

Abstract
Pyoverdine is a fluorescent nonribosomal peptide siderophore made by fluorescent pseudomonads. The Pseudomonas aeruginosa nonribosomal peptide synthetase (NRPS) PvdD contains two modules that each incorporate an l-threonine residue at the C-terminal end of pyoverdine. In an attempt to generate modified pyoverdine peptides, we substituted alternative-substrate-specifying adenylation (A) and peptide bond-catalyzing condensation (C) domains into the second module of PvdD. When just the A domain was substituted, the resulting strains produced only wild-type pyoverdine—at high levels if the introduced A domain specified threonine or at trace levels otherwise. The high levels of pyoverdine synthesis observed whenever the introduced A domain specified threonine indicated that these nonnative A domains were able to communicate effectively with the PvdD C domain. Moreover, the unexpected observation that non-threonine-specifying A domains nevertheless incorporated threonine into pyoverdine suggests that the native PvdD C domain exhibited stronger selectivity than these A domains for the incorporated amino acid substrate (i.e., misactivation of a threonine residue by the introduced A domains was more frequent than misincorporation of a nonthreonine residue by the PvdD C domain). In contrast, substitution of both the C and A domains of PvdD generated high yields of rationally modified pyoverdines in two instances, these pyoverdines having either a lysine or a serine residue in place of the terminal threonine. However, C-A domain substitution more commonly yielded a truncated peptide product, likely due to stalling of synthesis on a nonfunctional recombinant NRPS template.
INTRODUCTION
Nonribosomal peptide synthetases (NRPS) are microbial enzymes that can assemble diverse products from a pool of over 500 possible amino acid substrates (1). According to the most recent published report, there are 1,164 different nonribosomal peptides currently known (2), many of which are medically and industrially relevant (e.g., antibiotics, immunosuppressants, and anticancer agents) (3). The diversity of products is possible due to a modular enzymatic assembly line-like mechanism wherein, at least in linear syntheses, each NRPS module selects and incorporates a specific substrate into the final product.
With their complex structures often precluding effective in vitro synthesis, many clinical derivatives of natural products are created by semisynthesis, a process whereby the natural product is chemically modified after isolation from a biological source (4–6). However, the modular nature of NRPS enzymes offers enticing prospects for targeted modification of products at a biosynthetic level by altering the substrate specificity of individual modules. This could allow the creation of altered scaffolds for drug development or open up the possibility to create de novo NRPS pathways.
During NRPS-mediated peptide synthesis, discrete adenylation (A), thiolation (T), and condensation (C) domains act in substrate activation, transfer, and condensation, respectively (Fig. 1). Additional accessory domains can be present to alter monomers, such as an epimerization (E) domain that can be present between the T and C domains of adjacent modules and that catalyzes epimerization of the donor substrate to a D isomer prior to condensation by the C domain (7). The final product is typically released by a thioesterase (TE) domain associated with the terminal module in the NRPS complex.
FIG 1.
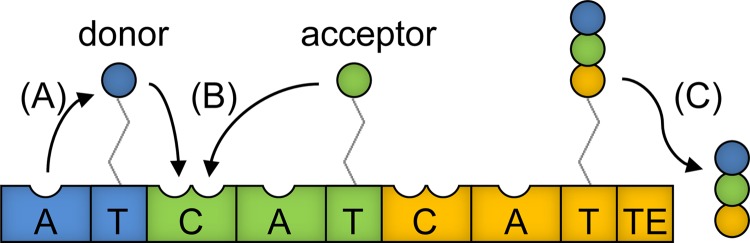
Domain arrangement within a hypothetical three-module NRPS comprised of an initiation (blue), an elongation (green), and a termination (orange) module. Numerous elongation modules may be present, with each incorporating an additional residue into the peptide product. Each module is activated by posttranslational attachment of a 4′-phosphopantetheine (4′-pp) cofactor to the T domain (also referred to as a peptidyl carrier protein [PCP] domain). (A) The first domain to act within each module is an A domain, which activates and tethers a specific monomer via a thioester bond to the 4′-pp prosthetic group of the immediately downstream T domain. (B) Starting at the first elongation module, the C domain condenses the upstream donor substrate onto the downstream acceptor substrate. This reaction breaks the upstream thioester bond, resulting in the newly formed peptide being attached to the downstream T domain, and this peptide serves as the donor substrate for the C domain of the next module. (C) In this way, the growing peptide chain is passed down the modules, and following addition of a final monomer by the termination module, the peptide product is released by a TE domain. Detailed analyses of the structures and functions of individual NRPS domains are provided in several reviews (3, 37–39).
Previous efforts to generate novel peptide products in vivo by NRPS manipulation have involved either (i) replacement of the A domain by one that activates an alternative substrate, (ii) alteration of the substrate binding pocket of the A domain, or (iii) substitutions that treat C-A domains as inseparable pairs (8). However, in nearly all cases, these manipulations have resulted in little to no product being made. In vitro observations have indicated that A domain manipulation strategies may be limited by the upstream C domain exhibiting specificity for the acceptor substrate (Fig. 1B) (9, 10), and it has also been inferred from structural studies that A domain substitutions may disrupt an essential C-A domain interface (11). C-A domain substitutions avoid both of these issues and yet have also enjoyed very little success.
This work uses the enzyme PvdD as a model to better understand constraints to NRPS domain substitution in vivo. PvdD from Pseudomonas aeruginosa PAO1 is a bimodular NRPS that incorporates the final two threonine residues into pyoverdine (12), a product synthesized from four NRPS enzymes comprising 12 modules (Fig. 2A). Pyoverdine consists of a conserved chromophore attached to a variable peptide chain (13) (Fig. 2C). The variable peptide chains of pyoverdines are highly diverse between different strains of fluorescent pseudomonads (14), an observation that provided this work with a range of evolutionarily related modules for domain substitutions (Fig. 2A and B). Other major advantages of this model system are that the pyoverdine chromophore is easily detectable in vivo due to characteristic absorbance and fluorescence and that, as a siderophore, an absolute requirement for pyoverdine synthesis can be imposed by growing the P. aeruginosa host strain in an iron-restricted environment.
FIG 2.

NRPS enzymes, labeled from A to I, with modules used for domain substitutions highlighted. (A) Domain architecture of the four NRPS enzymes involved in P. aeruginosa PAO1 pyoverdine synthesis, based on previous annotation (15). (B) Domain architecture of the NRPS enzymes involved in synthesis of the pyoverdine variable peptide chains from three other Pseudomonas strains. An additional NRPS, PvdL (as shown in panel A), is conserved in each strain and incorporates the residues that later form the chromophore. The figure is based on previous annotation by Owen and Ackerley (40) (P. syringae 1448a), Moon et al. (41) (P. fluorescens SBW25), and Ravel and Cornelis (42; adapted with permission of the publisher) (P. putida KT2440). (C) Structure of pyoverdine from P. aeruginosa PAO1 (43, 44) with the succinimide group highlighted in pink, the chromophore in green, the peptide backbone in red, and the side chains of the threonine residues incorporated by PvdD in blue. Dab, 2,4-diaminobutyric acid; hfOrn, l-N5-formyl-N5-hydroxyornithine; hOrn, N5-hydroxyornithine.
Previous domain substitution experiments in the first module of pvdD found that, consistent with A domain substitutions being limited by C domain acceptor site specificity, A domain substitutions were successful when the introduced A domain specified threonine but not otherwise (15). In contrast, C-A domain substitutions were all nonfunctional (15). However, the previous work had several limitations, including the facts that the sample size of substituted domains was small, the substituted domains were of both eukaryotic and prokaryotic origin, and the substitutions were introduced into the first module of pvdD. With regard to the last point, because this module must interact with the upstream NRPS enzyme in trans, it is unknown whether critical interactions between PvdJ and PvdD may have been disrupted by the C-A domain substitution. Another hypothesis for the lack of activity observed with C-A domain substitutions was that C domains might also exhibit some level of donor site specificity, so that the introduced C domain could prove incompatible with the incoming residue supplied by the upstream NRPS module, or that a newly incorporated amino acid substrate might not be recognized at the donor site of the C domain immediately downstream (15).
In this study, we used instead the second module of PvdD for domain substitution. This approach avoided the possibility that C-A domain substitution in the first module might disrupt critical interactions between PvdD and PvdJ. Moreover, targeting the final module of the NRPS template meant that introduction of a novel amino acid into a growing peptide chain could not cause donor site incompatibility with the C domain immediately downstream. By conducting a large number of carefully selected C-A domain substitutions and analyzing their abilities to generate a pyoverdine product relative to the corresponding A domain substitutions, we were able not only to create rationally modified pyoverdines with a high yield, but also to gain insight into factors that restrict the functionality of NRPS enzymes with domains substituted in vivo.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Escherichia coli DH5α, P. aeruginosa PAO1, and Pseudomonas putida KT2440 were sourced from existing Ackerley laboratory stocks. Pseudomonas syringae pv. phaseolicola 1448A was generously provided by John Mansfield (Imperial College, London, United Kingdom) and Pseudomonas fluorescens SBW25 by Paul Rainey (Massey University, Auckland, New Zealand). All strains were grown in LB medium with shaking at 200 rpm at 28°C for P. syringae pv. phaseolicola 1448A and 37°C for the other strains. For maintenance of plasmids, tetracycline was added to a final concentration of 15 μg/ml for E. coli and 100 μg/ml for P. aeruginosa PAO1.
DNA methods.
The PCR primers and plasmids used in this study are shown in Tables 1 and 2, respectively. The percent amino acid identity and similarity shared between each introduced A domain and the module 2 A domain of PvdD is listed in Table 3. Primers were designed to amplify pyoverdine NRPS domains from the genomes of fluorescent pseudomonads based on the annotation detailed in Fig. 2, as well as the polyketide synthase (PKS)/NRPS analysis tools available online (http://nrps.igs.umaryland.edu/nrps/; 16). PCRs used Phusion DNA polymerase (Finnzymes, Espoo, Finland). Plasmids were constructed from pSW196 (17) in E. coli DH5α and then transformed into a previously characterized pvdD deletion mutant (15) for single-copy integration at the attB locus (18).
TABLE 1.
Oligonucleotide primers used in this study
| Function | Name | Sequence (5′→3′)b |
|---|---|---|
| Primers for pSMC | CATfwd | CCCCGAATTCCATATGCAAGCACTCATAGAGAAGGTG |
| CATrev | CCCCActagTCAATCCCTGGGCGAACGC | |
| TTEFwd | CGCCCAGGGATTGTCTAGAGCGGCCGCGTCGCAGCAGGCCTATCGAGCGC | |
| TTERev | CCCCGAGCTCTCAGCGCCCGGCACGCTCCAGG | |
| Primers for pSMA | Thr-WT_CAfwd | GGGGTCTAGAACGACGGATGCGGTCTCGACGA |
| CATCrev | ATAGGCCTGCTGCGACGCGGCCGCTCTAGAATCAGCGCAACCGCCTGT | |
| Forward primers for C-A domain substitutions | Thr-A_CAfwd | CCCCTCTAGAGGTGTCAATCTCTTCGAG |
| Ser-B_CAfwd | CCCTCTAGAGCGGCGAGCGCTGCCCCTGTGCT | |
| Lys-C_CAfwd | CCCTCTAGATTCGCCCGCCTGCCGATCCCGC | |
| Thr-D_CAfwda | CCCCTCTAGAACGACGGATGCGGTCTCGACGATACCGCTTGCCGATCGGCAGGAAGATATCTACCCGCTGTCGC | |
| Thr-E_CAfwd | CCCCTCTAGAAATCTCTACGGGGTCACACCGA | |
| Ser-F_CAfwd | CCCCTCTAGAGGGCAGGGCAATGCTGCG | |
| Asp-G_CAfwd | CCCTCTAGAGATTTCGCGCTGTTGCCGATCGCG | |
| Gly-H_CAfwd | CCTCTAGAAACCTCTACGGGGTGACGCGCAT | |
| Ser-I_CAfwd | CCCTCTAGAGACTTTTCCCGGTTTCCGATTCC | |
| Forward primers for A domain substitutions | Thr-WT_Afwd | CCCCTCTAGACGAACAACGGTTGAGCTA |
| Thr-A_Afwd | CCCCTCTAGACGAACAACGGTTGAGCTA | |
| Ser-B_Afwd | CCCCTCTAGAGGAACGCCTGGACTACGCCGAG | |
| Lys-C_Afwd | CCTCTAGAGGGGCAGGCCTTGAGCTACGCC | |
| Thr-D_Afwd | CCCCTCTAGATGGCGAGCAATTGAGATA | |
| Thr-E_Afwd | CCCCTCTAGATGGCGAGCAATTGAGCTA | |
| Ser-F_Afwd | CCCCTCTAGAGGCTGAACAACTGAGCTA | |
| Asp-G_Afwd | CCCTCTAGACGATGGCTCGCTCAGTTACGGC | |
| Gly-H_Afwd | CCCTCTAGACGAGCAGACCTTGAGCTACGCCG | |
| Ser-I_Afwd | CCCTCTAGATGCAACCACGCTGACCTACGCCC | |
| Reverse primers for CA domain and A domain substitutions | Thr-WT_Rev | CCCCGCGGCCGCATCCGGTTGCGGCAACGCCTGC |
| Thr-A_Rev | CCCCGCGGCCGCATCCGGTTGCGGCAACGCCTGC | |
| Ser-B_Rev | CCCCGCGGCCGCTTGCGGTCGCGGCAGCGCCTTG | |
| Lys-C_Rev | CCCCGCGGCCGCATCCGGCGCGGGCAGCGCCTTG | |
| Ser-F_Rev | CCCCGCGGCCGCGTCTGGTGTCGGCAGGGC | |
| Asp-G_Rev | CCCCCGCGGCCGCGTCCGGCAGCGGCAATCTGGCC | |
| Gly-H_Rev | CCCCGCGGCCGCGTCAGGCGCCGGCAGCGCCTTG | |
| Ser-I_Rev | CCCCGCGGCCGCATCCGGCAATGGCAAGGCCTTG |
Due to lower sequence identity of DCL domains (C domains that receive a d-amino acid at their donor site), a linker of 42 nucleotides was inserted to shift the recombination site further downstream.
Restriction sites are underlined.
TABLE 2.
Plasmids used in this study
| Plasmid | Brief description | Reference |
|---|---|---|
| pSW196 | Integration-proficient vector; PBAD promoter | 17 |
| pSMC | pSW196-based staging plasmid allowing the substitution of C-A domains into pvdD | This study |
| pSMC::ThrWT | pSMC with C-A domains from the second module of pvdD | This study |
| pSMC::CAThrA | pSMC with C-A domains from module A | This study |
| pSMC::CASerB | pSMC with C-A domains from module B | This study |
| pSMC::CALysC | pSMC with C-A domains from module C | This study |
| pSMC::CAThrD | pSMC with C-A domains from module D | This study |
| pSMC::CAThrE | pSMC with C-A domains from module E | This study |
| pSMC::CASerF | pSMC with C-A domains from module F | This study |
| pSMC::CAAspG | pSMC with C-A domains from module G | This study |
| pSMC::CAGlyH | pSMC with C-A domains from module H | This study |
| pSMC::CASerI | pSMC with C-A domains from module I | This study |
| pSMA | pSW196-based staging plasmid allowing the substitution of A domains into pvdD | This study |
| pSMA::ThrWT | pSMC with C domain from the second module of pvdD | This study |
| pSMA::ThrA | pSMA with A domain from module A | This study |
| pSMA::SerB | pSMA with A domain from module B | This study |
| pSMA::LysC | pSMA with A domain from module C | This study |
| pSMA::ThrD | pSMA with A domain from module D | This study |
| pSMA::ThrE | pSMA with A domain from module E | This study |
| pSMA::SerF | pSMA with A domain from module F | This study |
| pSMA::AspG | pSMA with A domain from module G | This study |
| pSMA::GlyH | pSMA with A domain from module H | This study |
| pSMA::SerI | pSMA with A domain from module I | This study |
TABLE 3.
Amino acid sequence homology of introduced A domains aligned against the module 2 PvdD A domain
| Substitutiona | % identity | % similarity | % gaps |
|---|---|---|---|
| Thr-A | 99.6 | 99.8 | 0.0 |
| Ser-B | 50.6 | 64.1 | 7.3 |
| Lys-C | 52.3 | 64.9 | 5.9 |
| Thr-D | 64.7 | 77.4 | 2.3 |
| Thr-E | 65.1 | 77.3 | 3.5 |
| Ser-F | 47.6 | 61.5 | 5.7 |
| Asp-G | 40.7 | 56.3 | 5.3 |
| Gly-H | 47.1 | 62.3 | 6.8 |
| Ser-I | 46.8 | 61.7 | 6.9 |
| Thr-snbC m1 | 53.2 | 64.5 | 4.5 |
| Thr-syrB m9 | 44.1 | 60.2 | 5.4 |
| Cys-acvA m2 | 36.2 | 54.0 | 11.8 |
| Ser-pvdI m1 | 51.4 | 64.8 | 5.7 |
| Val-acvA m3 | 36.4 | 54.7 | 10.8 |
Substitutions created in this study are aligned with the A domain from Thr-WT. Substitutions from previous work (15) are in boldface. The nomenclature for A domain substitutions from the previous work shows the substrate activated by the module followed by the gene the module was sourced from and the number of the module within the named gene (15).
To generate the substitution plasmid pSMC (Fig. 3B), the first module of pvdD was PCR amplified using primers CATfwd and CATrev and ligated into pSW196 (17); then, the T-TE domains from the second module of pvdD were amplified using primers TTEFwd and TTERev and inserted downstream of the first module. pSMC contained SpeI and NotI restriction sites between the C-A-T and T-TE domains to enable addition of C-A domains. To create pSMA (Fig. 3A), the C domain from the second module of pvdD was amplified using primers Thr-WT_CAfwd and CATCrev and ligated into the SpeI and NotI restriction sites of pSMC using compatible XbaI and NotI restriction sites added by the PCR primers. This destroyed the SpeI restriction site immediately upstream of the C domain so that a new SpeI site added by CATCrev would be downstream of the introduced C domain. A and C-A domains were then amplified from genomic DNA and ligated into the corresponding plasmids pSMA and pSMC using XbaI and NotI restriction sites. Plasmids created in this work were sequence verified by Macrogen Inc. (Seoul, South Korea).
FIG 3.

Domain arrangement within substitution plasmids pSMA (A) and pSMC (B) used in this work. The domains colored blue and red were derived from the first and second modules of pvdD, respectively. The domains in green were absent in the substitution plasmids and indicate where alternative domains would be added. X refers to a SpeI restriction site that was destroyed when ligating A and C-A domains into the plasmids via a compatible XbaI sticky end.
Measurement of pyoverdine production.
To detect pyoverdine production on solid media, strains were inoculated onto iron-limiting King's B agar plates (20 g/liter Bacto peptone, 1.5 g/liter K2HPO4, 1% [vol/vol] glycerol, 6.1 mM MgSO4, and 1.5% [wt/vol] agar) in both the presence and absence of the iron chelator ethylenediamine-N,N′-bis(2-hydroxyphenylacetic acid) (EDDHA; final concentration, 200 µg/ml). In the presence of EDDHA, mutants deficient in pyoverdine production are unable to grow due to their inability to passively take up iron (15). Plates were incubated for 24 h at 37°C, and then growth was assessed visually and the presence or absence of pyoverdine was determined by fluorescence under UV light.
To measure pyoverdine levels in liquid media, strains were grown in 200 μl of LB medium in a 96-well plate for 24 h at 37°C. This starter culture was used to inoculate M9 medium [6 g/liter K2HPO4, 3 g/liter KH2PO4, 1 g/liter (NH4)2SO4, and 6.1 mM MgSO4] containing 0.2% (wt/vol) l-arabinose and 4 g/liter succinate (pH 7.0) at a 20× dilution with a total volume of 200 μl. After 24 h of growth, the cultures were centrifuged to pellet bacteria, and then 100 μl of supernatant was transferred to a fresh 96-well plate. The supernatant was diluted 2 times in fresh M9 medium to give a total volume of 200 μl. Fluorescence (excitation, 400 nm; emission, 440 nm) and absorbance (400 nm) were measured using an EnSpire 2300 Multilabel Reader (PerkinElmer, Waltham, MA, USA).
Mass spectrometry.
Samples were mixed with matrix solution (500 μl acetonitrile, 500 μl ultrapure water, 1 μl trifluoroacetic acid, 10 μg α-cyano-4-hydroxycinnamic acid) in a volumetric sample-to-matrix ratio ranging from 1:5 to 1:20. Aliquots of 1 μl from each sample were spotted in duplicate onto an Opti-TOF 384-well matrix-assisted laser desorption ionization (MALDI) plate (Applied Biosystems, Foster City, CA) and allowed to dry at room temperature. The spots were analyzed using a MALDI-time of flight (TOF)/TOF 5800 mass spectrometer (Applied Biosystems) in positive-ion mode. Each spot was externally calibrated using cal2 calibration mixture (Applied Biosystems). Peaks in spectra were labeled in Data Explorer (Applied Biosystems).
RESULTS
Selection of alternative A and C-A domains for substitution experiments.
To create domain substitution variants of pvdD, A and C-A domains were sourced from the nine modules labeled A to I in Fig. 2A and B. These modules are involved in pyoverdine synthesis by four different fluorescent Pseudomonas species. The modules were selected to provide a range of activated substrates (threonine, serine, glycine, aspartate, and lysine), as well as modules that receive peptide chains with different C-terminal amino acids (arginine, N5-formyl-N5-hydroxyornithine, aspartate, threonine, and diaminobutyric acid) into the donor site of their C domains. All selected modules, with the exception of module D, receive an l-amino acid at the C domain donor site in their native contexts.
Generation and phenotypic evaluation of A domain substitution strains.
Based on previous results (15), we reasoned that substituted A domains might be functional in an autonomous sense but limited by C domain acceptor site specificity within the recombinant NRPS environment. To test this, A domain substitutions were created by individual PCR amplifications of each of the A domains highlighted in Fig. 2, followed by ligation into a module 2 A domain deletion construct of pvdD in the plasmid pSMA (a complementation plasmid that integrates into the P. aeruginosa chromosome using the integrase ϕCTX [18]) (Fig. 3A). Recombination junctions in pSMA were chosen based on the equivalent location having previously been shown to be permissive of A domain substitutions in the first module of PvdD (15). Once created, A domain substitution variants were transformed into a previously characterized pvdD deletion mutant (15). The resulting substitution strains are referred to in terms of the substrate specified by the new A domain followed by the letter of the module designated in Fig. 2; for example, the variant containing the A domain from the first module of pvdD is referred to as strain Thr-A. In addition to the A domain substitution variants, a positive-control strain (Thr-WT) complemented by the wild-type pvdD module 2 A domain (with introduced restriction sites as shown in Fig. 3A) was generated and demonstrated to synthesize pyoverdine at levels similar to those for wild-type P. aeruginosa PAO1. This indicated that the introduced restriction sites did not impair function and that complementation of pvdD by a single gene copy expressed from the genome-integrated pSMA plasmid did not reduce pyoverdine production, as had been seen in previous work using a high-copy-number complementation plasmid (15).
As initial qualitative tests for pyoverdine production, the Thr-WT positive control, nine A domain substitution strains, and a pyoverdine negative control (a pvdD deletion strain containing the empty pSW196 plasmid from which pSMA was derived) were streaked on iron-limiting King's B agar plates in both the presence and absence of EDDHA. The three threonine-specifying A domain substitution strains (Thr-A, Thr-D, and Thr-E) each exhibited increased levels of fluorescence compared to the pyoverdine negative control and were also viable in the presence of EDDHA (Fig. 4A). The strain Thr-E had substantially reduced fluorescence and viability relative to Thr-A, Thr-D, or Thr-WT, indicating reduced pyoverdine synthesis, but was still clearly viable. In contrast, none of the nonthreonine A domain substitution strains were viable in the presence of EDDHA and all appeared to have levels of fluorescence similar to that of the negative control. However, due to the subjective nature of this test, it was difficult to ascertain whether there might have been small increases in fluorescence for these strains.
FIG 4.

Detection of pyoverdine production by A domain substitution variants. (A) Fluorescence of A domain substitution strains on King's B agar plates in the absence (−) or presence (+) of EDDHA. The plates were inoculated with the positive-control strain Thr-WT (WT), A domain substitution strains A to I (Fig. 2), and the pvdD deletion pyoverdine negative control (Del) and then incubated for 24 h at 37°C. The photographs were taken under UV light. (B) Percentages of pyoverdine production from A domain substitution strains grown in liquid media. The values are expressed as percentages of absorbance (400 nm) or fluorescence (excitation, 400 nm; emission, 440 nm) relative to the Thr-WT strain and were zeroed against the background levels recorded for the pvdD deletion mutant. The data are the means of 6 independent replicates, and the error bars indicate 1 standard deviation.
To more accurately quantify levels of pyoverdine, all A domain substitution strains were grown in liquid media, and their pyoverdine levels were measured by both absorbance (400 nm) and fluorescence (excitation, 400 nm; emission, 440 nm) of the supernatant (Fig. 4B). Both measurements were used because initial tests of pyoverdine serial dilutions found that absorbance followed a linear trend whereas fluorescence became saturated (see Fig. S1 in the supplemental material). This meant absorbance was generally more accurate in terms of quantifying pyoverdine levels than fluorescence; however, fluorescence was more sensitive and was able to detect pyoverdine at much lower levels.
The three threonine-specifying A domain substitution strains exhibited increased absorbance and fluorescence compared to all the other strains. The two substitution strains that were most viable in the presence of EDDHA (Thr-A and Thr-D) each had levels of fluorescence and absorbance similar to those of the Thr-WT strain. The strain Thr-E had absorbance levels at approximately 29% relative to Thr-WT, confirming lower levels of pyoverdine synthesis. In contrast, the remaining (non-threonine-specifying) A domain substitution strains all had levels of absorbance similar to that of the pvdD deletion mutant, with a possible small increase in absorbance observed for strain Ser-F. Surprisingly, each of these non-threonine-specifying A domain substitution strains appeared to have a small increase in fluorescence compared to the pvdD deletion strain. This suggested the non-threonine-specifying A domain substitution strains might be producing, at very low levels, an unknown pyoverdine-like product.
Detection of A domain substitution products by mass spectrometry.
The synthesis of pyoverdine by the A domain substitution strains was confirmed using mass spectrometry. For each of the A domain substitution strains, an ion was detected with an m/z ratio within 0.05% of 1,333.6 (see Fig. S2 in the supplemental material); this was the expected m/z for pyoverdine containing a succinimide acyl group (19). This product was confirmed to be wild-type pyoverdine by collision-induced dissociation (CID) (see Fig. S3 and Table S1 in the supplemental material) and was not detected from the pyoverdine-negative pvdD deletion strain. These results confirm that both the threonine- and non-threonine-specifying A domain substitution strains synthesized wild-type pyoverdine, the former efficiently and the latter at only trace levels.
Generation and testing of C-A domain substitution strains.
Substituting C-A domain pairs together bypasses the potential restrictions of C domain acceptor site specificity and avoids disruption of the C-A domain interface. However, four C-A domain substitutions attempted previously in the first module of pvdD all yielded nonfunctional hybrid enzymes (15). To more rigorously test whether C-A domain substitutions could provide a route to new pyoverdines, nine C-A domain substitutions corresponding to each of the highlighted modules in Fig. 2 were created by ligation of PCR-amplified C-A domains into a pvdD module 2 C-A domain deletion construct in the plasmid pSMC (Fig. 3B). The recombination junction between the T and C domains in pSMC was selected to be 37 amino acid residues downstream of the conserved serine residue of the T domain, a linker region shown to be tolerant of modifications in other NRPS enzymes (20, 21). Complementation strains were generated by integration of these C-A domain substitution constructs into the pvdD mutant and were named according to the convention used for the A domain substitution strains but with a CA prefix (for example, the variant containing the C-A domains from the first module of pvdD is referred to as strain CAThr-A). The positive-control strain, CAThr-WT, showed no reduction in pyoverdine synthesis compared to wild-type P. aeruginosa, indicating that the introduction of restriction sites had not impaired function.
When the C-A domain substitution strains were grown on solid King's B medium in the absence of EDDHA, only strains CAThr-A and CALys-C had levels of fluorescence that were visibly higher than the background of the pyoverdine negative control (Fig. 5A). However, in the presence of EDDHA, CAThr-A, CALys-C, and CASer-F all yielded visible levels of growth (Fig. 5A). Interestingly, in contrast to the A domain substitutions, only one threonine-specifying C-A domain substitution was functional. Of the two nonfunctional threonine-specifying C-A domain substitutions, strain CAThr-D contained a C domain that receives a d-amino acid from the upstream module in its native context; due to the stereospecificity of C domain donor sites (10), it was not expected to be able to accommodate the l-threonine at the C terminus of the peptide chain received from the upstream module of PvdD. However, strain CAThr-E, which contained not only an A domain that was functional in the A domain substitution experiments, but also a C domain that “expects” in its native context to receive l-threonine at its donor site, also failed to produce detectable levels of fluorescence.
FIG 5.

Detection of pyoverdine production by C-A domain substitution variants. (A) Fluorescence of C-A domain substitutions strains on King's B agar plates in the absence (−) or presence (+) of EDDHA. The plates were inoculated with the positive-control strain Thr-WT (WT), C-A domain substitution strains A to I (Fig. 2), and the pvdD deletion pyoverdine negative control (Del) and then incubated for 24 h at 37°C. The photographs were taken under UV light. (B) Percentages of pyoverdine production from C-A domain substitution strains grown in liquid media. The values are expressed as percentages of absorbance (400 nm) or fluorescence (excitation, 400 nm; emission, 440 nm) relative to the CAThr-WT strain and were zeroed against the background levels recorded for the pvdD deletion mutant. The data are the means of 6 independent replicates, and the error bars indicate 1 standard deviation.
When the pyoverdine absorbances of the supernatants of C-A domain substitution strains grown in liquid media were measured (Fig. 5B, blue bars), strains CAThr-A and CALys-C gave high levels of absorbance at 83% and 76% of the levels of the CAThr-WT positive control. Strain CASer-F, which exhibited low levels of growth on solid King's B medium containing EDDHA (Fig. 5A), had much lower absorbance at 18% of CAThr-WT levels. The other C-A domain substitution strains all had much lower levels of absorbance. However, when the fluorescence of the supernatant was measured (Fig. 5B, red bars), all C-A domain substitution strains except strain CAAsp-G still had elevated levels of fluorescence relative to the negative control.
Detection of pyoverdine from C-A domain substitution strains by mass spectrometry.
The synthesis of wild-type pyoverdine by strain CAThr-A and incorporation of lysine and serine into pyoverdine by strains CALys-C and CASer-F were confirmed by mass spectrometry. Assuming that cyclization of the peptide was still possible, replacement of the terminal threonine of pyoverdine with lysine or serine was expected to change the mass of pyoverdine by +27 Da and −14 Da, respectively. Consistent with this, products of m/z 1,333.6 from CAThr-A, 1,360.7 from CALys-C, and 1,319.6 from CASer-F were detected by mass spectrometry (see Fig. S4 in the supplemental material). From the CID spectra of the m/z 1,360.7 and 1,319.6 pyoverdine species (see Fig. S5A and Table S2 in the supplemental material), ions expected to contain the new lysine and serine residues had mass changes consistent with the presence of the altered residue. These results confirm that the CALys-C and CASer-F domain substitution strains incorporated new residues into pyoverdine.
In contrast, no full-length pyoverdine was detected from the other C-A domain substitution strains. Instead, a product of only m/z 991.5 was detected from all these strains except strain CAAsp-G, for which no pyoverdine was detected (see Fig. S4 in the supplemental material). The m/z 991.5 product was consistent with pyoverdine missing the three terminal residues and has been detected previously as a breakdown product when the final l-threonine residue was not incorporated onto pyoverdine (15). Ions produced by CID were also consistent with this product (see Fig. S5B and Table S3 in the supplemental material). Interestingly, the m/z 991.5 product was also detected as a minor peak during mass spectrometry of the CAThr-A, CALys-C, and CASer-F C-A domain substitution strains (see Fig. S5B and Table S3 in the supplemental material). This product was not detected for any of the A domain substitution strains above the signal/noise ratio of 20 set for CID, although a smaller peak did appear to be present for the strain Lys-C and may have been present but buried in the spectra for other A domain substitution strains (see Fig. S2 in the supplemental material).
DISCUSSION
In this study, the pvdD mutant strains expressing PvdD variants with A domain substitutions that specify threonine (n = 3) all synthesized pyoverdine relatively effectively. In contrast, non-threonine-specifying A domains (n = 6) yielded only trace levels of pyoverdine. It is possible that higher levels of sequence identity shared between the introduced threonine-specifying A domains and the PvdD module 2 A domain contributed in part to this successful outcome. The PvdD module 1 A domain in strain Thr-A shares 99.6% amino acid identity with the module 2 A domain, so it is not at all surprising that it proved a successful replacement, while the A domains substituted into Thr-D and Thr-E shared on average 17.3% greater amino acid identity with the native module 2 A domain than did the six inactive, non-threonine-specifying A domains (Table 3). Nonetheless, the observation that 3/3 threonine-specifying A domains were active while 6/6 non-threonine-specifying A domains were inactive provides strong evidence that substrate specificity plays a major role in determining whether a construct with an A domain substitution will be active.
This observation is consistent with an earlier pilot study conducted in the first module of pvdD, where 2/2 threonine-specifying A domains were active (despite being derived from different bacterial genera and sharing substantially lower levels of amino acid identity than the A domains used in this study), whereas 3/3 non-threonine-specifying A domains were inactive (15). Collectively, these results provide strong evidence in support of the hypothesis that C domain acceptor site substrate specificity plays a major role in determining whether an introduced A domain will be active in a nonnative context (9, 10), whereas the disruption of a specialized C-A domain interface that evolution has optimized (hypothesized to be a potential limiting factor [11]) was not a major factor restricting A domain substitution in our system.
The C domain acceptor site specificity hypothesis receives additional support from our novel observation that the non-threonine-specifying A domain substitution strains nevertheless produced low levels of wild-type pyoverdine. A likely scenario to explain the production of wild-type pyoverdine by these recombinant PvdD enzymes is low-level “leaky” activation of noncognate substrates, including threonine, followed by exclusive selection for threonine within the C domain acceptor site. Low-level promiscuous activities have frequently been observed for A domains in vitro (22–26). However, our result was particularly interesting in that it allowed a direct comparison of the relative strengths of the substrate specificities of the A domain and the acceptor site of the C domain. If the substrate specificities of all the domains were approximately equivalent, then both modified and wild-type pyoverdines should be produced (that is, the alternative substrate preferred by the introduced A domain would be accepted at low levels by a leaky C domain). As no modified pyoverdine species were observed, it can be concluded that the C domain proofreading is very stringent in PvdD. It has been suggested that strong specificity within the C domain acceptor site in conjunction with release of incorrect amino acids by type II thioesterases is of great value in ensuring correct peptide assembly, as it is more energetically favorable to release monomers before condensation than to produce an entire nonfunctional peptide chain (27).
Further support for stringent C domain acceptor site proofreading was provided by the C-A domain substitutions, which confirmed that at least two of the non-threonine-specifying A domains had the potential to be active in the PvdD context. When substituted along with their native C domain partners, the lysine- and serine-specifying A domains from PvdJ of P. aeruginosa and an unnamed P. syringae pyoverdine NRPS exhibited high-level activation of their natural substrates. This successful rational introduction of new amino acid residues into pyoverdine confirms that novel pyoverdine peptides can be potentially generated at a high yield via C-A domain substitution.
In contrast, two of the A domains that had previously been functional in the A domain substitution strains Thr-D and Thr-E no longer yielded a full-length pyoverdine when substituted as a C-A domain pairing, suggesting that the newly introduced C domain impaired function. We considered that this might have been due to selectivity constraints at the donor site of the C domain. In contrast with the evidence that C domains exhibit stereospecificity but not side chain specificity at the donor site (9, 10), Stein et al. found that the second C domain of the tyrocidine NRPS system showed a degree of side chain specificity for the terminal amino acid of dipeptides artificially loaded onto the upstream T domain (28). However, our results were not consistent with donor site side chain specificity being a major limiting factor. If this were the case, then the C domain introduced into strain CAThr-E should have been an effective replacement for the native C domain of PvdD module 2, as both C domains receive l-threonine at their donor and acceptor sites in their native contexts. More significantly, neither C domain in the functional C-A substitution strains CAThr-A and CA-LysC should have been able to tolerate l-threonine, as each receives l-N5-formyl-N5-hydroxyornithine as the C-terminal residue at the donor site in its native context. Finally, strain CASer-F, containing a substituted C domain that does normally receive l-threonine at the donor site, was the least active of the functional C-A domain substitution strains.
Instead, the detection of only truncated pyoverdine resulting from the nonfunctional C-A domain substitutions suggests that the inability to catalyze an effective condensation reaction causes peptide synthesis to stall. One possible explanation for this is that C domains in a new context may be unable to receive some incoming peptides due to steric constraints. The crystal structures of C domains show the catalytic center covered by a lid region within a channel between two subdomains (11, 29–31), and these features may contribute to some incoming peptide chains being physically blocked from reaching the catalytic center. It is also important to note that the previous in vitro studies of C domain donor site specificity focused on single residues (9, 10), or at most a tetrapeptide (28), rather than the longer peptide chains that substituted C domains further down an NRPS assembly line might encounter. Further tests of C domain donor site specificity using peptides rather than single amino acids are needed to more thoroughly interrogate the tolerance of C domain donor sites for alternative peptide substrates.
An alternative reason for the loss of activity observed with most C-A domain substitutions is that introduced C-A domains may not communicate appropriately with the neighboring PvdD T domains. T domains function to transfer substrates between multiple domains during a catalytic cycle. For example, the T domain from the first module of PvdD receives a threonine residue from the A domain and must pass it between the upstream and downstream C domains. Structural studies have shown that T domains adopt multiple distinct conformational states, and this conformational plasticity is hypothesized to aid in interacting with different domains (11, 32), which may themselves be highly dynamic (33, 34). Although our work here has shown that the PvdD T domains are generally able to communicate effectively with a new A domain introduced immediately upstream, in other work, we and others have seen that T domains can show some specificity for the type of domain immediately downstream; in particular, directed evolution has been required in several instances to enable an introduced T domain to communicate effectively with a downstream TE domain (35, 36; J. G. Owen, M. J. Calcott, K. J. Robins, and D. F. Ackerley, unpublished data). It may be that, in analogous fashion, a newly introduced C domain is often unable to communicate effectively with the T domain immediately upstream. This issue could potentially be resolved by treating NRPS modules as inseparable T-C-A domain units and substituting them accordingly. There is at least one example where dipeptides were created via T-C-A domain substitution (22). In ongoing work, we are testing T-C-A domain substitution in the PvdD model with the aim of keeping T-C domain communication intact, as well as examining how generally promiscuous T domains are in their interactions with other domains, in particular C domains, and whether this may be a primary factor inhibiting the success of C-A domain substitution strategies.
The pyoverdine NRPS system is not only ideally suited to targeted domain recombination studies, it also holds great potential for directed evolution, in particular of C domain regions. Excitingly, our discovery that certain C-A domain substitutions were active while the corresponding A domain substitutions were not opens the way for directed-evolution studies targeting the acceptor site of the native PvdD C domain. Relaxation of the stringent proofreading requirements of the acceptor site would open the door to generation of novel peptides via simple A domain substitution or recoding. The ability of pyoverdine to provide conditional viability selection (e.g., only pyoverdine-synthesizing strains will grow on media containing EDDHA) will enable high-throughput analysis of gene libraries derived from variants of pvdD with A domain substitutions to achieve this goal.
Supplementary Material
ACKNOWLEDGMENTS
We thank Lois Martin from the University of Otago for assistance with pyoverdine purification and analysis. We also thank Bill Jordan, Jonathan Dunne, and Danyl McLauchlan at the Victoria University of Wellington Centre for Biodiscovery for assistance with mass spectrometry.
This work was supported by the Royal Society of New Zealand Marsden Fund (contract number VUW0901) and the Victoria University Research Fund.
Footnotes
Published ahead of print 11 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01453-14.
REFERENCES
- 1.Caboche S, Leclere V, Pupin M, Kucherov G, Jacques P. 2010. Diversity of monomers in nonribosomal peptides: towards the prediction of origin and biological activity. J. Bacteriol. 192:5143–5150. 10.1128/JB.00315-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caradec T, Pupin M, Vanvlassenbroeck A, Devignes MD, Smaïl-Tabbone M, Jacques P, Leclère V. 2014. Prediction of monomer isomery in florine: a workflow dedicated to nonribosomal peptide discovery. PLoS One 9:e85667. 10.1371/journal.pone.0085667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Felnagle EA, Jackson EE, Chan YA, Podevels AM, Berti AD, McMahon MD, Thomas MG. 2008. Nonribosomal peptide synthetases involved in the production of medically relevant natural products. Mol. Pharm. 5:191–211. 10.1021/mp700137g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirschning A, Hahn F. 2012. Merging chemical synthesis and biosynthesis: a new chapter in the total synthesis of natural products and natural product libraries. Angew. Chem. Int. Ed. Engl. 51:4012–4022. 10.1002/anie.201107386 [DOI] [PubMed] [Google Scholar]
- 5.O'Connell KM, Hodgkinson JT, Sore HF, Welch M, Salmond GP, Spring DR. 2013. Combating multidrug-resistant bacteria: current strategies for the discovery of novel antibacterials. Angew. Chem. Int. Ed. Engl. 52:10706–10733. 10.1002/anie.201209979 [DOI] [PubMed] [Google Scholar]
- 6.Walsh C. 2003. Where will new antibiotics come from? Nat. Rev. Microbiol. 1:65–70. 10.1038/nrmicro727 [DOI] [PubMed] [Google Scholar]
- 7.Walsh CT, Chen H, Keating TA, Hubbard BK, Losey HC, Luo L, Marshall CG, Miller DA, Patel HM. 2001. Tailoring enzymes that modify nonribosomal peptides during and after chain elongation on NRPS assembly lines. Curr. Opin. Chem. Biol. 5:525–534. 10.1016/S1367-5931(00)00235-0 [DOI] [PubMed] [Google Scholar]
- 8.Stevens BW, Joska TM, Anderson AC. 2006. Progress toward re-engineering non-ribosomal peptide synthetase proteins: a potential new source of pharmacological agents. Drug Dev. Res. 66:9–18. 10.1002/ddr.20041 [DOI] [Google Scholar]
- 9.Belshaw PJ, Walsh CT, Stachelhaus T. 1999. Aminoacyl-CoAs as probes of condensation domain selectivity in nonribosomal peptide synthesis. Science 284:486–489. 10.1126/science.284.5413.486 [DOI] [PubMed] [Google Scholar]
- 10.Ehmann DE, Trauger JW, Stachelhaus T, Walsh CT. 2000. Aminoacyl-SNACs as small-molecule substrates for the condensation domains of nonribosomal peptide synthetases. Chem. Biol. 7:765–772. 10.1016/S1074-5521(00)00022-3 [DOI] [PubMed] [Google Scholar]
- 11.Tanovic A, Samel SA, Essen L-O, Marahiel MA. 2008. Crystal structure of the termination module of a nonribosomal peptide synthetase. Science 321:659–663. 10.1126/science.1159850 [DOI] [PubMed] [Google Scholar]
- 12.Ackerley DF, Caradoc-Davies TT, Lamont IL. 2003. Substrate specificity of the nonribosomal peptide synthetase PvdD from Pseudomonas aeruginosa. J. Bacteriol. 185:2848–2855. 10.1128/JB.185.9.2848-2855.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mossialos D, Ochsner U, Baysse C, Chablain P, Pirnay JP, Koedam N, Budzikiewicz H, Fernández DU, Schäfer M, Ravel J, Cornelis P. 2002. Identification of new, conserved, non-ribosomal peptide synthetases from fluorescent pseudomonads involved in the biosynthesis of the siderophore pyoverdine. Mol. Microbiol. 45:1673–1685. 10.1046/j.1365-2958.2002.03120.x [DOI] [PubMed] [Google Scholar]
- 14.Meyer J-M, Gruffaz C, Raharinosy V, Bezverbnaya I, Schäfer M, Budzikiewicz H. 2008. Siderotyping of fluorescent Pseudomonas: molecular mass determination by mass spectrometry as a powerful pyoverdine siderotyping method. Biometals 21:259–271. 10.1007/s10534-007-9115-6 [DOI] [PubMed] [Google Scholar]
- 15.Ackerley DF, Lamont IL. 2004. Characterization and genetic manipulation of peptide synthetases in Pseudomonas aeruginosa PAO1 in order to generate novel pyoverdines. Chem. Biol. 11:971–980. 10.1016/j.chembiol.2004.04.014 [DOI] [PubMed] [Google Scholar]
- 16.Bachmann BO, Ravel J. 2009. Chapter 8. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 458:181–217. 10.1016/S0076-6879(09)04808-3 [DOI] [PubMed] [Google Scholar]
- 17.Baynham PJ, Ramsey DM, Gvozdyev BV, Cordonnier EM, Wozniak DJ. 2006. The Pseudomonas aeruginosa ribbon-helix-helix DNA-binding protein AlgZ (AmrZ) controls twitching motility and biogenesis of type IV pili. J. Bacteriol. 188:132–140. 10.1128/JB.188.1.132-140.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, Xiong G, Lutz F. 1995. Site-specific integration of the phage ΦCTX genome into the Pseudomonas aeruginosa chromosome: characterization of the functional integrase gene located close to and upstream of attP. Mol. Gen. Genet. 246:72–79. 10.1007/BF00290135 [DOI] [PubMed] [Google Scholar]
- 19.Hannauer M, Schäfer M, Hoegy F, Gizzi P, Wehrung P, Mislin GLA, Budzikiewicz H, Schalk IJ. 2012. Biosynthesis of the pyoverdine siderophore of Pseudomonas aeruginosa involves precursors with a myristic or a myristoleic acid chain. FEBS Lett. 586:96–101. 10.1016/j.febslet.2011.12.004 [DOI] [PubMed] [Google Scholar]
- 20.Doekel S, Coëffet-Le Gal M-F, Gu J-Q, Chu M, Baltz RH, Brian P. 2008. Non-ribosomal peptide synthetase module fusions to produce derivatives of daptomycin in Streptomyces roseosporus. Microbiology 154:2872–2880. 10.1099/mic.0.2008/020685-0 [DOI] [PubMed] [Google Scholar]
- 21.Mootz HD, Schwarzer D, Marahiel MA. 2000. Construction of hybrid peptide synthetases by module and domain fusions. Proc. Natl. Acad. Sci. U. S. A. 97:5848–5853. 10.1073/pnas.100075897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doekel S, Marahiel MA. 2000. Dipeptide formation on engineered hybrid peptide synthetases. Chem. Biol. 7:373–384. 10.1016/S1074-5521(00)00118-6 [DOI] [PubMed] [Google Scholar]
- 23.Eppelmann K, Stachelhaus T, Marahiel MA. 2002. Exploitation of the selectivity-conferring code of nonribosomal peptide synthetases for the rational design of novel peptide antibiotics. Biochemistry 41:9718–9726. 10.1021/bi0259406 [DOI] [PubMed] [Google Scholar]
- 24.Mootz HD, Marahiel MA. 1997. The tyrocidine biosynthesis operon of Bacillus brevis: complete nucleotide sequence and biochemical characterization of functional internal adenylation domains. J. Bacteriol. 179:6843–6850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stevens BW, Lilien RH, Georgiev I, Donald BR, Anderson AC. 2006. Redesigning the PheA domain of gramicidin synthetase leads to a new understanding of the enzyme's mechanism and selectivity. Biochemistry 45:15495–15504. 10.1021/bi061788m [DOI] [PubMed] [Google Scholar]
- 26.Villiers BRM, Hollfelder F. 2009. Mapping the limits of substrate specificity of the adenylation domain of TycA. Chembiochem 10:671–682. 10.1002/cbic.200800553 [DOI] [PubMed] [Google Scholar]
- 27.Yeh E, Kohli RM, Bruner SD, Walsh CT. 2004. Type II thioesterase restores activity of a NRPS module stalled with an aminoacyl-S-enzyme that cannot be elongated. Chembiochem 5:1290–1293. 10.1002/cbic.200400077 [DOI] [PubMed] [Google Scholar]
- 28.Stein DB, Linne U, Marahiel MA. 2005. Utility of epimerization domains for the redesign of nonribosomal peptide synthetases. FEBS J. 272:4506–4520. 10.1111/j.1742-4658.2005.04871.x [DOI] [PubMed] [Google Scholar]
- 29.Bloudoff K, Rodionov D, Schmeing TM. 2013. Crystal structures of the first condensation domain of CDA synthetase suggest conformational changes during the synthetic cycle of nonribosomal peptide synthetases. J. Mol. Biol. 425:3137–3150. 10.1016/j.jmb.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keating TA, Marshall CG, Walsh CT, Keating AE. 2002. The structure of VibH represents nonribosomal peptide synthetase condensation, cyclization and epimerization domains. Nat. Struct. Mol. Biol. 9:522–526. 10.1038/nsb810 [DOI] [PubMed] [Google Scholar]
- 31.Samel SA, Schoenafinger G, Knappe TA, Marahiel MA, Essen L-O. 2007. Structural and functional insights into a peptide bond-forming bidomain from a nonribosomal peptide synthetase. Structure 15:781–792. 10.1016/j.str.2007.05.008 [DOI] [PubMed] [Google Scholar]
- 32.Lai JR, Koglin A, Walsh CT. 2006. Carrier protein structure and recognition in polyketide and nonribosomal peptide biosynthesis. Biochemistry 45:14869–14879. 10.1021/bi061979p [DOI] [PubMed] [Google Scholar]
- 33.Gulick AM. 2009. Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol. 4:811–827. 10.1021/cb900156h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sundlov JA, Shi C, Wilson DJ, Aldrich CC, Gulick AM. 2012. Structural and functional investigation of the intermolecular interaction between NRPS adenylation and carrier protein domains. Chem. Biol. 19:188–198. 10.1016/j.chembiol.2011.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Owen JG, Robins KJ, Parachin NS, Ackerley DF. 2012. A functional screen for recovery of 4′-phosphopantetheinyl transferase and associated natural product biosynthesis genes from metagenome libraries. Environ. Microbiol. 14:1198–1209. 10.1111/j.1462-2920.2012.02699.x [DOI] [PubMed] [Google Scholar]
- 36.Zhou Z, Lai JR, Walsh CT. 2007. Directed evolution of aryl carrier proteins in the enterobactin synthetase. Proc. Natl. Acad. Sci. U. S. A. 104:11621–11626. 10.1073/pnas.0705122104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hur GH, Vickery CR, Burkart MD. 2012. Explorations of catalytic domains in non-ribosomal peptide synthetase enzymology. Nat. Prod. Rep. 29:1074–1098. 10.1039/c2np20025b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marahiel MA, Stachelhaus T, Mootz HD. 1997. Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem. Rev. 97:2651–2674. 10.1021/cr960029e [DOI] [PubMed] [Google Scholar]
- 39.Sieber SA, Marahiel MA. 2005. Molecular mechanisms underlying nonribosomal peptide synthesis: approaches to new antibiotics. Chem. Rev. 105:715–738. 10.1021/cr0301191 [DOI] [PubMed] [Google Scholar]
- 40.Owen JG, Ackerley DF. 2011. Characterization of pyoverdine and achromobactin in Pseudomonas syringae pv. phaseolicola 1448a. BMC Microbiol. 11:218. 10.1186/1471-2180-11-218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moon C, Zhang X-X, Matthijs S, Schafer M, Budzikiewicz H, Rainey P. 2008. Genomic, genetic and structural analysis of pyoverdine-mediated iron acquisition in the plant growth-promoting bacterium Pseudomonas fluorescens SBW25. BMC Microbiol. 8:7. 10.1186/1471-2180-8-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ravel J, Cornelis P. 2003. Genomics of pyoverdine-mediated iron uptake in pseudomonads. Trends Microbiol. 11:195–200. 10.1016/S0966-842X(03)00076-3 [DOI] [PubMed] [Google Scholar]
- 43.Briskot G, Taraz K, Budzikiewicz H. 1989. Bacterial constituents. XXXVII. Pyoverdin-type siderophores from Pseudomonas aeruginosa. Liebigs Ann. Chem. 1989:375–384 [Google Scholar]
- 44.Demange P, Wendenbaum S, Linget C, Mertz C, Cung MT, Dell A, Abdallah MA. 1990. Bacterial siderophores: structure and NMR assignment of pyoverdins Pa, siderophores of Pseudomonas aeruginosa ATCC 15692. Biometals 3:155–170 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.