

ABSTRACT
The entry of enveloped viruses into host cells is preceded by membrane fusion, which in Epstein-Barr virus (EBV) is thought to be mediated by the refolding of glycoprotein B (gB) from a prefusion to a postfusion state. In our current studies, we characterized a gB C-terminal tail domain (CTD) mutant truncated at amino acid 843 (gB843). This truncation mutant is hyperfusogenic as monitored by syncytium formation and in a quantitative fusion assay and is dependent on gH/gL for fusion activity. gB843 can rescue the fusion function of other glycoprotein mutants that have null or decreased fusion activity in epithelial and B cells. In addition, gB843 requires less gp42 and gH/gL for fusion, and can function in fusion at a lower temperature than wild-type gB, indicating a lower energy requirement for fusion activation. Since a key step in fusion is the conversion of gB from a prefusion to an active postfusion state by gH/gL, gB843 may access this activated gB state more readily. Our studies indicate that the gB CTD may participate in the fusion function by maintaining gB in an inactive prefusion form prior to activation by receptor binding.
IMPORTANCE Diseases resulting from Epstein-Barr virus (EBV) infection in humans range from the fairly benign disease infectious mononucleosis to life-threatening cancer. As an enveloped virus, EBV must fuse with a host cell membrane for entry and infection by using glycoproteins gH/gL, gB, and gp42. Among these glycoproteins, gB is thought to be the protein that executes fusion. To further characterize the function of the EBV gB cytoplasmic C-terminal tail domain (CTD) in fusion, we used a previously constructed CTD truncation mutant and studied its fusion activity in the context of other EBV glycoprotein mutants. From these studies, we find that the gB CTD regulates fusion by altering the energy requirements for the triggering of fusion mediated by gH/gL or gp42. Overall, our studies may lead to a better understanding of EBV fusion and entry, which may result in novel therapies that target the EBV entry step.
INTRODUCTION
Epstein-Barr virus (EBV) is an enveloped, double-stranded DNA virus belonging to the family Herpesviridae. EBV infects more than 90% of the world population (1). EBV infection during childhood is asymptomatic. In adolescents, however, infection can result in the development of infectious mononucleosis (1). Numerous malignancies, such as Burkitt lymphoma, Hodgkin lymphoma, and nasopharyngeal carcinoma, are associated with EBV (1). In addition, EBV also causes lymphoproliferative disorders in AIDS patients with immune dysfunction or in transplant patients undergoing immune suppression (1). Four viral-membrane-associated proteins are responsible for the entry of the virus into the host cell: glycoprotein 42 (gp42), gH, gL, and gB. gp42 binds to the HLA class II molecules of B cells and determines the EBV infection tropism; it is required for B cell fusion and inhibits epithelial-cell fusion (2–7). gH is a receptor binding protein that interacts with gp42 or integrins, receptors that are important for epithelial-cell fusion (8, 9). gL is essential for the correct folding of gH and its transport to the cell surface (10), and gB is a class III viral fusogen (6, 11–15). Upon activation, gB is thought to insert fusion loops into target cell membranes and then to be refolded from a prefusion to a postfusion form to drive membrane fusion (6, 11, 15). gB is thought to be activated through interaction with gH/gL (16), yet whether the activation by gH/gL is the same for epithelial-cell entry and B cell entry is not known. Mutagenesis studies of gH/gL suggest that there are differing mechanisms in different cell types, since some gH/gL mutants behave distinctively in epithelial-cell fusion compared to B cell fusion (17–19).
gB is the most conserved of all the herpesvirus envelope glycoproteins. EBV gB is an 857-amino-acid protein containing three domains. The ectodomain contains nine potential N-linked glycosylation sites and a predicted 22-amino-acid cleavable signal sequence and is separated from the C-terminal tail domain (CTD) by a transmembrane domain (TMD) (amino acids 733 to 753) (11, 20). The crystal structure of the EBV gB ectodomain revealed structural homology to herpes simplex virus 1 (HSV-1) gB, vesicular stomatitis virus glycoprotein G (VSV G), and baculovirus gp64 (11–15). HSV-1 gB and EBV gB share 29% sequence identity and 43% similarity (11, 20). For HSV-1, a cryoelectron tomography study showed individual glycoprotein complexes in transitional conformations during pore formation and dilation, revealing potential fusion intermediates and structural changes that occur during herpesvirus entry (21). Although there is no direct evidence of EBV gB refolding, a model of the prefusion and postfusion states of gB has been proposed, and it is hypothesized that gB may undergo major conformational changes during the fusion process (11).
The gB ectodomain is composed of five domains in an elongated rod-like structure (11). For the structurally homologous VSV G to convert from a prefusion to a postfusion conformation, domain III undergoes significant refolding. Cys 25 and Pro 26 in VSV G are located at a junction of secondary structural changes. EBV gB has Cys 68 and Pro 69 in similar positions. In VSV G, the long αF domain is broken into two shorter helices in the prefusion form at amino acids 272 to 274. The analogous region in EBV gB contains Gly at position 477, which could destabilize the helical structure. Thus, these amino acids of EBV gB may also function as switch residues for gB conformational changes that are induced upon membrane fusion (11).
Although there are no crystal structures of the herpesvirus gB CTD, and the refolding mechanism that triggers EBV gB activation is still unclear, analysis of a comprehensive library of EBV gB CTD truncation mutants indicates that the gB tail plays important roles in gB-mediated fusion (22). To gain further insight into the gB CTD and its regulation of EBV fusion, we chose gB843 as a representative hyperfusogenic mutant identified in our previous studies (22). In our current studies, we found that gB843 rescues the function of other glycoprotein mutants that have null or decreased fusion activity in epithelial cell and B cell fusion. This rescue function may work by lowering the energy requirement for fusion activation, since gB843 allows fusion to occur more efficiently at lower temperatures than wild-type (wt) gB.
MATERIALS AND METHODS
Cell culture.
Chinese hamster ovary cells (CHO-K1) were grown in Ham's F-12 medium (Corning) containing 10% heat-inactivated fetal bovine serum (FBS) complex (Corning) and 1% penicillin-streptomycin (100 U penicillin/ml, 100 μg streptomycin/ml; Sigma). The Daudi 29 cell line (for B cell fusion) and human embryonic kidney 293 (HEK 293) cells (for epithelial-cell fusion) stably expressing T7 RNA polymerase (23, 24) were grown in RPMI 1640 medium (Corning) with 900 μg/ml G418 (Sigma) and in Dulbecco's modified Eagle medium (DMEM) with 100 μg/ml Zeocin (Invitrogen), respectively, containing 10% heat-inactivated FBS and 1% penicillin-streptomycin.
Transfection.
CHO-K1 cells, grown to approximately 80% confluence, were transiently transfected with plasmids expressing the mutants, other glycoproteins essential for fusion, including gB (0.8 μg), gH (0.5 μg), gL (0.5 μg), and gp42 (0.8 μg), and a luciferase reporter plasmid with a T7 promoter (0.8 μg) by using Lipofectamine 2000 transfection reagent (Invitrogen) in Opti-MEM (Gibco-Life Technologies), as described previously (25). Equal amounts of each glycoprotein or glycoprotein mutant DNA were used in each experiment.
Fusion assay.
The virus-free cell-based fusion assay was performed as described previously (25). Briefly, CHO-K1 effector cells were transfected as described above. Twenty-four hours posttransfection, the cells were detached, counted, and mixed 1:1 with target cells expressing T7 polymerase (Daudi 29 B cells or HEK 293 T7 cells; 0.25 × 106 target cells per sample) into a 24-well plate in 1 ml Ham's F-12 medium with 10% heat-inactivated FBS. Twenty-four hours later, the cells were washed once with phosphate-buffered saline (PBS) and were lysed with 100 μl of Passive Lysis Buffer (Promega). Luciferase was quantified in duplicate by transferring 20 μl of lysed cells to a 96-well plate, adding 100 μl of luciferase assay reagent (Promega), and measuring luminescence on a Victor plate reader (Perkin-Elmer).
Syncytium formation assay.
CHO-K1 cells in 6-well plates were transfected with 0.8 μg wt gB or gB843 with gH (0.5 μg) and gL (0.5 μg). Twenty-four hours posttransfection, the cells were detached, counted, and mixed 1:1 with 0.25 × 106 HEK 293 target cells in 10% heat-inactivated FBS on glass coverslips in 12-well plates. Twenty-four hours later, the cells were washed once with PBS, incubated with the primary antibody E1D1 for 30 min, and then fixed with 2% paraformaldehyde. The cells were washed twice with PBST (0.2% Tween 20 in PBS) and were incubated with Alexa Fluor 488-conjugated goat anti mouse IgG (Invitrogen) in 5% bovine serum albumin (BSA) in PBS at room temperature for 1 h. After additional washes with PBST and H2O, coverslips were mounted on glass slides using the Prolong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes). Fluorescence images were captured with a Leica fluorescence microscope. The sizes of the syncytia were determined with ImageJ from NIH.
Duolink assay.
A Duolink assay (Olink Bioscience) was performed according to the manufacturer's instructions. Briefly, CHO-K1 cells in 6-well plates were transfected with 0.8 μg wt gB or gB843 with Flag-tagged gH (0.5 μg) and gL (0.5 μg). Eighteen hours posttransfection, the cells were detached, and 2 × 104 cells were seeded onto 16-well chamber slides (Lab-Tek) for 24 h. The cells were then fixed with methanol and were blocked in 5% goat serum in PBS for 1 h. The slides were then incubated with mouse anti-gB antibody CL55, provided by Lindsey Hutt-Fletcher, and a rabbit anti-Flag antibody (Sigma), diluted at a 1:500 ratio in 5% goat serum in PBS overnight at 4°C. The slides were washed twice in PBS and were incubated with diluted PLA probes in a preheated humidity chamber for 1 h at 37°C. After two washes with wash buffer A, the slides were incubated with the ligation-ligase buffer in a preheated humidity chamber for 30 min at 37°C. After another two washes with wash buffer A, the slides were incubated with an amplification-polymerase solution in a preheated humidity chamber for 100 min at 37°C. The slides were washed with wash buffer B, incubated with Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen) in 5% BSA in PBS at room temperature for 1 h, and then washed twice with wash buffer B and once with 0.01× wash buffer B. The slides were then dried and were mounted on glass slides using the Prolong Gold antifade reagent with DAPI (Molecular Probes). Fluorescence images were captured with a Leica fluorescence microscope.
CELISA.
The expression of the various mutants in the plasma membrane was determined by a cell enzyme-linked immunosorbent assay (CELISA) as described in previous reports (25). CHO-K1 cells were transfected with various glycoproteins and mutants. Transfected cells were split for use in the fusion assay (described above) and CELISA. Twenty-four hours posttransfection, 4 × 104 cells/well were transferred to a 96-well plate and were incubated for another 24 h. The expression of each glycoprotein (including mutants) was evaluated using the conformation-specific antibody E1D1 for gH/gL or CL55 for gB. After incubation with the primary antibody for 30 min and fixation with 2% formaldehyde–0.2% glutaraldehyde in PBS for 15 min, a biotin-labeled anti-mouse secondary antibody was added at a 1:500 dilution and was incubated for 30 min. After washing, streptavidin-labeled horseradish peroxidase (dilution, 1:20,000) was further incubated with the fixed cells for 30 min. A peroxidase substrate was added, and the amount of cell surface staining was determined by measurement at 380 nm with a Victor plate reader (Perkin-Elmer).
SDS-PAGE migration assay for gB.
CHO-K1 cells were transfected with a control, gB, or gB843. After 24 h, transfected cells in 6-well plates were collected and were lysed in 200 μl lysis buffer (20 mM Tris-HCl [pH 7.4], 100 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 1% Triton X-100, and Calbiochem's 1× protease inhibitor cocktail set I). The cell lysates were cleared of debris by centrifugation. One hundred microliters of the cleared lysates was mixed with 100 μl 2× SDS loading buffer (60 mM Tris-Cl [pH 6.8], 0.2% SDS, 25% glycerol, 0.01% bromophenol blue). The samples were loaded onto a Bio-Rad 7.5% mini-Protean TGX gel for Western blotting. After electrophoresis, proteins were transferred to nitrocellulose membranes (Schleicher & Schuell, Keene, NH). The blots were blocked with 5% nonfat dry milk in TBS buffer (20 mM Tris-HCl [pH 7.6], 137 mM NaCl) for 2 h at room temperature (RT). The blots were washed with TBS and were incubated with primary antibodies (against gB or glyceraldehyde-3-phosphate dehydrogenase [GAPDH]) overnight at 4°C. An infrared dye 800 (IRDye 800)-conjugated anti-rabbit or IRDye 680-conjugated anti-mouse secondary antibody (Li-Cor Biosciences, Lincoln, NE) was added to the membrane at a dilution ratio of 1:10,000, and incubation was continued for 1 h at RT. The protein bands on the membrane were visualized with the Odyssey Fc Western blotting imager using Image Studio, version 2.0 (Li-Cor Biosciences, Lincoln, NE).
RESULTS
The hyperfusogenicity of gB843 is gH/gL dependent.
The cellular localization, expression, and fusion function of a library of EBV CTD truncation mutants have been characterized by our laboratory previously (22). The gB CTD truncation mutants were categorized into three different classes on the basis of cell surface expression and fusion function (22). The class II mutants had a level of gB expression similar to that of wt gB. The gB843 mutant from this class was particularly interesting because it was hyperfusogenic (22). We chose to investigate gB843 further in order to gain mechanistic insight into how the gB CTD regulates fusion. The fusion phenotype of the gB843 mutant was determined previously by quantitative virus-free, cell-cell fusion assays monitored by luciferase activity. Studies of the cytoplasmic tail of HSV-1 revealed different results by syncytium counting and the induction of luciferase expression following cell-cell fusion (26). Thus, in the current study, we wanted to confirm the hyperfusogenicity phenomenon using two different methods. As in our previous results, gB843 increased fusion activity for epithelial cells and Daudi B cells about 4-fold and 1.5-fold, respectively (Fig. 1A and B), as measured in the luciferase cell-cell fusion assay. There was a very modest increase in the expression of gB843 on the cell surface, as reported previously (22), whereas the expression levels of gH/gL were similar for wt gB and gB843 (Fig. 1C and D).
FIG 1.

The EBV CTD truncation mutant gB843 has increased fusion activity. (A) CHO-K1 cells were transiently transfected with the T7 luciferase plasmid and a vector plasmid (control) or with the T7 luciferase plasmid and EBV gH and gL, together with gB or gB843. Twenty-four hours posttransfection, transfected CHO-K1 cells were overlaid with epithelial cells expressing T7 polymerase. Luciferase activity was monitored 24 h after the overlay and was normalized to the activity in cells with wt gB and gH/gL, which was set at 100%. (B) CHO-K1 cells were transiently transfected with the T7 luciferase plasmid and a vector plasmid (control) or with the T7 luciferase plasmid and EBV gH, gL, and gp42, together with gB or the gB843 mutant. Twenty-four hours posttransfection, CHO-K1 cells were overlaid with Daudi B cells expressing T7 polymerase. Luciferase activity was monitored 24 h after the overlay and was normalized to the activity in cells with wt gB and gH/gL, set at 100%. The data are means plus standard errors of the means for three independent experiments. (C and D) Expression of gB and gH/gL. Twenty-four hours posttransfection, 4 × 104 cells were seeded into 96-well plates in triplicate. CELISA was performed with anti-gH/gL monoclonal antibody E1D1 or anti-gB monoclonal antibody CL55.
In addition, we examined syncytium formation by gB843 compared with that by wt gB in epithelial-cell fusion. Syncytia were stained with anti-gH/gL antibody E1D1, and the areas of syncytia were quantified using ImageJ. We found that gB843 formed much larger syncytia than wt gB (Fig. 2A and B). The mean syncytium area for gB843 is three times that for wt gB (Fig. 2C).
FIG 2.

Syncytium formation with gB843 is enhanced over that with wt gB. (A and B) CHO-K1 cells were transfected with wt gB or gB843 along with gH/gL and were overlaid with HEK293 cells. Syncytium formation was visualized by surface staining of the cells with antibody E1D1 against the gH/gL complex. (C) The areas of the individual syncytia were quantified using ImageJ. The asterisk indicates a significant difference (P < 0.05) by the Student t test.
Our laboratory previously identified EBV gB CTD mutants with truncations at amino acids 798 and 801, which mediate fusion at ∼30% of wt levels in the absence of EBV gH/gL (27, 28). However, these mutants do not have hyperfusogenic activity in the presence of gH/gL (22, 28). We tested whether the gB843 mutant could mediate fusion in the absence of EBV gH/gL. As expected, in the absence of gH/gL, wt gB had background levels of fusion (Fig. 3A). However, unlike those of the gB798 and gB801 mutants, the gB843 fusion function was still gH/gL dependent; in the absence of gH/gL, fusion activity with gB843 was comparable to that for control transfections with just wt gB (Fig. 3A). All of the gB mutants were expressed on the cell surface (Fig. 3B). We confirmed the absence of gH/gL expression when no gH/gL was included in the transfection (Fig. 3C). These data together demonstrate that the hyperfusogenic phenomenon of gB843 is dependent on gH/gL.
FIG 3.

gH/gL-independent epithelial-cell fusion. CHO-K1 target cells were transfected with the indicated expression constructs, including a T7-driven luciferase reporter construct. (A) The cells were overlaid with indicator epithelial cells expressing T7 polymerase, and cell-cell fusion was monitored by luciferase expression. (B) The expression of gB on the cell surface was monitored by CELISA using monoclonal antibody CL55, directed at gB. (C) The expression of gH/gL on the cell surface was monitored by CELISA using monoclonal antibody E1D1, directed against gH/gL.
gB843 does not alter gB oligomerization or the association of gB with gH/gL.
EBV gB and HSV-1 gB are known to form trimers, as observed both by size exclusion chromatography and in the relevant crystal structures (11, 29–31). Previous studies have suggested that a region within the gB CTD may play a role in trimer formation (30). gB oligomerization is important for fusion in B cells and epithelial cells, as evidenced by the fact that linker insertion mutants of gB defective in oligomerization fail to drive fusion (27). Interactions between EBV gB and gH/gL also play an important role in fusion activation (16). Given these observations, we hypothesized that the gB843 mutant could alter gB oligomerization or the interaction with gH/gL to mediate the hyperfusogenic phenomenon. To test these possibilities, we utilized the Duolink assay to examine the interaction of wt gB or gB843 with gL and Flag-tagged gH. Previous studies have shown that Flag-tagged gH and gL are functional in fusion (32). In the Duolink assay, cells are fixed, and two proteins of interest are bound with primary antibodies from two different species. The primary antibodies are then bound by species-specific secondary antibodies conjugated with oligonucleotides. If the oligonucleotides are in close proximity (<40 nm), they circularize and are amplified after the addition of complementary oligonucleotides. The interaction is visualized by adding fluorescently labeled nucleotides during amplification. We confirmed that the staining pattern for wt gB is relatively diffuse, whereas the gB843 staining pattern is more punctate (Fig. 4, top), as we reported previously (22). The patterns of red staining, indicating an interaction between gH/gL and gB, were similar for wt gB and gB843 (Fig. 4, center), showing that there was no dramatic change in the association of gB with gH/gL. Nuclei were stained with DAPI (bottom).
FIG 4.

Association of gB and gB843 with gH/gL. CHO-K1 cells were transfected with EBV gB or gB843 together with Flag-gH/gL. After 18 h of transfection, CHO-K1 cells were seeded in 16-well chamber slides. The chamber slides were then fixed with methanol after 24 h of culture. The proximity of gB to gH/gL was examined using a Duolink kit (Olink) with monoclonal antibody CL55 and the rabbit anti-Flag antibody from Sigma (center), and nuclei were stained with DAPI (bottom). gB was further stained using the Alexa Fluor 488-conjugated anti-mouse secondary antibody (Invitrogen) after Duolink antibodies were added (top).
To examine gB oligomerization, we compared the migration of wt gB with that of gB843 under nonreducing SDS-PAGE conditions. There was no difference in the larger oligomerized gB band between cells expressing wt gB and those expressing gB843 (Fig. 5). These data suggest that the hyperfusogenic phenotype of gB843 is not due to increased association with gH/gL or to a difference in oligomerization between gB843 and wt gB.
FIG 5.
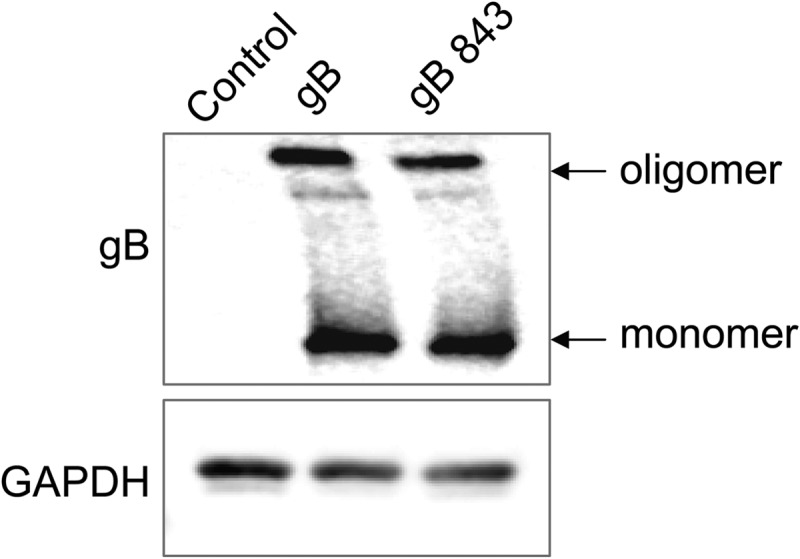
Oligomerization of gB and gB843. CHO-K1 cells were transfected with an EBV control plasmid or with a wt gB or gB843 expression plasmid, as indicated. After 24 h of transfection, 1 × 106 cells were first lysed in 100 μl lysis buffer (20 mM Tris-HCl [pH 7.4], 100 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 1% Triton X-100, 1× protease inhibitor) and then centrifuged at 13,000 rpm for 5 min. Equal amounts of 2× SDS sample buffer with a low level of SDS (0.2%) were added to the supernatant. The samples were then analyzed on Bio-Rad 7.5% mini-Protean TGX gels. The data are representative of results from three independent experiments.
gB843 allows fusion with other EBV glycoprotein mutants that have decreased or no fusion function.
We previously identified mutants with alterations in gH/gL and gp42 that have decreased or no detectable fusion activity in epithelial and Daudi B cells (17, 24, 25, 33, 34). We wanted to determine whether the hyperfusogenic activity of gB843 might rescue the function of these gH/gL and gp42 mutants. Using our cell-cell fusion assay, we first investigated if gB843 can rescue fusion function with EBV gH mutants gH-R152A, gH-G49C, and gH-AAA, all of which have 10 to 20% of the fusion activity of wt gH/gL in the presence of wt gB (17, 25). The gH-R152A and gH-G49C mutants are located at the large groove between domain I and domain II of gH/gL (17). The gH-AAA mutant is within the integrin binding KGD motif located on the surface of domain II (25). Interestingly, in the presence of gB843, the fusion activities of the gH-R152A, gH-G49C, and gH-AAA mutants increased to 60, 100, and 200%, respectively, of the fusion activity with wt gH/gL and wt gB (Fig. 6A).
FIG 6.

gB843 rescues a variety of gH/gL and gp42 mutants. (A) The fusion of epithelial cells expressing T7 polymerase was monitored using CHO-K1 effector cells expressing wt gB or the gB843 mutant along with wt gH/gL or gH/gL mutant expression constructs and the T7 luciferase plasmid. (B) The fusion of Daudi B cells with wt gB or the gB843 mutant, along with gH/gL expression constructs, the T7 luciferase reporter plasmid, and wt gp42 or gp42 mutants, was tested. M-gp42, membrane-bound gp42 mutant.
We had previously generated a panel of gp42 linker insertion and point mutants (24, 35) and tested these for function with gB843. The gp42 linker insertion mutants were categorized into four groups: unaffected mutants, core distant mutants that may alter gp42 structure, class II mutants that do not bind to the HLA class II receptor, and mutants with mutations within the gp42 hydrophobic pocket, which may participate in binding to gH/gL (24, 35). Except for the unaffected mutants, all the mutants exhibited decreased B cell fusion (24, 35). In addition to these mutants, we also tested site-specific mutants with alterations within gp42 that block HLA class II binding (24, 34) and a gp42 proteolytic cleavage mutant that is membrane bound (36). In our cell-cell fusion-based assay, the gB843 mutant rescued the activities of the membrane-bound gp42 mutant and four hydrophobic-pocket mutants from about 5 to 25% of wt fusion levels in the presence of wt gB to almost 100% of wt levels in the presence of gB843 (Fig. 6B). However, the core/distant and HLA class II gp42 mutants were poorly rescued by gB843, with no fusion levels greater than 25% of wt levels (Fig. 6B). These findings indicate that the overall gp42 structure and binding with the HLA class II receptor are particularly important for the function of gp42 in fusion and that fusion function is not restored by hyperfusogenic gB843.
We have also observed that rhesus gL does not function together with EBV gH, gB, and gp42 due to species-specific sequence differences in gB and gL. Therefore, we examined whether gB843 can bypass this block (Fig. 7A and B). In agreement with our previous data, EBV gB does not induce fusion in the presence of rhesus gL (Fig. 7A and B). However, in the presence of gB843, rhesus gL mediated levels of fusion similar to, or higher than, that with wt EBV gB and gL (Fig. 7A and B). This indicates that the gB843 mutant can bypass the species-specific nature of the dependence of gB on gL.
FIG 7.

gB843 rescues fusion when EBV gL is replaced by rhesus gL. CHO-K1 cells were transiently transfected with the T7 luciferase plasmid and a vector plasmid (control) or with the T7 luciferase plasmid, gH, and EBV gB or gB843 together with EBV gL or rhesus gL in the presence (A) or absence (B) of gp42. Transfected CHO-K1 cells were overlaid with Daudi B cells (A) or epithelial cells (B) expressing T7 polymerase and were monitored for luciferase activity after 24 h. Luciferase activity was normalized to that of cells with wt EBV gH/gL levels, which was set at 100%. The data are means plus standard errors of the means for three independent experiments.
Time and temperature dependence of the fusion activity of the gB843 mutant.
Previous studies have found that mutant forms of viral fusion proteins can exhibit a decrease in the energy required for fusion activation (37, 38). Our data demonstrate that gB843 is hyperfusogenic and rescues most of the gH/gL and gp42 mutant glycoproteins that have lower fusion activity. Therefore, we hypothesized that gB843 may also have a lower energy requirement for protein activation and fusion. To analyze whether the hyperfusogenic phenotype of gB843 was due to an alteration in the energy threshold for fusion activation, the fusion activities of gB843 at different temperatures were investigated and compared to that of wt gB. Since the hyperfusogenic phenomenon is more apparent in epithelial-cell fusion, we studied temperature dependence in these cells. We first compared the fusion activities of wt gB and gB843 at different temperatures. The fusion activity of wt gB at 37°C was set at 100%. We found that even at 25°C, gB843 had 50% of the fusion activity of wt gB at 37°C (Fig. 8A). At 28°C, the fusion activity of gB843 was about 300% of the level for wt gB at 37°C. These data confirmed our hypothesis that gB843 requires less energy for fusion than wt gB (Fig. 8A).
FIG 8.

Time and temperature dependence of fusion with gB843. (A) CHO-K1 cells were transiently transfected with the T7 luciferase plasmid and a vector plasmid (control) or with the T7 luciferase plasmid, gH/gL, and EBV gB or gB843, as indicated. Transfected CHO-K1 cells were overlaid with epithelial cells at different temperatures, as indicated, for 24 h, and luciferase activity was measured. The data are means plus standard errors of the means for three independent experiments. (B) CHO-K1 cells were transiently transfected with the T7 luciferase plasmid and a vector plasmid (control) or with the T7 luciferase plasmid, gH/gL, and EBV gB or gB843, as indicated. Transfected CHO-K1 cells were overlaid with epithelial cells, and luciferase activity was measured after different incubation times, as indicated. The data are means plus standard errors of the means for three independent experiments.
In addition to temperature dependence, the time dependence of fusion in the presence of wt gB or gB843 was also analyzed. The hyperfusogenic phenotype of gB843 was maintained at all the times assayed. At the shortest time assayed (2 h), gB843 induced about 12% of the wt fusion level at 24 h; after a 6-h overlay, gB843 mediated about 50% of wt gB fusion activity (Fig. 8B).
To further explore whether the gB843 mutant requires reduced amounts of gp42 or gH/gL for fusion, we varied the amount of gp42 or gH/gL and tested fusion in the presence of wt gB or gB843 (Fig. 9). For B cell fusion, we tested many concentrations of soluble gp42, since it can be added exogenously to cells expressing gB and gH/gL (Fig. 9A). We had shown previously that when a soluble form of gp42 is added exogenously, fusion with B cells is readily induced (2, 36). We observed that with 7.8 μl of soluble gp42, wt gB was unable to induce fusion whereas gB843 induced about 50% of the fusion activity exhibited by wt gB in the presence of the maximum amount of gp42 tested (Fig. 9A), indicating that gB843 requires less gp42 than wt gB. With as little as 1.95 μl soluble gp42, gB843 could induce fusion activity at about 13% of the level for wt gB with 250 μl of soluble gp42 added. In contrast, fusion with wt gB was detected only after 15.6 μl soluble gp42 was added (Fig. 9A). Since soluble gH/gL does not efficiently mediate fusion with epithelial cells (39), we transfected varying amounts of gH/gL with a constant amount of wt gB or gB843. As observed for gp42, gB843 had more fusion activity even with the smallest amount of gH/gL tested (0.25 μg) than wt gB had with the largest amount of gH/gL tested (1.0 μg) (Fig. 9B).
FIG 9.

Dose dependence of gB or gB843 requirement for gH/gL or gp42. (A) CHO-K1 cells were transiently transfected with the T7 luciferase plasmid and a vector plasmid (control) or with the T7 luciferase plasmid and EBV gH and gL, together with wt gB or gB843. Transfected CHO-K1 cells were overlaid with Daudi B cells along with an additional 250 μl of supernatants containing different amounts of gp42, as indicated. Twenty-four hours after the overlay, luciferase activity was measured. The data are means plus standard errors of the means for three independent experiments. (B) CHO-K1 cells were transfected either with the T7 luciferase plasmid and a vector plasmid (open bars), with the T7 luciferase plasmid, wt gB, and different amounts of gH/gL, as indicated (filled bars), or with the T7 luciferase plasmid, gB843, and different amounts of gH/gL, as indicated (stippled bars). Twenty-four hours posttransfection, CHO-K1 cells were overlaid with epithelial cells expressing T7 polymerase. Luciferase activity was monitored after a 24-h overlay and was normalized to the activity of cells with wt gB and wt gH/gL levels (0.5 μg), which was set at 100%. The data are means plus standard errors of the means for three independent experiments.
Together, our data indicate that the gB843 truncation decreases the energy required for fusion activation. The possibility that the ectodomain of gB843 is in a conformation more easily transformed to the postfusion state is compatible with the finding that for both HSV and EBV, removal of the gB TMD results in a spontaneous postfusion conformation in the crystal structure. Studies with Nipah virus and paramyxovirus using different monoclonal antibodies also indicate that removal of the cytoplasmic tail of the F protein may affect the ectodomain conformation, in turn resulting in faster or slower six-helix-bundle formation (38, 40).
DISCUSSION
The entry of enveloped viruses into a host cell is preceded by membrane fusion, which in EBV is triggered either by gp42 binding to HLA class II molecules or by gH/gL binding to integrins (8, 41). After receptor binding, the fusion protein gB induces the fusion of the virus envelope with the cell membrane. The activation of gB and the subsequent insertion of gB fusion loops are thought to be regulated by gH/gL upon binding to an epithelial-cell receptor or by the interaction of gp42 with B cells. Once activated, gB likely undergoes a large conformational change from a metastable prefusion state to a highly stable postfusion structure (6). Comparative analysis of domains III of EBV gB and VSV G indicates that gB residues Gly 477, Cys 68, and Pro 69 may participate in a conformational switch during virus entry by destabilizing helical structures in domain III, breaking them into 2 shorter helices in the prefusion form (11). The EBV and HSV gB CTD and TMD are not present in the crystal structures, since they were deleted to generate a soluble form of gB used for purification and subsequent crystallization. Removal of the gB TMD and CTD may be responsible in part for the spontaneous adoption of a postfusion conformation, as observed previously for other fusion proteins, such as the class III baculovirus protein gp64 (14) and the class I paramyxovirus F protein (42). Studies of the CTDs of paramyxovirus fusion proteins and the HSV-1 gB CTD, as well as our study of the EBV gB CTD, indicate that this domain has an important regulatory role in fusion activation. Both hyperfusogenic and hypofusogenic mutations can be found within the CTD (22, 43–49). A recent study with the HSV-1 gB CTD suggests that the hyperfusion phenotype is associated with diminished membrane interactions (29). However, hyperfusogenic HSV-1 gB CTD point mutants have either no change or increased membrane interactions, suggesting that other, unknown regulatory mechanisms may also exist (46). The HSV-1 gB study also suggested that gB CTD deletion mutants or point mutants result in local rather than global conformational changes (29, 46). It has also been shown that mutations in the CTDs of Nipah virus and paramyxovirus fusion proteins modulated membrane fusion by inside-out signaling, by altering the ectodomain conformation as mapped by different monoclonal antibodies (38, 40). It is possible that the cytoplasmic tail of EBV gB may also regulate the conformation of the EBV gB ectodomain and keep it in an inactive prefusion form before fusion is triggered by receptor binding. Several possibilities that are not mutually exclusive may explain the hyperfusogenicity of gB843: (i) the gB843 mutant may alter gB oligomerization; (ii) the gB843 mutant may alter the interaction of gB with gH/gL; and (iii) the amino acids beyond gB amino acid 843 may play a role in the stabilization of the prefusion conformation.
HSV gB has been shown to oligomerize in vivo and in the gB crystal structure (15, 50). Like that of HSV gB, the EBV gB ectodomain also forms long, spike-like trimers in the crystal structure, and oligomerization has been shown to play an important role in fusion (27). In addition, previous studies have shown that the CTDs of EBV gB and HSV-1 gB elute as trimers in size exclusion chromatography (29, 30), and only linker insertion mutants with alterations in the ectodomain, not in the cytoplasmic domain, abolished EBV gB oligomerization (27). Similarly, we observed no alteration in the oligomerization of gB in the gB843 mutant (Fig. 5). Thus, it is unlikely that changes in gB oligomerization are responsible for the hyperfusogenic phenotype we observe with the gB843 mutant.
Our laboratory also showed, by using split yellow fluorescent protein (YFP) bimolecular complementation, that gB residues 456 to 807 play an important role in the association with gH/gL. The region from amino acid 456 to 807 contains both the ectodomain and the CTD, suggesting that both regions may be required for interaction with gH/gL and for fusion function (16). In order to investigate if gB843 strengthens the interactions with gH/gL to increase fusion activity, we examined the association of gB843 with gH/gL by using the Duolink assay. Our data showed no significant difference between wt gB and gB843 in gH/gL association (Fig. 4), a finding compatible with the conclusion that no regions important for gH/gL association are contained within the gB CTD.
Our laboratory found previously that the region comprising amino acids 802 to 816 in the gB CTD is necessary for productive membrane fusion and that amino acids 817 to 841 constitute a domain that reduces gB-mediated membrane fusion (49). The amino acids beyond 843 may alter the energetics of the conversion of prefusion gB to postfusion gB, and the gB843 mutant might alter the prefusion conformation, allowing gB to acquire the postfusion state more readily in the absence of those amino acids. There are instances of virus glycoprotein mutants that can function without receptor binding or without other glycoproteins. For example, the Eisenberg and Cohen group identified novel gH mutants that can function in the absence of gD and a cellular receptor (51). Our group also identified gB mutants, gB801 and gB798, that can function without gH/gL in epithelial cells (28). To our surprise, unlike the gB801 mutant, which mediates epithelial-cell fusion in the absence of gH/gL at a level equivalent to ∼40% that of wt gB in the presence of gH/gL, the gB843 mutant does not have gH/gL-independent epithelial-cell fusion activity (Fig. 3). gB801 is expressed at much higher levels than other forms of gB; thus, gH/gL-independent fusion activity may be primarily a result of the higher cell surface expression of the gB801 mutant (28).
One interesting observation is that gB843 can compensate for a variety of gH/gL or gp42 mutants with low or no fusion activity (Fig. 6). We found previously that if we mutated the gH/gL KGD motif, which plays an important role in epithelial-cell fusion, to triple alanines, epithelial-cell fusion was greatly decreased (25). However, in the presence of gB843, epithelial-cell fusion was rescued to 200% of the levels with wt gH/gL (Fig. 6A). These data indicate that gB843 may overcome the requirement of gH/gL binding to the receptor or that greatly reduced levels of gH/gL binding are still sufficient when gB has enhanced fusion function. This is similar to the finding by the Eisenberg and Cohen laboratory that novel mutations in gB and gH circumvent the requirement for known gD receptors in HSV-1 entry and cell-to-cell spread (52). There are a variety of possible explanations that accommodate our current results. Our gB843 mutant may mimic or access a transition state of gB that responds efficiently to weak signals from gH/gL by becoming activated and mediating fusion. The situation is different for gp42 mutants that cannot bind to HLA class II molecules, since gB843 cannot compensate for their function (Fig. 6B). In contrast, mutants with alterations within the gp42 hydrophobic pocket are rescued by gB843. Finally, the gB843 mutant showed a hyperfusogenic phenotype at all fusion temperatures tested. Interestingly, the fusion activities at 31°C and 34°C were higher than those at 37°C for both gB843 and wt gB (Fig. 8). Taken together, our data suggest that the gB843 mutation lowers the energy requirement for the transition of gB from a prefusion to a postfusion form and that as a result, gB843 can be triggered to mediate fusion with the weaker signals provided by gH/gL or gp42 mutants.
In summary, the gB CTD plays an important role in regulating fusion activity for both epithelial-cell and B cell fusion and serves as an important regulator of EBV-induced membrane fusion. The gB843 mutant may compensate for the function of the other glycoproteins, a phenomenon related to its decreasing the energy barrier for the proper conformational change during fusion.
ACKNOWLEDGMENTS
This research was supported by grant AI076183 (to R.L. and T.S.J.) from the National Institute of Allergy and Infectious Diseases, by grant CA117794 (to R.L. and T.S.J.) from the National Cancer Institute, and by postdoctoral fellowships 12POST9380013 (to J.C.) and 14POST18600021 (to J.C.) from the American Heart Association, Midwest Affiliate. This work was supported in part by the Katten Muchin Rosenman Travel Scholarship Program and the Robert H. Lurie Comprehensive Cancer Center of Northwestern University.
We appreciate the help and advice of members of the Jardetzky and Longnecker laboratories, especially Nicholas J. Garcia and Nanette Susmarski, for the completion of these studies. We thank Lindsey Hutt-Fletcher for kindly providing the monoclonal antibodies used in these studies.
Footnotes
Published ahead of print 6 August 2014
REFERENCES
- 1. Longnecker R, Kieff E, Cohen JI. 2013. Chapter 61. Epstein-Barr virus, p 1898–1959 In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, 6th ed, vol 2 Lippincott, Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Kirschner AN, Omerovic J, Popov B, Longnecker R, Jardetzky TS. 2006. Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J. Virol. 80:9444–9454. 10.1128/JVI.00572-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mullen MM, Haan KM, Longnecker R, Jardetzky TS. 2002. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell 9:375–385. 10.1016/S1097-2765(02)00465-3 [DOI] [PubMed] [Google Scholar]
- 4. Borza CM, Hutt-Fletcher LM. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 8:594–599. 10.1038/nm0602-594 [DOI] [PubMed] [Google Scholar]
- 5. Wang X, Kenyon WJ, Li Q, Mullberg J, Hutt-Fletcher LM. 1998. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J. Virol. 72:5552–5558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9:369–381. 10.1038/nrmicro2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang X, Hutt-Fletcher LM. 1998. Epstein-Barr virus lacking glycoprotein gp42 can bind to B cells but is not able to infect. J. Virol. 72:158–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chesnokova LS, Nishimura SL, Hutt-Fletcher LM. 2009. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins αvβ6 or αvβ8. Proc. Natl. Acad. Sci. U. S. A. 106:20464–20469. 10.1073/pnas.0907508106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li Q, Turk SM, Hutt-Fletcher LM. 1995. The Epstein-Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of EBV and carries an epitope critical to infection of B cells but not of epithelial cells. J. Virol. 69:3987–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Q, Buranathai C, Grose C, Hutt-Fletcher LM. 1997. Chaperone functions common to nonhomologous Epstein-Barr virus gL and varicella-zoster virus gL proteins. J. Virol. 71:1667–1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 106:2880–2885. 10.1073/pnas.0810530106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roche S, Albertini AA, Lepault J, Bressanelli S, Gaudin Y. 2008. Structures of vesicular stomatitis virus glycoprotein: membrane fusion revisited. Cell. Mol. Life Sci. 65:1716–1728. 10.1007/s00018-008-7534-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roche S, Bressanelli S, Rey FA, Gaudin Y. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191. 10.1126/science.1127683 [DOI] [PubMed] [Google Scholar]
- 14. Kadlec J, Loureiro S, Abrescia NG, Stuart DI, Jones IM. 2008. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 15:1024–1030. 10.1038/nsmb.1484 [DOI] [PubMed] [Google Scholar]
- 15. Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. 10.1126/science.1126548 [DOI] [PubMed] [Google Scholar]
- 16. Plate AE, Reimer JJ, Jardetzky TS, Longnecker R. 2011. Mapping regions of Epstein-Barr virus (EBV) glycoprotein B (gB) important for fusion function with gH/gL. Virology 413:26–38. 10.1016/j.virol.2010.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen J, Jardetzky TS, Longnecker R. 2013. The large groove found in the gH/gL structure is an important functional domain for Epstein-Barr virus fusion. J. Virol. 87:3620–3627. 10.1128/JVI.03245-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu L, Borza CM, Hutt-Fletcher LM. 2005. Mutations of Epstein-Barr virus gH that are differentially able to support fusion with B cells or epithelial cells. J. Virol. 79:10923–10930. 10.1128/JVI.79.17.10923-10930.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu L, Hutt-Fletcher LM. 2007. Point mutations in EBV gH that abrogate or differentially affect B cell and epithelial cell fusion. Virology 363:148–155. 10.1016/j.virol.2007.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pellett PE, Biggin MD, Barrell B, Roizman B. 1985. Epstein-Barr virus genome may encode a protein showing significant amino acid and predicted secondary structure homology with glycoprotein B of herpes simplex virus 1. J. Virol. 56:807–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maurer UE, Sodeik B, Grunewald K. 2008. Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proc. Natl. Acad. Sci. U. S. A. 105:10559–10564. 10.1073/pnas.0801674105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Garcia NJ, Chen J, Longnecker R. 2013. Modulation of Epstein-Barr virus glycoprotein B (gB) fusion activity by the gB cytoplasmic tail domain. mBio 4(1):e00571-12. 10.1128/mBio.00571-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Omerović J, Lev L, Longnecker R. 2005. The amino terminus of Epstein-Barr virus glycoprotein gH is important for fusion with epithelial and B cells. J. Virol. 79:12408–12415. 10.1128/JVI.79.19.12408-12415.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Silva AL, Omerović J, Jardetzky TS, Longnecker R. 2004. Mutational analyses of Epstein-Barr virus glycoprotein 42 reveal functional domains not involved in receptor binding but required for membrane fusion. J. Virol. 78:5946–5956. 10.1128/JVI.78.11.5946-5956.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen J, Rowe CL, Jardetzky TS, Longnecker R. 2012. The KGD motif of Epstein-Barr virus gH/gL is bifunctional, orchestrating infection of B cells and epithelial cells. mBio 3(1):e00290-11. 10.1128/mBio.00290-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Silverman JL, Heldwein EE. 2013. Mutations in the cytoplasmic tail of herpes simplex virus 1 gH reduce the fusogenicity of gB in transfected cells. J. Virol. 87:10139–10147. 10.1128/JVI.01760-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Reimer JJ, Backovic M, Deshpande CG, Jardetzky T, Longnecker R. 2009. Analysis of Epstein-Barr virus glycoprotein B functional domains via linker insertion mutagenesis. J. Virol. 83:734–747. 10.1128/JVI.01817-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McShane MP, Longnecker R. 2004. Cell-surface expression of a mutated Epstein-Barr virus glycoprotein B allows fusion independent of other viral proteins. Proc. Natl. Acad. Sci. U. S. A. 101:17474–17479. 10.1073/pnas.0404535101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chowdary TK, Heldwein EE. 2010. Syncytial phenotype of C-terminally truncated herpes simplex virus type 1 gB is associated with diminished membrane interactions. J. Virol. 84:4923–4935. 10.1128/JVI.00206-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park SJ, Seo MD, Lee SK, Lee BJ. 2008. Membrane binding properties of EBV gp110 C-terminal domain; evidences for structural transition in the membrane environment. Virology 379:181–190. 10.1016/j.virol.2008.06.031 [DOI] [PubMed] [Google Scholar]
- 31. Silverman JL, Sharma S, Cairns TM, Heldwein EE. 2010. Fusion-deficient insertion mutants of herpes simplex virus type 1 glycoprotein B adopt the trimeric postfusion conformation. J. Virol. 84:2001–2012. 10.1128/JVI.01791-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Omerović J, Longnecker R. 2007. Functional homology of gHs and gLs from EBV-related gamma-herpesviruses for EBV-induced membrane fusion. Virology 365:157–165. 10.1016/j.virol.2007.03.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kirschner AN, Lowrey AS, Longnecker R, Jardetzky TS. 2007. Binding-site interactions between Epstein-Barr virus fusion proteins gp42 and gH/gL reveal a peptide that inhibits both epithelial and B-cell membrane fusion. J. Virol. 81:9216–9229. 10.1128/JVI.00575-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kirschner AN, Sorem J, Longnecker R, Jardetzky TS. 2009. Structure of Epstein-Barr virus glycoprotein 42 suggests a mechanism for triggering receptor-activated virus entry. Structure 17:223–233. 10.1016/j.str.2008.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shaw PL, Kirschner AN, Jardetzky TS, Longnecker R. 2010. Characteristics of Epstein-Barr virus envelope protein gp42. Virus Genes 40:307–319. 10.1007/s11262-010-0455-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sorem J, Jardetzky TS, Longnecker R. 2009. Cleavage and secretion of Epstein-Barr virus glycoprotein 42 promote membrane fusion with B lymphocytes. J. Virol. 83:6664–6672. 10.1128/JVI.00195-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Paterson RG, Russell CJ, Lamb RA. 2000. Fusion protein of the paramyxovirus SV5: destabilizing and stabilizing mutants of fusion activation. Virology 270:17–30. 10.1006/viro.2000.0267 [DOI] [PubMed] [Google Scholar]
- 38. Waning DL, Russell CJ, Jardetzky TS, Lamb RA. 2004. Activation of a paramyxovirus fusion protein is modulated by inside-out signaling from the cytoplasmic tail. Proc. Natl. Acad. Sci. U. S. A. 101:9217–9222. 10.1073/pnas.0403339101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rowe CL, Connolly SA, Chen J, Jardetzky TS, Longnecker R. 2013. A soluble form of Epstein-Barr virus gH/gL inhibits EBV-induced membrane fusion and does not function in fusion. Virology 436:118–126. 10.1016/j.virol.2012.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aguilar HC, Matreyek KA, Choi DY, Filone CM, Young S, Lee B. 2007. Polybasic KKR motif in the cytoplasmic tail of Nipah virus fusion protein modulates membrane fusion by inside-out signaling. J. Virol. 81:4520–4532. 10.1128/JVI.02205-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Q, Spriggs MK, Kovats S, Turk SM, Comeau MR, Nepom B, Hutt-Fletcher LM. 1997. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol. 71:4657–4662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U. S. A. 102:9288–9293. 10.1073/pnas.0503989102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Samal S, Khattar SK, Paldurai A, Palaniyandi S, Zhu X, Collins PL, Samal SK. 2013. Mutations in the cytoplasmic domain of the Newcastle disease virus fusion protein confer hyperfusogenic phenotypes modulating viral replication and pathogenicity. J. Virol. 87:10083–10093. 10.1128/JVI.01446-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zokarkar A, Lamb RA. 2012. The paramyxovirus fusion protein C-terminal region: mutagenesis indicates an indivisible protein unit. J. Virol. 86:2600–2609. 10.1128/JVI.06546-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sergel T, Morrison TG. 1995. Mutations in the cytoplasmic domain of the fusion glycoprotein of Newcastle disease virus depress syncytia formation. Virology 210:264–272. 10.1006/viro.1995.1343 [DOI] [PubMed] [Google Scholar]
- 46. Silverman JL, Greene NG, King DS, Heldwein EE. 2012. Membrane requirement for folding of the herpes simplex virus 1 gB cytodomain suggests a unique mechanism of fusion regulation. J. Virol. 86:8171–8184. 10.1128/JVI.00932-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Baghian A, Huang L, Newman S, Jayachandra S, Kousoulas KG. 1993. Truncation of the carboxy-terminal 28 amino acids of glycoprotein B specified by herpes simplex virus type 1 mutant amb1511-7 causes extensive cell fusion. J. Virol. 67:2396–2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gage PJ, Levine M, Glorioso JC. 1993. Syncytium-inducing mutations localize to two discrete regions within the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B. J. Virol. 67:2191–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haan KM, Lee SK, Longnecker R. 2001. Different functional domains in the cytoplasmic tail of glycoprotein B are involved in Epstein-Barr virus-induced membrane fusion. Virology 290:106–114. 10.1006/viro.2001.1141 [DOI] [PubMed] [Google Scholar]
- 50. Claesson-Welsh L, Spear PG. 1986. Oligomerization of herpes simplex virus glycoprotein B. J. Virol. 60:803–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Atanasiu D, Cairns TM, Whitbeck JC, Saw WT, Rao S, Eisenberg RJ, Cohen GH. 2013. Regulation of herpes simplex virus gB-induced cell-cell fusion by mutant forms of gH/gL in the absence of gD and cellular receptors. mBio 4(2):e00046-13. 10.1128/mBio.00046-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Uchida H, Chan J, Shrivastava I, Reinhart B, Grandi P, Glorioso JC, Cohen JB. 2013. Novel mutations in gB and gH circumvent the requirement for known gD receptors in herpes simplex virus 1 entry and cell-to-cell spread. J. Virol. 87:1430–1442. 10.1128/JVI.02804-12 [DOI] [PMC free article] [PubMed] [Google Scholar]