

Abstract
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide, and is responsible for a quarter of a million deaths annually. The survival rate for HNSCC patients is poor, showing only minor improvement in the last three decades. Despite new surgical techniques and chemotherapy protocols, tumor resistance to chemotherapy remains a significant challenge for HNSCC patients. Numerous mechanisms underlie chemoresistance, including genetic and epigenetic alterations in cancer cells that may be acquired during treatment and activation of mitogenic signaling pathways, such as nuclear factor kappa-light-chain-enhancer-of activated B cell, that cause reduced apoptosis. In addition to dysfunctional molecular signaling, emerging evidence reveals involvement of cancer stem cells (CSCs) in tumor development and in tumor resistance to chemotherapy and radiotherapy. These observations have sparked interest in understanding the mechanisms involved in the control of CSC function and fate. Post-translational modifications of histones dynamically influence gene expression independent of alterations to the DNA sequence. Recent findings from our group have shown that pharmacological induction of post-translational modifications of tumor histones dynamically modulates CSC plasticity. These findings suggest that a better understanding of the biology of CSCs in response to epigenetic switches and pharmacological inhibitors of histone function may directly translate to the development of a mechanism-based strategy to disrupt CSCs. In this review, we present and discuss current knowledge on epigenetic modifications of HNSCC and CSC response to DNA methylation and histone modifications. In addition, we discuss chromatin modifications and their role in tumor resistance to therapy.
Keywords: Head and neck squamous cell carcinoma, Chromatin remodeling, Histone deacetylases inhibitor, Histone acetylation, Cancer-initiating cell, Epigenetic target, Epigenetic marker, Oral squamous cell carcinoma, Tumor resistance
Core tip: Stem cells are long-lived, therefore their genome is subject to more stress from genetic mutations and epigenetic factors than their short-lived, differentiated progeny. Recent evidence strongly indicates that a subpopulation of tumor initiating cells, termed “cancer stem cells”, play a fundamental role in tumor heterogeneity, growth, and preservation. Cancer stem cell behavior is influenced by epigenetic events comprised primarily of DNA methylation and histone modifications that dynamically regulate gene expression and silencing.
INTRODUCTION
There are approximately 560000 cases of head and neck cancer diagnosed worldwide each year and approximately 300000 deaths annually. This cancer type occurs in the head and neck region, involves the nasal and oral cavity, pharynx, and larynx and primarily occurs as squamous cell carcinoma (HNSCC)[1-4]. Although HNSCC has well recognized risk factors, including tobacco use, excess alcohol consumption, and infection by high risk papillomaviruses[5,6], we do not fully understand the mechanisms underlying its malignant progression[5]. Our understanding of the molecular biology of HNSCC has significantly improved in the last few decades, contributing to the development of novel therapies targeted against pro-survival signaling circuitries, including the epidermal growth factor receptor (EGFR), vascular endothelial growth factor, receptor tyrosine kinases, interleukins, and phosphoinositide 3-kinase (PI3K) pathways, among others. Unfortunately, the long-term survival rate for HNSCC patients, which is 50% at five years after diagnosis, has remained consistent over the past thirty years[3,7-9]. The incidence of HNSCC is much higher in developing nations, where it is the third most common malignancy in Asian countries compared to the sixth most common malignancy in Western countries[10-12]. This discrepancy in incidence of HNSCC is associated with varying risk factors, such as chewing Betel quid in the Asia-Pacific region compared to consumption of tobacco and alcohol and/or human papillomavirus infection outside Asia[1,13-17].
The poor long-term survival rates in HNSCC patients may be due to diagnosis of disease at an advanced-stage and development of chemoresistance[8,18]. Numerous mechanisms underlie chemoresistance, including genetic and epigenetic alterations in cancer cells that may be acquired during treatment[19,20] and the activation of mitogenic signaling pathways, such as nuclear factor kappa-light-chain-enhancer-of activated B cell (NFκB), that result in reduced apoptosis[21]. Furthermore, the recurrence of cancers depend on a subpopulation of cancer stem cells (CSCs) that possess the unique and exclusive ability to self-renew and differentiate into nontumorigenic heterogenous cell types that maintain the tumor[7,22-24]. Therefore, many factors play a critical role in the maintenance of tumor heterogeneity and CSC behavior, including the tumor microenvironment, genomic instability and the effect of genetic mutations and epigenetic changes on gene expression[22,25-27].
In a significant number of HNSCC, tumor progression results from mutations in genes, such as TP53, CDKN2A, HRAS, PTEN, and PIK3CA. This causes alterations in cell signaling cascades (e.g., PI3K/mTOR, NFκB, ERK, p53), resulting in aberrant cell growth, migration, and survival[3,8,23,28,29]. Epigenetic changes also play a key role in regulating gene expression through histone modifications, DNA methylation, miRNA silencing and DNA repair mechanisms [HMT (Histone methyltransferases), HAT (Histone acetyltransferases), HDAC (Histone deacteylases) ncRNA (non-coding RNA), and lncRNA (long non-coding RNA)][30-33]. Consequently, by identifying the molecular mechanisms that drive progression and recurrence of HNSCC, novel cancer therapeutics can be developed to improve the effectiveness of treatment and the rate of long-term survival in patients. In this review, we highlight the current understanding on cancer stem cells and the effects of epigenetic modifications on tumor behavior. We also discuss the latest findings on pharmacological manipulation of epigenetic circuitries that may result in the development of novel therapeutic strategies that target cancer stem cells.
CANCER STEM CELLS
Because normal stem cells are long-lived, their genome is subject to more stress from genetic mutations and epigenetic factors than their short-lived, differentiated progeny. The majority of oncogenic mutations in stem cells perturb central cellular processes that regulate cellular division, DNA damage repair, and signal transduction pathways[24,25,34]. Certain HNSCC-related phenotypes that arise from mutations in oncogenes and tumor suppressor genes, such as PIK3CA, TP63, PTEN, EGFR, and MET, result in limitless replication potential, insensitivity to apoptotic signals, angiogenesis, invasion and metastasis[28,35-38]. Therefore, tumors arise when stem cells lose their ability to regulate and maintain tissue form and function and when they show reduced control over apoptosis, cellular senescence and cellular proliferation. Additionally, although tumors are a population of malfunctioning cells, they are commonly characterized by histological features that resemble normal tissue[39]. Similarly, hematopoietic cancers are comprised of identical neoplastic cells, but solid tumors from HNSCC consist of non-identical cells, resulting in phenotypic heterogeneity[25,27,40-42]. Within the polyclonal tumor, there is a cellular hierarchy in which a small subpopulation of neoplastic cells with the highest potential for tumorigenesis and self-renewal are positioned at the top. The remaining bulk of the tumor primarily consists of well-differentiated non-tumorigenic cells that are susceptible to chemotherapy and radiation[43-45]. In addition to HNSCC, solid tumors of the breast, brain, colon, lung and prostate also demonstrate a diverse array of cellular heterogeneity that increases genomic instability and adaptability of the tumor to its microenvironment[25,46,47]. Recent evidence strongly indicates that a subpopulation of tumor initiating cells, termed “cancer stem cells” play a fundamental role in tumor heterogeneity, growth, and preservation[25,44,48,49]. The cancer stem cell hypothesis, first conceptualized by Bonnet et al[44] in 1997, established that a subpopulation of human leukemic cells, positive for CD34 and negative for CD38 cell surface markers, initiates human acute myeloid leukemia in Non-obese diabetic/Severe combined immunodeficient (NOD/SCID) mice. The following observations support the cancer stem cell hypothesis: (1) only a subpopulation of tumor cells within a tumor mass grow in immunodeficient mice; (2) the subpopulation of tumor cells generate both CSCs and heterogeneous nontumorigenic cancer cells; and (3) cancer stem cells self-renew, as revealed by serial transplantation assays[22,44,50]. The frequency of CSCs is relatively low in HNSCC, lung squamous cell carcinoma, lung adenocarcinoma, and human pancreatic adenocarcinoma, but xenotransplantation assays greatly increase their frequency[51].
Cancer stem cell surface markers
CSCs were first discovered in solid tumors in 2003[52], and the isolation of CSCs in HNSCC, based on the CD44+ cell surface marker, occurred in 2007[18]. In that study, approximately 70% of NOD/SCID mice receiving CD44+ tumor cell xenografts showed tumor formation compared to 1% of mice receiving CD44- xenografts. In addition to their association with CSCs in HNSCC[53-56], CD44+ cells also play a role in chemoresistance. Genes associated with chemoresistance, including ABCB1, ABCG2, CYP2C8 and TERT, are upregulated in CD44+ cells compared to CD44- cells[57]. Furthermore, CD44+ HNSCC cells express high levels of B lymphoma Mo-MLV insertion region 1 homolog (Bmi-1), a self-renewal and oncogenic protein associated with poor survival and tumor aggressiveness[18,58-62]. Different isoforms of CD44 differentially modify the behavior of HNSCC. For instance, the v3, v6, and v10 isoforms of CD44 promote HNSCC tumor migration, invasion, and metastasis[63,64] and confer chemoresistance in other solid tumors, attributes commonly associated with the chemo- and radio-resistant fractions of cancer stem cells[65]. Therefore, CD44 is used to identify CSCs, and it promotes many of the biological characteristics associated with cancer “stemness”. These characteristics include tumorsphere formation in suspension, unrestricted cellular proliferation, enhanced migration, tumor invasion, and resistance to chemotherapy and ionizing radiation therapy. CD24 and CD133 (also known as Prominin 1) are also CSC cell surface markers[66-68].
The increased enzymatic activity of aldehyde dehydrogenase 1 (ALDH1) is commonly used to identify normal pluripotent cells and tumor cells harboring “stemness” potential in various solid tumors, including HNSCC[51,69-75]. ALDH is a detoxifying enzyme involved in the oxidation of intracellular aldehydes and was initially described for its role in hematopoietic stem cell self-renewal via reduction of retinoic acid activity[76,77]. The presence of ALDH1-positive tumor cells correlates with poor clinical outcome in breast cancer[69], ovarian cancer[78], papillary thyroid carcinoma[79], and pancreatic adenocarcinoma[80], among other solid tumors[70,81-83].
It is believed that HNSCC progression and invasion, in addition to resistance to non-surgical therapies, may be regulated by the rare population of CSCs[18,43,84,85]. Therefore, to effectively treat this type of cancer, we must develop a therapy that can target and eliminate CSCs.
EPIGENETICS OF HEAD AND NECK CANCER AND ITS STEM CELLS
Basic concepts of epigenetic regulation
DNA methylation: When exploring the molecular mechanisms underlying cancer, DNA methylation is the most commonly studied epigenetic alteration[86-88]. DNA methylation patterns occur in early and precancerous stages and most frequently discovered in tumors compared to normal tissues[89,90]. Methylation occurs sporadically and is globally distributed in mammals throughout the genome at cytosine-phospho-guanine (CpG) dinucleotide sequences, as revealed by immunofluorescent labeled 5-methylcytosine. Without considering CpG-rich islands (approximately 1 kilobase in length), there is a low, but global level of methylation in specific CpG sequences throughout the entire mammalian genome[26,91]. Therefore, aberrant DNA methylation of these CpG islands or specific sequences can lead to oncogenic activation via silencing of tumor suppressor gene expression[92,93]. Hypomethylation is associated with activation of oncogenes, while hypermethylation is associated with the silencing of tumor suppressor genes. Both mechanisms induce genomic instability and play a dominant role in tumor initiation and progression[90,94]. The most common types of DNA methylation in tumors are hypermethylation of CpG islands and global hypomethylation[89]. Hypermethylated CpG islands are often associated with gene promoters; thus, methylation results in a transcriptionally inactive gene. In contrast, methylation of DNA sequences further from promoter sequences has less of an effect on transcription[26].
Histone modifications: In addition to DNA methylation, the chromatin architecture can be remodeled by a network of protein mediators called histones that play an important role in gene regulation by compacting DNA. Histones can be post-translationally modified at the amino-terminal ends by acetylation, methylation, phosphorylation, sumoylation, ubiquitination, and ADP-ribosylation[95]. These modifications result in gene transcription through the uncoiling of chromatin or gene silencing through compacting DNA[96]. HAT, HMT, and HDAC are key co-factors that modify histones and produce the epigenetic changes observed in cancer. Histone acetylation, deacetylation and methylation are the major marks associated with transcriptional activity. Histone acetylation results in chromatin decondensation, promotion of transcription, and inhibition of DNA methylation, and is often correlated with the formation of euchromatin. In contrast, histone deacetylation is the predominant epigenetic influence in transcriptional gene silencing[95,97,98]. In general, histone modifications modulate a diverse array of biological processes, including gene regulation, DNA repair, mitosis and meiosis via chromosome remodeling[99].
Histone acetylation and deacetylation: Dysregulation of the exquisite interplay between acetylation and deacetylation controlled by HAT and HDAC is coupled to the initiation and progression of cancer, cellular plasticity, inflammation, and dynamic transformation in metabolic cascades[100,101]. In addition to the histone substrate peptides described in[102], HAT is associated with non-histone proteins, transcription co-factors, such as p53, p65, c-MYC, NFκB, STAT3 (signal transducer and activator of transcription 3) and BRCA1 (breast cancer 1), among others[30,103]. In particular, acetylation of the p53 tumor suppressor and pro-apoptotic protein by the CBP (CREB-binding protein)/p300 family of HATs has been extensively reviewed in[104,105]. Modification of p53 is associated with increased DNA binding affinity, transcriptional activity[106,107] and protein stability[108]. Similar to p53, CBP/p300 is associated with the pro-proliferative and oncoproteins previously listed, and its expression impacts a variety of human diseases, such as leukemia[109,110], lung cancer[111], colon cancer[112], bladder cancer[113] and prostate cancer[114-116]. CBP/p300 is also associated with transcription factors involved in heart disease[117,118], diabetes[119,120] and neurological disorders[121,122].
Histone methylation: Histone methylation is the third major epigenetic process that affects transcriptional activation via chromatin remodeling. Similar to previously described post-translational histone modifications, methylation and demethylation of amino acids at different sites on histones either promotes or prevents transcriptional activity[123]. For example, methylation of lysine residues is associated with transcription and DNA repair, but methylation of arginine residues is only associated with transcription[95,124,125]. Histone H3 is methylated at different lysine sites, including K4, K9, K27, K36, and K79, that experience various methylated states, including monomethylated, dimethylated, and trimethylated. Therefore, the epigenetic modification of the chromatin depends on the location and state of methylation[126,127]. K9 and K27 methylation is associated with heterochromatin formation and inactive transcription. In contrast, K4 methylation is associated with euchromatin formation and active transcription[128,129].
HAT and HDAC inhibitors: The development of HAT inhibitors (HATi) are in the early stages of preclinical studies. Although drugs that regulate HDAC activity are being used for cancer treatment, there is great interest in developing HAT inhibitors as a potential treatment for cancer and other human diseases[130]. Several natural compounds effectively inhibit HAT activity. For example, Marcu et al[131] demonstrated that curcumin inhibits HAT activity by promoting proteasome-dependent degradation of CBP/p300 in both prostate cancer cells and in HDAC inhibitor-induced peripheral blood lymphocytes. In addition, epigallocatechin-3-gallate and plumbagin are selective inhibitors of CBP/p300[132-134]. The potential for HDAC inhibitors (HDACi) to serve as cancer chemotherapeutics has been examined in clinical trials due to the role of HDAC in genome stability, proliferation, differentiation, apoptosis, and metabolism. A current list of HDACi under clinical investigation can be found in a review by Li et al[135] that focuses on HDAC and its clinical implications in cancer therapy.
In summary, epigenetic modifications constitute the next frontier in tumor biology research. Post-translational modification of histones dynamically influences gene expression independent of alterations to the DNA sequence. These mechanisms are often mediated by histone linkers, proteins associated with the recruitment of DNA-binding proteins, HDAC I and III interacting proteins and transcriptional activators, coactivators or corepressors. Therefore, histones are molecular markers of epigenetic changes[136].
Epigenetic regulation of HNSCC
In HNSCC and other carcinomas, the combination of genetic and epigenetic factors affect gene expression, resulting in altered downstream cellular signaling pathways that regulate tumor growth, anti-apoptosis, DNA repair, resistance to extrinsic factors, angiogenesis, and epithelial-mesenchymal transition (EMT)[31,137-140]. Although both genetics and epigenetics may affect the initiation and progression of HNSCC, epigenetic factors regulate gene expression in the absence of genomic mutations[19,141,142]. Therefore, epigenetics is defined as a stable heritable phenotype passed on through either mitosis or meiosis, resulting in changes in chromosome characteristics without inducing genome alterations, as proposed by Conrad Waddington in the early 1940s[143-145].
Tumor development is a multi-stage process that requires the accumulation of numerous genetic mutations and often results in gain-of-function in oncogenes and loss-of-function in tumor suppressor genes[146-150]. In addition to genetic mutations, tumor development and progression is extensively influenced by changes in gene expression independent of alterations in the DNA sequence, a mechanism known as epigenetic modification. Epigenetic events are comprised primarily of DNA methylation and histone modifications that dynamically regulate gene expression and silencing[19,31,141,142,151]. These dynamic processes occur within the chromatin that is packed into the nucleus through interactions with core histone proteins.
The effect of chromatin on cellular behavior depends on how tightly DNA is spooled around H2A, H2B, H3 and H4 core histones[152]. Together, histones and DNA form nucleosomes, the fundamental units of chromatin. Gene expression is driven by the ability of chromatin to fold and unfold in a process that requires rapid acetylation/deacetylation of the histone core, resulting in alterations in the cellular response to environmental cues[153].
DNA methylation in HNSCC: In Demokan et al[89] extensive review[89] of DNA methylation in head and neck cancers, they provide a list of the most frequently methylated genes. In this list, the hypermethylated genes include the following: (1) Adenomatous polyposis coli (APC), which is the most common gene methylated in HNSCC[154,155]; (2) p16, a cell cycle controller encoded by the CDKN2A gene, which plays a critical role in inducing cellular senescence in tumor cells and is downregulated via promoter hypermethylation[156-167]; and (3) p14, also known as ARF, that in combination with p16 is involved in regulating the cell cycle and in activating the p53 tumor suppressor gene by inhibiting MDM2[168]. Surprisingly, in 96 human samples of oral squamous cell carcinoma, methylation of p14ARF is associated with a good prognosis, methylation of MINT1 and MINT31 is associated with poor prognosis, and DCC methylation is associated with increased bone invasion by squamous cell carcinoma from the gingiva[169]. Notably, Carvalhoet al[159] and Ogi et al[169] also identified methylated MINT31 as an independent predictor of outcome and showed its association with the T4 disease group, according to the Union for International Cancer Control classification. RASSF1A is a tumor suppressor gene that is frequently silenced in tumors, including HNSCC. RASSF1A is involved in the maintenance of genomic stability and is highly mutated in poorly differentiated HNSCC compared to moderate and well-differentiated HNSCC[154,159,160,163,165,167,170,171]. RASSF2 is a novel Ras-associated protein that negatively regulates Ras signaling[172]. RASSF2 binds directly to K-Ras in a GTP-dependent manner promoting apoptosis and cell cycle arrest; however, RASSF2 weakly interacts with H-Ras. In solid tumors, including human colorectal cancer and HNSCC, RASSF2 is frequently silenced by DNA methylation at 5′ CpG islands[167,173].
Other interesting genes methylated in head and neck cancer include EDNRB, a member of the G protein-coupled receptor family that encodes endothelin receptor type B protein; EDNRB is methylated in 97% of primary HNSCC tissues[174]. EDNRB is involved in the development and function of blood vessels, cellular growth and mitosis[174]. Another gene methylated in HNSCC is RARB, which encodes retinoic acid receptor beta and restricts cell growth by altering gene expression. Hypermethylation of RARB results in loss of function and reduced control of transcription[154,162,163,167,175,176]. Currently, only a few methylated genes can predict the clinical outcome of HNSCC patients. It is unknown how methylated genes correlate with cancer therapy, patient response and tumor progression and behavior. Methylation analysis techniques have revealed that methylation patterns are not affected by external factors and are increased during cancer progression. Therefore, as with stem cell surface markers, increased sensitivity and specificity of quantitative methodologies for DNA methylation analyses will allow scientists to develop prognostic tools for clinical evaluation of head and neck cancer.
Histone methylation in HNSCC: Mancuso et al[177] showed that the level of H3K4 methylation is significantly different in normal mucosa compared to oral squamous cell carcinoma (OSCC) tissues, with dimethylated K4 increased and trimethylated K4 decreased. A similar trend was observed in oral leukoplakias compared to the pathological sample[177]. H3K9 and H3K27 are targets for methylation by enhancer of zeste homolog 2 (EZH2), a member of the Polycomb-group family, resulting in gene silencing via chromatin condensation[178-181]. Interestingly, overexpression of EZH2 is associated with malignancy and prognosis of a variety of cancers, including breast[182,183], prostate[184-186], gastric[187], hepatic[188], bladder[189,190] and oral squamous cell carcinoma[129,191]. Wei et al showed that increased expression of EZH2 is associated with dysplasia and malignant transformation. Similarly, Kidani et al[191] revealed that overexpression of EZH2 is associated with tumor progression, malignancy and poor prognosis in OSCC. Collectively, these data reveal that different histone methylation patterns can greatly influence gene expression in cancer, thereby affecting malignant behavior.
Histone acetylation in HNSCC: Early evidence suggested that histones and their modifiers are involved in sophisticated processes that modulate tumor behavior and cellular phenotype. We recently reported that chromatin folding in HNSCC during tumor response to environmental cues dynamically modulates tumor behavior and cellular phenotype[151]. We found that HNSCC cell lines are hypoacetylated compared to normal mucosa controls (Figure 1A). Furthermore, we found that endothelial cell-secreted factors, but not fibroblast cell-secreted factors, are able to trigger the acetylation of histones in tumor cells (also referred to as tumor histones) (Figure 1B). In fact, paracrine-induced histone modifications resulted in enhanced expression of Bmi-1, a transcriptional repressor upregulated in a variety of cancers and associated with tumor aggressiveness, and poor survival along with the expression of vimentin, a canonical marker of EMT (Figure 1B)[192-199]. Similar to our in vitro findings, human HNSCC samples presented coexpression of acetylated histone 3 and vimentin in the proximity of normal endothelial cells (Figure 1C-white dashed line) next to the tumor invasion front in human HNSSC samples (Figure 1C-yellow dashed line). Therefore, acetylation of tumor histones are associated to changes in cellular behavior, phenotype and associated to increased invasion. In fact, malignant tumors derived from epithelial cells (carcinomas) are known to undergo EMT that precedes local invasion and metastasis of cancer cells[200-204]. EMT is characterized by the loss of cell adhesion, increased motility, aggressive behavior, acquisition of an elongated fibroblastoid morphology and expression of vimentin[200,205,206], similar to what we observe with pharmacological inhibition of HDAC in HNSCC cell lines (Figure 2-HN6 and HN13 cells). Interestingly, cellular morphology is not altered and vimentin is not induced in normal epithelial cells (NOK-SI) treated with HDAC inhibitors, suggesting that hyperacetylation of chromatin differentially modulates normal and neoplastic cells (Figure 2). However, changes in the acetylation of HNSCC chromatin also triggered an unexpected phenotype, which was the loss of CSCs. HNSCC treated with Trichostatin A, a histone deacetylase inhibitor, lose the ability to generate and maintain tumor spheres and experience rapid reduction in the enzymatic activity of ALDH1 (Figure 3)[151]. It has been suggested that epigenetic signals play a major role in stem cell control through deacetylation of histones, which promotes chromatin condensation and reactivation of stem cell-like transcription programs[34]. These striking findings suggest that chromatin acetylation selectively disrupts the physiological requirements for maintenance of CSC. Indeed, chromatin acetylation has long been known to induce cellular differentiation and restrict cellular transformation of normal cells[34,207,208].
Figure 1.
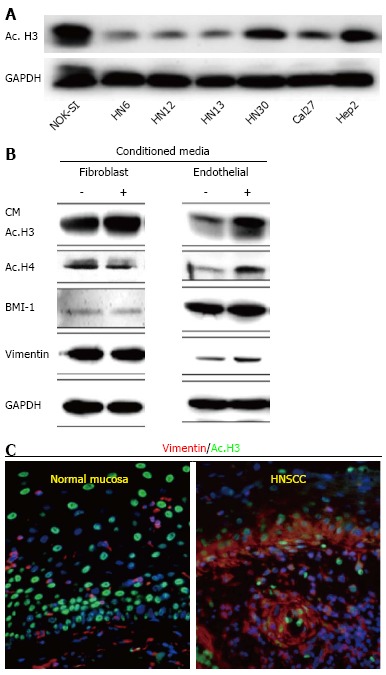
Data represents acetylation status of histone 3 in Head and Neck Squamous Cell Carcinoma by Giudice et al[151]. A: Tumor cells present hypoacetylation of histone 3 (ac.H3) in a panel of Head and Neck Squamous Cell Carcinoma (HNSCC) compared to control cells (NOK-SI); B: Endothelial cell-secreted factors are capable of inducing ac.H3 while fibroblast cell-secreted factors cannot. Also, endothelial cell-secreted factors induce increased expression of BMI-1 and vimentin compared to the fibroblast counterpart; C: Representative examples of human samples of normal oral mucosa and HNSCC. Note acetylated tumor cells (Ac. H3-FITC) with high levels of the epithelial-mesenchymal transition marker vimentin (TRICT) are localized at the invasion front of HNSCC (arrow). Normal mucosa display acetylated cells distributed throughout the epidermis but do not express vimentin. ac. H3: Acetyl histone 3; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; CM: Conditioned Media; BMI-1: B lymphoma Mo-MLV insertion region 1 homolog; NOK-SI: Normal oral epithelial keratinocytes.
Figure 2.
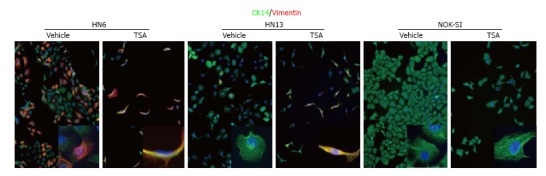
Figure from Giudice et al[151] depicting chemically-induced chromatin acetylation leading to activation of the epithelial-mesenchymal transition phenotype. Inhibition of HDAC induces vimentin expression in HNSCC cells and EMT. Vehicle treated HNSCC cells (HN6 and HN13) present an epithelioid shape and express CK14. Administration of TSA result in acquisition of a fusiform morphology and expression of vimentin. Normal keratinocytes (NOK-SI) are not sensitive to EMT upon administration of TSA. TSA: Trichostatin A; CK14: Cytokeratin 14; EMT: Epithelial-mesenchymal transition; HDAC: Histone deacteylases; HNSCC: Head and Neck Squamous Cell Carcinoma; NOK-SI: Normal oral epithelial keratinocytes.
Figure 3.
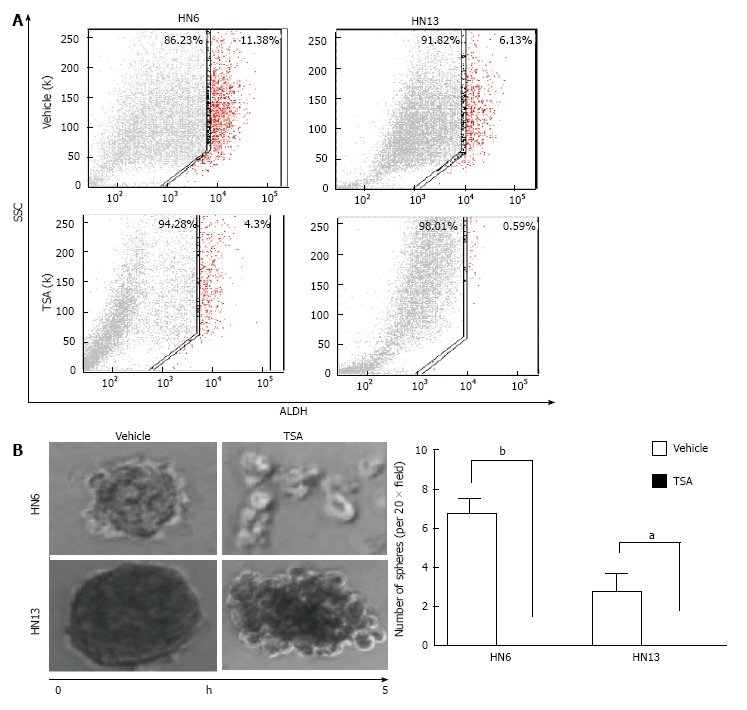
Data from Giudice et al[151] showing the impact of histone deacetylases inhibitor on the population of CICs. A: Fluorescence-activated cell sorting of ALDH+ cells demonstrates that HNSCC cell lines have a high number CICs, and that administration of TSA reduced total number of ALDH+ cells; B: HDACi (TSA) disrupts tumor spheres as depicted in representative images of tumor spheres and by quantification of spheres (HN6 bP < 0.01, HN13 aP < 0.05). TSA: Trichostatin A; ALDH: Aldehyde dehydrogenase; SCC: Side scatter of light; HDACi: Histone deacteylases inhibitors; HNSCC: Head and neck squamous cell carcinoma.
In summary, histone modifications via methylation, acetylation and deacetylation play a critical role in transcriptional activation and gene expression. Aside from the physiological maintenance of cellular homeostasis, aberrant alterations in histone methylation proteins and/or an imbalance in the HAT/HDAC network results in dysfunctions in cellular processes, such as proliferation, differentiation, DNA repair and apoptosis. Importantly, post-translational histone modification and DNA methylation can have similar patterns in the same cancer type. For example, a study by Piyathilake et al[209] revealed that patterns of global DNA and histone methylation are similar in different human mucosal tissues (e.g., normal, dysplastic and squamous cell carcinoma). Using immunohistochemical analysis, they also found that global DNA methylation and H3 methylation at lysine 4 and lysine 9 are significantly higher in dysplastic lesions and carcinoma cells compared to normal oral epithelium[209]. Therefore, when developing methods and techniques for identifying epigenetic markers in premalignant cells, we must consider analyzing both global DNA and histone methylation levels concurrently in the progression of cancer. In conclusion, the previously described epigenetic alterations are closely associated with tumorigenesis and malignancy in many types of cancers. As a result, genomic instability affects numerous intracellular signaling cascades. We will discuss the NFκB signaling pathway in the next section.
TUMOR HISTONE MODIFICATIONS: EVIDENCE FOR AN EPIGENETIC MECHANISM RESPONSIBLE FOR ACQUIRED TUMOR RESISTANCE TO THERAPY
NFκB is an epigenetic modifier that plays a major role in malignant transformation[210], and this pathway serves as a target for epigenetic drugs[211-213]. We, along with others, have previously reported that constitutive activation of NFκB signaling is often observed in HNSCC, suggesting a common epigenetic mechanism in HNSCC biology[214,215]. Indeed, activation of NFκB signaling in HNSCC induced chromatin compaction and acquisition of resistance to chemotherapy[216]. NFκB is active following its translocation to the nucleus, a process that is regulated by the IκB kinase (IKK) complex. IκB proteins are targeted for degradation by phosphorylation, which permits nuclear translocation. Nuclear NFκB binds to target DNA sequences and modulates the expression of target genes involved in immune response, cell growth, and cell survival[217]. Targeted inhibition of NFκB through IKKα and IKKβ silencing resulted in disrupted accumulation of nuclear phospho-p65, increased acetylation of histone 3 and accumulation of BRCA1. Collectively, we showed that NFκB epigenetically modulates chromatin organization and recruits BRCA1 to the nucleus. Indeed, histone 3 is acetylated following loss of NFκB, resulting in decondensation of tumor chromatin and sensitization of head and neck tumors to chemotherapy. This indicates that the effect of NFκB on chromatin organization directly influences tumor response to therapy. As proof of concept, administration of HDAC inhibitors recapitulate the effects of NFκB targeted inhibition by promoting chromatin decondensation and sensitizing tumor cells to chemotherapy, resulting in increased sensitivity of tumor cells to chemotherapy (Figure 4).
Figure 4.
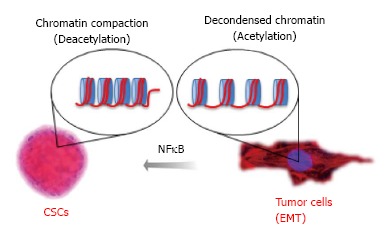
Data from Almeida et al proposing the mechanism for nuclear factor kappa-light-chain-enhancer of activated B cells driven resistance to chemotherapy in head and neck squamous cell carcinoma. Chromatin undergo normal compaction and decondensation through the acetylation of core histones organized in nucleosomes. Acetylation of tumor histones driven by expression of NFκB influences tumor behavior and plasticity of Cancer Stem Cells. NFκB: Nuclear factor kappa-light-chain-enhancer of activated B cells; EMT: Epithelial to mesenchymal transition.
In addition to chemoresistance, activation of NFκB signaling increases the number of tumor spheres, indicating a broader role of NFκB as an epigenetic switch in CSCs. Notably, NFκB signaling is required for the development of tumor spheres in breast, cervical and head and neck cancers[218] (Almeida and Castilho, submitted). We established that by controlling tumor histones, we can dynamically regulate the behavior and number of HNSCC and its CSCs[151]. Epigenetic signals may play a major role in stem cell control through deacetylation of histones, which promotes chromatin condensation and reactivation of stem cell-like transcription programs[34]. Aligned with previous reports[219-221], we showed that HNSCC tumor cell lines have a subpopulation of CSC, as detected by elevated ALDH activity, and clonogenic potential[151]. This subpopulation of CSCs is highly tumorigenic and can self-renew, as observed by serial transplantation assays[37]. By inhibiting HDAC and inducing acetylation of tumor histones, we found that CSCs lose their “stemness”, as evidenced by a reduction in ALDH+ cells and progressive disruption of tumor spheres. These findings indicate that HDAC inhibition disrupts the physiological requirements for CSC maintenance. Indeed, chromatin acetylation induces cellular differentiation and restricts cellular transformation[207,208].
Altogether, HNSCC behavior appears dependent on dynamic changes in chromatin organization and subsequent gene transcription. Unlike stable DNA modifications mediated by methylation, acetylation of histones dynamically alters gene expression, thereby influencing tumor behavior following changes in the microenvironment as observed during administration of secreted factor from endothelial cells[151] and expression of tumor aggressiveness markers[222-225].
CONCLUSION
The role of epigenetic modifications in HNSCC warrants further investigation. Compared to histone modifications, the role of DNA methylation in regulating gene expression is better characterized. Nonetheless, recent studies have correlated the effects of histone acetylation in the dynamic process of tumor adaptation to its microenvironment and the acquisition of a resistant phenotype[151]. The identification of the NFκB signaling pathway as an epigenetic modulator of tumor behavior and resistance to chemotherapy further improved our knowledge in the intricate molecular mechanism of HNSCC and further clarified our understanding of the NFκB signaling pathway[216]. Novel therapeutic strategies can now be developed that target epigenetic alterations driven by histone modifications, and the NFκB signaling may serve as an ideal coadjuvant target for therapy. The development of personalized therapies specific for tumor subtypes, in this case tumors with active NFκB signaling, holds the promise of preventing tumor resistance and sensitizing tumors to chemotherapy. Recent advances in genome sequencing, including next-generation sequencing (NGS), have also improved our understanding of altered molecular signaling in HNSCC. NGS was used to identify single-base changes and larger structural variants characterized by insertions, deletions, translocations and viral insertions in HNSCC[3,226]. Interestingly, NGS also revealed that HNSCC have a significant number of mutations in histones, histone modifiers, transcriptional activators and coactivators, and transcription regulators, further emphasizing the complexity of tumor signaling[30]. Collectively, emerging knowledge about tumor behavior and how it correlates with dynamic changes in gene expression mediated by epigenetic events have substantially clarified the concept that successful therapeutic strategies will require targeting of both genetic and epigenetic pathways.
ACKNOWLEDGMENTS
We thank Dr. Luciana Almeida Oliveira for the CSC images used to illustrate Figure 4.
Footnotes
P- Reviewer: Chen LY, Holan V S- Editor: Tian YL L- Editor: A E- Editor: Lu YJ
Supported by University of Michigan, School of Dentistry startup
References
- 1.Boyle P, Bernard L. World cancer report 2008. Lyon: IARC Press, International Agency for Research on Cancer; 2008. [Google Scholar]
- 2.Franceschi S, Bidoli E, Herrero R, Muñoz N. Comparison of cancers of the oral cavity and pharynx worldwide: etiological clues. Oral Oncol. 2000;36:106–115. doi: 10.1016/s1368-8375(99)00070-6. [DOI] [PubMed] [Google Scholar]
- 3.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 5.Mao L, Hong WK, Papadimitrakopoulou VA. Focus on head and neck cancer. Cancer Cell. 2004;5:311–316. doi: 10.1016/s1535-6108(04)00090-x. [DOI] [PubMed] [Google Scholar]
- 6.Forastiere A, Koch W, Trotti A, Sidransky D. Head and neck cancer. N Engl J Med. 2001;345:1890–1900. doi: 10.1056/NEJMra001375. [DOI] [PubMed] [Google Scholar]
- 7.Mackenzie IC. Cancer stem cells. Ann Oncol. 2008;19 Suppl 5:v40–v43. doi: 10.1093/annonc/mdn306. [DOI] [PubMed] [Google Scholar]
- 8.Molinolo AA, Amornphimoltham P, Squarize CH, Castilho RM, Patel V, Gutkind JS. Dysregulated molecular networks in head and neck carcinogenesis. Oral Oncol. 2009;45:324–334. doi: 10.1016/j.oraloncology.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Partridge M, Li SR, Pateromichelakis S, Francis R, Phillips E, Huang XH, Tesfa-Selase F, Langdon JD. Detection of minimal residual cancer to investigate why oral tumors recur despite seemingly adequate treatment. Clin Cancer Res. 2000;6:2718–2725. [PubMed] [Google Scholar]
- 10.Parkin DM, Läärä E, Muir CS. Estimates of the worldwide frequency of sixteen major cancers in 1980. Int J Cancer. 1988;41:184–197. doi: 10.1002/ijc.2910410205. [DOI] [PubMed] [Google Scholar]
- 11.Palme CE, Gullane PJ, Gilbert RW. Current treatment options in squamous cell carcinoma of the oral cavity. Surg Oncol Clin N Am. 2004;13:47–70. doi: 10.1016/S1055-3207(03)00123-6. [DOI] [PubMed] [Google Scholar]
- 12.Takiar R, Nadayil D, Nandakumar A. Projections of number of cancer cases in India (2010-2020) by cancer groups. Asian Pac J Cancer Prev. 2010;11:1045–1049. [PubMed] [Google Scholar]
- 13.Blot WJ, McLaughlin JK, Winn DM, Austin DF, Greenberg RS, Preston-Martin S, Bernstein L, Schoenberg JB, Stemhagen A, Fraumeni JF. Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. 1988;48:3282–3287. [PubMed] [Google Scholar]
- 14.Johnson NW. Orofacial neoplasms: global epidemiology, risk factors and recommendations for research. Int Dent J. 1991;41:365–375. [PubMed] [Google Scholar]
- 15.Weinberg RA. Mutistep tumorigenesis. The biology of cancer. New York: Garland Science, Taylor & Francis Group LLC; 2014. pp. 439–506. [Google Scholar]
- 16.Graham S, Dayal H, Rohrer T, Swanson M, Sultz H, Shedd D, Fischman S. Dentition, diet, tobacco, and alcohol in the epidemiology of oral cancer. J Natl Cancer Inst. 1977;59:1611–1618. doi: 10.1093/jnci/59.6.1611. [DOI] [PubMed] [Google Scholar]
- 17.Dayal PK, Mani NJ, Bhargava K. Prevalence of oral cancer and precancerous lesions in ‘pan’/’supari’ chewers. Indian J Public Health. 1978;22:234–245. [PubMed] [Google Scholar]
- 18.Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, Weissman IL, Clarke MF, Ailles LE. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci USA. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song SH, Han SW, Bang YJ. Epigenetic-based therapies in cancer: progress to date. Drugs. 2011;71:2391–2403. doi: 10.2165/11596690-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 20.Khan O, La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012;90:85–94. doi: 10.1038/icb.2011.100. [DOI] [PubMed] [Google Scholar]
- 21.Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nat Immunol. 2011;12:715–723. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- 22.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 23.Weinberg RA. Maintenance of genomic integrity and the development of cancer. The biology of cancer. New York: Garland Science, Taylor and Francis Group, LLC; 2014. pp. 511–572. [Google Scholar]
- 24.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 25.Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675–699. doi: 10.1146/annurev.cellbio.22.010305.104154. [DOI] [PubMed] [Google Scholar]
- 26.Weinberg RA. Tumor suppressor genes. The biology of cancer New York: Garland Science, Taylor & Francis Group, LLC; 2014. pp. 231–272. [Google Scholar]
- 27.Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822–829. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 28.Squarize CH, Castilho RM, Abrahao AC, Molinolo A, Lingen MW, Gutkind JS. PTEN deficiency contributes to the development and progression of head and neck cancer. Neoplasia. 2013;15:461–471. doi: 10.1593/neo.121024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stadler ME, Patel MR, Couch ME, Hayes DN. Molecular biology of head and neck cancer: risks and pathways. Hematol Oncol Clin North Am. 2008;22:1099–1124, vii. doi: 10.1016/j.hoc.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martins MD, Castilho RM. Histones: Controlling tumor signaling circuitry. J Carcinogene Mutagene. 2013 doi: 10.4172/2157-2518.S5-001. Available from: http: //omicsonline.Org/histones-controlling-tumor-signaling-circuitry-2157-2518-s5-001.Php?Aid=19129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.González-Ramírez I, Soto-Reyes E, Sánchez-Pérez Y, Herrera LA, García-Cuellar C. Histones and long non-coding RNAs: the new insights of epigenetic deregulation involved in oral cancer. Oral Oncol. 2014;50:691–695. doi: 10.1016/j.oraloncology.2014.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esteller M. Epigenetic changes in cancer. F1000 Biol Rep. 2011;3:9. doi: 10.3410/B3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 35.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 36.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 37.Krishnamurthy S, Dong Z, Vodopyanov D, Imai A, Helman JI, Prince ME, Wicha MS, Nör JE. Endothelial cell-initiated signaling promotes the survival and self-renewal of cancer stem cells. Cancer Res. 2010;70:9969–9978. doi: 10.1158/0008-5472.CAN-10-1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neiva KG, Zhang Z, Miyazawa M, Warner KA, Karl E, Nör JE. Cross talk initiated by endothelial cells enhances migration and inhibits anoikis of squamous cell carcinoma cells through STAT3/Akt/ERK signaling. Neoplasia. 2009;11:583–593. doi: 10.1593/neo.09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weinberg RA. The nature of cancer. The biology of cancer. New York: Garland Science, Taylor & Francis Group, LLC; 2014. pp. 31–67. [Google Scholar]
- 40.Park CH, Bergsagel DE, McCulloch EA. Mouse myeloma tumor stem cells: a primary cell culture assay. J Natl Cancer Inst. 1971;46:411–422. [PubMed] [Google Scholar]
- 41.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 42.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szafarowski T, Szczepanski MJ. Cancer stem cells in head and neck squamous cell carcinoma. Otolaryngol Pol. 2014;68:105–111. doi: 10.1016/j.otpol.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 44.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 45.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grady WM, Markowitz S. Genomic instability and colorectal cancer. Curr Opin Gastroenterol. 2000;16:62–67. doi: 10.1097/00001574-200001000-00012. [DOI] [PubMed] [Google Scholar]
- 47.Heng HH, Stevens JB, Liu G, Bremer SW, Ye KJ, Reddy PV, Wu GS, Wang YA, Tainsky MA, Ye CJ. Stochastic cancer progression driven by non-clonal chromosome aberrations. J Cell Physiol. 2006;208:461–472. doi: 10.1002/jcp.20685. [DOI] [PubMed] [Google Scholar]
- 48.Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003;36 Suppl 1:59–72. doi: 10.1046/j.1365-2184.36.s.1.6.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang T, Rycaj K, Liu ZM, Tang DG. Cancer stem cells: constantly evolving and functionally heterogeneous therapeutic targets. Cancer Res. 2014;74:2922–2927. doi: 10.1158/0008-5472.CAN-14-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prince ME, Ailles LE. Cancer stem cells in head and neck squamous cell cancer. J Clin Oncol. 2008;26:2871–2875. doi: 10.1200/JCO.2007.15.1613. [DOI] [PubMed] [Google Scholar]
- 51.Ishizawa K, Rasheed ZA, Karisch R, Wang Q, Kowalski J, Susky E, Pereira K, Karamboulas C, Moghal N, Rajeshkumar NV, et al. Tumor-initiating cells are rare in many human tumors. Cell Stem Cell. 2010;7:279–282. doi: 10.1016/j.stem.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baumann M, Krause M. CD44: a cancer stem cell-related biomarker with predictive potential for radiotherapy. Clin Cancer Res. 2010;16:5091–5093. doi: 10.1158/1078-0432.CCR-10-2244. [DOI] [PubMed] [Google Scholar]
- 54.Chikamatsu K, Ishii H, Takahashi G, Okamoto A, Moriyama M, Sakakura K, Masuyama K. Resistance to apoptosis-inducing stimuli in CD44+ head and neck squamous cell carcinoma cells. Head Neck. 2012;34:336–343. doi: 10.1002/hed.21732. [DOI] [PubMed] [Google Scholar]
- 55.Perez A, Neskey DM, Wen J, Pereira L, Reategui EP, Goodwin WJ, Carraway KL, Franzmann EJ. CD44 interacts with EGFR and promotes head and neck squamous cell carcinoma initiation and progression. Oral Oncol. 2013;49:306–313. doi: 10.1016/j.oraloncology.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Faber A, Barth C, Hörmann K, Kassner S, Schultz JD, Sommer U, Stern-Straeter J, Thorn C, Goessler UR. CD44 as a stem cell marker in head and neck squamous cell carcinoma. Oncol Rep. 2011;26:321–326. doi: 10.3892/or.2011.1322. [DOI] [PubMed] [Google Scholar]
- 57.Okamoto A, Chikamatsu K, Sakakura K, Hatsushika K, Takahashi G, Masuyama K. Expansion and characterization of cancer stem-like cells in squamous cell carcinoma of the head and neck. Oral Oncol. 2009;45:633–639. doi: 10.1016/j.oraloncology.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 58.Dalley AJ, Pitty LP, Major AG, Abdulmajeed AA, Farah CS. Expression of ABCG2 and Bmi-1 in oral potentially malignant lesions and oral squamous cell carcinoma. Cancer Med. 2014;3:273–283. doi: 10.1002/cam4.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Major AG, Pitty LP, Farah CS. Cancer stem cell markers in head and neck squamous cell carcinoma. Stem Cells Int. 2013;2013:319489. doi: 10.1155/2013/319489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang P, Zhang Y, Mao L, Zhang Z, Chen W. Side population in oral squamous cell carcinoma possesses tumor stem cell phenotypes. Cancer Lett. 2009;277:227–234. doi: 10.1016/j.canlet.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 61.Yamazaki H, Mori T, Yazawa M, Maeshima AM, Matsumoto F, Yoshimoto S, Ota Y, Kaneko A, Tsuda H, Kanai Y. Stem cell self-renewal factors Bmi1 and HMGA2 in head and neck squamous cell carcinoma: clues for diagnosis. Lab Invest. 2013;93:1331–1338. doi: 10.1038/labinvest.2013.120. [DOI] [PubMed] [Google Scholar]
- 62.Nör C, Zhang Z, Warner KA, Bernardi L, Visioli F, Helman JI, Roesler R, Nör JE. Cisplatin induces Bmi-1 and enhances the stem cell fraction in head and neck cancer. Neoplasia. 2014;16:137–146. doi: 10.1593/neo.131744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Franzmann EJ, Weed DT, Civantos FJ, Goodwin WJ, Bourguignon LY. A novel CD44 v3 isoform is involved in head and neck squamous cell carcinoma progression. Otolaryngol Head Neck Surg. 2001;124:426–432. doi: 10.1067/mhn.2001.114674. [DOI] [PubMed] [Google Scholar]
- 64.Wang SJ, Wong G, de Heer AM, Xia W, Bourguignon LY. CD44 variant isoforms in head and neck squamous cell carcinoma progression. Laryngoscope. 2009;119:1518–1530. doi: 10.1002/lary.20506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bates RC, Edwards NS, Burns GF, Fisher DE. A CD44 survival pathway triggers chemoresistance via lyn kinase and phosphoinositide 3-kinase/Akt in colon carcinoma cells. Cancer Res. 2001;61:5275–5283. [PubMed] [Google Scholar]
- 66.Yang JP, Liu Y, Zhong W, Yu D, Wen LJ, Jin CS. Chemoresistance of CD133+ cancer stem cells in laryngeal carcinoma. Chin Med J (Engl) 2011;124:1055–1060. [PubMed] [Google Scholar]
- 67.Pellacani D, Oldridge EE, Collins AT, Maitland NJ. Prominin-1 (CD133) Expression in the Prostate and Prostate Cancer: A Marker for Quiescent Stem Cells. Adv Exp Med Biol. 2013;777:167–184. doi: 10.1007/978-1-4614-5894-4_11. [DOI] [PubMed] [Google Scholar]
- 68.Meng X, Li M, Wang X, Wang Y, Ma D. Both CD133+ and CD133- subpopulations of A549 and H446 cells contain cancer-initiating cells. Cancer Sci. 2009;100:1040–1046. doi: 10.1111/j.1349-7006.2009.01144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clay MR, Tabor M, Owen JH, Carey TE, Bradford CR, Wolf GT, Wicha MS, Prince ME. Single-marker identification of head and neck squamous cell carcinoma cancer stem cells with aldehyde dehydrogenase. Head Neck. 2010;32:1195–1201. doi: 10.1002/hed.21315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Keysar SB, Jimeno A. More than markers: biological significance of cancer stem cell-defining molecules. Mol Cancer Ther. 2010;9:2450–2457. doi: 10.1158/1535-7163.MCT-10-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sano A, Kato H, Sakurai S, Sakai M, Tanaka N, Inose T, Saito K, Sohda M, Nakajima M, Nakajima T, et al. CD24 expression is a novel prognostic factor in esophageal squamous cell carcinoma. Ann Surg Oncol. 2009;16:506–514. doi: 10.1245/s10434-008-0252-0. [DOI] [PubMed] [Google Scholar]
- 73.Lim SC. CD24 and human carcinoma: tumor biological aspects. Biomed Pharmacother. 2005;59 Suppl 2:S351–S354. doi: 10.1016/s0753-3322(05)80076-9. [DOI] [PubMed] [Google Scholar]
- 74.Neuzil J, Stantic M, Zobalova R, Chladova J, Wang X, Prochazka L, Dong L, Andera L, Ralph SJ. Tumour-initiating cells vs. cancer ‘stem’ cells and CD133: what’s in the name? Biochem Biophys Res Commun. 2007;355:855–859. doi: 10.1016/j.bbrc.2007.01.159. [DOI] [PubMed] [Google Scholar]
- 75.Chen YC, Chen YW, Hsu HS, Tseng LM, Huang PI, Lu KH, Chen DT, Tai LK, Yung MC, Chang SC, et al. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem Biophys Res Commun. 2009;385:307–313. doi: 10.1016/j.bbrc.2009.05.048. [DOI] [PubMed] [Google Scholar]
- 76.Sophos NA, Vasiliou V. Aldehyde dehydrogenase gene superfamily: the 2002 update. Chem Biol Interact. 2003;143-144:5–22. doi: 10.1016/s0009-2797(02)00163-1. [DOI] [PubMed] [Google Scholar]
- 77.Chute JP, Muramoto GG, Whitesides J, Colvin M, Safi R, Chao NJ, McDonnell DP. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc Natl Acad Sci USA. 2006;103:11707–11712. doi: 10.1073/pnas.0603806103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Silva IA, Bai S, McLean K, Yang K, Griffith K, Thomas D, Ginestier C, Johnston C, Kueck A, Reynolds RK, et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011;71:3991–4001. doi: 10.1158/0008-5472.CAN-10-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xing Y, Luo DY, Long MY, Zeng SL, Li HH. High ALDH1A1 expression correlates with poor survival in papillary thyroid carcinoma. World J Surg Oncol. 2014;12:29. doi: 10.1186/1477-7819-12-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rasheed ZA, Yang J, Wang Q, Kowalski J, Freed I, Murter C, Hong SM, Koorstra JB, Rajeshkumar NV, He X, et al. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J Natl Cancer Inst. 2010;102:340–351. doi: 10.1093/jnci/djp535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tirino V, Desiderio V, Paino F, De Rosa A, Papaccio F, La Noce M, Laino L, De Francesco F, Papaccio G. Cancer stem cells in solid tumors: an overview and new approaches for their isolation and characterization. FASEB J. 2013;27:13–24. doi: 10.1096/fj.12-218222. [DOI] [PubMed] [Google Scholar]
- 82.Ucar D, Cogle CR, Zucali JR, Ostmark B, Scott EW, Zori R, Gray BA, Moreb JS. Aldehyde dehydrogenase activity as a functional marker for lung cancer. Chem Biol Interact. 2009;178:48–55. doi: 10.1016/j.cbi.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sun S, Wang Z. ALDH high adenoid cystic carcinoma cells display cancer stem cell properties and are responsible for mediating metastasis. Biochem Biophys Res Commun. 2010;396:843–848. doi: 10.1016/j.bbrc.2010.04.170. [DOI] [PubMed] [Google Scholar]
- 84.Sayed SI, Dwivedi RC, Katna R, Garg A, Pathak KA, Nutting CM, Rhys-Evans P, Harrington KJ, Kazi R. Implications of understanding cancer stem cell (CSC) biology in head and neck squamous cell cancer. Oral Oncol. 2011;47:237–243. doi: 10.1016/j.oraloncology.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 85.Allegra E, Trapasso S. Cancer stem cells in head and neck cancer. Onco Targets Ther. 2012;5:375–383. doi: 10.2147/OTT.S38694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O’Sullivan E, Goggins M. DNA methylation analysis in human cancer. Methods Mol Biol. 2013;980:131–156. doi: 10.1007/978-1-62703-287-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 88.Beck S, Rakyan VK. The methylome: approaches for global DNA methylation profiling. Trends Genet. 2008;24:231–237. doi: 10.1016/j.tig.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 89.Demokan S, Dalay N. Role of DNA methylation in head and neck cancer. Clin Epigenetics. 2011;2:123–150. doi: 10.1007/s13148-011-0045-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 91.Phillips T. The role of methylation in gene expression. Available from: http: //www.nature.com/scitable/topicpage/the-role-of-methylation-in-gene-expression-1070.
- 92.Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol. 2007;213:384–390. doi: 10.1002/jcp.21224. [DOI] [PubMed] [Google Scholar]
- 93.Weber M, Schübeler D. Genomic patterns of DNA methylation: targets and function of an epigenetic mark. Curr Opin Cell Biol. 2007;19:273–280. doi: 10.1016/j.ceb.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 94.Momparler RL, Bovenzi V. DNA methylation and cancer. J Cell Physiol. 2000;183:145–154. doi: 10.1002/(SICI)1097-4652(200005)183:2<145::AID-JCP1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 95.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 96.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. DNA, chromosomes, and genomes. Molecular biology of the cell. New York: Garland Science, Taylor and Francis Group, LLC; 2008. pp. 222–226. [Google Scholar]
- 97.Kim JK, Samaranayake M, Pradhan S. Epigenetic mechanisms in mammals. Cell Mol Life Sci. 2009;66:596–612. doi: 10.1007/s00018-008-8432-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Eberharter A, Ferreira R, Becker P. Dynamic chromatin: concerted nucleosome remodelling and acetylation. Biol Chem. 2005;386:745–751. doi: 10.1515/BC.2005.087. [DOI] [PubMed] [Google Scholar]
- 99.Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol. 2013;20:259–266. doi: 10.1038/nsmb.2470. [DOI] [PubMed] [Google Scholar]
- 100.Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther. 2009;8:1409–1420. doi: 10.1158/1535-7163.MCT-08-0860. [DOI] [PubMed] [Google Scholar]
- 101.Dekker FJ, Haisma HJ. Histone acetyl transferases as emerging drug targets. Drug Discov Today. 2009;14:942–948. doi: 10.1016/j.drudis.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 102.Furdas SD, Kannan S, Sippl W, Jung M. Small molecule inhibitors of histone acetyltransferases as epigenetic tools and drug candidates. Arch Pharm (Weinheim) 2012;345:7–21. doi: 10.1002/ardp.201100209. [DOI] [PubMed] [Google Scholar]
- 103.Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 104.Brooks CL, Gu W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell. 2011;2:456–462. doi: 10.1007/s13238-011-1063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang F, Marshall CB, Ikura M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell Mol Life Sci. 2013;70:3989–4008. doi: 10.1007/s00018-012-1254-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 107.Luo J, Li M, Tang Y, Laszkowska M, Roeder RG, Gu W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci USA. 2004;101:2259–2264. doi: 10.1073/pnas.0308762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, Yao TP. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21:6236–6245. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shima Y, Kitabayashi I. Deregulated transcription factors in leukemia. Int J Hematol. 2011;94:134–141. doi: 10.1007/s12185-011-0905-9. [DOI] [PubMed] [Google Scholar]
- 110.Kida A, Kahn M. Hypoxia selects for a quiescent, CML stem/leukemia initiating-like population dependent on CBP/catenin transcription. Curr Mol Pharmacol. 2013;6:204–210. doi: 10.2174/1874467207666140219121219. [DOI] [PubMed] [Google Scholar]
- 111.Gao Y, Geng J, Hong X, Qi J, Teng Y, Yang Y, Qu D, Chen G. Expression of p300 and CBP is associated with poor prognosis in small cell lung cancer. Int J Clin Exp Pathol. 2014;7:760–767. [PMC free article] [PubMed] [Google Scholar]
- 112.Ionov Y, Matsui S, Cowell JK. A role for p300/CREB binding protein genes in promoting cancer progression in colon cancer cell lines with microsatellite instability. Proc Natl Acad Sci USA. 2004;101:1273–1278. doi: 10.1073/pnas.0307276101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Takeuchi A, Shiota M, Tatsugami K, Yokomizo A, Tanaka S, Kuroiwa K, Eto M, Naito S. p300 mediates cellular resistance to doxorubicin in bladder cancer. Mol Med Rep. 2012;5:173–176. doi: 10.3892/mmr.2011.593. [DOI] [PubMed] [Google Scholar]
- 114.Debes JD, Sebo TJ, Lohse CM, Murphy LM, Haugen DA, Tindall DJ. p300 in prostate cancer proliferation and progression. Cancer Res. 2003;63:7638–7640. [PubMed] [Google Scholar]
- 115.Ianculescu I, Wu DY, Siegmund KD, Stallcup MR. Selective roles for cAMP response element-binding protein binding protein and p300 protein as coregulators for androgen-regulated gene expression in advanced prostate cancer cells. J Biol Chem. 2012;287:4000–4013. doi: 10.1074/jbc.M111.300194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bouchal J, Santer FR, Höschele PP, Tomastikova E, Neuwirt H, Culig Z. Transcriptional coactivators p300 and CBP stimulate estrogen receptor-beta signaling and regulate cellular events in prostate cancer. Prostate. 2011;71:431–437. doi: 10.1002/pros.21257. [DOI] [PubMed] [Google Scholar]
- 117.Davidson SM, Townsend PA, Carroll C, Yurek-George A, Balasubramanyam K, Kundu TK, Stephanou A, Packham G, Ganesan A, Latchman DS. The transcriptional coactivator p300 plays a critical role in the hypertrophic and protective pathways induced by phenylephrine in cardiac cells but is specific to the hypertrophic effect of urocortin. Chembiochem. 2005;6:162–170. doi: 10.1002/cbic.200400246. [DOI] [PubMed] [Google Scholar]
- 118.Chen G, Zhu J, Lv T, Wu G, Sun H, Huang X, Tian J. Spatiotemporal expression of histone acetyltransferases, p300 and CBP, in developing embryonic hearts. J Biomed Sci. 2009;16:24. doi: 10.1186/1423-0127-16-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Stanojevic V, Habener JF, Thomas MK. Pancreas duodenum homeobox-1 transcriptional activation requires interactions with p300. Endocrinology. 2004;145:2918–2928. doi: 10.1210/en.2003-1188. [DOI] [PubMed] [Google Scholar]
- 120.He L, Naik K, Meng S, Cao J, Sidhaye AR, Ma A, Radovick S, Wondisford FE. Transcriptional co-activator p300 maintains basal hepatic gluconeogenesis. J Biol Chem. 2012;287:32069–32077. doi: 10.1074/jbc.M112.385864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2010;107:22687–22692. doi: 10.1073/pnas.1012851108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rouaux C, Loeffler JP, Boutillier AL. Targeting CREB-binding protein (CBP) loss of function as a therapeutic strategy in neurological disorders. Biochem Pharmacol. 2004;68:1157–1164. doi: 10.1016/j.bcp.2004.05.035. [DOI] [PubMed] [Google Scholar]
- 123.Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkühler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17:195–211. doi: 10.1038/sj.cr.7310149. [DOI] [PubMed] [Google Scholar]
- 124.Di Lorenzo A, Bedford MT. Histone arginine methylation. FEBS Lett. 2011;585:2024–2031. doi: 10.1016/j.febslet.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Völkel P, Angrand PO. The control of histone lysine methylation in epigenetic regulation. Biochimie. 2007;89:1–20. doi: 10.1016/j.biochi.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 126.Binda O. On your histone mark, SET, methylate! Epigenetics. 2013;8:457–463. doi: 10.4161/epi.24451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lachner M, Jenuwein T. The many faces of histone lysine methylation. Curr Opin Cell Biol. 2002;14:286–298. doi: 10.1016/s0955-0674(02)00335-6. [DOI] [PubMed] [Google Scholar]
- 129.Cao W, Younis RH, Li J, Chen H, Xia R, Mao L, Chen W, Ren H. EZH2 promotes malignant phenotypes and is a predictor of oral cancer development in patients with oral leukoplakia. Cancer Prev Res (Phila) 2011;4:1816–1824. doi: 10.1158/1940-6207.CAPR-11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Manzo F, Tambaro FP, Mai A, Altucci L. Histone acetyltransferase inhibitors and preclinical studies. Expert Opin Ther Pat. 2009;19:761–774. doi: 10.1517/13543770902895727. [DOI] [PubMed] [Google Scholar]
- 131.Marcu MG, Jung YJ, Lee S, Chung EJ, Lee MJ, Trepel J, Neckers L. Curcumin is an inhibitor of p300 histone acetylatransferase. Med Chem. 2006;2:169–174. doi: 10.2174/157340606776056133. [DOI] [PubMed] [Google Scholar]
- 132.Choi KC, Jung MG, Lee YH, Yoon JC, Kwon SH, Kang HB, Kim MJ, Cha JH, Kim YJ, Jun WJ, et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009;69:583–592. doi: 10.1158/0008-5472.CAN-08-2442. [DOI] [PubMed] [Google Scholar]
- 133.Ravindra KC, Selvi BR, Arif M, Reddy BA, Thanuja GR, Agrawal S, Pradhan SK, Nagashayana N, Dasgupta D, Kundu TK. Inhibition of lysine acetyltransferase KAT3B/p300 activity by a naturally occurring hydroxynaphthoquinone, plumbagin. J Biol Chem. 2009;284:24453–24464. doi: 10.1074/jbc.M109.023861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yuan H, Marmorstein R. Histone acetyltransferases: Rising ancient counterparts to protein kinases. Biopolymers. 2013;99:98–111. doi: 10.1002/bip.22128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li Z, Zhu WG. Targeting histone deacetylases for cancer therapy: from molecular mechanisms to clinical implications. Int J Biol Sci. 2014;10:757–770. doi: 10.7150/ijbs.9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bártová E, Krejcí J, Harnicarová A, Galiová G, Kozubek S. Histone modifications and nuclear architecture: a review. J Histochem Cytochem. 2008;56:711–721. doi: 10.1369/jhc.2008.951251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Towle R, Garnis C. Methylation-mediated molecular dysregulation in clinical oral malignancy. J Oncol. 2012;2012:170172. doi: 10.1155/2012/170172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.García MP, García-García A. Epigenome and DNA methylation in oral squamous cell carcinoma. Methods Mol Biol. 2012;863:207–219. doi: 10.1007/978-1-61779-612-8_12. [DOI] [PubMed] [Google Scholar]
- 139.Mascolo M, Siano M, Ilardi G, Russo D, Merolla F, De Rosa G, Staibano S. Epigenetic disregulation in oral cancer. Int J Mol Sci. 2012;13:2331–2353. doi: 10.3390/ijms13022331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lingen MW, Pinto A, Mendes RA, Franchini R, Czerninski R, Tilakaratne WM, Partridge M, Peterson DE, Woo SB. Genetics/epigenetics of oral premalignancy: current status and future research. Oral Dis. 2011;17 Suppl 1:7–22. doi: 10.1111/j.1601-0825.2011.01789.x. [DOI] [PubMed] [Google Scholar]
- 141.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37–50. doi: 10.1038/nrd1930. [DOI] [PubMed] [Google Scholar]
- 143.Van Speybroeck L. From epigenesis to epigenetics: the case of C. H. Waddington. Ann N Y Acad Sci. 2002;981:61–81. [PubMed] [Google Scholar]
- 144.Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 145.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.ARMITAGE P, DOLL R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8:1–12. doi: 10.1038/bjc.1954.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Day NE, Brown CC. Multistage models and primary prevention of cancer. J Natl Cancer Inst. 1980;64:977–989. [PubMed] [Google Scholar]
- 148.Stenbäck F, Peto R, Shubik P. Initiation and promotion at different ages and doses in 2200 mice. I. Methods, and the apparent persistence of initiated cells. Br J Cancer. 1981;44:1–14. doi: 10.1038/bjc.1981.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.NORDLING CO. A new theory on cancer-inducing mechanism. Br J Cancer. 1953;7:68–72. doi: 10.1038/bjc.1953.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Luebeck EG, Moolgavkar SH. Multistage carcinogenesis and the incidence of colorectal cancer. Proc Natl Acad Sci USA. 2002;99:15095–15100. doi: 10.1073/pnas.222118199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Giudice FS, Pinto DS, Nör JE, Squarize CH, Castilho RM. Inhibition of histone deacetylase impacts cancer stem cells and induces epithelial-mesenchyme transition of head and neck cancer. PLoS One. 2013;8:e58672. doi: 10.1371/journal.pone.0058672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 153.Karlic R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci USA. 2010;107:2926–2931. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chen K, Sawhney R, Khan M, Benninger MS, Hou Z, Sethi S, Stephen JK, Worsham MJ. Methylation of multiple genes as diagnostic and therapeutic markers in primary head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2007;133:1131–1138. doi: 10.1001/archotol.133.11.1131. [DOI] [PubMed] [Google Scholar]
- 155.Worsham MJ, Chen KM, Meduri V, Nygren AO, Errami A, Schouten JP, Benninger MS. Epigenetic events of disease progression in head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2006;132:668–677. doi: 10.1001/archotol.132.6.668. [DOI] [PubMed] [Google Scholar]
- 156.Rayess H, Wang MB, Srivatsan ES. Cellular senescence and tumor suppressor gene p16. Int J Cancer. 2012;130:1715–1725. doi: 10.1002/ijc.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Shaw RJ, Liloglou T, Rogers SN, Brown JS, Vaughan ED, Lowe D, Field JK, Risk JM. Promoter methylation of P16, RARbeta, E-cadherin, cyclin A1 and cytoglobin in oral cancer: quantitative evaluation using pyrosequencing. Br J Cancer. 2006;94:561–568. doi: 10.1038/sj.bjc.6602972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ai L, Stephenson KK, Ling W, Zuo C, Mukunyadzi P, Suen JY, Hanna E, Fan CY. The p16 (CDKN2a/INK4a) tumor-suppressor gene in head and neck squamous cell carcinoma: a promoter methylation and protein expression study in 100 cases. Mod Pathol. 2003;16:944–950. doi: 10.1097/01.MP.0000085760.74313.DD. [DOI] [PubMed] [Google Scholar]
- 159.Carvalho AL, Jeronimo C, Kim MM, Henrique R, Zhang Z, Hoque MO, Chang S, Brait M, Nayak CS, Jiang WW, et al. Evaluation of promoter hypermethylation detection in body fluids as a screening/diagnosis tool for head and neck squamous cell carcinoma. Clin Cancer Res. 2008;14:97–107. doi: 10.1158/1078-0432.CCR-07-0722. [DOI] [PubMed] [Google Scholar]
- 160.Hasegawa M, Nelson HH, Peters E, Ringstrom E, Posner M, Kelsey KT. Patterns of gene promoter methylation in squamous cell cancer of the head and neck. Oncogene. 2002;21:4231–4236. doi: 10.1038/sj.onc.1205528. [DOI] [PubMed] [Google Scholar]
- 161.Koscielny S, Dahse R, Ernst G, von Eggeling F. The prognostic relevance of p16 inactivation in head and neck cancer. ORL J Otorhinolaryngol Relat Spec. 2007;69:30–36. doi: 10.1159/000096714. [DOI] [PubMed] [Google Scholar]
- 162.Maruya S, Issa JP, Weber RS, Rosenthal DI, Haviland JC, Lotan R, El-Naggar AK. Differential methylation status of tumor-associated genes in head and neck squamous carcinoma: incidence and potential implications. Clin Cancer Res. 2004;10:3825–3830. doi: 10.1158/1078-0432.CCR-03-0370. [DOI] [PubMed] [Google Scholar]
- 163.Okami K, Sakai A, Onuki J, Hamano T, Iida M, Takahashi M. [Promoter hypermethylation of tumor-associated genes in head and neck cancer] Nihon Jibiinkoka Gakkai Kaiho. 2005;108:207–213. doi: 10.3950/jibiinkoka.108.207. [DOI] [PubMed] [Google Scholar]
- 164.Puri SK, Si L, Fan CY, Hanna E. Aberrant promoter hypermethylation of multiple genes in head and neck squamous cell carcinoma. Am J Otolaryngol. 2005;26:12–17. doi: 10.1016/j.amjoto.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 165.Righini CA, de Fraipont F, Timsit JF, Faure C, Brambilla E, Reyt E, Favrot MC. Tumor-specific methylation in saliva: a promising biomarker for early detection of head and neck cancer recurrence. Clin Cancer Res. 2007;13:1179–1185. doi: 10.1158/1078-0432.CCR-06-2027. [DOI] [PubMed] [Google Scholar]
- 166.Rosas SL, Koch W, da Costa Carvalho MG, Wu L, Califano J, Westra W, Jen J, Sidransky D. Promoter hypermethylation patterns of p16, O6-methylguanine-DNA-methyltransferase, and death-associated protein kinase in tumors and saliva of head and neck cancer patients. Cancer Res. 2001;61:939–942. [PubMed] [Google Scholar]
- 167.Steinmann K, Sandner A, Schagdarsurengin U, Dammann RH. Frequent promoter hypermethylation of tumor-related genes in head and neck squamous cell carcinoma. Oncol Rep. 2009;22:1519–1526. doi: 10.3892/or_00000596. [DOI] [PubMed] [Google Scholar]
- 168.Weber A, Bellmann U, Bootz F, Wittekind C, Tannapfel A. INK4a-ARF alterations and p53 mutations in primary and consecutive squamous cell carcinoma of the head and neck. Virchows Arch. 2002;441:133–142. doi: 10.1007/s00428-002-0637-6. [DOI] [PubMed] [Google Scholar]
- 169.Ogi K, Toyota M, Ohe-Toyota M, Tanaka N, Noguchi M, Sonoda T, Kohama G, Tokino T. Aberrant methylation of multiple genes and clinicopathological features in oral squamous cell carcinoma. Clin Cancer Res. 2002;8:3164–3171. [PubMed] [Google Scholar]
- 170.Hogg RP, Honorio S, Martinez A, Agathanggelou A, Dallol A, Fullwood P, Weichselbaum R, Kuo MJ, Maher ER, Latif F. Frequent 3p allele loss and epigenetic inactivation of the RASSF1A tumour suppressor gene from region 3p21.3 in head and neck squamous cell carcinoma. Eur J Cancer. 2002;38:1585–1592. doi: 10.1016/s0959-8049(01)00422-1. [DOI] [PubMed] [Google Scholar]
- 171.Dong SM, Sun DI, Benoit NE, Kuzmin I, Lerman MI, Sidransky D. Epigenetic inactivation of RASSF1A in head and neck cancer. Clin Cancer Res. 2003;9:3635–3640. [PubMed] [Google Scholar]
- 172.Vos MD, Ellis CA, Elam C, Ulku AS, Taylor BJ, Clark GJ. RASSF2 is a novel K-Ras-specific effector and potential tumor suppressor. J Biol Chem. 2003;278:28045–28051. doi: 10.1074/jbc.M300554200. [DOI] [PubMed] [Google Scholar]
- 173.Akino K, Toyota M, Suzuki H, Mita H, Sasaki Y, Ohe-Toyota M, Issa JP, Hinoda Y, Imai K, Tokino T. The Ras effector RASSF2 is a novel tumor-suppressor gene in human colorectal cancer. Gastroenterology. 2005;129:156–169. doi: 10.1053/j.gastro.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 174.Demokan S, Chang X, Chuang A, Mydlarz WK, Kaur J, Huang P, Khan Z, Khan T, Ostrow KL, Brait M, et al. KIF1A and EDNRB are differentially methylated in primary HNSCC and salivary rinses. Int J Cancer. 2010;127:2351–2359. doi: 10.1002/ijc.25248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Yalniz Z, Demokan S, Suoglu Y, Ulusan M, Dalay N. Simultaneous methylation profiling of tumor suppressor genes in head and neck cancer. DNA Cell Biol. 2011;30:17–24. doi: 10.1089/dna.2010.1090. [DOI] [PubMed] [Google Scholar]
- 176.Youssef EM, Lotan D, Issa JP, Wakasa K, Fan YH, Mao L, Hassan K, Feng L, Lee JJ, Lippman SM, et al. Hypermethylation of the retinoic acid receptor-beta(2) gene in head and neck carcinogenesis. Clin Cancer Res. 2004;10:1733–1742. doi: 10.1158/1078-0432.ccr-0989-3. [DOI] [PubMed] [Google Scholar]
- 177.Mancuso M, Matassa DS, Conte M, Colella G, Rana G, Fucci L, Piscopo M. H3K4 histone methylation in oral squamous cell carcinoma. Acta Biochim Pol. 2009;56:405–410. [PubMed] [Google Scholar]
- 178.Lund AH, van Lohuizen M. Polycomb complexes and silencing mechanisms. Curr Opin Cell Biol. 2004;16:239–246. doi: 10.1016/j.ceb.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 179.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 180.Müller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O’Connor MB, Kingston RE, Simon JA. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 181.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Raaphorst FM, Meijer CJ, Fieret E, Blokzijl T, Mommers E, Buerger H, Packeisen J, Sewalt RA, Otte AP, van Diest PJ. Poorly differentiated breast carcinoma is associated with increased expression of the human polycomb group EZH2 gene. Neoplasia. 2003;5:481–488. doi: 10.1016/s1476-5586(03)80032-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 185.Rhodes DR, Sanda MG, Otte AP, Chinnaiyan AM, Rubin MA. Multiplex biomarker approach for determining risk of prostate-specific antigen-defined recurrence of prostate cancer. J Natl Cancer Inst. 2003;95:661–668. doi: 10.1093/jnci/95.9.661. [DOI] [PubMed] [Google Scholar]
- 186.Xiao Y. Enhancer of zeste homolog 2: A potential target for tumor therapy. Int J Biochem Cell Biol. 2011;43:474–477. doi: 10.1016/j.biocel.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 187.Matsukawa Y, Semba S, Kato H, Ito A, Yanagihara K, Yokozaki H. Expression of the enhancer of zeste homolog 2 is correlated with poor prognosis in human gastric cancer. Cancer Sci. 2006;97:484–491. doi: 10.1111/j.1349-7006.2006.00203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sudo T, Utsunomiya T, Mimori K, Nagahara H, Ogawa K, Inoue H, Wakiyama S, Fujita H, Shirouzu K, Mori M. Clinicopathological significance of EZH2 mRNA expression in patients with hepatocellular carcinoma. Br J Cancer. 2005;92:1754–1758. doi: 10.1038/sj.bjc.6602531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Arisan S, Buyuktuncer ED, Palavan-Unsal N, Caşkurlu T, Cakir OO, Ergenekon E. Increased expression of EZH2, a polycomb group protein, in bladder carcinoma. Urol Int. 2005;75:252–257. doi: 10.1159/000087804. [DOI] [PubMed] [Google Scholar]
- 190.Weikert S, Christoph F, Köllermann J, Müller M, Schrader M, Miller K, Krause H. Expression levels of the EZH2 polycomb transcriptional repressor correlate with aggressiveness and invasive potential of bladder carcinomas. Int J Mol Med. 2005;16:349–353. [PubMed] [Google Scholar]
- 191.Kidani K, Osaki M, Tamura T, Yamaga K, Shomori K, Ryoke K, Ito H. High expression of EZH2 is associated with tumor proliferation and prognosis in human oral squamous cell carcinomas. Oral Oncol. 2009;45:39–46. doi: 10.1016/j.oraloncology.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 192.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 193.Kim JH, Yoon SY, Jeong SH, Kim SY, Moon SK, Joo JH, Lee Y, Choe IS, Kim JW. Overexpression of Bmi-1 oncoprotein correlates with axillary lymph node metastases in invasive ductal breast cancer. Breast. 2004;13:383–388. doi: 10.1016/j.breast.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 194.Song LB, Zeng MS, Liao WT, Zhang L, Mo HY, Liu WL, Shao JY, Wu QL, Li MZ, Xia YF, et al. Bmi-1 is a novel molecular marker of nasopharyngeal carcinoma progression and immortalizes primary human nasopharyngeal epithelial cells. Cancer Res. 2006;66:6225–6232. doi: 10.1158/0008-5472.CAN-06-0094. [DOI] [PubMed] [Google Scholar]
- 195.Wang H, Pan K, Zhang HK, Weng DS, Zhou J, Li JJ, Huang W, Song HF, Chen MS, Xia JC. Increased polycomb-group oncogene Bmi-1 expression correlates with poor prognosis in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2008;134:535–541. doi: 10.1007/s00432-007-0316-8. [DOI] [PubMed] [Google Scholar]
- 196.Qin ZK, Yang JA, Ye YL, Zhang X, Xu LH, Zhou FJ, Han H, Liu ZW, Song LB, Zeng MS. Expression of Bmi-1 is a prognostic marker in bladder cancer. BMC Cancer. 2009;9:61. doi: 10.1186/1471-2407-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zhang XW, Sheng YP, Li Q, Qin W, Lu YW, Cheng YF, Liu BY, Zhang FC, Li J, Dimri GP, et al. BMI1 and Mel-18 oppositely regulate carcinogenesis and progression of gastric cancer. Mol Cancer. 2010;9:40. doi: 10.1186/1476-4598-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005;115:1503–1521. doi: 10.1172/JCI23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Leung C, Lingbeek M, Shakhova O, Liu J, Tanger E, Saremaslani P, Van Lohuizen M, Marino S. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428:337–341. doi: 10.1038/nature02385. [DOI] [PubMed] [Google Scholar]
- 200.Thiery JP. [Epithelial-mesenchymal transitions in cancer onset and progression] Bull Acad Natl Med. 2009;193:1969–1978; discussion 1969-1978. [PubMed] [Google Scholar]
- 201.Birchmeier C, Birchmeier W, Brand-Saberi B. Epithelial-mesenchymal transitions in cancer progression. Acta Anat (Basel) 1996;156:217–226. doi: 10.1159/000147848. [DOI] [PubMed] [Google Scholar]
- 202.Huber MA, Kraut N, Beug H. Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 203.Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 204.Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66:8319–8326. doi: 10.1158/0008-5472.CAN-06-0410. [DOI] [PubMed] [Google Scholar]
- 205.Mandal M, Myers JN, Lippman SM, Johnson FM, Williams MD, Rayala S, Ohshiro K, Rosenthal DI, Weber RS, Gallick GE, et al. Epithelial to mesenchymal transition in head and neck squamous carcinoma: association of Src activation with E-cadherin down-regulation, vimentin expression, and aggressive tumor features. Cancer. 2008;112:2088–2100. doi: 10.1002/cncr.23410. [DOI] [PubMed] [Google Scholar]
- 206.Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68:3033–3046. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Yoshida M, Nomura S, Beppu T. Effects of trichostatins on differentiation of murine erythroleukemia cells. Cancer Res. 1987;47:3688–3691. [PubMed] [Google Scholar]
- 208.Sugita K, Koizumi K, Yoshida H. Morphological reversion of sis-transformed NIH3T3 cells by trichostatin A. Cancer Res. 1992;52:168–172. [PubMed] [Google Scholar]
- 209.Piyathilake CJ, Bell WC, Jones J, Henao OL, Heimburger DC, Niveleau A, Grizzle WE. Patterns of global DNA and histone methylation appear to be similar in normal, dysplastic and neoplastic oral epithelium of humans. Dis Markers. 2005;21:147–151. doi: 10.1155/2005/285134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lanzillotta A, Pignataro G, Branca C, Cuomo O, Sarnico I, Benarese M, Annunziato L, Spano P, Pizzi M. Targeted acetylation of NF-kappaB/RelA and histones by epigenetic drugs reduces post-ischemic brain injury in mice with an extended therapeutic window. Neurobiol Dis. 2012;49C:177–189. doi: 10.1016/j.nbd.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 212.Aurora AB, Biyashev D, Mirochnik Y, Zaichuk TA, Sánchez-Martinez C, Renault MA, Losordo D, Volpert OV. NF-kappaB balances vascular regression and angiogenesis via chromatin remodeling and NFAT displacement. Blood. 2010;116:475–484. doi: 10.1182/blood-2009-07-232132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Larsen L, Tonnesen M, Ronn SG, Størling J, Jørgensen S, Mascagni P, Dinarello CA, Billestrup N, Mandrup-Poulsen T. Inhibition of histone deacetylases prevents cytokine-induced toxicity in beta cells. Diabetologia. 2007;50:779–789. doi: 10.1007/s00125-006-0562-3. [DOI] [PubMed] [Google Scholar]
- 214.Squarize CH, Castilho RM, Sriuranpong V, Pinto DS, Gutkind JS. Molecular cross-talk between the NFkappaB and STAT3 signaling pathways in head and neck squamous cell carcinoma. Neoplasia. 2006;8:733–746. doi: 10.1593/neo.06274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Ondrey FG, Dong G, Sunwoo J, Chen Z, Wolf JS, Crowl-Bancroft CV, Mukaida N, Van Waes C. Constitutive activation of transcription factors NF-(kappa)B, AP-1, and NF-IL6 in human head and neck squamous cell carcinoma cell lines that express pro-inflammatory and pro-angiogenic cytokines. Mol Carcinog. 1999;26:119–129. doi: 10.1002/(sici)1098-2744(199910)26:2<119::aid-mc6>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 216.Almeida LO, Abrahao AC, Rosselli-Murai LK, Giudice FS, Zagni C, Leopoldino AM, Squarize CH, Castilho RM. NFκB mediates cisplatin resistance through histone modifications in head and neck squamous cell carcinoma (HNSCC) FEBS Open Bio. 2014;4:96–104. doi: 10.1016/j.fob.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kim HJ, Hawke N, Baldwin AS. NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006;13:738–747. doi: 10.1038/sj.cdd.4401877. [DOI] [PubMed] [Google Scholar]
- 218.Gallardo-Pérez JC, Espinosa M, Ceballos-Cancino G, Daniel A, Rodríguez-Enríquez S, Aviles A, Moreno-Sánchez R, Melendez-Zajgla J, Maldonado V. NF-kappa B is required for the development of tumor spheroids. J Cell Biochem. 2009;108:169–180. doi: 10.1002/jcb.22237. [DOI] [PubMed] [Google Scholar]
- 219.Locke M, Heywood M, Fawell S, Mackenzie IC. Retention of intrinsic stem cell hierarchies in carcinoma-derived cell lines. Cancer Res. 2005;65:8944–8950. doi: 10.1158/0008-5472.CAN-05-0931. [DOI] [PubMed] [Google Scholar]
- 220.Mackenzie IC. Retention of stem cell patterns in malignant cell lines. Cell Prolif. 2005;38:347–355. doi: 10.1111/j.1365-2184.2005.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mackenzie IC. Stem cell properties and epithelial malignancies. Eur J Cancer. 2006;42:1204–1212. doi: 10.1016/j.ejca.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 222.Li W, Li Y, Tan Y, Ma K, Cui J. Bmi-1 is critical for the proliferation and invasiveness of gastric carcinoma cells. J Gastroenterol Hepatol. 2010;25:568–575. doi: 10.1111/j.1440-1746.2009.06045.x. [DOI] [PubMed] [Google Scholar]
- 223.Li H, Song F, Chen X, Li Y, Fan J, Wu X. Bmi-1 regulates epithelial-to-mesenchymal transition to promote migration and invasion of breast cancer cells. Int J Clin Exp Pathol. 2014;7:3057–3064. [PMC free article] [PubMed] [Google Scholar]
- 224.Zhang Y, Zhang YL, Chen HM, Pu HW, Ma WJ, Li XM, Ma H, Chen X. Expression of Bmi-1 and PAI-1 in esophageal squamous cell carcinoma. World J Gastroenterol. 2014;20:5533–5539. doi: 10.3748/wjg.v20.i18.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Li X, Yang Z, Song W, Zhou L, Li Q, Tao K, Zhou J, Wang X, Zheng Z, You N, et al. Overexpression of Bmi-1 contributes to the invasion and metastasis of hepatocellular carcinoma by increasing the expression of matrix metalloproteinase (MMP)-2, MMP-9 and vascular endothelial growth factor via the PTEN/PI3K/Akt pathway. Int J Oncol. 2013;43:793–802. doi: 10.3892/ijo.2013.1992. [DOI] [PubMed] [Google Scholar]
- 226.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]