

ABSTRACT
High-risk human papillomaviruses (HPVs), including HPV-16 and HPV-18, are the causative agents of cervical carcinomas and are linked to several other tumors of the anogenital and oropharyngeal regions. The majority of HPV-induced tumors contain integrated copies of the normally episomal HPV genome that invariably retain intact forms of the two HPV oncogenes E6 and E7. E6 induces degradation of the cellular tumor suppressor p53, while E7 destabilizes the retinoblastoma (Rb) protein. Previous work has shown that loss of E6 function in cervical cancer cells induces p53 expression as well as downstream effectors that induce apoptosis and cell cycle arrest. Similarly, loss of E7 allows increased Rb expression, leading to cell cycle arrest and senescence. Here, we demonstrate that expression of a bacterial Cas9 RNA-guided endonuclease, together with single guide RNAs (sgRNAs) specific for E6 or E7, is able to induce cleavage of the HPV genome, resulting in the introduction of inactivating deletion and insertion mutations into the E6 or E7 gene. This results in the induction of p53 or Rb, leading to cell cycle arrest and eventual cell death. Both HPV-16- and HPV-18-transformed cells were found to be responsive to targeted HPV genome-specific DNA cleavage. These data provide a proof of principle for the idea that vector-delivered Cas9/sgRNA combinations could represent effective treatment modalities for HPV-induced cancers.
IMPORTANCE Human papillomaviruses (HPVs) are the causative agents of almost all cervical carcinomas and many other tumors, including many head and neck cancers. In these cancer cells, the HPV DNA genome is integrated into the cellular genome, where it expresses high levels of two viral oncogenes, called E6 and E7, that are required for cancer cell growth and viability. Here, we demonstrate that the recently described bacterial CRISPR/Cas RNA-guided endonuclease can be reprogrammed to target and destroy the E6 or E7 gene in cervical carcinoma cells transformed by HPV, resulting in cell cycle arrest, leading to cancer cell death. We propose that viral vectors designed to deliver E6- and/or E7-specific CRISPR/Cas to tumor cells could represent a novel and highly effective tool to treat and eliminate HPV-induced cancers.
INTRODUCTION
Cervical carcinomas arise as a result of infection by human papillomavirus (HPV), most commonly high-risk HPV serotype HPV-16 or HPV-18 (1–4). Tumorigenesis results primarily from the deregulated expression of two viral oncoproteins, termed E6 and E7, which induce the degradation of the host cell tumor suppressors p53 and retinoblastoma (Rb) protein, respectively (2–7). This suggests that inactivation of either E6 or E7 expression in cervical carcinoma cells should result in the induction of p53 or Rb, respectively, as these proteins normally remain fully intact in cervical carcinoma cells. In fact, repression of E6 expression not only induces p53 but also upregulates the downstream effectors of p53 function, including proapoptotic proteins and the cyclin-dependent kinase inhibitor p21, resulting in cell cycle arrest, senescence, and apoptosis (5, 6, 8, 9). Similarly, inhibition of E7 expression induces the expression of Rb and the formation of Rb/E2F complexes, which again leads to cell cycle arrest and senescence (5, 8, 9). Therefore, agents that can repress E6 and/or E7 expression in HPV-induced cervical carcinomas, as well as other HPV-induced cancers such as head and neck tumors, represent a potential treatment for these types of cancer in humans.
Previous research investigating methods to selectively block the expression of HPV E6 and/or E7 in cervical carcinoma cells in culture has demonstrated effective repression by ectopic expression of the HPV or bovine papillomavirus E2 protein, which blocks E6 and E7 mRNA transcription (5, 10–12), and by RNA interference directed against the mRNA encoding E6 or E7 (8, 13). However, both of these approaches have technical difficulties that have so far made them unsuitable for use in a clinical setting, including the fact that both approaches leave the HPV E6 and E7 genes intact, thus potentially allowing escape from repression. An alternative approach to the treatment of HPV-induced cancers would be to permanently inactivate the viral E6 and/or E7 gene by targeted mutagenesis, which is predicted to result in the death of HPV-transformed cells. This goal has now become feasible with the rapid development of bacterially derived RNA-guided DNA endonucleases (RGNs) derived from the type II bacterial CRISPR (clustered regulatory interspaced short palindromic repeats)/Cas system of adaptive antiviral immunity (14).
Type II CRISPR/Cas systems rely on a single effector protein, called Cas9, that is normally guided to a specific DNA sequence, generally of bacteriophage origin, by two small RNA molecules called the tracrRNA and the crRNA (14, 15). The demonstration that the tracrRNA and the crRNA could be combined into a single guide RNA (sgRNA) (16, 17) represented a major breakthrough in the development of CRISPR/Cas systems into effective gene editing tools. To this point, most research has focused on the Cas9 protein from Streptococcus pyogenes, which uses a 20-nucleotide (nt) guide sequence flanked at the 3′ end by a so-called protospacer-adjacent motif (PAM), with the sequence 5′-NGG-3′, that is an invariant part of the DNA target but is not present in the sgRNA (14–17). Using transfection or lentiviral transduction, it is possible to induce the cleavage of specific sequences in the human genome by the expression of S. pyogenes Cas9 and an sgRNA molecule transcribed by an RNA polymerase III promoter (16, 17). Gene inactivation using this system is highly efficient, and S. pyogenes Cas9 has in fact been used for high-throughput genetic screens in human cells in culture (18–20) as well as for the generation of multigene knockouts in mice (21).
We therefore reasoned that the S. pyogenes Cas9 protein, in combination with sgRNAs designed to selectively target and inactivate the HPV E6 or E7 gene, might represent a powerful way to permanently inactivate these viral oncogenes in cervical carcinoma cells and induce their subsequent death. This would provide a proof of principle for the idea that inactivation of HPV E6 and E7 by viral vector-delivered S. pyogenes Cas9/sgRNA combinations has significant clinical potential in the treatment of HPV-induced cancers in vivo. Here, we demonstrate that S. pyogenes Cas9/sgRNA combinations are indeed able to efficiently mutationally inactivate HPV E6 and E7 protein function in both HPV-16- and HPV-18-transformed cervical carcinoma cells, resulting in cell cycle arrest and tumor cell death.
MATERIALS AND METHODS
CRISPR/Cas9 constructs and sgRNA design.
Two pairs of sgRNAs were designed by using the ZiFit Web application (http://zifit.partners.org/) to target the amino-terminal regions of the HPV-18 E6 and E7 open reading frames (ORFs). RGNs were constructed by cloning HPV-specific sgRNAs into the px330 vector (Addgene) expressing S. pyogenes Cas9 (16). sgRNAs were also cloned into the px458 vector, an alternative version of px330 containing a gfp marker useful for flow cytometric analysis (22). RGN function was tested by generating a vector containing either HPV-18 E6- or E7-derived viral DNA targets inserted in frame between an HIV-1 rev gene fragment encoding amino acids 1 to 59 of Rev (23) and a 3′ gfp indicator gene. Following cotransfection of the reporter plasmid with a S. pyogenes Cas9/sgRNA expression construct, function was determined by detecting the specific loss of Rev and green fluorescent protein (GFP) expression by Western blotting and flow cytometry, respectively. HPV-16-specific sgRNAs targeting the HPV-16 E6 and E7 ORFs integrated into the SiHa cell line were designed and tested by using a similar approach. The following gene-specific sgRNA sequences were used and constructed, as outlined previously (16): HPV-18 E6t1 (GGCGCTTTGAGGATCCAACA), HPV-18 E6t2 (GAAGCTACCTGATCTGTGCA), HPV-18 E7t1 (GGAGCAATTAAGCGACTCAG), HPV-18 E7t2 (GAAGAAAACGATGAAATAGA), HPV-16 E6t1 (GCAACAGTTACTGCGACGTG), and HPV-16 E7t1 (GCCAGCTGGACAAGCAGAAC) (nucleotides in boldface type indicate mismatched 5′ G residues required for transcription initiation from a U6 promoter).
Cell culture.
HeLa, 293T, and SiHa cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM antibiotic-antimycotic (Gibco Cell Culture), and 50 μg/ml gentamicin (LifeTechnologies) at 37°C.
Reporter assays.
Reporter assays with 293T cells were performed by using the calcium phosphate transfection method. 293T cells were plated at ∼1.25 × 105 cells per well in 12-well plates and transfected with a 4:1 ratio of the RGN expression plasmid to the indicator plasmid. 293T cells were then assayed by Western blotting to detect Rev expression and by flow cytometry to determine the positive fraction and mean fluorescence intensity (MFI) of enhanced GFP (eGFP)-positive cells at 3 days posttransfection.
Surveyor assays and mutagenesis spectra.
HeLa cells were plated at 2.5 × 105 cells per well in 6-well plates and transfected by using Fugene6 with a 3-to-1 ratio of the RGN expression vector to pL/CMV/eGFP (pLCE) (24), which expresses eGFP, to determine transfection efficiency. Genomic DNA was extracted at 48 h posttransfection by using a DNeasy kit (Qiagen) according to the manufacturer's protocol. The genomic region surrounding the viral target sites was PCR amplified by using the GoTaq cocktail (catalog no. 9PIM300; Promega) and then purified. PCR products were then denatured and reannealed to enable DNA heteroduplex formation as follows: 95°C for 10 min, 95°C to 85°C with ramping at −1°C/s, 85°C to 25°C with ramping at −0.25°C/s, and a hold at 25°C for 1 min. After reannealing, products were treated with Surveyor nuclease and Surveyor enhancer S (Transgenomics) according to the manufacturer's protocol and analyzed on a 2% agarose gel. To determine the sequence spectra of the mutations introduced by HPV-18 E6- and E7-specific sgRNAs, we designed primers with unique restriction enzyme sites that bound sequences flanking the predicted RGN cleavage locations. The mutagenized HeLa genomic DNA was PCR amplified 48 h after transfection, cloned, sequenced, and aligned to the HeLa genome.
Western blots.
Phenotypic analysis of RGNs in HeLa cells was performed by using the Fugene6 transfection reagent according to the manufacturer's protocol. Cells were plated at 2.5 × 105 cells per well in 6-well plates and transfected with a 3-to-1 ratio of the RGN expression vector to pLCE (a GFP-expressing plasmid included to determine transfection efficiency). SiHa cells were transfected with HPV-16-specific RGN expression vectors by using Lipofectamine3000 (Clontech) according to the manufacturer's protocol. Cells were then harvested and lysed in SDS–β-mercaptoethanol protein lysis buffer at 48 h posttransfection. Lysates were subjected to electrophoresis on 4 to 20% SDS-polyacrylamide gels (Bio-Rad) and transferred onto nitrocellulose membranes. The membranes were then probed in 5% milk–PBS-T (phosphate-buffered saline, 0.1% Tween 20, 0.5% bovine serum albumin) with the following antibodies: anti-Flag (catalog no. F1804; Sigma), a rabbit Rev polyclonal antiserum (23), anti-β-actin (catalog no. SC-47778; Santa Cruz), anti-p53 (catalog no. SC-126; Santa Cruz), anti-p21 (catalog no. SC-397; Santa Cruz), and anti-Rb (catalog no. 554136; BD Pharmingen). The membranes were washed in PBS-T, incubated with a species-specific secondary antibody, and then washed again in PBS-T. The membranes were incubated with a Western Bright Sirius Western blot detection kit (Advansta), and signals were captured by using GeneSnap and quantified by using GeneTools (Syngene). In order to determine the specificity of RGN cleavage, we generated a pcDNA3 construct expressing an E6 mutant harboring synonymous mutations in the Cas9 “seed” region. These experiments were conducted by transfecting HeLa cells in 6-well plates at 30 to 40% confluence with a 3-to-1 ratio of an RGN expression vector to a mutant E6 expression plasmid with Fugene6. Western blots were conducted in triplicate at 48 h posttransfection, as previously described (23).
Growth inhibition and cell cycle analysis.
To determine growth effects of RGNs, HeLa cells were plated into 12-well plates at 105 cells per well and transfected with 750 ng of an HPV-18-specific RGN expression vector in triplicate. Flow cytometric analysis measuring the percentage of eGFP-positive cells by using FACSCanto software was performed at 48, 72, and 96 h posttransfection. Data were normalized to data for a px458-transfected culture. To examine cell cycle progression, 106 HeLa cells were plated into 6-cm plates and cotransfected with HPV-18 E6- or E7-specific RGN expression vectors and an eGFP expression plasmid, at a 3-to-1 ratio, by using Fugene6. Exponentially growing cells were treated at 48 h posttransfection with 10 μg/ml 5-bromodeoxyuridine (BrdU; Calbiochem) for 1 h. Cells were then trypsinized, washed with PBS, and fixed with 2% paraformaldehyde for 1 h at 25°C. Cells were washed with PBS and permeabilized with 70% ethanol overnight at 4°C. After washing with PBS, DNA was denatured by treating the cells with 2 M HCl for 30 min at 25°C and then washing the cells twice with PBS-T. Cells were resuspended in 100 μl PBS-T and 2.5 μl Alexa Fluor 647 anti-BrdU antibody (catalog no. 560209; BD Biosciences) for 20 min at 25°C. Cells were washed once with PBS and resuspended in 200 μl propidium iodide (PI)-RNase staining buffer (BD Biosciences). Cells were then analyzed by flow cytometry using a BD FACSCanto II instrument and FlowJo software. Separate flow plots comparing BrdU and PI staining were generated to compare transfected (GFP-positive) and nontransfected (GFP-negative) HeLa cells.
Cell survival assay.
To measure HeLa cell transduction efficiency, we employed the lentiviral LCE eGFP expression vector (24). This allowed us to determine that 89.3% of the HeLa cells in culture were transduced, consistent with an initial multiplicity of infection (MOI) of ∼2.2. The S. pyogenes Cas9/sgRNA-expressing lentiviral vectors used were based on lentiCRISPR (20), with the appropriate sgRNAs inserted 3′ to a U6 promoter. HeLa cells (5 × 104) were transduced with these lentiviral vectors, and fresh medium was added 24 h after infection. The cells were then cultivated for 10 days, and cell numbers were quantified at 2-day intervals.
In a second experiment, HeLa cells were again transduced with lentiCRISPR-based vectors expressing S. pyogenes Cas9 and a nonspecific sgRNA, E6 sgRNA1 or E7 sgRNA1, or with LCE. In the second experiment, we used a higher predicted MOI of ∼37, which should result in the infection of essentially every cell. After cultivation for 10 days, the cells were stained with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). MTT was eluted from cells with isopropanol containing 0.04 M HCl, and the absorbance at 590 nm was determined by using a Fluostar Omega instrument (BMG Labtech) with a reference filter of 620 nm. Survival was calculated relative to the survival of mock-infected cells.
RESULTS
To target the integrated genomic copies of HPV-18 present in HeLa cells for DNA cleavage, we designed sgRNAs complementary to nucleotides 5 to 24 and 36 to 55 of the HPV-18 E6 open reading frame (ORF) and nucleotides 84 to 103 and 106 to 125 of the HPV-18 E7 ORF. These sgRNAs were expressed under the control of a U6 RNA polymerase III promoter present in the px330 expression vector, which also expresses the S. pyogenes Cas9 protein (16). To assess the functionality of these sgRNAs, we cloned cognate HPV-18 E6- or E7-derived target sequences in frame between an amino-terminal region encoding the first 59 amino acids of the HIV-1 Rev protein (23) and a carboxy-terminal gfp indicator gene (Fig. 1A). If an sgRNA is indeed functional, it should inhibit the expression of the predicted Rev-target-GFP fusion protein determined by either GFP fluorescence or Western blot analysis using a Rev-specific polyclonal antiserum. Indeed, as shown in Fig. 1B, both E6-specific sgRNAs greatly reduced both the number of GFP-positive cells, when the RGN expression vector was cotransfected into 293T cells along with the indicator vector, as well as the average mean fluorescence intensity (MFI) of the remaining GFP-positive cells. Similarly, both E7-specific sgRNAs dramatically inhibited GFP expression from their cognate indicator plasmids in cotransfected cells (Fig. 1C). Furthermore, analysis of Rev-target-GFP fusion protein expression in these same cotransfected 293T cells by Western blotting (Fig. 1D) revealed an almost total loss of indicator protein expression. We therefore concluded that all four HPV-18 E6- or E7-specific sgRNAs can effectively silence the expression of a cognate target gene.
FIG 1.
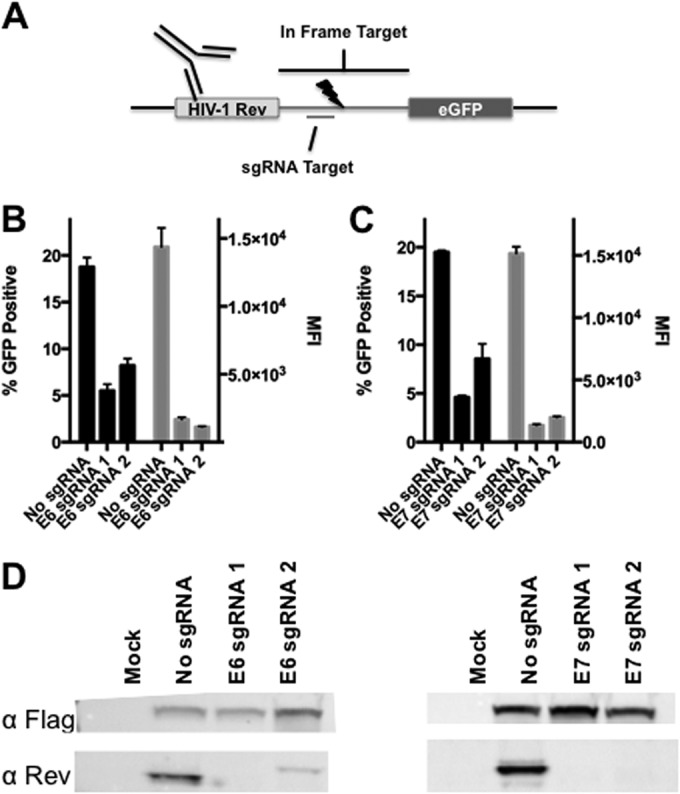
S. pyogenes Cas9 HPV-18-specific sgRNA screening. Two sgRNAs were designed to target the amino terminus of the HPV-18 E6 or E7 protein and screened to identify the most effective candidate. (A) Schematic depicting the fusion protein-based reporter assay, which includes an amino-terminal HIV-1 Rev fragment that acts as an epitope tag, an in-frame HPV-18-derived target sequence, and a carboxy-terminal eGFP ORF (described in detail in Materials and Methods). (B) Graph showing eGFP expression data for 293T cells cotransfected with plasmids expressing the Flag-tagged S. pyogenes Cas9 protein and sgRNAs specific for the HPV-18 E6 gene, or a control construct, and their cognate indicator plasmids. Transfected cells were processed for flow cytometry at 72 h. The number of GFP-positive cells and the mean fluorescence intensity (MFI) of these cells are indicated. Averages and standard deviations of data from three independent experiments are indicated. (C) Similar to panel B but with two HPV-18 E7-specific sgRNAs. (D) Western blot using an HIV-1 Rev-specific antiserum to detect expression of the Rev-GFP indicator fusion protein, thus demonstrating sgRNA efficacy and specificity.
We next asked if we could inactivate the endogenous HPV-18 E6 and E7 genes integrated into the HeLa cell genome. As all four sgRNAs appeared to function equivalently, we focused our subsequent research on E6 sgRNA1 and E7 sgRNA1. To confirm that these sgRNAs were indeed inducing the expected DNA cleavage, we used the Surveyor assay to determine if we could detect RGN-induced indels at the predicted Cas9 cleavage site. For this purpose, we transfected HeLa cells with a px330-based vector encoding S. pyogenes Cas9 (16) and an E6- or E7-specific sgRNA. At 48 h posttransfection, we isolated HeLa cell genomic DNA and PCR amplified the regions of the HPV-18 genome containing either the E6 or E7 target site for Cas9. The resultant PCR fragment was then isolated, denatured, and reannealed to produce a population of DNA heteroduplexes that were digested by using Surveyor nuclease, which is predicted to cleave at any sites of Cas9-induced mutagenesis. As shown in Fig. 2A, we indeed readily detected the presence of indels in both the HPV-18 E6 and E7 genes at the predicted Cas9 target site. To further characterize these mutations, we cloned the same PCR fragments derived from the HPV-18 E6 or E7 gene and then performed DNA sequencing across the predicted cleavage site located 3 bp 5′ to the Cas9 PAM sequence (17). Analysis of the HPV-18 E6 gene gave 234 DNA sequence reads, of which 19 bore an assortment of different deletion mutations and 34 contained the same insertion mutation of a single “A” residue located 3 bp 5′ to the S. pyogenes Cas9 PAM motif (Fig. 2B). The remaining 181 samples analyzed contained the wild-type E6 sequence. Similarly, in the case of the HPV-18 E7 gene, we recovered 232 DNA sequences, of which 6 represent deletion mutations adjacent to the predicted S. pyogenes Cas9 cleavage site and 46 represent insertions of 1 or 2 bp, again at a site 3 bp 5′ to the PAM motif, with by far the most common being a single “T” nucleotide insertion (Fig. 2C). Interestingly, we also recovered one sequence containing a large insertion mutation that introduced a sequence derived from human chromosome 8 (Fig. 2C). Therefore, for both the HPV-18 E6 and E7 genes, we were able to recover multiple mutations at the predicted E6 or E7 RGN cleavage site, almost all of which are predicted to disrupt the E6 or E7 ORF.
FIG 2.

HPV-18 E6- and E7-specific S. pyogenes Cas9 sgRNAs induce mutagenesis at the predicted cleavage site in the HPV genome. (A) E6 and E7 sgRNA and S. pyogenes Cas9 expression constructs were transfected, and the Surveyor assay was performed. The predicted size of the Surveyor cleavage product is indicated by an arrow. DNA markers (left lane) are indicated in base pairs. WT, wild type. (B) Alignment of the DNA sequence of the targeted region of the HPV-18 E6 gene isolated from HeLa cells expressing an E6-specific RGN. The sgRNA target and PAM for the HPV-18 E6 locus are indicated. (C) Similar to panel B but with the HPV-18 E7 gene.
Both E6 and E7 expressions are known to be required for HeLa cell growth and survival (4–8), and we therefore next asked if targeting E6 and E7 with an RGN would indeed inactivate E6 and E7 function. As noted above, the HPV E6 protein functions to repress the expression of the host p53 tumor suppressor so that a loss of E6 function is expected to result in the activation of not only p53 expression but also downstream effectors of p53, including the cyclin-dependent kinase inhibitor p21 (2, 3, 5, 7). As shown in Fig. 3A, we indeed observed the specific induction of both p53 and p21 expression in cells expressing the E6 sgRNA but not in control cells or in cells expressing the E7 sgRNA.
FIG 3.

HPV-18 E6- and E7-specific RGNs induce tumor suppressor gene expression in HeLa cells. HeLa cells were transfected with an S. pyogenes Cas9 expression vector and HPV-18-specific sgRNAs, as indicated, and were processed for Western blotting. These data are representative of data from 3 biological replicates. (A) The lysate was probed for p53 and p21 expression, with endogenous β-actin being used as a loading control. The Cas9 protein was detected by an antibody specific for the Flag epitope tag. (B) Similar to panel A except that the lysate was probed for Rb. (C) Sequence of a mutant E6 expression construct designed to be resistant to cleavage by S. pyogenes Cas9 in the presence of E6 sgRNA1. The mutations are in lowercase type, and the PAM is underlined. (D) Expression of the cleavage-resistant E6 gene shown in panel C reveals trans-complementation of p53 protein repression in the presence of S. pyogenes Cas9 and E6 sgRNA1. N.S., nonspecific.
The HPV-18 E7 protein functions to repress the function of the host cell retinoblastoma (Rb) protein by binding to the hypophosphorylated form of Rb, thereby inducing Rb degradation and preventing the formation of Rb/E2F complexes that would block cell cycle progression (2–5). As shown in Fig. 3B, we did detect an increase in Rb expression that was, however, fairly modest (1.4-fold ± 0.03-fold) when normalized to the expression of the β-actin internal control. We note, however, that disruption of E7 expression results in cell cycle arrest (see below) so that cells bearing a disrupted E7 gene are expected to be selectively lost from the transfected culture. Indeed, we observed that expression of the Flag-tagged S. pyogenes Cas9 protein was specifically reduced in cells expressing an sgRNA specific for either E6 or E7 (Fig. 3B). If this decrease is taken into account, the increase in the Rb expression level is a more substantial 5.54-fold ± 1.42-fold over that of the control, which is statistically significant (P = 0.011).
It could be argued that the activation of p53 in HeLa cells expressing S. pyogenes Cas9 and E6-specific sgRNA1 is a nonspecific effect resulting from an off-target effect, such as promiscuous DNA damage. However, analysis of potential target sites in the human genome for E6 sgRNA1, by a BLAST search of the NCBI human genome database, failed to identify any targets with perfect complementarity to the 13-nt E6 sgRNA1 seed sequence in combination with the S. pyogenes Cas9 PAM 5′-NGG-3′ (data not shown). We did identify a single perfect human genome target site for E7 sgRNA1 located at nucleotide position 164032096 on the long arm of chromosome 2 that is not believed to be transcribed. In addition, we detected a number of potential target sites for E6 sgRNA1 and E7 sgRNA1 that differed from a perfect target site at a single base pair and therefore might also represent potential off-target cleavage sites in the human genome.
Off-target genomic DNA damage would be predicted to activate p53, and we were particularly interested in demonstrating that the activation of p53 by E6 sgRNA1 resulted exclusively from cleavage of the HPV E6 gene. We therefore generated a mutant form of the HPV-18 E6 gene in which we introduced mutations that would be predicted to block sgRNA cleavage but that did not alter the underlying E6 amino acid sequence (Fig. 3C). As shown in Fig. 3D, cotransfection of an expression vector encoding this variant form of E6 entirely blocked the activation of p53 expression by the E6-specific RGN, which strongly suggests that p53 induction in the presence of E6 sgRNA1 indeed reflects the selective loss of E6 expression.
Previous work has shown that the loss of either E6 or E7 function in HeLa cells, and in other HPV-transformed cells, results in senescence, cell cycle arrest, and, in the case of E6 repression, also apoptosis (2, 3, 5). To test whether RGNs specific for E6 or E7 would exert the predicted phenotypic effect, we analyzed growth in culture of cells expressing S. pyogenes Cas9 and either E6 sgRNA1 or E7 sgRNA1 compared to that of cells expressing a control sgRNA. As shown in Fig. 4A, while the control cells continued to replicate, the number of HeLa cells expressing the E6- or E7-specific sgRNA strongly decreased over time. We next analyzed the cell cycle progression of control cells relative to that of E6 sgRNA1- or E7 sgRNA-expressing cells by culturing the cells in the presence of BrdU, which is incorporated into newly synthesized DNA, and then staining the cells with propidium iodide (PI), which allows measurement of the total DNA level per cell. As shown by flow cytometry (Fig. 4C), the control culture showed 57% of cells in G1, 34% in S phase, and 9.9% in G2. In contrast, the culture expressing E6 sgRNA1 showed 77% of cells in G1, 17% in S phase, and 6.0% in G2, while the culture expressing E7 sgRNA1 showed 79% of cells in G1, 15% in S phase, and 6.6% in G2. A bar graph summarizing data derived from four separate BrdU-PI labeling experiments, shown in Fig. 4B, confirms that both the E6- and E7-specific sgRNAs induce cell cycle arrest in G1 while strongly reducing the number of cells in S phase or entering G2, as expected for inhibitors of E6 or E7 expression (2, 3, 5, 6).
FIG 4.

RGN-directed mutagenesis of either the E6 or E7 locus induces cell cycle arrest in G1. (A) S. pyogenes Cas9-mediated disruption of either the HPV-18 E6 or E7 gene results in the expected inhibition of HeLa cell growth. The number of GFP-positive cells transfected with vectors encoding S. pyogenes Cas9, a control or HPV-specific sgRNA, and the gfp gene is shown. (B) Cell cycle analysis of HeLa cells expressing E6- or E7-specific RGNs using BrdU incorporation and PI staining followed by fluorescence-activated cell sorter analysis. Results from four separate biological replicates of the transfected eGFP-positive HeLa cell population are shown. (C) Representative flow cytometry plots are shown for the sgRNAs indicated. The percentage of GFP-positive HeLa cells in each phase of the cell cycle was quantitated and is indicated. NS, nonspecific; APC, allophycocyanin.
As noted above, repression of E6 or E7 expression in cervical carcinoma cells is predicted to induce not only cell cycle arrest, as reproduced here in Fig. 4, but also senescence and/or apoptosis (2, 3, 5, 6, 8). Therefore, it is expected that the expression of Cas9/sgRNA combinations specific for E6 or E7 would initially inhibit cell growth, followed by induction of cell death. To validate this prediction, we transduced HeLa cells with lentiviral vectors encoding Cas9 and either a control or E6- or E7-specific sgRNA, with a similar lentiviral vector encoding GFP, or mock transduced the cells. Using fluorescence-activated cell sorting, we observed that 89.3% of the cells transduced with the GFP-expressing lentivirus were GFP positive, consistent with an initial MOI of ∼2.2. We then counted the cells in culture at 2-day intervals for 10 days (Fig. 5A). The cultures transduced with Cas9/sgRNA combinations specific for HPV-18 E6 or E7 had ∼10-fold fewer cells than the control cultures at all time points, thus suggesting that the growing cells were likely derived from ∼10% of the initial culture that was not transduced.
FIG 5.
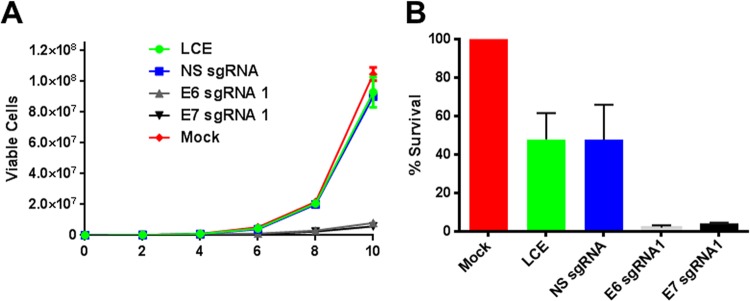
Lentiviral vectors expressing S. pyogenes Cas9 and sgRNAs specific for the HPV-18 E6 and E7 genes induce the death of cervical carcinoma cells. HeLa cells were transduced with a lentiviral vector expressing eGFP, to control for lentiviral toxicity (LCE), or a lentiviral vector expressing S. pyogenes Cas9 and a nonspecific sgRNA or E6- or E7-specific sgRNAs. (A) HeLa cells were transduced with the lentiviral vector at an MOI of ∼2.2, resulting in transduction of ∼90% of the cells in culture. Cell growth was then monitored over a 10-day period. Data from a representative experiment, derived from three similar independent experiments, are presented. (B) HeLa cells were transduced with the lentiviral vector at an MOI of ∼37, which is predicted to transduce >99% of the cells in culture. The cells were then assayed for viability by MTT staining on day 10. Averages of data from two independent experiments normalized to values for a mock-transduced culture are shown. N.S., nonspecific.
To determine if expression of Cas9/sgRNA combinations specific for HPV-18 E6 or E7 would indeed induce HeLa cell death, we repeated the above-described transduction experiment using a higher titer of the lentiviral vector (MOI of ∼37) that is predicted to transduce almost every HeLa cell in the culture. After 10 days in culture, the cells were stained with MTT, and the percentage of viable cells was determined (Fig. 5B). Surprisingly, in this experiment, the control lentiviral vectors resulted in an ∼2-fold decrease in cell viability that was clearly not due to S. pyogenes Cas9 expression, as the same effect was observed with the GFP-expressing lentiviral vector. In contrast, both the E6- and E7-specific sgRNAs, in the presence of S. pyogenes Cas9, induced the almost complete demise of the transduced HeLa cell culture. Therefore, targeted disruption of HPV E6 or E7 has the potential to induce the specific elimination of HPV-transformed cells.
All the work described thus far was done by using the HPV-18-transformed cervical carcinoma cell line HeLa. To extend these studies, we also analyzed the human SiHa cervical carcinoma cell line, which is transformed by HPV-16 (25), using sgRNAs specific for HPV-16 E6 and E7, which differ in sequence from HPV-18 E6 and E7. In this experiment, we therefore used E6 sgRNA1, which is specific for HPV-18 E6, as our “nonspecific” sgRNA. Targeting of the HPV-16 gene in SiHa cells using the S. pyogenes Cas9 protein and an HPV-16 E6-specific sgRNA resulted, as expected, in the specific induction of the p53 effector protein p21 (Fig. 6A), as also seen in HeLa cells after targeting of E6 (Fig. 3A). However, HPV-18-specific E6 sgRNA1 failed to induce p21 expression, thus further confirming that activation is due to E6 cleavage and is not due to off-target DNA cleavage. Similarly, targeting of the HPV-16 E7 gene in SiHa cells resulted in a modest but significant enhancement in the expression of the cell Rb protein (Fig. 6B), as also seen in HeLa cells (Fig. 3B). Therefore, RGNs targeted to either E6 or E7 are able to trigger the p53 or Rb pathway, respectively, in not only HPV-18- but also HPV-16-transformed cervical carcinoma cells.
FIG 6.
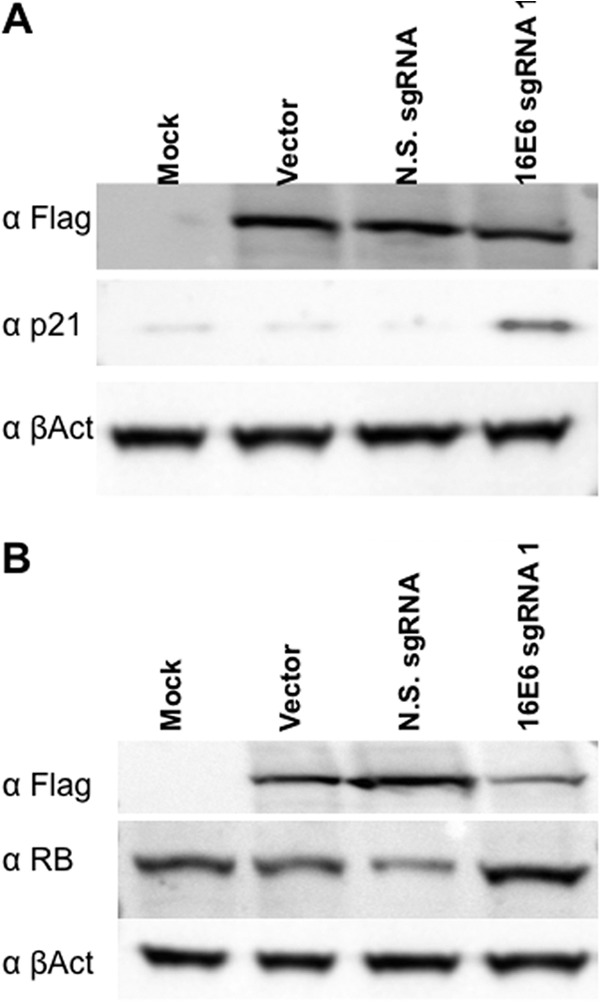
HPV-16 E6 and E7 RGNs rescue p21 and Rb expression in SiHa cells. SiHa cells were transfected with an S. pyogenes Cas9 expression vector and the HPV-16-specific sgRNA constructs indicated and processed for Western blotting. These data are representative of data from 3 biological replicates. (A) Lysate probed for the Flag-tagged Cas9 protein, the p53 effector protein p21, and endogenous β-actin (B). Similar to panel A but with the lysate being probed for Rb expression. The N.S. (nonspecific) sgRNA used as a control is E6 sgRNA1, which is specific for the HPV-18 E6 gene but is not predicted to recognize the HPV-16 E6 gene.
DISCUSSION
High-risk HPVs, including HPV-16, HPV-18, and HPV-31, are etiological agents of cervical carcinoma as well as several other cancers of the anogenital and oropharyngeal regions (1–4). In many HPV-induced carcinomas, the normally episomal HPV DNA genome is integrated into the host cell DNA in a manner that disrupts the viral E2 gene, which normally acts to repress E6 and E7 expression, while leaving the viral E6 and E7 genes intact (5, 10–12). While expression of HPV E6 and E7 is not sufficient to induce cell transformation, ample evidence indicates that the continued expression of E6 and E7 is required for the continued viability and growth of HPV-transformed cancer cells (2–6, 8–10). This requirement reflects the ability of E6 and E7 to serve as potent inhibitors of cellular tumor suppressor pathways. In particular, E6 binds to, and induces the degradation of, the host cell tumor suppressor p53, thus preventing the activation of downstream effectors that induce cell cycle arrest and apoptosis. In contrast, E7 binds to and destabilizes the host cell Rb tumor suppressor, thus preventing the formation of Rb/E2F complexes that block cell cycle progression and induce senescence (2–5, 7).
The importance of both E6 and E7 for the continued growth and viability of HPV-transformed cervical carcinoma cells has been demonstrated previously by introduction of an intact HPV or bovine papillomavirus E2 protein, which inhibits E6 and E7 transcription, into these cells or by using small interfering RNAs specific for the E6 or E7 mRNA (5, 6, 8–13). Here, we extend those previous studies by demonstrating that bacterial RNA-guided endonucleases of the CRISPR/Cas variety are able to efficiently cleave and inactivate either the E6 or E7 gene in HPV-18- or HPV-16-transformed cells in culture. As expected, inactivation of HPV E6 expression induced the expression of not only p53 but also downstream effectors, such as p21, while inactivation of the HPV E7 gene resulted in enhanced Rb expression (Fig. 3 and 6). This in turn initially induced cell cycle arrest followed by the eventual death of essentially all of these HPV-transformed cells (Fig. 4 and 5). Although the precise mechanism of cell death induction remains to be elucidated, it is conceivable that both the disruption of E6 and E7 expression as well as the targeted induction of specific DNA double-stranded breaks within the E6 and E7 genes by CRISPR/Cas9 contribute to the killing of HPV-transformed cells.
While ectopic expression of the HPV E2 protein and of small interfering RNAs specific for E6 or E7 both represent potential approaches to the elimination of HPV-transformed cells (5, 6, 8–10, 13), neither strategy is able to permanently inactivate the E6 or E7 gene in a way that makes the death of the HPV-transformed cell inevitable. In contrast, we hypothesized that the combination of a Cas9 protein and an E6- or E7-specific sgRNA would be capable of permanently preventing E6 or E7 expression, thus potentially clearing all HPV-transformed cells from the body. Here, we present proof-of-concept experiments in support of this hypothesis by using HPV-transformed cell lines and Cas9/sgRNA expression vectors delivered by transfection or lentiviral transduction. However, we believe that the effective elimination of HPV-induced cancers in vivo will require the development of vectors based on adeno-associated virus (AAV) that deliver both a Cas9 and an sgRNA expression cassette(s). AAV vectors have been approved previously for gene therapy in humans, are able to grow to extremely high titers, and have a low risk of insertional mutagenesis compared to lentiviral or retroviral vectors (26). Unfortunately, they also have a smaller packaging capacity of only ∼4.6 kb, which renders them too small to deliver the S. pyogenes Cas9 gene and an sgRNA expression cassette. However, recent research has identified a number of smaller Cas9 proteins, and one of these, the Cas9 protein encoded by Neisseria meningitidis, is able to effectively cleave DNA targets in human cells in culture despite its smaller size of ∼3.25 kb (27, 28). Moreover, a number of other Cas9 genes in the ∼3.2-kb size range have been identified and would also be able to fit into an AAV vector together with one or two sgRNA expression cassettes.
We therefore believe that it will be possible to develop AAV-based vectors that express Cas9/sgRNA combinations and that are able to transduce HPV-transformed cells in vivo with high efficiency. Expression of one or more sgRNAs specific for conserved DNA regions found in the high-risk HPV variant present in these transformed cells should then result in their selective elimination without affecting normal cells located in the vicinity of the tumor. Furthermore, dual-nickase approaches pioneered by others could be employed to further reduce any potential off-target DNA cleavage and enhance the specificity and safety of this proposed future therapy (22). Whether this approach will prove to be both effective and safe in vivo will clearly need to be addressed by future in vivo experiments assessing the antitumor potential and safety of HPV-specific AAV-based Cas9/sgRNA expression vectors.
Footnotes
Published ahead of print 6 August 2014
REFERENCES
- 1. zur Hausen H, de Villiers EM. 1994. Human papillomaviruses. Annu. Rev. Microbiol. 48:427–447. 10.1146/annurev.mi.48.100194.002235 [DOI] [PubMed] [Google Scholar]
- 2. Mighty KK, Laimins LA. 2014. The role of human papillomaviruses in oncogenesis. Recent Results Cancer Res. 193:135–148. 10.1007/978-3-642-38965-8_8 [DOI] [PubMed] [Google Scholar]
- 3. McLaughlin-Drubin ME, Münger K. 2009. Oncogenic activities of human papillomaviruses. Virus Res. 143:195–208. 10.1016/j.virusres.2009.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Howley PM. 1991. Role of the human papillomaviruses in human cancer. Cancer Res. 51:5019s–5022s [PubMed] [Google Scholar]
- 5. DeFilippis RA, Goodwin EC, Wu L, DiMaio D. 2003. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J. Virol. 77:1551–1563. 10.1128/JVI.77.2.1551-1563.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goodwin EC, DiMaio D. 2000. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc. Natl. Acad. Sci. U. S. A. 97:12513–12518. 10.1073/pnas.97.23.12513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. 1990. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63:1129–1136. 10.1016/0092-8674(90)90409-8 [DOI] [PubMed] [Google Scholar]
- 8. Hall AH, Alexander KA. 2003. RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in HeLa cells. J. Virol. 77:6066–6069. 10.1128/JVI.77.10.6066-6069.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Horner SM, DeFilippis RA, Manuelidis L, DiMaio D. 2004. Repression of the human papillomavirus E6 gene initiates p53-dependent, telomerase-independent senescence and apoptosis in HeLa cervical carcinoma cells. J. Virol. 78:4063–4073. 10.1128/JVI.78.8.4063-4073.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goodwin EC, Yang E, Lee CJ, Lee HW, DiMaio D, Hwang ES. 2000. Rapid induction of senescence in human cervical carcinoma cells. Proc. Natl. Acad. Sci. U. S. A. 97:10978–10983. 10.1073/pnas.97.20.10978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thierry F, Howley PM. 1991. Functional analysis of E2-mediated repression of the HPV18 P105 promoter. New Biol. 3:90–100 [PubMed] [Google Scholar]
- 12. Romanczuk H, Thierry F, Howley PM. 1990. Mutational analysis of cis elements involved in E2 modulation of human papillomavirus type 16 P97 and type 18 P105 promoters. J. Virol. 64:2849–2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fujii T, Saito M, Iwasaki E, Ochiya T, Takei Y, Hayashi S, Ono A, Hirao N, Nakamura M, Kubushiro K, Tsukazaki K, Aoki D. 2006. Intratumor injection of small interfering RNA-targeting human papillomavirus 18 E6 and E7 successfully inhibits the growth of cervical cancer. Int. J. Oncol. 29:541–548. 10.3892/ijo.29.3.541 [DOI] [PubMed] [Google Scholar]
- 14. Hsu PD, Lander ES, Zhang F. 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278. 10.1016/j.cell.2014.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barrangou R, Marraffini LA. 2014. CRISPR-Cas systems: prokaryotes upgrade to adaptive immunity. Mol. Cell 54:234–244. 10.1016/j.molcel.2014.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, Wei W. 2014. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509:487–491. 10.1038/nature13166 [DOI] [PubMed] [Google Scholar]
- 19. Wang T, Wei JJ, Sabatini DM, Lander ES. 2014. Genetic screens in human cells using the CRISPR-Cas9 system. Science 343:80–84. 10.1126/science.1246981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84–87. 10.1126/science.1247005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. 2013. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154:1370–1379. 10.1016/j.cell.2013.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. 2013. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154:1380–1389. 10.1016/j.cell.2013.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Malim MH, Bohnlein S, Hauber J, Cullen BR. 1989. Functional dissection of the HIV-1 Rev trans-activator—derivation of a trans-dominant repressor of Rev function. Cell 58:205–214. 10.1016/0092-8674(89)90416-9 [DOI] [PubMed] [Google Scholar]
- 24. Zhang J, Jima DD, Jacobs C, Fischer R, Gottwein E, Huang G, Lugar PL, Lagoo AS, Rizzieri DA, Friedman DR, Weinberg JB, Lipsky PE, Dave SS. 2009. Patterns of microRNA expression characterize stages of human B-cell differentiation. Blood 113:4586–4594. 10.1182/blood-2008-09-178186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seedorf K, Oltersdorf T, Krammer G, Rowekamp W. 1987. Identification of early proteins of the human papilloma viruses type 16 (HPV 16) and type 18 (HPV 18) in cervical carcinoma cells. EMBO J. 6:139–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Logan GJ, Alexander IE. 2012. Adeno-associated virus vectors: immunobiology and potential use for immune modulation. Curr. Gene Ther. 12:333–343. 10.2174/156652312802083639 [DOI] [PubMed] [Google Scholar]
- 27. Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, Thomson JA. 2013. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. U. S. A. 110:15644–15649. 10.1073/pnas.1313587110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. 2013. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat. Methods 10:1116–1121. 10.1038/nmeth.2681 [DOI] [PMC free article] [PubMed] [Google Scholar]