

Abstract
OBJECTIVE
Given the high rates of cardiovascular disease (CVD) and associated mortality in individuals with type 2 diabetes, identifying and understanding predictors of CVD events and mortality could help inform clinical management in this high-risk group. Recent large-scale genetic studies may provide additional tools in this regard.
RESEARCH DESIGN AND METHODS
Genetic risk scores (GRSs) were constructed in 1,175 self-identified European American (EA) individuals comprising the family-based Diabetes Heart Study based on 1) 13 single nucleotide polymorphisms (SNPs) and 2) 30 SNPs with previously documented associations with CVD in genome-wide association studies. Associations between each GRS and a self-reported history of CVD, coronary artery calcified plaque (CAC) determined by noncontrast computed tomography scan, all-cause mortality, and CVD mortality were examined using marginal models with generalized estimating equations and Cox proportional hazards models.
RESULTS
The weighted 13-SNP GRS was associated with prior CVD (odds ratio [OR] 1.51 [95% CI 1.22–1.86]; P = 0.0002), CAC (β-coefficient [β] 0.22 [0.02–0.43]; P = 0.04) and CVD mortality (hazard ratio [HR] 1.35 [1.10–1.81]; P = 0.04) when adjusting for the other known CVD risk factors: age, sex, type 2 diabetes affection status, BMI, current smoking status, hypertension, and dyslipidemia. The weighted 30-SNP GRS was also associated with prior CVD (OR 1.33 [1.08–1.65]; P = 0.008), CAC (β 0.29 [0.08–0.50]; P = 0.006), all-cause mortality (HR 1.28 [1.05–1.56]; P = 0.01), and CVD mortality (HR 1.46 [1.08–1.96]; P = 0.01).
CONCLUSIONS
These findings support the utility of two simple GRSs in examining genetic associations for adverse outcomes in EAs with type 2 diabetes.
Introduction
As the major cause of mortality in Western industrialized countries, cardiovascular disease (CVD) accounts for ∼35% of all-cause mortality (1). A greater burden of CVD is seen in individuals with type 2 diabetes mellitus (T2DM), who experience a twofold increased risk for coronary artery disease compared with nondiabetic individuals and in whom up to 65% of all-cause mortality is attributed to CVD (1,2). Identifying predictors of CVD events and mortality in high-risk populations with T2DM would aid in risk stratification and improve clinical management.
Recently, attempts have been made to use genetic information in the assessment of whether cumulative genetic risk conferred across multiple loci is a more robust tool for examining disease risk than associations of single nucleotide polymorphisms (SNPs). This approach has been applied in the context of the prediction of CVD events among individuals with T2DM using risk scores generated from SNPs associated with T2DM risk (3,4). A recent genome-wide association study (GWAS) (5) in smokers found that a genetic risk score (GRS) constructed from 24 replicated CVD-associated SNPs was associated with vascular calcification. In addition, Thanassoulis et al. (6), constructed a 13-SNP GRS, also using a subset of GWAS-significant CVD-associated SNPs, which was associated with both incident CVD and coronary artery calcified plaque (CAC) in the Framingham Heart Study. Given that CAC is an established predictor of both CVD events (7,8) and mortality (9,10), and considering the excess of CVD risk factors in individuals with T2DM, we explored whether the genetic associations reported by Thanassoulis et al. (6) were also applicable in a T2DM-enriched population with high risk for CVD. We extended the 13-SNP model reported by Thanassoulis et al. (6) by examining additional CVD-associated SNPs and also tested for associations with all-cause and CVD mortality.
Research Design and Methods
This investigation included 1,220 self-identified European American (EA) individuals from 475 families enrolled in the Diabetes Heart Study (DHS) cohort. Briefly, the DHS includes siblings concordant for T2DM, but without advanced renal insufficiency. When possible, unaffected siblings of T2DM-affected individuals were also recruited. T2DM was clinically defined as diabetes developing after the age of 35 years and treated with insulin and/or oral agents, in the absence of historical evidence of ketoacidosis. Diagnoses were confirmed by baseline measurement of fasting blood glucose and glycosylated hemoglobin A1c. Ascertainment and recruitment have been previously described in detail (11,12).
Study protocols were approved by the Institutional Review Board at Wake Forest School of Medicine, and all participants provided written informed consent. Participant examinations were conducted in the General Clinical Research Center of the Wake Forest Baptist Medical Center, and the examinations included interviews for medical history and health behaviors, anthropometric measures, resting blood pressure, electrocardiography, and fasting blood sampling for laboratory analyses. Subclinical CVD was assessed by measurement of CAC using fast-gated helical computed tomography scanners, with calcium scores calculated as previously described (13,14). Not all measurements were available for all participants. Individuals self-reported a history of prior CVD based on prior events (e.g., angina, myocardial infarction, stroke) and/or interventions (e.g., coronary angioplasty/stenting, coronary artery bypass grafting, carotid endarterectomy). Individuals were classified as hypertensive if they had been prescribed antihypertensive medication or if blood pressure measurements exceeded 140 mmHg (systolic) or 90 mmHg (diastolic); and as dyslipidemic based on the criteria established in the Third Report of the National Cholesterol Education Program Expert Panel Detection, Evaluation and Treatment in Adults (15).
Vital Status
Vital status was determined for all participants from the National Social Security Death Index maintained by the U.S. Social Security Administration. For participants confirmed as deceased, length of follow-up was determined from the date of the initial study visit to the date of death. For all other participants, the length of follow-up was determined from the date of the initial study visit to the end of 2011. For deceased participants, copies of death certificates were obtained from relevant county vital records offices to determine cause of death. Cause of death was categorized based on information contained in death certificates as CVD-related (e.g., myocardial infarction, congestive heart failure, cardiac arrhythmia, sudden cardiac death, peripheral vascular disease, and stroke) or cancer, infection, end-stage renal disease, accidental, or other (including obstructive pulmonary disease, pulmonary fibrosis, liver failure, and Alzheimer dementia).
Genetic Analysis
Total genomic DNA was purified from whole-blood samples using the PUREGENE DNA isolation kit (Gentra, Inc., Minneapolis, MN). DNA concentration was quantified using standardized fluorometric readings on a DyNA Quant 200 fluorometer (Hoefer Pharmacia Biotech Inc., San Francisco, CA). Genotyping was completed in two stages, as follows: 1) using the Affymetrix (Santa Clara, CA) Genome-Wide Human SNP Array 5.0 (GWAS); and 2) using the Illumina (San Diego, CA) Infinium Human Exome BeadChip version 1.0 (Exome).
For the GWAS, genotype calling was completed using the BRLLM-P algorithm in Genotyping Console version 4.0 (Affymetrix). Samples failing to meet an intensity quality control threshold (n = 4) were not included for genotype calling, and those failing to meet a minimum acceptable call rate of 95% (n = 3) were excluded from further analyses. For the Exome, genotype calling was completed using Genome Studio Software version 1.9.4 (Illumina). Samples failing to meet a minimum acceptable call rate of 98% (n = 3) were excluded from further analyses. Blind duplicate samples were included in both GWAS (n = 39) and Exome (n = 58) with average concordance rates of 99.0 ± 0.72% (mean ± SD) and 99.9 ± 0.0001%, respectively.
After genotype calling, samples identified with poor-quality genotype calls, sex errors, or unclear/unexpected sibling relationships were excluded from further analysis; a total of 1,175 samples with available genotype data in both the GWAS and Exome were included in subsequent analysis. The following additional exclusion criteria based on SNP performance were included for the GWAS: call rate <95% (n = 11,085), Hardy-Weinberg equilibrium P value <1 × 10−6 (n = 332), and minor allele frequency <0.01 (n = 57,382); and for the Exome: call rate <99% (n = 972), monomorphic SNPs (n = 157,754), and Hardy-Weinberg equilibrium P value <1 × 10−6 (n = 26).
For SNPs where direct genotyping data were unavailable, genotype data were obtained from GWAS imputed data. Imputation of 1,000 Genome Project SNPs was completed using the program IMPUTE2 (http://mathgen.stats.ox.ac.uk/impute/impute_v2.html) and Phase I version 2, cosmopolitan (integrated) reference panel, build 37 (16). SNPs used for imputation were required to have low levels of missingness and to show no significant departure from Hardy-Weinberg expectations. To maximize the quality of imputation, the samples were not prephased. Only imputed SNPs with a confidence score >0.90 and an information score >0.50 were used.
GRS Calculation
A 13 SNP GRS (GRS13) was calculated to replicate the previously established 13-SNP score reported by Thanassoulis et al. (6). The score was constructed initially as an unweighted score, calculated by adding the number of risk alleles across each SNP. Considering the range in the previously reported effect sizes (odds ratio [OR] 1.07–1.92) for each SNP (6), a weighted score was also constructed in which each risk allele was weighted by the previously documented effect size. Details for SNPs included in the GRS13 have been provided in Supplementary Table 1A and B. A 30-SNP GRS (GRS30) was also constructed based on the inclusion of an additional 17 SNPs with compelling and/or replicated associations with CVD in other published genetic association studies (Supplementary Table 1A and B) constructed in the same way; again, both unweighted and weighted scores were derived.
For individuals missing genotype data for a particular SNP, the mean risk allele count in the DHS for that specific SNP was assigned; such an approach has been suggested previously by Fontaine-Bisson et al. (17). The GRSs were used to ascribe individuals to one of three genetic risk groups (low, moderate, or high) based on the number of risk alleles. Given the potentially narrow distribution of GRS13, the low- and high-risk groups were designed to capture extremes of the risk score distribution (∼20%) to allow for assessment of the impacts of very high risk allele loads on disease risk. Groupings differed slightly for GRS13 and GRS30, and for unweighted and weighted GRSs based on the underlying distributions of risk allele load.
Statistical Analysis
Continuous outcomes (i.e., CAC) were log-transformed prior to analysis to approximate conditional normality. Initial analysis included examining the association between both the unweighted and weighted GRSs and outcomes of interest to determine the extent of association per unit increase in the GRS. GRSs were then considered as an ordinal variable (based on ascribed risk groups: low, moderate, and high). Exploratory tests for trend across increasing GRS groups were performed to examine the association between both the unweighted and weighted GRSs and history of CVD and CAC using marginal models. Generalized estimating equations were used to account for familial correlation. Cox proportional hazards models with sandwich-based variance estimation were used to examine the association between the GRS and all-cause and CVD mortality. All models were then partially adjusted (age, sex, and T2DM-affected status) and fully adjusted (age, sex, T2DM-affected status, BMI, hypertension, dyslipidemia, and current smoking) for other known CVD risk factors. To further quantify risk across the ascribed GRS groups, risk for outcome (prior CVD, CAC, all-cause mortality, and CVD mortality) was determined for the moderate- and high-risk groups relative to the low-risk group. These analyses were also completed by restricting participants to T2DM-affected individuals only.
Receiver operating characteristic curves were computed for models containing traditional CVD risk factors (as used in the fully adjusted models cited above) and with the addition of either 13 SNP or 30 SNP genetic risk groups. Area under the curve (AUC) analysis was performed to compare the ability of various models to predict outcome. The net reclassification improvement (NRI) was also determined to measure the degree to which risk for prior CVD, all-cause mortality, or CVD mortality was reclassified using the fully adjusted models with the addition of the genetic risk groups. The percentage of the sample reclassified (either into higher of lower risk groups) was reported.
All analyses were performed in SAS version 9.3 (SAS Institute, Cary, NC). Statistical significance was accepted at P < 0.05.
Results
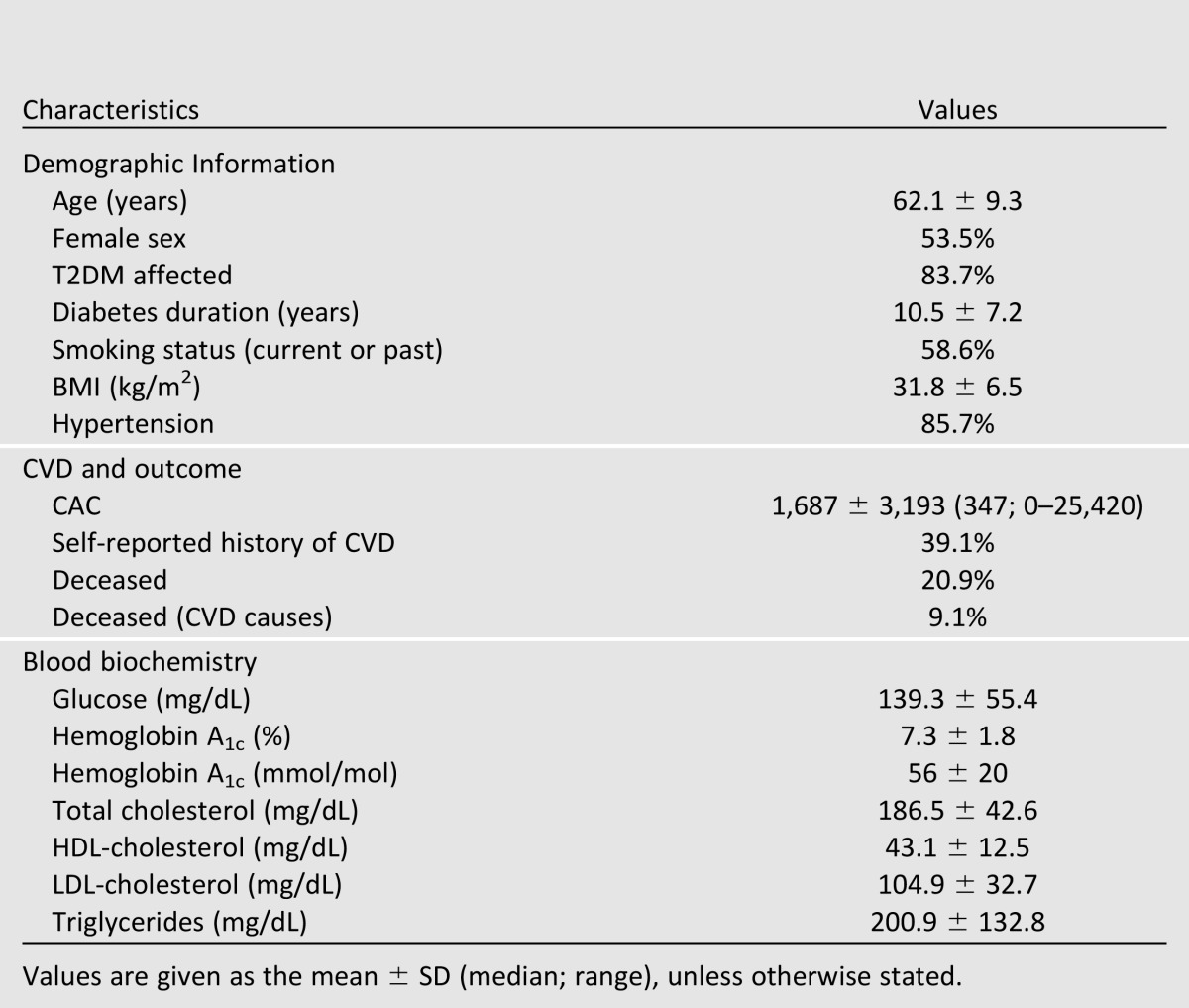
The demographic and clinical characteristics of the DHS cohort are presented in Table 1. As anticipated in a T2DM-enriched sample, a predominance of traditional CVD risk factors, including high BMI, hypertension, dyslipidemia, and prior CVD events, was evident. This cohort was observed for a mean (± SD) time of 8.3 ± 2.6 years, and over that time 246 participants (20.9%) died, 107 (9.1%) from CVD causes.
Table 1.
Demographic characteristics of the 1,175 DHS participants with available genotype data
As anticipated, the distributions of unweighted and weighted scores differed slightly for both GRS13 and GRS30. The unweighted GRS13 ranged from 5 to 20 risk alleles (12.6 ± 2.1, mean ± SD), and the rounded weighted GRS13 ranged from 4 to 17 risk alleles (10.0 ± 1.8). The unweighted GRS30 ranged from 17 to 40 risk alleles (29.8 ± 3.5), and the rounded weighted GRS30 ranged from 16 to 39 risk alleles (27.9 ± 3.5). Association results from preliminary analysis indicated small (4–11%), but significant (0.003 < P < 0.05), increases in risk for adverse outcomes with each unit increase in both GRS13 and GRS30 (Supplementary Table 1).
Ascribed risk groupings for the unweighted GRS13 were low (≤11 risk alleles, 28.9% of the sample), moderate (12–14 risk alleles, 52.6% of the sample), and high (≥15 risk alleles, 18.6% of the sample). The ascribed risk groupings for the weighted GRS13 were low (≤8 risk alleles, 18.4% of the sample), moderate (9–11 risk alleles, 62.2% of the sample), and high (≥12 risk alleles, 19.3% of the sample). The ascribed risk groupings for the unweighted GRS30 were low (≤26 risk alleles, 16.4% of the sample), moderate (27–33 risk alleles, 70% of the sample), and high (≥34 risk alleles, 13.5% of the sample). The ascribed risk groupings for the weighted GRS30 were low (≤24 risk alleles, 16.4% of the sample), moderate (25–30 risk alleles, 60.2% of the sample), and high (≥31 risk alleles 23.5% of the sample).
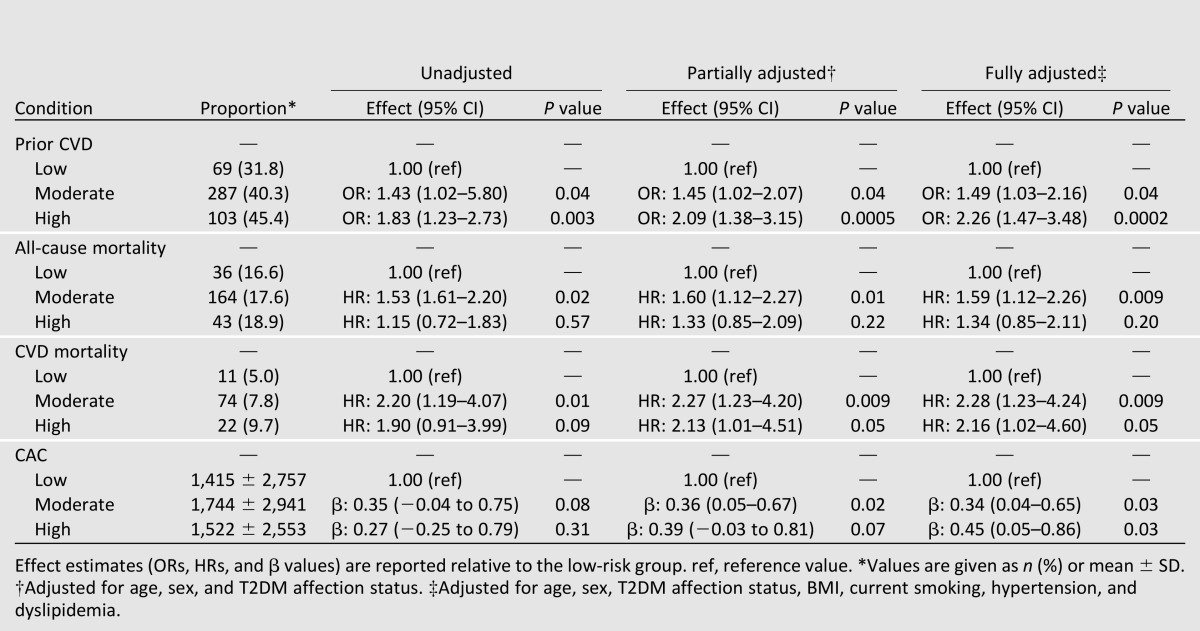
When considered as an ordinal measure, both the unweighted and weighted GRS13 were associated with a history of CVD in the DHS, both prior to and after adjustment for other known CVD risk factors (Table 2). There were no clear associations between the GRS13 and all-cause mortality; however, significant associations with CVD mortality and CAC were evident when using the weighted GRS13 (Table 2). Given the range in the previously reported effect sizes for this set of SNPs (OR 1.08–1.92) (Supplementary Table 1A and B) risk for outcome between the GRS13 groups was further quantified using the weighted GRS13 (results for the unweighted GRS13 are reported in Supplementary Table 1). Relative to the low-GRS13 group, individuals in the moderate-GRS13 group were ∼1.5 times more likely, and individuals in the high-GRS13 group were greater than 2 times more likely, to have a history of CVD when accounting for other known CVD risk factors (Table 3). Interestingly, a progressive increase in risk for either CVD mortality or CAC was not observed between moderate- and high-GRS13 groups (Table 3). Relative to the low-GRS13 group, the moderate- and high-GRS13 groups were at ∼2.2-fold greater risk for CVD mortality; however, hazard ratios (HRs) were not significantly different between the two groups (P = 0.93 from the z statistic, accounting for the covariance between the two regression coefficients); for the moderate- and high-GRS13 groups the combined HR was 2.25 (95% CI 1.22–4.17; P = 0.01). Similarly, associations with CAC were not significantly different between the moderate- and high-GRS13 groups (P = 0.97); for the moderate- and high-GRS13 groups, the combined β-coefficient (β) was 0.36 (95% CI 0.07–0.66; P = 0.02).
Table 2.
Association of ordinal measure (low, moderate, high) of GRS13 and GRS30 with history of CVD, all-cause mortality, CVD mortality, and CAC in EAs (n = 1,175) from the DHS
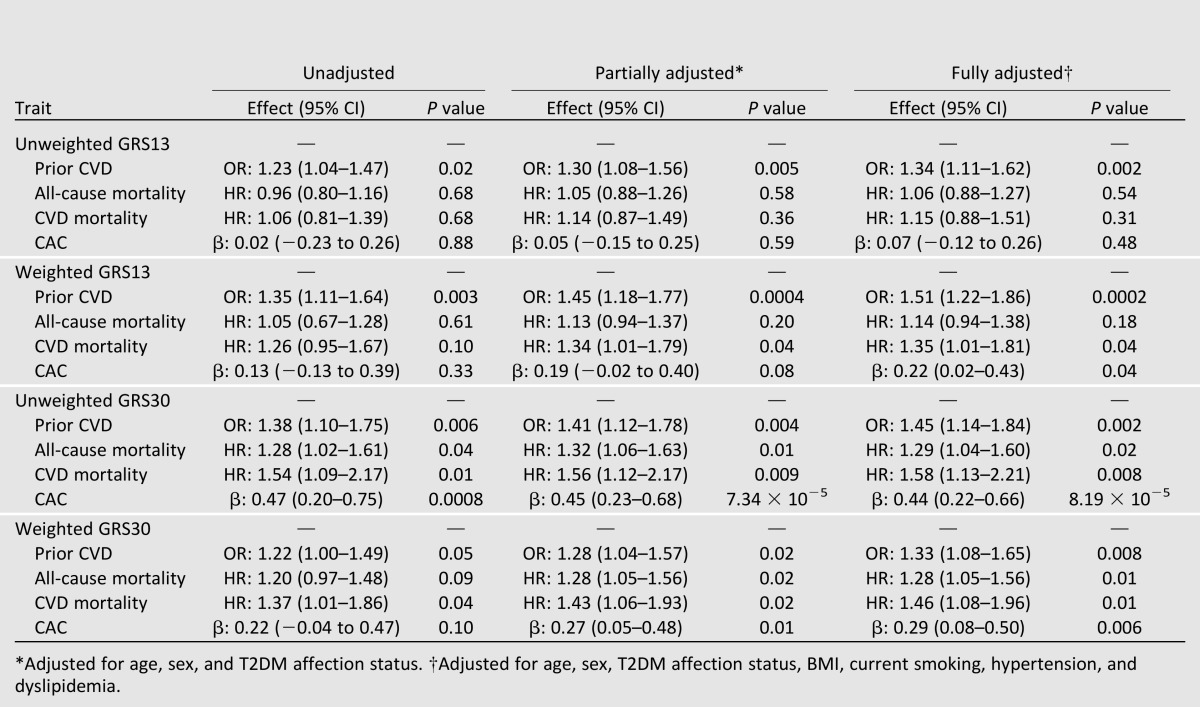
Table 3.
Association between weighted GRS13 low (n = 217), moderate (n = 713), and high (n = 227) groups and history of CVD, all-cause mortality, CVD mortality, and CAC in EAs from the DHS
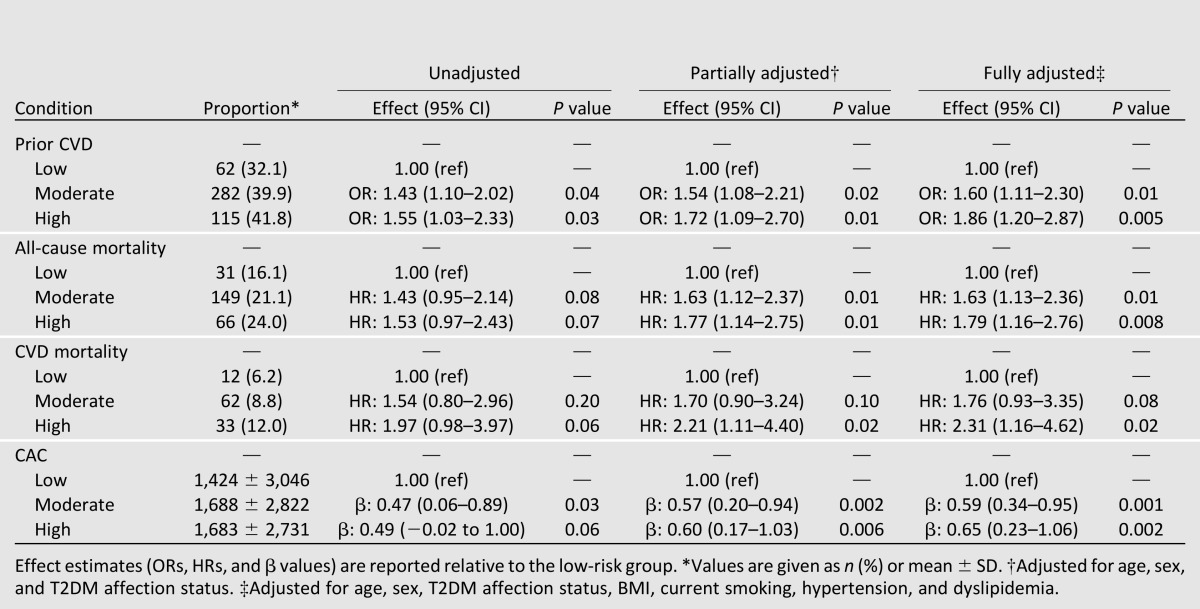
When considered as an ordinal measure, both the unweighted and weighted GRS30 were associated with a history of CVD in the DHS both prior to and after adjustment for other known CVD risk factors (Table 2). The GRS30 was also significantly associated with all-cause mortality, CVD mortality, and CAC (Table 2). Again, to account for the different effect sizes of this set of SNPs (OR 1.06–1.92) (Supplementary Table 1A and B), risk for outcome between the GRS30 groups was further quantified using the weighted GRS (results for the unweighted model are reported in Supplementary Table 1). Relative to the low-GRS30 group, individuals in the moderate-GRS30 group were ∼1.6 times more likely, and individuals in the high-GRS30 group were 1.8 times more likely, to have a history of CVD when accounting for other known CVD risk factors (Table 4). Similarly, the risk for all-cause mortality was 1.8 times greater and the risk for CVD mortality was 2.3 times greater in the high-GRS30 group (Table 4). Finally, associations with CAC were also significant in both the moderate- and high-GRS30 groups (Table 4). Results were essentially unchanged when analyses were repeated, restricting to the T2DM-affected individuals only (Supplementary Tables 5–8).
Table 4.
Association between weighted GRS30 low (n = 193), moderate (n = 823), and high (n = 159) groups and history of CVD, all-cause mortality, CVD mortality, and CAC in EAs from the DHS
The area under the curve for the prediction of outcome was 0.71 for prior CVD, 0.73 for all-cause mortality, and 0.69 for CVD mortality using fully adjusted models. AUC analysis indicated that the addition of the various GRS risk groups to fully adjusted models did not substantially improve model prediction (ΔAUC for prior CVD = 0.004–0.006; ΔAUC all-cause mortality = 0.001–0.005; ΔAUC CVD mortality = −0.006 to 0.014). Broadly speaking, the NRI index also suggested that the addition of the GRS risk groups did not substantially improve risk prediction (Supplementary Table 1). While the NRI indicated that between 4 and 20% of the sample was reclassified, depending on the model, it was only when predicting prior CVD using the 13-SNP unweighted risk group (NRI index 0.041; P = 0.04), and mortality using both the 13-SNP weighted (NRI index 0.040; P = 0.03) and the 30-SNP weighted risk groups (NRI index 0.055; P = 0.02) that there were significant improvements in prediction.
Conclusions
The current study explored the utility of two GRSs, based on previously reported CVD-associated SNPs, in predicting subclinical CVD, prevalent CVD, all-cause mortality, and CVD mortality in the DHS, a study enriched for individuals with T2DM. Associations of the GRS13 with prevalent CVD, CAC, and CVD mortality were evident when SNPs were weighted based on previously documented effects on CVD risk. In addition, the GRS30 was also significantly associated with all-cause mortality. These findings extend the report by Thanassoulis et al. (6) in the Framingham Heart Study and demonstrate that relatively simple GRS models are effective tools for considering genetic risk for CVD in EAs with T2DM.
Given that the selection of the SNPs included in both the GRS13 and GRS30 was based on prior associations with CVD from the GWAS, it is not surprising that the GRS strongly replicated associations with prevalent CVD. Indeed, the SNPs included in both GRSs are some of the most well-characterized CVD risk SNPs and have been associated with incident CVD in a range of prior studies (18–23); the verification of results from large meta-analyses in smaller community-based cohorts remains a critical step for successful translation of outcomes from large genetic studies. In the DHS, these SNPs do not show compelling associations with CAC, prevalent CVD, all-cause mortality, or CVD mortality in single SNP analyses (Supplementary Table 1A and B), and we hypothesize that the underlying metabolic disturbance and excess burden of CVD risk factors in our T2DM-enriched sample confound some of the underlying genetic risk. However, using a multiple-SNP GRS approach, we were able to replicate the associations with CVD reported by Thanassoulis et al. (6) supporting the use of multiple loci and a derived GRS when considering genetic risk for complex disease. Further, similar to the recent study by van Setten et al. (5) using a 24-SNP GRS, we also found that our GRS30 based on CVD-associated SNPs was associated with CAC. While here the use of multiple loci in a GRS was advantageous in the context of risk stratification, we acknowledge that such an approach may not be as beneficial if the objective is to identify and isolate functional mechanisms/pathways underpinning disease risk. Likewise, given the proximity of clinical biomarkers to outcome along the causal pathway, GRS approaches are unlikely to replace traditional risk factors for prediction of outcome in individuals with advanced disease, as was suggested by the AUC and NRI analysis performed here.
In contrast to the clear increase in risk for prevalent CVD observed across the GRS groups (both GRS13 and GRS30 models), the significant overall associations with CVD mortality and CAC (both GRS13 and GRS30 models), and the graded increased in risk for both CVD mortality and CAC across the GRS30 groups, the same graded increase in risk between the moderate- and high-GRS groups was not observed for the GRS13 model. For CVD mortality, increased mortality rates were observed across the GRS13 groups (5.0%, 7.8%, and 9.7%), and the threshold effect in the reported HR may simply be related to the sample size. In contrast, the average CAC score was actually slightly lower in the high-GRS13 group compared with the moderate-GRS13 group, which would explain why a further increase in the HR was not observed in the high-GRS13 group. The association of CVD variants with CAC has been recently reported by others (5), and it is possible that the greater number of SNPs in the GRS30 model accounts for a greater proportion of the phenotypic variance, which is why these associations were observed with the GRS30 model but not the GRS13 model. Indeed, the precision of the effect size estimates is also better for the GRS30 when examining the associations based on a per unit increase in the GRS, further supporting the likelihood that the greater number of SNPs accounts for a greater proportion of the phenotypic variance. Given that the SNPs were selected based on prior association with incident CVD and not subclinical CVD, the GRS30 may be preferable to the GRS13 when assessing associations with other CVD-related phenotypes.
In the current study, we chose to create GRS groups to more easily quantify risk for outcome among individuals at the ends of the GRS distribution; groupings were constructed to capture ∼20% of the sample in both the low-GRS and high-GRS groups. Differences in the composition of the risk groups between unweighted and weighted scores may be one explanation for the varying performance of unweighted and weighted models as the associations based on a per unit increase in the GRS are broadly similar between unweighted and weighted models. Likewise, grouping structures based on cut points other than the 20% that were used here may have produced different results. Further to this point, the GRS13 resulted in a narrower distribution compared with the GRS30, and as such it was more difficult to generate distinct GRS groups. While the GRS30 appears to perform more favorably based on the strength of reported associations, the report by Thanassoulis et al. (6) also evaluated a more extensive 102-SNP GRS and found that it performed no better than the GRS13 described here. Obviously, construction of a GRS based on a smaller number of SNPs is more practical if application to clinical or personalized medicine settings is favored; the GRS30 score constructed in the DHS may offer a feasible alternative from a practical standpoint. Finally, it is also worth acknowledging that in the current models SNPs were weighted based on effects in European-derived populations, and it remains unclear whether this score would translate to other ethnic groups.
These results extend the report by Thanassoulis et al. (6) in which a 13-SNP GRS was shown to be associated with incident CVD and CAC in the Framingham Heart Study. Here we observed that GRSs derived from 13 and 30 SNPs were associated with prevalent CVD and CAC in the T2DM-enriched DHS sample, and were able to extend these findings to also show association with all-cause and CVD mortality. Overall, these findings provide further evidence supporting the genetic underpinnings of adverse outcomes in high-risk individuals with T2DM.
Supplementary Material
Article Information
Acknowledgments. The authors thank the other investigators, the staff, and the participants of the Diabetes Heart Study for their valuable contributions.
Funding. This study was supported in part by National Institutes of Health grants R01-HL-67348, R01-HL-092301, and R01-NS-058700 (to D.W.B.), and General Clinical Research Center of the Wake Forest School of Medicine grants M01-RR-07122 and F32-HL-085989.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. A.J.C. collected data, performed statistical analysis, and wrote the manuscript. F.-C.H. contributed to the statistical analysis, and reviewed and edited the manuscript. M.C.Y.N. contributed to the management of the genetic data and reviewed the manuscript. C.D.L. contributed to the management of the genetic data and reviewed the manuscript. B.I.F. was involved in the initial design of the Diabetes Heart Study, contributed to patient ascertainment and clinical evaluation, and reviewed and edited the manuscript. J.J.C. was involved in the initial design of the Diabetes Heart Study, contributed to patient ascertainment and clinical evaluation, and reviewed the manuscript. D.W.B. leads the Diabetes Heart Study, and reviewed and edited the manuscript. D.W.B. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://care.diabetesjournals.org/lookup/suppl/doi:10.2337/dc13-1514/-/DC1.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 2012;125:e2–e220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention Diabetes Data & Trends. Washington, D.C., Centers for Disease Control and Prevention, Department of Health and Human Services, 2009 [Google Scholar]
- 3.Qi Q, Meigs JB, Rexrode KM, Hu FB, Qi L. Diabetes genetic predisposition score and cardiovascular complications among patients with type 2 diabetes. Diabetes Care 2013;36:737–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pfister R, Barnes D, Luben RN, Khaw KT, Wareham NJ, Langenberg C. Individual and cumulative effect of type 2 diabetes genetic susceptibility variants on risk of coronary heart disease. Diabetologia 2011;54:2283–2287 [DOI] [PubMed] [Google Scholar]
- 5.van Setten J, Isgum I, Smolonska J, et al. Genome-wide association study of coronary and aortic calcification implicates risk loci for coronary artery disease and myocardial infarction. Atherosclerosis 2013;228:400–405 [DOI] [PubMed] [Google Scholar]
- 6.Thanassoulis G, Peloso GM, Pencina MJ, et al. A genetic risk score is associated with incident cardiovascular disease and coronary artery calcium: the Framingham Heart Study. Circ Cardiovasc Genet 2012;5:113–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Detrano R, Guerci AD, Carr JJ, et al. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med 2008;358:1336–1345 [DOI] [PubMed] [Google Scholar]
- 8.Folsom AR, Kronmal RA, Detrano RC, et al. Coronary artery calcification compared with carotid intima-media thickness in the prediction of cardiovascular disease incidence: the Multi-Ethnic Study of Atherosclerosis (MESA). Arch Intern Med 2008;168:1333–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agarwal S, Cox AJ, Herrington DM, et al. Coronary calcium score predicts cardiovascular mortality in diabetes: Diabetes Heart Study. Diabetes Care 2013;36:972–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agarwal S, Morgan T, Herrington DM, et al. Coronary calcium score and prediction of all-cause mortality in diabetes: the Diabetes Heart Study. Diabetes Care 2011;34:1219–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bowden DW, Cox AJ, Freedman BI, et al. Review of the Diabetes Heart Study (DHS) family of studies: a comprehensively examined sample for genetic and epidemiological studies of type 2 diabetes and its complications. Rev Diabet Stud 2010;7:188–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bowden DW, Lehtinen AB, Ziegler JT, et al. Genetic epidemiology of subclinical cardiovascular disease in the Diabetes Heart Study. Ann Hum Genet 2008;72:598–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carr JJ, Nelson JC, Wong ND, et al. Calcified coronary artery plaque measurement with cardiac CT in population-based studies: standardized protocol of Multi-Ethnic Study of Atherosclerosis (MESA) and Coronary Artery Risk Development in Young Adults (CARDIA) study. Radiology 2005;234:35–43 [DOI] [PubMed] [Google Scholar]
- 14.Carr JJ, Crouse JR, 3rd, Goff DC, Jr, D’Agostino RB, Jr, Peterson NP, Burke GL. Evaluation of subsecond gated helical CT for quantification of coronary artery calcium and comparison with electron beam CT. AJR Am J Roentgenol 2000;174:915–921 [DOI] [PubMed] [Google Scholar]
- 15.Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of igh blood cholesterol in adults (Adult Treatment Panel III). JAMA 2001;285:2486–2497 [DOI] [PubMed] [Google Scholar]
- 16.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fontaine-Bisson B, Renström F, Rolandsson O, et al. MAGIC Evaluating the discriminative power of multi-trait genetic risk scores for type 2 diabetes in a northern Swedish population. Diabetologia 2010;53:2155–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erdmann J, Grosshennig A, Braund PS, et al. Italian Atherosclerosis, Thrombosis, and Vascular Biology Working Group. Myocardial Infarction Genetics Consortium. Wellcome Trust Case Control Consortium. Cardiogenics Consortium New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet 2009;41:280–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kathiresan S, Voight BF, Purcell S, et al. Myocardial Infarction Genetics Consortium. Wellcome Trust Case Control Consortium Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants [published correction appears in Nat Genet 2009;41:762]. Nat Genet 2009;41:334–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke R, Peden JF, Hopewell JC, et al. PROCARDIS Consortium Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med 2009;361:2518–2528 [DOI] [PubMed] [Google Scholar]
- 21.Gudbjartsson DF, Bjornsdottir US, Halapi E, et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet 2009;41:342–347 [DOI] [PubMed] [Google Scholar]
- 22.Samani NJ, Erdmann J, Hall AS, et al. WTCCC and the Cardiogenics Consortium Genomewide association analysis of coronary artery disease. N Engl J Med 2007;357:443–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schunkert H, König IR, Kathiresan S, et al. Cardiogenics. CARDIoGRAM Consortium Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 2011;43:333–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.