

Abstract
Bifidobacteria represent one of the dominant microbial groups that are present in the gut of various animals, being particularly prevalent during the suckling stage of life of humans and other mammals. However, the overall genome structure of this group of microorganisms remains largely unexplored. Here, we sequenced the genomes of 42 representative (sub)species across the Bifidobacterium genus and used this information to explore the overall genetic picture of this bacterial group. Furthermore, the genomic data described here were used to reconstruct the evolutionary development of the Bifidobacterium genus. This reconstruction suggests that its evolution was substantially influenced by genetic adaptations to obtain access to glycans, thereby representing a common and potent evolutionary force in shaping bifidobacterial genomes.
INTRODUCTION
Bifidobacteria represent one of the dominant microbial groups that occur in the gut of various animals, including warm-blooded mammals and social insects (1, 2). In these environments, bifidobacteria reach a particularly high relative abundance as part of the infant gut microbiota (3–5), and this early life prevalence supports their purported role as modulators of various metabolic and immune activities of their immature host (1). Various members of the genus Bifidobacterium have attracted substantial scientific and commercial interest due to various professed beneficial health effects that they exert on their human host (6–10). Currently, the genus Bifidobacterium includes 47 taxa, involving 38 species and 9 subspecies (2, 11–14). Genomics has been crucial in revealing the evolutionary development as well as the biology of any taxonomical group of bacteria and thus in understanding the genetic forces that sustain specific adaptations to an ecological niche (15). However, representatives of only 10 of the 47 currently recognized bifidobacterial sub/species have been genomically decoded (1). Here, we describe the genome analysis of representatives of all 47 (sub)species that are currently assigned to the Bifidobacterium genus. Based on the generated genome information, we hypothesize that the bifidobacterial genome coevolved with its animal host via gene loss and, in particular, genetic acquisition events, of which the latter events appear to be responsible for species-specific adaptations to a glycan-rich environment.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All Bifidobacterium strains were cultivated in an anaerobic atmosphere (2.99% H2, 17.01% CO2, and 80% N2) in a chamber (Concept 400, Ruskin) on De Man-Rogosa-Sharp (MRS) broth (Scharlau Chemie, Barcelona, Spain) supplemented with 0.05% (wt/vol) l-cysteine hydrochloride and were incubated at 37°C. Bacterial cultures were subjected to DNA extraction using a previously described protocol (4).
Genome sequencing and bioinformatics analyses.
The genome sequences of all studied Bifidobacterium species were determined by GenProbio srl (Parma, Italy) using an Ion Torrent PGM platform (Life Technologies, Carlsbad, CA). A genomic library was generated using 1 μg of genomic DNA and an Ion Xpress Plus fragment library kit and employing the Ion Shear chemistry according to the user guide. After a dilution to 2.66 × 107 molecules/μl, 4.5 × 108 molecules were used as the template for clonal amplification on Ion Sphere particles during the emulsion PCR according to an Ion Xpress Template 400 kit manual. The quality of the amplification was estimated, and the amplification product was loaded onto an Ion 316 chip and was subsequently sequenced using 125 sequencing cycles according to an Ion Sequencing 400 kit user guide. A total of 125 sequencing cycles resulted in an average read length of approximately 400 nucleotides. The MIRA program (version 3.4.0) was used for de novo assembly of each bifidobacterial genome sequence (16). The number of contigs generated by MIRA was further subjected to manual inspection and alignment using SeqMan (Lasergene) software in order to identify putative overlaps between contig ends. These overlaps were validated by PCR, thus reducing the number of gaps in each bacterial chromosome.
Sequence annotation.
The analyzed genomes consisted of five complete and publicly available bifidobacterial genome sequences plus, as part of this study, 42 newly sequenced genomes. In order to ensure that identical sequence quality standards were applied to all investigated genomes, the five publicly available nucleotide sequences that we used as part of this study were reanalyzed using common software and parameters (see below). Overall DNA analyses of the similarities between the bifidobacterial genomes were carried out using BLASTN (17) and Artemis (18). Protein-encoding open reading frames (ORFs) were predicted using a combination of Prodigal (19) and BLASTX (17) for comparative analysis. Results of the gene-finder program were combined manually with data from BLASTP (20) analysis of a nonredundant protein database provided by the National Center for Biotechnology Information. The combined results were inspected by Artemis, which was used for a manual editing effort to verify and, if necessary, to redefine the start of each predicted coding region or to remove or add coding regions.
Assignment of protein functions to predicted coding regions of the bifidobacterial genomes was performed manually. Moreover, the revised gene/protein set was searched using the Swiss-Prot (www.expasy.ch/sprot/)/TrEMBL, PRIAM (http://priam.prabi.fr/), protein family (Pfam, http://pfam.sanger.ac.uk/), TIGRFam (http://www.jcvi.org/cms/research/projects/tigrfams/overview/), Interpro (INTERPROSCAN; http://www.ebi.ac.uk/Tools/InterProScan/), Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/), and COG (http://www.ncbi.nlm.nih.gov/COG/) databases, in addition to BLASTP (17). Functional assignments were defined by manual processing of the combined results. Manual corrections of automated functional assignments were completed on an individual gene-by-gene basis as needed.
Additional bioinformatic analyses included the following: identification of tRNA genes using tRNAscan-SE (21) and detection of rRNA genes using RNAmmer (http://www.cbs.dtu.dk/services/RNAmmer/) followed by manual annotation on the basis of BLASTN searches and Enzyme Commission (EC)/Gene Onthology (GO) annotation of ORFs using annot8r (22).
Insertion sequence (IS) families were assigned using ISFinder (http://www-is.biotoul.fr/), restriction-modification systems were searched using the REBASE database (23), transporter classification was performed according to the Transporter Classification Database scheme (24), and ORF attribution to a specific COG family of clusters of orthologous genes (COGs) was made by searching the COG database (http://www.ncbi.nlm.nih.gov/COG/).
Pan-genome and extraction of shared and unique genes.
For all bifidobacterial genomes used in this study, a pan-genome calculation was performed using the PGAP pipeline (25); the ORF content of all genomes was organized in functional gene clusters using the GF (Gene Family) method involving comparison of each protein to all other proteins using BLAST analysis (cutoff E value of 1 × 10−4 and 50% identity over at least 50% of both protein sequences), followed by clustering into protein families, named Bifidobacterium-specific clusters of orthologous genes (BifCOGs), using MCL (graph-theory-based Markov clustering algorithm) (26). A pan-genome profile was built using an optimized algorithm incorporated in PGAP software, based on a presence/absence matrix that included all identified bifCOGs in the analyzed genomes. Following this, the unique protein families for each of the 47 bifidobacterial genomes were classified. Protein families shared between all genomes, named core BifCOGs, were defined by selecting the families that contained at least one single protein member for each genome.
The PGAP pipeline calculation was performed again with the inclusion of the remaining members of the family Bifidobacteriaceae in order to predict the pan-genome and core COGs of the entire family.
Each set of orthologous proteins constituting core COGs with one member per genome was aligned using MAFFT (27), and phylogenetic trees were constructed using the neighbor-joining method in Clustal W version 2.1 (28). The supertree was built using FigTree (http://tree.bio.ed.ac.uk/software/figtree/). PhyloPhlAn (29) was used to construct an additional phylogenetic tree based on >400 proteins optimized from among 3,737 bacterial genomes. This method measures the sequence diversity of all clades, classifies genomes from deep-branching candidate divisions through closely related subspecies, and improves the consistency of the phylogenetic and taxonomic groupings based on the 400 most conserved bacterial proteins.
Prediction of gene acquisition and loss.
Prediction and tree visualization of gene acquisition and loss were performed with BlastGraph (30). Data from BLASTP (17) comparisons of all the deduced proteins derived from the pan-genome to each other were used as the input, and the clustering cutoff was set at 50% identity over at least 50% of both protein sequences.
Prediction of the mobilome of bifidobacteria.
The identification of the so-called bifidobacterial “mobilome” (i.e., the genes that may have been acquired by horizontal gene transfer [HGT]) was achieved by merging results from DarkHorse v1.5 (31) and suite COLOMBO v3.8 implemented with the program SIGI-HMM (32). DarkHorse was run with default parameters, and only results with an E value of <1e-30 were retained, while COLOMBO was run with a sensitivity value of 0.4.
Identification of CRISPR.
Clustered regularly interspaced short palindromic repeats (CRISPR) were identified using the CRISPR Finder software (33). Once CRISPR were identified, flanking coding sequences were analyzed and mined for the presence of cas genes. Once cas genes were identified, the universal cas1 gene, in combination with the signature genes for type I, type II, and type III CRISPR-associated proteins (Cas) systems, namely, cas3, cas9, and cas10, respectively, were used for CRISPR type assignment. Furthermore, CRISPR locus orientations were determined using the widely applicable codirectional transcription pattern of cas genes with the CRISPR-spacer array. Once the orientation of CRISPR was determined and the corresponding sequence established, CRISPR within a locus were identified, and interspacing sequences were established as spacers.
Data deposition.
The sequences reported in this paper have been deposited in the GenBank database under the accession numbers indicated in Table 1.
TABLE 1.
General features of the Bifidobacterium genomesa
| Genome | Bifidobacterium strain | Fold coverage | Genome status (no. of contigs)b | Approximate genome size (nt) | GC content | No. of ORFs | No. of ORFs with an assigned function | No. of tRNAs | No. of complete CRISPR loci | No. of partial CRISPR locic | Avg length of ORFs (nt) | ORF region (%) | Isolation source | GenBank accession no. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | B. actinocoloniiforme DSM 22766 | 88.61 | Draft (4) | 1,823,388 | 62.71 | 1,484 | 1,190 | 46 | 1 | 1,066.01 | 86.99 | Bumblebee digestive tract | JGYK00000000 | |
| 2 | B. adolescentis ATCC 15703 | Complete | 2,089,645 | 59.18 | 1,649 | 1,419 | 54 | 1 | 1,093.98 | 86.33 | Intestine of adult | AP009256.1 | ||
| 3 | B. angulatum LMG 11039 | 84.47 | Draft (6) | 2,003,806 | 59.41 | 1,523 | 1,314 | 48 | 1 | 1,139.59 | 86.61 | Human feces | JGYL00000000 | |
| 4 | B. animalis subsp. animalis LMG 10508 | 61.16 | Draft (13) | 1,915,007 | 60.47 | 1,527 | 1,254 | 52 | 1 | 1,081.93 | 86.27 | Rat feces | JGYM00000000 | |
| 5 | B. animalis subsp. lactis DSM 10140 | Complete | 1,938,606 | 60.48 | 1,518 | 1,242 | 52 | 1 | 1,100.61 | 86.18 | Fermented milk | CP001606.1 | ||
| 6 | B. asteroides LMG 10735 (PRL2011) | Complete | 2,167,304 | 60.05 | 1,653 | 1,363 | 44 | 1 | 1,138.82 | 86.86 | Honeybee hindgut | CP003325.1 | ||
| 7 | B. biavatii DSM 23969 | 45.11 | Draft (56) | 3,252,147 | 63.1 | 2,557 | 2,068 | 61 | 1,095.68 | 86.15 | Feces of tamarin | JGYN00000000 | ||
| 8 | B. bifidum LMG 11041 | 118.97 | Draft (2) | 2,208,468 | 62.67 | 1,704 | 1,270 | 53 | 1,088.44 | 83.98 | Feces from breast-fed infant | JGYO00000000 | ||
| 9 | B. bohemicum DSM 22767 | 140.24 | Draft (5) | 2,052,470 | 57.45 | 1,632 | 1,271 | 47 | 1 | 1 | 1,049.90 | 83.48 | Bumblebee digestive tract | JGYP00000000 |
| 10 | B. bombi DSM 19703 | 103.8 | Draft (4) | 1,895,239 | 56.08 | 1,454 | 1,121 | 48 | 1 | 1,081.75 | 82.76 | Bumblebee digestive tract | ATLK00000000 | |
| 11 | B. boum LMG 10736 | 71.1 | Draft (18) | 2,171,356 | 59.31 | 1,726 | 1,412 | 49 | 1 | 1,075.57 | 85.50 | Bovine rumen | JGYQ00000000 | |
| 12 | B. breve LMG 13208 | 21.14 | Draft (31) | 2,263,780 | 58.88 | 1,887 | 1,506 | 53 | 2 | 1,036.17 | 86.42 | Infant intestine | JGYR00000000 | |
| 13 | B. callitrichos DSM 23973 | 65.86 | Draft (33) | 2,887,313 | 63.52 | 2,364 | 1,970 | 58 | 1 | 1,051.77 | 86.11 | Feces of common marmoset | JGYS00000000 | |
| 14 | B. catenulatum LMG 11043 | 31.21 | Draft (11) | 2,082,756 | 56.11 | 1,664 | 1,396 | 55 | 1 | 1,072.31 | 85.67 | Adult intestine | JGYT00000000 | |
| 15 | B. choerinum LMG 10510 | 107.33 | Draft (20) | 2,096,123 | 65.53 | 1,672 | 1,397 | 55 | 1 | 1,074.54 | 85.71 | Piglet feces | JGYU00000000 | |
| 16 | B. coryneforme LMG 18911 | 182.57 | Complete | 1,755,151 | 60.51 | 1,364 | 1,133 | 56 | 1,130.49 | 87.85 | Honeybee hindgut | CP007287 | ||
| 17 | B. crudilactis LMG 23609 | 90.52 | Draft (6) | 2,362,816 | 57.72 | 1,883 | 1,606 | 45 | 1 | 1,089.40 | 86.82 | Raw cow milk | JHAL00000000 | |
| 18 | B. cuniculi LMG 10738 | 120.38 | Draft (41) | 2,531,592 | 64.87 | 2,194 | 1,661 | 63 | 3 | 994.86 | 86.22 | Rabbit feces | JGYV00000000 | |
| 19 | B. dentium LMG 11045 (Bd1) | Complete | 2,636,367 | 58.54 | 2,129 | 1,625 | 55 | 2 | 2 | 1,067.06 | 86.17 | Oral cavity | CP001750.1 | |
| 20 | B. gallicum LMG 11596 | 109.79 | Draft (12) | 2,004,594 | 57.61 | 1,507 | 1,293 | 58 | 2 | 1,116.18 | 83.91 | Adult intestine | JGYW00000000 | |
| 21 | B. gallinarum LMG 11586 | 244.47 | Draft (10) | 2,160,836 | 64.22 | 1,654 | 1,384 | 53 | 1,131.86 | 86.64 | Chicken cecum | JGYX00000000 | ||
| 22 | B. indicum LMG 11587 | 280.81 | Complete | 1,734,546 | 60.49 | 1,352 | 1,141 | 47 | 1,129.67 | 88.05 | Insect | CP006018 | ||
| 23 | B. kashiwanohense DSM 21854 | 63.96 | Draft (30) | 2,307,960 | 56.2 | 1,948 | 1,618 | 53 | 1,023.36 | 86.37 | Infant feces | JGYY00000000 | ||
| 24 | B. longum subsp. infantis ATCC 15697 | Complete | 2,832,748 | 59.86 | 2,500 | 1,939 | 79 | 974.00 | 85.96 | Intestine of infant | AP010889.1 | |||
| 25 | B. longum subsp. longum LMG 13197 | 34.84 | Draft (8) | 2,384,703 | 60.33 | 1,899 | 1,556 | 71 | 1,083.59 | 86.29 | Adult intestine | JGYZ00000000 | ||
| 26 | B. longum subsp. suis LMG 21814 | 70.88 | Draft (36) | 2,335,832 | 59.96 | 1,955 | 1,675 | 55 | 1 | 1,027.46 | 86.04 | Pig feces | JGZA00000000 | |
| 27 | B. magnum LMG 11591 | 80.25 | Draft (13) | 1,822,476 | 58.72 | 1,507 | 1,234 | 56 | 1 | 1,060.59 | 87.64 | Rabbit feces | JGZB00000000 | |
| 28 | B. merycicum LMG 11341 | 78.12 | Draft (16) | 2,280,236 | 60.33 | 1,741 | 1,413 | 53 | 1 | 1 | 1,105.42 | 84.45 | Bovine rumen | JGZC00000000 |
| 29 | B. minimum LMG 11592 | 231.93 | Draft (18) | 1,892,860 | 62.73 | 1,590 | 1,356 | 53 | 1 | 1,032.77 | 86.75 | Sewage | JGZD00000000 | |
| 30 | B. mongoliense DSM 21395 | 128.8 | Draft (43) | 2,170,490 | 62.78 | 1,798 | 1,514 | 47 | 1,040.02 | 86.15 | Fermented mare's milk | JGZE00000000 | ||
| 31 | B. pseudocatenulatum LMG 10505 | 52 | Draft (10) | 2,283,767 | 56.36 | 1,771 | 1,527 | 53 | 1 | 1,112.53 | 86.27 | Infant feces | JGZF00000000 | |
| 32 | B. pseudolongum subsp. globosum LMG 11569 | 151.96 | Draft (26) | 1,935,255 | 63.39 | 1,574 | 1,367 | 52 | 1,091.36 | 88.76 | Bovine rumen | JGZG00000000 | ||
| 33 | B. pseudolongum subsp. pseudolongum LMG 11571 | 85.19 | Draft (11) | 1,898,684 | 63.06 | 1,495 | 1,310 | 52 | 2 | 1,111.81 | 87.54 | Swine feces | JGZH00000000 | |
| 34 | B. psychraerophilum LMG 21775 | 72.17 | Draft (11) | 2,615,078 | 58.75 | 2,122 | 1,809 | 45 | 1,080.93 | 87.71 | Pig cecum | JGZI00000000 | ||
| 35 | B. pullorum LMG 21816 | 99.94 | Draft (11) | 2,153,559 | 64.22 | 1,691 | 1,466 | 53 | 1 | 1,097.52 | 86.18 | Chicken feces | JGZJ00000000 | |
| 36 | B. reuteri DSM 23975 | 61.47 | Draft (28) | 2,847,572 | 60.45 | 2,149 | 1,747 | 53 | 2 | 1,127.08 | 85.06 | Feces of common marmoset | JGZK00000000 | |
| 37 | B. ruminantium LMG 21811 | 102.57 | Draft (23) | 2,249,807 | 59.18 | 1,832 | 1,433 | 50 | 1 | 5 | 1,068.51 | 87.01 | Bovine rumen | JGZL00000000 |
| 38 | B. saeculare LMG 14934 | 68.91 | Draft (14) | 2,263,283 | 63.75 | 1,857 | 1,524 | 48 | 1,079.55 | 88.58 | Rabbit feces | JGZM00000000 | ||
| 39 | B. saguini DSM 23967 | 189.92 | Draft (33) | 2,787,036 | 56.35 | 2,321 | 1,853 | 59 | 1 | 1,055.87 | 87.93 | Feces of tamarin | JGZN00000000 | |
| 40 | B. scardovii LMG 21589 | 68.58 | Draft (34) | 3,141,793 | 64.63 | 2,480 | 2,098 | 55 | 1 | 1 | 1,070.48 | 84.50 | Blood | JGZO00000000 |
| 41 | B. stellenboschense DSM 23968 | 108.97 | Draft (40) | 2,812,864 | 65.34 | 2,202 | 1,810 | 59 | 1 | 1,100.08 | 86.12 | Feces of tamarin | JGZP00000000 | |
| 42 | B. stercoris DSM 24849 | 173.24 | Draft (15) | 2,304,613 | 59.38 | 1,891 | 1,548 | 54 | 1 | 1,070.70 | 87.85 | Adult feces | JGZQ00000000 | |
| 43 | B. subtile LMG 11597 | 123.73 | Draft (27) | 2,790,088 | 60.92 | 2,260 | 1,881 | 47 | 3 | 1,027.75 | 83.25 | Sewage | JGZR00000000 | |
| 44 | B. thermacidophilum subsp. porcinum LMG 21689 | 130.58 | Draft (3) | 2,079,368 | 60.2 | 1,738 | 1,229 | 40 | 1 | 1,009.07 | 84.34 | Piglet feces | JGZS00000000 | |
| 45 | B. thermacidophilum subsp. thermacidophilum LMG 21395 | 75.19 | Draft (8) | 2,233,072 | 60.38 | 1,823 | 1,339 | 48 | 1 | 1,021.36 | 83.38 | Anaerobic digester | JGZT00000000 | |
| 46 | B. thermophilum JCM 1207 | 84.48 | Draft (12) | 2,099,496 | 59.91 | 1,700 | 1,305 | 44 | 2 | 1,065.02 | 86.24 | Swine feces | JGZV00000000 | |
| 47 | B. tsurumiense JCM 13495 | 89.67 | Draft (25) | 2,164,426 | 52.84 | 1,629 | 1,403 | 46 | 2 | 1,114.56 | 83.88 | Hamster dental plaque | JGZU00000000 |
nt, nucleotides.
The genome sequences were fragmented into various numbers of contigs; the numbers are provided within parentheses.
CRISPR partial loci represent a CRISPR-Cas system without the full complement of cas genes.
RESULTS AND DISCUSSION
General features of Bifidobacterium genomes.
Genome sequences were determined for 42 distinct bifidobacterial strains, while an additional five bifidobacterial genome sequences were retrieved from the NCBI public database, together representing the neotype for each of the currently described 47 species and subspecies within the Bifidobacterium genus (34). The sequencing and assembly statistics of the 42 newly determined bifidobacterial genomes are summarized in Table 1. The approximate Bifidobacterium genome size ranged from 1.73 Mb (Bifidobacterium indicum) to 3.25 Mb (Bifidobacterium biavatii), corresponding to 1,352 and 2,557 predicted protein-encoding open reading frames, respectively (Table 1). Given the close phylogenetic relationship between bifidobacteria, such a substantial size difference suggests that bifidobacterial genomes have evolved as a result of many gene loss and/or acquisition events (35). Genome features of the sequenced bifidobacterial strains are presented in Table 1; functional annotations were assigned for 81.9% of the predicted ORFs identified in the analyzed members of the Bifidobacterium genus, representing the Bifidobacterium pan-genome (see below). The remaining 18.1% ORFs were assigned as proteins with an unknown function. Results from BLASTP searches of the NCBI database show that 17.7% of these ORFs of “unknown function” (corresponding to 3.2% of the total Bifidobacterium pan-genome) have homologs in other bacterial genera within the Bifidobacteriaceae family (i.e., members of the genera Scardovia, Parascardovia, Metascardovia, and Gardnerella). It is noteworthy that approximately 12.4% of the annotated ORFs were attributed to carbohydrate metabolism. These data are a genetic reflection of the metabolic commitment of bifidobacteria to a saccharolytic life style, a notion observed for other bacteria of the human gut microbiota (36).
The pan-genome, core genome, and variome of the Bifidobacterium genus.
Genome sequences from each of the 47 Bifidobacterium (sub)species were used to analyze the corresponding pan-genome, the core genome, and the variome (variable genome sequences), determined as described previously (37). A total of 18,181 BifCOGs (Bifidobacterium-specific clusters of orthologous genes), of which 6,464 had members present in at least two genomes, and which together represent the pan-genome of the Bifidobacterium genus, were identified in the 47 bifidobacterial genomes. The pan-genome size, when plotted versus the number of included genomes, clearly shows that the power trend line has yet to reach a plateau (Fig. 1). Nevertheless, the number of new genes discovered by sequential addition of genome sequences was reduced from 770 to 588 BifCOGs in the first three genome additions to 252 to 249 BifCOGs in the final three additions, indicating the existence of an open pan-genome within the Bifidobacterium genus. These findings suggest that additional sequencing efforts are needed in order to identify (essentially) all genes of members of this genus. Analysis of the set of predicted BifCOGs allowed the identification of 551 COGs shared by all 47 Bifidobacterium (sub)species, thereby representing the core of bifidobacterial genomic coding sequences (core BifCOGs). Plotting the identified number of core BifCOGs as a function of the included number of genomes shows that the core BifCOG set is not expected to be significantly reduced in number by the addition of further genomes since the exponential trendline essentially reached a plateau (Fig. 1). Inclusion of available genome sequences of other members of the family Bifidobacteriaceae (i.e., Gardnerella vaginalis 409-05, Metascardovia criceti DSM 17774, Parascardovia denticolens DSM 10105, Scardovia inopinata F0304, and Scardovia wiggsiae F0424) generated a core COG set of the family Bifidobacteriaceae consisting of 451 members. This relatively high number of members of the conserved genetic arsenal within the Bifidobacteriaceae is indicative of a close evolutionary relationship between members of this family (38). Examination of the functional annotation of the core BifCOGs, based on the updated COG database (39), suggests, as anticipated, that most of the conserved core genes specify housekeeping functions or functions related to adaptation to or interaction with a particular environment, such as carbohydrate metabolism, cell envelope biogenesis, amino acid biosynthesis and transport, or nucleotide biosynthesis and transport (see Fig. S1 in the supplemental material). Notably, only 5.5% of the core genome is involved in carbohydrate metabolism (see Fig. S1), whereas the carbohydrate metabolism functional family is the most highly represented COG family within the Bifidobacterium pan-genome (13.7%) (see Fig. S1). This indicates that a strong selective pressure exists with respect to the acquisition and retention of accessory (novel) genes for carbohydrate utilization by bifidobacteria in order for them to be competitive in the particular ecological niche in which they reside. The pan-genome analysis also allowed the identification of the variome, which includes truly unique genes (TUGs), i.e., genes present in just one of the examined bifidobacterial genomes. Predicted TUGs were validated by BLASTx searches in the analyzed genomes in order to avoid false positives imputable to the gene-calling algorithm. The numbers of TUGs range from 47 for B. indicum to 595 for Bifidobacterium cuniculi LMG10738 in the 47 bifidobacterial genomes analyzed (see Fig. S1). The mean number of TUGs found in the Bifidobacterium genome data set is 249. The large deviation from the mean is indicative of a high degree of genome diversity within members of the genus Bifidobacterium, which is typical for related species that have individually adapted to different environments (40). As expected, the majority (54.1%) of TUGs have no functional annotation (see Fig. S1). Nevertheless, 13.2% of TUGs can be attributed to a COG family representing proteins involved in carbohydrate metabolism, including glycosyl hydrolases (GH) and proteins involved in carbohydrate uptake. TUG identification in bifidobacteria may serve to identify targets for functional studies on adaptive abilities, in particular, studies on host interactions and metabolism of (saccharidic) host/diet-derived components (1).
FIG 1.
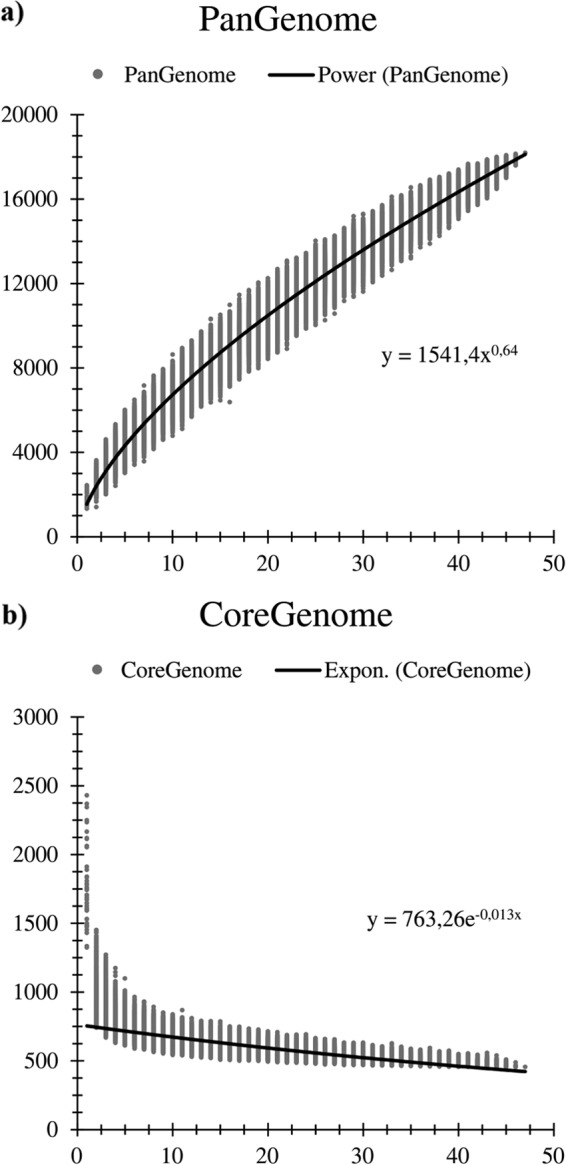
Pan-genome and core genome of the genus Bifidobacterium. The pan-genome (panel a) and core genome (panel b) are represented as variations of the sizes of their gene pools upon sequential addition of the 47 bifidobacterial genomes. The x axes represent the numbers of genomes, whereas the y axes represent the numbers of genes. Expon., exponential.
Phylogenomics of Bifidobacterium genus.
The availability of genome sequences for all members of the genus Bifidobacterium and for five members of the Bifidobacteriaceae family allows an in-depth analysis of the projected evolutionary development of this genus and family. A phylogenetic supertree was constructed based on the concatenated protein sequences of 404 identified Bifidobacteriaceae core COGs, excluding paralogs from the same genome (Fig. 2), an approach that increases the robustness of phylogenetic analyses (41). A consistent phylogeny was obtained using PhyloPhlAn (29), whereas certain discrepancies in the branching of the various bifidobacterial (sub)species were noticed in comparisons of the bifidobacterial core COG-based tree with the 16S rRNA gene-based tree. This observation reveals evolutionary development within the Bifidobacterium genus that is somewhat different from that previously reported, although it did confirm that bifidobacteria represent the deepest branch separating them from other genera within this family (34) (Fig. 2). Furthermore, the Bifidobacterium asteroides phylogenetic group is positioned close to the root in the core genome-based supertree, suggesting a close relationship of members of this group to the Bifidobacterium ancestor, as was previously noticed for the genome of B. asteroides PRL2011 (42).
FIG 2.

Phylogenomic overview of the family Bifidobacteriaceae. A supertree based on the alignment of 404 core COGs (with a single representative identified for each genome of members of the family Bifidobacteriaceae) was constructed in order to obtain a robust phylogenetic reconstruction. Phylogenetic clusters are highlighted with similarly colored branches, and nodes with bootstrap values higher than 70% are marked with a purple dot. The phylogenetic clusters close to the root of the tree may represent species that are most closely related to the ancestor of the Bifidobacterium genus. Circles surrounding the tree represent the approximate genome sizes (in blue), numbers of TUGs (in red), percentages of genes predicted to have undergone horizontal gene transfer (in green), and percentages of genes predicted to be subject to horizontal gene transfer and carbohydrate metabolism and transport (in orange). The outermost layer represents the numbers of the complete predicted degradation pathways. E. coli, Escherichia coli; met., metabolism.
Evolution of bifidobacterial genomes.
Evolution by gene acquisition and loss of the genus Bifidobacterium following speciation from a common ancestor of all Bifidobacteriaceae can be reconstructed through BlastGraph (30), thereby generating a tree based on information regarding the presence or absence of COGs in every taxa of this family and on the use of the maximum-parsimony algorithm (43) (Fig. 3). The observed difference in species clustering as revealed by this tree compared to that shown by the core COG-based supertree (Fig. 2) is highly informative with respect to possible horizontal gene transfer (HGT) events (44). Such analyses predict that the genome of the common ancestor of the genus Bifidobacterium consisted of approximately 1,048 COGs. This putative ancestor possessed just 179 fewer COGs than the number harbored by the B. indicum genome and as many as 1,091 fewer COGs than the B. biavatii chromosome, representing the smallest and largest genomes, respectively. Thus, the evolution of current bifidobacterial species appears to have involved a relatively limited number of ancestral gene loss incidences but an extensive number of gene acquisition events (Fig. 3). This contrasts with other bacteria, for example, the genomes of genera belonging to the lactic acid bacteria, which are believed to have undergone extensive simplification (45). Various changes identified at this stage of evolution may be linked to the transition to life in an environment characterized by high complexity and abundance of microbial communities. In this context, the acquisition of genes required for the utilization of diet/host-derived carbohydrates provided a clear competitive advantage in a complex microbial community such as the ecological niches of bifidobacteria. An example of this evolutionary trend is represented by the milk-adapted Streptococcus thermophilus and the closest phylogenetic neighbor Streptococcus salivarius. S. salivarius is an inhabitant of the oral cavity of mammals, and, despite the high-level phylogenetic relationship with S. thermophilus, the two species show extremely different carbohydrate utilization patterns, with only a few sugars utilized by the latter (46). The predicted Bifidobacterium ancestor would have been a microaerophile or facultative aerobe, which is reflected by the loss of the genes specifying the electron chain transport cytochrome bd subunits and particular enzymes (i.e., catalase and superoxide dismutase), which allow removal of toxic products that arise as a result of oxygen-mediated respiration (predicted to be present in members of the B. asteroides phylogenetic group). Gain of new gene families that originated either by lineage-specific gene duplication or by acquisition of paralogous genes through HGT seems to be a prevailing trend in the evolution of the genus Bifidobacterium (Fig. 3) and is different from what is observed in other bacterial lineages, e.g., the genus Lactobacillus (45). The evolution of the genome of lactobacilli is thought to have involved ancestral gene decay and metabolic simplification but also a substantial number of duplications and acquisition of unique genes, most of which are predicted to code for peptidases or proteases (45). Lineage-specific gene acquisition appears to have been extensive within the Bifidobacterium genus, as illustrated by the B. biavatii and Bifidobacterium longum subsp. infantis taxa (showing acquisition of 1,091 and 1,092 COGs compared to the presumed ancestral Bifidobacterium taxon, respectively), while these two species seem to have undergone relatively limited genome decay (Fig. 3). Gene acquisition events occurring in the course of evolution of microbial genomes are believed to support adaptation to a new ecological niche or acquisition of increased competitiveness in an existing ecological niche (40). Analysis of the gene families putatively involved in acquisition events indicates that adaptation to growth in environments rich in complex carbohydrates, such as the animal gut, has been the main driving force responsible for retention of gene duplications and HGT-acquired genes during the speciation of Bifidobacterium. An intriguing finding supporting this hypothesis is the presence of a large arsenal of genes encoding enzymes involved in carbohydrate metabolism, especially glycosyl hydrolases, many of which are predicted to have been duplicated or acquired at different times throughout the evolution of this genus (Fig. 4). GHs feed cell bioenergetics, i.e., ATP-producing pathways, which is known to be under high selection pressure during evolution (47), thus representing a strong driving force in genome shaping. Notably, we identified eight COGs predicted to encompass GH43 family members, which are GHs crucial for the degradation of plant polysaccharides (48) and appear to have been acquired early in the evolution of bifidobacteria, while seven COGs encompass members of the large GH13 family, representing α-amylases (48), and appear to have been acquired during the evolution of Bifidobacteriaceae and prior to the GH43 member acquisition (Fig. 4). Furthermore, several presumably acquired genes were identified that encode proteins with predicted carbohydrate uptake functions, including ATP-binding cassette (ABC) transporters, phosphoenolpyruvate-phosphotransferase system (PEP-PTS) transporters, and major facilitator superfamily (MFS) transporters. This supports the hypothesis that bifidobacteria selectively acquired new metabolic capabilities which allowed them access to a larger number of carbon and energy sources. While it seems clear that gene gain was and is the main driving force of bifidobacterial evolution (Fig. 3 and 4), gene decay and metabolic simplification may still be very important for niche-specific adaptation. Various gene loss events, in particular, loss of those encoding biosynthetic enzymes, were detected in the main phylogenetic groups of the genus Bifidobacterium, which presumably reflects analogous environmental pressures. Regarding GHs, it was observed that GH43 family members involved in the degradation of plant polysaccharides appear to have been largely lost in more recent times by a subgroup of 18 Bifidobacterium species (Fig. 3 and 4), while most GH13 family members encompassing α-amylases seem to have been deleted (with respect to the predicted Bifidobacteriaceae ancestor) from the genomes of the clade encompassing bifidobacteria isolated from honeybees and bumblebees (B. asteroides, Bifidobacterium actinocoloniiforme, B. indicum, Bifidobacterim coryneforme, Bifidobacterium bombi, and Bifidobacterium bohemicum), perhaps because these metabolic abilities became obsolete due to the particular diet of their arthropod hosts (Fig. 4).
FIG 3.

Gene gain and loss events in a reconstruction of data representing the family Bifidobacteriaceae. A tree was constructed using information related to the presence or absence of COGs for the whole Bifidobacteriaceae pan-genome. Each node is represented by a pie diagram showing the acquired COGs (in black) and the COGs derived from the previous node (in gray). Furthermore, additional information is displayed at each node as follows: number of acquired genes/number of lost genes/total number of COGs. The predicted Bifidobacterium ancestor is highlighted with thick black circle surrounding the pie diagram.
FIG 4.

Reconstruction of gene gain and loss events regarding genes encoding members of the GH3, GH13, and GH43 families in the family Bifidobacteriaceae. A tree was constructed using information related to the presence or absence of COGs for the whole Bifidobacteriaceae pan-genome. Each node is marked by a pie diagram showing the acquired COGs (in black) and the COGs derived from the previous node (in gray). Furthermore, the number of members of the glycosyl hydrolase families GH3, GH13, and GH43 that had been acquired (in black) or lost (in gray) is indicated close to each diagram.
Mobilome of bifidobacterial genomes.
The identification of genes that may have been acquired by HGT (the so-called mobilome) was performed using the software suite COLOMBO v3.8 implemented with the program SIGI-HMM (32) and DarkHorse software (31). The obtained results were merged, and the identified percentages of predicted alien genes, compared to the total number of ORFs, were shown to range from 6.1% in B. indicum to 26.5% in Bifidobacterium saguini (Table 2). Predicting the donors of these putative alien genes indicated a preferential origin from other members of the Actinobacteria class (28.5%), followed by Bacillus (11.7%), Gammaproteobacteria (8.7%), Clostridium (8.7%), and Alphaproteobacteria (5.9%) (Table 3). It is noteworthy that members of these donor classes are also widespread in the gut environment (49). These data are supportive of the idea that HGT events are the major driver for evolutionary development in members of the Bifidobacterium genus.
TABLE 2.
Predicted horizontal gene transfer in the Bifidobacterium genus
| Bifidobacterium strain | No. of native genes | Putative no. of alien genes | Native genes (%) | Putative alien genes (%) |
|---|---|---|---|---|
| B. actinocoloniiforme DSM 22766 | 1,230 | 258 | 82.7 | 17.3 |
| B. adolescentis ATCC 15703 | 1,475 | 174 | 89.4 | 10.6 |
| B. angulatum LMG 11039 | 1,420 | 103 | 93.2 | 6.8 |
| B. animalis subsp. animalis LMG 10508 | 1,366 | 161 | 89.5 | 10.5 |
| B. animalis subsp. lactis DSM 10140 | 1,373 | 145 | 90.4 | 9.6 |
| B. asteroides LMG 10735 (PRL2011) | 1,227 | 426 | 74.2 | 25.8 |
| B. biavatii DSM 23969 | 1,918 | 639 | 75.0 | 25.0 |
| B. bifidum LMG 11041 | 1,499 | 205 | 88.0 | 12.0 |
| B. bohemicum DSM 22767 | 1,388 | 244 | 85.0 | 15.0 |
| B. bombi DSM 19703 | 1,278 | 176 | 87.9 | 12.1 |
| B. boum LMG 10736 | 1,532 | 194 | 88.8 | 11.2 |
| B. breve LMG 13208 | 1,563 | 324 | 82.8 | 17.2 |
| B. callitrichos DSM 23973 | 1,921 | 443 | 81.3 | 18.7 |
| B. catenulatum LMG 11043 | 1,540 | 124 | 92.5 | 7.5 |
| B. choerinum LMG 10510 | 1,467 | 205 | 87.7 | 12.3 |
| B. coryneforme LMG 18911 | 1,264 | 100 | 92.7 | 7.3 |
| B. crudilactis LMG 23609 | 1,476 | 407 | 78.4 | 21.6 |
| B. cuniculi LMG 10738 | 1,700 | 494 | 77.5 | 22.5 |
| B. dentium LMG 11045 (Bd1) | 1,831 | 298 | 86.0 | 14.0 |
| B. gallicum LMG 11596 | 1,339 | 168 | 88.9 | 11.1 |
| B. gallinarum LMG 11586 | 1,365 | 289 | 82.5 | 17.5 |
| B. indicum LMG 11587 | 1,269 | 83 | 93.9 | 6.1 |
| B. kashiwanohense DSM 21854 | 1,703 | 245 | 87.4 | 12.6 |
| B. longum subsp. infantis ATCC 15697 | 1,845 | 655 | 73.8 | 26.2 |
| B. longum subsp. longum LMG 13197 | 1,648 | 251 | 86.8 | 13.2 |
| B. longum subsp. suis LMG 21814 | 1,635 | 321 | 83.6 | 16.4 |
| B. magnum LMG 11591 | 1,346 | 161 | 89.3 | 10.7 |
| B. merycicum LMG 11341 | 1,506 | 236 | 86.5 | 13.5 |
| B. minimum LMG 11592 | 1,342 | 248 | 84.4 | 15.6 |
| B. mongoliense DSM 21395 | 1,444 | 354 | 80.3 | 19.7 |
| B. pseudocatenulatum LMG 10505 | 1,578 | 193 | 89.1 | 10.9 |
| B. pseudolongum subsp. globosum LMG 11569 | 1,413 | 161 | 89.8 | 10.2 |
| B. pseudolongum subsp. pseudolongum LMG 11571 | 1,360 | 135 | 91.0 | 9.0 |
| B. psychraerophilum LMG 21775 | 1,574 | 548 | 74.2 | 25.8 |
| B. pullorum LMG 21816 | 1,363 | 328 | 80.6 | 19.4 |
| B. reuteri DSM 23975 | 1,791 | 358 | 83.3 | 16.7 |
| B. ruminantium LMG 21811 | 1,608 | 224 | 87.8 | 12.2 |
| B. saeculare LMG 14934 | 1,427 | 430 | 76.8 | 23.2 |
| B. saguini DSM 23967 | 1,707 | 614 | 73.5 | 26.5 |
| B. scardovii LMG 21589 | 1,858 | 622 | 74.9 | 25.1 |
| B. stellenboschense DSM 23968 | 1,865 | 337 | 84.7 | 15.3 |
| B. stercoris DSM 24849 | 1,716 | 175 | 90.7 | 9.3 |
| B. subtile LMG 11597 | 1,692 | 568 | 74.9 | 25.1 |
| B. thermacidophilum subsp. porcinum LMG 21689 | 1,572 | 166 | 90.4 | 9.6 |
| B. thermacidophilum subsp. thermacidophilum LMG 21395 | 1,571 | 252 | 86.2 | 13.8 |
| B. thermophilum JCM 1207 | 1,441 | 259 | 84.8 | 15.2 |
| B. tsurumiense JCM 13495 | 1,416 | 213 | 86.9 | 13.1 |
TABLE 3.
HGT in the Bifidobacterium pan-genome
| Putative donor | %a |
|---|---|
| Actinobacteria | 28.5 |
| Alphaproteobacteria | 5.9 |
| Bacilli | 11.7 |
| Bacteroides | 0.2 |
| Bacteroidia | 0.6 |
| Betaproteobacteria | 3.4 |
| Chlorobia | 1.1 |
| Chloroflexi | 0.2 |
| Clostridia | 8.7 |
| Deltaproteobacteria | 0.9 |
| Erysipelotrichia | 0.4 |
| Flavobacteria | 2.0 |
| Gammaproteobacteria | 8.7 |
| Halobacteria | 0.7 |
| Methanopyri | 0.4 |
| Negativicutes | 0.7 |
| Nitrospira | 1.1 |
Percentage of the total HGT sequences identified in bifidobacteria corresponding to the various taxa.
The predicted bifidobacterial mobilome, with exclusion of prophage-associated and transposase-encoding genes or genes with no known function, was analyzed through COG assignment, revealing that the most highly represented (15.3%) functional class is that of carbohydrate metabolism and transport (Table 4). Notably, HGT events encompassing genes involved in carbohydrate metabolism and transport include genes encoding key enzymes such as GHs (representing 3.4% of the predicted mobilome) and genes predicted to specify glycosyl transferases (GTs) and carbohydrate transporters (ABC, MFS, and PTS classes), which constitute 2.6% and 4.0% of the predicted mobilome, respectively, while genes involved in exopolysaccharide (EPS) biosynthesis (with the partial inclusion of GTs) correspond to 3.7% of the predicted mobilome. Interestingly, the GH families that appear most affected by HGT events are GH43 and GH3, representing, respectively, 10.7% and 8.7% of the total pool of GHs involved in HGT. Members of GH43 and GH3 families have been shown to be involved in the breakdown of polysaccharides encompassing arabinose and xylose residues (50), thus supporting the hypothesis that the ability to utilize plant polysaccharides has been acquired by HGT in recent ancestors or actual members (e.g., Bifidobacterium reuteri, B. biavatii, and Bifidobacterium scardovii) of the genus Bifidobacterium rather than by vertical evolution.
TABLE 4.
COG function
| Category of cluster of orthologous genes | %a |
|---|---|
| Translation, ribosomal structure, and biogenesis | 3.8 |
| RNA processing and modification | 0.0 |
| Transcription | 10.7 |
| Replication, recombination, and repair | 5.7 |
| Chromatin structure and dynamics | 0.0 |
| Cell cycle control, cell division, chromosome partitioning | 1.2 |
| Nuclear structure | 0.0 |
| Defense mechanisms | 7.4 |
| Signal transduction mechanisms | 3.3 |
| Cell wall/membrane/envelope biogenesis | 7.5 |
| Cell motility | 0.1 |
| Cytoskeleton | 0.0 |
| Extracellular structures | 0.0 |
| Intracellular trafficking, secretion, and vesicular transport | 0.6 |
| Posttranslational modification, protein turnover, chaperones | 1.8 |
| Energy production and conversion | 3.6 |
| Carbohydrate transport and metabolism | 15.3 |
| Amino acid transport and metabolism | 9.4 |
| Nucleotide transport and metabolism | 2.3 |
| Coenzyme transport and metabolism | 2.2 |
| Lipid transport and metabolism | 2.2 |
| Inorganic ion transport and metabolism | 4.9 |
| Secondary metabolites biosynthesis, transport, and catabolism | 1.1 |
| General function prediction only | 12.8 |
| Function unknown | 4.0 |
Percentage of coding sequences for each category.
In silico analysis of the bifidobacterial pan-genome highlights an abundance of prophage-like elements (3.2% of the total pan-genome size, representing about 7.6% of the predicted bifidobacterial mobilome) and a rich arsenal of insertion sequences (IS), belonging to 16 IS families, with an abundance of IS3, IS21, IS256, ISL3, and IS200/IS605 family members, constituting approximately 3.2% of the total predicted mobilome.
Additional putative mobile elements identified in the Bifidobacterium pan-genome are represented by CRISPR loci. CRISPR and CRISPR-associated proteins (Cas) constitute the CRISPR-Cas system, which provides adaptive immunity against exogenous genetic elements in bacteria and archaea (51). Typically, DNA from invasive elements is captured in CRISPR loci and subsequently transcribed into small interfering RNAs that guide Cas nucleases for sequence-specific targeting and cleavage of cDNA (52). We identified the three main types of CRISPR-Cas systems, namely, type I, type II, and type III, in the genomes of bifidobacteria and observed 43, 6, and 7 systems, respectively (Table 1). Overall, we identified 56 distinct loci in 35 genomes, and the high level of occurrence of type I systems (43 loci with a type I CRISPR and 29 loci with a cas3 signature gene) is consistent with their prevalent distribution in bacteria (53). Interestingly, we observed 6 type II systems and identified 5 cas9 signature genes. Lastly, we observed remnants of 7 putative type III loci, including several cmr genes. Overall, a diversity of CRISPR-Cas systems occurs in bifidobacteria, at a frequency (35/47 genomes, 75%) much higher than that generally observed in the genomes of bacteria, of which just 46% contain CRISPR loci (54). Beyond diversity at the CRISPR-Cas system type level, we further observed diversity in terms of locus size with loci ranging from 4 to 172 CRISPR spacers, with an average of 60 spacers, which is also unusually high. It is noteworthy that we observed CRISPR loci in all the major phylogenetic groups of bifidobacteria, indicating that these systems are evolutionarily widespread throughout this genus. This is consistent with previous analyses reporting their occurrence in various Bifidobacterium species (55, 56), and matches between CRISPR spacer sequences and those of bacteriophages and plasmids suggest these widespread loci may also provide adaptive immunity against viruses and plasmids in most bifidobacteria.
In silico analyses of central metabolism.
In order to depict an overview of the metabolic capabilities of the entire genus Bifidobacterium, we conducted a prediction of complete metabolic pathways in every species through the use of Pathway tools software. Homologs of all enzymes necessary for the fermentation of glucose and fructose to lactic acid and acetate through the characteristic “fructose-6-phosphate shunt” (57), as well as a partial Embden-Meyerhoff pathway, were annotated in the Bifidobacterium core genome. These metabolic pathways are important for generation of pyruvate and oxidation of NADH, as well as for synthesis of an additional ATP molecule per glucose during the conversion of pyruvate to acetate, producing a higher energetic yield than lactic acid bacteria (58).
Genes encoding complete biosynthetic pathways for amino acids, purines, and pyrimidines from glutamine were variously present within the genus Bifidobacterium, with generally fewer of such pathways in the genomes of bifidobacteria isolated from insects (Fig. 5).
FIG 5.

Prediction of complete amino acid, vitamin, and cofactor biosynthesis pathways and non-carbohydrate degradation pathways. Panel a shows a heatmap of the amino acid, vitamin, and cofactor biosynthetic pathways present in the analyzed bifidobacterial genomes. Panel b displays a heatmap that shows all complete non-carbohydrate degradation pathways found in the genus Bifidobacterium. Panel c shows a heatmap illustrating glycodeoxycholate and taurodeoxycholate degradation capabilities, along with the presence of a bile salt hydrolase gene, in the analyzed bifidobacterial species. Each bifidobacterial genome analyzed is numbered according to the numbering of the species displayed in Table 1. Black and gray squares in panels a to c represent the absence and presence of genes.
Similarly, homologs for pathways to produce the vitamins riboflavin (B2), tetrahydrofolate (B9), thiamine (B10), and pyridoxal 5′-phosphate (B6) are also variously distributed in the genomes of this bacterial genus (Fig. 5). Interestingly, while tetrahydrofolate is not produced by mammals (59), it is predicted to be synthetized by all the analyzed species of bifidobacteria isolated from humans (with the sole exception of B. gallicum) or other primates (with the sole exception of B. biavatii). This suggests that tetrahydrofolate production by gut bacteria represents an important source of vitamin B11 for the host and a clear example of microbe-host coevolution (Fig. 5). Additionally, an intermediate in the riboflavin biosynthetic pathway has been shown to be involved in activation of mucosa-associated invariant T (MAIT) cells (60). Notably, four bifidobacterial species are predicted to possess a complete riboflavin biosynthesis pathway (Fig. 5), which may represent an additional mechanism for microbe-host interaction by stimulation of the host's immune system (Fig. 5).
Notably, a hierarchical representation of these biosynthetic pathways highlighted a closer coclustering of those species isolated from insects as well as from rabbit and poultry (Fig. 5), suggesting specialization with respect to these ecological niches following an adaptation to their host diet. Other metabolic capabilities of the genus Bifidobacterium predicted by our in silico analyses are displayed in Fig. 5. Interestingly, B. asteroides, B. indicum, B. coryneforme, B. actinoloniiforme, B. bohemicum, and B. bombi, isolated from various insect guts and with a small genome size compared to those of other members of the Bifidobacterium genus, possess narrow repertoires of biosynthetic pathways, while B. callitrichos and B. stellenboschense, possessing two of the largest genomes within the Bifidobacteriaceae, seem to have retained a much broader biosynthetic inventory.
Furthermore, none of the currently described taxa belonging to the Bifidobacterium genus possess a complete mevalonate pathway for isoprenoid biosynthesis, except for seven members of the most ancient branches of the core COG tree encompassing the B. actinocoloniiforme, B. bohemicum, B. bombi, B. crudilactis, B. mongoliense, Bifidobacterium psychraerophilum, and B. subtile taxa (Fig. 5). With the exception of B. psychraerophilum and B. crudilactis, this pathway was displaced by the alternative, non-mevalonate 2-C-methyl-d-erythritol 4-phosphate/1-deoxy-d-xylulose 5-phosphate pathway (MEP/DOXP pathway) for isoprenoid biosynthesis. Interestingly, the intermediate HMB-PP [(E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate] is an activator for human Vγ9/Vδ2 T cells, the major γδ T cell population in peripheral blood (61, 62), playing an important (even if not fully understood) role in the initial training and subsequent regulation of the mucosal immune system.
Pathways for degradation of alcohols (2,3-butanediol, ethanol, and glycerophosphodiester), amines and polyamines (4-aminobutyrate [GABA]), N-acetylglucosamine, and urea as well as allophanate, gluconate, phospholipids, 2-aminoethyl phosphonate, nucleotides, and xylitol are widely distributed among bifidobacterial species (Fig. 5). Notably, only B. actinocoloniiforme and B. bohemicum are predicted to possess a complete citrate degradation pathway and a complete d-glucarate degradation pathway. With respect to nitrogen metabolism, only the genome of B. callitrichos appears to encompass the nitrate reduction VI (assimilatory) pathway, which is predicted to be involved in nitrogen assimilation (63). Interestingly, the genetic locus encompassing the nitrite reductase also includes a gene encoding ferredoxin-NADP reductase, a flavodoxin-encoding gene, and a gene encoding an ABC-type nitrate/nitrite porter. Other intriguing bifidobacterial metabolic properties here identified involved the presence of a complete pathway for degradation of d-glucuronate, one of the main constituents of proteoglycans, which are present only in the genomes of B. asteroides, B. indicum, B. coryneforme, and B. biavatii. Proteoglycans have an important role in the physiology of insect gut since they constitute the peritrophic matrix, a physical barrier that plays a role analogous to that of mucous secretions of the vertebrate digestive tract (64). Thus, the presence of a degradation pathway for d-glucuronate in genomes of bifidobacterial species isolated from honey bees (B. asteroides, B. indicum, and B. coryneforme) and isolated from the gut of the insect-feeding tamarin monkey (B. biavatii) may represent a key example of strict genetic adaptation of bifidobacteria to the gut of insects. The genomes of the species isolated from insect gut such as B. asteroides, B. indicum, B. coryneforme, B. actinocoloniiforme, B. bohemicum, and B. bombi, in addition to B. mongoliense and B. subtile, highlighted the presence of a complete electron transfer chain consisting of four complexes (complex I, NADH dehydrogenase, flavin mononucleotide, and iron-sulfur cluster-containing protein; complex II, succinate dehydrogenase; complex III, cytochrome d oxidase; and complex IV, F1F0-ATPase), which suggests that these species have the option of operating a simplified respiratory metabolism (65).
The metabolic potential of Bifidobacterium is complemented by its predicted transport capabilities. In particular, ABC transporters that represent putative sugar uptake systems are present in greater numbers than those that represent predicted amino acid, peptide, and metal uptake systems. Among the detected carbohydrate uptake systems, those predicted to be specific for oligosaccharides and glycosides outnumber transporters for free sugars.
Conclusions.
This report represents an extensive comparative analysis of the genomes of all representative species belonging to the Bifidobacterium genus, revealing a distinct saccharolytic genotype. An extensive gene acquisition trend over the course of evolutionary development of bifidobacteria through HGT events seems to have allowed the enrichment of metabolic traits sustaining the utilization of a vast array of carbohydrates, in terms of both transport and degradation. In the ancestral bifidobacteria that are believed to closely resemble the current members of the B. asteroides phylogenetic group, carbohydrate metabolism is centered on the use of simple sugars commonly identified in plant cells. Furthermore, subsequent specialization of bifidobacterial taxa associated with the mammalian gut seems to have been subject to Darwinian selection that led the acquisition of genetic pathways indispensable for the metabolism of complex carbohydrates found in the mammalian diet.
Lastly, the results of the comparative genomic analyses provided here also indicate that a revision of the taxonomy of the currently distinguished Bifidobacterium species may be necessary, as these analyses revealed very close phylogenetic relatedness of bifidobacterial taxa that are currently considered separate species.
Supplementary Material
ACKNOWLEDGMENTS
We thank GenProbio srl for financial support of the Laboratory of Probiogenomics. This work was financially supported by a FEMS Jensen Award to F.T. and by a Ph.D. fellowship (Spinner 2013, Regione Emilia Romagna) to S.D. D.V.S., F.B., and F.T. are members of The Alimentary Pharmabiotic Centre, while D.V.S. is also a member of the Alimentary Glycoscience Research Cluster, both funded by Science Foundation Ireland (SFI) through the Irish Government's National Development Plan (grant numbers SFI/12/RC/2273 and 08/SRC/B1393, respectively). B.S. was the recipient of a Ramón y Cajal contract from MINECO.
Footnotes
Published ahead of print 1 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02308-14.
REFERENCES
- 1.Ventura M, Turroni F, O'Connell Motherway M, MacSharry J, van Sinderen D. 2012. Host-microbe interactions that facilitate gut colonization by commensal bifidobacteria. Trends Microbiol. 20:467–476. 10.1016/j.tim.2012.07.002 [DOI] [PubMed] [Google Scholar]
- 2.Bergey DH, Goodfellow M, Whitman WB, Parte AC. 2012. The actinobacteria, p 1 In Bergey's manual of systematic bacteriology, vol 5, 2nd ed. Springer, New York, NY [Google Scholar]
- 3.Turroni F, Peano C, Pass DA, Foroni E, Severgnini M, Claesson MJ, Kerr C, Hourihane J, Murray D, Fuligni F, Gueimonde M, Margolles A, De Bellis G, O'Toole PW, van Sinderen D, Marchesi JR, Ventura M. 2012. Diversity of bifidobacteria within the infant gut microbiota. PLoS One 7:e36957. 10.1371/journal.pone.0036957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Milani C, Hevia A, Foroni E, Duranti S, Turroni F, Lugli GA, Sanchez B, Martin R, Gueimonde M, van Sinderen D, Margolles A, Ventura M. 2013. Assessing the fecal microbiota: an optimized ion torrent 16S rRNA gene-based analysis protocol. PLoS One 8:e68739. 10.1371/journal.pone.0068739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turroni F, Foroni E, Pizzetti P, Giubellini V, Ribbera A, Merusi P, Cagnasso P, Bizzarri B, de'Angelis GL, Shanahan F, van Sinderen D, Ventura M. 2009. Exploring the diversity of the bifidobacterial population in the human intestinal tract. Appl. Environ. Microbiol. 75:1534–1545. 10.1128/AEM.02216-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chenoll E, Casinos B, Bataller E, Astals P, Echevarria J, Iglesias JR, Balbarie P, Ramon D, Genoves S. 2011. Novel probiotic Bifidobacterium bifidum CECT 7366 strain active against the pathogenic bacterium Helicobacter pylori. Appl. Environ. Microbiol. 77:1335–1343. 10.1128/AEM.01820-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shirasawa Y, Shibahara-Sone H, Iino T, Ishikawa F. 2010. Bifidobacterium bifidum BF-1 suppresses Helicobacter pylori-induced genes in human epithelial cells. J. Dairy Sci. 93:4526–4534. 10.3168/jds.2010-3274 [DOI] [PubMed] [Google Scholar]
- 8.Khailova L, Mount Patrick SK, Arganbright KM, Halpern MD, Kinouchi T, Dvorak B. 2010. Bifidobacterium bifidum reduces apoptosis in the intestinal epithelium in necrotizing enterocolitis. Am. J. Physiol. Gastrointest. Liver Physiol. 299:G1118–G1127. 10.1152/ajpgi.00131.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guglielmetti S, Mora D, Gschwender M, Popp K. 2011. Randomised clinical trial: Bifidobacterium bifidum MIMBb75 significantly alleviates irritable bowel syndrome and improves quality of life—a double-blind, placebo-controlled study. Aliment. Pharmacol. Ther. 33:1123–1132. 10.1111/j.1365-2036.2011.04633.x [DOI] [PubMed] [Google Scholar]
- 10.Malago JJ, Tooten PC, Koninkx JF. 2010. Anti-inflammatory properties of probiotic bacteria on Salmonella-induced IL-8 synthesis in enterocyte-like Caco-2 cells. Benef. Microbes 1:121–130. 10.3920/BM2009.0021 [DOI] [PubMed] [Google Scholar]
- 11.Ventura M, Turroni F, Lugli GA, van Sinderen D. 2014. Bifidobacteria and humans: our special friends, from ecological to genomic perspectives. J. Sci. Food Agric. 94:163–168. 10.1002/jsfa.6356 [DOI] [PubMed] [Google Scholar]
- 12.Biavati B, Mattarelli P. 1991. Bifidobacterium ruminantium sp. nov. and Bifidobacterium merycicum sp. nov. from the rumens of cattle. Int. J. Syst. Bacteriol. 41:163–168. 10.1099/00207713-41-1-163 [DOI] [PubMed] [Google Scholar]
- 13.Endo A, Futagawa-Endo Y, Schumann P, Pukall R, Dicks LM. 2012. Bifidobacterium reuteri sp. nov., Bifidobacterium callitrichos sp. nov., Bifidobacterium saguini sp. nov., Bifidobacterium stellenboschense sp. nov. and Bifidobacterium biavatii sp. nov. isolated from faeces of common marmoset (Callithrix jacchus) and red-handed tamarin (Saguinus midas). Syst. Appl. Microbiol. 35:92–97. 10.1016/j.syapm.2011.11.006 [DOI] [PubMed] [Google Scholar]
- 14.Killer J, Kopecny J, Mrazek J, Koppova I, Havlik J, Benada O, Kott T. 2011. Bifidobacterium actinocoloniiforme sp. nov. and Bifidobacterium bohemicum sp. nov., from the bumblebee digestive tract. Int. J. Syst. Evol. Microbiol. 61(Pt 6):1315–1321. 10.1099/ijs.0.022525-0 [DOI] [PubMed] [Google Scholar]
- 15.Ventura M, O'Flaherty S, Claesson MJ, Turroni F, Klaenhammer TR, van Sinderen D, O'Toole PW. 2009. Genome-scale analyses of health-promoting bacteria: probiogenomics. Nat. Rev. Microbiol. 7:61–71. 10.1038/nrmicro2047 [DOI] [PubMed] [Google Scholar]
- 16.Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Muller WE, Wetter T, Suhai S. 2004. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 14:1147–1159. 10.1101/gr.1917404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gish W, States DJ. 1993. Identification of protein coding regions by database similarity search. Nat. Genet. 3:266–272. 10.1038/ng0393-266 [DOI] [PubMed] [Google Scholar]
- 18.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. 2000. Artemis: sequence visualization and annotation. Bioinformatics 16:944–945. 10.1093/bioinformatics/16.10.944 [DOI] [PubMed] [Google Scholar]
- 19.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 21.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmid R, Blaxter ML. 2008. annot8r: GO, EC and KEGG annotation of EST datasets. BMC Bioinformatics 9:180. 10.1186/1471-2105-9-180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts RJ, Vincze T, Posfai J, Macelis D. 2010. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 38:D234–D236. 10.1093/nar/gkp874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Busch W, Saier MH., Jr 2002. The transporter classification (TC) system, 2002. Crit. Rev. Biochem. Mol. Biol. 37:287–337. 10.1080/10409230290771528 [DOI] [PubMed] [Google Scholar]
- 25.Zhao Y, Wu J, Yang J, Sun S, Xiao J, Yu J. 2012. PGAP: pan-genomes analysis pipeline. Bioinformatics 28:416–418. 10.1093/bioinformatics/btr655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Enright AJ, Van Dongen S, Ouzounis CA. 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30:1575–1584. 10.1093/nar/30.7.1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30:3059–3066. 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- 29.Segata N, Bornigen D, Morgan XC, Huttenhower C. 2013. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4:2304. 10.1038/ncomms3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye Y, Wei B, Wen L, Rayner S. 2013. BlastGraph: a comparative genomics tool based on BLAST and graph algorithms. Bioinformatics 29:3222–3224. 10.1093/bioinformatics/btt553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Podell S, Gaasterland T. 2007. DarkHorse: a method for genome-wide prediction of horizontal gene transfer. Genome Biol. 8:R16. 10.1186/gb-2007-8-2-r16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waack S, Keller O, Asper R, Brodag T, Damm C, Fricke WF, Surovcik K, Meinicke P, Merkl R. 2006. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7:142. 10.1186/1471-2105-7-142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35:W52–W57. 10.1093/nar/gkm360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ventura M, Turroni F, Lugli GA, van Sinderen D. 2014. Bifidobacteria and humans: our special friends, from ecological to genomics perspectives. J. Sci. Food Agric. 94:163–168. 10.1002/jsfa.6356 [DOI] [PubMed] [Google Scholar]
- 35.Koskiniemi S, Sun S, Berg OG, Andersson DI. 2012. Selection-driven gene loss in bacteria. PLoS Genet. 8:e1002787. 10.1371/journal.pgen.1002787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, Bertalan M, Borruel N, Casellas F, Fernandez L, Gautier L, Hansen T, Hattori M, Hayashi T, Kleerebezem M, Kurokawa K, Leclerc M, Levenez F, Manichanh C, Nielsen HB, Nielsen T, Pons N, Poulain J, Qin J, Sicheritz-Ponten T, Tims S, Torrents D, Ugarte E, Zoetendal EG, Wang J, Guarner F, Pedersen O, de Vos WM, Brunak S, Dore J, MetaHIT Consortium, Antolin M, Artiguenave F, Blottiere HM, Almeida M, Brechot C, Cara C, Chervaux C, Cultrone A, et al. 2011. Enterotypes of the human gut microbiome. Nature 473:174–180. 10.1038/nature09944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit y Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, Khouri H, Radune D, Dimitrov G, Watkins K, O'Connor KJ, Smith S, Utterback TR, White O, Rubens CE, Grandi G, Madoff LC, Kasper DL, Telford JL, Wessels MR, Rappuoli R, Fraser CM. 2005. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.” Proc. Natl. Acad. Sci. U. S. A. 102:13950–13955. 10.1073/pnas.0506758102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Callister SJ, McCue LA, Turse JE, Monroe ME, Auberry KJ, Smith RD, Adkins JN, Lipton MS. 2008. Comparative bacterial proteomics: analysis of the core genome concept. PLoS One 3:e1542. 10.1371/journal.pone.0001542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. 2003. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. 10.1186/1471-2105-4-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Philippot L, Andersson SG, Battin TJ, Prosser JI, Schimel JP, Whitman WB, Hallin S. 2010. The ecological coherence of high bacterial taxonomic ranks. Nat. Rev. Microbiol. 8:523–529. 10.1038/nrmicro2367 [DOI] [PubMed] [Google Scholar]
- 41.Ventura M, Canchaya C, Del Casale A, Dellaglio F, Neviani E, Fitzgerald GF, van Sinderen D. 2006. Analysis of bifidobacterial evolution using a multilocus approach. Int. J. Syst. Evol. Microbiol. 56:2783–2792. 10.1099/ijs.0.64233-0 [DOI] [PubMed] [Google Scholar]
- 42.Bottacini F, Milani C, Turroni F, Sanchez B, Foroni E, Duranti S, Serafini F, Viappiani A, Strati F, Ferrarini A, Delledonne M, Henrissat B, Coutinho P, Fitzgerald GF, Margolles A, van Sinderen D, Ventura M. 2012. Bifidobacterium asteroides PRL2011 genome analysis reveals clues for colonization of the insect gut. PLoS One 7:e44229. 10.1371/journal.pone.0044229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mirkin BG, Fenner TI, Galperin MY, Koonin EV. 2003. Algorithms for computing parsimonious evolutionary scenarios for genome evolution, the last universal common ancestor and dominance of horizontal gene transfer in the evolution of prokaryotes. BMC Evol. Biol. 3:2. 10.1186/1471-2148-3-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snipen L, Ussery DW. 2010. Standard operating procedure for computing pangenome trees. Stand. Genomic Sci. 2:135–141. 10.4056/sigs.38923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Makarova K, Slesarev A, Wolf Y, Sorokin A, Mirkin B, Koonin E, Pavlov A, Pavlova N, Karamychev V, Polouchine N, Shakhova V, Grigoriev I, Lou Y, Rohksar D, Lucas S, Huang K, Goodstein DM, Hawkins T, Plengvidhya V, Welker D, Hughes J, Goh Y, Benson A, Baldwin K, Lee JH, Diaz-Muniz I, Dosti B, Smeianov V, Wechter W, Barabote R, Lorca G, Altermann E, Barrangou R, Ganesan B, Xie Y, Rawsthorne H, Tamir D, Parker C, Breidt F, Broadbent J, Hutkins R, O'Sullivan D, Steele J, Unlu G, Saier M, Klaenhammer T, Richardson P, Kozyavkin S, Weimer B, Mills D. 2006. Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. U. S. A. 103:15611–15616. 10.1073/pnas.0607117103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schleifer ME, Krusch KHU, Neve H. 1991. Revival of the species Streptococcus thermophilus (ex Orla-Jensen, 1919) nom. rev. Syst. Appl. Microbiol. 14:386–388. 10.1016/S0723-2020(11)80314-0 [DOI] [Google Scholar]
- 47.Pfeiffer T, Schuster S, Bonhoeffer S. 2001. Cooperation and competition in the evolution of ATP-producing pathways. Science 292:504–507. 10.1126/science.1058079 [DOI] [PubMed] [Google Scholar]
- 48.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42:D490–D495. 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cho I, Blaser MJ. 2012. The human microbiome: at the interface of health and disease. Nat. Rev. Genet. 13:260–270. 10.1038/nrg3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeBoy RT, Mongodin EF, Fouts DE, Tailford LE, Khouri H, Emerson JB, Mohamoud Y, Watkins K, Henrissat B, Gilbert HJ, Nelson KE. 2008. Insights into plant cell wall degradation from the genome sequence of the soil bacterium Cellvibrio japonicus. J. Bacteriol. 190:5455–5463. 10.1128/JB.01701-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. 10.1126/science.1138140 [DOI] [PubMed] [Google Scholar]
- 52.Barrangou R. 2013. CRISPR-Cas systems and RNA-guided interference. Wiley Interdiscip. Rev. RNA 4:267–278. 10.1002/wrna.1159 [DOI] [PubMed] [Google Scholar]
- 53.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. 2011. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9:467–477. 10.1038/nrmicro2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Makarova KS, Koonin EV. 2007. Evolutionary genomics of lactic acid bacteria. J. Bacteriol. 189:1199–1208. 10.1128/JB.01351-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Loquasto JR, Barrangou R, Dudley EG, Roberts RF. 2011. Short communication: the complete genome sequence of Bifidobacterium animalis subspecies animalis ATCC 25527T and comparative analysis of growth in milk with B. animalis subspecies lactis DSM 10140T. J. Dairy Sci. 94:5864–5870. 10.3168/jds.2011-4499 [DOI] [PubMed] [Google Scholar]
- 56.Ventura M, Turroni F, Lima-Mendez G, Foroni E, Zomer A, Duranti S, Giubellini V, Bottacini F, Horvath P, Barrangou R, Sela DA, Mills DA, van Sinderen D. 2009. Comparative analyses of prophage-like elements present in bifidobacterial genomes. Appl. Environ. Microbiol. 75:6929–6936. 10.1128/AEM.01112-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Vries W, Stouthamer AH. 1969. Factors determining the degree of anaerobiosis of Bifidobacterium strains. Arch. Mikrobiol. 65:275–287. 10.1007/BF00407109 [DOI] [PubMed] [Google Scholar]
- 58.Scardovi V, Trovatelli LD. 1965. The fructose-6-phosphate shunt as a peculiar pattern of hexose degradation in the genus Bifidobacterium. Ann. Microbiol. 15:19–29 [Google Scholar]
- 59.Camilo E, Zimmerman J, Mason JB, Golner B, Russell R, Selhub J, Rosenberg IH. 1996. Folate synthesized by bacteria in the human upper small intestine is assimilated by the host. Gastroenterology 110:991–998. 10.1053/gast.1996.v110.pm8613033 [DOI] [PubMed] [Google Scholar]
- 60.Corbett AJ, Eckle SB, Birkinshaw RW, Liu L, Patel O, Mahony J, Chen Z, Reantragoon R, Meehan B, Cao H, Williamson NA, Strugnell RA, Van Sinderen D, Mak JY, Fairlie DP, Kjer-Nielsen L, Rossjohn J, McCluskey J. 2014. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 509:361–365. 10.1038/nature13160 [DOI] [PubMed] [Google Scholar]
- 61.Heuston S, Begley M, Davey MS, Eberl M, Casey PG, Hill C, Gahan CG. 2012. HmgR, a key enzyme in the mevalonate pathway for isoprenoid biosynthesis, is essential for growth of Listeria monocytogenes EGDe. Microbiology 158:1684–1693. 10.1099/mic.0.056069-0 [DOI] [PubMed] [Google Scholar]
- 62.Eberl M, Hintz M, Reichenberg A, Kollas AK, Wiesner J, Jomaa H. 2003. Microbial isoprenoid biosynthesis and human γδ T cell activation. FEBS Lett. 544:4–10. 10.1016/S0014-5793(03)00483-6 [DOI] [PubMed] [Google Scholar]
- 63.Lin JT, Stewart V. 1998. Nitrate assimilation by bacteria. Adv. Microb. Physiol. 39:1–30, 379 [DOI] [PubMed] [Google Scholar]
- 64.Kuraishi T, Binggeli O, Opota O, Buchon N, Lemaitre B. 2011. Genetic evidence for a protective role of the peritrophic matrix against intestinal bacterial infection in Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. 108:15966–15971. 10.1073/pnas.1105994108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borisov VB, Gennis RB, Hemp J, Verkhovsky MI. 2011. The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta 1807:1398–1413. 10.1016/j.bbabio.2011.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.