

ABSTRACT
The type II transmembrane serine protease (TTSP) TMPRSS2 cleaves and activates the influenza virus and coronavirus surface proteins. Expression of TMPRSS2 is essential for the spread and pathogenesis of H1N1 influenza viruses in mice. In contrast, H3N2 viruses are less dependent on TMPRSS2 for viral amplification, suggesting that these viruses might employ other TTSPs for their activation. Here, we analyzed TTSPs, reported to be expressed in the respiratory system, for the ability to activate influenza viruses and coronaviruses. We found that MSPL and, to a lesser degree, DESC1 are expressed in human lung tissue and cleave and activate the spike proteins of the Middle East respiratory syndrome and severe acute respiratory syndrome coronaviruses for cell-cell and virus-cell fusion. In addition, we show that these proteases support the spread of all influenza virus subtypes previously pandemic in humans. In sum, we identified two host cell proteases that could promote the amplification of influenza viruses and emerging coronaviruses in humans and might constitute targets for antiviral intervention.
IMPORTANCE Activation of influenza viruses by host cell proteases is essential for viral infectivity and the enzymes responsible are potential targets for antiviral intervention. The present study demonstrates that two cellular serine proteases, DESC1 and MSPL, activate influenza viruses and emerging coronaviruses in cell culture and, because of their expression in human lung tissue, might promote viral spread in the infected host. Antiviral strategies aiming to prevent viral activation might thus need to encompass inhibitors targeting MSPL and DESC1.
INTRODUCTION
Influenza A viruses (FLUAVs) and the emerging pathogens severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) are respiratory viruses that pose a significant threat to human health. Annual influenza epidemics cause 250,000 to 500,000 deaths worldwide (1), and intermittent pandemics can have even more severe consequences, as exemplified by the devastating Spanish influenza of 1918, which is estimated to be responsible for 30 to 50 million deaths (2). The SARS-CoV emerged in southern China in 2002, and its subsequent spread, mainly in Asia, was responsible for 774 deaths (3). A related virus, the MERS-CoV, emerged in the Middle East in 2012 (4), and new cases of MERS-CoV infection continue to be reported in July 2014, with the total number of cases amounting to 834, of which 288 took a fatal course (5). Therefore, the development of novel strategies to combat FLUAV and emerging CoVs is urgently required, and host cell factors essential for viral spread but dispensable for cellular survival are attractive targets.
The viral hemagglutinin (HA) and spike (S) surface proteins are responsible for host cell entry of FLUAV (6) and CoVs (7, 8), respectively. Both proteins use their N-terminal surface units, termed HA1 (FLUAV) and S1 (CoVs), to engage cellular receptors, while the C-terminal transmembrane units, termed HA2 (FLUAV) and S2 (CoVs), facilitate fusion of the viral membrane with a host cell membrane—processes that are essential for infectious entry (6–8). Notably, the HA and S proteins are synthesized as inactive precursors in infected cells and acquire the ability to drive membrane fusion only upon activation by host cell proteases (9, 10). Activation refers to proteolytic separation of the surface and transmembrane units, which is essential for viral infectivity. Consequently, the proteases responsible for HA and S protein activation are potential therapeutic targets.
It has been suggested that several proteases secreted in the lung lumen can activate FLUAV (11–13). However, examination of cultured human respiratory epithelium revealed a key role for membrane-associated proteases (14), and work by Böttcher and colleagues identified the transmembrane serine proteases TMPRSS2 and HAT as potent activators of FLUAV (15), at least upon engineered expression in cell lines. Subsequent studies showed that endogenous expression of TMPRSS2 in cell lines can support trypsin-independent FLUAV spread (16, 17), and coexpression of TMPRSS2 and 2,6-linked sialic acid has been demonstrated in most parts of the human airways (18), suggesting that TMPRSS2 might support FLUAV spread in the infected host. Indeed, work by Hatesuer and colleagues demonstrated that knockout of Tmprss2 in mice largely abrogates the spread of FLUAV and prevents viral pathogenesis (19) and similar findings were subsequently reported by other groups (20, 21). Moreover, TMPRSS2 was shown to activate the SARS- (22–24) and MERS- (25, 26) CoVs for entry into target cells in which the activity of cathepsin L, another protease able to activate SARS-CoV S (27) and MERS-CoV S (25, 26), was blocked by inhibitors. Thus, FLUAV, highly pathogenic CoVs, and several other respiratory viruses (28–30) can be activated by TMPRSS2.
An essential role for TMPRSS2 in FLUAV spread and pathogenesis in mice has been demonstrated with viruses of the H1N1 and H7N9 subtypes (19–21). Dependence on TMPRSS2 was also reported for a virus of the H3N2 subtype (20). In contrast, a different H3N2 virus tested in a separate study was much less dependent on TMPRSS2 expression (19), suggesting that certain H3N2 viruses might be able to usurp another protease to ensure their activation, and members of the type II transmembrane serine protease (TTSP) family, which comprises more than 20 proteins (31), are interesting candidates. Here, we assessed seven TTSPs previously reported to be expressed in the lung for the ability to activate the surface proteins of FLUAV, MERS-CoV, and SARS-CoV. We show that MSPL and, to a lesser degree, DESC1 are expressed in the human lung and activate FLUAV and CoV surface glycoproteins, suggesting that these proteases could contribute to viral spread in the host.
MATERIALS AND METHODS
Plasmid construction.
Expression plasmids for SARS-CoV S (32), MERS-CoV S (25), FLUAV H1 HA (15, 33), ACE2 (34), and CD26 (DPP4) (25) have been described previously. Plasmids for H2 and H3 (15) HA of FLUAV were kindly provided by Mikhail Matrosovich, Institute of Virology, Marburg, Germany. Plasmids encoding the human transmembrane proteases TMPRSS2, HAT, TMPRSS3, TMPRSS9, and TMPRSS10 were also published earlier (16, 18, 35–37). The sequences encoding human DESC1, MSPL, TMPRSS11F, prostasin, and TMPRSS11B were amplified by reverse transcription (RT)-PCR with mRNA prepared from Caco2 (DESC1, MSPL), H1299 (TMPRSS11F), or LNCaP (prostasin, TMPRSS11B) cells as the template. The protease sequences were inserted into plasmid pCAGGS by using the appropriate restriction sites. The resulting plasmids encode human TTSPs with amino acid sequences identical to those deposited in the GenBank database, i.e., DESC1 (AF064819.1), MSPL (NM_001077263.2), TMPRSS11F (NM_207407.2), prostasin (NM_002773.3), and TMPRSS11B (NM_182502.3). Expression plasmids for TTSPs with an N-terminal myc tag were generated by PCR by using the above-described plasmids as templates as reported previously (38). The integrity of all PCR-amplified sequences was verified by automated sequence analysis.
Cell culture.
Human embryonal kidney 293T cells were propagated in Dulbecco's modified Eagle's medium (DMEM), and MDCK cells were grown in minimum essential medium. All media were supplemented with 10% fetal bovine serum, penicillin, and streptomycin. The cells were maintained in a humidified atmosphere containing 5% CO2.
Analysis of TTSP expression.
For analysis of TTSP expression by Western blotting, 293T cells were seeded into six-well plates at a density of 2.2 × 105/well, cultivated for 24 h, and then transfected with plasmids encoding proteases equipped with an N-terminal myc tag or transfected with an empty plasmid as a control. At 16 h posttransfection, the medium was replaced with fresh DMEM, and at 48 h posttransfection, the cells were washed with phosphate-buffered saline (PBS) and detached with 100 μl of 2× sodium dodecyl sulfate (SDS) loading buffer per well. All samples were denatured for 30 min at 95°C, separated by SDS-PAGE, and blotted onto a nitrocellulose membrane (Hartenstein). The proteins were detected by using a mouse anti-myc antibody (Biomol) as the primary antibody and a horseradish peroxidase (HRP)-coupled anti-mouse antibody (Dianova) as the secondary antibody. For analysis of TTSP expression by flow cytometry, 293T cells transfected with TTSP-encoding plasmids were detached, washed with PBS, incubated with ice-cold ethanol for 10 min, and then stained with a mouse anti-myc antibody (Biomol) diluted in 0.1% saponin. Mouse IgG1 (R&D Systems) was used as an isotype-matched control. After 30 min of incubation with primary antibodies at 4°C, cells were washed twice with PBS and incubated for 30 min at 4°C with DyLight 647-coupled anti-mouse secondary antibodies (Dianova) diluted in 0.1% saponin. After two final washing steps, cells were fixed with 2% paraformaldehyde and staining was analyzed with an LSR II flow cytometer (BD Biosciences).
TaqMan-based quantitative RT-PCR analysis of protease expression in the lung.
Two pools of commercially available RNA (Agilent Technologies), each prepared from normal human lung tissue obtained from four male donors, were used to quantify protease transcripts. Total RNA (1 μg) was reverse transcribed with 50 U of BioScript RNase H Low reverse transcriptase (BIO-27036; Bioline) in 20-μl reaction mixtures. The enzyme was finally inactivated for 10 min at 70°C. cDNA aliquots (1 μl) corresponding to 50 ng total RNA equivalents were used for real-time PCR in triplicate 10-μl reaction mixtures with the ABI 7500 Fast real-time PCR system (Applied Biosystems). Specific amplification was ensured by using TaqMan gene expression assays according to the manufacturer's recommendations. The specific assays used were Hs00237175_m1 (TMPRSS2), Hs01070171_m1 (DESC1 = TMPRSS11E), Hs00361060_m1 (MSPL = TMPRSS13), and Hs99999908_m1 (GUSB). The average cycle threshold (CT) for each individual assay was calculated from triplicate measurements by means of the instrument's software in “auto CT” mode (ABI 7500 Fast system software, version 1.3.0). Average CT values calculated for TMPRSS2, DESC1, and MSPL were normalized by subtraction from the CT values obtained for GUSB (housekeeping reference). Template-free cDNA reaction mixtures prepared in triplicate were analyzed in parallel with all of the TaqMan assays, and no specific signal was detected in any of these negative controls.
Cleavage of SARS-CoV S, MERS-CoV S, and influenza virus HA by TTSPs.
For the detection of SARS-CoV S, MERS-CoV S, and FLUAV HA cleavage by TTSPs, 293T cells were seeded into six-well plates at a density of 2.2 × 105/well and cotransfected with an expression plasmid for SARS-CoV S with a C-terminal V5 tag, MERS-CoV S with a C-terminal V5 tag, or FLUAV HA of the H1, H2, or H3 subtype and either plasmids encoding the specified proteases or an empty plasmid. The medium was replaced with fresh DMEM at 8 to 16 h posttransfection. At 48 h posttransfection, the cells were harvested in 1 ml of PBS, treated with PBS or tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin (Sigma) (SARS-CoV S-positive cells were treated with 100 μg/ml TPCK-trypsin, while MERS-CoV S- and HA-expressing cells were treated with 250 μg/ml TPCK-trypsin) for 10 min at room temperature and lysed in SDS loading buffer. The lysates were separated by SDS-PAGE and blotted onto nitrocellulose membranes. The SARS- and MERS-CoV S proteins with a C-terminal V5 antigenic tag (25, 32) were detected by staining with mouse monoclonal antibody specific for the V5 tag (Invitrogen), followed by incubation with an HRP-coupled anti-mouse secondary antibody (Dianova), respectively. The expression of FLUAV HA of the H1 subtype was detected by staining with a mouse monoclonal antibody reactive against the HA of the 1918 H1N1 FLUAV (39), followed by incubation with an HRP-coupled anti-mouse secondary antibody (Dianova). The expression of FLUAV HA of the H2 subtype was detected with a goat anti-FLUAV polyclonal antibody (Millipore) and an HRP-coupled anti-goat antibody (Dianova). For detection of expression of FLUAV HA of the H3 subtype, a rabbit anti-H3 HA serum (Immune Technology) and an HRP-coupled anti-rabbit antibody were used. As a loading control, expression of β-actin was detected with an anti-β-actin antibody (Sigma). Staining was detected with the ECL Prime Western blotting detection reagent (Amersham) as specified by the manufacturer and with a ChemoCam Imager (Intas).
Analysis of proteolytic activation of the SARS- and MERS-CoV S proteins for cell-cell fusion.
For analysis of SARS- and MERS-CoV S activation for cell-cell fusion, a previously described cell-cell fusion assay was used (40). In brief, 293T effector cells, seeded into six-well plates at 1.3 × 105/well, were calcium phosphate transfected with either the empty pCAGGS plasmid or pCAGGS encoding SARS- or MERS-CoV S in combination with plasmid pGAL4-VP16, which encodes the herpes simplex virus VP16 transactivator fused to the DNA binding domain of the yeast transcription factor GAL4. In parallel, 293T target cells were seeded into 48-well plates at 0.8 × 105/well and transfected with the empty pcDNA3 plasmid or the expression plasmids for ACE2 or CD26 jointly with protease expression plasmids and plasmid pGal5-luc, which carries the firefly luciferase reporter gene under the control of a promoter containing five GAL4 binding sites. The day after transfection, effector cells were detached, diluted in fresh medium, and added to the target cells. For trypsin treatment, medium from target cells was completely removed and effector cells in medium supplemented with 100 ng/ml TPCK-treated trypsin (Sigma) or PBS were added. After 6 h of incubation time, the culture media were replaced with fresh medium without trypsin. Cell-cell fusion was quantified by determination of luciferase activities in cell lysates at 48 h after cocultivation with a commercially available kit (Promega).
Production of lentiviral pseudotypes and transduction experiments.
The generation of lentiviral vectors was carried out as described previously (16, 25, 41). In brief, 293T cells were transiently cotransfected with pNL4-3-LucR−E− (42) and expression plasmids for SARS-CoV S, MERS-CoV S, HA and neuraminidase (NA) of the 1918 H1N1 FLUAV, or vesicular stomatitis virus glycoprotein (VSV-G) or the empty plasmid pCAGGS. For analysis of HA activation by TTSPs, the proteases analyzed were coexpressed with the viral components specified above during the generation of lentiviral pseudotypes. The culture medium was replaced at 16 h posttransfection, and supernatants were harvested at 48 h posttransfection. The supernatants were passed through 0.45-μm filters, aliquoted, and stored at −80°C. For analysis of MERS- and SARS-CoV S-mediated transduction, 293T cells were cotransfected with expression plasmids for ACE2 or CD26 and the TTSPs indicated or an empty plasmid. The culture medium was replaced with fresh medium at 8 h posttransfection, and the cells were seeded into 96-well plates at 24 h posttransfection. At 48 h posttransfection, cells were preincubated with dimethyl sulfoxide (DMSO) or 10 μM cathepsin inhibitor MDL 28170 (Calbiochem) for 1 h and then incubated with equal volumes of infectivity-normalized pseudotypes for 8 h. Thereafter, the medium was changed and the luciferase activities in cell lysates were determined with a commercially available kit (Promega, Madison, WI) at 72 h postinfection. For the analysis of HA-mediated transduction, 293T cells were either directly seeded into 96-well plates at a density of 0.8 × 104/well or first transfected with protease-encoding plasmids and then seeded into 96-well plates. Subsequently, the cells were incubated with equal volumes of infectivity-normalized vector preparations pretreated with PBS or TPCK-trypsin. At 12 h posttransduction, fresh medium was added, and at 72 h posttransduction, the cells were lysed and the luciferase activities in cell lysates were determined.
Production of influenza viruses and infection experiments.
A/PR/8/34 (H1N1) (19) and A/Panama/2007/99 (H3N2) (43) were propagated in the chorioallantoic cavities of 10-day-old embryonated hen eggs (Harlan Winkelman, Germany) for 48 h at 37°C. A/Panama/2007/99 (H3N2) was reconstituted from an eight-plasmid system (43) before amplification in eggs. For infection experiments, 293T cells were seeded into six-well plates at a density of 2.8 × 105/well. After 24 h, the cells were transfected with 6 μg of expression plasmids encoding the indicated TTSPs or an empty plasmid by the calcium phosphate transfection method. The transfection medium was replaced with fresh medium after overnight incubation. At 24 h posttransfection, the culture medium was removed and the cells were incubated with infection medium (DMEM supplemented with 0.2% bovine serum albumin [BSA]) containing A/PR/8/34 (H1N1) at a multiplicity of infection (MOI) of 0.01 or A/Panama/2007/99 (H3N2) at an MOI of 0.1. After 1 h of incubation at 37°C, the infection medium was removed, the cells were gently washed with PBS, and fresh infection medium (again without trypsin) was added. Culture supernatants were collected at 48 h postinfection. The amount of infectious units within the culture supernatants was determined by focus formation assay as described previously by us (44). In brief, serial 10-fold dilutions of samples were prepared in infection medium (DMEM with 1% penicillin-streptomycin and 0.1% BSA) and added to MDCK cells. After 1 h of incubation, the medium was replaced with infection medium containing an Avicel overlay and 2.5 μg/ml N-acetylated trypsin (Sigma), and the cells were incubated for 24 h. Subsequently, the cells were fixed with 4% formalin in PBS and incubated for 1 h with a goat anti-influenza virus nucleocapsid NP polyclonal antibody (Virostat), followed by 1 h of incubation with HRP-conjugated anti-goat antibodies (Dianova) and 10 min incubation with True Blue substrate (KPL). Foci were counted, and viral titers were calculated as numbers of focus-forming units per milliliter of culture supernatant.
Analysis of HA and TTSP colocalization.
To determine the cellular localization of FLUAV HA and TTSPs, we seeded COS-7 cells onto coverslips in six-well plates at a density of 0.6 × 105/well. The cells were calcium phosphate transfected with plasmids encoding TMPRSS2, TMPRSS3, DESC1, and MSPL, all containing an N-terminal myc antigenic tag. Cells transfected with an empty plasmid served as a negative control. At 16 h posttransfection, the medium was replaced with fresh DMEM, and at 24 h posttransfection, the cells were washed with PBS and infected with FLUAV A/PR/8/34 at an MOI of 1. At 1 h postinfection, the medium was replaced with DMEM supplemented with 1 μg/ml TPCK-trypsin. At 24 h postinfection, the cells were treated with ice-cold methanol (7.5 min at 4°C), blocked with 3% BSA for 1 h, and then incubated with mouse anti-myc and rabbit anti-PR8 HA antibodies (Biomol and Sino Biological Inc., respectively). After 1 h of incubation with primary antibodies at room temperature, cells were washed three times with PBS and incubated for 1 h at room temperature with Rhodamine Red-X-coupled anti-mouse and fluorescein isothiocyanate (FITC)-coupled anti-rabbit secondary antibodies (Dianova). After three final washing steps, cells were treated with Vectashield 4′,6-diamidino-2-phenylindole mounting medium (Vector Laboratories) and analyzed with a Zeiss LSM 5 laser scanning microscope. Image capture was performed with Pascal Software (Zeiss), and further image analysis, including calculation of the Pearson correlation coefficient (PCC), was done with ImageJ with Just Another Colocalization Plugin (45).
RESULTS
TTSPs are expressed in transfected 293T cells.
For all known human TTSPs, expression in the lung has been reported but expression levels differ profoundly (31) and for many of these enzymes it is unknown if they can cleave and activate viral glycoproteins. In order to address this question, we first amplified the sequences of seven TTSPs not previously characterized in the context of virus infection and then examined the expression of the cloned protease sequences in transiently transfected 293T cells. For this, the protease sequences were fused to an N-terminal myc tag and protease expression was detected by Western blotting (Fig. 1A). The expression of most proteases was readily detectable, with the expression of TMPRSS2 and MSPL being particularly prominent and the expression of HAT and TMPRSS11F being relatively weak. In contrast, signals obtained for prostasin-expressing cells were close to (data not shown) or within the background range. The molecular weights of the proteases studied roughly matched the predicted ones, and bands expected upon autocatalytic activation were seen for all of the proteases except TMPRSS9 (a faint signal was consistently observed for TMPRSS11B).
FIG 1.
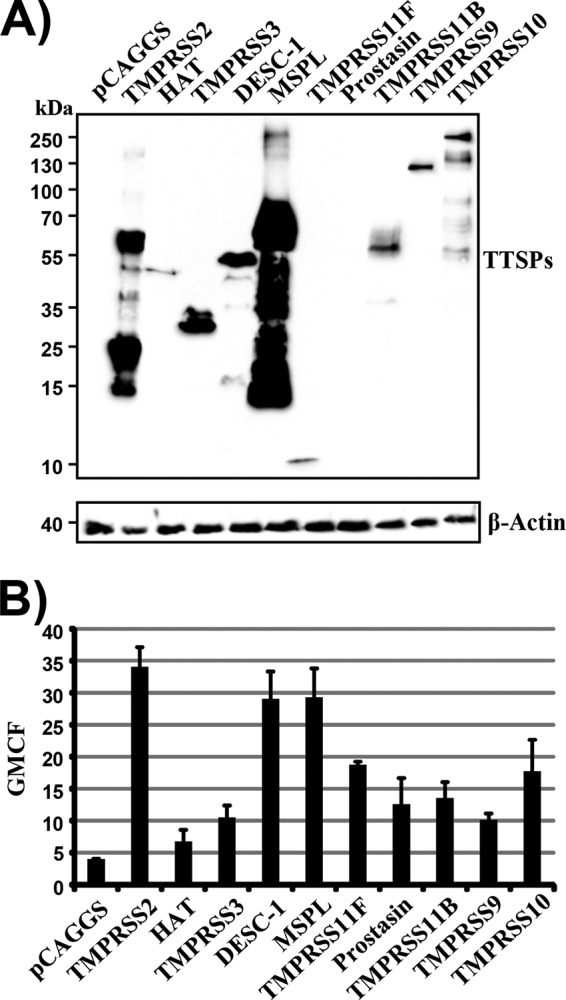
Expression of TTSPs in 293T cells. (A) Plasmids encoding the indicated proteases equipped with an N-terminal myc tag were transiently transfected into 293T cells. Transfection of an empty plasmid (pCAGGS) served as a negative control. Protease expression in cell lysates was detected by Western blotting with an anti-myc antibody. Detection of β-actin served as a loading control. Similar results were obtained in three separate experiments. (B) The experiment was conducted as described for panel A, but TTSP expression was detected by FACS. The geometric mean channel fluorescence (GMCF) measured in a representative experiment performed with triplicate samples is shown. Error bars indicate standard deviations. Similar results were obtained in three independent experiments.
In order to obtain further information on TTSP expression, we also analyzed TTSP levels by fluorescence-activated cell sorting (FACS) of stained permeabilized cells, which allows the detection of intracellular and surface-expressed TTSPs. Again, prominent signals were measured in TMPRSS2- and MSPL-expressing cells (Fig. 1B) and similar expression of DESC1 was noted. Specific and roughly comparable signals were measured in the cells expressing the remaining proteases, indicating that all of the proteases examined were produced in transiently transfected cells, although at different levels.
The TTSPs DESC1 and MSPL cleave the S proteins of the SARS- and MERS-CoVs.
We first assessed if the TTSPs studied were able to cleave the S proteins of the emerging SARS- and MERS-CoVs. Cleavage of MERS-CoV S was readily detected upon the coexpression of TMPRSS2 and HAT or upon the treatment of S-protein-expressing cells with trypsin (Fig. 2A), as expected (22–25, 35). Proteolysis of MERS-CoV S was also observed upon the coexpression of DESC1, MSPL, and TMPRSS10 (Fig. 2A), although differences in the size and number of cleavage fragments were observed. Similarly, cleavage of SARS-CoV S was observed in cells coexpressing DESC1 and MSPL (Fig. 2B). In contrast, TMPRSS10 did not facilitate the cleavage of SARS-CoV S. Thus, DESC1 and MSPL can process the MERS- and SARS-CoV S proteins while TMPRSS10 processes exclusively MERS-CoV S.
FIG 2.
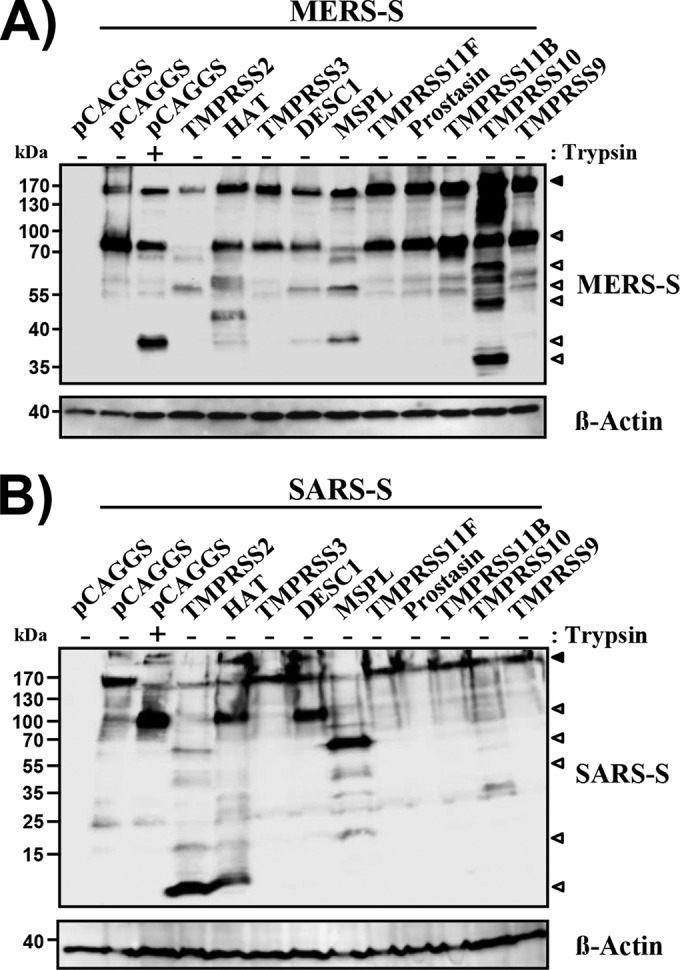
DESC1 and MSPL cleave the MERS- and SARS-CoV S proteins. 293T cells were cotransfected with plasmids encoding MERS- (A) or SARS- (B) CoV S with a C-terminal V5 tag and plasmids encoding the proteases indicated. Transfection of empty plasmid pCAGGS served as a negative control. At 48 h posttransfection, cells were either left untreated or treated with trypsin and lysates were analyzed by Western blotting with a V5-specific antibody. Expression of β-actin in cell lysates was determined as a loading control. The results are representative of three independent experiments with different plasmid preparations. Black-filled arrowheads, uncleaved S protein; gray-filled arrowheads, S2 subunit; white-filled arrowheads, C-terminal cleavage fragments.
DESC1 and MSPL activate the S proteins of the SARS- and MERS-CoVs.
In order to examine whether S-protein cleavage results in activation, we assessed the impact of protease expression on S-protein-driven cell-cell fusion by using a previously reported cell-cell fusion assay (40). Expression of MERS-CoV S in effector cells allowed fusion with target cells transfected with a CD26-encoding plasmid but not a control plasmid (Fig. 3A), as expected. Notably, the directed expression of CD26 on target cells was not required for robust MERS-CoV S-driven cell-cell fusion when the effector and target cell mixture was treated with trypsin (Fig. 3A), in keeping with the finding that 293T cells express small amounts of endogenous CD26 (46). These observations indicate that both receptor and protease expression levels limit MERS-CoV S-driven cell-cell fusion, a scenario similar to that previously reported for SARS-CoV S-mediated cell-cell fusion (47). Importantly, expression of TMPRSS2, HAT, DESC1, and MSPL activated MERS-CoV S with the same efficiency as trypsin (Fig. 3A), indicating that cleavage of MERS-CoV S by these proteases results in S protein activation. In contrast, TMPRSS10 expression failed to activate MERS-CoV S (Fig. 3A). Similar results were obtained upon the analysis of SARS-CoV S-driven cell-cell fusion, which was promoted by the expression of TMPRSS2, HAT, DESC1, and MSPL but not TMPRSS10 (Fig. 3B). Thus, DESC1 and MSPL can activate the MERS- and SARS-CoV S proteins and could contribute to syncytium formation in infected patients.
FIG 3.
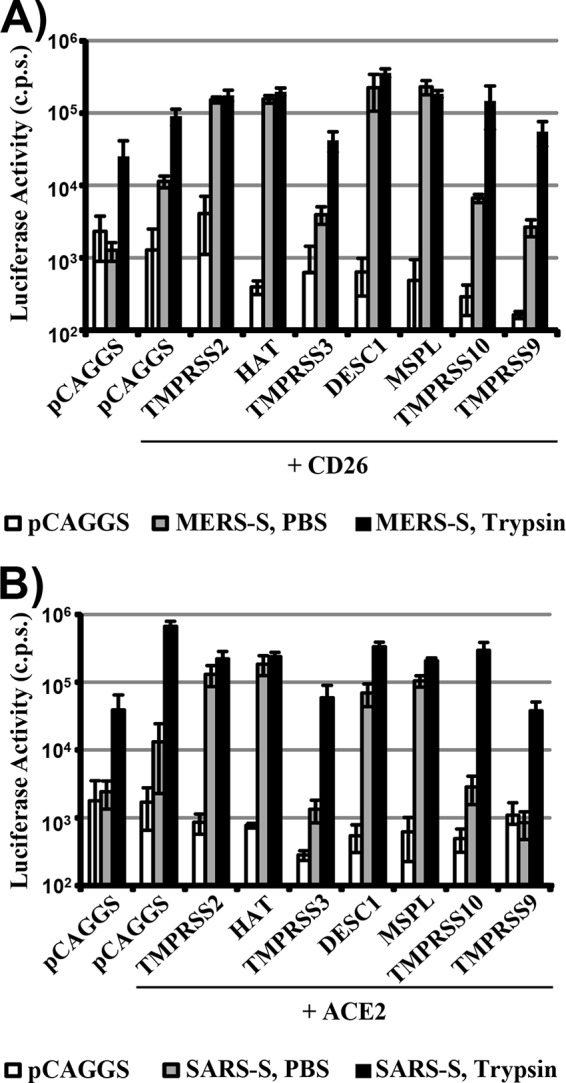
DESC1 and MSPL activate the MERS- and SARS-CoV S proteins for cell-cell fusion. For the analysis of S protein-driven cell-cell fusion, effector 293T cells were cotransfected with the pGAL4-VP16 expression plasmid and either the empty plasmid or a MERS- (A) or SARS- (B) CoV S expression plasmid. Subsequently, the effector cells were mixed with 293T target cells transfected with plasmids encoding the indicated receptors and proteases and a plasmid encoding luciferase under the control of a promoter with multiple GAL4 binding sites. The cell mixtures were then treated with either PBS or trypsin, and the luciferase activities in cell lysates were quantified at 48 h after cell mixing. The results of a representative experiment performed with triplicate samples are shown. Error bars indicate standard deviations. Similar results were observed in two independent experiments. c.p.s., counts per second.
The cell-cell fusion assay allows the interaction of large cell surfaces on which large amounts of receptor and protease are expressed. Therefore, the assay is highly sensitive but might not fully mirror S-protein-driven virus-cell fusion. To assess whether the TTSPs studied can activate virion-associated S proteins, we analyzed whether TTSP expression rescues S-protein-driven virus-cell fusion from blockade by an inhibitor of cathepsin B/L, proteases known to activate the MERS- and SARS-CoV S proteins for host cell entry (25–27). For this, we used lentiviral vectors pseudotyped with the SARS- and MERS-CoV S proteins, as previously described (22, 25). We found that incubation of 293T target cells with the cathepsin B/L inhibitor MDL 28170 reduced both MERS-CoV S- (Fig. 4A) and SARS-CoV S-driven virus-cell fusion (Fig. 4B), as expected (25, 27). Expression of TMPRSS2 fully rescued MERS-CoV S- and SARS-CoV S-dependent virus-cell fusion from inhibition by MDL 28170, again in keeping with previously published results (22–25). In addition, MERS-CoV S-driven transduction was rescued by the expression of HAT, DESC1, and MSPL while similar effects were not measured for SARS-CoV S-driven transduction. Collectively, these results indicate that DESC1 and MSPL can activate the MERS- and SARS-CoV S proteins for cell-cell fusion and could thus contribute to syncytium formation in patients. In addition, DESC1 and MSPL can activate MERS-CoV S for virus-cell fusion and may facilitate viral spreading in infected tissues. Why these proteases fail to activate SARS-CoV S for virus-cell fusion, at least in the system tested here, is unclear but most likely reflects a need for higher protease expression levels for SARS-CoV S activation than for MERS-CoV S activation.
FIG 4.
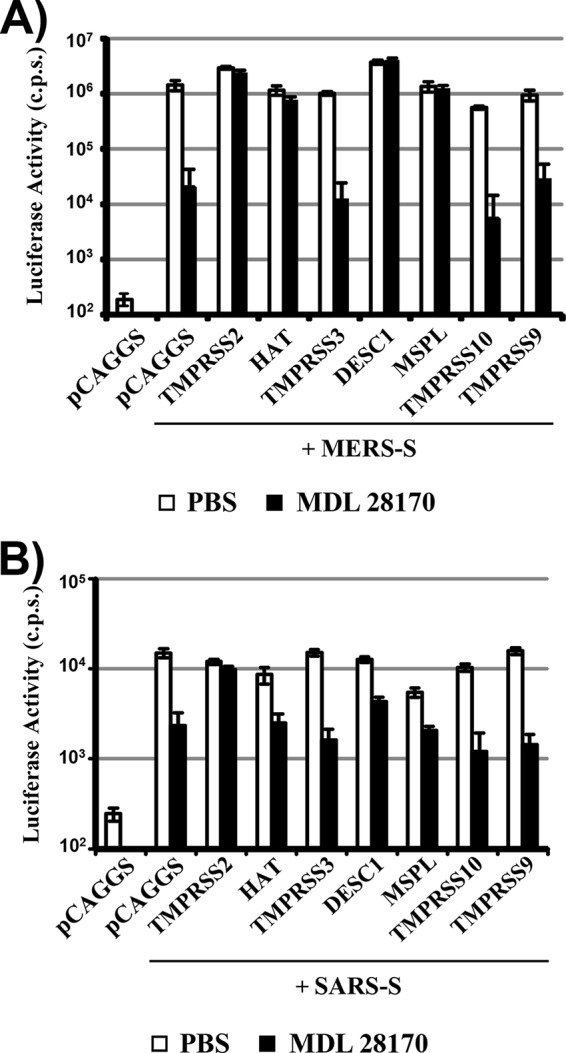
DESC1 and MSPL activate MERS-CoV S for virus-cell fusion. For analysis of S protein-driven virus-cell fusion, the proteases indicated were coexpressed in 293T cells with CD26 (for MERS-CoV S-driven transduction, panel A) or ACE2 (for SARS-CoV S-driven transduction, panel B) and the cells were pretreated with medium containing DMSO or 10 μM cathepsin B/L inhibitor MDL 28170. Subsequently, the cells were transduced with pseudotypes bearing MERS- (A) or SARS- (B) CoV S in the absence or presence of the inhibitor. The infection medium was replaced with fresh medium without inhibitor at 8 h posttransduction, and the luciferase activities in cell lysates were analyzed at 72 h posttransduction. The results of a representative experiment performed with triplicate samples are shown; error bars indicate standard deviations. Similar results were obtained in two to three independent experiments. c.p.s., counts per second.
DESC1 and MSPL cleave influenza virus HA of the H1, H2, and H3 subtypes and colocalize with HA in infected cells.
In order to determine whether DESC1 and MSPL can activate FLUAV, we assessed their ability to cleave the HA proteins of viruses previously pandemic in humans. For this, the proteases were coexpressed with the HA proteins of A/South Carolina/1/1918 (H1N1), A/Singapore/1/57 (H2N2), and A/Hong Kong/1/1968 (H3N2) in transfected 293T cells. Cleavage of the HA precursor protein HA0 (as indicated by production of the N-terminal cleavage product HA1) was observed upon the coexpression of TMPRSS2 and HAT (Fig. 5), in keeping with previously published data (15). In addition, the HA1 processing product was observed upon the coexpression of DESC1 and MSPL (Fig. 5). In contrast, none of the other proteases tested facilitated HA cleavage.
FIG 5.
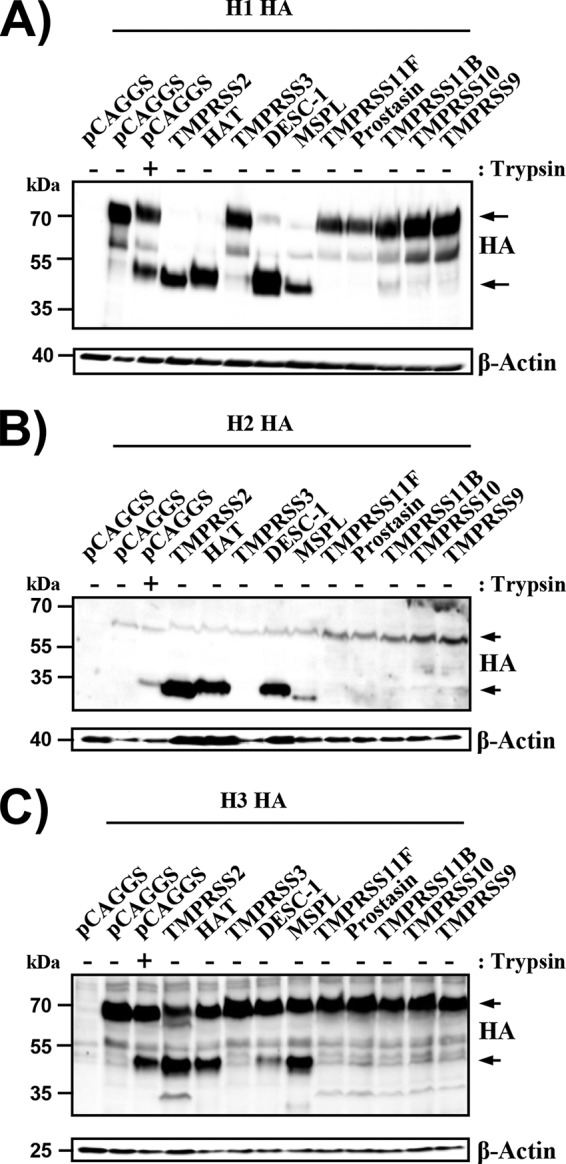
DESC1 and MSPL cleave the FLUAV HA. Expression plasmids encoding influenza virus HA subtypes H1 (A), H2 (B), and H3 (C) and the indicated proteases or empty plasmid pCAGGS were transiently cotransfected into 293T cells. The cells were then treated with PBS or trypsin, and HA cleavage was detected by Western blotting of cell lysates. Similar results were obtained in three independent experiments. The HA0 (upper arrow) and HA1 (lower arrow) subunits are indicated.
In order to further determine whether DESC1 and MSPL can activate FLUAV, we next investigated whether these proteases colocalize with HA in infected cells. Immunofluorescence staining and visual inspection of COS-7 cells transfected to express TTSPs and infected with FLUAV revealed extensive colocalization of HA with TMPRSS2, DESC1, or MSPL (Fig. 6A). In contrast, TMPRSS3 and HA barely colocalized, although the signals were close. This assessment was confirmed upon determination of the PCC for the HA and protease signals (PCC of 1, perfect positive correlation; PCC of −1, perfect negative correlation). The HA signals correlated well with those of TMPRSS2, DESC1, and MSPL, indicating extensive colocalization, while little correlation was measured for the HA and TMPRSS3 signals (Fig. 6B). Thus, the cellular localizations of HA and DESC1 or MSPL overlap extensively, suggesting that these proteases could cleave HA in infected cells.
FIG 6.
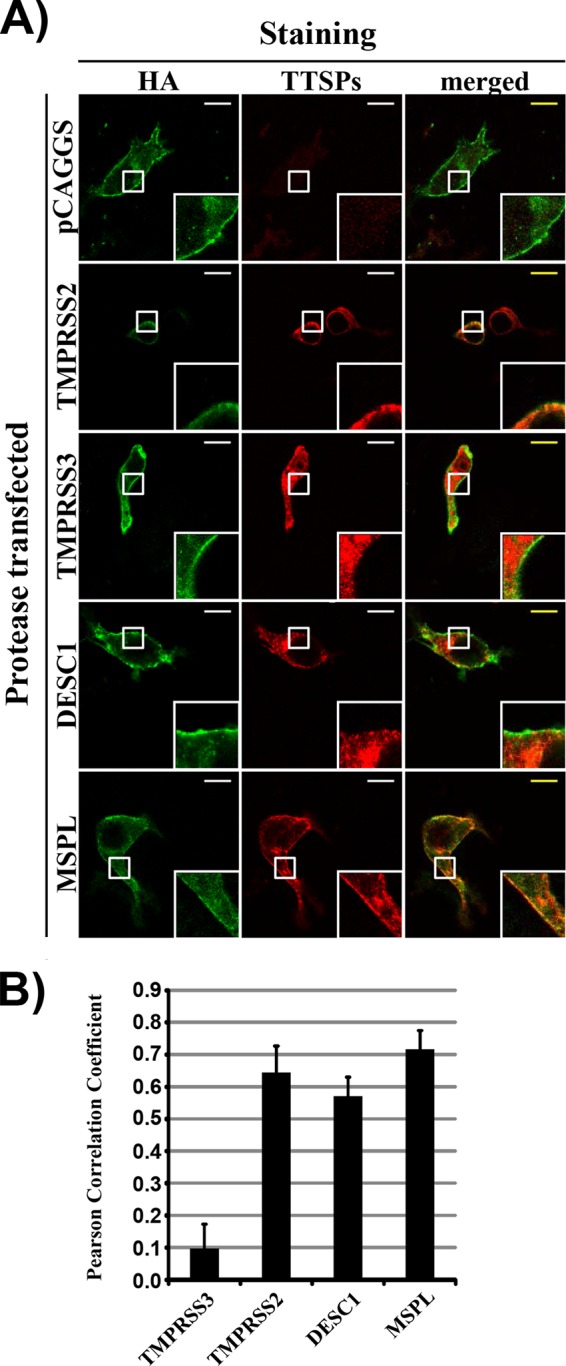
Analysis of protease and HA colocalization. (A) COS7 cells were transfected with plasmids encoding the indicated TTSPs or transfected with an empty plasmid (control). Subsequently, the cells were infected with FLUAV A/PR/8/34 (H1N1) at an MOI of 1. At 24 h postinfection, the cells were fixed with ice-cold methanol and influenza virus HA was detected by immunostaining with rabbit-anti-HA and FITC-conjugated anti-rabbit antibodies (green). In parallel, proteases were detected with mouse anti-myc and Rhodamine Red-X-conjugated anti-mouse antibodies (red signal). White squares indicate examples of colocalization of HA and TTSPs (yellow signal) that were magnified ×4. Similar results were obtained in three separate experiments. (B) The experiment was conducted as described for panel A, and colocalization of TTSPs and HA was determined by analyzing images with ImageJ software in combination with Just Another Colocalization Plugin, which allows calculation of the PCC, a measure of colocalization. The average PCC measured for three to five cells from separate experiments is shown, error bars indicate the standard errors of the means.
DESC1 and MSPL activate the HA proteins of influenza viruses.
Cleavage of a viral glycoprotein must not necessarily result in activation, as observed for MERS-CoV S cleavage by TMPRSS10 (Fig. 2 to 4). We therefore determined whether DESC1 and MSPL expression facilitates HA activation. First, we assessed if these proteases activate HA in the context of a lentiviral vector system. For this, a plasmid encoding the env-deficient lentiviral vector, FLUAV HA and NA, or the indicated proteases or an empty plasmid was transfected into 293T cells and the supernatants were analyzed for the transduction of target cells. All HA-bearing vectors were able to efficiently and comparably transduce target cells upon treatment with trypsin, while trypsin treatment did not affect transduction mediated by VSV-G, as expected (33). Similarly, expression of TMPRSS2 and HAT rendered pseudotypes infectious, again in keeping with previously published data (16, 33), and the same effects were observed upon the expression of DESC1 and MSPL (Fig. 7A), indicating that these proteases can activate HA when expressed in virus-producing cells. In contrast, no activation of HA was observed when the proteases studied were expressed in viral target cells (data not shown), suggesting that DESC1 and MSPL fail to activate HA during viral entry. To address the activation of authentic FLUAV, we next examined the spreading of A/PR/8/34 and A/Panama/2007/99 in 293T cells transfected to express the proteases under study. In the absence of protease expression, viral spread was dependent on the presence of trypsin in the culture medium (Fig. 7B). If TMPRSS2, HAT, DESC1, and MSPL were produced in the target cells, efficient viral spread was measured (Fig. 7B), indicating that these proteases can activate HA in the context of infection with authentic FLUAV.
FIG 7.
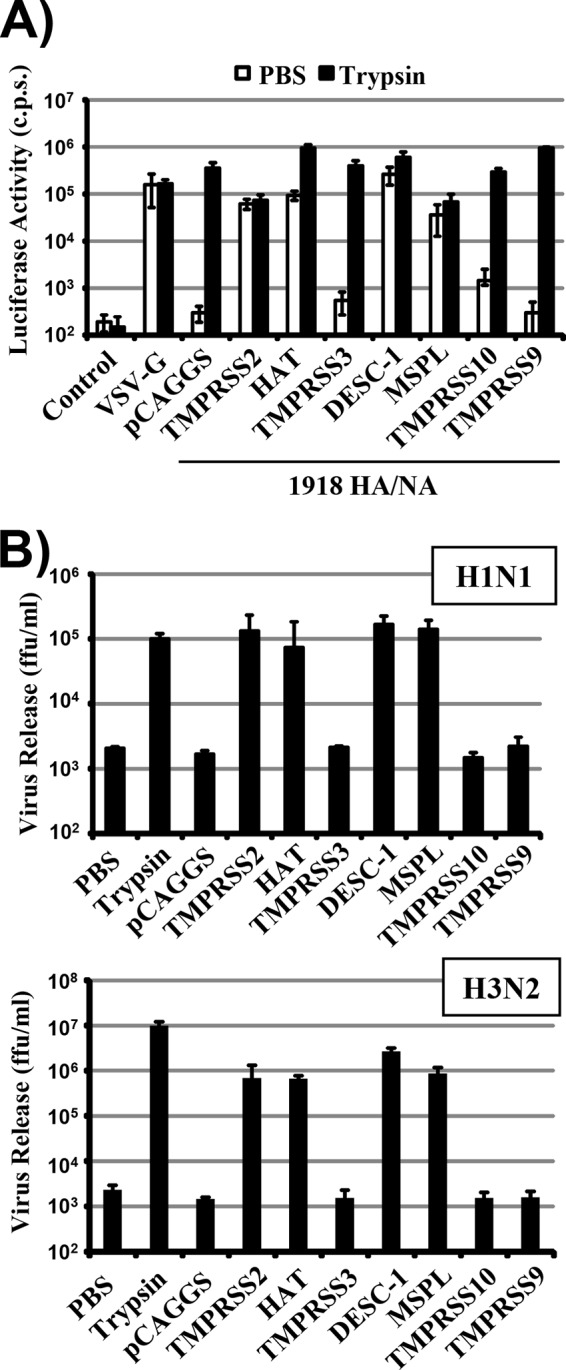
DESC1 and MSPL activate the FLUAV HA. (A) Lentiviral vectors bearing the HA and NA proteins of the H1N1 1918 influenza virus (1918 HA/NA) or VSV-G were generated in 293T cells coexpressing the proteases indicated or transfected with empty plasmid pCAGGS. The culture supernatants were harvested, treated with PBS or trypsin, and used for transduction of 293T target cells. Luciferase activities in the cell lysates were determined at 72 h posttransduction. The results of a representative experiment performed with triplicate samples are shown and were confirmed in two independent experiments. Error bars indicate standard deviations. (B) The proteases indicated were transiently expressed in 293T cells, and the cells were infected with FLUAV A/PR/8/34 (H1N1) at an MOI of 0.01 (top) or FLUAV A/Panama/2007/1999 (H3N2) at an MOI of 0.1 (bottom) and treated with either trypsin or PBS. At 48 h postinfection, viral spread was quantified as the release of infectious particles into the culture supernatants, as measured by a focus formation assay. The results of representative experiments performed with triplicate samples are shown. Error bars indicate standard deviations. Similar results were obtained in two separate experiments. c.p.s., counts per second; ffu, focus-forming units.
DESC1 and MSPL are expressed in the human lung.
Activation of FLUAV, SARS-CoV, and MERS-CoV by DESC1 and MSPL in infected patients requires these proteases to be expressed in the human lung, the major viral target organ. In order to determine the expression of these proteases, we performed a quantitative RT-PCR analysis of two pools of RNA, each generated from normal lung tissue of four human male donors. TMPRSS2, MSPL, and DESC1 transcripts were detected in both pools although in different amounts. The expression of TMPRSS2 transcripts was most robust, but MSPL transcripts were also readily detectable. In contrast, expression of DESC1 mRNA was very low and thus might be confined to a limited subset of lung cells (Fig. 8). These results suggest that TMPRSS2, MSPL, and, to a lower degree, DESC1 are expressed in the lung and could thus facilitate the pulmonary spread of FLUAV and CoVs in human patients.
FIG 8.
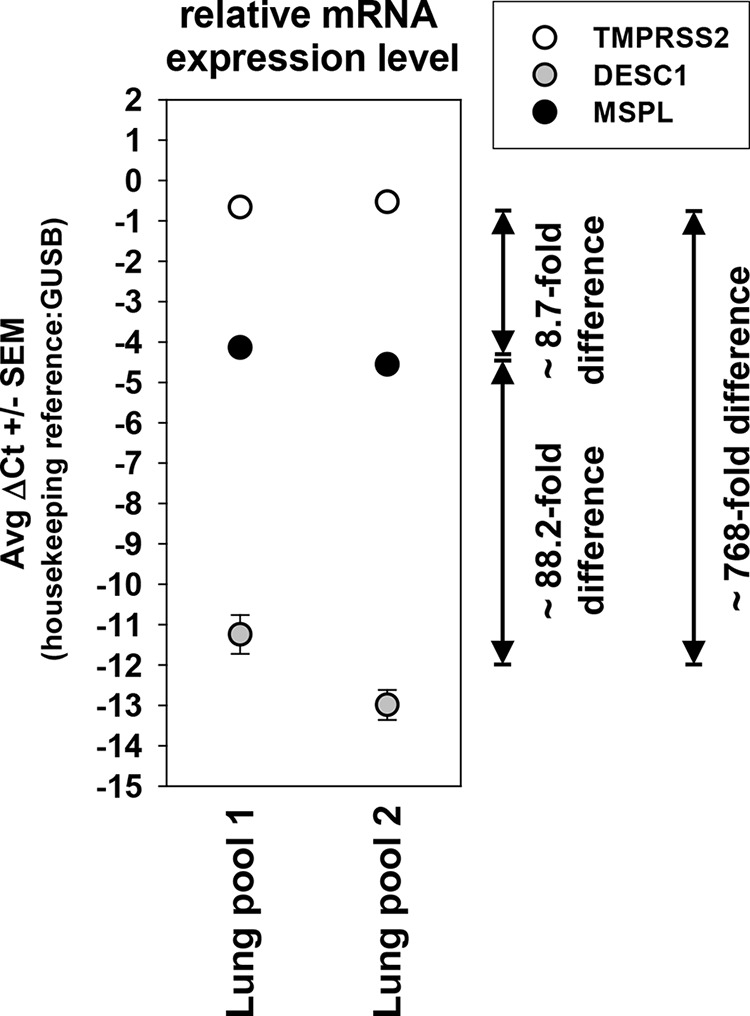
DESC1 and MSPL transcripts are expressed in the human lung. The presence of TMPRSS2, DESC1, and MSPL transcripts in two RNA pools each prepared from normal lung tissue of four human donors was assessed by quantitative PCR. Average ΔCT values from triplicate measurements and the standard errors of the means are shown. The relative difference in the abundance of protease transcripts was estimated and is depicted as fold difference (an estimated 1.78-fold increase in amplified material per PCR cycle formed the basis for this calculation). Similar results were obtained in a separate experiment.
DISCUSSION
FLUAVs and emerging CoVs pose a significant threat to public health, and novel strategies for antiviral intervention are called for. TMPRSS2, a TTSP, is a potential target, since this enzyme activates FLUAV (15, 33) and CoVs (22–24, 26, 28, 29) in cell culture and was recently shown to be essential for FLUAV (H1N1, H7N9) spread and pathogenesis in mice (19–21). In contrast, the absence of TMPRSS2 had a much less profound effect in the context of infection with an H3N2 virus (19), suggesting that certain FLUAVs might employ TTSPs other than TMPRSS2 to ensure their activation in the host. Here, we show that DESC1 and MSPL are potential candidates, since these proteases are expressed in the human lung and activate the HA proteins of all of the FLUAV subtypes previously pandemic in humans. In addition, we demonstrate that DESC1 and MSPL activate the emerging MERS-CoV and might thus also be exploited by this highly pathogenic agent for spread in the infected host.
It was observed in the early 1970s that the FLUAV HA is cleaved by host cell proteases and that cleavage is essential for viral infectivity (48–50). Subsequently, a variety of secreted proteases that cleave and activate HA in cell culture was identified (12, 13, 51, 52). These findings suggested that FLUAV might exploit multiple proteolytic systems present in the lung lumen to ensure its activation in the host. In contrast, recent studies indicate that FLUAV activation is cell associated (14) and mediated by TTSPs (15), with a single enzyme, TMPRSS2, being essential for the spread of the H1N1 and H7N9 FLUAV subtypes (19–21), while certain H3N2 viruses show little TMPRSS2 dependence. In fact, a recent study reported that an HA of the H3 subtype derived from avian FLUAV was fully resistant to cleavage by TMPRSS2 and HAT (53). The subtype or strain specificity of TMPRSS2 dependence is perhaps not unexpected, given the known differences in sequence and spatial presentation of the respective cleavage sites (54), and suggests that some viruses might use TTSPs other than TMPRSS2 to ensure their activation. In order to identify such enzymes, we focused our analysis on TTSPs known to be expressed in the murine and/or human lung, which comprise DESC1, MSPL, TMPRSS11F, prostasin, TMPRSS11B, TMPRSS9, and TMPRSS10 (31).
All of the TTSPs studied were expressed in transfected 293T cells, although substantial differences in expression efficiency were noted. Therefore, the possibility cannot be excluded that proteases found to be inactive in our study are able to activate viral glycoproteins when expressed at higher levels. TTSPs are synthesized as zymogens, and several can transit into an active form upon autocatalytic activation (31, 55). Activation of TTSPs results in the production of an N-terminal cleavage fragment (linked to the C-terminal fragment via a disulfide bond), which was amenable to detection in our analysis because of the presence of an N-terminal myc antigenic tag. This fragment was detected upon the expression of most of the proteases studied, indicating that the failure of certain proteases to activate viral glycoproteins was not due to a failure of these enzymes to transit into their catalytically active forms. The only exceptions were TMPRSS9, for which no evidence of activation was observed. Whether the absence of N-terminal cleavage products of TMPRSS9 in protease-expressing cells was due to instability of the respective fragments or to a genuine lack of protease activation remains to be determined.
TMPRSS2 was previously shown to activate the SARS- and MERS-CoV S proteins for cathepsin B/L-independent host cell entry (22–26), presumably by cleaving the S proteins at or close to the cell surface. Cleavage of the MERS- and SARS-CoV S proteins by HAT has also been reported (25, 35). While the functional consequences of MERS-CoV S cleavage by HAT were unknown, SARS-CoV S cleavage by this protease was shown to activate the S protein for cell-cell fusion (35). The present study demonstrates that HAT can activate MERS-CoV S for entry into target cells in which cathepsin B/L activity has been blocked. In addition, our work shows that MSPL and DESC1 activate both the MERS- and SARS-CoV S proteins for cell-cell fusion and MERS-CoV S for virus-cell fusion. Why MSPL, DESC1, and HAT failed to promote SARS-CoV S-driven virus-cell fusion is unclear. One explanation could be that activation of virion-associated SARS-CoV S requires higher protease expression levels than activation of MERS-CoV S. Alternatively, cleavage of SARS-CoV S at sites recognized by TMPRSS2, HAT, DESC1, and MSPL might be sufficient to activate the S protein for cell-cell fusion, while processing at one or more additional sites recognized only by TMPRSS2 might be required for virus-cell fusion. Which of the proteases able to activate the SARS- and MERS-CoV S proteins in cell culture contributes to viral spread in the infected host is unclear. A previous report demonstrated the coexpression of ACE2, the SARS-CoV receptor, and TMPRSS2/HAT in human respiratory epithelia (18), arguing for a role for these proteases in SARS-CoV infection. Whether DESC1 and MSPL are also present in viral target cells remains to be determined. Finally, the analysis of knockout mice, which have been reported for several TTSPs, including TMPRSS2 (56) and HAT (57), might help to define the relative contributions of the above-discussed enzymes to viral spread.
The ability of several TTSPs to cleave and activate the SARS- and MERS-CoV S proteins suggests a certain uniformity of the activation process. However, the analysis of the cleavage fragments indicates that this might not be the case. Thus, each protease produced different cleavage fragments, although the S protein fragments obtained upon MERS-CoV S and SARS-CoV S proteolysis by trypsin, HAT, and DESC1 exhibited a certain similarity. This is in keeping with the published observation that both trypsin and HAT cleave SARS-CoV S at R667 (35). Whether DESC1 requires the presence of the same residue for S protein cleavage remains to be determined, and also the sequences cleaved by TMPRSS2 and MSPL are unknown. In this context, it should be emphasized that although different S protein-activating proteases produced different cleavage fragments, the robust, multiple cleavage of MERS-CoV S by TMPRSS10 did not result in S protein activation, underlining the existing but limited plasticity of the cleavage-activation process. Furthermore, it should be noted that S protein cleavage was analyzed in a cis format (i.e., protease and S protein were expressed in the same cell [Fig. 2]), which allows efficient detection of cleavage fragments, while S protein activation was determined in a trans format (protease and S protein were expressed in different cells [Fig. 3 and 4]). Our previous work demonstrated that cis and trans cleavage of SARS-CoV S by TMPRSS2 produces comparable, if not identical, S protein fragments (22), indicating that the cleavage products observed in the present study should mirror those produced upon S protein activation. Finally, it has been suggested that binding of SARS-CoV S to its receptor might induce subtle conformational changes in the S protein that increase its protease sensitivity (27, 58) and MERS-CoV S binding to CD26 might have a similar effect. In the present study, S protein cleavage was assessed with cells expressing low, endogenous levels of receptor and we can thus not exclude the possibility that slightly different S protein processing products would be generated in a cell system expressing high levels of receptor.
TMPRSS2 and HAT were initially identified as FLUAV-activating proteases (15) and were subsequently shown to activate CoVs (22–24, 29, 35). Our results show that the novel S-protein-activating enzymes DESC1 and MSPL also activate FLUAV, further strengthening the concept that different respiratory viruses might have adapted to use the same host cell enzymes to accomplish activation. Furthermore, the observation that MSPL and DESC1, like TMPRSS2 and HAT (15), can activate HA proteins of all of the FLUAV subtypes responsible for pandemics indicates a potential role in viral spread in vivo. Are MSPL and DESC1 the elusive proteases that allowed an H3N2 virus to spread in a TMPRSS2 knockout mouse? The observation that MSPL and DESC1 can activate H1N1 and H3N2 viruses seems to argue against this possibility. However, it is conceivable that only certain H3N2 viruses, but not H1N1 viruses, can target DESC1- and MSPL-positive cells in the lung. Another potential candidate for activation of H3N2 viruses is matriptase, a TTSP recently reported to activate FLUAV (59–61). However, cell culture studies demonstrated that this protease activates certain HAs of the H1 subtype while activation of HAs of the H2 and H3 subtypes was not observed (60, 61). Finally, it cannot be disregarded that although several TTSPs cleave HA, the cleavage rates might be different and might determine whether a protease can support viral spread in the host.
Here, we demonstrate that DESC1 and MSPL, the latter of which is known to activate certain FLUAVs with a multibasic cleavage site (62), activate the S proteins of emerging CoVs and human FLUAV. Whether DESC1 and MSPL contribute to viral spread in the infected host remains to be determined, and knockout mice, as well as specific protease inhibitors, might be useful tools for these endeavors. In this regard, it should be mentioned that camostat, an inhibitor of TMPRSS2, also blocks DESC1 and MSPL (data not shown) and can thus not be used for differential protease inhibition. Therefore, the generation of more specific inhibitors, as recently reported for TMPRSS2 (63), will be an important task. Finally, it is worth noting that FLUAV-activating TTSPs colocalized with HA, while an inactive TTSP did not, despite robust expression in the cellular system analyzed. It is therefore conceivable that the cellular localization of a TTSP, apart from its substrate specificity, might determine whether the protease can activate HA and other viral glycoproteins, a possibility that deserves further investigation.
ACKNOWLEDGMENTS
We thank Mikhail Matrosovich, Volker Czudai-Matwich, and Jan Baumann for H2 and H3 HA plasmids; Klaus Schughart and Thorsten Wolff for influenza viruses; Heike Hofmann-Winkler for critically reading the manuscript; and Oliver Dittrich-Breiholz for supervision of quantitative RT-PCR experiments.
This work was supported by the DFG (PO 716/6-1), the BMBF (01KI1005C), the Leibniz Graduate School for Emerging Infectious Diseases, and the Göttingen Graduate School for Neurosciences, Biophysics, and Molecular Biosciences (DFG grants GSC 226/1 and GSC 226/2).
Footnotes
Published ahead of print 13 August 2014
REFERENCES
- 1.World Health Organization. 2014. Influenza (seasonal). World Health Organization, Geneva, Switzerland: http://www.who.int/mediacentre/factsheets/fs211/en/index.html [Google Scholar]
- 2. Johnson NP, Mueller J. 2002. Updating the accounts: global mortality of the 1918-1920 “Spanish” influenza pandemic. Bull. Hist. Med. 76:105–115. 10.1353/bhm.2002.0022 [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization. 2014. Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003. World Health Organization, Geneva, Switzerland: http://www.who.int/csr/sars/country/table2004_04_21/en/index.html [Google Scholar]
- 4. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367:1814–1820. 10.1056/NEJMoa1211721 [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization. 14 July 2014. Middle East respiratory syndrome coronavirus (MERS-CoV)—update. World Health Organization, Geneva, Switzerland: http://www.who.int/csr/don/2014_07_14_mers/en/ [Google Scholar]
- 6. Sun X, Whittaker GR. 2013. Entry of influenza virus. Adv. Exp. Med. Biol. 790:72–82. 10.1007/978-1-4614-7651-1_4 [DOI] [PubMed] [Google Scholar]
- 7. Belouzard S, Millet JK, Licitra BN, Whittaker GR. 2012. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 4:1011–1033. 10.3390/v4061011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heald-Sargent T, Gallagher T. 2012. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 4:557–580. 10.3390/v4040557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bertram S, Glowacka I, Steffen I, Kühl A, Pöhlmann S. 2010. Novel insights into proteolytic cleavage of influenza virus hemagglutinin. Rev. Med. Virol. 20:298–310. 10.1002/rmv.657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simmons G, Zmora P, Gierer S, Heurich A, Pöhlmann S. 2013. Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antiviral Res. 100:605–614. 10.1016/j.antiviral.2013.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kido H, Okumura Y, Takahashi E, Pan HY, Wang S, Chida J, Le TQ, Yano M. 2008. Host envelope glycoprotein processing proteases are indispensable for entry into human cells by seasonal and highly pathogenic avian influenza viruses. J. Mol. Genet. Med. 3:167–175. 10.4172/1747-0862.1000029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murakami M, Towatari T, Ohuchi M, Shiota M, Akao M, Okumura Y, Parry MA, Kido H. 2001. Mini-plasmin found in the epithelial cells of bronchioles triggers infection by broad-spectrum influenza A viruses and Sendai virus. Eur. J. Biochem. 268:2847–2855. 10.1046/j.1432-1327.2001.02166.x [DOI] [PubMed] [Google Scholar]
- 13. Towatari T, Ide M, Ohba K, Chiba Y, Murakami M, Shiota M, Kawachi M, Yamada H, Kido H. 2002. Identification of ectopic anionic trypsin I in rat lungs potentiating pneumotropic virus infectivity and increased enzyme level after virus infection. Eur. J. Biochem. 269:2613–2621. 10.1046/j.1432-1033.2002.02937.x [DOI] [PubMed] [Google Scholar]
- 14. Zhirnov OP, Ikizler MR, Wright PF. 2002. Cleavage of influenza A virus hemagglutinin in human respiratory epithelium is cell associated and sensitive to exogenous antiproteases. J. Virol. 76:8682–8689. 10.1128/JVI.76.17.8682-8689.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Böttcher E, Matrosovich T, Beyerle M, Klenk HD, Garten W, Matrosovich M. 2006. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 80:9896–9898. 10.1128/JVI.01118-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bertram S, Glowacka I, Blazejewska P, Soilleux E, Allen P, Danisch S, Steffen I, Choi SY, Park Y, Schneider H, Schughart K, Pöhlmann S. 2010. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells. J. Virol. 84:10016–10025. 10.1128/JVI.00239-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Böttcher-Friebertshäuser E, Stein DA, Klenk HD, Garten W. 2011. Inhibition of influenza virus infection in human airway cell cultures by an antisense peptide-conjugated morpholino oligomer targeting the hemagglutinin-activating protease TMPRSS2. J. Virol. 85:1554–1562. 10.1128/JVI.01294-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bertram S, Heurich A, Lavender H, Gierer S, Danisch S, Perin P, Lucas JM, Nelson PS, Pöhlmann S, Soilleux EJ. 2012. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS One 7(4):e35876. 10.1371/journal.pone.0035876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hatesuer B, Bertram S, Mehnert N, Bahgat MM, Nelson PS, Pohlman S, Schughart K. 2013. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog. 9(12):e1003774. 10.1371/journal.ppat.1003774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sakai K, Ami Y, Tahara M, Kubota T, Anraku M, Abe M, Nakajima N, Sekizuka T, Shirato K, Suzaki Y, Ainai A, Nakatsu Y, Kanou K, Nakamura K, Suzuki T, Komase K, Nobusawa E, Maenaka K, Kuroda M, Hasegawa H, Kawaoka Y, Tashiro M, Takeda M. 2014. The host protease TMPRSS2 plays a major role in in vivo replication of emerging H7N9 and seasonal influenza viruses. J. Virol. 88:5608–5616. 10.1128/JVI.03677-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tarnow C, Engels G, Arendt A, Schwalm F, Sediri H, Preuss A, Nelson PS, Garten W, Klenk HD, Gabriel G, Böttcher-Friebertshäuser E. 2014. TMPRSS2 is a host factor that is essential for pneumotropism and pathogenicity of H7N9 influenza A virus in mice. J. Virol. 88:4744–4751. 10.1128/JVI.03799-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Glowacka I, Bertram S, Muller MA, Allen P, Soilleux E, Pfefferle S, Steffen I, Tsegaye TS, He Y, Gnirss K, Niemeyer D, Schneider H, Drosten C, Pöhlmann S. 2011. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 85:4122–4134. 10.1128/JVI.02232-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. 2010. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 84:12658–12664. 10.1128/JVI.01542-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. 2011. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 85:873–882. 10.1128/JVI.02062-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gierer S, Bertram S, Kaup F, Wrensch F, Heurich A, Kramer-Kuhl A, Welsch K, Winkler M, Meyer B, Drosten C, Dittmer U, HT von, Simmons G, Hofmann H, Pöhlmann S. 2013. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J. Virol. 87:5502–5511. 10.1128/JVI.00128-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shirato K, Kawase M, Matsuyama S. 2013. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J. Virol. 87:12552–12561. 10.1128/JVI.01890-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. 2005. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A. 102:11876–11881. 10.1073/pnas.0505577102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bertram S, Dijkman R, Habjan M, Heurich A, Gierer S, Glowacka I, Welsch K, Winkler M, Schneider H, Hofmann-Winkler H, Thiel V, Pöhlmann S. 2013. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J. Virol. 87:6150–6160. 10.1128/JVI.03372-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S. 2012. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 86:6537–6545. 10.1128/JVI.00094-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shirogane Y, Takeda M, Iwasaki M, Ishiguro N, Takeuchi H, Nakatsu Y, Tahara M, Kikuta H, Yanagi Y. 2008. Efficient multiplication of human metapneumovirus in Vero cells expressing the transmembrane serine protease TMPRSS2. J. Virol. 82:8942–8946. 10.1128/JVI.00676-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Antalis TM, Bugge TH, Wu Q. 2011. Membrane-anchored serine proteases in health and disease. Prog. Mol. Biol. Transl. Sci. 99:1–50. 10.1016/B978-0-12-385504-6.00001-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. 2004. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl. Acad. Sci. U. S. A. 101:4240–4245. 10.1073/pnas.0306446101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chaipan C, Kobasa D, Bertram S, Glowacka I, Steffen I, Tsegaye TS, Takeda M, Bugge TH, Kim S, Park Y, Marzi A, Pöhlmann S. 2009. Proteolytic activation of the 1918 influenza virus hemagglutinin. J. Virol. 83:3200–3211. 10.1128/JVI.02205-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hofmann H, Geier M, Marzi A, Krumbiegel M, Peipp M, Fey GH, Gramberg T, Pöhlmann S. 2004. Susceptibility to SARS coronavirus S protein-driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem. Biophys. Res. Commun. 319:1216–1221. 10.1016/j.bbrc.2004.05.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bertram S, Glowacka I, Muller MA, Lavender H, Gnirss K, Nehlmeier I, Niemeyer D, He Y, Simmons G, Drosten C, Soilleux EJ, Jahn O, Steffen I, Pöhlmann S. 2011. Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin-like protease. J. Virol. 85:13363–13372. 10.1128/JVI.05300-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cal S, Quesada V, Garabaya C, Lopez-Otin C. 2003. Polyserase-I, a human polyprotease with the ability to generate independent serine protease domains from a single translation product. Proc. Natl. Acad. Sci. U. S. A. 100:9185–9190. 10.1073/pnas.1633392100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liao X, Wang W, Chen S, Wu Q. 2007. Role of glycosylation in corin zymogen activation. J. Biol. Chem. 282:27728–27735. 10.1074/jbc.M703687200 [DOI] [PubMed] [Google Scholar]
- 38. Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pöhlmann S. 2014. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 88:1293–1307. 10.1128/JVI.02202-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Glaser L, Stevens J, Zamarin D, Wilson IA, Garcia-Sastre A, Tumpey TM, Basler CF, Taubenberger JK, Palese P. 2005. A single amino acid substitution in 1918 influenza virus hemagglutinin changes receptor binding specificity. J. Virol. 79:11533–11536. 10.1128/JVI.79.17.11533-11536.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hofmann H, Simmons G, Rennekamp AJ, Chaipan C, Gramberg T, Heck E, Geier M, Wegele A, Marzi A, Bates P, Pöhlmann S. 2006. Highly conserved regions within the spike proteins of human coronaviruses 229E and NL63 determine recognition of their respective cellular receptors. J. Virol. 80:8639–8652. 10.1128/JVI.00560-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Simmons G, Reeves JD, Grogan CC, Vandenberghe LH, Baribaud F, Whitbeck JC, Burke E, Buchmeier MJ, Soilleux EJ, Riley JL, Doms RW, Bates P, Pöhlmann S. 2003. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 305:115–123. 10.1006/viro.2002.1730 [DOI] [PubMed] [Google Scholar]
- 42. Connor RI, Chen BK, Choe S, Landau NR. 1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206:935–944. 10.1006/viro.1995.1016 [DOI] [PubMed] [Google Scholar]
- 43. Matthaei M, Budt M, Wolff T. 2013. Highly pathogenic H5N1 influenza A virus strains provoke heterogeneous IFN-alpha/beta responses that distinctively affect viral propagation in human cells. PLoS One 8(2):e56659. 10.1371/journal.pone.0056659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Winkler M, Bertram S, Gnirß K, Nehlmeier I, Gawanbacht A, Kirchhoff F, Ehrhardt C, Ludwig S, Kiene M, Moldenhauer AS, Goedecke U, Karsten CB, Kuhl A, Pöhlmann S. 2012. Influenza A virus does not encode a tetherin antagonist with Vpu-like activity and induces IFN-dependent tetherin expression in infected cells. PLoS One 7(8):e43337. 10.1371/journal.pone.0043337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bolte S, Cordelieres FP. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224:213–232. 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
- 46. Wilson CH, Abbott CA. 2012. Expression profiling of dipeptidyl peptidase 8 and 9 in breast and ovarian carcinoma cell lines. Int. J. Oncol. 41:919–932. 10.3892/ijo.2012.1522 [DOI] [PubMed] [Google Scholar]
- 47. Simmons G, Bertram S, Glowacka I, Steffen I, Chaipan C, Agudelo J, Lu K, Rennekamp AJ, Hofmann H, Bates P, Pöhlmann S. 2011. Different host cell proteases activate the SARS-coronavirus spike-protein for cell-cell and virus-cell fusion. Virology 413:265–274. 10.1016/j.virol.2011.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Klenk HD, Rott R. 1973. Formation of influenza virus proteins. J. Virol. 11:823–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Klenk HD, Rott R, Orlich M, Blodorn J. 1975. Activation of influenza A viruses by trypsin treatment. Virology 68:426–439. 10.1016/0042-6822(75)90284-6 [DOI] [PubMed] [Google Scholar]
- 50. Lazarowitz SG, Compans RW, Choppin PW. 1973. Proteolytic cleavage of the hemagglutinin polypeptide of influenza virus. Function of the uncleaved polypeptide HA. Virology 52:199–212 [DOI] [PubMed] [Google Scholar]
- 51. Kido H, Yokogoshi Y, Sakai K, Tashiro M, Kishino Y, Fukutomi A, Katunuma N. 1992. Isolation and characterization of a novel trypsin-like protease found in rat bronchiolar epithelial Clara cells. A possible activator of the viral fusion glycoprotein. J. Biol. Chem. 267:13573–13579 [PubMed] [Google Scholar]
- 52. Kido H, Okumura Y, Yamada H, Le TQ, Yano M. 2007. Proteases essential for human influenza virus entry into cells and their inhibitors as potential therapeutic agents. Curr. Pharm. Des. 13:405–414. 10.2174/138161207780162971 [DOI] [PubMed] [Google Scholar]
- 53. Galloway SE, Reed ML, Russell CJ, Steinhauer DA. 2013. Influenza HA subtypes demonstrate divergent phenotypes for cleavage activation and pH of fusion: implications for host range and adaptation. PLoS Pathog. 9(2):e1003151. 10.1371/journal.ppat.1003151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lu X, Shi Y, Gao F, Xiao H, Wang M, Qi J, Gao GF. 2012. Insights into avian influenza virus pathogenicity: the hemagglutinin precursor HA0 of subtype H16 has an alpha-helix structure in its cleavage site with inefficient HA1/HA2 cleavage. J. Virol. 86:12861–12870. 10.1128/JVI.01606-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bugge TH, Antalis TM, Wu Q. 2009. Type II transmembrane serine proteases. J. Biol. Chem. 284:23177–23181. 10.1074/jbc.R109.021006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim TS, Heinlein C, Hackman RC, Nelson PS. 2006. Phenotypic analysis of mice lacking the Tmprss2-encoded protease. Mol. Cell. Biol. 26:965–975. 10.1128/MCB.26.3.965-975.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sales KU, Hobson JP, Wagenaar-Miller R, Szabo R, Rasmussen AL, Bey A, Shah MF, Molinolo AA, Bugge TH. 2011. Expression and genetic loss of function analysis of the HAT/DESC cluster proteases TMPRSS11A and HAT. PLoS One 6(8):e23261. 10.1371/journal.pone.0023261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Matsuyama S, Ujike M, Morikawa S, Tashiro M, Taguchi F. 2005. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. U. S. A. 102:12543–12547. 10.1073/pnas.0503203102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Baron J, Tarnow C, Mayoli-Nussle D, Schilling E, Meyer D, Hammami M, Schwalm F, Steinmetzer T, Guan Y, Garten W, Klenk HD, Böttcher-Friebertshäuser E. 2013. Matriptase, HAT, and TMPRSS2 activate the hemagglutinin of H9N2 influenza A viruses. J. Virol. 87:1811–1820. 10.1128/JVI.02320-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Beaulieu A, Gravel E, Cloutier A, Marois I, Colombo E, Desilets A, Verreault C, Leduc R, Marsault E, Richter MV. 2013. Matriptase proteolytically activates influenza virus and promotes multicycle replication in the human airway epithelium. J. Virol. 87:4237–4251. 10.1128/JVI.03005-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hamilton BS, Gludish DW, Whittaker GR. 2012. Cleavage activation of the human-adapted influenza virus subtypes by matriptase reveals both subtype and strain specificities. J. Virol. 86:10579–10586. 10.1128/JVI.00306-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Okumura Y, Takahashi E, Yano M, Ohuchi M, Daidoji T, Nakaya T, Böttcher E, Garten W, Klenk HD, Kido H. 2010. Novel type II transmembrane serine proteases, MSPL and TMPRSS13, proteolytically activate membrane fusion activity of the hemagglutinin of highly pathogenic avian influenza viruses and induce their multicycle replication. J. Virol. 84:5089–5096. 10.1128/JVI.02605-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Meyer D, Sielaff F, Hammami M, Böttcher-Friebertshäuser E, Garten W, Steinmetzer T. 2013. Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochem. J. 452:331–343. 10.1042/BJ20130101 [DOI] [PubMed] [Google Scholar]