled PCR analysis of Abcd2 mRNA expression in primary peritoneal macrophages from two Abcd2 +/+ and two Abcd2 −/− mice. Abcd2-specific primers amplified the expected 1,051-bp product in Abcd2 +/+ but not in Abcd2 −/− macrophages. As a positive control, amplification of Iba1 cDNA (310-bp fragment) was confirmed in all samples.
Generation of Abcd2-deficient mice
Mouse RI embryonic stem (ES) cells [51], kindly provided by A. Nagy (Toronto), were transfected by electroporation using 50 µg of the NotI-linearized targeting vector. Colonies were isolated after G418/gancyclovir selection as previously described [50], [52]. Double-resistant clones resulting from homologous recombination were identified by PCR amplification of a 3.1-kb genomic Abcd2-neo junction fragment from DNA of heat-lysed cells using Abcd2 forward primer (5′-AGGATCTGCTTAAGAGTTCCACT-3′) at position P1, flanking the homology region, and neo reverse primer (5′-CCATCTTGTTCAATGGCCGATC-3′) at position P2 (as indicated in Fig. 1A). The initial denaturation at 94°C for 3 min was followed by 32 cycles of PCR (denaturation at 94°C for 30 s, annealing at 61°C for 30 s, and extension at 72°C for 2 min) and final extension at 72°C for 2 min. Individual PCR-positive clones were expanded and homologous recombination was confirmed by Southern blot analysis of ScaI-digested DNA (8 µg/sample) using a digoxygenin-labelled hybridization probe (indicated in Fig. 1A) derived from the 5′-flanking region, upstream from the neo insertion.
Microinjection of selected ES cells into C57BL/6J blastocysts and embryo transfer to foster mothers were performed by standard procedures. Resulting chimeras were mated with C57BL/6J mice and germ line-transmission was obtained from one founder. Heterozygous (Abcd2+/−) F1 mice were again crossed with C57BL/6J mice and F2 heterozygous mutants were interbred to generate homozygous (Abcd2−/−) mice in the F3 generation. The Abcd2 genotype of these mice was determined by allele-specific PCR and Southern blot analyses using genomic DNA (Fig. 1B) as outlined for ES cells.
RNA analyses for verification of functional disruption of Abcd2
Northern blot analysis was used to confirm the absence of functional Abcd2 gene expression in Abcd2−/− mutant mice. Total RNA was isolated from flash frozen tissues [53] of 2.5-months-old Abcd2 +/+ and Abcd2 −/− littermates. For each sample, the polyA+-RNA fraction (approximately 2 µg) obtained by oligo(dT) selection of 100 µg total RNA was size-separated on formaldehyde-agarose gels to prepare northern blots as previously described [54]. A gene-specific, [α-32P]dCTP-labelled hybridization probe covering exon 1 was generated by random priming with Abcd2 cDNA as a template. As a control of loading and transfer, the blot was stripped and reprobed with β-actin cDNA. All blots were washed to high stringency (at 68°C in 0.2 x SSC, 0.2% SDS) and monitored with a phoshorimager (Molecular Imager FX System, Bio-Rad).
For end point RT-PCR analysis of Abcd2 mRNA expression in MPMФ, total cellular RNA was isolated and reverse transcribed as described for the qRT-PCR analysis (see below). Diluted cDNA, corresponding to 4 ng of total RNA, was applied for 35 cycles of PCR using GoTaq polymerase (Promega) with Abcd2 forward primer (5′-GAAGCCTCGGACTTTCATCATC-3′) and reverse primer (5′-GTGTAATTATGGGAACATTTTCAC-3′) from exon 1 and exon 5, respectively, at an annealing temperature of 59°C, to generate a 1,051-bp product in Abcd2+/+ cDNA samples. No amplicon was observed when reverse transcriptase was omitted or when genomic DNA was used as template. As a positive control, the macrophage marker ionized calcium-binding adapter molecule 1 (Iba1), also known as allograft inflammatory factor 1 (Aif1), was amplified from the same cDNA samples under identical conditions, except that annealing was at 54°C, with Iba1 forward primer (5′-AGAAGAGACTGGGGAGCTGGT-3′) from exon 3 and reverse primer (5′-CCAAGTTTCTCCAGCATTCGC -3′) from exon 7 (based on GenBank™ Accession No. NM 019467.2) generating a specific 310-bp PCR product.
Isolation and Cell Culture of Mouse Peritoneal Macrophages
Primary mouse peritoneal macrophages (MPMΦ) were isolated from wild-type and knock-out mice as described in [55] with some minor modifications. Briefly, mice were injected intraperitoneally with 2 ml of 4% thioglycollate medium (Sigma). Four days later, mice were sacrificed for peritoneal lavage with 10 ml of complete medium [RMPI-1640 medium (PAA) supplemented with 10% fetal bovine serum (PAA), 100 units/ml each of penicillin and streptomycin (Lonza) and 2.5 µg/ml Fungizone (Gibco)] using a syringe with a 23G needle. The recovered cells were centrifuged at 300 × g at 4°C for 10 min; the supernatant was aspirated and the cell pellet resuspended and washed twice in 10 ml complete RPMI-1640 medium. Finally, cells were counted and the required numbers were plated and cultured at 37°C with 5% CO2 for 4 h, during which the MPMΦ attach to the cell culture plate (CELLSTAR, Greiner Bio-One) while the vast majority of other cell types from the peritoneal lavage remain in suspension and are removed by aspiration. After this incubation period, the attached MPMΦ were washed with phosphate buffered saline (PBS) and used according to the experimental requirements. Unless stated otherwise, the cells were maintained for 20 h in complete medium at 37°C with 5% CO2 before harvesting.
RNA isolation and quantitative reverse transcription-coupled PCR (qRT-PCR)
MPMΦ (3×105 cells/well) were seeded in 6-well cell culture plates. At harvest, the attached MPMΦ were washed with PBS and total RNA was isolated using the RNeasy Mini kit (Qiagen), including on-column DNA digestion with RNase-free DNase (Qiagen), according to the manufacturer’s instructions. RNA concentrations were determined using a NanoDrop spectrophotometer (PEQlab). Total RNA (80 ng) was reverse transcribed into cDNA in 20 µl final volume using the iScript™ cDNA synthesis kit (Bio-Rad) at 25°C for 5 min, 42°C for 30 min and 85°C for 5 min and holding at 4°C. The cDNA was diluted 1∶5 and 5-µl aliquots were applied for two-step qRT-PCR (denaturation at 95°C, annealing/extension at 60°C) using SsoFast EvaGreen Supermix (Bio-Rad) and the CFX96 Real-Time PCR Detection System (Bio-Rad) according to the manufacturer’s recommendations together with the following gene specific primer combinations: Hprt, forward 5′-ACTTCAGGGATTTGAATCACGTT-3′ and reverse 5′-GCAGATGGCCACAGGACTAGA-3′, product size 154 bp; Abcd1, forward 5′-GCTGTGACCTCCTACACTCTCC-3′ and reverse 5′-AGTAGTGCCAGTTCCACCTCA-3′, product size 251 bp; Abcd2, forward 5′-GAACTACCCCTCAGCGACAC-3′ and reverse 5′- ATGGCCTCTGTGGAATATAGAAC-3′, product size 280 bp; and Elovl1, forward 5′-ATTGAGCTGATGGACACAGTGAT-3′ and reverse 5′-GACCAGGACAAACTGGATCAGC-3′, product size 279 bp.
The absolute quantification of each mRNA was based on standard curves, which were generated from serial dilutions of known copy numbers of linearized plasmids containing the corresponding cDNA region of murine Abcd1, Abcd2, Elovl1 or Hprt. The quantity obtained for each mRNA after reverse transcription to cDNA (Table S1 and Table S2) was normalized to that of Hprt in order to compensate for variation in amount of total RNA and/or cDNA conversion between samples.
VLCFA measurements
Deuterium-labelled VLCFA standards: [3,3,5,5-2H4]-hexacosanoic acid (2H4-C26∶0), [3,3,5,5-2H4]-tetracosanoic acid (2H4-C24∶0) and [3,3,5,5-2H4]-docosanoic acid (2H4-C22∶0) were obtained from Dr. Herman J. ten Brink (Free University Hospital, Amsterdam, The Netherlands); and [7,7,8,8-2H4]-palmitic acid (2H4-C16∶0) was from Cambridge Isotope Laboratories Inc. (Andover, MA, USA); and the unlabelled fatty acids C26∶0, C24∶0, C22∶0, and C16∶0 were from Sigma-Aldrich.
For each sample, 5−7×106 peritoneal cells were seeded in one culture plate and maintained in complete medium for either 1 day or 5 days. At harvest, the MPMΦ were washed three times with PBS and pelleted. Cell pellets were suspended in distilled water and sonicated three times for 30 s on ice. Protein concentration was determined by the method of Lowry using bovine serum albumin (Sigma-Aldrich) as standard [56]. Extraction and quantitative analysis of fatty acids by gas chromatography-mass spectrometry (GC-MS) were carried out as previously described [57]. GC-MS measurements were carried out in a TRACE MS Plus gas chromatograph single quadrupol mass spectrometer (Thermo Fisher Scientific) equipped with a J&W Scientific DB-1ms capillary column (30 m x 0.25 mm I.D., film thickness 0.25 µm; Agilent Technologies). Data were analyzed with the Finnigan Xcalibur™ software package (Thermo Fisher Scientific) using calibration curves obtained from unlabelled fatty acids in the concentration range from 0.005 to 0.2 µg/ml for C26∶0, from 0.25 to 2.5 µg/ml for C22∶0 and C24∶0, and from 5 to 100 µg/ml for C16∶0.
β-Oxidation of 1-14C-labelled fatty acids
The MPMΦ obtained after selective adherence of 2×106 peritoneal cells, seeded in one culture plate and maintained in complete medium for 20 h, were washed with PBS before harvesting and resuspending in 200 µl of buffer (250 mM sucrose, 10 mM Tris-Cl, pH 8.0). The protein concentration was measured in an aliquot of each sample using the CBQCA Protein Quantitation Kit (Molecular Probes). To determine the β-oxidation activity for C26∶0 and C16∶0, aliquots of 100 µl and 33 µl were used, respectively.
Radiolabelled 1-14C palmitic acid (C16∶0; ARC 0172A) and 1-14C hexacosanoic acid (C26∶0; ARC 1253) were obtained from American Radiolabeled Chemicals. Free fatty acids in ethanol were aliquoted into glass reaction tubes, dried under a stream of nitrogen and solubilized in 10 mg/ml α-cyclodextrin by ultrasonication. β-Oxidation of labelled fatty acids to acetate was carried out according to [58] with modifications as described previously [7]. Briefly, for each sample the reaction mix of 250 µl contained 4 µM radiolabelled fatty acid (either C16∶0 or C26∶0), 2 mg/ml α-cyclodextrin, 250 mM sucrose, 30 mM KCl, 20 mM Tris-Cl (pH 8.0), 8.5 mM ATP, 8.5 mM MgCl2, 2.5 mM l-carnitine, 1 mM DTT, 1 mM NAD+, 0.5 mM malate, 0.2 mM EDTA, 0.17 mM FAD, 0.16 mM CoA and cell preparation (as described above). Reactions were started by addition of cellular protein, incubated for 1 h at 37°C and stopped by adding KOH and heating at 60°C for 1 h. After protein precipitation by HClO4, a Folch partition was carried out and the amount of 14C-acetate in the aqueous phase was determined in a scintillation counter.
Statistical analyses
Statistical analyses were carried out either with two-tailed t-test or one-way analysis of variance (ANOVA) with appropriate post-hoc test. Statistical analyses for VLCFA were performed on log-transformed data using one-way ANOVA followed by Tukey’s post-hoc test. Thereafter, these data were back-transformed to obtain and represent the geometric means with error bars showing the asymmetrical standard deviations. Statistical analyses for β-oxidation assays were performed using one-way ANOVA followed by Tukey’s post-hoc test. Data from β-oxidation assays and qRT-PCR are represented as arithmetic means ± standard deviation (SD). Differences in mean values were considered statistically significant at p<0.05.
Results
Targeted inactivation of the murine Abcd2 gene
To study the compensatory role of Abcd2 in tissues and distinct cell types of X-ALD mice, we generated mice with a null mutation in the Abcd2 gene by applying a similar strategy as previously used for the targeted inactivation of Abcd1 [15]. Briefly, the targeting vector for homologous recombination in mouse ES cells was constructed from isogenic (129Sv) DNA encompassing the 5′ region of the murine Abcd2 gene (Fig. 1A). Between the “short arm” (3.5 kb of DNA upstream from exon 1) and the “long arm” (6 kb from the 3′ end of exon 1 through intron 3) of the targeting vector, 1,238 bp of the Abcd2 gene containing 250 bp of the promoter region and most of exon 1 were replaced by a neo gene cassette, which also served as a marker for positive selection. This deletion eliminates the transcription and translation start sites and the coding capacity for the 272 amino-terminal amino acids of ABCD2 [59]. A thymidine kinase gene cassette was inserted 5′ to the homology region for negative selection against random integration events. ES cells were transfected with the targeting vector and double (G418, gancyclovir)-resistant recombinant clones selected, of which 10% were identified by PCR to result from homologous recombination. Correct gene targeting was confirmed by Southern blot analysis discriminating between wild-type (Abcd2 +) and disrupted (Abcd2 −) alleles based on the loss of a diagnostic ScaI restriction site in exon 1 (Fig. 1A).
Germline chimeras were obtained from a correctly targeted ES clone injected into C57BL/6 blastocysts. The progeny obtained after breeding of this founder and subsequent generations with C57BL/6J mice gave rise to a permanent mouse line (B6.129-Abcd2tm1Sfp) carrying the disrupted Abcd2 gene (here referred to as Abcd2-deficient, Abcd2 −/−, or Abcd2 knock-out). The genotype was initially determined by PCR (data not shown) and Southern blot analysis (Fig. 1B) to confirm the gene structure expected after a unique homologous recombination event. Heterozygous and homozygous animals were obtained at the expected Mendelian ratios and were viable, fertile and developed into adults with normal home cage behaviour and healthy appearance. Starting around 12 months of age, Abcd2 −/− mice developed a sensory–motor impairment (data not shown), which has been reported previously in an independent Abcd2-deficient mouse strain [31], [33]. This ataxic phenotype developed with a similar age-of-onset also in Abcd1/Abcd2 double-deficient mice.
Loss of Abcd2 mRNA expression in gene targeted mice
To verify that homologous recombination had inactivated functional expression of Abcd2, we assessed the presence of Abcd2 mRNA by Northern blot analysis in brain and skeletal muscle, two tissues in which Abcd2 is normally abundantly expressed [60]. The Abcd2-specific cDNA probe detected the major 4.2 kb Abcd2 mRNA, as well as the minor 5.5 and 2.8 kb transcripts arising from alternative polyadenylation sites, in both tissues of wild-type but not Abcd2 −/− mice (Fig. 1C). The loading control, β-actin, gave similar signals in corresponding wild-type and knock-out tissues.
The absence of Abcd2 mRNA was also verified in primary mouse peritoneal macrophages (MPMФ) isolated from Abcd2−/− mice by end point RT-PCR using total cellular RNA. In wild-type cells, RT-PCR with Abcd2-specific primers robustly amplified the expected product, whereas no signal was obtained in Abcd2 −/− MPMФ (Fig. 1D). As a positive control for the quality of the RNA/cDNA preparations, the macrophage marker Iba1 could be easily amplified in all cDNA samples. Taken together, these expression studies confirm the absence of functional Abcd2 mRNA in cells and tissues of the Abcd2 knock-out mice, validating that the gene disruption generated a null mutation.
By cross-breeding the Abcd1- and Abcd2-deficient mutant strains, we generated mice with single and combined Abcd1/Abcd2-deficiencies and appropriate wild-type littermate controls, all on the C57BL/6J background. Mice of the different allelic combinations appeared at the expected frequencies, were viable and reached adulthood without overt phenotypical abnormalities. In the present study only young male mice were used as donors of thioglycollate-elicited peritoneal macrophages, because in X-linked ALD, predominantly the male (hemizygous) patients are severely affected.
Abcd2 mRNA is expressed at half the level of Abcd1 mRNA in mouse peritoneal macrophages
First, we explored the basal expression of the Abcd1 and Abcd2 genes in MPMΦ collected from wild-type mice and then compared these with the levels in Abcd1 and Abcd2-deficient MPMΦ. The absolute quantity of each mRNA was determined by qRT-PCR analysis and normalized to the level of Hprt mRNA (Fig. 2A and Table S1). In wild-type cells, both mRNAs were detected at low abundance; however, the Abcd2 mRNA was present at 58% of the level of the Abcd1 mRNA (Fig. 2A). Because we had previously found Abcd2 mRNA expression to be altered in hepatic tissue of Abcd1-deficient mice [61], we determined the Abcd2 mRNA levels in Abcd1-deficient MPMΦ in order to uncover any potential compensatory dysregulation. However, there was no statistically significant difference between the Abcd2 mRNA levels of wild-type and Abcd1-deficient MPMΦ (Fig. 2B). Vice versa, the Abcd1 mRNA remained at wild-type level in MPMΦ from Abcd2-deficient mice (Fig. S1).
Figure 2. Endogenous Abcd1 and Abcd2 mRNA levels in peritoneal macrophages of wild-type and Abcd1-deficient mice.

(A) The Abcd1 and Abcd2 mRNA copy numbers in C57BL/6J wild-type MPMΦ were determined by qRT-PCR. (B) Comparison of Abcd2 mRNA levels in wild-type (WT) and Abcd1-deficient (KO) MPMΦ. The numbers of samples (n) are indicated below the graphs. The graphs indicate mean values ± SD after normalization to the level of Hprt mRNA in each sample; n.s., no statistically significant difference.
Inactivation of Abcd2 alone has no impact on VLCFA levels of MPMΦ but a strong synergistic effect in combined Abcd1/Abcd2 deficiency
MPMΦ isolated from mice with single or combined Abcd1 and Abcd2 deficiency or from wild-type littermates were cultured for either one or five days before harvesting for fatty acid analysis. The amounts of VLCFA (C26∶0, C24∶0, C22∶0) and LCFA (C16∶0) were measured by GC-MS. First, we established the extent of accumulation of saturated VLCFA in Abcd1-deficient MPMΦ. Compared with wild-type levels, we found a two-fold increase in the ratio C26∶0/C22∶0 (Fig. 3A). There was no statistically significant difference in the C26∶0/C22∶0 ratio of Abcd2-deficient MPMΦ when compared with wild-type. However, we noticed strikingly increased accumulation of C26∶0 in MPMΦ from Abcd1/Abcd2 double-deficient mice, as judged by the ratio C26∶0/C22∶0 (Fig. 3A) as well as the absolute amounts (Fig. S2). The difference in accumulation (C26∶0/C22∶0) was statistically highly significant in comparison with wild-type or Abcd2-deficient or even with Abcd1-deficient samples (p<0.001). We observed similar C26∶0/C22∶0 profiles in MPMΦ harvested after five days (Fig. 3A) or after one day (Fig. S3) in cell culture. Because the level of C22∶0 remains unaltered or even drops somewhat in X-ALD, we also established the levels of C16∶0 in all samples, as an additional reference for comparisons of the different VLCFA species, in addition to their absolute values normalized to the amount of protein in the cell homogenate (Fig. S2). The C16∶0 levels were comparable across the different genotypes when normalizing to protein (Fig. S2D). The ratio C26∶0/C16∶0 (Fig. 3B) was increased in MPMΦ from Abcd1-deficient mice to a similar extent as that of C26∶0/C22∶0 (Fig. 3A). Also the marked increase in C26∶0 levels of Abcd1/Abcd2 double-deficient compared with Abcd1-deficient cells was obvious in the C26∶0/C16∶0 ratios (p<0.01) (Fig. 3B). Whereas the level of C24∶0 was only slightly elevated upon Abcd1 deficiency, also this fatty acid accumulated substantially in the Abcd1/Abcd2 double-deficient MPMΦ, as indicated by the C24∶0/C22∶0 ratio (Fig. 3C) as well as the absolute values (Fig. S2B). In contrast, the levels of C22∶0 were slightly lower in MPMΦ from Abcd1-deficient mice, both when expressed as ratio to C16∶0 (Fig. 3D) or after normalizing to protein amounts (Fig. S2C), but the reduction did not reach statistical significance. Interestingly, there was even a further, statistically significant decrease in the C22∶0/C16∶0 ratios in Abcd1/Abcd2 double-deficient MPMΦ when compared with wild-type, Abcd1-deficient and Abcd2-deficient MPMΦ (Fig. 3D).
Figure 3. VLCFA levels in wild-type, Abcd1 and Abcd2 single-deficient and Abcd1/Abcd2 double-deficient mouse peritoneal macrophages.

The concentrations of the VLCFA species C26∶0, C24∶0 and C22∶0 and the LCFA C16∶0 were determined by GC-MS. The relative amounts of fatty acids, expressed as ratios: (A) C26∶0/C22∶0; (B) C26∶0/C16∶0; (C) C24∶0/C22∶0 and (D), C22∶0/C16∶0 were analyzed in mouse peritoneal macrophages of wild-type (WT), Abcd1-deficient (Abcd1 KO), Abcd2-deficient (Abcd2 KO) and Abcd1/Abcd2 double-deficient (DOKO) mice after 5 days in culture (n = 3). The graphs indicate geometric means ± SD (asymmetrical). Statistically significant differences are indicated: * p<0.05, ** p<0.01, *** p<0.001; n.s., no statistically significant difference.
Elovl1 mRNA levels of MPMΦ are not affected by Abcd1 or Abcd2 deficiency
ELOVL1 is responsible for the carbon-chain elongation of VLCFA (C22∶0, C24∶0, C26∶0). As the marked increase in C26∶0 accumulation in Abcd1/Abcd2 double-deficient MPMΦ compared with isolated Abcd1 deficiency could be partially, or entirely, due to increased elongation of VLCFA, we measured the Elovl1 mRNA levels by qRT-PCR in wild-type and Abcd1/Abcd2 single- and double-deficient MPMΦ. After normalization to Hprt mRNA levels, we observed no difference in Elovl1 mRNA expression in any of the mutant genotypes (Fig. 4 and Table S2). Thus, at least at the level of gene expression, Elovl1, encoding the rate limiting enzyme for elongation of VLCFA, was not upregulated.
Figure 4. Elovl1 mRNA levels in wild-type, Abcd1-, Abcd2-, and Abcd1/Abcd2 double-deficient mouse peritoneal macrophages.
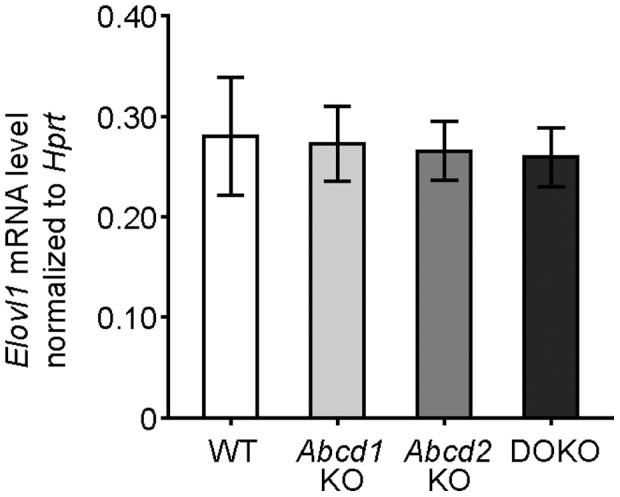
The Elovl1 mRNA copy numbers were determined by qRT-PCR in total RNA from mouse peritoneal macrophages of wild-type (WT), Abcd1-deficient (Abcd1 KO), Abcd2-deficient (Abcd2 KO) and Abcd1/Abcd2 double-deficient (DOKO) mice. The graphs indicate mean values ± SD after normalization to the level of Hprt mRNA in each sample (n = 3).
Peroxisomal β-oxidation of C26∶0 is markedly more impaired in Abcd1/Abcd2 double-deficient MPMΦ than in Abcd1 single deficiency
To establish whether the degradation of VLCFA is impaired in the different mutant macrophages, we determined the rate of peroxisomal β-oxidation in MPMΦ from wild-type, Abcd1-, Abcd2- and Abcd1/Abcd2 double-deficient mice using radiolabelled C26∶0 as substrate. As a control, and for normalization of the overall activity of each cell preparation, we measured the rate of β-oxidation for the LCFA C16∶0, which in mammals is primarily degraded in mitochondria. We found a highly significant (p<0.001) decrease (38%) in the relative β-oxidation activity for C26∶0 (depicted as ratio to C16∶0) in Abcd1-deficient MPMΦ compared with wild-type values (Fig. 5). Isolated Abcd2 deficiency had no statistically significant effect on the capacity for β-oxidation of either substrate. However, there was a highly significant (p<0.001) further reduction in the β-oxidation activity for C26∶0 in Abcd1/Abcd2 double-deficient MPMΦ when compared with Abcd1-deficient (54%) as well as wild-type (71%) and Abcd2-deficient (65%) MPMΦ (Fig. 5). Similar results were obtained when considering the absolute values of the β-oxidation rates for C26∶0 normalized to the amount of cellular protein in the MPMΦ samples (Fig. S4A). The β-oxidation activity for C16∶0 was similar in MPMΦ of all different genotypes (Fig. S4B).
Figure 5. β-Oxidation activity for C26∶0 in wild-type, Abcd1-, Abcd2-, and Abcd1/Abcd2 double-deficient mouse peritoneal macrophages.
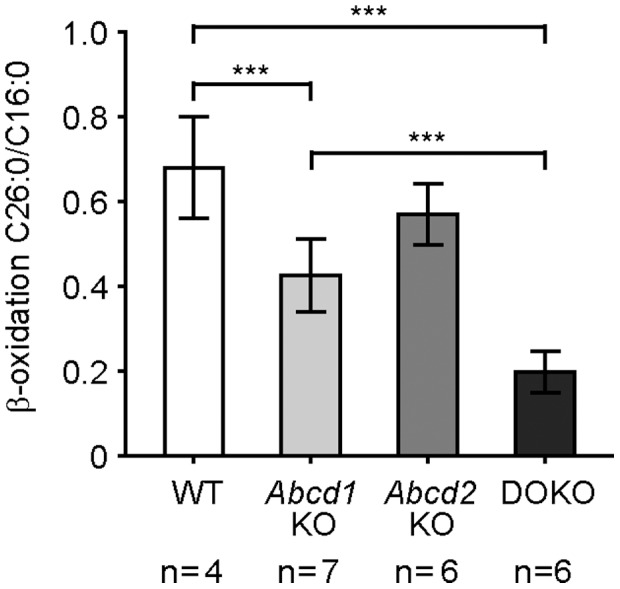
The rates of β-oxidation for radiolabelled C26∶0 and C16∶0 fatty acid substrates were measured in wild-type (WT), Abcd1-deficient (Abcd1 KO), Abcd2-deficient (Abcd2 KO) and Abcd1/Abcd2 double-deficient (DOKO) cell preparations. The results are shown as mean values ± SD of the ratio between C26∶0 and C16∶0. The numbers of samples (n) are indicated below the graphs. Statistically significant differences are indicated; *** p<0.001.
Discussion
Our results show that primary macrophages from Abcd1/Abcd2 double-deficient mice have a considerably more severe metabolic phenotype than those with single Abcd1 deficiency, as documented by a markedly reduced rate of peroxisomal β-oxidation for C26∶0 leading to the strongly increased accumulation of saturated VLCFA (C26∶0 and C24∶0). Inactivation of Abcd2 alone has no significant effect on the levels of saturated VLCFA in most murine tissues including brain, spinal cord and sciatic nerve, which are affected by the pathology in X-ALD [31], which is also the case in our Abcd2 −/− mouse strain (Forss-Petter, unpublished observation). One notable exception are the dorsal root ganglia [33], which may explain the earlier onset of a sensory motor phenotype in Abcd1/Abcd2 double-deficient and Abcd2 single-deficient mice compared with Abcd1-deficient mice [31]. Considering that ubiquitous transgenic overexpression of the murine ABCD2 protein in Abcd1-deficient mice rescued the metabolic impairment and the motor-behavioural phenotype [31], a more dramatic phenotype might have been anticipated in Abcd1/Abcd2 double-mutant mice. However, the additive but not amplified severity of the metabolic and behavioural deficits might be partially explained by the rather complementary expression patterns of these two peroxisomal ABC transporters [41], [60], [62].
In MPMΦ, we found both the Abcd1 and Abcd2 mRNAs to be expressed at relatively low abundance (i.e. estimated to represent less than 0.01% of the total mRNA molecules and lower than the level of Hprt mRNA). By qRT-PCR, the steady-state level of the Abcd1 mRNA was about twice that of Abcd2. Thus, MPMΦ constitute a good model system to evaluate the level of Abcd2 expression required to prevent a severe metabolic phenotype upon Abcd1 deficiency. Our results revealed about twofold higher levels of VLCFA in Abcd1-deficient MPMΦ, as judged by the ratio C26∶0/C22∶0, in agreement with previous observations [27]. This is similar to the extent of VLCFA storage in modestly affected tissues like the liver of X-ALD mice [63]. Interestingly, in Abcd1/Abcd2 double-deficient MPMΦ, we found an additional sixfold increase in accumulation of C26∶0 when compared with Abcd1 single-deficiency, which amounted to approximately twelvefold higher than wild-type levels. In brain and adrenal glands, the pathologically affected tissues in human X-ALD patients, the C26∶0/C22∶0 ratio is elevated up to fourfold in Abcd1-deficient mice [15], [63]. The extent of VLCFA accumulation in myelin-enriched tissues, such as spinal cord and sciatic nerve, was reported to be five to sixfold in Abcd1-deficient mice and was not further elevated in Abcd1/Abcd2 double-deficient mice [31]. Thus, the twelvefold accumulation of C26∶0 that we observed here in Abcd1/Abcd2 double-deficient MPMΦ is indeed remarkably strong. These results indicate that ABCD2 provides an alternative transport route across the peroxisomal membrane for saturated VLCFA destined for β-oxidation, thereby counteracting a more dramatic accumulation in MPMΦ lacking functional ABCD1 and rendering the levels of VLCFA only modestly higher than in wild-type macrophages.
The increased accumulation of saturated VLCFA in MPMΦ from Abcd1/Abcd2 double-deficient mice most likely results from their strongly decreased capacity (29% of wild-type activity) to catabolize VLCFA via the peroxisomal β-oxidation pathway. Thus, also in murine monocyte-derived macrophages, the primary peroxisomal transporter for saturated VLCFA-CoA is ABCD1. However, in contrast to human monocytes [41] or human fibroblasts [7], in the absence of ABCD1, sufficient amounts of endogenous ABCD2 are present to maintain a substantial β-oxidation capacity for VLCFA substrates (62% of wild-type β-oxidation activity for C26∶0, Fig. 5). The most likely candidate for mediating the residual β-oxidation activity (29%) for C26∶0 in the Abcd1/Abcd2 double-deficient MPMΦ is the third peroxisomal ABC transporter, ABCD3, in similarity to human X-ALD fibroblasts [7] and monocytes [41]. Unsaturated long- branched-chain and dicarboxylic fatty acids were recently shown to be the preferred substrates for human ABCD3 in a heterologous expression system, while low activity was obtained with C26∶0 [64].
Another possibility to promote accumulation of VLCFA would be through increased elongation of fatty acids boosting the biosynthesis of C26∶0. The chain length-specific enzyme in this process is ELOVL1 [12]. In a model based on knock-down of Abcd1 mRNA in rat B12 oligodendrocytes, elevated Elovl1 mRNA levels were reported to coincide with increased VLCFA levels [65] indicating an association between VLCFA levels and the regulation of Elovl1 expression. However, we found no difference from wild-type levels of Elovl1 mRNA in any of the mutant MPMΦ by qRT-PCR. This indicates that regulation at the level of Elovl1 gene expression is unlikely to account for the increased accumulation of VLCFA in Abcd1/Abcd2 double-deficient MPMΦ. Although we cannot exclude that altered enzymatic activity or substrate availability for ELOVL1 plays a role, a major contribution appears unlikely when considering the strongly impaired rate of degradation of VLCFA.
Taken together, these results suggest that the additional loss of Abcd2 in Abcd1 deficiency results in a strikingly more severe metabolic phenotype in mouse peritoneal macrophages. Although the Abcd2 mRNA is of low abundance and was present at about only half the level of the Abcd1 mRNA, in Abcd1 deficiency this modest endogenous level is apparently sufficient to prevent the dramatic metabolic phenotype that we observed in Abcd1/Abcd2 double-mutant macrophages. In contrast to primary mouse peritoneal macrophages, primary human monocytes/macrophages virtually lack ABCD2 gene expression [40], [41]. Accordingly, in human X-ALD monocytes, the metabolic defects such as accumulation of VLCFA and their degradation by peroxisomal β-oxidation are comparable to those found in Abcd1/Abcd2 double-deficient rather than Abcd1-deficient peritoneal macrophages. In MPMФ, the Abcd2 gene is apparently not underlying the strong repression or silencing that was observed for ABCD2 in human monocytes. Recently, we obtained evidence showing that the feasibility to induce ABCD2 depends on the differentiation state in the human monocyte/macrophage lineage [66]. Whereas ABCD2 was unresponsive to retinoids in primary CD14+ monocytes, 13-cis-retinoic acid produced a fourfold induction after in vitro differentiation into macrophages [66].
The difference between human ABCD2 and murine Abcd2 expression in macrophages could potentially be one contributing factor to the absence of a cerebral inflammatory phenotype in the mouse model of X-ALD. However, as also Abcd1/Abcd2 double-deficient mice do not develop demyelinating brain inflammation (Forss-Petter, unpublished observation) and [31], additional species differences or other unknown triggers must play a critical role.
In conclusion, these results provide proof of principle that even a modest endogenous expression of Abcd2 in macrophages can largely compensate for Abcd1 deficiency and significantly reduce the amount of C26∶0 in Abcd1-deficient macrophages. Although these observations cannot be directly translated to human perivascular or parenchymal macrophages or activated microglia cells in brain lesions of an X-ALD patient, the results are encouraging for therapeutic strategies aiming at upregulating expression of ABCD2. The use of Abcd1 and Abcd2 single and double-deficient macrophages provides a unique model to discriminate whether ABCD2 is necessary for any observed effects of treatment in the macrophage lineage. The marked additional decrease in the capacity for β-oxidation of VLCFA upon inactivation of Abcd2 in Abcd1 deficiency implies a strong synergistic effect of ABCD1 and ABCD2 in this cell type.
Supporting Information
Abcd1 mRNA levels in mouse peritoneal macrophages from wild-type and Abcd2 -deficient mice. The Abcd1 and Hprt mRNA copy numbers were determined by qRT-PCR in wild-type (WT) and Abcd2-deficient (Abcd2 KO) cells. The graphs indicate mean values ± SD for Abcd1 mRNA after normalization to the level of Hprt mRNA in each sample (n = 3); n.s., no statistically significant difference.
(TIF)
Absolute fatty acid levels in wild-type, Abcd1 -, Abcd2- and Abcd1/Abcd2 double-deficient mouse peritoneal macrophages. The concentrations of the VLCFA species C26∶0, C24∶0 and C22∶0 and the LCFA C16∶0 were determined by GC-MS in mouse peritoneal macrophages of wild-type (WT), Abcd1-deficient (Abcd1 KO), Abcd2-deficient (Abcd2 KO) and Abcd1/Abcd2 double-deficient (DOKO) mice after 5 days in culture (n = 3). The amounts of fatty acids: (A) C26∶0; (B) C24∶0; (C) C22∶0 and (D) C16∶0 were normalized to the protein content of each sample. The graphs indicate geometric means ± SD (asymmetrical). Statistically significant differences are indicated: * p<0.05, ** p<0.01; n.s., no statistically significant difference.
(TIF)
Relative C26∶0 levels in wild-type, Abcd1- , Abcd2- and Abcd1/Abcd2 double-deficient peritoneal macrophages cultured for 1-day. The concentrations of C26∶0 and C22∶0 were determined by GC-MS in wild-type (WT), Abcd1-deficient (Abcd1 KO), Abcd2-deficient (Abcd2 KO) and Abcd1/Abcd2 double-deficient (DOKO) mouse peritoneal macrophages after 1 day in culture. The relative amounts of C26∶0 are expressed as C26∶0/C22∶0 ratio. The graphs indicate geometric means ± SD (asymmetrical). Statistically significant differences are indicated: * p<0.05, *** p<0.001; (n = 3).
(TIF)
β-Oxidation activity towards C26∶0 and C16∶0 in wild-type, Abcd1-, Abcd2, and Abcd1/Abcd2 double-deficient peritoneal macrophages. The absolute rates of β-oxidation of [14C]-labelled fatty acid substrates for: (A) C26∶0 and (B) C16∶0 were measured in wild-type (WT), Abcd1-deficient (Abcd1 KO), Abcd2-deficient (Abcd2 KO) and Abcd1/Abcd2 double-deficient (DOKO) cells. The results are shown as the mean values ± SD of the rate (pmol/min) of released, water-soluble [14C]-acetyl-CoA normalized to the protein content (mg). The numbers of samples (n) are indicated below the graphs. Statistically significant differences are indicated: * p<0.05, ** p<0.01, *** p<0.001.
(TIF)
Absolute and normalized mRNA copy numbers of Abcd1 , Abcd2 and Hprt determined by qRT-PCR.
(DOCX)
Absolute and normalized mRNA copy numbers of Elovl1 and Hprt determined by qRT-PCR.
(DOCX)
Acknowledgments
We are grateful to Klaus-Armin Nave for providing support, tools and facilities for the generation of knock-out mice. We thank Manuela Haberl, Gerhard Zeitler and Regina Sundt for excellent technical assistance.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by grants from the European Union (FP7-HEALTH-2009) project No. 241622, “LEUKOTREAT” (URL: http://cordis.europa.eu/fp7/home_en.html) and from the Austrian Science Fund (FWF) project P26112-B19 (URL: http://www.fwf.ac.at). ZM was supported by a PhD scholarship from the Higher Education Commission of Pakistan (URL: http://www.hec.gov.pk/Pages/HECMain.aspx). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Berger J, Gartner J (2006) X-linked adrenoleukodystrophy: clinical, biochemical and pathogenetic aspects. Biochim Biophys Acta 1763: 1721–1732. [DOI] [PubMed] [Google Scholar]
- 2. Kemp S, Berger J, Aubourg P (2012) X-linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochim Biophys Acta 1822: 1465–1474. [DOI] [PubMed] [Google Scholar]
- 3. Moser HW (1997) Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain 120 (Pt 8): 1485–1508. [DOI] [PubMed] [Google Scholar]
- 4. Engelen M, Barbier M, Dijkstra IM, Schur R, de Bie RM, et al. (2014) X-linked adrenoleukodystrophy in women: a cross-sectional cohort study. Brain 137: 693–706. [DOI] [PubMed] [Google Scholar]
- 5. Mosser J, Douar AM, Sarde CO, Kioschis P, Feil R, et al. (1993) Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 361: 726–730. [DOI] [PubMed] [Google Scholar]
- 6. van Roermund CW, Visser WF, Ijlst L, Waterham HR, Wanders RJ (2011) Differential substrate specificities of human ABCD1 and ABCD2 in peroxisomal fatty acid beta-oxidation. Biochim Biophys Acta 1811: 148–152. [DOI] [PubMed] [Google Scholar]
- 7. Wiesinger C, Kunze M, Regelsberger G, Forss-Petter S, Berger J (2013) Impaired very long-chain acyl-CoA beta-oxidation in human X-linked adrenoleukodystrophy fibroblasts is a direct consequence of ABCD1 transporter dysfunction. J Biol Chem 288: 19269–19279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moser HW, Moser AB, Frayer KK, Chen W, Schulman JD, et al. (1981) Adrenoleukodystrophy: increased plasma content of saturated very long chain fatty acids. Neurology 31: 1241–1249. [DOI] [PubMed] [Google Scholar]
- 9. Kemp S, Valianpour F, Denis S, Ofman R, Sanders RJ, et al. (2005) Elongation of very long-chain fatty acids is enhanced in X-linked adrenoleukodystrophy. Mol Genet Metab 84: 144–151. [DOI] [PubMed] [Google Scholar]
- 10. Tsuji S, Sano T, Ariga T, Miyatake T (1981) Increased synthesis of hexacosanoic acid (C23:0) by cultured skin fibroblasts from patients with adrenoleukodystrophy (ALD) and adrenomyeloneuropathy (AMN). J Biochem 90: 1233–1236. [DOI] [PubMed] [Google Scholar]
- 11. Kishimoto Y, Moser HW, Kawamura N, Platt M, Pallante SL, et al. (1980) Adrenoleukodystrophy: evidence that abnormal very long chain fatty acids of brain cholesterol esters are of exogenous origin. Biochem Biophys Res Commun 96: 69–76. [DOI] [PubMed] [Google Scholar]
- 12. Jakobsson A, Westerberg R, Jacobsson A (2006) Fatty acid elongases in mammals: their regulation and roles in metabolism. Prog Lipid Res 45: 237–249. [DOI] [PubMed] [Google Scholar]
- 13. Ofman R, Dijkstra IM, van Roermund CW, Burger N, Turkenburg M, et al. (2010) The role of ELOVL1 in very long-chain fatty acid homeostasis and X-linked adrenoleukodystrophy. EMBO Mol Med 2: 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Singh I, Moser AE, Moser HW, Kishimoto Y (1984) Adrenoleukodystrophy: impaired oxidation of very long chain fatty acids in white blood cells, cultured skin fibroblasts, and amniocytes. Pediatr Res 18: 286–290. [DOI] [PubMed] [Google Scholar]
- 15. Forss-Petter S, Werner H, Berger J, Lassmann H, Molzer B, et al. (1997) Targeted inactivation of the X-linked adrenoleukodystrophy gene in mice. J Neurosci Res 50: 829–843. [DOI] [PubMed] [Google Scholar]
- 16. Kobayashi T, Shinnoh N, Kondo A, Yamada T (1997) Adrenoleukodystrophy protein-deficient mice represent abnormality of very long chain fatty acid metabolism. Biochem Biophys Res Commun 232: 631–636. [DOI] [PubMed] [Google Scholar]
- 17. Lu JF, Lawler AM, Watkins PA, Powers JM, Moser AB, et al. (1997) A mouse model for X-linked adrenoleukodystrophy. Proc Natl Acad Sci U S A 94: 9366–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kemp S, Wanders R (2010) Biochemical aspects of X-linked adrenoleukodystrophy. Brain Pathol 20: 831–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ho JK, Moser H, Kishimoto Y, Hamilton JA (1995) Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J Clin Invest 96: 1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Knazek RA, Rizzo WB, Schulman JD, Dave JR (1983) Membrane microviscosity is increased in the erythrocytes of patients with adrenoleukodystrophy and adrenomyeloneuropathy. J Clin Invest 72: 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bizzozero OA, Zuniga G, Lees MB (1991) Fatty acid composition of human myelin proteolipid protein in peroxisomal disorders. J Neurochem 56: 872–878. [DOI] [PubMed] [Google Scholar]
- 22. Fourcade S, Lopez-Erauskin J, Galino J, Duval C, Naudi A, et al. (2008) Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum Mol Genet 17: 1762–1773. [DOI] [PubMed] [Google Scholar]
- 23. Powers JM, Pei Z, Heinzer AK, Deering R, Moser AB, et al. (2005) Adreno-leukodystrophy: oxidative stress of mice and men. J Neuropathol Exp Neurol 64: 1067–1079. [DOI] [PubMed] [Google Scholar]
- 24. Galino J, Ruiz M, Fourcade S, Schluter A, Lopez-Erauskin J, et al. (2011) Oxidative damage compromises energy metabolism in the axonal degeneration mouse model of X-adrenoleukodystrophy. Antioxid Redox Signal 15: 2095–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lopez-Erauskin J, Galino J, Ruiz M, Cuezva JM, Fabregat I, et al. (2013) Impaired mitochondrial oxidative phosphorylation in the peroxisomal disease X-linked adrenoleukodystrophy. Hum Mol Genet 22: 3296–3305. [DOI] [PubMed] [Google Scholar]
- 26. Gilg AG, Singh AK, Singh I (2000) Inducible nitric oxide synthase in the central nervous system of patients with X-adrenoleukodystrophy. J Neuropathol Exp Neurol 59: 1063–1069. [DOI] [PubMed] [Google Scholar]
- 27. Yanagisawa N, Shimada K, Miyazaki T, Kume A, Kitamura Y, et al. (2008) Enhanced production of nitric oxide, reactive oxygen species, and pro-inflammatory cytokines in very long chain saturated fatty acid-accumulated macrophages. Lipids Health Dis 7: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holzinger A, Kammerer S, Berger J, Roscher AA (1997) cDNA cloning and mRNA expression of the human adrenoleukodystrophy related protein (ALDRP), a peroxisomal ABC transporter. Biochem Biophys Res Commun 239: 261–264. [DOI] [PubMed] [Google Scholar]
- 29. Kemp S, Wei HM, Lu JF, Braiterman LT, McGuinness MC, et al. (1998) Gene redundancy and pharmacological gene therapy: implications for X-linked adrenoleukodystrophy. Nat Med 4: 1261–1268. [DOI] [PubMed] [Google Scholar]
- 30. Netik A, Forss-Petter S, Holzinger A, Molzer B, Unterrainer G, et al. (1999) Adrenoleukodystrophy-related protein can compensate functionally for adrenoleukodystrophy protein deficiency (X-ALD): implications for therapy. Hum Mol Genet 8: 907–913. [DOI] [PubMed] [Google Scholar]
- 31. Pujol A, Ferrer I, Camps C, Metzger E, Hindelang C, et al. (2004) Functional overlap between ABCD1 (ALD) and ABCD2 (ALDR) transporters: a therapeutic target for X-adrenoleukodystrophy. Hum Mol Genet 13: 2997–3006. [DOI] [PubMed] [Google Scholar]
- 32. van Roermund CW, Ijlst L, Wagemans T, Wanders RJ, Waterham HR (2014) A role for the human peroxisomal half-transporter ABCD3 in the oxidation of dicarboxylic acids. Biochim Biophys Acta 1841: 563–568. [DOI] [PubMed] [Google Scholar]
- 33. Ferrer I, Kapfhammer JP, Hindelang C, Kemp S, Troffer-Charlier N, et al. (2005) Inactivation of the peroxisomal ABCD2 transporter in the mouse leads to late-onset ataxia involving mitochondria, Golgi and endoplasmic reticulum damage. Hum Mol Genet 14: 3565–3577. [DOI] [PubMed] [Google Scholar]
- 34. Fourcade S, Ruiz M, Camps C, Schluter A, Houten SM, et al. (2009) A key role for the peroxisomal ABCD2 transporter in fatty acid homeostasis. Am J Physiol Endocrinol Metab 296: E211–221. [DOI] [PubMed] [Google Scholar]
- 35. Berger J, Pujol A, Aubourg P, Forss-Petter S (2010) Current and future pharmacological treatment strategies in X-linked adrenoleukodystrophy. Brain Pathol 20: 845–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aubourg P, Blanche S, Jambaque I, Rocchiccioli F, Kalifa G, et al. (1990) Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation. N Engl J Med 322: 1860–1866. [DOI] [PubMed] [Google Scholar]
- 37. Peters C, Charnas LR, Tan Y, Ziegler RS, Shapiro EG, et al. (2004) Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood 104: 881–888. [DOI] [PubMed] [Google Scholar]
- 38. Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, et al. (2009) Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 326: 818–823. [DOI] [PubMed] [Google Scholar]
- 39. Cartier N, Aubourg P (2010) Hematopoietic stem cell transplantation and hematopoietic stem cell gene therapy in X-linked adrenoleukodystrophy. Brain Pathol 20: 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Langmann T, Mauerer R, Schmitz G (2006) Human ATP-binding cassette transporter TaqMan low-density array: analysis of macrophage differentiation and foam cell formation. Clin Chem 52: 310–313. [DOI] [PubMed] [Google Scholar]
- 41. Weber FD, Wiesinger C, Forss-Petter S, Regelsberger G, Einwich A, et al. (2014) X-linked adrenoleukodystrophy: very long-chain fatty acid metabolism is severely impaired in monocytes but not in lymphocytes. Hum Mol Genet 23: 2542–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eichler FS, Ren JQ, Cossoy M, Rietsch AM, Nagpal S, et al. (2008) Is microglial apoptosis an early pathogenic change in cerebral X-linked adrenoleukodystrophy? Ann Neurol 63: 729–742. [DOI] [PubMed] [Google Scholar]
- 43. Berger J, Forss-Petter S, Eichler FS (2014) Pathophysiology of X-linked adrenoleukodystrophy. Biochimie 98: 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hubbard WC, Moser AB, Tortorelli S, Liu A, Jones D, et al. (2006) Combined liquid chromatography-tandem mass spectrometry as an analytical method for high throughput screening for X-linked adrenoleukodystrophy and other peroxisomal disorders: preliminary findings. Mol Genet Metab 89: 185–187. [DOI] [PubMed] [Google Scholar]
- 45. Cohn ZA (1978) Activation of mononuclear phagocytes: fact, fancy, and future. J Immunol 121: 813–816. [PubMed] [Google Scholar]
- 46. Leijh PC, van Zwet TL, ter Kuile MN, van Furth R (1984) Effect of thioglycolate on phagocytic and microbicidal activities of peritoneal macrophages. Infect Immun 46: 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Misharin AV, Saber R, Perlman H (2012) Eosinophil contamination of thioglycollate-elicited peritoneal macrophage cultures skews the functional readouts of in vitro assays. J Leukoc Biol 92: 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ghosn EE, Cassado AA, Govoni GR, Fukuhara T, Yang Y, et al. (2010) Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc Natl Acad Sci U S A 107: 2568–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Saijo K, Glass CK (2011) Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol 11: 775–787. [DOI] [PubMed] [Google Scholar]
- 50. Mansour SL, Thomas KR, Capecchi MR (1988) Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature 336: 348–352. [DOI] [PubMed] [Google Scholar]
- 51. Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC (1993) Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci U S A 90: 8424–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Klugmann M, Schwab MH, Puhlhofer A, Schneider A, Zimmermann F, et al. (1997) Assembly of CNS myelin in the absence of proteolipid protein. Neuron 18: 59–70. [DOI] [PubMed] [Google Scholar]
- 53. Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156–159. [DOI] [PubMed] [Google Scholar]
- 54. Forss-Petter S, Danielson PE, Catsicas S, Battenberg E, Price J, et al. (1990) Transgenic mice expressing beta-galactosidase in mature neurons under neuron-specific enolase promoter control. Neuron 5: 187–197. [DOI] [PubMed] [Google Scholar]
- 55.Zhang X, Goncalves R, Mosser DM (2008) The isolation and characterization of murine macrophages. Curr Protoc Immunol Chapter 14: Unit 14 11. [DOI] [PMC free article] [PubMed]
- 56. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275. [PubMed] [Google Scholar]
- 57. Unterberger U, Regelsberger G, Sundt R, Bernheimer H, Voigtlander T (2007) Diagnosis of X-linked adrenoleukodystrophy in blood leukocytes. Clin Biochem 40: 1037–1044. [DOI] [PubMed] [Google Scholar]
- 58. Watkins PA, Ferrell EV Jr, Pedersen JI, Hoefler G (1991) Peroxisomal fatty acid beta-oxidation in HepG2 cells. Arch Biochem Biophys 289: 329–336. [DOI] [PubMed] [Google Scholar]
- 59. Lombard-Platet G, Savary S, Sarde CO, Mandel JL, Chimini G (1996) A close relative of the adrenoleukodystrophy (ALD) gene codes for a peroxisomal protein with a specific expression pattern. Proc Natl Acad Sci U S A 93: 1265–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Berger J, Albet S, Bentejac M, Netik A, Holzinger A, et al. (1999) The four murine peroxisomal ABC-transporter genes differ in constitutive, inducible and developmental expression. Eur J Biochem 265: 719–727. [DOI] [PubMed] [Google Scholar]
- 61. Weinhofer I, Forss-Petter S, Kunze M, Zigman M, Berger J (2005) X-linked adrenoleukodystrophy mice demonstrate abnormalities in cholesterol metabolism. FEBS Lett 579: 5512–5516. [DOI] [PubMed] [Google Scholar]
- 62. Troffer-Charlier N, Doerflinger N, Metzger E, Fouquet F, Mandel JL, et al. (1998) Mirror expression of adrenoleukodystrophy and adrenoleukodystrophy related genes in mouse tissues and human cell lines. Eur J Cell Biol 75: 254–264. [DOI] [PubMed] [Google Scholar]
- 63. McGuinness MC, Lu JF, Zhang HP, Dong GX, Heinzer AK, et al. (2003) Role of ALDP (ABCD1) and mitochondria in X-linked adrenoleukodystrophy. Mol Cell Biol 23: 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Van Roermund CW, Ijlst L, Wagemans T, Wanders RJ, Waterham HR (2013) A role for the human peroxisomal half-transporter ABCD3 in the oxidation of dicarboxylic acids. Biochim Biophys Acta. [DOI] [PubMed]
- 65. Singh J, Khan M, Singh I (2013) Caffeic acid phenethyl ester induces adrenoleukodystrophy (Abcd2) gene in human X-ALD fibroblasts and inhibits the proinflammatory response in Abcd1/2 silenced mouse primary astrocytes. Biochim Biophys Acta 1831: 747–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Weber FD, Weinhofer I, Einwich A, Forss-Petter S, Muneer Z, et al. (2014) Evaluation of retinoids for induction of the redundant gene ABCD2 as an alternative treatment option in X-linked adrenoleukodystrophy. PLoS ONE 9(7): e103742. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Abcd1 mRNA levels in mouse peritoneal macrophages from wild-type and Abcd2 -deficient mice. The Abcd1 and Hprt mRNA copy numbers were determined by qRT-PCR in wild-type (WT) and Abcd2-deficient (Abcd2 KO) cells. The graphs indicate mean values ± SD for Abcd1 mRNA after normalization to the level of Hprt mRNA in each sample (n = 3); n.s., no statistically significant difference.
(TIF)
Absolute fatty acid levels in wild-type, Abcd1 -, Abcd2- and Abcd1/Abcd2 double-deficient mouse peritoneal macrophages. The concentrations of the VLCFA species C26∶0, C24∶0 and C22∶0 and the LCFA C16∶0 were determined by GC-MS in mouse peritoneal macrophages of wild-type (WT), Abcd1-deficient (Abcd1 KO), Abcd2-deficient (Abcd2 KO) and Abcd1/Abcd2 double-deficient (DOKO) mice after 5 days in culture (n = 3). The amounts of fatty acids: (A) C26∶0; (B) C24∶0; (C) C22∶0 and (D) C16∶0 were normalized to the protein content of each sample. The graphs indicate geometric means ± SD (asymmetrical). Statistically significant differences are indicated: * p<0.05, ** p<0.01; n.s., no statistically significant difference.
(TIF)
Relative C26∶0 levels in wild-type, Abcd1- , Abcd2- and Abcd1/Abcd2 double-deficient peritoneal macrophages cultured for 1-day. The concentrations of C26∶0 and C22∶0 were determined by GC-MS in wild-type (WT), Abcd1-deficient (Abcd1 KO), Abcd2-deficient (Abcd2 KO) and Abcd1/Abcd2 double-deficient (DOKO) mouse peritoneal macrophages after 1 day in culture. The relative amounts of C26∶0 are expressed as C26∶0/C22∶0 ratio. The graphs indicate geometric means ± SD (asymmetrical). Statistically significant differences are indicated: * p<0.05, *** p<0.001; (n = 3).
(TIF)
β-Oxidation activity towards C26∶0 and C16∶0 in wild-type, Abcd1-, Abcd2, and Abcd1/Abcd2 double-deficient peritoneal macrophages. The absolute rates of β-oxidation of [14C]-labelled fatty acid substrates for: (A) C26∶0 and (B) C16∶0 were measured in wild-type (WT), Abcd1-deficient (Abcd1 KO), Abcd2-deficient (Abcd2 KO) and Abcd1/Abcd2 double-deficient (DOKO) cells. The results are shown as the mean values ± SD of the rate (pmol/min) of released, water-soluble [14C]-acetyl-CoA normalized to the protein content (mg). The numbers of samples (n) are indicated below the graphs. Statistically significant differences are indicated: * p<0.05, ** p<0.01, *** p<0.001.
(TIF)
Absolute and normalized mRNA copy numbers of Abcd1 , Abcd2 and Hprt determined by qRT-PCR.
(DOCX)
Absolute and normalized mRNA copy numbers of Elovl1 and Hprt determined by qRT-PCR.
(DOCX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.