

Abstract
Methane is a potent greenhouse gas that is generated and consumed in anaerobic environments through the energy metabolism of methanogens and anaerobic methanotrophic archaea (ANME), respectively. Coenzyme F430 is essential for methanogenesis, and a structural variant of F430, 172-methylthio-F430 (F430-2), is found in ANME and is presumably essential for the anaerobic oxidation of methane. Here we use liquid chromatography–high-resolution mass spectrometry to identify several new structural variants of F430 in the cell extracts of selected methanogens and ANME. Methanocaldococcus jannaschii and Methanococcus maripaludis contain an F430 variant (denoted F430-3) that has an M+ of 1,009.2781. This mass increase of 103.9913 over that of F430 corresponds to C3H4O2S and is consistent with the addition of a 3-mercaptopropionate moiety bound as a thioether followed by a cyclization. The UV absorbance spectrum of F430-3 was different from that of F430 and instead matched that of an F430 derivative where the 173 keto moiety had been reduced. This is the first report of a modified F430 in methanogens. In a search for F430-2 and F430-3 in other methanogens and ANME, we have identified a total of nine modified F430 structures. One of these compounds may be an abiotic oxidative product of F430, but the others represent naturally modified versions of F430. This work indicates that F430-related molecules have additional functions in nature and will inspire further research to determine the biochemical role(s) of these variants and the pathways involved in their biosynthesis.
INTRODUCTION
Methanogens are a diverse group of anaerobic archaeal organisms with an energy metabolism dependent on one-carbon biochemistry to reduce CO2, CO, formate, methyl group-containing compounds, and/or acetate to generate methane (see Fig. S1 in the supplemental material) (1). Several specialized coenzymes are required for this process. One such molecule, coenzyme F430 (Fig. 1), is a nickel-containing hydroporphinoid which is chemically most closely related to vitamin B12 and siroheme. It was originally characterized as a prosthetic group of methyl coenzyme M (CoM) reductase (MCR), the terminal key enzyme in methanogenesis (2). MCR catalyzes the reduction of the methyl group of methyl-CoM to generate methane in the final step of methanogenesis. The mechanism and role of F430 in this reaction are still unclear, but the Ni(I) center of F430 is thought to initiate methane formation by one of two mechanisms. The first involves an organometallic methyl-Ni(III) intermediate (3, 4), and the second involves the formation of a methyl radical (5, 6). The structure of F430 was determined on the basis of biosynthetic incorporation experiments, chemical stabilities, and nuclear magnetic resonance (NMR) spectroscopy (7–9).
FIG 1.
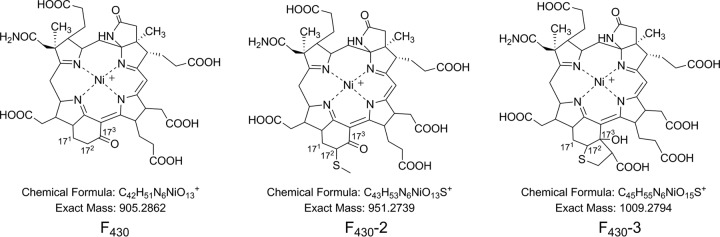
Structures of coenzyme F430 and F430-2 and the proposed structure of F430-3 described in this work.
The anaerobic oxidation of methane (AOM) is a microbially mediated process which consumes approximately 90% of the methane produced in marine sediments and, hence, is of importance for the global budget of this greenhouse gas (10). The organisms carrying out this difficult biochemical reaction consist of anaerobic methanotrophic archaea (ANME) and sulfate-reducing bacteria found near methane seeps in the ocean, which together convert methane and sulfate to carbonate and hydrogen sulfide (11, 12). ANME can be classified into at least three groups which are phylogenetically related to methanogens (13–15). Group 1 ANME (ANME-1) are likely most closely related to the Methanomicrobiales, while ANME-2 and ANME-3 are affiliated with the Methanosarcinales. The chemically challenging activation of methane by ANME occurs via a reversal of the MCR-catalyzed step in methanogenesis (16). Metagenomic data have shown that ANME possess homologs of MCR as well as most genes normally associated with methanogenic archaea, indicating that these organisms oxidize methane using a pathway which is essentially methanogenesis in reverse (15, 17). A modified form of F430, F430-2 (Fig. 1), was identified in samples collected from cold methane seeps in the Black Sea enriched with ANME-1 (18). The structure of this modified F430 was determined by mass spectrometry and NMR spectroscopy and was shown to contain a methylthio group at the 172 position (19).
Here, we describe the presence of a new modified F430 in two Methanococcales organisms. This is the first report of a modified F430 occurring in methanogens. Based on high-resolution mass spectral data coupled with traditional biochemical methods, we have assigned the structure shown in Fig. 1 (F430-3). This discovery led us to expand our search for additional modified F430 molecules in cell extracts from three Methanococcales species as well as extracts from samples containing ANME. Multiple F430 variants were identified in each of the samples that we analyzed. One of the compounds is likely an abiotic oxidative product of F430, but the others could not have been produced during the extraction procedure and therefore must be biologically relevant.
MATERIALS AND METHODS
Chemicals.
All chemicals and reagents were purchased from Sigma-Aldrich.
Source of methanogenic cells.
Methanocaldococcus jannaschii (JAL-1, DSM 2661) was grown on an inorganic salts medium as previously described (20), and cell pellets were stored at −80°C. Methanococcus maripaludis (S2, DSM 2067 [JJ]) was grown on mineral medium containing 10 mM sodium acetate, 2% Casamino Acids, and 1% vitamin mixture (McCA) as previously described (21) and was supplied by William B. Whitman. Methanococcus vannielii (DSM 1224 [SB]) was grown on a defined salts medium containing formate (22).
Source of samples of ANME.
Black Sea mat material was sampled from methane-derived microbial chimneys during RVPoseidon cruise 317-2 in August 2004 using the submersible Jago (44°46′N, 31°60′E). Microbial methane oxidation was maintained by a methane headspace and repeatedly exchanging the marine sulfate reducer medium at 4°C. The thermophilic AOM enrichment was derived from the Guaymas Basin (Gulf of California, Mexico; 27°00.437′N, 111°24.548′W). Samples were obtained during the RV Atlantis cruise AT15-56 in November and December 2009 with the submersible Alvin. Cored material was incubated at 50°C with marine seawater medium and a methane headspace. The samples were repeatedly diluted, and medium was exchanged when sulfide concentrations exceeded 12 mM until sediment-free cultures were retrieved. Samples were taken from the living cultures within the last year and pooled. Samples are dominated by ANME-1 and HotSeep-1 partner bacteria. The sample Caldera derives from methane-rich sediments of the Nile Deep Sea Fan (32°07′N, 28°12′E) and was sampled in 2003 during the RV l'Atalante (expedition NAUTINIL). The sample Hydrate Ridge was retrieved from seeps in the Cascadia Margin (northeast Pacific; 44°34.2′N, 125°08.7′W). Cored material was incubated at 20°C (Caldera) or 12°C (Hydrate Ridge) with marine seawater medium and a methane headspace, and due to proliferation and repeated dilution, the material has been sediment free for several years. All samples possessed a methane-dependent sulfide production of approximately 100 to 250 μmol liter−1 sulfide per day before sampling.
Preparation of methanogen extracts.
Frozen cell pellets (1 g [wet weight]) were added to a sample tube and suspended in 5 ml water. Six milliliters of methanol was added to this suspension, and the sample was shaken to obtain an even suspension of material. The tube was then sealed, and the sample was heated with shaking for 15 min at 100°C. This heating ensured that all proteins were denatured so as to release all bound cofactors. After cooling, the samples were centrifuged and the pellets were reextracted in the same manner a second time. The resulting combined extracts were evaporated to 1 ml with a stream of nitrogen, and large molecules were removed from the sample using an Amicon Ultra 0.5-ml centrifugal filter with a molecular mass cutoff of 3 kDa. Filtrate was evaporated to 0.1 ml prior to analysis.
Preparation of extracts of ANME.
The dried lyophilized cells (100 mg) were suspended in 1 ml of water and mixed thoroughly. One and a half milliliters of methanol was added, and the sample was heated at 100°C for 5 min. The sample was then centrifuged, and the resulting extract was separated from the pellet. The pellet was reextracted by the same procedure. The resulting combined extracts were evaporated to 1 ml with a stream of nitrogen, and large molecules were removed from the sample using an Amicon Ultra 0.5-ml centrifugal filter with a molecular mass cutoff of 3 kDa. Filtrate was evaporated to 0.1 ml prior to analysis.
Liquid chromatography–high-resolution mass spectrometry (LC–HR-MS) analysis of cell extracts.
The cell extracts were analyzed directly on a Waters SYNAPT G2-S high-definition mass spectrometer connected to a Waters Acquity UPLC I-class system with an Acquity UPLC BEH C18 column (2.1 mm by 75 mm; particle size, 1.7 μm; Waters). Solvent A was water with 0.1% formic acid, and solvent B was acetonitrile. The flow rate was 0.2 ml/min, and gradient elution was employed in the following manner (time [min], percent solvent B): (0.01, 5), (6, 15), (21, 35), and (23, 65). Ten microliters of sample was injected. The mass spectral data were collected in high-resolution MSe continuum mode (nonselective MS/MS acquisition mode). A lock spray scan (function 3) was collected every 20 s for calibration, and the lock spray analyte used was leucine-enkephalin. Parameters were a 2.8-kV capillary voltage, a 125°C source temperature, a 350°C desolvation temperature, a 35-V sampling cone, 50-liter/h cone gas flow, a 500-liter/h desolvation gas flow, and a 6-liter/h nebulizer gas flow. The collision energies for the low-energy scans (function 1) were 4 V and 2 V in the trap region and the transfer region, respectively. Collision energies for the high-energy scans (function 2) were ramped from 25 to 45 V in the trap region and 2 V in the transfer region. Data were analyzed using MassLynx software (Waters).
Isolation of F430-3 from M. jannaschii.
A portion of the cell extract from M. jannaschii was purified by chromatographic separation using a Shimadzu Prominence ultrafast LC system equipped with a photodiode array detector (PDA) and a fluorescence detector. A strong anion-exchange column (8.0 by 75 mm; particle size, 12 μm; Shodex QA-825) was used at a flow rate of 1 ml/min, and the elution profile consisted of a 5-min water wash followed by a 40-min linear gradient to 2 M ammonium bicarbonate. The injection volume was 0.1 ml, and 1-ml fractions were collected. In this chromatographic system, F430-3 eluted in a fraction (fraction 19) after the F430 isomers, which were observed in a single fraction (fraction 16).
Liquid chromatography-UV/visible light spectrophotometry-mass spectrometry (LC-UV-MS) analysis of metabolites.
In order to simultaneously obtain the absorbance spectra and mass spectral data for coenzyme F430 derivatives, analyses were performed using an Agilent 1200 Series liquid chromatograph with a Zorbax Eclipse XDB-C18 column (4.6 by 50 mm; particle size, 1.8 μm; Agilent) equipped with a PDA detector interfaced with an AB Sciex 3200 Q TRAP electrospray ionization mass spectrometer. Fractions from the Shodex anion-exchange purification were evaporated 3 times with a stream of nitrogen gas to remove ammonium bicarbonate and resuspended in 0.1 ml water for LC-UV-MS analysis. Solvent A was water with 0.1% formic acid, and solvent B was methanol with 0.1% formic acid. The flow rate was 0.5 ml/min, and gradient elution was employed in the following manner: (time [min], percent solvent B): (0.01, 5), (10, 65), and (15, 65). Ten microliters of sample was injected. The mass spectral data were collected in positive mode, and electrospray ionization was employed at 4,500 V and a temperature of 400°C. The curtain gas was set at 35, and gas source 1 and gas source 2 were 60 and 50, respectively. Analyst software (MDS SCIEX; Applied Biosystems) was used for system operation and data processing.
Preparation of the methyl esters of F430 and F430-3.
The M. jannaschii cell extract (50 μl) was placed in a vial, and the water was removed by evaporation. The sample was dissolved in 200 μl of methanol and evaporated a second time to remove any remaining water. The sample was then placed in 200 μl of methanol with ∼0.2 mg of p-toluene sulfonic acid, and the solution was stored at room temperature in the dark overnight. The methanol was removed by evaporation, 100 μl of 0.1 M NaClO4 was added, and the sample was extracted with chloroform (2 times with 300 μl each time). The combined extracts were dried by passing through a small amount of cotton, the chloroform was evaporated, and the extract was dissolved in 60 μl of 40% methanol for LC-MS analysis.
RESULTS
High-resolution mass spectral analysis of M. jannaschii extracts.
Analysis of M. jannaschii cell extracts showed five isomers of coenzyme F430 eluting at 7.79, 8.08, 8.35, 8.46, and 8.76 min (Fig. 2A, middle). The relative ratios of the peaks were 5, 80, 100, 7, and 95, respectively, on the basis of the absorbance at 430 nm and the intensity of their M+ ions. The interconversion of F430 to different, more thermodynamically stable isomers has been observed previously (23–25) and has been studied by empirical force field analysis (26). Each peak showed the same molecular ion M+ at 905.2867 (calculated mass = 905.2862 for C42H51N6NiO13+) and confirmed the presence of F430 in the sample, as was expected. Each peak had the same isotopic signatures (Fig. 2B) with the following exact masses (and isotopomeric intensities): 905.2867 (100), 906.2927 (37), 907.2961 (40), 908.2865 (15), 909.2875 (7), and 910.2891 (2). The M+ ion is specifically observed as a result of the net single positive charge from the presence of Ni(II). The data are the same as those previously reported by others (27) and confirmed the presence of Ni in the molecule. The increase in the intensity of the M+ + 2 ion is indicative of Ni58 and Ni60 isotopes (Ni58 = 57.9353 [68.27%], Ni60 = 59.9308 [26.22%]). The doubly charged MH+2 at 453.1464 was also observed for each of the isomers, further supporting the identity of coenzyme F430. We have previously observed the same ions and isomers for F430 isolated from Methanobacterium thermoautotrophicum ΔH (unpublished results).
FIG 2.

High-resolution mass spectral analysis of M. jannaschii cell extract. (A) Total ion chromatogram of M. jannaschii cell extract (top); extracted ion chromatogram of M+ of 905.286 for coenzyme F430 showing isomers eluting at 7.79, 8.08, 8.35, 8.46, and 8.76 min (middle); extracted ion chromatogram of M+ of 1009.287 for F430-3 (bottom). The peaks at 6.26, 7.37, and 7.52 min represent the different isomers of F430-3. (B) Molecular ion isotope peaks for F430. (C) Molecular ion isotope peaks for F430-3.
In addition to the expected F430 peaks, three peaks eluting at 6.26, 7.37, and 7.52 min at relative ratios of 6, 37, and 100 were also observed. These peaks contained an F430 derivative that will be referred to as F430-3. The most intense of these peaks, the one eluting at 7.52 min, was similar in intensity to the most intense F430 peak (Fig. 2A, bottom). Each peak had the same molecular ion M+ at 1,009.2781 (calculated exact mass = 1,009.2794 for C45H55N6NiO15S+). Each peak also had the same isotopic signatures (Fig. 2C) with the following exact masses (and relative isotopomeric intensities): 1,009.2781 (100), 1,010.2847 (43), 1,011.2780 (46), 1,012.2786 (23), 1,013.2728 (11), and 1,014.2744 (4). The doubly charged MH+2 at 505.1452 was also observed with the following isotope peaks (and relative isotopomeric intensities): 505.1452 (100), 505.6462 (51), 506.1425 (60), 506.6469 (25), 507.1407 (13), and 507.6426 (5). The calculated ratios for the six isotope peaks with nominal masses of 1,009 to 1,014 are 100, 52.9, 59.8, 28.4, 15.9, and 5.9, which is consistent with the observed isotope peaks. These same peaks for F430-3 were also observed in negative ion mode at the same elution positions. The major ion observed was the (M+ − 2H)− at 1,007.2595 (calculated exact mass = 1,007.2649 for C45H53N6NiO15S−) for each of the isomers and the (M+ -3H)−2 at 503.1265 (calculated exact mass = 503.1288 for C45H52N6NiO15S2−) for the doubly negatively charged molecule.
Characterization of the coenzyme F430 variant from M. jannaschii.
Coenzymes F430 and F430-3 from the M. jannaschii cell extract were partially purified by strong anion-exchange chromatography. F430-3 eluted after F430 in the chromatographic system. In order to obtain absorbance data simultaneously with mass spectral data, the fractions containing F430 and F430-3 were assayed by high-pressure liquid chromatography interfaced with a photodiode array detector and a mass spectrometer (LC-UV-MS). The UV absorbance spectrum of F430 was as expected (Fig. 3, top), while the UV spectrum of the F430-3 was surprisingly quite different (Fig. 3, bottom) and did not have a 430-nm absorbance band. The major isomer of F430-3 showed absorbances at 224, 281, ∼330, and ∼365 nm, and the minor isomer showed absorbances at 209, 285, ∼317, and ∼373 nm. Each absorbance peak corresponded to the peaks in the F430-3 1,009 M+ ion elution profile. Neither of these UV spectra is like any of the spectra for the eight different tetrahydro- and hexahydrocorphinoid Ni(II) complexes used to model the F430 structure (28–30). The spectra are not consistent with a change in the α-axial ligands to the Ni(III), since such ligation is reported to affect the absorbance spectrum to only a small extent (31). The spectrum that we observed for F430-3 was, however, comparable to the UV spectrum of the F330 produced by the reduction of the exocyclic keto group at the 173 position of the tetrahydrocorphin with borohydride (32). This reduction produces a 100-nm shift in the absorbance and a decrease in the absorbance intensity at the higher wavelength, similar to what we observed. We propose that the same hydroxyl is present in F430-3, and the elimination of this π bond decreases the overall π-bond order. Reduction of the keto group, however, would increase the mass of the molecule by 2 mass units. Since this was not the case, another site of desaturation not conjugated to the absorbing system must be in the structure. We propose that this results from the introduction of the tetrahydrothiophene ring in F430-3 as opposed to the presence of a linear 3-mercaptopropionate moiety.
FIG 3.
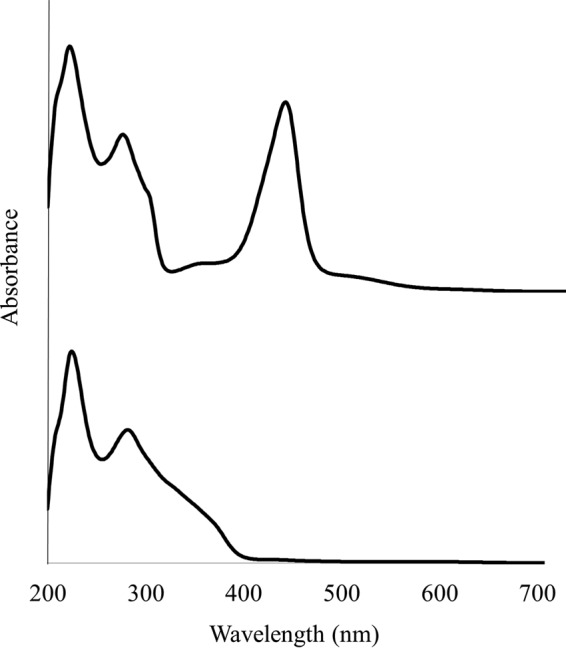
UV-visible spectra of F430 (top) and F430-3 (bottom) isolated from M. jannaschii.
The UV data, retention times, and mass spectral data all indicate that the F430-3 from M. jannaschii is the structure depicted in Fig. 1. The difference in the masses between the M+ for F430 at 905.2867 and the modified F430-3 at 1,009.2781 is 103.9914, corresponding to C3H4O2S (calculated exact mass = 103.9932). This represents the addition of either 2- or 3-mercaptopropionate to the F430 structure. The addition of a carboxylic acid to the F430 structure is consistent with the increased polarity of F430-3, as evidenced by the retention time compared to that of F430 on reverse-phase and anion-exchange columns. Confirmation of this addition was obtained by preparing a methyl ester derivative, which was found to contain six methyl groups with an M+ of 1,093 [1,009 + (6 × 14)]. This is 118 mass units larger than that observed for the pentamethylated F430 M+ of 975 [905 + (5 × 14)], consistent with the addition of one more methyl group to F430-3 compared to the number of methyl groups on F430. On the basis of the observed exact mass of F430-3, the mercaptopropionate moiety would have to be added to the F430 structure not as an amide, ester, or thioester but as an insertion of the sulfur into a C—H bond. A precedent for this can be found in the structure of the first F430-2 variant shown in Fig. 1, which was isolated from ANME-1 (19). In this structure, a CH3S group is added to the 172 carbon. We propose that this is the case here for addition of the mercaptopropionate modification. The addition of such a group at 172, however, would not explain the change in the UV absorbance spectrum that was observed, as described above. Instead, this can be resolved by an intramolecular Claisen condensation of the introduced group with the carbonyl group at the 173 position. This would produce the desired mass, and an alcohol in place of a carbonyl at the 173 position would alter the UV to be consistent with what we observed (32). Additional support for the F430-3 structure is the observation of a M − 104 fragment in the high-energy collision-induced dissociation (CID) mass spectral analysis, which corresponds to the loss of a thiirane-2-carboxylic acid moiety (Fig. 4). An equivalent fragment was not seen in the F430 high-energy CID spectrum.
FIG 4.

Observed molecular ion and CID fragment ions for F430-3. Analogous fragments were observed for F430, except for the M+ − 104 fragment.
Identification of additional F430 variants in methanogens and ANME.
Having found and characterized F430-3, we then searched for the presence of this molecule in two other methanogens, Methanococcus maripaludis and Methanococcus vannielii, as well as in samples containing archaea of the ANME type. The samples containing a mixture of ANME-1 and ANME-2 were derived from two specific enrichments from the Black Sea (33). The ANME-1 sample was from the Guaymas Basin (Gulf of California). To date, enrichments from the Guaymas Basin are the only cultures of ANME-1 that proliferate under laboratory conditions (34). The samples of ANME-2 were derived from enrichments obtained from Hydrate Ridge (35) and Caldera (Mediterranean seep).
F430-3 was present in M. maripaludis but absent from the other organisms that we analyzed (Table 1). In the process of our analysis, we consistently observed a wide assortment of additional compounds that contained Ni on the basis of the clear isotopic signature and had masses within 50 m/z units of the mass observed for F430. When the UV data could be obtained, most of these additional molecules had absorbance spectra consistent with those for the F430 core structure. The results of our search for additional F430 variants are reported in an abbreviated format in Table 1 and with further details in Tables S1 to S5 in the supplemental material. We observed a total of 10 F430-related coenzymes, including the canonical F430 and the previously described F430-2 coenzyme. The proposed structures (Fig. 5) were deduced from the high-resolution mass measurements and the assumption that they are all derived biosynthetically from F430. Since F430 has a wide distribution in deep sea sediments (27) and was detected in all the samples and since the new F430 structures can be explained by biochemically feasible alterations of the original F430 structure, it is very likely that all of these variants are in fact derived from F430.
TABLE 1.
Occurrence of coenzyme F430 and its variants in methanogens and ANMEa
| Sample | Occurrence of the following variants: |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| F430 | F430-2 | F430-3 | F430-4 | F430-5 | F430-6 | F430-7 | F430-8 | F430-9 | F430-10 | |
| M. jannaschii | +++ | − | +++ | − | − | − | − | − | − | + |
| M. maripaludis | +++ | − | +++ | +++ | ++ | − | − | ++ | − | − |
| M. vannielii | +++ | − | − | ++ | + | − | − | − | − | − |
| Black Sea ANME-1 and ANME-2 | +++ | +++ | − | +++ | ++ | − | − | + | − | − |
| Guaymas Basin ANME-1 | +++ | +++ | − | − | − | + | ++ | − | + | − |
| Caldera ANME-2 | +++ | +++ | − | +++ | ++ | − | − | − | − | − |
| Hydrate Ridge ANME-2 | +++ | +++ | − | +++ | ++ | − | − | − | − | − |
The corresponding structures are shown in Fig. 5. +, ++, and +++, the relative amount of each coenzyme detected; −, coenzyme not detected.
FIG 5.

Proposed structures for the F430 variants based on mass spectral data and how they are chemically related to F430. The red portions indicate the proposed structural modifications.
Coenzyme F430-2 was not detected in the three methanogens but was detected in all ANME samples. 12,13-Didehydro-F430 (Fig. 5, F430-4) was detected in all samples except the M. jannaschii and Guaymas Basin samples. This molecule has been reported and is considered to be an oxidative degradation product of F430 (24). However, since the samples in this study were all extracted by the same method yet this compound was not detected in all the samples, F430-4 may not simply be produced by an air oxidation of F430 but could be a functioning coenzyme in some of these organisms. Additional support for the possibility that F430-4 is biologically relevant is that we did not detect a didehydro form of F430-2 or F430-3, even though they should also be susceptible to air oxidation, just as was observed with F430.
An intriguing F430 derivative that was detected in some samples of both methanogens and ANME has a mass of 859.2836, consistent with oxidative decarboxylation of one of the propionic acid side chains of F430 to generate vinyl-F430 (Fig. 5, F430-5). There are three possible positions for the vinyl modification, which we cannot yet distinguish. A derivative of F430-2 containing a vinyl side chain was also observed in the sample of ANME-1 from Guaymas Basin (Fig. 5, F430-6). These F430 derivatives with a vinyl group can be compared to heme, which contains two propionic acid-derived vinyl groups.
Another possible modified form of F430-2 was detected in samples containing ANME-1. The mass spectral data for this molecule, designated F430-7 (Fig. 5), is consistent with the presence of a methyl sulfoxide in place of the methyl thiol. We also detected various oxidized F430s with the addition of either an extra keto group (F430-8), a hydroxyl group (F430-9), or two hydroxyl groups (F430-10). We cannot yet confirm the position of these modifications; however, we propose that the modifications are present at the 172 or 173 position.
DISCUSSION
Here we report the identification of several previously unknown modified coenzyme F430 molecules in methanogens and ANME. Our ability to identify these compounds was based on two primary factors. First, Ni-containing F430 molecules are readily identified in a complex mixture by mass spectrometry due to the unique isotopic signature and exact mass of Ni. Second, the Ni acts as a built-in single positive charge which allows the detection of molecular ions for the different F430 molecules using low-energy electrospray ionization. The presence of this single positive charge greatly increases the sensitivity of detection. These two factors, coupled with high-resolution mass measurements and fragmentation information, have allowed us to propose structures to the compounds and possible biosynthetic pathways for their production. Although this work represents a special case of metabolite identification, it demonstrates the power of current analytical methods to obtain likely chemical structures with only trace amounts of metabolites.
Since the distribution of the modified F430 coenzymes varies considerably among the organisms analyzed, while the canonical coenzyme F430 is present in all the samples, it is very unlikely that the new F430 variants result from abiotic transformations of F430. Additionally, the chemistry required for all transformations, except the didehydro derivative (F430-4), could not occur spontaneously. The presence of these modified coenzymes in these organisms indicates that they must be biologically relevant and brings up two important questions. First, what is the purpose of these modifications, and second, how are they generated biosynthetically?
The previously described F430-2 was originally characterized from samples collected from the Black Sea (18). This modified F430 was shown to function in an MCR homolog from ANME that presumably functions in vivo to oxidize methane instead of generate methane (16). This distinct functional role of F430-2 may be the reason for the presence of the 172 methyl thiol group; however, the role of the modification has not been elucidated. A recent study suggested that F430-2 was associated only with ANME-1 and not ANME-2 (36). However, we observed F430-2 in both the Hydrate Ridge and Caldera samples, which contain only ANME-2. This indicates that F430-2 and not F430 is exclusively used by the MCR homolog to perform the first step in AOM in ANME-1 and ANME-2. Additionally, our studies indicate that the presence of F430-2 can possibly be used as a marker for both groups of ANME.
Since the only known function of F430 is as a coenzyme for MCR, the presence of multiple modified F430 coenzymes is unexpected and suggests that F430 may be involved in many other reactions. Another possibility is that the modifications of F430 modulate the activity of MCR under various environmental conditions. We are currently carrying out studies to determine the protein that binds F430-3 in M. jannaschii and M. maripaludis. Our current results indicate that F430-3 is not associated with MCR. When the total cellular proteins present in M. jannaschii were fractionated via anion-exchange chromatography, F430, but not F430-3, was identified in the fraction containing MCR. F430-3 was instead present in a fraction containing other proteins.
F430-5 and F430-6, which contain a vinyl side chain in place of a carboxylate, are reminiscent of heme. The vinyl modification allows the heme to bind to hydrophobic protein cores and, more importantly, act as a site for its covalent attachment to the protein through cysteine residues (37, 38). We are presently performing studies to determine whether this modification allows the covalent attachment of F430 to MCR and/or other F430-dependent enzymes that have not yet been discovered.
Model reactions (39) indicate that the Ni in F430 can function in a manner analogous to Co in vitamin B12-related coenzymes found in the methanogens (40). This indicates that enzymes with these modified F430s as coenzymes have the potential to function in a wide assortment of different reactions, as seen with vitamin B12 biochemistry (41). Thus, F430 variants may be involved in methyl group transfer to CoM, as seen in the methanol:CoM-methyltransferase (42), or they may be involved in the formation or breakdown of acetate, as seen in acetyl coenzyme A synthase/decarboxylase (43). The possibilities are vast and will likely reveal new coenzyme F430-dependent chemistry.
The biosyntheses of F430-2 and F430-3 likely involve similar chemistries. Thio methyl group biosynthesis is well-known in many different molecules. In the biosynthesis of caldariellaquinone in Sulfolobus spp. (44), both the sulfur and the methyl groups are derived from methionine but not as an intact subunit (45). The enzyme(s) that catalyzes this reaction has not yet been described. Other examples involve enzymes belonging to the radical S-adenosyl-l-methionine (SAM) superfamily termed methylthiotransferases (MTTases). Radical SAM enzymes use a reduced [Fe-4S]+1 cluster (cluster I) and SAM to generate a 5′-deoxyadenosyl radical (Ado-CH2·) (46). Ado-CH2· then abstracts a hydrogen atom from a generally unreactive site to activate a substrate for further chemistry. MTTases, exemplified by MiaB and RimO, catalyze the generation of an —SCH3 group in place of a hydrogen on either tRNA or a ribosomal protein substrate, respectively, resulting in the formation of a methyl thioether (47). The source of the sulfur for this reaction is still unclear, but it has been proposed to arise from a sulfide coordinated by an auxiliary [4Fe-4S] cluster (cluster II) (48, 49). The methyl group is derived from one of the two SAM molecules required for the reaction (47). A series of reactions analogous to the chemistry performed by radical SAM MTTases provides an excellent mechanism for the biosynthesis of F430-2, as shown in Fig. 6 (48). In the first part of the reaction, the methyl group of SAM is transferred to a sulfur coordinated by the [4Fe-4S] cluster II. A radical is then formed at the 172 position of F430 via hydrogen atom abstraction with typical radical SAM chemistry. The substrate radical attacks the sulfur of the methylated sulfur atom to generate 172-methylthio-F430.
FIG 6.

Possible pathway for the biosynthesis of F430-2. SAH, S-adenosylhomocysteine.
Biosynthesis of F430-3 likely also involves radical SAM-dependent chemistry and may also require two molecules of SAM, as proposed for F430-2 biosynthesis. In our first proposed biosynthetic scheme (Fig. 7A), sulfide bound by the [4Fe-4S] cluster II attacks the C-4 carbon of SAM, resulting in the transfer of the aminocarboxypropyl (ACP) portion of SAM and yielding a homocysteine bound to the [4Fe-4S] cluster II. This use of the ACP portion of SAM through the exploitation of the electrophilicity of the C-4 carbon has precedent in N-acylhomoserine lactone production and in tRNA modification (50). Next, a radical SAM-generated 172 radical attacks the sulfur of the homocysteine to yield an F430 intermediate containing a homocysteine in place of the thiomethyl group seen for F430-2. The amino acid could then be converted to the keto acid through a transamination reaction followed by an oxidative decarboxylation and an intramolecular Claisen condensation to produce F430-3 (Fig. 7A). Alternatively, sulfur insertion may be the first step in the biosynthesis of F430-3 (not shown here), catalyzed by an enzyme similar to the radical SAM sulfur insertion enzymes biotin synthase and lipoyl synthase (51, 52). If this were the case, the newly added sulfide at the 172 position would react with SAM and the remaining biosynthetic scheme in Fig. 7A would proceed and would be catalyzed by additional enzymes. A more straightforward mechanism in which the 172 radical reacts directly with 3-mercaptopropionate coordinated to the [4Fe-4S] cluster II followed by a Claisen condensation to produce the final F430-3 is also possible (Fig. 7B). 3-Mercaptopropionate is highly prevalent in anoxic marine environments and is synthesized from methionine, homocysteine, or acrylate and dimethyl sulfide (53, 54). If any of these proposed mechanisms is occurring, the enzyme catalyzing this reaction is likely to harbor two [4Fe-4S] clusters, one for radical SAM chemistry (cluster I) and the other to bind the sulfur-containing substrate or provide the sulfur for the reaction (cluster II).
FIG 7.

Possible pathways for the biosynthesis of F430-3.
F430-5 and F430-6, which contain a vinyl side chain, are likely derived from the oxidative decarboxylation of the propionic acid side chain via a mechanism similar to that for the well-characterized radical SAM enzyme anaerobic coproporphyrinogen III oxidase (HemN) (55).
The identification of these modified F430s in methanogens and ANME opens up exciting new areas for future investigations. Coenzyme F430 may be involved in several biochemical reactions that await characterization, and the reactions/enzymes required for generation of the modifications will reveal unique biochemistry. Further work will also be necessary to analyze a variety of other methanogens for the presence of these and other modified F430 coenzymes. Importantly, this work further strengthens the biochemical similarity between methanogens and ANME.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Science Foundation grant MCB0722787 awarded to R.H.W. G.W. was supported by the DFG excellence cluster MARUM and a DFG Leibniz grant to Antje Boetius. The mass spectrometry resources are maintained by the Virginia Polytechnic Institute and State University Mass Spectrometry Incubator, a facility operated in part through funding by the Fralin Life Science Institute at Virginia Polytechnic Institute and State University and by the Agricultural Experiment Station Hatch Program (CRIS project number VA-135981).
We thank Walter Niehaus for valuable discussion and W. Keith Ray for obtaining the high-resolution mass spectrometry data. We thank Antje Boetius and Friedrich Widdel for providing the AOM enrichments and Thomas Holler, Ramona Appel, and Susanne Menger for long-standing AOM culture maintenance.
Footnotes
Published ahead of print 8 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02202-14.
REFERENCES
- 1.Costa KC, Leigh JA. 2014. Metabolic versatility in methanogens. Curr. Opin. Biotechnol. 29C:70–75. 10.1016/j.copbio.2014.02.012 [DOI] [PubMed] [Google Scholar]
- 2.Ermler U, Grabarse W, Shima S, Goubeaud M, Thauer RK. 1997. Crystal structure of methyl-coenzyme M reductase: the key enzyme of biological methane formation. Science 278:1457–1462. 10.1126/science.278.5342.1457 [DOI] [PubMed] [Google Scholar]
- 3.Dey M, Li X, Kunz RC, Ragsdale SW. 2010. Detection of organometallic and radical intermediates in the catalytic mechanism of methyl-coenzyme M reductase using the natural substrate methyl-coenzyme M and a coenzyme B substrate analogue. Biochemistry 49:10902–10911. 10.1021/bi101562m [DOI] [PubMed] [Google Scholar]
- 4.Ragsdale SW. 2007. Nickel and the carbon cycle. J. Inorg. Biochem. 101:1657–1666. 10.1016/j.jinorgbio.2007.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen SL, Blomberg MR, Siegbahn PE. 2012. How is methane formed and oxidized reversibly when catalyzed by Ni-containing methyl-coenzyme M reductase? Chemistry (Easton) 18:6309–6315. 10.1002/chem.201200274 [DOI] [PubMed] [Google Scholar]
- 6.Scheller S, Goenrich M, Thauer RK, Jaun B. 2013. Methyl-coenzyme M reductase from methanogenic archaea: isotope effects on label exchange and ethane formation with the homologous substrate ethyl-coenzyme M. J. Am. Chem. Soc. 135:14985–14995. 10.1021/ja4064876 [DOI] [PubMed] [Google Scholar]
- 7.Hausinger RP, Ormejohnson WH, Walsh C. 1984. Nickel tetrapyrrole cofactor F430: comparison of the forms bound to methyl S-coenzyme M reductase and protein free in cells of Methanobacterium thermoautotrophicum ΔH. Biochemistry 23:801–804. 10.1021/bi00300a003 [DOI] [Google Scholar]
- 8.Pfaltz A, Jaun B, Fassler A, Eschenmoser A, Jaenchen R, Gilles HH, Diekert G, Thauer RK. 1982. Factor F430 from methanogenic bacteria: structure of the porphinoid ligand system. Helv. Chim. Acta 65:828–865. 10.1002/hlca.19820650320 [DOI] [Google Scholar]
- 9.Livingston DA, Pfaltz A, Schreiber J, Eschenmoser A, Ankelfuchs D, Moll J, Jaenchen R, Thauer RK. 1984. Factor F430 from methanogenic bacteria: structure of the protein-free factor. Helv. Chim. Acta 67:334–351. 10.1002/hlca.19840670141 [DOI] [Google Scholar]
- 10.Boetius A, Wenzhofer F. 2013. Seafloor oxygen consumption fuelled by methane from cold seeps. Nat. Geosci. 6:725–734. 10.1038/ngeo1926 [DOI] [Google Scholar]
- 11.Holler T, Widdel F, Knittel K, Amann R, Kellermann MY, Hinrichs KU, Teske A, Boetius A, Wegener G. 2011. Thermophilic anaerobic oxidation of methane by marine microbial consortia. ISME J. 5:1946–1956. 10.1038/ismej.2011.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kellermann MY, Wegener G, Elvert M, Yoshinaga MY, Lin YS, Holler T, Mollar XP, Knittel K, Hinrichs KU. 2012. Autotrophy as a predominant mode of carbon fixation in anaerobic methane-oxidizing microbial communities. Proc. Natl. Acad. Sci. U. S. A. 109:19321–19326. 10.1073/pnas.1208795109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knittel K, Boetius A. 2009. Anaerobic oxidation of methane: progress with an unknown process. Annu. Rev. Microbiol. 63:311–334. 10.1146/annurev.micro.61.080706.093130 [DOI] [PubMed] [Google Scholar]
- 14.Lösekann T, Knittel K, Nadalig T, Fuchs B, Niemann H, Boetius A, Amann R. 2007. Diversity and abundance of aerobic and anaerobic methane oxidizers at the Haakon Mosby Mud Volcano, Barents Sea. Appl. Environ. Microbiol. 73:3348–3362. 10.1128/AEM.00016-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meyerdierks A, Kube M, Lombardot T, Knittel K, Bauer M, Glöckner FO, Reinhardt R, Amann R. 2005. Insights into the genomes of archaea mediating the anaerobic oxidation of methane. Environ. Microbiol. 7:1937–1951. 10.1111/j.1462-2920.2005.00844.x [DOI] [PubMed] [Google Scholar]
- 16.Scheller S, Goenrich M, Boecher R, Thauer RK, Jaun B. 2010. The key nickel enzyme of methanogenesis catalyses the anaerobic oxidation of methane. Nature 465:606–608. 10.1038/nature09015 [DOI] [PubMed] [Google Scholar]
- 17.Hallam SJ, Putnam N, Preston CM, Detter JC, Rokhsar D, Richardson PM, DeLong EF. 2004. Reverse methanogenesis: testing the hypothesis with environmental genomics. Science 305:1457–1462. 10.1126/science.1100025 [DOI] [PubMed] [Google Scholar]
- 18.Krüger M, Meyerdierks A, Glöckner FO, Amann R, Widdel F, Kube M, Reinhardt R, Kahnt J, Bocher R, Thauer RK, Shima S. 2003. A conspicuous nickel protein in microbial mats that oxidize methane anaerobically. Nature 426:878–881. 10.1038/nature02207 [DOI] [PubMed] [Google Scholar]
- 19.Mayr S, Latkoczy C, Krüger M, Günther D, Shima S, Thauer RK, Widdel F, Jaun B. 2008. Structure of an F430 variant from archaea associated with anaerobic oxidation of methane. J. Am. Chem. Soc. 130:10758–10767. 10.1021/ja802929z [DOI] [PubMed] [Google Scholar]
- 20.Mukhopadhyay B, Johnson EF, Wolfe RS. 1999. Reactor-scale cultivation of the hyperthermophilic methanarchaeon Methanococcus jannaschii to high cell densities. Appl. Environ. Microbiol. 65:5059–5065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin W, Whitman WB. 2004. The importance of porE and porF in the anabolic pyruvate oxidoreductase of Methanococcus maripaludis. Arch. Microbiol. 181:68–73. 10.1007/s00203-003-0629-1 [DOI] [PubMed] [Google Scholar]
- 22.Jones JB, Stadtman TC. 1977. Methanococcus vannielii: culture and effects of selenium and tungsten on growth. J. Bacteriol. 130:1404–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keltjens JT, Hermans JM, Rijsdijk GJ, van der Drift C, Vogels GD. 1988. Interconversion of F430 derivatives of methanogenic bacteria. Antonie Van Leeuwenhoek 54:207–220. 10.1007/BF00443579 [DOI] [PubMed] [Google Scholar]
- 24.Pfaltz A, Livingston AD, Juan B, Diekert G, Thauer RK, Eschenmoser A. 1985. Zur Kenntnis des Faktors F430 aus methanogenen Bakterien: Über die Natur der Isolierungsartefakte von F430, ein Beitrag zur Chemie von F430 und zur konformationellen Stereochemie der Ligandperipherie von hydroporphinoiden Nickel(II)-Komplexen. Helv. Chim. Acta 68:1338–1358. 10.1002/hlca.19850680527 [DOI] [Google Scholar]
- 25.Diekert G, Konheiser U, Piechulla K, Thauer RK. 1981. Nickel requirement and factor F430 content of methanogenic bacteria. J. Bacteriol. 148:459–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zimmer M. 1993. Empirical force field analysis of the revised structure of coenzyme F430. Epimerization and geometry of the corphinoid tetrapyrrole. J. Biomol. Struct. Dyn. 11:203–214 [DOI] [PubMed] [Google Scholar]
- 27.Takano Y, Kaneko M, Kahnt J, Imachi H, Shima S, Ohkouchi N. 2013. Detection of coenzyme F430 in deep sea sediments: a key molecule for biological methanogenesis. Org. Geochem. 58:137–140. 10.1016/j.orggeochem.2013.01.012 [DOI] [Google Scholar]
- 28.Fassler A, Pfaltz A, Muller PM, Farooq S, Kratky C, Krautler B, Eschenmoser A. 1982. Preparation and properties of some hydrocorphinoid nickel(II)-complexes. Helv. Chim. Acta 65:812–827. 10.1002/hlca.19820650319 [DOI] [Google Scholar]
- 29.Fassler A, Pfaltz A, Krautler B, Eschenmoser A. 1984. Chemistry of corphinoids: synthesis of a nickel(II) complex containing the chromophore system of coenzyme F430. J. Chem. Soc. Chem. Commun. 1984:1365–1367. 10.1039/C39840001365 [DOI] [Google Scholar]
- 30.Kratky C, Fassler A, Pfaltz A, Krautler B, Jaun B, Eschenmoser A. 1984. Chemistry of corphinoids: structural properties of corphinoid nickel(II) complexes related to coenzyme F430. J. Chem. Soc. Chem. Commun. 1984:1368–1371. 10.1039/C39840001368 [DOI] [Google Scholar]
- 31.Bauer C, Jaun B. 2003. Derivatives of coenzyme F430 with a covalently attached alpha-axial ligand. Part II. Partial synthesis of the five coenzyme F430 tetramethyl esters and of a derivative with a coordinating NTT–methyl-l-histidine ligand covalently attached to the side chain at C(3) of F430 via a peptidic linker. Helv. Chim. Acta 86:4254–4269. 10.1002/hlca.200390348 [DOI] [Google Scholar]
- 32.Dey M, Kunz RC, Van Heuvelen KM, Craft JL, Horng YC, Tang Q, Bocian DF, George SJ, Brunold TC, Ragsdale SW. 2006. Spectroscopic and computational studies of reduction of the metal versus the tetrapyrrole ring of coenzyme F430 from methyl-coenzyme M reductase. Biochemistry 45:11915–11933. 10.1021/bi0613269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wegener G, Niemann H, Elvert M, Hinrichs KU, Boetius A. 2008. Assimilation of methane and inorganic carbon by microbial communities mediating the anaerobic oxidation of methane. Environ. Microbiol. 10:2287–2298. 10.1111/j.1462-2920.2008.01653.x [DOI] [PubMed] [Google Scholar]
- 34.Holler T, Wegener G, Niemann H, Deusner C, Ferdelman TG, Boetius A, Brunner B, Widdel F. 2012. Carbon and sulfur back flux during anaerobic microbial oxidation of methane and coupled sulfate reduction. Proc. Natl. Acad. Sci. U. S. A. 109:21170–21173. 10.1073/pnas.1218683109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knittel K, Lösekann T, Boetius A, Kort R, Amann R. 2005. Diversity and distribution of methanotrophic archaea at cold seeps. Appl. Environ. Microbiol. 71:467–479. 10.1128/AEM.71.1.467-479.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaneko M, Takano Y, Chikaraishi Y, Ogawa NO, Asakawa S, Watanabe T, Shima S, Krüger M, Matsushita M, Kimura H, Ohkouchi N. 2014. Quantitative analysis of coenzyme F430 in environmental samples: a new diagnostic tool for methanogenesis and anaerobic methane oxidation. Anal. Chem. 86:3633–3638. 10.1021/ac500305j [DOI] [PubMed] [Google Scholar]
- 37.Bali S, Palmer DJ, Schroeder S, Ferguson SJ, Warren MJ. 2014. Recent advances in the biosynthesis of modified tetrapyrroles: the discovery of an alternative pathway for the formation of heme and heme d1. Cell. Mol. Life Sci. 71:2837–2863. 10.1007/s00018-014-1563-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kranz RG, Richard-Fogal C, Taylor JS, Frawley ER. 2009. Cytochrome c biogenesis: mechanisms for covalent modifications and trafficking of heme and for heme-iron redox control. Microbiol. Mol. Biol. Rev. 73:510–528. 10.1128/MMBR.00001-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dey M, Li X, Zhou Y, Ragsdale SW. 2010. Evidence for organometallic intermediates in bacterial methane formation involving the nickel coenzyme F430. Met. Ions Life Sci. 7:71–110 [DOI] [PubMed] [Google Scholar]
- 40.Stupperich E, Krautler B. 1988. Pseudo vitamin-B12 or 5-hydroxybenzimidazolyl-cobamide are the corrinoids found in methanogenic bacteria. Arch. Microbiol. 149:268–271. 10.1007/BF00422016 [DOI] [Google Scholar]
- 41.Frey PA. 2010. Cobalamin coenzymes in enzymology, p 501–546 In Comprehensive natural products chemistry and biology, vol 7 Elsevier, Oxford, United Kingdom [Google Scholar]
- 42.Zydowsky LD, Zydowsky TM, Haas ES, Brown JW, Reeve JN, Floss HG. 1987. Stereochemical course of methyl transfer from methanol to methyl coenzyme M in cell-free extracts of Methanosarcina barkeri. J. Am. Chem. Soc. 109:7922–7923. 10.1021/ja00259a073 [DOI] [Google Scholar]
- 43.Boer JL, Mulrooney SB, Hausinger RP. 2014. Nickel-dependent metalloenzymes. Arch. Biochem. Biophys. 544:142–152. 10.1016/j.abb.2013.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou D, White RH. 1989. Biosynthesis of caldariellaquinone in Sulfolobus spp. J. Bacteriol. 171:6610–6616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou D, White RH. 1990. Biosynthesis of the methylthio side-chain of caldariellaquinone. J. Chem. Soc. Perkin Trans. 1 1990:2346–2348 [Google Scholar]
- 46.Frey PA, Hegeman AD, Ruzicka FJ. 2008. The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 43:63–88. 10.1080/10409230701829169 [DOI] [PubMed] [Google Scholar]
- 47.Atta M, Mulliez E, Arragain S, Forouhar F, Hunt JF, Fontecave M. 2010. S-Adenosylmethionine-dependent radical-based modification of biological macromolecules. Curr. Opin. Struct. Biol. 20:684–692. 10.1016/j.sbi.2010.09.009 [DOI] [PubMed] [Google Scholar]
- 48.Landgraf BJ, Arcinas AJ, Lee KH, Booker SJ. 2013. Identification of an intermediate methyl carrier in the radical S-adenosylmethionine methylthiotransferases RimO and MiaB. J. Am. Chem. Soc. 135:15404–15416. 10.1021/ja4048448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Forouhar F, Arragain S, Atta M, Gambarelli S, Mouesca JM, Hussain M, Xiao R, Kieffer-Jaquinod S, Seetharaman J, Acton TB, Montelione GT, Mulliez E, Hunt JF, Fontecave M. 2013. Two Fe-S clusters catalyze sulfur insertion by radical-SAM methylthiotransferases. Nat. Chem. Biol. 9:333–338. 10.1038/nchembio.1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fontecave M, Atta M, Mulliez E. 2004. S-Adenosylmethionine: nothing goes to waste. Trends Biochem. Sci. 29:243–249. 10.1016/j.tibs.2004.03.007 [DOI] [PubMed] [Google Scholar]
- 51.Booker SJ. 2009. Anaerobic functionalization of unactivated C—H bonds. Curr. Opin. Chem. Biol. 13:58–73. 10.1016/j.cbpa.2009.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fugate CJ, Jarrett JT. 2012. Biotin synthase: insights into radical-mediated carbon-sulfur bond formation. Biochim. Biophys. Acta 1824:1213–1222. 10.1016/j.bbapap.2012.01.010 [DOI] [PubMed] [Google Scholar]
- 53.Kiene RP, Taylor BF. 1988. Biotransformations of organosulfur compounds in sediments via 3-mercaptopropionate. Nature 332:148–150. 10.1038/332148a0 [DOI] [Google Scholar]
- 54.Hu HY, Mylon SE, Benoit G. 2006. Distribution of the thiols glutathione and 3-mercaptopropionic acid in Connecticut lakes. Limnol. Oceanogr. 51:2763–2774. 10.4319/lo.2006.51.6.2763 [DOI] [Google Scholar]
- 55.Layer G, Kervio E, Morlock G, Heinz DW, Jahn D, Retey J, Schubert WD. 2005. Structural and functional comparison of HemN to other radical SAM enzymes. Biol. Chem. 386:971–980. 10.1515/BC.2005.113 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.