

Abstract
Marine red macroalgae have emerged to be renewable biomass for the production of chemicals and biofuels, because carbohydrates that form the major component of red macroalgae can be hydrolyzed into fermentable sugars. The main carbohydrate in red algae is agarose, and it is composed of d-galactose and 3,6-anhydro-l-galactose (AHG), which are alternately bonded by β1-4 and α1-3 linkages. In this study, a novel β-galactosidase that can act on agarooligosaccharides (AOSs) to release galactose was discovered in a marine bacterium (Vibrio sp. strain EJY3); the enzyme is annotated as Vibrio sp. EJY3 agarolytic β-galactosidase (VejABG). Unlike the lacZ-encoded β-galactosidase from Escherichia coli, VejABG does not hydrolyze common substrates like lactose and can act only on the galactose moiety at the nonreducing end of AOS. The optimum pH and temperature of VejABG on an agarotriose substrate were 7 and 35°C, respectively. Its catalytic efficiency with agarotriose was also similar to that with agaropentaose or agaroheptaose. Since agarotriose lingers as the unreacted residual oligomer in the currently available saccharification system using β-agarases and acid prehydrolysis, the agarotriose-hydrolyzing capability of this novel β-galactosidase offers an enormous advantage in the saccharification of agarose or agar in red macroalgae for its use as a biomass feedstock for fermentable sugar production.
INTRODUCTION
Red algae (Rhodophyta) could be considered a potential source of renewable biomass for the production of fuels and chemicals because the monomeric sugars obtained by the hydrolysis of algal carbohydrates can be fermented by microorganisms (1, 2). The main constituent of red algae is agarose, which is also a cell wall component (3). Agarose consists of d-galactose and 3,6-anhydro-l-galactose (AHG), which are alternately bonded by β1-4 and α1-3 linkages (3). Agarose-derived oligosaccharides are classified into agarooligosaccharides (AOSs) and neoagarooligosaccharides (NAOSs) (Fig. 1). The nonreducing end of AOSs is the galactose unit, whereas that of NAOSs is the AHG unit (3, 4).
FIG 1.

Chemical structures of agarose-derived oligosaccharides. (A) d-Galactose (Gal); (B) 3,6-anhydro-l-galactose (AHG); (C) agarooligosaccharide (AOS); (D) neoagarooligosaccharide (NAOS); (E) agarotriose. The nonreducing end of agarooligosaccharide is the galactose unit, whereas that of neoagarooligosaccharide is the AHG unit. Agarotriose is the smallest odd-numbered agarooligosaccharide.
Three methods have primarily been used for agarose hydrolysis: acid hydrolysis, enzymatic hydrolysis, and enzymatic saccharification combined with acid prehydrolysis. Acid hydrolysis using a strong acid at a high temperature is a simple, less expensive, and rapid method (5, 6); however, this method produces sugar degradation products such as 5-hydroxymethylfurfural (6–9), which can inhibit fermentative microorganisms (10, 11).
This problem is circumvented by the enzymatic hydrolysis method because it is performed at a milder temperature. The β-agarase system that is commonly used by marine agarolytic bacteria has been well established for agarose hydrolysis (7, 8, 12–14). This system is capable of producing monomeric sugars from agarose and consists of β-agarases I and II (EC 3.2.1.81) and a neoagarooligosaccharide hydrolase (NAOS hydrolase) or a neoagarobiose hydrolase (NABH; EC 3.2.1.159), which was discovered in several agarolytic bacteria, including Saccharophagus degradans 2-40T and Zobellia galactanivorans (12, 14). β-Agarases I and II cleave the β1-4 linkages of agarose to release NAOS and neoagarobiose, respectively, whereas NAOS hydrolase degrades neoagarobiose into AHG and galactose (15–17).
Agarose hydrolysis using enzymes alone results in a low yield of monomeric sugars owing to the low solubility of agarose in water as the substrate for the enzyme reaction (18). Therefore, enzymatic hydrolysis combined with weak acid prehydrolysis was recently developed for agarose saccharification (18–20), in which agarose is predominantly prehydrolyzed into even-numbered AOSs with galactose at the nonreducing ends since α1-3 linkages are preferentially cleaved by a weak acid. The AOSs are then hydrolyzed into monomeric sugars by a β-agarase II and an NABH. A major disadvantage of this method is that the smallest odd-numbered AOS, agarotriose [galactose-β1-4-AHG-α1-3-galactose] (Fig. 1), produced by a β-agarase II, cannot be further hydrolyzed into monomers by using typical β-agarases and a NAOS hydrolase (or an NABH) (20). For the cost-effective use of agarose as a biomass feedstock, a high yield of saccharification that does not leave unreacted oligomeric sugars is highly desirable. In our previous study (20), when an NABH was combined with the crude enzyme of Vibrio sp. strain EJY3, which was previously isolated from the coast of the West Sea of Korea (21), the crude enzyme was found to hydrolyze agarotriose into AHG and galactose, thus increasing the final yield of monomeric sugars compared to that achieved in experiments that did not include the crude enzyme. Therefore, we hypothesized that such an enzyme capable of cleaving the β linkage of agarotriose may be expressed from the genes that encode three putative β-galactosidases (EC 3.2.1.22) in Vibrio sp. EJY3, the nucleotide sequences of which were obtained from UniProt (22). Such an enzyme that can act on agarose-derived galactan to produce galactose is considered a novel β-galactosidase capable of hydrolyzing agarose in agarooligosaccharides.
In this study, we describe the identification, expression, and biochemical characterization of this novel β-galactosidase isolated from Vibrio sp. EJY3 and its role in the agar degradation process. This novel agarolytic enzyme is extremely important, not only due to its novelty but also by virtue of its industrial application. This enzyme can supplement the missing function of the currently available β-agarase system in the saccharification of agarose by completely hydrolyzing the entrapped, odd-numbered short-chain AOSs.
MATERIALS AND METHODS
Cloning and expression of putative genes of agarolytic β-galactosidase.
Vibrio sp. EJY3, which was previously isolated from the coast of the West Sea of Korea (21), was grown in a sea salt minimal medium containing 23 g of synthetic sea salt (Aquarium Systems), 50 mmol Tris-HCl, 2 g agarose (Invitrogen), 1 g yeast extract, and 0.5 g ammonium chloride in 1 liter of water at 30°C for 4 h (20, 21). The genomic DNA was extracted using a commercial DNA isolation kit (Qiagen). Eight putative genes annotated as encoding an α- or β-galactosidase (VEJY3_05670 [UniProt accession no. H2IA24], VEJY3_09135 [UniProt accession no. H2IFD0], VEJY3_09170 [UniProt accession no. H2IFD7], VEJY3_09535 [UniProt accession no. H2IG01], VEJY3_21461 [UniProt accession no. H2IN31], VEJY3_22581 [UniProt accession no. H2ILJ6], VEJY3_22596 [UniProt accession no. H2ILJ9], and VEJY3_24051 [UniProt accession no. H2INB0]) were amplified using Phusion thermostable polymerase (Thermo Scientific). The primers used for the amplification of these genes are described in Table 1. The restriction sites of BamHI and NotI were added at the 5′ and 3′ regions of the N- and C-terminal ends, respectively. The PCR product and pET21a (Novagen) were digested by BamHI and NotI and ligated together using T4 DNA ligase (BioLabs). The resulting pET21a vectors harboring each putative gene were transformed into Escherichia coli BL21(DE3) (Novagen).
TABLE 1.
Primers used for cloning the putative genes of α- and β-galactosidases from Vibrio sp. EJY3
| Putative function | Gene name | Sequence (5′ →3′) |
|
|---|---|---|---|
| Forward primer | Reverse primer | ||
| β-Galactosidase | VEJY3_09170 | GCGGGATCCATGCACAATTCACCGAGAAGTAC | GCGGCGGCCGCAAGTAGTGAAAAAGAGGCTTTCAC |
| VEJY3_22596 | GCGGGATCCATGGCATTTTCAGATATTATTCAACG | GCGGCGGCCGCAGCGGGCTGAAACGATACTC | |
| VEJY3_24051 | GCGGGATCCATGGCATCACAGATAGAAAAGAC | GCGGCGGCCGCGAAATGAAATTCGCCACAATCTG | |
| α-Galactosidase | VEJY3_22581 | GCGGTCGACATGACGGACAAAACGCTGATTG | GCGGCGGCCGCTTCGTTTACGCGTTCAAACTTCAC |
| VEJY3_09535 | GCGGGATCCATGAATAATTTTGTACATCTGCAAAGT | GCGGCGGCCGCTATTTTTTGTAAATGAACAATCAGTGC | |
| VEJY3_21461 | GCGGTCGACATGAAAGAAAAAAAGTTAGTTGAGTTAAG | GCGGCGGCCGCATTTATCTTCTCGACTTTAATTAACATC | |
| VEJY3_09135 | GCGGGATCCATGCAAAGAATCGTTCACCTAAAATC | GCGGCGGCCGCAAGTTTTTTCAGGTGAACAATAAGCG | |
| VEJY3_05670 | GCGGGATCCATGACTCAAACCTTTCAGTTCGC | GCGGCGGCCGCTACCGTGACGATGGCTTTTGC | |
To produce recombinant proteins from each gene, cells were grown at 37°C in Luria-Bertani broth (BD) containing 50 μg/ml of ampicillin until the mid-exponential phase and then induced by adding 0.1 mM isopropyl-β-d-thiogalactopyranoside (Sigma) at 16°C for 16 h. These cells were harvested by centrifugation at 5,000 × g for 15 min at 4°C and resuspended in ice-cold lysis buffer, composed of 20 mM sodium phosphate and 500 mM NaCl (pH 7.4). Each cell suspension was disrupted by a sonicator (Branson), and the supernatant was collected by centrifugation at 16,000 × g for 1 h at 4°C. The recombinant proteins were purified by two consecutive chromatographic separations using a His-trap column (GE Healthcare) and an anion-exchange column (GE Healthcare). The purified proteins were concentrated using an Amicon ultrafiltration membrane (Millipore), and the protein concentration was determined using a bicinchoninic acid protein assay kit (Pierce).
Determination of enzymatic activity of agarolytic β-galactosidase.
To determine the enzyme activity of the agarolytic β-galactosidase from Vibrio sp. EJY3 (VejABG) on agarotriose, 9.14 pmol of protein was incubated in 100 μl of 20 mM Tris-HCl buffer (pH 7) containing 2.5 mM agarotriose (Beijing Ad Hoc International Technologies) at 30°C for 25 min. After the reaction was stopped by boiling the reaction mixture, galactose was quantified by high-performance liquid chromatography (HPLC) as described later in the section “Analyses of the enzymatic reaction products by TLC and HPLC.” A commercial β-galactosidase from E. coli (i.e., lacZ-encoded β-galactosidase; catalog no. G4155; Sigma) (23) was also tested using agarotriose as the substrate. One unit of agarolytic β-galactosidase on agarotriose was defined as the amount of enzyme required to produce 1 μmol of galactose per min using the conditions described above.
Screening for substrate specificity of agarolytic β-galactosidase.
To examine the substrate specificity of VejABG, a total of 7 substrates were used. The first substrate group was composed of 3 AOSs, agarotriose, agaropentaose, and agaroheptaose (Beijing Ad Hoc International Technologies), whereas the second group consisted of common substrates of general β-galactosidases, lactose, lactulose (Sigma), lacto-N-neotetraose (LNT; Dextra Laboratories), and galactobiose (galactose-β1-4-galactose; Dextra Laboratories). To determine the enzymatic activity on the even-numbered AOSs, acetic acid-prehydrolyzed agarose was used as the substrate, which was prepared by dissolving agarose (Invitrogen) in a 3 N acetic acid solution to a final concentration of 7% (wt/vol), using a microwave digester (Milestone) at 130°C for 10 min. The acid-prehydrolyzed agarose was then lyophilized using a freeze dryer for 24 h. To screen for the substrate specificity of VejABG, 9.14 pmol of the purified recombinant protein was incubated in 100 μl of 20 mM Tris-HCl buffer (pH 7) containing 2.5 mM each substrate.
Characterization of agarolytic β-galactosidase.
To study the temperature optimum of VejABG, this enzyme was incubated with 2.5 mM agarotriose at 10 to 50°C in 20 mM Tris-HCl (pH 7) for 25 min. The optimum pH of VejABG was also examined by incubation with 2.5 mM agarotriose contained in buffers of different pHs, such as 50 mM citric acid (pH 5 to 6), 20 mM Tris-HCl (pH 7 to 8), and glycine-NaOH (pH 9 to 10), at 35°C for 25 min. The kinetic parameters (Vmax and Km) of the enzyme for the different odd-numbered AOSs were determined from the Lineweaver-Burk plot, in which the enzyme activity was measured using agarotriose, agaropentaose, and agaroheptaose at concentrations ranging from 0.625 mM to 40 mM.
To verify whether it is possible to completely saccharify AOSs into monomeric sugars using VejABG and the NAOS hydrolase from Saccharophagus degradans 2-40T (SdNABH), 91.4 nmol of each enzyme was incubated in 100 μl of 20 mM Tris-HCl buffer (pH 7) containing 2.5 mM agarotriose, agaropentaose, or agaroheptaose at 35°C for 24 h.
Analyses of the enzymatic reaction products by TLC and HPLC.
To analyze the reaction products of VejABG by thin-layer chromatography (TLC), a 1-μl aliquot from each reaction mixture was loaded onto silica gel 60 TLC plates (Merck), and the plates were developed with n-butanol–ethanol–water (3:1:1, by volume). The plates loaded with sample were visualized with 10% (vol/vol) H2SO4 and 0.2% (wt/vol) naphthoresorcinol in ethanol (19).
The final products from the combined treatment of VejABG and SdNABH were analyzed using an Agilent 1100 HPLC system (Agilent Technologies) equipped with a gel permeation column (KS-802; Shodex) and a refractive index detector. Analysis was conducted at 80°C using distilled water as the mobile phase at a flow rate of 0.5 ml/min. Molecular size markers (Waters) ranging from 106 Da to 4,270 Da were used to distinguish the degrees of polymerization (DPs) of products in the range of 1 to 7.
Analyses of the enzymatic reaction products by GC-MS and MALDI-TOF/TOF MS.
To analyze monomeric sugars obtained from the enzymatic reaction on acid-prehydrolyzed agarose, gas chromatography-mass spectrometry (GC-MS) was performed according to the previously described methods (24). Briefly, the aldehyde group of the sample was derivatized by methoxyamination using 50 μl of 20 mg/ml methoxyamine hydrochloride in pyridine (Sigma) at 75°C for 30 min. The volatility of the sample was increased by adding 80 μl of N-methyl-N-(trimethylsilyl)trifluoroacetamide (Fluka). An Agilent 7890A GC/5975C MSD system (Agilent Technologies) equipped with a DB5-MS column (30 m by 0.25 mm [inner diameter], 0.25-μm film thickness; Agilent Technologies) was used. For the GC-MS analysis, the column temperature of the GC was initially set at 100°C for 3.5 min, increased to 160°C at 15°C/min, and maintained at 160°C for 20 min. The temperature was then increased to 200°C at 20°C/min, this temperature was maintained for 15 min, and finally, it was increased to 280°C at 20°C/min and held at 280°C for 5 min. The mass spectra were recorded at 70 eV of electron impact, with the temperature of the ion source of the MS being 230°C. Mass spectra were recorded in the range of 50 to 700 m/z.
The matrix-assisted laser desorption ionization–tandem time of flight mass spectrometry (MALDI-TOF/TOF MS) analysis was performed in the positive ion reflectron mode using an ultrafleXtreme system (Bruker Daltonics). Purified reaction products were dissolved in water, and 1 μl of the solubilized reaction products was spotted onto a stainless steel target plate, followed by the addition of 0.3 μl of 0.01 M NaCl and 0.5 μl of 50 mg/ml 2,5-dihydroxybenzoic acid in 50% acetonitrile. The spot was rapidly dried under vacuum for homogeneous crystallization. Each acquired spectrum represented the combined signal from 800 laser shots at each of 3 random locations on the spot, totaling 2,400 laser shots, using a 1-kHz laser. The laser attenuator offset and range were set at 68% and 15%, respectively, and the laser focus was set at 50%. Mass spectra were recorded over the range of 0 to 5,100 m/z. To obtain high-resolution data, the detector sampling rate was set at a maximum rate of 4.00 gigasamples/s, and the detector gain was set at 4.0×. Mass spectra were externally calibrated using maltooligosaccharides isolated from commercial beer. A ladder of hexose polymers, spaced 1 hexose unit (162.053 Da) apart, provided comprehensive coverage of the entire mass acquisition range, enabling the accurate mass calibration of the MALDI-TOF/TOF MS just prior to the sample analysis. Raw MS data were processed with FlexAnalysis software (version 3.3; Bruker Daltonics). MS peaks were filtered with a signal-to-noise ratio of 3.0 and manually inspected to detect the formation of sodium, potassium, or other common adducts. All peaks were then deconvoluted, and a list of all neutral masses in the samples was generated, with the abundances represented by mass spectral peak intensities.
Phylogenetic analysis.
To determine the amino acid sequence homology between VejABG and other β-galactosidases from glycoside hydrolase family 2 (GH2), a phylogenetic tree was drawn using the MEGA (version 5) program with the neighbor-joining method (25), and the GH2 β-galactosidases were selected from the CAZy database (26).
RESULTS
Identification of a β-galactosidase that acts on agarotriose.
We previously showed that agarotriose, consisting of two units of galactose and one unit of AHG in alternating β1-4 and α1-3 glycosidic linkages, can be depolymerized using a NAOS hydrolase and the crude enzyme of Vibrio sp. EJY3 (20). To search for the enzyme responsible for this β-galactosidase activity against agarotriose, eight putative genes encoding α- or β-galactosidases in Vibrio sp. EJY3 were cloned, expressed, and tested for the activities of their recombinant enzymes. Among these, the crude enzyme obtained from E. coli BL21(DE3) harboring a plasmid containing the VEJY3_09170 gene was found to have the ability to hydrolyze agarotriose into galactose and neoagarobiose (data not shown). Therefore, the purified enzyme with a theoretical mass of 93 kDa (Fig. 2), encoded by VEJY_09170, was named agarolytic β-galactosidase (VejABG) and was then examined as the potential β-galactosidase acting an agarotriose.
FIG 2.
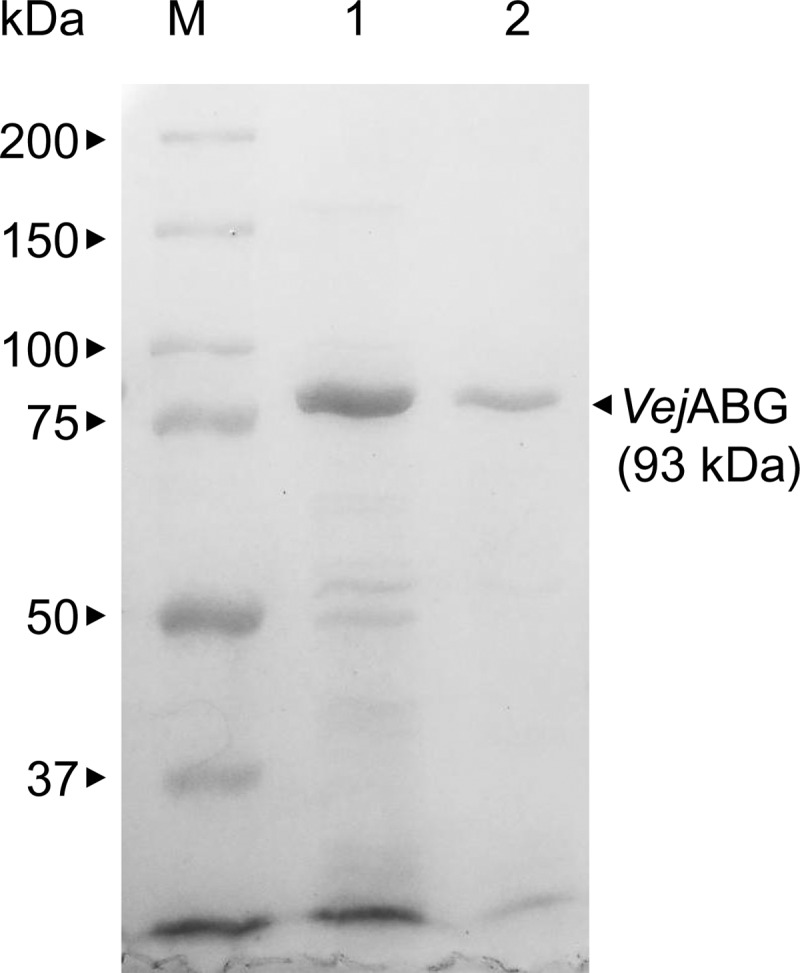
SDS-PAGE analysis of the purified recombinant VejABG. Lanes: M, protein markers; 1, VejABG purified by His-tag affinity chromatography; 2, VejABG purified by His-tag and anion-exchange chromatography.
Substrate specificity of agarolytic β-galactosidase.
The specificity of VejABG was tested on 7 different substrates. VejABG was found to release galactose from odd-numbered AOSs, such as agarotriose, agaropentaose, and agaroheptaose (Fig. 3A). However, lactose, lacto-N-neotetraose, lactulose, and galactobiose, which are the common substrates hydrolyzable by the general β-galactosidases, such as the lacZ-encoded β-galactosidase of E. coli, were not hydrolyzed by VejABG (Fig. 3B). Although the lacZ-encoded β-galactosidase did not hydrolyze the AOSs with DP3, DP5, and DP7, which indicate agarotriose, agaropentaose, and agaroheptaose, respectively, it was capable of hydrolyzing all the general substrates (data not shown).
FIG 3.
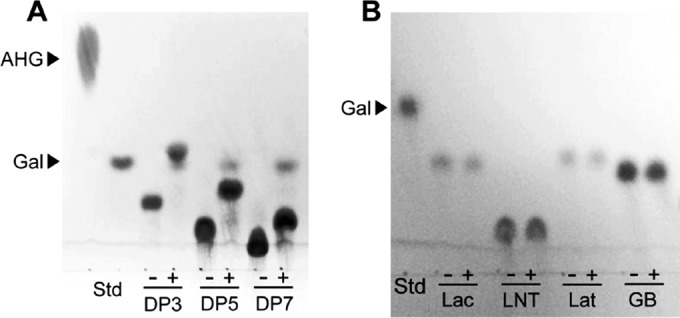
Substrate specificity of VejABG. TLC analysis of the reaction products of recombinant VejABG incubated with agarooligosaccharides with different DPs (A) and the general substrates of β-galactosidase (B). −, substrate only; +, substrate incubated with VejABG for 12 h. DP3, agarotriose; DP5, agaropentaose; DP7, agaroheptaose; Lac, lactose; LNT, lacto-N-neotetraose; Lat, lactulose; GB, galactobiose. Galactose (Gal) and AHG were used as markers for the reaction products. Std, standard.
Optimum pH and temperature of VejABG for agarotriose hydrolysis.
To determine the optimum pH and temperature of VejABG for the hydrolysis of agarotriose, 2.5 mM agarotriose was incubated with VejABG at different temperatures (10 to 50°C) and pHs (pH 4 to 10). The enzyme showed the highest activity at pH 7 and maintained approximately 70% of its maximum activity over a broad pH range from 6 to 9 (Fig. 4A). The optimum temperature for VejABG was determined to be approximately 35°C in 20 mM Tris-HCl buffer (pH 7), and its activity decreased significantly at temperatures higher or lower than 35°C (Fig. 4B).
FIG 4.

Effect of pH and temperature on the activity of VejABG. The optimum pH (A) and temperature (B) of VejABG in the hydrolysis of agarotriose were determined. The enzymatic reactions were performed using buffers of different pHs (pH 4 to 10) and different temperature (10 to 50°C) conditions for 25 min.
Kinetic parameters of VejABG.
The kinetic parameters of VejABG were determined from the Lineweaver-Burk plot by using agarotriose, agaropentaose, and agaroheptaose as the substrates at pH 7 and 35°C. To determine the kinetic parameters of VejABG for these AOSs, the amount of galactose produced was measured by HPLC. The Km for agarotriose was approximately 2 times higher than the Kms for agaropentaose and agaroheptaose. However, the Vmax value for agarotriose was 1.9 and 2.3 times higher than the Vmax values for agaropentaose and agaroheptaose, respectively. As a result, the kcat/Km values were similar for all three substrates (Table 2).
TABLE 2.
Kinetic parameters of VejABG in the hydrolysis of different types of agarooligosaccharidesb
| Substrate | Km (mM) | Vmax (Ua/mg) | kcat (s−1) |
|---|---|---|---|
| Agarotriose (DP3) | 3.25 ± 0.38 | 9.12 ± 0.54 | 14.14 ± 0.83 |
| Agaropentaose (DP5) | 1.73 ± 0.13 | 4.63 ± 0.06 | 7.17 ± 0.10 |
| Agaroheptaose (DP7) | 1.76 ± 0.24 | 3.92 ± 0.16 | 6.07 ± 0.25 |
One unit of VejABG was defined as the amount of enzyme that releases 1 μmol/min of galactose.
Experiments were performed in triplicate, and data are shown as means ± standard deviations.
Mode of action of VejABG toward AOSs and NAOSs.
MALDI-TOF/TOF MS analysis showed that even-numbered AOSs were predominantly produced by the acid prehydrolysis of agarose due to the preferential cleavage action on α1-3 linkages, which did not affect the β1-4 linkages in agarose (6) (Fig. 5A). The sum of the abundances of agarotetraose (DP4), agarohexaose (DP6), and agarooctaose (DP8) was more than 70% of the abundance of all the AOSs from the acid prehydrolysate of agarose (Fig. 5A). Incubation of the AOSs with VejABG clearly showed that VejABG primarily hydrolyzed them into galactose and NAOSs (Fig. 5B). The AOSs with a wide DP range containing galactose at their nonreducing ends were hydrolyzed by VejABG, thus producing the corresponding NAOSs with one less galactose unit and the addition of one water molecule (Fig. 5B). The GC-MS analysis over the range of 50 to 700 m/z showed that the amount of galactose increased after the incubation of AOSs prepared by acid prehydrolysis with VejABG (Fig. 6B) compared to the amount obtained before the reaction with VejABG (Fig. 6A). However, the amount of AHG did not change after the reaction with VejABG.
FIG 5.

MALDI-TOF/TOF MS of AOSs before and after reaction with VejABG. (A) AOSs were prepared by acid prehydrolysis of agarose; (B) AOSs were further hydrolyzed predominantly to NAOSs by VejABG.
FIG 6.

GC-MS of agarooligosaccharides before and after reaction with VejABG. Agarooligosaccharides were prepared by acid prehydrolysis of agarose (A) and were further hydrolyzed using VejABG (B).
The possibility of the complete hydrolysis of agarose-derived oligosaccharides into monomeric sugars by combining VejABG and a NAOS hydrolase (i.e., SdNABH) was examined. When agarotriose (Fig. 7A), agaropentaose (Fig. 7B), or agaroheptaose (Fig. 7C) was incubated with VejABG and SdNABH in a single reaction, only AHG and galactose were formed, without any significant amount of residual AOSs or NAOSs being found.
FIG 7.

Reaction products from various agarooligosaccharides treated with VejABG and SdNABH. Agarotriose (DP3) (A), agaropentaose (DP5) (B), and agaroheptaose (DP7) (C) were treated with VejABG alone and with VejABG and SdNABH. Control, substrate only; +VejABG, substrate incubated with VejABG for 12 h; +VejABG and +SdNABH, substrate incubated with VejABG and SdNABH for 12 h. The reaction products were analyzed by gel permeation chromatography.
DISCUSSION
In this study, we discovered a novel agarolytic β-galactosidase, VejABG, originating from Vibrio sp. EJY3. VejABG is capable of cleaving the β1-4 linkage at the nonreducing end of AOS to release galactose and NAOS, as β-galactosidases can cleave β1-4 linkages to release galactose as a product (27). VejABG is the first β-galactosidase identified that is capable of hydrolyzing AOSs. This novel enzyme acts on the β1-4 linkage only at the nonreducing end of AOSs, as shown by the release of galactose only from the odd-numbered (Fig. 3A) or even-numbered (Fig. 5 and 6) AOSs. However, VejABG cannot cleave the β1-4 linkage at the reducing end and is unable to release AHG. Therefore, the hydrolysis of AOSs by VejABG did not increase the amount of AHG (Fig. 6).
The activity of VejABG can explain several heretofore unknown mechanisms of agar degradation in agarolytic bacteria. The first role of VejABG is in the conversion of AOSs into NAOSs, by which the α-agarase can be linked to the β-agarase system in the hydrolysis of agarose into monomeric sugars. AOSs produced by α-agarases (28, 29) can be converted to NAOSs by VejABG, and the NAOSs can then be hydrolyzed by a β-agarase II into neoagarobiose and AHG (Fig. 8A).
FIG 8.

Schematic diagram representing the key modes of action of VejABG in the enzymatic hydrolysis of agarose. (A) The α-agarase system for producing monomeric sugars from agar is relatively unclear. VejABG converts agarooligosaccharides (AOSs) into neoagarooligosaccharides (NAOSs), thereby linking the α-agarase and β-agarase systems. (B) Some NAOSs, such as neoagarotetraose, remain unhydrolyzed in the β-agarase system and are further hydrolyzed at the α1-3 and β1-4 linkages into monomers by treatment with a NAOS hydrolase and VejABG, respectively. (C) In the saccharification of agarose for producing monomeric sugars using acid prehydrolysis and enzymatic hydrolysis, agarotriose remains unhydrolyzed. This problem can be resolved by the additional application of VejABG.
The second function of VejABG is the direct production of monomeric sugars from NAOSs through treatment of NAOSs with a combination of a NAOS hydrolase and the β-agarase system (Fig. 8B). Neoagarohexaose and neoagarotetraose are predominantly produced from agarose by a β-agarase I (30–32) and then primarily hydrolyzed into neoagarobiose by a β-agarase II. However, in some cases, residual NAOSs, such as neoagarotetraose, persist in the reaction (33); therefore, the additional degradation of these residual NAOSs is necessary. By the combined action of a NAOS hydrolase (or an NABH) and VejABG on the α1-3 and β1-4 linkages at the nonreducing end of a neoagarotetraose molecule, respectively, two molecules of AHG and galactose are successively produced (Fig. 8B).
Although VejABG was putatively annotated as a β-galactosidase and classified into glycoside hydrolase family 2 (GH2) on the basis of its amino acid sequence, this study revealed that its functional properties are significantly different from those of general GH2 β-galactosidases. In the phylogenetic analysis of the β-galactosidases belonging to GH2, VejABG locates distantly from other β-galactosidases and does not belong to any other group of previously characterized β-galactosidases (Fig. 9). While the commercial lacZ-encoded β-galactosidase from E. coli hydrolyzed all the general substrates of a β-galactosidase, including lactose and lactulose, VejABG did not hydrolyze these substrates (Fig. 3B). Conversely, AOSs were hydrolyzed by VejABG (Fig. 3A) but not by the E. coli β-galactosidase. Based on these results, we speculated that the differential substrate specificity of VejABG and the E. coli lacZ-encoded β-galactosidase is attributed to the difference in their ability to recognize galactose and AHG linked by the β1-4 bond located at the nonreducing end of AOSs. AHG is a rare sugar with a bicyclic structure that is not observed among the general substrates of β-galactosidases. Therefore, the unique mode of action of VejABG may partly be due to the presence of AHG in AOSs.
FIG 9.

Phylogenetic tree of the host microorganisms of VejABG and other functionally characterized β-galactosidases in the GH2 family. Genetic information was obtained from the CAZy database and UniProt (22). The amino acid sequences of the enzymes were aligned by the ClustalW program, and the tree was constructed using the MEGA (version 5) program (25). The UniProt entries are indicated after the organism names.
In the industrial application of red algae as the biomass feedstock for fuel and chemical production, the complete hydrolysis of agarose into monomeric sugars is important (20). Acid-prehydrolyzed agarose can be further enzymatically hydrolyzed into neoagarobiose, agarotriose, and AHG by the action of a β-agarase II (Fig. 8C). Among these products, neoagarobiose can be hydrolyzed into monomeric sugars by a NAOS hydrolase, but agarotriose cannot be further hydrolyzed by either β-agarases or a NAOS hydrolase (20). Additional application of VejABG in this case leads to the hydrolysis of agarotriose into monomeric sugars by the combined action of VejABG and a NAOS hydrolase or an NABH (i.e., SdNABH; Fig. 8C). VejABG not only is a novel β-galactosidase with a unique function but also is industrially important for producing monomeric sugars from agar.
In conclusion, VejABG (agarolytic β1-4-d-galactosidase), isolated from Vibrio sp. EJY3, is a novel β-galactosidase with a distinct substrate specificity, unlike all other previously characterized β-galactosidases. This novel enzyme cleaves the β1-4 linkage between galactose and AHG only at the nonreducing end of an AOS. This reaction can link the products of the α-agarase system to the β-agarase system. Moreover, this enzyme can also be used to increase the yield of monomeric sugars in the saccharification process by using agar or red algae as biomass.
ACKNOWLEDGMENTS
This research was supported by the Fishery Commercialization Technology Development Program of the Ministry of Oceans and Fisheries (2012100788). This work was also supported by a grant from the Advanced Biomass R&D Center of Korea (2011-0031353), funded by the Ministry of Science, ICT & Future Planning. Page charges were supported by the School of Life Sciences and Biotechnology for BK21 PLUS, Korea University.
Experiments were performed at the Korea University Food Safety Hall for the Institute of Biomedical Science and Food Safety.
Footnotes
Published ahead of print 18 July 2014
REFERENCES
- 1.Kim SR, Ha S-J, Wei N, Oh EJ, Jin Y-S. 2012. Simultaneous co-fermentation of mixed sugars: a promising strategy for producing cellulosic ethanol. Trends Biotechnol. 30:274–282. 10.1016/j.tibtech.2012.01.005 [DOI] [PubMed] [Google Scholar]
- 2.Goh CS, Lee KT. 2010. A visionary and conceptual macroalgae-based third-generation bioethanol (TGB) biorefinery in Sabah, Malaysia as an underlay for renewable and sustainable development. Renew. Sust. Energ. Rev. 14:842–848. 10.1016/j.rser.2009.10.001 [DOI] [Google Scholar]
- 3.Araki C. 1956. Structure of the agarose constituent of agar-agar. Bull. Chem. Soc. Jpn. 29:543–544. 10.1246/bcsj.29.543 [DOI] [Google Scholar]
- 4.Knutsen SH, Myslabodski DE, Larsen B, Usov AI. 1994. A modified system of nomenclature for red algal galactans. Bot. Mar. 37:163–170 [Google Scholar]
- 5.Chen H-M, Zheng L, Yan X-J. 2005. The preparation and bioactivity research of agaro-oligosaccharides. Food Technol. Biotechnol. 43:29–36 [Google Scholar]
- 6.Yang B, Yu G, Zhao X, Jiao G, Ren S, Chai W. 2009. Mechanism of mild acid hydrolysis of galactan polysaccharides with highly ordered disaccharide repeats leading to a complete series of exclusively odd-numbered oligosaccharides. FEBS J. 276:2125–2137. 10.1111/j.1742-4658.2009.06947.x [DOI] [PubMed] [Google Scholar]
- 7.Kim C, Ryu HJ, Kim SH, Yoon J-J, Kim HS, Kim YJ. 2010. Acidity tunable ionic liquids as catalysts for conversion of agar into mixed sugars. Bull. Korean Chem. Soc. 31:511–514. 10.5012/bkcs.2010.31.02.511 [DOI] [Google Scholar]
- 8.Kim HT, Lee S, Lee D, Kim H-S, Bang W-G, Kim KH, Choi I-G. 2010. Overexpression and molecular characterization of Aga50D from Saccharophagus degradans 2-40: an exo-type β-agarase producing neoagarobiose. Appl. Microbiol. Biotechnol. 86:227–234. 10.1007/s00253-009-2256-5 [DOI] [PubMed] [Google Scholar]
- 9.Stevenson TT, Furneaux RH. 1991. Chemical methods for the analysis of sulphated galactans from red algae. Carbohydr. Res. 210:277–298. 10.1016/0008-6215(91)80129-B [DOI] [PubMed] [Google Scholar]
- 10.Larsson S, Palmqvist E, Hahn-Hägerdal B, Tengborg C, Stenberg K, Zacchi G, Nilvebrant N-O. 1999. The generation of fermentation inhibitors during dilute acid hydrolysis of softwood. Enzyme Microb. Technol. 24:151–159. 10.1016/S0141-0229(98)00101-X [DOI] [Google Scholar]
- 11.Palmqvist E, Hahn-Hägerdal B. 2000. Fermentation of lignocellulosic hydrolysates. I. Inhibition and detoxification. Bioresour. Technol. 74:17–24. 10.1016/S0960-8524(99)00160-1 [DOI] [Google Scholar]
- 12.Ekborg NA, Taylor LE, Longmire AG, Henrissat B, Weiner RM, Hutcheson SW. 2006. Genomic and proteomic analyses of the agarolytic system expressed by Saccharophagus degradans 2-40. Appl. Environ. Microbiol. 72:3396–3405. 10.1128/AEM.72.5.3396-3405.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vera J, Alvarez R, Murano E, Slebe JC, Leon O. 1998. Identification of a marine agarolytic Pseudoalteromonas isolate and characterization of its extracellular agarase. Appl. Environ. Microbiol. 64:4378–4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hehemann J-H, Correc G, Thomas F, Bernard T, Barbeyron T, Jam M, Helbert W, Michel G, Czjzek M. 2012. Biochemical and structural characterization of the complex agarolytic enzyme system from the marine bacterium Zobellia galactanivorans. J. Biol. Chem. 287:30571–30584. 10.1074/jbc.M112.377184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugano Y, Kodama H, Terada I, Yamazaki Y, Noma M. 1994. Purification and characterization of a novel enzyme, α-neoagarooligosaccharide hydrolase (α-NAOS hydrolase), from a marine bacterium, Vibrio sp. strain JT0107. J. Bacteriol. 176:6812–6818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michel G, Nyval-Collen P, Barbeyron T, Czjzek M, Helbert W. 2006. Bioconversion of red seaweed galactans: a focus on bacterial agarases and carrageenases. Appl. Microbiol. Biotechnol. 71:23–33. 10.1007/s00253-006-0377-7 [DOI] [PubMed] [Google Scholar]
- 17.Ha SC, Lee S, Lee J, Kim HT, Ko H-J, Kim KH, Choi I-G. 2011. Crystal structure of a key enzyme in the agarolytic pathway, α-neoagarobiose hydrolase from Saccharophagus degradans 2-40. Biochem. Biophys. Res. Commun. 412:238–244. 10.1016/j.bbrc.2011.07.073 [DOI] [PubMed] [Google Scholar]
- 18.Kim HT, Lee S, Kim KH, Choi I-G. 2012. The complete enzymatic saccharification of agarose and its application to simultaneous saccharification and fermentation of agarose for ethanol production. Bioresour. Technol. 107:301–306. 10.1016/j.biortech.2011.11.120 [DOI] [PubMed] [Google Scholar]
- 19.Yun EJ, Lee S, Kim JH, Kim BB, Kim HT, Lee SH, Pelton JG, Kang NJ, Choi I-G, Kim KH. 2013. Enzymatic production of 3,6-anhydro-l-galactose from agarose and its purification and in vitro skin whitening and anti-inflammatory activities. Appl. Microbiol. Biotechnol. 97:2961–2970. 10.1007/s00253-012-4184-z [DOI] [PubMed] [Google Scholar]
- 20.Kim HT, Yun EJ, Wang D, Chung JH, Choi I-G, Kim KH. 2013. High temperature and low acid pretreatment and agarase treatment of agarose for the production of sugar and ethanol from red seaweed biomass. Bioresour. Technol. 136:582–587. 10.1016/j.biortech.2013.03.038 [DOI] [PubMed] [Google Scholar]
- 21.Roh H, Yun EJ, Lee S, Ko H-J, Kim S, Kim B-Y, Song H, Lim K, Kim KH, Choi I-G. 2012. Genome sequence of Vibrio sp. strain EJY3, an agarolytic marine bacterium metabolizing 3,6-anhydro-l-galactose as a sole carbon source. J. Bacteriol. 194:2773–2774. 10.1128/JB.00303-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The UniProt Consortium. 2013. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 41:D43–D47. 10.1093/nar/gks1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Craven GR, Steers E, Jr, Anfinsen CB. 1965. Purification, composition, and molecular weight of the β-galactosidase of Escherichia coli K12. J. Biol. Chem. 240:2468–2477 [PubMed] [Google Scholar]
- 24.Yun EJ, Shin MH, Yoon J-J, Kim YJ, Choi I-G, Kim KH. 2011. Production of 3,6-anhydro-l-galactose from agarose by agarolytic enzymes of Saccharophagus degradans 2-40. Process Biochem. 46:88–93. 10.1016/j.procbio.2010.07.019 [DOI] [Google Scholar]
- 25.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37:D233–D238. 10.1093/nar/gkn663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacob F, Monod J. 1961. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 3:318–356. 10.1016/S0022-2836(61)80072-7 [DOI] [PubMed] [Google Scholar]
- 28.Ohta Y, Hatada Y, Miyazaki M, Nogi Y, Ito S, Horikoshi K. 2005. Purification and characterization of a novel α-agarase from a Thalassomonas sp. Curr. Microbiol. 50:212–216. 10.1007/s00284-004-4435-z [DOI] [PubMed] [Google Scholar]
- 29.Potin P, Richard C, Rochas C, Kloareg B. 1993. Purification and characterization of the α-agarase from Alteromonas agarlyticus (Cataldi) comb. nov., strain GJ1B. Eur. J. Biochem. 214:599–607. 10.1111/j.1432-1033.1993.tb17959.x [DOI] [PubMed] [Google Scholar]
- 30.Sugano Y, Matsumoto T, Kodama H, Noma M. 1993. Cloning and sequencing of agaA, a unique agarase 0107 gene from a marine bacterium, Vibrio sp. strain JT0107. Appl. Environ. Microbiol. 59:3750–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Temuujin U, Chi W-J, Lee S-Y, Chang Y-K, Hong S-K. 2011. Overexpression and biochemical characterization of DagA from Streptomyces coelicolor A3(2): an endo-type β-agarase producing neoagarotetraose and neoagarohexaose. Appl. Microbiol. Biotechnol. 92:749–759. 10.1007/s00253-011-3347-7 [DOI] [PubMed] [Google Scholar]
- 32.Oh C, Nikapitiya C, Lee Y, Whang I, Kim S-J, Kang D-H, Lee J. 2010. Cloning, purification and biochemical characterization of beta agarase from the marine bacterium Pseudoalteromonas sp. AG4. J. Ind. Microbiol. Biotechnol. 37:483–494. 10.1007/s10295-010-0694-9 [DOI] [PubMed] [Google Scholar]
- 33.Fu XT, Lin H, Kim SM. 2008. Purification and characterization of a novel β-agarase, AgaA34, from Agarivorans albus YKW-34. Appl. Microbiol. Biotechnol. 78:265–273. 10.1007/s00253-007-1303-3 [DOI] [PubMed] [Google Scholar]