

Abstract
Microbial abundance is central to most investigations in microbial ecology, and its accurate measurement is a challenging task that has been significantly facilitated by the advent of molecular techniques over the last 20 years. Fluorescence in situ hybridization (FISH) is considered the gold standard of quantification techniques; however, it is expensive and offers low sample throughput, both of which limit its wider application. Quantitative PCR (qPCR) is an alternative that offers significantly higher throughput, and it is used extensively in molecular biology. The accuracy of qPCR can be compromised by biases in the DNA extraction and amplification steps. In this study, we compared the accuracy of these two established quantification techniques to measure the abundance of a key functional group in biological wastewater treatment systems, the ammonia-oxidizing bacteria (AOB), in samples from a time-series experiment monitoring a set of laboratory-scale reactors and a full-scale plant. For the qPCR analysis, we tested two different sets of AOB-specific primers, one targeting the 16SrRNA gene and one targeting the ammonia monooxygenase (amoA) gene. We found that there was a positive linear logarithmic relationship between FISH and the amoA gene-specific qPCR, where the data obtained from both techniques was equivalent at the order of magnitude level. The 16S rRNA gene-specific qPCR assay consistently underestimated AOB numbers.
INTRODUCTION
Measurement, and its corollary quantification, is generally regarded as one of the most important defining features of the natural sciences. Quantification lends objectivity to the sciences and thus has unparalleled power and prestige in the modern world (1). The quantification of microbial communities has always proved very challenging (2, 3); however, the introduction of molecular methods in the last 20 years has brought forward new techniques that can improve our ability to observe and predict the composition of microbial communities in natural and engineered systems. For instance, in biological wastewater treatment systems, quantification can benefit both the researcher and the practitioner. In research, quantification is essential for the determination of microbial growth and substrate consumption kinetics (e.g., cell yields and growth rates) and of the population size of specific communities that is essential in theoretical modeling (e.g., resource ratio/Monod kinetics and island biogeography) and practical ecology. In real systems, quantification could enable practitioners to monitor the abundance of key organisms and anticipate and obviate failure.
Fluorescence in situ hybridization (FISH) was one of the first quantitative methods of the molecular age which enabled the identification and quantification of specific functional groups. In essence a phylogenetic “stain,” FISH involves the detection and enumeration of individual cells of specific microbial populations (4, 5). It has been called the “gold standard” of quantification (6), because it enables the direct counting of individuals, which is one of the fundamental units of ecology, and can be readily converted to other units, e.g., mass (5–7). For this reason, the accuracy of FISH is thought to be superior to that of other conventional quantification methods (6), such as cultivation-based methods (e.g., most probable number [8]), immunological methods (9), and DNA amplification-based methods (10, 11). However, FISH suffers from some disadvantages that limit its wider application. In particular, it is slow, it has low throughput, and it requires the use of expensive microscopes to obviate problems of background fluorescence and resolution for accurate quantification in many types of sample (12). In our experience, it takes a few days to obtain statistically valid counts for a single type of microbe in just a few samples. This is a clear obstacle to understanding the ecology of organisms whose populations can change on an hourly basis. In addition, the sensitivity of this technique is compromised in environments where microorganisms are not very active, since the signal from the target cells is likely to be low and therefore swamped by background fluorescence (12, 13). This problem can be circumvented by the use of a variation on FISH: catalyzed reporter deposition fluorescence in situ hybridization (CARD-FISH [14]), which greatly improves the detection and quantification of target microbial communities but increases sample processing time.
More recently, other faster quantification methods have been developed. Most notably, quantitative real-time PCR (qPCR) has greatly simplified the quantification of nucleic acids and has been extensively used across a wide range of disciplines and environments (5, 10, 15, 16). Quantitative PCR offers many putative advantages beyond rapid sample processing, such as a linear range exceeding 4 orders of magnitude (13, 15, 17), high precision (<2% standard deviation [15]), and high sensitivity (<5 copies [15]). In addition, the specificity of the amplification reaction (e.g., the domain level down to the species level) and the gene to be targeted (e.g., a taxonomic or a functional gene) can be easily controlled by the choice of oligonucleotides (12, 17, 18). Using TaqMan probes instead of SYBR green further increases the specificity and sensitivity of qPCR (13, 17). However, the need for an additional oligonucleotide complicates the design of primer and probe combinations that target the sequence of interest and, in some cases, can make it impossible to design such primer-probe systems to target taxa at a broader resolution (16, 17).
The simplicity and versatility of qPCR have made it an attractive option for the quantification of microbial populations and have contributed to its widespread application. However, it suffers from many of the biases associated with PCR, only measures gene copy numbers (not cell numbers), and is relatively expensive (5, 10, 13, 15, 18, 19). In addition, and most importantly, the quality and accuracy of the data are dependent on many factors other than the amplification reaction, such as sample preparation, DNA extraction procedure, quality of the standards, and the choice of target gene and amplification primers and probes (15, 17). Guidelines have been developed to promote the accuracy and consistency of qPCR data (20), but these guidelines are not always followed. Therefore, qPCR data are often viewed with caution.
Validation of qPCR against a more widely acceptable quantification method would be of great benefit to microbial quantification in environmental samples, and yet research in this area is rare (see, for example, references 21 and 22). In the present study, we calibrated qPCR against FISH by using both methods to measure the general community of ammonia-oxidizing bacteria (AOB) in serial dilutions of two environmental samples from the biological stage of a full-scale nitrifying wastewater treatment plant and in random samples from a full-scale and laboratory-scale time-series reactor study, looking at the effect of temperature variation on the microbial communities treating predominantly domestic wastewater. We then evaluated the nature of the correlation between the qPCR and the FISH data.
MATERIALS AND METHODS
Origin of samples for this study. (i) Serial dilution samples.
Serial dilutions were performed on two samples—one collected in July 2011 and the other one collected in February 2014—from the biological stage of the Tudhoe Mill wastewater treatment plant (WWTP) in County Durham, United Kingdom, a full-scale nitrifying activated sludge plant treating mostly domestic wastewater. For the sample collected in July 2011, four different dilutions were prepared: neat, 10−1, 10−2, and 10−3. For the sample collected in February 2014, nine different dilutions were prepared: neat, 5 × 10−1, 10−1, 5 × 10−2, 10−2, 5 × 10−3, 10−3, 5 × 10−4, and 10−4.
(ii) Samples from time-series study.
In this experiment, a total of 18 samples were collected from the reactors in a time-series study monitoring the changes in the heterotrophic and AOB communities in the biological reactor of a full-scale WWTP and a set of 12 laboratory-scale reactors. The general AOB communities in these samples were quantified by using FISH and qPCR.
The full-scale study took place between July 2011 and July 2012. During this time, samples were collected on a weekly basis from the aeration basin (3,600 m3) of a conventional nitrifying domestic wastewater treatment plant, situated at Tudhoe Mill, County Durham, United Kingdom. The aeration basin had a mean hydraulic retention time of 10 h, a mean sludge age of 12 days, and a mean mixed-liquor suspended solids (MLSS) concentration of 2,700 mg liter−1. The mean compositions of the influent with respect to COD, ammonium, nitrate, and phosphate were 230, 33, 3.6, and 12 mg liter−1, respectively. A sample was collected for FISH and qPCR analysis every 4 weeks for the duration of the study (except for January and February 2012), resulting in a total of 12 samples.
The 200-day laboratory-scale study was run between June and December 2011 and involved the operation and sampling of 12 identical continuous flow bioreactors (CFBs). The CFBs, with a working volume of 950 ± 26 ml and a sludge age of 4 days, were fed with settled sewage from Tudhoe Mill WWTP at an average rate of 10.2 ml h−1, which is equivalent to a hydraulic retention time of approximately 4 days. The CFBs were seeded with activated sludge from the aeration basin of the Tudhoe Mill WWTP. The mean concentrations of COD, ammonium, nitrate, and phosphate in the influent were 218, 38, 1.3, and 19 mg liter−1, respectively, and the mean MLSS concentration in the reactors was 134 mg liter−1. All CFBs were initially operated under steady-state conditions for a period of 76 days, the acclimatization phase, during which the temperature was maintained at 14.5°C, which was the temperature of the Tudhoe Mill aeration basin when the “seed” was collected. Once stable conditions were achieved, the temperature in 6 reactors (“test reactors”) was varied in a sine wave mode with temperatures ranging from 8°C to 21°C to mimic the annual variation in the Tudhoe Mill WWTP. The temperature in the remaining six reactors (“control reactors”) stayed constant at 14.5°C. All other operational and environmental conditions in both sets of reactors were identical during the entire study. Samples for FISH and qPCR analysis were collected every 4 weeks from a reactor chosen at random, resulting in a total of six samples.
Sample collection and storage.
All full-scale samples destined for qPCR analysis were collected into 50-ml sterile polypropylene vials, transported to the laboratory at approximately 4°C and stored at −20°C until DNA extraction was carried out, which happened no later than a week after sample collection. Samples for FISH analysis were collected into 50-ml sterile polypropylene vials containing absolute ethanol. The volume of sample collected was such that the ratio of the volume of sample to volume of absolute ethanol was 1:1. Samples were transported to the laboratory at approximately 4°C and fixed immediately as briefly described below for FISH, after which they were stored at −20°C for up to 6 months. For the laboratory-scale reactors, 15 ml of mixed liquor was collected for qPCR analysis, centrifuged at 13,000 × g for 15 min, and frozen at −20°C until further processing which, as for the full-scale samples, happened no later than a week after sample collection. One milliliter of sample was collected for FISH analysis, immediately fixed, and stored at −20°C.
DNA extraction.
For the full-scale WWTP, DNA was extracted from 250 μl and, for the reactor samples, 15 ml were collected and centrifuged at 3,392 × g for 15 min, and the supernatant was removed down to a volume of 250 μl, which was then used for the DNA extraction. A volume of 244.5 μl of sodium phosphate buffer and a 30.5-μl portion of MT buffer from the FastDNA Spin kit for soil (Qbiogene, Cambridge, United Kingdom) were added to the samples, which were subsequently transferred to a Lysing Matrix E tube (also from the kit). Samples were then lysed in the FastPrep instrument (MP Biomedicals, Santa Ana, CA) at 6.5 ms−1 for 30 s, after which they were centrifuged at 14,000 × g for 15 min. DNA purification was carried out automatically in a MagNA Pure LC 2.0 (Roche, Burgess Hill, United Kingdom) using a MagNA Pure LC DNA isolation kit III (Bacteria, Fungi). Thus, 250 μl of the supernatant from the centrifuged samples was transferred to a Roche sample plate, and the instrument was set up with plasticware and reagents according to the manufacturer's instructions.
Quantitative PCR.
Two different sets of primers were used in the analysis of the general AOB communities in the samples: one specific to the 16S rRNA gene (CTO189f and CTO654r [23]) and the other specific to the ammonia monooxygenase (amoA) gene (amoA-1F* [24] and amoA-2R [25]). The reaction conditions used for the 16S rRNA gene-specific primers were: 98°C for 3 min for 1 cycle; and 98°C for 5 s, followed by 64°C for 5 s for 49 cycles. The conditions used for the amoA gene-specific primers were identical, except that the annealing temperature was 56°C. For simplicity, qPCR data generated using the set of primers specific to the 16S rRNA gene are referred to here as 16S qPCR data, and those generated using the set of primers specific to the amoA gene are described as amoA qPCR data.
Each sample contained 3 μl of template DNA, 0.5 μl of forward and reverse primer (10 pmol/μl), 5 μl of SsoFast EvaGreen Supermix (Bio-Rad, Hemel Hempstead, United Kingdom), and 1 μl of molecular-grade water. Sample preparation was carried out in a biosafety level 2+ class II microbiological safety cabinet (Envair, Lancashire, England) in order to minimize contamination of the samples. PCR amplification reactions were performed using a CFX96 real-time PCR detection system (Bio-Rad). Samples were run in triplicate. Every qPCR included a set of standards with concentrations ranging between 102 and 108 fragment copies per μl and a blank (where the sample was replaced with water), both run in triplicate. Standards were generated from circular plasmids containing the target fragment of DNA. Most of the AOB in the full-scale and the laboratory-scale reactors were Nitrosomonas-like organisms (unpublished data) and, therefore, we assumed that our AOB communities contained, on average, one copy of the 16S rRNA gene and two copies of the amoA gene (10, 26). All qPCRs had an R2 of 0.99. The efficiencies of the amoA qPCRs for the serial dilution and the time-series samples were 103% (standard curve: slope = −3.26; intercept = 40.7) and 102% (standard curve: slope = −3.27; intercept = 40.3), and those for the 16S qPCRs were 96% (standard curve: slope = −3.42; intercept = 38.2) and 100% (standard curve: slope = −3.79; intercept = 45.8), respectively. Melting-curve peaks for the standards and samples amplified using the amoA gene-specific primers and 16S rRNA gene-specific primers occurred at temperatures ranging between 83.0 to 85.5°C and 85.5 to 86.5°C, respectively, with no other minor peaks being detected indicating the absence of nonspecific binding.
Fluorescence in situ hybridization.
AOB cells were fixed using a 4% paraformaldehyde (PFA) solution as described by Amann et al. (27). After fixation, samples were stored at −20°C in a suspension of 1 volume of phosphate-buffered saline to 1 volume of absolute ethanol until they were hybridized. Whole-cell hybridization was carried out on the PFA-fixed samples as described by Amann et al. (28), except in solution (6), using the AOB-specific probes Nso1225, NEU, and 6a192 and the competitive probes CTE and c6a192 (29–31). Hybridizations were carried out at 46°C for all of the probes. The hybridization buffer used is the one described by Amann et al. (28). The hybridization buffer for probes NEU and CTE contained 40% formamide, while the hybridization buffers for all other probes contained 35% formamide. Each sample was divided into four subsamples: a negative control, to which no probe was added with the purpose of checking for autofluorescence in the sample; a second negative control, where a nonsense probe (Anti-Bact338) was added with the purpose of checking for nonspecific binding in the sample; a positive control, consisting of a mixture of the three universal bacterial probes (Bact338I, Bact338II, and Bact338III), added to check the validity of the technique to be used in the samples from the present study and to obtain a measure of the total number of bacterial cells in the sample; and a fourth sample, where the bacterial probes in the positive control, AOB-specific probes Nso1225, NEU, and 6a192 and the competitive probes CTE and c6a192 were added to obtain an estimate of the number of AOB cells in the sample. All oligonucleotide probes were purchased from Thermo Scientific (Thermo Electron Gmbh, Ulm, Germany). Table 1 provides information on the probes used for the different subsamples. The fixed and hybridized samples were visualized at ×630 magnification (×63 objective lens) using a Leica TCS SP2UV confocal laser scanning microscope (CLSM; Leica Microsystems [UK], Ltd., Milton Keynes, United Kingdom), and images were captured and analyzed with the aid of the LEICA TCS software version 2.00 build 0770. Images of each randomly selected field of view (FOV) were acquired using a transect method across an image spot (33), and they were collected at 1-μm intervals on the z axis. Ten FOV were acquired per sample.
TABLE 1.
Description of the oligonucleotide probes used for the FISH analysis
| Probe | Positiona | Probe sequence (5′–3′) | Target | Reference(s) |
|---|---|---|---|---|
| Bac338I | 338–355 | GCTGCCTCCCGTAGGAGT | Domain Bacteria (positive control) | 28, 32 |
| Bac338II | 338–355 | GCAGCCACCCGTAGGTGT | Domain Bacteria (positive control) | 32 |
| Bac338III | 338–355 | GCTGCCACCCGTAGGTGT | Domain Bacteria (positive control) | 32 |
| Anti-Bac338 | 338–355 | ACTCCTACGGGAGGCAGC | None (negative control) | 32 |
| Nso1225 | 1225–1244 | CGCCATTGTATTACGTGTGA | Ammonia-oxidizing betaproteobacteria | 29 |
| NEU | 653–670 | CCCCTCTGCTGCACTCTA | Halophilic and halotolerant members of the genus Nitrosomonas | 30 |
| CTE | 659–676 | TTCCATCCCCCTCTGCCG | Competitor probe for NEU, unlabeled | 30 |
| 6a192 | 192–212 | CTTTCGATCCCCTACTTTCC | Nitrosomonas oligotropha lineage | 31 |
| c6a192 | 192–212 | CTTTCGATCCCCGACTTTCC | Competitor probe for 6a192, unlabeled | 31 |
Position in the 16S rRNA of E. coli (42).
Most AOB cells occurred in spherical or elliptical colonies, but some were present as individual cells. For the AOB colonies, the long and short diameters of every colony present in a FOV were registered and converted to AOB cell numbers using the relationship described in Coskuner et al. (6). For the quantification of individual cells, the number of cells present in each FOV was registered. For every sample, the numbers of cells in each of the 10 FOV were checked for normality and the number of cells per ml was calculated using the following formula:
| (1) |
where the average number of cells refers to the average number of cells per field of view, A is the area of sample spot, Af is the area of the field of view, dil is the sample dilution in ethanol upon collection (i.e., 1/2, as we collected our samples in ethanol at a 50:50 ratio), v0 is the starting volume of sample used in the hybridization, vf is the final volume of sample at the end of the hybridization step (prior to application onto the slide), and v is the volume of sample applied to the slide.
Statistical analysis of the qPCR and FISH data.
All statistical analysis was performed on AOB abundance data logged to the base 10, because previous studies have established that these types of data are log-normally distributed (see, for example, references 6 and 34). The best-fit models for all of the data, and associated R2 values where applicable, were generated using R (R Foundation for Statistical Computing, Vienna, Austria [http://www.R-project.org]). All quantification data sets for the serial dilution samples and for the time-series samples were compared using a standard linear model routine from R. The quantification data sets combining the serial dilution and the time-series samples were compared using the mixed-effects model routine (35) in R. The software produced estimates for the equation coefficients and intercepts, as well as the standard errors, P values, and 95% confidence intervals associated with the estimated values. Two-sample t tests were used for the comparison of the expected and observed quantification data for the serial dilution samples and were performed using Minitab software (Minitab, State College, PA). Expected AOB abundances were generated for each quantification data set by taking the AOB abundance in the undiluted samples and multiplying by the dilution factor. Analysis of variance (ANOVA), also performed with Minitab, was used to compare the ratios of AOB abundances obtained using the 16S qPCR with the other two methods.
RESULTS
Serial dilution data.
The AOB were quantified in two different serially diluted samples using the qPCR assays and FISH (Fig. 1a). The sample collected in July 2011 is referred to as the “first dilution,” and the one collected in February 2014 is referred to as the “second dilution.”
FIG 1.

Mean AOB cell abundances (cells/ml) obtained using FISH, amoA qPCR, and 16S qPCR for serial dilution (a) and time-series (b) samples.
Linear regression analysis of the three AOB abundance data sets versus the dilution factor had an R2 = 0.99 and produced the following three best-fit equations:
| (2) |
| (3) |
| (4) |
The variable “second” is 0 for the “first dilution” data and 1 for the “second dilution” data; ε is the error term. The slopes of the relationships between the AOB abundance data sets and the dilution factor (Fig. 2 and Table 2) were statistically indistinguishable from 1 for the FISH data sets and for the qPCR amoA “second dilution” data set but not for the others, corroborating the credibility of FISH as a quantification technique. Comparison of the expected (as described in Materials and Methods) and observed AOB abundance values for each of the three analytical methods showed that they were statistically indistinguishable (two-sample t test; 0.75 < P < 0.88), although the gap between the expected and observed abundance values generally increased with the dilution factor. In addition, the differences between expected and observed values were smallest for FISH and largest for 16S qPCR, although these differences were not statistically significant (ANOVA; P = 0.44).
FIG 2.
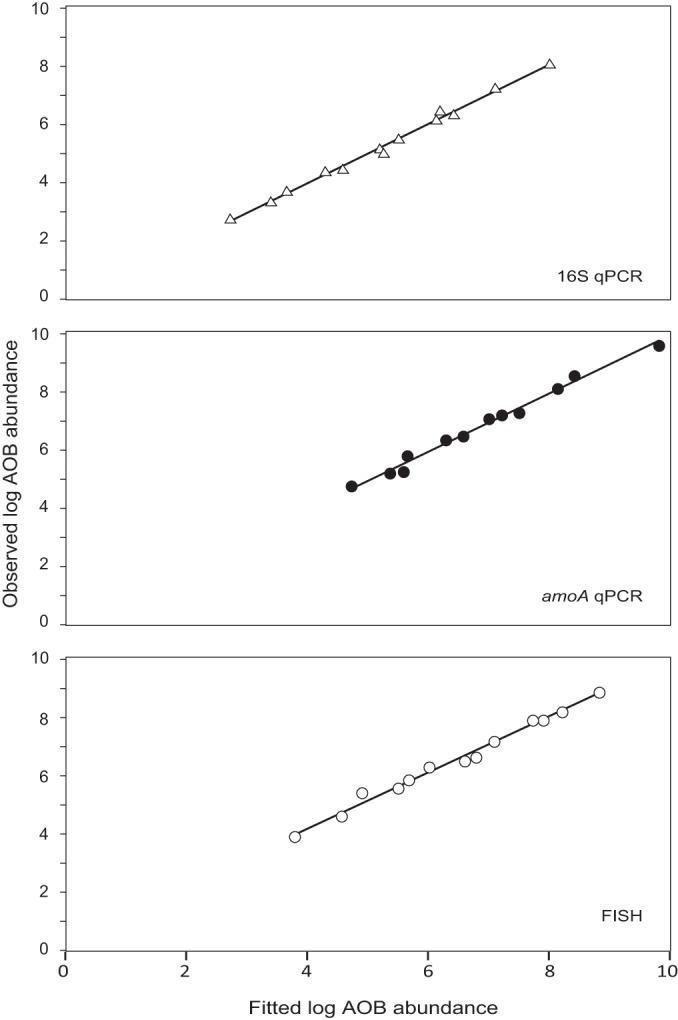
Fitted versus observed log AOB abundances in the serial dilution samples for comparison of the dilution factor with FISH, amoA qPCR, and 16S qPCR. Fitted values were calculated by using equations 2, 3, and 4.
TABLE 2.
Comparison of 16SrRNA-specific qPCR data, amoA-specific qPCR data, and FISH data with dilution factors for the serial dilution samplesa
| Model parameter | Estimate | SE | P | 95% CI (maximum, minimum) |
|---|---|---|---|---|
| First dilution | ||||
| 16S rRNA-specific qPCR | ||||
| Intercept | 8.0 | 0.078 | 0.000*** | 7.8, 8.2 |
| Coefficient of log qPCR 16S | 0.92 | 0.029 | 0.000*** | 0.86, 0.98 |
| amoA-specific qPCR | ||||
| Intercept | 9.8 | 0.14 | 0.000*** | 9.5, 10 |
| Coefficient of log qPCR amoA | 1.4 | 0.077 | 0.000*** | 1.3, 1.6 |
| FISH | ||||
| Intercept | 8.8 | 0.097 | 0.000*** | 8.6, 9.0 |
| Coefficient of log FISH | 1.1 | 0.036 | 0.000*** | 0.98, 1.1 |
| Second dilution | ||||
| 16S rRNA-specific qPCR | ||||
| Intercept | 6.4 | 0.079 | 0.000*** | 6.2, 6.5 |
| Coefficient of log qPCR 16S | 0.92 | 0.029 | 0.000*** | 0.86, 0.98 |
| amoA-specific qPCR | ||||
| Intercept | 8.4 | 0.18 | 0.000*** | 8.0, 8.8 |
| Coefficient of log qPCR amoA | 0.92 | 0.089 | 0.000*** | 0.75, 1.1 |
| FISH | ||||
| Intercept | 8.2 | 0.098 | 0.000*** | 8.0, 8.4 |
| Coefficient of log FISH | 1.1 | 0.036 | 0.000*** | 0.98, 1.1 |
Coefficients and corresponding standard errors, P values, and 95% confidence intervals (CI) for the model parameters of a comparison of 16S rRNA-specific qPCR data, amoA-specific qPCR data, and FISH data with dilution factors for the serial dilution samples are shown. ***, highly significant P value.
The AOB abundance data sets obtained from the three quantification methods were compared using statistical regression, resulting in three best-fit models: one for the comparison of the two qPCR assays (equation 5, R2 = 0.99), one for the comparison of 16S qPCR with FISH (equation 6, R2 = 0.99), and one for the comparison of the amoA qPCR with FISH (equation 7, R2 = 0.95; Fig. 3; Table 3), as shown below.
| (5) |
| (6) |
| (7) |
FIG 3.
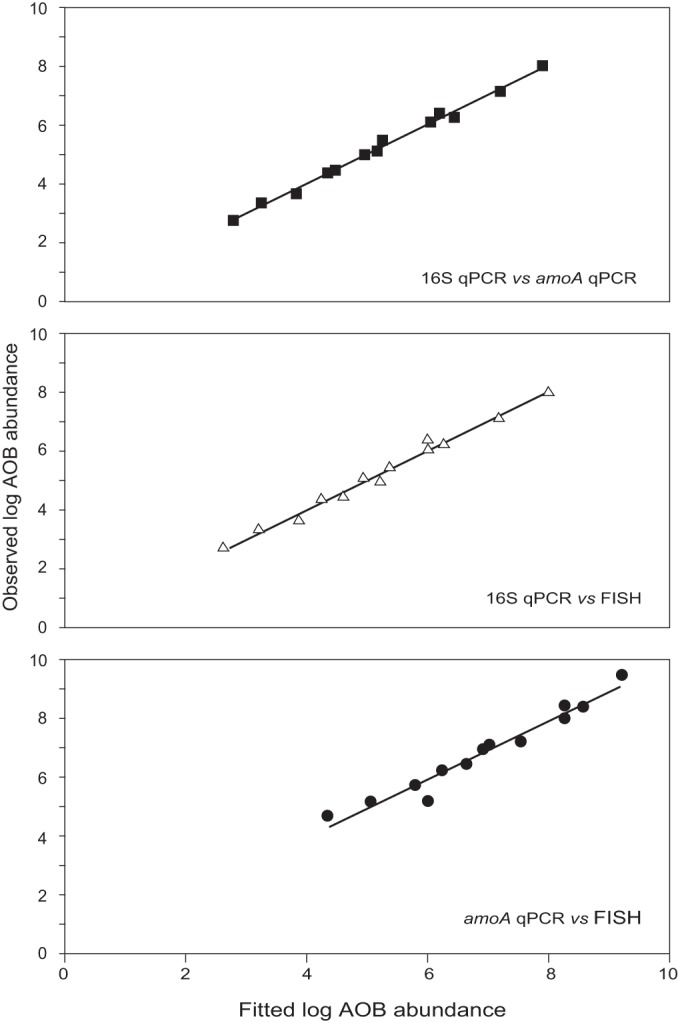
Fitted versus observed log AOB abundances in the serial dilution samples for the comparison of 16S and amoA qPCR, 16S qPCR and FISH, and amoA qPCR and FISH. Fitted values were calculated using equations 5, 6, and 7.
TABLE 3.
Comparison of 16S rRNA-specific qPCR data with amoA-specific qPCR data, 16S rRNA-specific qPCR data with FISH data, and amoA-specific qPCR data with FISH data for the serial dilution samplesa
| Model parameter | Estimate | SE | P | 95% CI (maximum, minimum) |
|---|---|---|---|---|
| First dilution | ||||
| 16S rRNA-specific qPCR | ||||
| Intercept | 1.4 | 0.31 | 0.001** | 0.83, 2.0 |
| Coefficient of log qPCR amoA | 0.68 | 0.040 | 0.000*** | 0.60, 0.76 |
| 16S rRNA-specific qPCR | ||||
| Intercept | 0.39 | 0.30 | 0.22 | −0.20, 0.98 |
| Coefficient of log FISH | 0.86 | 0.042 | 0.000*** | 0.78, 0.94 |
| amoA-specific qPCR | ||||
| Intercept | 0.45 | 0.43 | 0.32 | −0.39, 1.3 |
| Coefficient of log FISH | 0.99 | 0.060 | 0.000*** | 0.87, 1.1 |
| Second dilution | ||||
| 16S rRNA-specific qPCR | ||||
| Intercept | –1.8 | 0.41 | 0.000*** | −2.6, −1.0 |
| Coefficient of log qPCR amoA | 0.97 | 0.051 | 0.000*** | 0.87, 1.1 |
| 16S rRNA-specific qPCR | ||||
| Intercept | –0.78 | 0.12 | 0.000*** | −1.0, −0.54 |
| Coefficient of log FISH | 0.86 | 0.042 | 0.000*** | 0.78, 0.94 |
| amoA-specific qPCR | ||||
| Intercept | 0.45 | 0.43 | 0.32 | −0.39, 1.3 |
| Coefficient of log FISH | 0.99 | 0.060 | 0.000*** | 0.87, 1.1 |
Coefficients and corresponding standard errors, P values, and 95% confidence intervals (CI) for the model parameters of the comparison of the 16S rRNA-specific qPCR data with the amoA-specific qPCR data, 16S rRNA-specific qPCR data with the FISH data, and amoA-specific qPCR data with the FISH data for the serial dilution samples are shown. **, very significant P value; ***, highly significant P value.
The 95% confidence intervals for the slope coefficient and the intercept of the equation describing the relationship between the amoA qPCR and the FISH data included 1 and 0, respectively, implying that the logged AOB cell numbers obtained using these two quantification methods were comparable for both serial dilution samples. Comparison of the 16S qPCR data to that of the other two quantification methods produced relationships with slope coefficients significantly less than 1, suggesting that 16S qPCR underestimates AOB numbers in the serial dilution samples. In addition, the equations describing the relationship of the 16S qPCR data with the data from the other two quantification methods were different for each serial dilution sample, while the comparison of the amoA qPCR and the FISH data produced a single equation for both samples.
Time-series data.
Laboratory-scale and full-scale samples were collected over a period of six and 12 months, respectively, and the abundances of the AOB communities were analyzed using the three quantification methods described above (Fig. 1b). Separate analysis of the abundance data sets for the laboratory-scale samples and for the full-scale samples did not produce significant regressions (the P values for all intercepts and slope coefficients were >0.05). However, analysis of both sets of samples in combination produced significant regressions for all comparisons (Fig. 4; Table 4).
FIG 4.
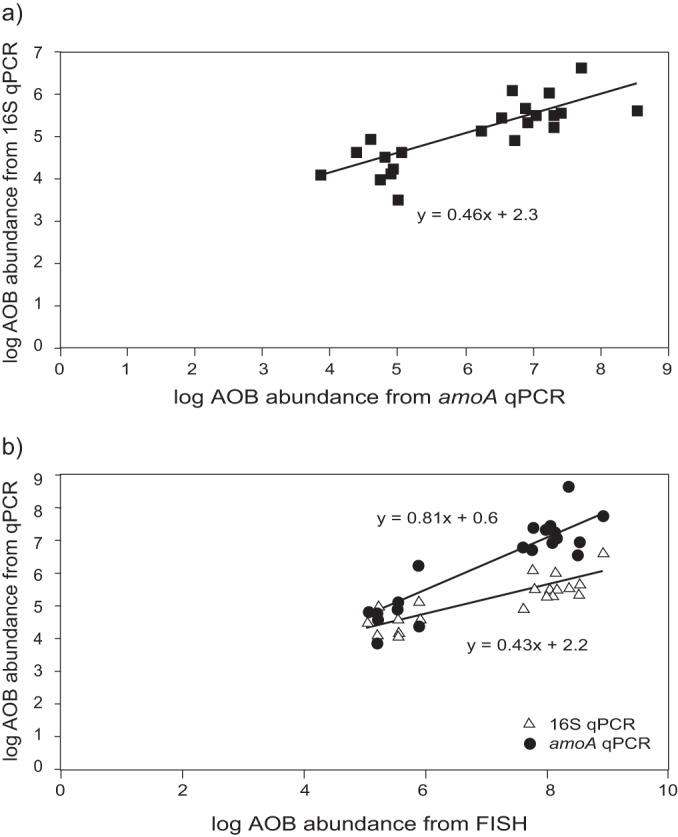
Graphic representation of the best-fit regression models for the time-series data set comparing 16S qPCR with amoA qPCR (a) and comparing 16S qPCR and amoA qPCR with FISH (b).
TABLE 4.
Comparison of 16S rRNA-specific qPCR data with amoA-specific qPCR data, 16S rRNA-specific qPCR data with FISH data, and amoA-specific qPCR data with FISH data for the time-series samplesa
| Model parameter | Estimate | SE | P | 95% CI (maximum, minimum) |
|---|---|---|---|---|
| Comparison of 16SrRNA-specific qPCR data with the amoA-specific qPCR | ||||
| Intercept | 2.3 | 0.47 | 0.000*** | 1.4, 3.2 |
| Coefficient of log qPCR amoA | 0.46 | 0.075 | 0.000*** | 0.31, 0.61 |
| Comparison of the 16S rRNA-specific qPCR data with the FISH data | ||||
| Intercept | 2.2 | 0.45 | 0.001** | 1.3, 3.1 |
| Coefficient of log FISH | 0.43 | 0.064 | 0.000*** | 0.30, 0.56 |
| Comparison of amoA-specific qPCR data with the FISH data | ||||
| Intercept | 0.60 | 0.59 | 0.794 | −0.56, 1.8 |
| Coefficient of log FISH | 0.81 | 0.097 | 0.000*** | 0.62, 1.0 |
Coefficients and corresponding standard errors, P values, and 95% confidence intervals (CI) for the model parameters of the comparison of the 16S rRNA-specific qPCR data with the amoA-specific qPCR data, the 16S rRNA-specific qPCR data with the FISH data, and the amoA-specific qPCR data with the FISH data for the time-series samples are shown. **, very significant P value; ***, highly significant P value.
The best-fit regression models generated for the abundance data obtained from the 16S rRNA qPCR and amoA qPCR (equation 8; R2 = 0.63), 16S qPCR with FISH (equation 8; R2 = 0.70), and amoA qPCR and FISH (equation 10; R2 = 0.84) are given below:
| (8) |
| (9) |
| (10) |
The slope coefficients for regressions of 16S qPCR with the other two methods (equations 8 and 9) were significantly lower than 1, suggesting that the AOB abundances obtained using 16S qPCR were generally significantly lower than the abundances obtained using the other two methods. The regression of the amoA qPCR data on the FISH data produced an equation with an intercept and a slope coefficient that were not statistically different from 0 and 1, respectively, indicating that the AOB abundances obtained using these two methods were comparable.
Combination of the serial dilution and time-series data.
We thought it would be important to incorporate the differences between the four sets of samples in the statistical analysis of our data. Simple linear regression could not be used for the statistical analysis of the data, as the model residuals were not normally distributed (the model assumes a normal distribution). The data were analyzed using the mixed-effects model routine in R, which is a hierarchical linear model that takes into account the different times and locations samples were collected from. The models generated for the comparisons between the three data sets using this statistical method are shown in Fig. 5 and are given by the following equations for the comparison of the two qPCR data sets (equation 11), of the 16S qPCR and FISH (equation 12), and of the amoA qPCR and FISH (equation 13), respectively:
| (11) |
| (12) |
| (13) |
FIG 5.
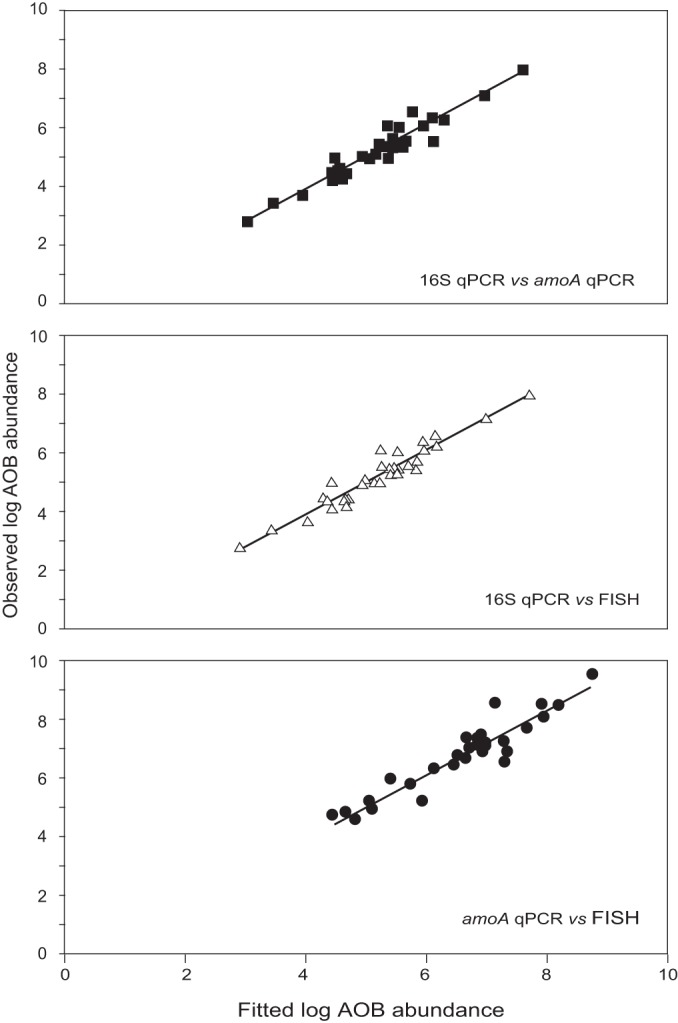
Graphic representation of the fitted versus the observed log abundance data obtained using the random effects models routine for all the samples for the 16S qPCR against the amoA qPCR data, the 16S qPCR against the FISH data, and the amoA qPCR against the FISH data. Fitted values were calculated using equations 11, 12, and 13.
The variable u(group) is a measurement of the error between different groups of samples (serial dilutions, laboratory-scale samples, and full-scale samples), which accounts for variation in the quantification data due to different sampling times and locations. The error term, ε, is the residual error associated with each individual measurement. Both these variables were normally distributed with a mean of 0. The variable “labscale” is 1 for the lab-scale data and 0 for the full-scale data, while the variable “fullscale” is 0 for the lab-scale data and 1 for the full-scale data. The values, standard errors, and P values for the parameters for the three models are given in Table 5.
TABLE 5.
Model parameters for mixed-effects models for comparisons between the two qPCR datasets and between each qPCR and FISH for the entire data seta
| Model parameter | Estimate | SE | P | 95% CI (maximum, minimum) |
|---|---|---|---|---|
| 16S rRNA vs amoA qPCR | ||||
| Intercept for first dilution | 1.4 | 0.50 | 0.012 | 0.42, 2.4 |
| Intercept for second dilution | –1.8 | 0.47 | 0.001** | −2.8, −0.92 |
| Intercept for full-scale and lab-scale | 2.3 | 0.55 | 0.001** | 1.2, 3.4 |
| Log amoA for first dilution | 0.68 | 0.050 | 0.001** | 0.59, 0.79 |
| Log amoA for second dilution | 0.97 | 0.040 | 0.000*** | 0.89, 1.0 |
| Log amoA for full-scale and lab-scale | 0.46 | 0.090 | 0.000*** | 0.28, 0.64 |
| SD u(group) | 0.40 | 0.27, 0.59 | ||
| SD ε | 0.13 | 0.082, 0.21 | ||
| 16S rRNA qPCR vs FISH | ||||
| Intercept for first dilution | 0.43 | 0.45 | 0.35 | 0.22, 2.1 |
| Intercept for second dilution | –0.74 | 0.42 | 0.096 | −0.45, 1.3 |
| Intercept for full-scale | –1.4 | 0.34 | 0.001** | −2.0, −0.69 |
| Intercept for lab-scale | –0.15 | 0.26 | 0.57 | −0.66, 0.36 |
| Log FISH | 0.86 | 0.040 | 0.000*** | 0.78, 0.94 |
| SD u(group) | 0.34 | 0.21, 0.54 | ||
| SD ε | 0.19 | 0.12, 0.29 | ||
| amoA qPCR vs FISH | ||||
| Intercept for first and second dilution | 0.53 | 0.54 | 0.34 | −0.53, 1.6 |
| Intercept for full-scale | –0.76 | 0.60 | 0.22 | −1.9, 0.42 |
| Intercept for lab-scale | –0.33 | 0.43 | 0.46 | −1.2, 0.51 |
| Log FISH | 0.98 | 0.070 | 0.000*** | 0.84, 1.1 |
| SD u(group) | 0.31 | 0.098, 0.98 | ||
| SD ε | 0.36 | 0.21, 0.59 |
Coefficients and corresponding standard errors, P values, and 95% confidence intervals (CI) for the model parameters for the mixed-effects models for the comparisons between the two qPCR datasets and between each qPCR and FISH for the entire data set are shown. **, very significant P value; ***, highly significant P value.
The mixed-effects models provided a very good fit to the data, despite the inherent variability in the measurements obtained from the different groups of samples, substantiating the inclusion of the different sampling times and locations in the comparative models as a random effect. Comparison of the amoA qPCR and the FISH data generated a model with a slope coefficient indistinguishable from 1 with intercepts that were not statistically significant from 0, indicating that the AOB abundances obtained using both methods were equivalent. The slope coefficients for the comparison of the 16S qPCR data with the other two methods were generally lower than 1 and, furthermore, different slope coefficients and intercepts were obtained for different sample groups, suggesting that 16S qPCR underestimates AOB numbers in an inconsistent manner across sample groups. In fact, there is a significant difference (ANOVA; P = 0.001) between the ratios of the mean AOB abundances obtained using the 16S qPCR and either of the other two methods for each group of samples, where the ratios for the first serial dilution are highest and those for the second serial dilution are lowest.
DISCUSSION
We conclude that qPCR targeting the ammonia monooxygenase gene can reliably detect order-of-magnitude differences in AOB cell abundances. We base this conclusion primarily on analysis of amoA qPCR data with FISH (equation 13): the slope being ∼1 (implying correspondence of both methods over the measurement range), the intercept being ∼0 (implying neither over- nor underestimation), and the positive correlation (implying most, but not all, of the variation in qPCR is explained by FISH). This finding increases our confidence in qPCR with these primers in this particular setting and, potentially, in qPCR in general. The need for caution is illustrated by the inferior agreement between FISH and the qPCRs targeting the 16S rRNA gene; the intercept was consistently significantly different from 0 and the slope was <1. It would appear that the 16S rRNA qPCR is not ideal for monitoring AOB communities in the environments tested here.
Robust evaluations of qPCR in microbial ecology are rare. An analogous study carried out by Einen et al. (21) found good agreement between qPCR and quantitative fluorescence microscopy in bacterial and archaeal communities in the glassy rind of seafloor basalts. Negative results tend to go unpublished (see, for example, reference 36). However, Matturro et al. (22) used qPCR and catalyzed reporter deposition-fluorescence in situ hybridization (CARD-FISH) to quantify Dehalococcoides mccartyi in the field, which contain a single copy of the 16S rRNA gene and were present as dispersed cells. These researchers found that qPCR underestimated cell concentrations by at least an order of magnitude.
It seems likely that qPCR protocols should be judged on a case by case basis. Moreover, in the absence of explicit corroboration, the results of a given protocol should be treated with circumspection. A wide range of factors can affect the accuracy of any given quantitative method, including qPCR. In the present study, the importance of the choice of gene is highlighted by the discrepancy in the abundance data obtained using primers targeting different genes, i.e., 16S rRNA gene-specific and amoA gene-specific primers. Functional genes are often thought to be better suited for the quantification of specific populations performing a defined function (25), a view which is corroborated by our data.
The 16S rRNA gene-specific primers underestimated abundances in all of the samples, and these underestimations were inconsistent between locations and sampling events (i.e., the first and second serial dilutions, laboratory-scale reactors, and full-scale plant). These inconsistencies are likely to be a result of the different coverages of the primers and probes used. The amoA gene-specific primers and the FISH probes used target organisms belonging to all known AOB betaproteobacterial lineages (5, 37, 38), while the set of primers targeting the 16S rRNA gene amplifies a few non-AOB sequences and misses some members from the Nitrosomonas communis and the Nitrosomonas oligotropha clusters (37, 39, 40). The fact that members from the N. oligotropha cluster were detected in the lab-scale and full-scale reactors sampled in the present study (sequencing data not shown) could have contributed to the lower cell numbers we obtained using these primers compared to the other two methods; additionally, members of both clusters have been previously detected in biological wastewater treatment plants in the United States, Germany, and Japan (41). If these groups of AOB were present at different abundances in the different sample groups, it would explain the different degrees of underestimation by these primers across sample groups.
The choice of a suitable statistical tool for data analysis is another crucial aspect in the development of a successful quantification protocol. Here, simple linear regression was used for most comparisons, but the combined data sets incorporating the serial dilution and the time-series data did not fit the model assumptions, requiring the use of another statistical tool whose assumptions could be met by our data and that produced a good fit. The mixed-effects model was found to be an appropriate choice, since it took into account the variations in AOB abundance caused by the different times and locations that the samples were collected from.
Despite the good agreement between the abundance data obtained using amoA gene-specific qPCR and FISH, linear regression analysis showed there was no correlation for the laboratory-scale reactor samples and for the full-scale reactor samples, which would suggest that at least one of the quantification methods was not accurately detecting AOB cells in those samples. However, the fitted amoA qPCR logged data for the laboratory and the full-scale reactor samples, calculated using the mixed-effects model equation, differed from the observed values by no more than a factor of 1 (mean variation = 0.30), which means that the qPCR values predicted using the model equation differed from the observed values by no more than an order of magnitude. This further illustrates the importance of selecting an appropriate statistical tool when developing a quantification protocol. The nature of the biases encountered in the present study highlights the importance of producing a calibration curve using samples collected at different times and/or locations and to include abundance data that spans several orders of magnitude (e.g., by using serial dilutions). Routine analysis of biological replicate samples might also be required for finer resolution, such as comparing samples from the same environment collected over a short period of time.
All quantification techniques have their inherent biases. FISH and other methods involving cell staining and microscopy remain the most unequivocal since they involve the direct detection and quantification of cells. However, the cost and time of such methods preclude their use for routine quantification of large numbers of samples. Moreover, microscopic methods have relatively high detection limits and may be subject to observer bias. Thus, the most suitable quantification strategy for a particular situation is dependent on the level of accuracy we are ready to accept. Nonetheless, the approach used here, involving the use of two quantification techniques—one for routine use and the other one to be tested on random samples—proved to be successful. The rigorous evaluation of quantification protocols may appear to be arcane, even boring. However, the complexities of the microbial world are unlikely to be elucidated by a qualitative approach. Moreover, any quantitative approach will never yield the nuanced and predictive understanding we require, if it is undermined by inadequate methodologies.
ACKNOWLEDGMENTS
This study was carried out as part of standard research grant EP/H012133/1 supported by the Engineering and Physical Sciences Research Council.
We acknowledge the help of Trevor Booth with the use of the CLSM and the time and insights of anonymous reviewers.
Footnotes
Published ahead of print 7 July 2014
REFERENCES
- 1.Levy E. 2001. Quantification, mandated science, and judgment. Stud. Hist. Philos. Sci. 32:723–737. 10.1016/S0039-3681(01)00019-X [DOI] [Google Scholar]
- 2.Jones JG. 1979. A guide to methods for estimating microbial numbers and biomass in fresh water. Document SP39 Freshwater Biological Association, Cumbria, United Kingdom [Google Scholar]
- 3.Madigan MT, Martinko JM, Stahl DA, Clark DP. 2012. Brock: biology of microorganisms, 13th ed. Pearson Education, Cranbury Township, NJ [Google Scholar]
- 4.Koops H-P, Pommerening-Röser A. 2001. Distribution and ecophysiology of the nitrifying bacteria emphasizing cultured species. FEMS Microbiol. Ecol. 37:1–9. 10.1111/j.1574-6941.2001.tb00847.x [DOI] [Google Scholar]
- 5.Koops H-P, Purkhold U, Pommerening-Röser A, Timmermann G, Wagner M. 2006. The lithoautotrophic ammonia-oxidizing bacteria, p 778–811 In Dworkin M, Falkow S, Osenberg E, Schleifer K-H, Stackebrandt E. (ed), The prokaryotes, 3rd ed, vol 5 Springer, New York, NY [Google Scholar]
- 6.Coskuner G, Ballinger SJ, Davenport RJ, Pickering RL, Solera RR, Head IM, Curtis TP. 2005. Agreement between theory and measurement in quantification of ammonia-oxidizing bacteria. Appl. Environ. Microbiol. 71:6325–6334. 10.1128/AEM.71.10.6325-6334.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davenport RJ, Curtis TP. 2004. Quantitative fluorescence in situ hybridisation (FISH): statistical methods for valid cell counting, p 3389–3417 In Kowalchuk GA, Brujin FJ, Head IM, Akkermans AD, van Elsas JD. (ed), Molecular microbial ecology manual, 2nd ed, vol 2 Springer, Dordrecht, Netherlands [Google Scholar]
- 8.Mitulewich VA, Strom PF, Finstein MS. 1975. Length of incubation for enumerating nitrifying bacteria present in various environments. Appl. Microbiol. 29:265–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikuta H, Noda N, Ebie Y, Hirata A, Tsuneda S, Matsumura M, Inamori Y. 2000. The rapid quantification and detection of nitrifying bacteria by using monoclonal antibody method. Water Sci. Technol. 42:1–7 [Google Scholar]
- 10.Dionisi HM, Harms G, Layton AC, Gregory IR, Parker J, Hawkins SA, Robinson KG, Sayler GS. 2003. Power analysis for real-time PCR quantification of genes in activated sludge and analysis of the variability introduced by DNA extraction. Appl. Environ. Microbiol. 69:6597–6604. 10.1128/AEM.69.11.6597-6604.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harms G, Layton AC, Dionisi HM, Gregory IR, Garrett VM, Hawkins SA, Robinson KG, Sayler GS. 2003. Real-time PCR quantification of nitrifying bacteria in a municipal wastewater treatment plant. Environ. Sci. Technol. 37:343–351. 10.1021/es0257164 [DOI] [PubMed] [Google Scholar]
- 12.Kindaichi T, Kawano Y, Ito T, Satoh H, Okabe S. 2006. Population dynamics and in situ kinetics of nitrifying bacteria in autotrophic nitrifying biofilms as determined by real-time quantitative PCR. Biotechnol. Bioeng. 94:1111–1121. 10.1002/bit.20926 [DOI] [PubMed] [Google Scholar]
- 13.Suzuki M, Taylor LT, DeLong EF. 2000. Quantitative analysis of small-subunit rRNA genes in mixed microbial populations via 5′-nuclease assays. Appl. Environ. Microbiol. 66:4605–4614. 10.1128/AEM.66.11.4605-4614.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pernthaler A, Pernthaler J, Amann R. 2002. Fluorescence in situ hybridization and catalyzed reporter deposition for the identification of marine bacteria. Appl. Environ. Microbiol. 68:3094–3101. 10.1128/AEM.68.6.3094-3101.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klein D. 2002. Quantification using real-time PCR technology: applications and limitations. Trends Mol. Med. 8:257–260. 10.1016/S1471-4914(02)02355-9 [DOI] [PubMed] [Google Scholar]
- 16.Sekido T, Bodelier PLE, Shoji T, Suwa Y, Laanbroek HJ. 2008. Limitations of the use of group-specific primers in real-time PCR as appear from quantitative analyses of closely related ammonia-oxidizing species. Water Res. 42:1093–1101. 10.1016/j.watres.2007.08.024 [DOI] [PubMed] [Google Scholar]
- 17.Lim J, Do H, Shin SG, Seokhwan H. 2007. Primer and probe sets for group-specific quantification of the genera Nitrosomonas and Nitrosospira using real-time PCR. Biotechnol. Bioeng. 99:1374–1383. 10.1002/bit.21715 [DOI] [PubMed] [Google Scholar]
- 18.Smith CJ, Osborn AM. 2009. Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology. FEMS Microbiol. Ecol. 67:6–20. 10.1111/j.1574-6941.2008.00629.x [DOI] [PubMed] [Google Scholar]
- 19.Fogel GB, Collins CR, Brunk CF. 1999. Prokaryotic genome size and SSU rDNA copy number: estimation of microbial relative abundance from a mixed population. Microb. Ecol. 38:93–113. 10.1007/s002489900162 [DOI] [PubMed] [Google Scholar]
- 20.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55:611–622. 10.1373/clinchem.2008.112797 [DOI] [PubMed] [Google Scholar]
- 21.Einen J, Thorseth IH, Øvreås L. 2008. Enumeration of Archaea and Bacteria in seafloor basalt using real-time quantitative PCR and fluorescence microscopy. FEMS Microbiol. Lett. 282:182–187. 10.1111/j.1574-6968.2008.01119.x [DOI] [PubMed] [Google Scholar]
- 22.Matturro B, Heavner GL, Richardson RE, Rossetti S. 2013. Quantitative estimation of Dehalococcoides mccartyi at laboratory and field scale: comparative study between CARD-FISH and real-time PCR. J. Microbiol. Methods 93:127–133. 10.1016/j.mimet.2013.02.011 [DOI] [PubMed] [Google Scholar]
- 23.Kowalchuk GA, Stephen JR, De Boer W, Prosser JI, Embley TM, Woldendorp JW. 1997. Analysis of ammonia-oxidizing bacteria of the β subdivision of the class Proteobacteria in coastal sand dunes by denaturing gradient gel electrophoresis and sequencing of PCR-amplified 16S ribosomal DNA fragments. Appl. Environ. Microbiol. 63:1489–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stephen JR, Chang Y-J, MacNaughton SJ, Kowalchuk GA, Leung KT, Flemming CA, White DC. 1999. Effect of toxic metals on indigenous soil β-subgroup proteobacterium ammonia oxidizer community structure and protection against toxicity by inoculated metal-resistant bacteria. Appl. Environ. Microbiol. 65:95–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rotthawe J-H, Witzel K-P, Liesack W. 1997. The ammonia monooxygenase structural gene amoA as a functional marker: molecular fine-scale analysis of natural ammonia-oxidizing populations. Appl. Environ. Microbiol. 63:4704–4712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McTavish H, Fuchs JA, Hooper AB. 1993. Sequence of the gene coding for ammonia monooxygenase in Nitrosomonas europaea J. Bacteriol. 175:2436–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA. 1990. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56:1919–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amann RI, Krumholz L, Stahl DA. 1990. Fluorescent-oligonucleotide probing of whole cells for determinative phylogenetic and environmental studies in microbiology. J. Bacteriol. 172:762–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mobarry BK, Wagner M, Urbain V, Rittman BE, Stahl DA. 1996. Phylogenetic probes for analyzing abundance and spatial organization of nitrifying bacteria. Appl. Environ. Microbiol. 62:2156–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner M, Rath G, Amann R, Koops H-P, Schleifer K-H. 1995. In situ identification of ammonia-oxidizing bacteria. Syst. Appl. Microbiol. 18:251–264. 10.1016/S0723-2020(11)80396-6 [DOI] [Google Scholar]
- 31.Adamczyk J, Hesselsoe M, Iversen N, Horn M, Lehner A, Nielsen PH, Schloter M, Roslev P, Wagner M. 2003. The isotope array, a new tool that employs substrate-mediated labeling of rRNA for determination of microbial community structure and function. Appl. Environ. Microbiol. 69:6875–6887. 10.1128/AEM.69.11.6875-6887.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daims H, Brühl A, Amann R, Schleifer K-H, Wagner M. 1999. The domain-specific probe EUB338 is insufficient for the detection of all bacteria: development and evaluation of a more comprehensive probe set. Syst. Appl. Microbiol. 22:434–444. 10.1016/S0723-2020(99)80053-8 [DOI] [PubMed] [Google Scholar]
- 33.Lewis T, Taylor LR. 1967. Introduction to experimental ecology. Academic Press, New York, NY [Google Scholar]
- 34.Limpert E, Stahel WA, Abbt M. 2001. Log-normal distributions across the sciences: keys and clues. Bioscience 51:341–352. 10.1641/0006-3568(2001)051[0341:LNDATS]2.0.CO;2 [DOI] [Google Scholar]
- 35.Pinheiro JC, Bates DM. 2000. Mixed-effects models in S and S-PLUS. Springer-Verlag, New York, NY [Google Scholar]
- 36.Pickering R. 2008. How hard is the biomass working? Ph.D. thesis Newcastle University, Newcastle upon Tyne, United Kingdom [Google Scholar]
- 37.Lydmark P, Almstrand R, Samuelsson K, Mattsson A, Sörensson F, Lindgren P-E, Hermansson M. 2007. Effects of environmental conditions on the nitrifying population dynamics in a pilot wastewater treatment plant. Environ. Microbiol. 9:2220–2233. 10.1111/j.1462-2920.2007.01336.x [DOI] [PubMed] [Google Scholar]
- 38.Purkhold U, Pommerening-Röser A, Juretschko S, Schmid MC, Koops H-P, Wagner M. 2000. Phylogeny of all recognized species of ammonia oxidizers based on comparative 16S rRNA and amoA sequence analysis: implications for molecular diversity surveys. Appl. Environ. Microbiol. 66:5368–5382. 10.1128/AEM.66.12.5368-5382.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cébron A, Coci M, Garnier J, Laanbroek HJ. 2004. Denaturing gradient gel electrophoretic analysis of ammonia-oxidizing bacterial community structure in the lower Seine River: impact of Paris wastewater effluents. Appl. Environ. Microbiol. 70:6726–6737. 10.1128/AEM.70.11.6726-6737.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahmood S, Freitag TE, Prosser JI. 2006. Comparison of PCR primer-based strategies for characterization of ammonia oxidizer communities in environmental samples. FEMS Microbiol. Ecol. 56:482–493. 10.1111/j.1574-6941.2006.00080.x [DOI] [PubMed] [Google Scholar]
- 41.Wells GF, Park H-D, Yeung C-H, Eggleston B, Francis CA, Criddle CS. 2009. Ammonia-oxidizing communities in a highly aerated full-scale activated sludge bioreactor: betaproteobacterial dynamics and low relative abundance of Crenarchaea. Environ. Microbiol. 11:2310–2328. 10.1111/j.1462-2920.2009.01958.x [DOI] [PubMed] [Google Scholar]
- 42.Brosius J, Palmer ML, Kennedy PJ, Noller HF. 1978. Complete nucleotide sequence of a 16S ribosomal RNA gene from Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 75:4801–4805. 10.1073/pnas.75.10.4801 [DOI] [PMC free article] [PubMed] [Google Scholar]