

Abstract
There are close to 70,000 new cases of primary central nervous system tumors diagnosed annually in the United States. Meningiomas, gliomas, nerve sheath tumors and pituitary tumors account for 85% of them. There is abundant literature on these commonly occurring tumors but data from the literature on infrequently encountered tumors such as atypical teratoid/rhabdoid tumor, choroid plexus carcinoma, ganglioglioma, hemangiopericytoma, and pleomorphic xanthoastrocytoma are limited. This review provides an overview of the clinicopathologic and therapeutic aspects of these rare primary central nervous system tumors.
Key words: atypical teratoid/rhabdoid tumor, choroid plexus carcinoma, ganglioglioma, hemangiopericytoma, pleomorphic xanthoastrocytoma
Introduction
An estimated 69,720 new cases of primary central nervous system tumors are expected to be diagnosed in 2013 based on projection using the 2013 Central Brain Tumor Registry of the United States (CBTRUS) Statistical Report (www.cbtrus.org). Meningiomas, gliomas, nerve sheath tumors, and pituitary tumors together constitute more than 85% of all primary central nervous system tumors diagnosed in the US.1 There are some tumors which constitute less than 1% of all brain tumors. The purpose of this paper is to provide an overview of the clinical, pathologic and therapeutic aspects of some of the more commonly encountered rare primary brain tumors, including atypical teratoid/rhabdoid tumor, choroid plexus carcinoma, ganglioglioma, hemangiopericytoma, and pleomorphic xanthoastrocytoma.
Atypical teratoid/rhabdoid tumor
Epidemiology
Atypical teratoid/rhabdoid tumor (AT/RT) is a highly malignant tumor of the childhood. It accounts for 1-2% of all pediatric brain tumors and has a striking predilection for infants. Among children younger than 3 years of age, AT/RT accounts for up to 20% of brain tumors.2 The median age of diagnosis is 17 months. The male to female ratio is approximately 2:1.3
Histopathology and molecular features
Atypical teratoid/rhabdoid tumor is a high grade malignant embryonal tumor of the central nerves system, WHO grade IV. It contains sheets of rhabdoid cells, with additional components of small embryonal, mesenchymal and epithelial cells.4 AT/RT is the only pediatric malignancy that is uniquely associated with a tumor suppressor gene. In 1995, Rorke et al. first reported a link between AT/RT and chromosome 22.3 Further investigation identified the hSNF5/INI1 gene, which maps to chromosome 22q11.2. A homogyzous deletion or mutation of the hSNF5/INI1 gene occurs in over 90% of AT/RTs. The hSNF5/INI1 gene encodes for a component of the SWI/SNF ATP-dependent chromatin-remodeling complex. This complex interacts with histone deacetylase and the retinoblastoma protein. It plays an important role at the G1/S cell cycle checkpoint. The exact mechanism by which hSNF5/INI1 mediates tumorigenesis is not fully understood. Better understanding the functionality of this gene can help elucidate the molecular pathways and identify potential therapeutic targets for AT/RT.2
Imaging characteristics
A computed tomography (CT) scan is typically done during the initial work-up. AT/RTs are typically hyperdense on CT, and enhance intensely with contrast. Calcifications are uncommon. Cysts are more common in supratentorial lesions, and less common in infratentorial lesions. On T1-weighted MRI, tumors are isointense with frequent hyperintense foci consistent with intratumoral hemorrhage. Most tumors enhance strongly with gadolinium. On T2-weighted images, tumors are often heterogeneous reflecting a mixture of tumor, hemorrhage, necrosis and cysts.5
Clinical presentation
About 50% of AT/RTs arise in the posterior fossa. Up to 35-40% of patients have leptomeningeal disease at the time diagnosis. About 15% of patients have metastatic disease at presentation. Depending on the location, children may present with lethargy, nausea, vomiting, or cranial nerve palsies.2
Treatment options
Maximum safe resection is performed for most patients. Maximum safe resection can be either gross total or subtotal resections, based on what the neurosurgeon deems as the tumor volume that can be safely resected. It is estimated that total or near-total resection can be achieved in less than 1/3 of patients. Even though the extent of resection and its impact on survival has not been studied prospectively, retrospective analyses suggest improved survival with gross total resection.5-7
Adjuvant radiotherapy appears to have an impact on survival of AT/RT patients.6,8 Historically, for patients younger than 3 years, radiation was routinely withheld due to concern for long term neurocognitive toxicities. However, many chemotherapy regimens used in place of radiation, have not been shown to be effective. Therefore, the question of whether radiation should be given to children younger than 3 was raised.
Tekautz et al. reported a retrospective review of 31 patients treated at St. Jude Children’s Hospital.6 Twnty-two patients were younger than 3 years, and 9 were older than 3. All patients underwent surgery, 30 received adjuvant chemotherapy. The majority of the children 3 years or older received adjuvant cranial spinal radiation. The 2 year overall survival and event-free survival for children older than 3 years were 89% and 78%, significantly higher than those younger than 3 years, 17% and 11%, respectively. Among those younger than 3 years, two were long term survivors and both received radiotherapy as part of the initial treatment, suggesting administrating radiation earlier in the treatment course may be of benefit. A multi-institutional prospective study included a higher percentage of children younger than 3 years who received radiation (8 out of 15), and the disease free survival and overall survival rates were higher, compared to that from St. Jude.8 To further study the potential benefit and age cutoff of radiation in AT/RT, a phase III trial ACNS0333 by Children’s Oncology Group was carried out. Both standard and intensified chemotherapy regimens have been shown to be active in AT/RT.
The use of alkylating agents, high-dose methotrexate, or high dose chemotherapy followed by stem cell rescue, may be associated with improved survival.2 To determine the optimal adjuvant therapy for AT/RT, the Children’s Oncology Group conducted a prospective study, ACNS0333, which was recently closed in February 2014. This study was designed to determine whether surgery, intensive chemotherapy and early radiation improve survival compared to historical controls. In this study, children with post fossa tumors 6 months or older after induction chemotherapy were randomized to early radiation followed consolidative chemotherapy vs upfront consolidative chemotherapy and delayed radiation. Similarly, in supratentorial tumors, children 12 months or older were randomized to early vs. delay radiation after induction. Craniospinal radiation was administered in children with metastasis after chemotherapy and are at least 12 months of age. Neurotoxicity was closely monitored in the study. Results will be published in the future. http://www.childrensoncologygroup.org/
When administered, radiation therapy typically involves using a 3D conformal technique, targeting the operativebed and residual tumor with and without cranial spinal radiation. Craniospinal radiation is typically reserved for those with disseminated disease.
Clinical outcome
Atypical teratoid/rhabdoid tumor is an extremely aggressive tumor. A recent Surveillance, Epidemiology, and End Result (SEER) analysis reported a median survival of 10 months.9 Median survival and event-free survival of children enrolled in the AT/RT registry were 17 and 10 months, respectively.7
Future direction
Molecular targeted therapy has been an area of active research for AT/RT. It has been reported that in the absence of INI1, Cyclin D1 may drive rhabdoid tumor proliferation, bypassing the G1 cell cycle check point. Histone deacetylase (HDAC) inhibitors and Vitamin A analogs such as retinoids and rexinoids have been shown to reduce Cyclin D1 expressions in cell culture, making them attractive agents for AT/RT.2
INI1 has been implicated in the down regulation of the Aurora A gene transcription, suggesting the Aurora kinase pathway may be implicated in the pathogenesis of AT/RT. Aurora kinase inhibitors have been studied in clinical trials of other tumors. They may be reasonable candidates for AT/RT. Other pathways, such as the insulin-growth factor are also being investigated.2
Choroid plexus carcinoma
Epidemiology
Choroid plexus tumors are rare tumors arising from the choroid plexus epithelium. They include three histologies, choroid plexus papilloma (WHO grade I), atypical choroid plexus papilloma (WHO grade II) and choroid plexus carcinoma (WHO grade III). All together, they account for 0.4-0.6% of all brain tumors. Choroid plexus tumors occur more frequently in children, comprising approximately 4% of all pediatric brain tumors. Up to 20% of these tumors occur during the first year of life. 20% to 40% of choroid plexus tumors are choroid plexus carcinomas.10, 11
Histopathology and molecular features
On gross pathology, choroid plexus carcinomas have a papillary or cauliflower-like appearance. They often show areas of hemorrhage and necrosis, with tumor invasion into the periventricular brain parenchyma.10 On histologic examination, these tumors are characterized by increased cell density, increased mitotic figures (>5 per 10 high power field), nuclear pleomorphism and necrosis. On immunohistochemistry, choroid plexus carcinomas are almost always positive for cytokeratin, focally positive for synaptophysin, GFAP, EMA, CD 44, and CA 19-9. They are less likely to stain positive for S-100 and transthyretin. Histologically, choroid plexus carcinomas and AT/RT have overlapping features. However, the majority of choroid plexus carcinomas stain positive for INI1, whereas nuclear staining in AT/RT is almost always absent. This is a helpful distinguishing feature.12
Imaging characteristics
Choroid plexus tumors most commonly arise from the lateral ventricles (50%), followed by the fourth (40%) and the third ventricle (5%). Other locations are rare, including the cerebellopontine angle, supresellar region, brain parenchyma and the spine. On CT, choroid plexus tumors appear heterogeneous and isodense with calcifications and necrosis. On MRI, choroid plexus carcinomas show heterogeneous intensities on both T1 and T2, with irregular enhancement and edema. Flow voids are often seen reflecting the vascular nature of the tumor. Tumor hemorrhage and necrosis are commonly seen as well. On MR spectroscopy, choroid plexus carcinomas can be distinguished from choroid plexus papillomas by exhibiting lower levels of myoinositol and higher levels of choline.10,11
Clinical presentation
Patients present with signs and symptoms of cerebrospinal fluid obstruction, leading to increased intracranial pressure and hydrocephalus. Infants may have increased head circumference, bulging fontanelles, separate sutures, strabismus, vomiting or delayed development. Older children and adults may present with headaches, nausea, vomiting, lethargy, seizures, neurologic deficits or behavioral changes.11 Choroid plexus carcinomas can metastasize to the spine, therefore imaging of the entire neuraxis and CSF cytology are highly recommended. Approximately two thirds of the tumors disseminate throughout the entire CSF space. Extraneural metastases may also occur.10
Treatment options
Surgery
Surgery is the standard first line therapy for all choroid plexus carcinomas. Gross total resection is the most important predictor of outcome.12-18 However, it is only achievable in 40-50% of cases. A number of factors contribute to the difficulty in obtaining a complete resection, including young patient age, large tumor size and vascularity. Operating on choroid plexus carcinomas carries a high risk of intraoperative hemorrhage. Pre-operative angiography, often combined with embolization, is done to help facilitate a complete resection. In addition, pre-operative chemotherapy has also been used to reduce tumor size and vasculature.11,19,20 A retrospective analysis by Wrede et al. demonstrated that in patients with an initial subtotal resection, undergoing a second resection was associated with improved survival, compared with those who did not (overall survival 69% vs 30% at 2 years). This study underscores the importance of surgery in the overall management of this disease.14
Chemotherapy
The role of adjuvant chemotherapy in choroid plexus carcinoma is controversial. It is often used in patients with a subtotal resection and in infants who are too young to receive radiation. Wrede et al. conducted a meta-analysis that included 347 choroid plexus carcinomas reported in the literature prior to 2005.12 The use of chemotherapy was associated with improved survival in all patients (P=0.0004) and among those with incompletely resected tumors (55% vs 24% at 2 years). Multivariate analysis identified the use of chemotherapy as a significant prognostic factor (P=0.001).
Radiation therapy
The role of adjuvant radiotherapy has been studied in several retrospective series.12-18 Adjuvant radiotherapy is recommended in children and adults after a subtotal resection. Radiation is generally avoided in infants and children younger than 2-3 years.
In patients with a complete tumor resection, the role of adjuvant radiotherapy is controversial. Wolff et al. reported a cohort of 48 patients with gross total resection, half of whom received radiation. The 5 year OS was significantly higher in irradiated patients, compared with those without radiation (68% vs 14%).16 On the other hand, Fitzpatrick reported that in a cohort of 37 patients with a gross total resection, four patients did not receive adjuvant therapy, whereas the remaining 34 patients received either chemotherapy, radiation or both. The addition of chemotherapy and/or radiation after surgery did not seem to improve survival. However, the size and characteristics of the two groups (GTR with and without adjuvant therapy) were too imbalanced to draw any statistically meaningful conclusions.13 A recent SEER analysis (in abstract form) showed that radiation did not impact survival in patients with complete resection, suggesting a gross total resection may be sufficient in a subset of patients.21
The optimal field of radiation is also controversial. Given the high propensity for CSF seeding, some investigators have advocated for prophylactic cranial spinal irradiation without positive CSF cytology or MRI findings in the spine, whereas others use involved field radiation. A retrospective analysis of radiation fields by Mazloom et al. showed patients who received cranial spinal irradiation had improved survival compared to those who did not, 44% vs 15% at 5 years.17 Additional studies are needed to examine the patterns of failure and need for prophylactic cranial spinal irradiation.
Outcome
Five-year overall survival is about 40-50%. Gross total resection is the most consistent predictor of survival.
Ganglioglioma
Epidemiology
Gangliogliomas account for 0.4% of the all CNS tumors, 1.3% of brain tumors.22 The most commonly affected areas are the temporal lobe and the cerebellum. The least affected area is the spinal cord. When the spine is involved, the cervical spine is the most common site.23 Up to 80% of cases occur during the first three decades of life. There is a slight preponderance in males.24
Histopathology and molecular features
As its name suggests, gangliogliomas consist of a mixed population of ganglion and glial cells. Grading of gangliogliomas has typically been based on the characteristics of the glial component.22 In the current WHO classification, gangliogliomas are considered WHO grade I.25 Anaplastic gangliogliomas are considered WHO grade III and accounts for 5% of gangliogliomas. Malignant transformation has been reported in about 10% of cases. The transformation is mostly attributed to the changes of the glial component.26 The rate of transformation among spinal gangliogliomas is unknown.
Imaging characteristics
Computed tomography is often performed during initial work-up, showing hypodense, or mixed hypodense/isodense, solid or partially cystic masses. Calcifications are seen in 10-50% of cases.27 On MRI, tumors are usually isointense or hypointense on T1-weighted images and hyperintense on T2-weighted images. Contrast enhancement is seen and may be nodular, rim-like or entirely solid. Cysts are seen in about 50% of the cases and may be associated with mural nodules.22,28
Clinical presentation
For spine gangiogliomas, the most common presenting symptoms are back pain and extremity weakness.23 For brain gangliogliomas, the most common presenting symptom is chronic seizures, followed by neuroophthalmological findings, elevated ICP, and focal deficits.27 Gangliogliomas are highly epileptogenic.
Treatment options
Complete surgical resection is the treatment of choice, and is the single most important predictor of tumor control.
There is no clear benefit of adjuvant radiotherapy after gross total resection. For those with subtotal resection or biopsy, the role of radiation and/or chemotherapy is controversial. In a retrospective review from University of California San Francisco (UCSF), the recurrence rate after subtotal resection was three times higher in those with highly anaplastic features, compared to those with moderate anaplastic features, suggesting a role for adjuvant treatment in this setting.27 Because gangliogliomas predominantly affect young individuals, the benefit of radiation must be carefully weighed against long term toxicities of radiation, especially its impact on neuro-cognition.29,30 In addition, some have expressed concern that malignant degeneration can occur after radiation for low grade gangliogliomas.31 Adjuvant radiation after a subtotal resection should be reserved only for those with a high risk of recurrence. For recurrent disease, surgical resection is still the first line therapy. Adjuvant radiation may be considered, especially in those with anaplastic transformation. Chemotherapy may be considered as well, although the agent of choice or the optimal duration is unclear.27,32-34
Clinical outcome
Low grade gangliogliomas have favorable prognosis with 5 year overall survival over 90%.31 In a University of California Los Angeles (UCLA) series of 34 patients, including 25 low grade and 9 high grade tumors, progression free survival was 67% at 4 years, 77% after gross total resection and 51% after subtotal resection. Median time to progression was 14 months. Overall survival was 75%. Age, gender, tumor size, symptom duration, tumor grade, calcification, or enhancement had no impact on OS or PFS.28
In a SEER analysis of 58 patients with anaplastic ganglioglioma, overall survival was 63% at 5 years and the median overall survival was 28.5 months. Uni-focal disease and surgery predicted better outcome. Adjuvant radiation was not associated with improved survival.24
Hemangiopericytoma
Epidemiology
Hemangiopericytomas represent <1 % of all primary central nervous system tumors. Its incidence is about 1/50 of that of meningioma. The median age of onset is in the 40s. There is a slight male predominance.35
Histopathology and molecular features
Hemangiopericytoma was first described as a variant of meningioma, hence the term angioblastic meningioma. In 1993, it became recognized as a distinct entity by the World Health Organization grading system. It is now commonly classified as WHO grade II, with an anaplastic variant classified as WHO grade III.35,36 Grossly, hemangiopericytomas are solitary and well circumscribed tumors.37 They often have grossly visible vascular structures. Brain invasion is seen in some.38 Histologically, hemangiopericytomas are rare, highly vascularized mesenchymal tumors. They develop from malignant transformation of pericytes, which are contractile spindle cells surrounding the walls of capillaries and post-capillary venules.35 Morphologically, hemangiopericytomas are remarkably similar to meningioma. To distinguish the two tumors, a number of immunohistochemical and genetic markers have been developed. They include EMA, claudin-1, 1p, 14q, NF2, and 4.1B.39 EMA and claudin-1 staining are strong and diffuse in meningiomas, but weak and patchy in hemangiopericytomas. Deletions such as 14q, 1p, NF2, and protein 4.1B are typical of meningiomas, but are rare in hemangiopericytomas.
Imaging characteristics
On CT, hemangiopericytomas are well demarcated, contrast enhancing lesions with dural attachment, often found in the parasagittal sinus and falcine regions. Angiography reveals extensive vasculature, often with dual cortical and meningeal supplies. On MRI, these tumors are iso- to hyperintense on T1 and iso to hypointense on T2-weighted images. They show bright and often heterogeneous enhancement with gadolinium. Imaging differentials include meningioma and hemangioblastoma. In contrast to meningioma, hemangiopericytomas have a more narrow-based attachment to dura. Hemangioblastomas are more frequently infratentorial.38
Clinical presentation
Clinical presentation is largely dependent on lesion location. Patients may present with headache, seizures, focal weakness, visual changes, gait disturbances, or cognitive changes. Tumors infrequently involve the spine. In contrast to most primary brain tumors, hemangiopericytomas have a high propensity for extracranial metastasis. Common sites include bone, lung, liver and abdominal cavity.38 The incidence of metastases has been reported to be as high as 65% at 15 years.40 Therefore, it is important to perform staging work-up at the time of initial diagnosis and at recurrence.
Treatment
Surgery is the first line therapy for the treatment of hemangiopericytoma. Whenever feasible, gross total resection should be pursued. Resectability is largely dependent on tumor location. Because of the extreme vascular nature of the tumor, a major cause of operative mortality is uncontrolled bleeding.37 Reported rates of complete resection range from 30-70%.35,36,40-42 Greater extent of resection has been associated with improved survival.35 With surgery alone, local recurrence rate has been reported to be as high as 90%, and relapse can occur as early as 7 months after the initial treatment.35,43 Several retrospective series have shown that radiation plays an important role in preventing local recurrence. Both conventionally fractionated external beam radiotherapy and radiosurgery have been used in the post-operative setting.36,37,44,45
A radiation dose response for hemangiopericytoma has been reported. In a retrospective review from the University of Pittsburgh, all patients received adjuvant stereotactic radio-surgery. 19% of the lesions treated with a marginal dose ≥14 Gy developed an in-field recurrence/progression, whereas 50% of the lesions receiving <14 Gy progressed. In multivariate analysis, a marginal dose ≥14 Gy was significantly associated with improved progression free survival (P=0.02).44 Similarly, in external beam series, Bastin et al. reported 14% local recurrence rate for patients receiving more than 50 Gy, and 78% for those receiving less than 45 Gy.37 Mayo Clinic reported no recurrence in patients treated with 51-56 Gy, and 88% in those with <45 Gy.37,43 Based on this literature, a typical regimen is to deliver adjuvant radiation to 50-60 Gy in conventional fractionation to the operative bed and residual tumor. Figure 1 shows a case example. Radiosurgery can be used for small target volumes at a dose of 15 Gy or higher.
Figure 1.
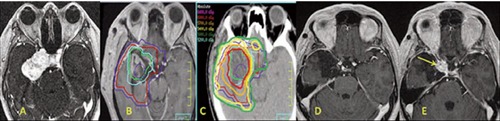
A 27-year-old patient was diagnosed with an atypical hemangiopericytoma and baseline magnetic resonance imaging (MRI) in A). Subtotal resection was performed. The post-operative radiation planning MRI is shown in B), GTV outlined as surgical bed (green), a 0.5 cm expansion on the GTV to create the PTV 60 (sky blue), a 1.5 cm CTV applied to the GTV to give a CTV 54 (red) and a 0.5 cm PTV margin applied to the CTV 54 to give a PTV 54 (blue). This patient was treated with a two phase technique to a total of 60 Gy in 30 fractions and isodose lines shown in C). The patient’s tumor was controlled for 5 years with clean images as shown in D). At 5 years, a recurrent tumor nodule developed at the region of the cavernous sinus edge (yellow arrow) and this is where relative under-dosing occurred to keep the optic chiasm to a point maximum dose of 54 Gy. The patient is now on bevacizumab.
The importance of radiation therapy in preventing local recurrence has been well established. It has also been noted that radiation does not reduce the rate of distant metastasis. Whether radiation therapy can improve survival is unclear. Stessin et al. conducted a SEER analysis of 76 patients. They found that radiation was associated with improved cause-specific survival and, in particular, in those who underwent subtotal resection.41
The role of chemotherapy in hemangiopericytoma is mostly limited to recurrent, surgery and radiation refractory tumors. Several regimens have been reported with modest efficacy, including cyclophosphamide, doxorubicin, and vincristine (CAV), alpha-interferon, ifosfamide, cisplatin and etoposide.46
Clinical outcome
Median survival for patients with intracranial hemangiopericytoma is 13 years. Ten yr OS and DFS are 60-70% and 20-30%, respectively. Even after aggressive upfront treatment, about 50% of patients develop recurrence. Median time to recurrence is about 5 years. Radiation is recommended after both gross total and subtotal resection.
Pleomorphic xanthoastrocytoma
Epidemiology
Pleomorphic xanthoastrocytomas (PXA) comprise of <1% of primary brain tumors. It was first described by Kepes et al. in 1979. It was incorporated into the WHO classification in 1993.47-49 PXA is a tumor of children and young adults, with a median age of diagnosis of 20.5 years. It most commonly involves the temporal lobe. The tumor occurs equally in males and females.
Histopathology and molecular features
Pleomorphic xanthoastrocytomas is composed of astrocytes with pleomorphic nuclei and multinucleated giant cells. Historically, these tumors were classified as giant cell glioblastomas due to its morphology. However, patients with PXAs have much longer disease free survival and overall survival, compared to those with glioblastomas. It typically presents as WHO grade II tumors. Transformation into Grade III and IV disease can occur in 15-20% of cases.49,50 BRAFV600E mutation and homozygous deletion of CDKN2A (p16) have been reported in PXA. Their roles in tumorigenesis have not been clearly defined.51
Imaging characteristics
On CT, PXA appears hypodense and enhances with contrast. On T1-weighted MRI, PXA can have variable intensity and variable degree of enhancement with gadolinium. It is typically hyperintense on T2. The most typical appearance of a PXA is a partially calcified, cystic or solid lesion in the superficial temporal cortex involving the leptomeninges. A key feature differentiating PXA from pilocystic astrocytoma is the minimal or no enhancement of the cyst wall. Radiologic differential diagnoses include meningioma, dysembryonic neuroectodermal tumor (DNET), ganglioglioma, and pilocystic astrocytoma.52
Clinical presentation
The most common presenting symptom is seizure, followed by headaches, nausea, vomiting consistent with increased intracranial pressure, or focal neurologic deficits.
Treatment options
Maximal safe surgical resection is the first line therapy for all patients. Greater extent of surgical resection has consistently been associated with improved survival.48,49,53 After gross total resection, adjuvant therapy is generally not recommended. Local recurrence can occur in 15-20% of cases after surgical resection. Anaplastic transformation has been reported at the time of recurrence.51
The role of adjuvant chemotherapy and radiation is not well established in PXA. Pahapill suggested a possible benefit of radiation in tumors without necrosis.53 McCaulay et al. reported no difference in overall survival among those treated or not treated with radiation, but the incidence of recurrence was slightly higher among those not receiving radiation (42% vs 30%).54 In a SEER analysis by Perkins et al., the use of radiation was associated with worse survival. However, a significantly higher percentage (55%) of grade III and IV tumors received radiation, whereas only 10% of patients with low grade tumors received radiation. Since their analysis did not include grade as a variable due to missing information among subjects, interpretation of this result is difficult. While there is no clear benefit of radiation, it is often used for grade III-IV tumor, recurrent disease or after subtotal resection.48
Clinical outcome
In a SEER analysis of 214 patients, five and ten year overall survival rates were 75% and 67%, respectively. For patients with WHO grade IV tumors, median survival was 45 months. Male gender, increasing age, high grade and incomplete surgical resection were associated with worse prognosis. PXA patients had worse survival than those with pilocystic astrocytoma and oligodendroglioma, but better outcomes compared to ependymoma and glioblastoma patients.48
Conclusions
Given the rarity of these primary central nervous system tumors, it is highly unlikely that large scale prospective trials evaluating different treatment approaches will be completed within a reasonable timeframe. Currently, one has to resort to small or fair-sized retrospective studies to gain knowledge of the clinical, pathologic, and therapeutic aspects of these tumors to guide clinical practice. Collaborative research through rare tumor network (http://www.rarecancer.net/), whichcan potentially collect information from a large number of patients from multiple cancer centers or population databases, may be the best means to enhance better understanding of the behaviors of these rare central nervous system tumors and to determine the best treatment strategies.
References
- 1.Ostrom QT, Gittleman H, Farah P, et al. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro Oncol 2013;15:1-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ginn KF, Gajjar A.Atypical teratoid rhabdoid tumor: current therapy and future directions. Front Oncol 2012;2:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rorke LB, Packer R, Biegel J.Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. J Neurooncol 1995;24:21-8 [DOI] [PubMed] [Google Scholar]
- 4.Adesina AM, Hunter J, Rorke-Adams L.Atypical teratoid rhabdoid tumor. Adesina AM, Tihan T, Fuller C, Young Poussaint T, Atlas of pediatric brain tumors. New York: Springer Publication; 2010. pp 95-107 [Google Scholar]
- 5.Packer RJ, Biegel JA, Blaney S, et al. , Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol 2002;24:337-42 [DOI] [PubMed] [Google Scholar]
- 6.Tekautz TM, Fuller CE, Blaney S, et al. Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 2005;2:1491-9 [DOI] [PubMed] [Google Scholar]
- 7.Hilden JM, Meerbaum S, Burger P, et al. , Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 2004;22:2877-84 [DOI] [PubMed] [Google Scholar]
- 8.Chi SN, Zimmerman MA, Yao X, et al. Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 2009;27:385-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buscariollo DL, Park HS, Roberts KB, Yu JB.Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a surveillance, epidemiology, and end results analysis. Cancer 2012;118:4212-9 [DOI] [PubMed] [Google Scholar]
- 10.Fuller CE, Narendra S, Tolocica I.Choroid plexus neoplasm. Adesina AM, Tihan T, Fuller C, Young Poussaint T, Atlas of pediatric brain tumors. New York: Springer Publication; 2010. pp 269-279 [Google Scholar]
- 11.Gopal P, Parker JR, Debski R, Parker JC., Jr.Choroid plexus carcinoma. Arch Pathol Lab Med 2008;132:1350-4 [DOI] [PubMed] [Google Scholar]
- 12.Wrede B, Liu P, Wolff JE.Chemotherapy improves the survival of patients with choroid plexus carcinoma: a meta-analysis of individual cases with choroid plexus tumors. J Neurooncol 2007;85:345-51 [DOI] [PubMed] [Google Scholar]
- 13.Fitzpatrick LK, Aronson LJ, Cohen KJ.Is there a requirement for adjuvant therapy for choroid plexus carcinoma that has been completely resected? J Neurooncol 2002;57:123-6 [DOI] [PubMed] [Google Scholar]
- 14.Wrede B, Liu P, Ater J, Wolff JE.Second surgery and the prognosis of choroid plexus carcinoma--results of a meta-analysis of individual cases. Anticancer Res 2005;25:4429-33 [PubMed] [Google Scholar]
- 15.Wolff JE, Sajedi M, Brant R, et al. Choroid plexus tumours. Br J Cancer 2002;87:1086-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wolff JE, Sajedi M, Coppes MJ, et al. , Radiation therapy and survival in choroid plexus carcinoma. Lancet 1999;353:2126. [DOI] [PubMed] [Google Scholar]
- 17.Mazloom A, Wolff JE, Paulino AC.The impact of radiotherapy fields in the treatment of patients with choroid plexus carcinoma. Int J Radiat Oncol Biol Phys 2010;78:79-84 [DOI] [PubMed] [Google Scholar]
- 18.Chow E, Reardon DA, Shah AB, et al. Pediatric choroid plexus neoplasms. Int J Radiat Oncol Biol Phys 1999;44:249-54 [DOI] [PubMed] [Google Scholar]
- 19.Souweidane MM, Johnson JH, Jr, Lis E.Volumetric reduction of a choroid plexus carcinoma using preoperative chemotherapy. J Neurooncol 1999;43:167-71 [DOI] [PubMed] [Google Scholar]
- 20.Razzaq AA, Cohen AR.Neoadjuvant chemotherapy for hypervascular malignant brain tumors of childhood. Pediatr Neurosurg 1997;27:296-303 [DOI] [PubMed] [Google Scholar]
- 21.Cannon DM, Mohindra P, Gondi V, et al. Impact of radiotherapy and extent of resection in choroid plexus carcinoma: an analysis of the Surveillance Epidemiology and End Results (SEER) Database. Int J Radiat Oncol Biol Phys 2012;84:S303 [Google Scholar]
- 22.Gupta N, Banerjee A, Haas-Kogan D, Pediatric CNS Tumors. 2nd ed Berlin: Springer Velrag; 2009 [Google Scholar]
- 23.Lotfinia I, Vahedi P.Intramedullary cervical spinal cord ganglioglioma, review of the literature and therapeutic controversies. Spinal Cord 2009;47:87-90 [DOI] [PubMed] [Google Scholar]
- 24.Selvanathan SK, Hammouche S, Salminen HJ, Jenkinson MD.Outcome and prognostic features in anaplastic ganglioglioma: analysis of cases from the SEER database. J Neurooncol 2011;105:539-45 [DOI] [PubMed] [Google Scholar]
- 25.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007;114:97-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamburger CA, Buttner A, Weis S.Ganglioglioma of the spinal cord: report of two rare cases and review of the literature. Neurosurgery 1997;41:1410-6 ; [DOI] [PubMed] [Google Scholar]
- 27.Krouwer HG, Davis RL, McDermott MW, et al. Gangliogliomas: a clinicopathological study of 25 cases and review of the literature. J Neurooncol 1993;17:139-54 [DOI] [PubMed] [Google Scholar]
- 28.Selch MT, Goy BW, Lee SP, et al. Gangliogliomas: experience with 34 patients and review of the literature. Am J Clin Oncol 1998;21:557-64 [DOI] [PubMed] [Google Scholar]
- 29.Hall WA, Yunis EJ, Albright AL.Anaplastic ganglioglioma in an infant: case report and review of the literature. Neurosurgery 1986;19:1016-20 [DOI] [PubMed] [Google Scholar]
- 30.Johnson JH, Jr, Hariharan S, Berman J, et al. Clinical outcome of pediatric gangliogliomas: ninety-nine cases over 20 years. Pediatr Neurosurg 1997;27:203-7 [DOI] [PubMed] [Google Scholar]
- 31.Luyken C, Blümcke I, Fimmers R, et al. Supratentorial gangliogliomas: histopathologic grading and tumor recurrence in 184 patients with a median follow-up of 8 years. Cancer 2004;101:146-55 [DOI] [PubMed] [Google Scholar]
- 32.DeMarchi R, Abu-Abed S, Munoz D, Loch Macdonald R.Malignant ganglioglioma: case report and review of literature. J Neurooncol 2011;101:311-8 [DOI] [PubMed] [Google Scholar]
- 33.Majores M, von Lehe M, Fassunke J, et al. Tumor recurrence and malignant progression of gangliogliomas. Cancer 2008;113:3355-63 [DOI] [PubMed] [Google Scholar]
- 34.Liauw SL, Byer JE, Yachnis AT, et al. Radiotherapy after subtotally resected or recurrent ganglioglioma. Int J Radiat Oncol Biol Phys 2007;67:244-7 [DOI] [PubMed] [Google Scholar]
- 35.Rutkowski MJ, Jian BJ, Bloch O, et al. Intracranial hemangiopericytoma: clinical experience and treatment considerations in a modern series of 40 adult patients. Cancer 2012;118:1628-36 [DOI] [PubMed] [Google Scholar]
- 36.Schiariti M, Goetz P, El-Maghraby H, et al. Hemangiopericytoma: long-term outcome revisited. Clinical article. J Neurosurg 2011;114:747-55 [DOI] [PubMed] [Google Scholar]
- 37.Bastin KT, Mehta MP.Meningeal hemangiopericytoma: defining the role for radiation therapy. J Neurooncol 1992;14:277-87 [DOI] [PubMed] [Google Scholar]
- 38.Fuller CE.Hemangiopericytoma and other mesenchymal tumors. Adesina AM, Tihan T, Fuller C, Young Poussaint T, Atlas of pediatric brain tumors. New York: Springer Publication; 2010. pp 125-132 [Google Scholar]
- 39.Rajaram V, Brat DJ, Perry A.Anaplastic meningioma versus meningeal hemangiopericytoma: immunohistochemical and genetic markers. Hum Pathol 2004;35: 1413-8 [DOI] [PubMed] [Google Scholar]
- 40.Galanis E, Buckner JC, Scheithauer BW, et al. Management of recurrent meningeal hemangiopericytoma. Cancer 1998;82: 1915-20 [PubMed] [Google Scholar]
- 41.Stessin AM, Sison C, Nieto J, et al. The role of postoperative radiation therapy in the treatment of meningeal hemangiopericytoma-experience from the SEER database. Int J Radiat Oncol Biol Phys 2013; 85:784-90 [DOI] [PubMed] [Google Scholar]
- 42.Rutkowski MJ, Sughrue ME, Kane AJ, et al. Predictors of mortality following treatment of intracranial hemangiopericytoma. J Neurosurg 2010;113:333-9 [DOI] [PubMed] [Google Scholar]
- 43.Guthrie BL, Ebersold MJ, Scheithauer BW, Shaw EG.Meningeal hemangiopericytoma: histopathological features, treatment, and long-term follow-up of 44 cases. Neurosurgery 1989;25:514-22 [PubMed] [Google Scholar]
- 44.Kano H, Niranjan A, Kondziolka D, et al. Adjuvant stereotactic radiosurgery after resection of intracranial hemangiopericytomas. Int J Radiat Oncol Biol Phys 2008;72:1333-9 [DOI] [PubMed] [Google Scholar]
- 45.Olson C, Yen CP, Schlesinger D, Sheehan J.Radiosurgery for intracranial hemangiopericytomas: outcomes after initial and repeat Gamma Knife surgery. J Neurosurg 2010;112:133-9 [DOI] [PubMed] [Google Scholar]
- 46.Chamberlain MC, Glantz MJ.Sequential salvage chemotherapy for recurrent intracranial hemangiopericytoma. Neurosurgery 2008;63:720-7 [DOI] [PubMed] [Google Scholar]
- 47.Kepes JJ, Rubinstein LJ, Eng FE.Pleomorphic xanthoastrocytoma: a distinctive meningocerebral glioma of young subjects with relatively favorable prognosis. A study of 12 cases. Cancer 1979;44:1839-52 [DOI] [PubMed] [Google Scholar]
- 48.Perkins SM, Mitra N, Fei W, Shinohara ET, Patterns of care and outcomes of patients with pleomorphic xanthoastrocytoma: a SEER analysis. J Neurooncol 2012;110:99-104 [DOI] [PubMed] [Google Scholar]
- 49.Giannini C, Scheithauer BW, Burger PC, et al. Pleomorphic xanthoastrocytoma: what do we really know about it? Cancer 1999;85:2033-45 [PubMed] [Google Scholar]
- 50.Nakajima T, Kumabe T, Shamoto H, et al. Malignant transformation of pleomorphic xanthoastrocytoma. Acta Neurochir (Wien) 2006;148:67-71 [DOI] [PubMed] [Google Scholar]
- 51.Merrel R, Norden AD.Uncommon gliomas in adults: brainstem, gliomas, pilocytic astrocytomas, and pleomorphic xanthoastrocytomas. Norden AD, Reardon DA, Wen PCY, Primary central nervous system tumors. New York: Humana Press; 2011. pp. 263-282 [Google Scholar]
- 52.Fuller CE.Pleomorphic xanthoastrocytoma. Adesina AM, Tihan T, Fuller C, Young Poussaint T, Atlas of pediatric brain tumors. New York: Springer Publication; 2010. pp 19-24 [Google Scholar]
- 53.Pahapill PA, Ramsay DA, Del Maestro DF.Pleomorphic xanthoastrocytoma: case report and analysis of the literature concerning the efficacy of resection and the significance of necrosis. Neurosurgery 1996;38:822-9 [PubMed] [Google Scholar]
- 54.Macaulay RJ, Jay V, Hoffman HJ, Becker LE.Increased mitotic activity as a negative prognostic indicator in pleomorphic xanthoastrocytoma. Case report. J Neurosurg 1993;79: 761-8 [DOI] [PubMed] [Google Scholar]