

ABSTRACT
Pathogen-specific antibodies (Abs) protect against respiratory infection with influenza A virus (IAV) and Streptococcus pneumoniae and are the basis of effective vaccines. Sequential or overlapping coinfections with both pathogens are common, yet the impact of coinfection on the generation and maintenance of Ab responses is largely unknown. We report here that the B cell response to IAV is altered in mice coinfected with IAV and S. pneumoniae and that this response differs, depending on the order of pathogen exposure. In mice exposed to S. pneumoniae prior to IAV, the initial virus-specific germinal center (GC) B cell response is significantly enhanced in the lung-draining mediastinal lymph node and spleen, and there is an increase in CD4+ T follicular helper (TFH) cell numbers. In contrast, secondary S. pneumoniae infection exaggerates early antiviral antibody-secreting cell formation, and at later times, levels of GCs, TFH cells, and antiviral serum IgG are elevated. Mice exposed to S. pneumoniae prior to IAV do not maintain the initially robust GC response in secondary lymphoid organs and exhibit reduced antiviral serum IgG with diminished virus neutralization activity a month after infection. Our data suggest that the history of pathogen exposures can critically affect the generation of protective antiviral Abs and may partially explain the differential susceptibility to and disease outcomes from IAV infection in humans.
IMPORTANCE Respiratory tract coinfections, specifically those involving influenza A viruses and Streptococcus pneumoniae, remain a top global health burden. We sought to determine how S. pneumoniae coinfection modulates the B cell immune response to influenza virus since antibodies are key mediators of protection.
INTRODUCTION
Respiratory tract (RT) coinfections with influenza A virus (IAV) and Streptococcus pneumoniae remain a major health problem worldwide (1–3). While IAV infection can increase susceptibility to secondary bacterial infections (1), much less is known about the immunological consequences that result when bacterial infections are present prior to IAV infection. This understudied problem is clinically relevant, since up to 60% of children and 10 to 40% of adults carry S. pneumoniae asymptomatically in the RT (4, 5).
Mouse models of IAV and S. pneumoniae coinfection revealed that the order of pathogen exposure critically determines disease outcome; McCullers et al. were the first to show that S. pneumoniae infection following a viral infection can exacerbate disease, whereas an S. pneumoniae infection preceding IAV can reduce morbidity (6). Many studies have investigated the impact of a secondary S. pneumoniae infection on host immune defenses after coinfection (reviewed in references 3 and 7). IAV can cause damage to the epithelial cell lining of the RT that then facilitates the adherence of bacteria. Furthermore, IAV induces suppression and apoptosis of macrophages and neutrophils, which lead to inhibition of bacterial clearance. Bacterial outgrowth then causes excessive cell infiltrations into the lung and an increase in the proinflammatory cytokines tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), IL-6, and gamma interferon (IFN-γ) as part of the “cytokine storm” that contributes to lung tissue pathology. In contrast, the impact of S. pneumoniae colonization on a subsequent IAV infection has been less well investigated. A recent study showed that colonization with Staphylococcus aureus promotes the generation of alternatively activated macrophages that suppress host response-mediated immune pathology to IAV (8). Despite progress on understanding the effects of viral and bacterial synergy on innate immune cells and the short-term disease outcomes, the impact of coinfection on adaptive immunity remains poorly defined (9).
The B cell response to single IAV infection has been well studied (reviewed in references 10–12). Following IAV infection, B cell activation initially occurs in the lung-draining mediastinal lymph node (medLN) (13–15). The induction and kinetics of germinal center (GC) B cells, virus-specific antibody (Ab)-secreting cells (ASCs), and memory B cells after IAV infection have been documented (16–21). GC B cells are present in medLN, spleen, lungs, and nasal-associated lymphoid tissue, where they peak around 3 weeks after infection (16). Likewise, recent studies have revealed much about the generation and maintenance of IAV-specific ASCs, including their isotype distribution at various sites, such as the medLN, lungs, spleen, and bone marrow (BM), where they can persist for several months (17–19). Anti-IAV memory B cells have not been investigated to the same extent but can also display organ- and isotype-specific distribution and contribution to protection (20, 21).
Optimal B cell responses are largely dependent on CD4+ T cell help. Thus, mice that lack CD4+ T cells (i.e., major histocompatibility complex class II-deficient [MHC II−/−] mice) or the costimulatory molecules CD40 and ICOS have severely impaired antiviral antibody (Ab) responses, although T-independent B cell responses can provide a certain degree of protection in response to IAV infection (15, 22–24). B cell help is provided by a subset of CD4+ T helper cells, termed T follicular helper (TFH) cells, that are essential for GC formation and maintenance (25–28) and produce the cytokines IL-6 and IL-21 in response to IAV or virus-like particles (29–31). Exaggerated and prolonged CD4+ T cell responses and hyperelevated levels of the cytokines IL-6, IL-21, and IFN-γ can dysregulate GC responses and humoral immunity (reviewed in references 28 and 32), such as in chronic lymphocytic choriomeningitis virus (LCMV) infection (33). Viral infections can also elicit antigen-independent B cell activation and production of non-virus-specific Abs that can result in hypergammaglobulinemia (34, 35), which has been associated with heightened immunopathology and immune complex-mediated disease (36, 37).
Because B cells integrate signals through the BCR and T helper cell-derived costimulation that synergize with signals from TLRs and cytokine receptors (38), we hypothesized that an altered milieu in the context of a bacterial coinfection with S. pneumoniae would affect the B cell response to IAV infection. Here, we established two models of RT coinfection to investigate how the presence of S. pneumoniae either prior to or following IAV infection impacts the generation and maintenance of antiviral Ab responses.
MATERIALS AND METHODS
Mice.
BALB/c mice (8- to 12-week-old females) were purchased from the National Cancer Institute and housed under specific-pathogen-free conditions in the Animal Facilities of the Wistar Institute and School of Medicine of the University of Pennsylvania. T cell receptor (TCR) transgenic TS1 mice (39), with CD4+ T cells specific for an I-Ed-restricted MHC-II peptide for influenza A virus PR8 hemagglutinin (HA111–119), and ICOS−/− mice (40) were housed and bred under the same conditions. All experiments were performed in accordance with Institutional Animal Care and Use Committee guidelines at the Wistar Institute and the University of Pennsylvania.
Pathogens and infections.
Mouse-adapted influenza virus PR8 (influenza virus A/Puerto Rico/8/34; H1N1; Mount Sinai strain) and the P1121 strain of Streptococcus pneumoniae (41) were used. P1121 was originally isolated from the nasopharynx of a subject enrolled in the human experimental carriage study (42) and is a derivative of the P833 strain, a type 23F isolate originally obtained from a child with otitis media.
PR8 was grown in the allantoic fluid of 10-day embryonated chicken eggs (B&E Eggs), and aliquots were stored at −80°C as described previously (19). P1121 was grown as static cultures in tryptic soy broth (TSB) at 37°C in 5% CO2 until the mid-log phase and an optical density at 620 nm of ∼0.6 to −0.7 was reached as previously described (41). All stocks were diluted in sterile phosphate-buffered saline (PBS) for infections.
For each experiment, bacterial titers in the inoculum were determined as described previously (41). In brief, serial dilutions were plated on tryptic soy agar (TSA) plates containing catalase (3,309 U/plate) (Worthington Biochemical Corporation) and neomycin (20 μg/ml) (Sigma). Bacteria were grown overnight at 37°C in 5% CO2, and colonies were counted and expressed as CFU.
For all infections, mice were immobilized with ketamine-xylazine (70 mg/10 mg/kg). Influenza virus PR8 was administered at 150 to 200 mean 50% tissue culture infectious doses (TCID50) in 30 μl, and S. pneumoniae P1121 was given at a dose of 106 to 107 CFU in 30 μl. For the coinfection models, S. pneumoniae P1121 was given either 10 days prior to infection with PR8 (Sp+PR8) or 5 days after PR8 infection (PR8+Sp).
Cell and sample preparations.
Tissues were collected and cells prepared as previously described (43). In brief, blood was collected through cardiac puncture. bronchoalveolar lavage (BAL) fluid was collected in 0.5 ml PBS–1% fetal bovine serum (PBS/FBS) through tracheal cannulation. Tissues were passed through metal wire mesh, and after red blood cell lysis (spleen and BM), cells were resuspended in Iscove's complete medium containing 10% FBS for enzyme-linked immunosorbent spot (ELISPOT) assays or in PBS/FBS for flow cytometric analysis. Live cell counts were obtained using a hemocytometer and trypan blue exclusion.
Serial bleeds for serum collection were obtained from the tail vein or by mandibular bleeds at 7- to 10-day intervals.
T cell transfer experiments.
Single cell suspensions from peripheral lymph nodes and spleens of TCR-transgenic TS1 mice were stained with CD4 and CD25 and CD4+ CD25− T cells isolated by sorting on a FACS-Aria fluorescence-activated cell sorter (FACS) (BD Biosciences) or MoFlo (DakoCytomation) cell sorter. Four × 106 to 5 × 106 CD4+ CD25− naive T cells were transferred by tail vein injection into sex-matched recipients 1 day prior to infection with PR8.
Flow cytometry.
For surface staining, cells were first incubated with Fc block (α-CD32/α-CD16), then incubated with various Abs. The following antibodies were purchased from eBioscience unless otherwise indicated: CD19-phycoerythrin (PE)-Cy7, IgDa-Pacific Blue, Fas-PE, GL-7–fluorescein isothiocyanate (FITC) or PNA-FITC (Sigma), CD45R/B220-PE-Texas Red (Caltag), or CD138-PE and -allophycocyanin (APC) (BD Biosciences). PE-labeled hemagglutinin (HA) probe (PR8-HA, H1) and APC-labeled HA probe (Hk68-HA, H3) were engineered to eliminate sialic acid binding residues responsible for nonspecific staining (44). For TCR transgenic T cell transfer experiments, cells were stained with CD4-peridinin chlorophyll protein (PerCP)-Cy5.5 and biotinylated Ab 6.5 specific for the TCR followed by streptavidin-APC or -Qdot 605. Analysis was performed on a LSRII (BD Bioscience). Data were processed using FlowJo software (Tree Star).
ELISA and ELISPOT for viral and bacterial antigens.
The enzyme-linked immunosorbent assay (ELISA) for determination of anti-PR8 Abs in the serum and BAL fluid was done as described previously (19, 45). A PR8-specific C12 idiotype (C12Id) ELISA was done as described previously (46). Relative virus-specific Ab concentrations were calculated based on a standard anti-C12 MAb (clone H35-C12.6.2, 23-Id) and expressed as relative units defined as equivalent to binding of 1 μg/ml of standard monoclonal antibody (MAb). Purified hemagglutinin (H1) from A/PR/8/34 was obtained from BEI Resources (NR-19240) and used at 1 μg/ml for coating of ELISA plates to determine PR8 HA-specific Ab levels. Results are expressed as relative units, defined as equivalent to binding of 1 μg/ml of standard anti-HA MAbs (mixture of H37-65-5 [IgG1], H36-4-5.2 [IgG2a], H36-12-3 [IgG2b], and H36-7-3.1 [IgG3]) in order to equalize for possible differences in isotype contributions.
ELISPOT assays for detection of virus-specific and total ASCs were performed as previously described (19).
S. pneumoniae-specific Abs were determined as described previously (41). In brief, whole P1121 and purified PspA39–189 (1 μg/ml) (42), the immunodominant portion of pneumococcal surface protein A (PspA), were used to coat 96-well Immulon II high-binding plates (Thermo Scientific). Plates were washed with 0.01% Brij-35–Tris-buffered saline (TBS), blocked with 1% bovine serum albumin (BSA) (Sigma) for 1 h at 37°C, and washed again. Serial dilutions of serum samples were added and then incubated for 2 h at room temperature. Bound Abs were detected with anti-IgG conjugated to alkaline phosphatase (AP) (Sigma) and developed with p-nitrophenyl phosphate (pNPP) (Sigma) for a standardized time of 30 min. The absorbance at 405 nm was recorded with a Versamax ELISA reader (Molecular Devices).
HI assay.
The hemagglutination inhibition (HI) assay was performed as described previously (47). Serum samples were serially diluted and mixed in equal volumes with purified PR8 virus. After 1 h, chicken red blood cells (RBCs) (B&E Eggs) were added, and the pattern of agglutination was recorded after another 0.5 h. The HI titer is expressed as the reciprocal of the highest serum dilution at which virus was inhibited from agglutinating RBCs.
Immunohistochemistry.
MedLN and spleen were snap-frozen in OCT medium (Tissue Tek). Sections were fixed in acetone and stained with GL7-FITC and biotinylated IgDa (BD Biosciences). Streptavidin-horseradish peroxidase (HRP) (Southern Biotech) and anti-FITC-AP (Millipore) were used as secondary Abs. All histology was examined and recorded on an upright Nikon E600 microscope with the image software Image Pro (Media Cybernetics).
Determination of viral titers.
Infectious virus in lungs was determined by titration in Madin-Darby canine kidney (MDCK) cell microcultures as described previously (48). Lung titers are expressed as the dilution at which 50% of the MDCK cultures revealed virus growth (TCID50/ml).
Cytokine and chemokine measurements in sera.
Sera were collected from naive mice and from infected mice (10 days after PR8 infection). Millipore Multiplex kits (MPXMCYTO-70k) and Luminex xMAP technology were used for detection of cytokines and chemokines. Assays were performed by the Human Immunology Core at the University of Pennsylvania.
Statistical analyses.
All data are from at least two independent experiments and are presented as the mean ± standard error of the mean (SEM). Student's t test or the Mann-Whitney test was used to calculate the statistical significance between two groups, and a one-way analysis of variance (ANOVA) or Kruskal-Wallis test was used for multiple group comparison unless stated otherwise. For viral titers of groups with a mixture of values above and below a detection limit, a two-part test was used by combining the statistics from the χ2 test (for comparing the proportions as percentages of values above/below the detection limit between two groups) and a t test for the values above detection. All statistical tests were performed using Prism software (GraphPad Software).
RESULTS
B cell differentiation in response to IAV infection is altered with bacterial coinfection.
First, we developed RT infection models with mouse-adapted influenza virus A/PR/8/34 (PR8) and the human isolate P1121 strain of Streptococcus pneumoniae (serotype 23F). Infection with PR8 alone elicits an acute RT infection that is associated with transient morbidity and virus clearance by day 10 (43). The P1121 S. pneumoniae isolate, on the other hand, does not induce signs of morbidity, consistent with an asymptomatic carrier model, and bacteria persist in the upper RT for up to 2 months after infection (data not shown) (41). Based on the time frames used by McCullers et al. (6), we coinfected mice with S. pneumoniae either 10 days prior (Sp+PR8) or 5 days after (PR8+Sp) PR8 infection to model two clinically relevant scenarios: the first represents individuals carrying S. pneumoniae prior to contracting an IAV infection, and the second is a model of a secondary bacterial infection. As has been shown previously (6), PR8+Sp-coinfected mice exhibited increased morbidity and exacerbated lung pathology, but strikingly, prior S. pneumoniae exposure protected mice from influenza-induced disease (49).
We next assessed the B cell response to infection with PR8 alone and in the context of S. pneumoniae coinfection. In response to single infection with PR8, GC (GL7+ Fas+ B220+) B cells and (CD138hi) ASCs were induced in the lung-draining medLN around day 5 postinfection (p.i.), with ASCs peaking at day 10 p.i. and GCs at day 15 p.i. (Fig. 1A and B), consistent with published observations (16, 19). S. pneumoniae infection alone did not elicit a marked GC or ASC response in the medLN and cervical LNs (Fig. 1A and B) (data not shown). Strikingly, mice colonized with S. pneumoniae prior to PR8 (Sp+PR8) revealed a higher percentage and number of GC B cells in the medLN 10 days after PR8 infection compared to mice infected with PR8 alone (Fig. 1C and D), whereas ASC numbers were not altered. In contrast, PR8+Sp-coinfected mice had an exaggerated ASC response, while GC numbers were not affected (Fig. 1C and D). Twenty days after PR8 infection, all groups had similar GC and ASC cell numbers in the medLN (Fig. 1D), but by day 34, GC numbers were reduced in the Sp+PR8 group, suggesting that prior S. pneumoniae infection impairs GC maintenance (Fig. 1D). The magnitude and kinetics of GCs were similarly altered in the spleen (data not shown), indicating that the impact of coinfection on the B cell response was not limited to the regional LN.
FIG 1.

Bacterial coinfection alters GC B cell and ASC responses in the medLN of influenza virus-infected mice. (A and B) Mice were infected with influenza virus A/PR8 or Streptococcus pneumoniae (Sp) P1121. (A) GL7+ Fas+ GC B cells and (B) CD138+ ASCs in the medLN at indicated times after infection (n = 3 to 13 mice/group). (C and D) Mice were infected with PR8 alone or coinfected with S. pneumoniae 5 days after PR8 (PR8+Sp) or 10 days prior to PR8 (Sp+PR8). (C) GC B cells (top row, gated on B220+ cells) and CD138+ ASCs (bottom row) in the medLN in infected/coinfected mice 10 days after PR8 infection. (D) Kinetics of GC B cells and CD138+ ASCs in medLN in PR8 singly infected and coinfected mice at indicated days (d10, d20, and d34) after PR8 infection. Significance was determined by one-way ANOVA with Tukey's posttest. ns, nonsignificant; *, P < 0.05; ***, P < 0.001.
To address whether virus-specific B cells are among the increased GC B cells in Sp+PR8-coinfected mice at day 10 p.i. with PR8, we used a fluorescently labeled PR8 (H1) HA probe. This probe was engineered so that the HA B cell epitopes are maintained, but nonspecific staining of lymphocytes is eliminated by a single-residue mutation of the sialic acid binding site (44). As a staining control, we also used an HA probe derived from an H3 virus. B cells from medLN of mice infected with PR8 did not stain with this non-cross-reactive H3 probe (Fig. 2A). Additional controls included B cells from medLN of naive mice or cervical LNs from PR8-infected mice, which are not the primary site of B cell activation (50) and which did not bind either the H1 or H3 probe (Fig. 2A) (data not shown). GC B cells from mice infected with X31, an H3N2 virus, also did not bind the H1 probe (Fig. 2B). In contrast, 3 to 8% of GC B cells from medLN of PR8-infected mice bound the H1 HA probe (Fig. 2A and B). As was the case for total GCs, the percentage and numbers of PR8 HA-specific GCs were significantly increased in Sp+PR8- but not in PR8+Sp-coinfected mice (Fig. 2B and C) (data not shown).
FIG 2.

Virus-specific GC B cell and ASC responses in the medLN of influenza virus-infected mice are altered by bacterial coinfection. (A) MedLN cells from naive and PR8-infected mice (10 days after PR8 infection) stained with GL7 for GC B cells (top row) and with CD138 for ASCs (bottom row). Cells were costained with a PE-labeled PR8 HA probe or APC-labeled H3 (from Hk68). Plots are shown gated on B220+ cells (top) and total live cells (bottom row). (B) MedLN from X31 (H3N2)-infected or PR8-infected and coinfected mice (10 days after PR8 infection) stained with GL7 for GC B cells and the PE-labeled PR8 HA probe. (C) Numbers of virus-specific HA+ GCs and HA+ ASCs in infected/coinfected mice (n = 4 to 8 mice/group; 10 days after PR8 infection). (D) PR8-specific IgG ASCs in medLN of indicated groups (n = 5 to 6 mice/group) were determined by ELISPOT assay (10 days after PR8 infection). (E) PR8-specific IgG ASCs in medLN and BM of the indicated groups (n = 11 to 18 mice/group) were determined by ELISPOT assay (34 to 53 days after PR8 infection). The results shown are from at least 3 independent experiments. Coinfected groups were analyzed for statistical significance relative to mice infected with PR8 alone using Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We next asked whether the increase in total ASCs in the medLN of PR8+Sp-coinfected mice translated into a boost in virus-specific ASCS. We used flow cytometry in conjunction with the HA probe to identify the HA-specific subset of ASCs and ELISPOT coated with PR8 virus to enumerate virus-specific ASCs. Compared to PR8 singly infected and Sp+PR8-coinfected mice, PR8+Sp-coinfected mice exhibited a marked increase in HA-specific ASCs (Fig. 2C) and total PR8-specific IgG and IgM ASCs (Fig. 2D) (data not shown). After 1 to 2 months of infection, PR8-specific IgG ASCs also remained increased in the medLN of PR8+Sp-coinfected mice compared to mice infected with PR8 alone (Fig. 2E). In the BM, this increase was not statistically significant; however, PR8-specific IgG ASC numbers were significantly lower in Sp+PR8-coinfected mice (Fig. 2E). Collectively, these data show that S. pneumoniae coinfection substantially modulated the generation of virus-specific and overall GC B cells and ASCs and that the outcome differed depending on the order of coinfection.
Modulation of TFH responses in the medLN correlates with GC responses.
TFH cells are essential for GC formation and maintenance (reviewed in references 26–28). Having established that antiviral GC and ASC responses in medLN are altered by coinfection, we hypothesized that the CD4+ TFH cell response would also be modified. In the medLN of naive and S. pneumoniae-infected mice, TFH cells were undetectable, consistent with a lack of GCs (Fig. 1A). In PR8-infected mice, TFH cells accounted for 3.5% of CD4+ T cells in the medLN 10 days after infection (Fig. 3A). Notably, we detected an increase in the percentage and number of TFH cells in Sp+PR8-coinfected mice, correlating with heightened GC numbers in that group (Fig. 3A and B). Likewise, by day 34 p.i., TFH cell numbers were highest in the PR8+Sp-coinfected group when GC numbers were highest (Fig. 3B).
FIG 3.

Bacterial coinfection alters T follicular helper cell responses in the medLN of influenza virus-infected mice. (A) Representative FACS profiles of CXCR5 and PD1 staining on CD4+ T cells in the medLN in indicated groups of mice. (B) CXCR5+ PD1+ CD4+ TFH cells in medLN of infected and coinfected mice at indicated days after PR8 infection. Data represent means ± SEM from n = 3 to 5 mice and are the results from 2 independent experiments. Significance was determined by Student's t test. NS, nonsignificant; *, P < 0.05. (C to E) PR8-HA-specific CD4+ T cells from TS1 mice were adoptively transferred 1 day prior to PR8 infection. (C) Representative FACS profile of medLN cells from a naïve mouse and a PR8-infected mouse (10 days p.i. with PR8) stained for CD4 and the TCR-specific Ab 6.5. (D) Frequency of ICOShi PD1hi cells of CD4+ 6.5+ T cells in the medLN of infected/coinfected mice 10 days after PR8 infection (n = 3 naive mice and n = 5 to 11 infected mice/group). n.d., not determined. (E) CD4+ 6.5+ T cell numbers in the medLN of naive and infected/coinfected mice 10 days and 34 to 35 days after PR8 infection. Significance was determined by Student's t test. ns, not significant; *, P < 0.05.
We next used an adoptive transfer system with CD4+ T cells specific for PR8 HA (39) to examine the impact of coinfection on virus-specific CD4+ TFH cells. We found that PR8 HA-specific CD4+ T cells identified by the clonotypic Ab 6.5 expanded about 200-fold in the medLN by day 10 after infection (Fig. 3C). Five percent of CD4+ 6.5+ T cells expressed a TFH phenotype (ICOS+ PD1hi) in PR8-infected mice that was further increased in Sp+PR8-coinfected mice (Fig. 3D). Five weeks after infection, higher numbers of HA-specific CD4+ T cells were maintained in the medLN of PR8+Sp-coinfected mice (Fig. 3E). Thus, as was the case with total CD4+ TFH cells (Fig. 3A and B), S. pneumoniae coinfection also induces changes in the magnitude of virus-specific TFH cell responses.
Bacterial coinfection modulates antiviral serum Ab levels but not anti-S. pneumoniae Abs.
Next, we analyzed Abs specific to IAV and S. pneumoniae in serial bleeds from singly infected and coinfected mice by ELISA. Antiviral serum IgG Abs showed marked modulation with bacterial coinfection (Fig. 4A). In PR8+Sp-coinfected mice, antiviral IgG levels were significantly elevated 3 weeks following PR8 infection, while Sp+PR8-coinfected mice had significantly reduced antiviral IgG responses compared to those in mice infected with PR8 alone. In contrast to antiviral serum IgG, S. pneumoniae-specific IgG measured against whole bacteria or purified pneumococcal surface protein A (PspA) was not significantly altered in the different coinfection groups (Fig. 4B) (data not shown).
FIG 4.

Bacterial coinfection modulates antiviral serum Abs but not anti-S. pneumoniae serum Abs. Serial bleeds from mice in indicated groups were collected and analyzed for PR8-specific IgG (A) and S. pneumoniae-specific IgG (B) by ELISA (n = 6 mice/group for infected mice and n = 2 naive mice). (C) Sera were collected on days 34 to 35 p.i. with PR8, and virus-neutralizing Abs were determined by HI assay. The kinetics of PR8-HA-specific serum IgG (D) and C12 idiotype (Id) Ig (E) in PR8- and coinfected mice. All results are from at least n = 6 mice/group. (F) PR8-specific IgG and IgA in BAL fluid of PR8 and coinfected mice (n = 5 to 12 mice/group) 10 days and 34 days after PR8 infection. Significance was determined by Student's t test. ns, nonsignificant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Consistent with a reduction in antiviral IgG in Sp+PR8-coinfected mice, Abs mediating hemagglutination inhibition (HI) were also significantly lower in this group (Fig. 4C). Surprisingly, despite the increase in virus-specific IgG in PR8+Sp-coinfected mice, there was no difference in HI titers. To test whether this is due to a shift away from HA-specific Abs that mediate HI activity, we performed ELISA with purified PR8 HA. However, this does not appear to be the case as the increase in virus-specific IgG serum Abs in PR8+Sp-coinfected mice was accompanied by an increase in HA-specific IgG (Fig. 4D). These data suggest that distinct HA-reactive B cells may be activated in the context of PR8+Sp coinfection producing Abs with lower HI activity.
Roughly 25% of anti-HA Abs generated in a primary response to PR8 can be identified by an idiotypic Ab termed C12 (C12Id) (46, 51, 52). These C12Id+ Abs have low HI capacity compared to other Abs directed to the same HA antigenic site (53). C12Id+ ASCs are short-lived, with serum Abs peaking during the first week after infection and then dropping rapidly thereafter (46, 51). We assessed whether PR8+Sp coinfection increased the production and/or maintenance of C12Id+ Abs and thus contributes to the overall reduced HI activity. While we detected significantly higher C12Id+ Ab levels at their peak in PR8+Sp-coinfected mice compared to PR8-infected mice (Fig. 4E), the levels diminished in all groups to the same extent, indicating that coinfection does not alter their transient nature.
To test the impact of coinfection on local RT antiviral Abs, PR8-specific IgG and IgA in the BAL fluid were determined by ELISA (Fig. 4F). Ten days after PR8 infection, there was little accumulation of PR8-specific IgG and IgA in all groups; nevertheless, we detected some statistical differences, as noted in Fig. 4F. At day 34, BAL fluid antiviral IgG was higher in PR8+Sp-coinfected mice, like it was in the serum (Fig. 4A). PR8-specific IgA was reduced in the Sp+PR8 group. Thus, the order of S. pneumoniae coinfection critically impacts the magnitude of both systemic and pulmonary antiviral Abs.
ICOS is required for antiviral Ab responses in the context of a secondary bacterial infection.
We hypothesized that signals in the medLN microenvironment of PR8+Sp-coinfected mice relax the stringent requirement for T cell help, in particular the dependence on the costimulatory molecule ICOS. In the absence of ICOS, mice have profound defects in GC responses that result in impaired Ab responses (40, 54). Similarly, GCs were absent in the spleens of ICOS−/− mice at all time points analyzed following PR8 infection (Fig. 5A and B) (data not shown). However, GCs were detectable in the medLN of ICOS−/− mice, albeit in reduced numbers (Fig. 5A to C) (data not shown). Initially at day 8 p.i., induction of PR8-specific IgM and IgG ASCs occurred comparably in the medLN of wild-type (WT) and ICOS−/− mice, but ASC numbers were significantly lower in the medLN and BM from ICOS−/− mice 4 weeks after infection (Fig. 5D). With or without a secondary bacterial infection, ICOS−/− mice had little detectable antiviral IgG in the serum (Fig. 5E). Together, these data indicate that signals in PR8+Sp-coinfected mice are unable to override the dependence on ICOS for optimal antiviral Ab production.
FIG 5.

Bacterial coinfection does not alter the dependence of antiviral Ab responses on ICOS. (A) Spleen and medLN from WT and ICOS−/− mice 33 days after PR8 infection stained with PNA and Fas to identify GC B cells. The numbers represent percentages of PNA+ Fas+ cells of CD19+ cells. (B) Immunohistochemistry of spleen and medLN sections as in panel A stained with GL7 (brown) and IgDa (blue). Original magnification, 10×. (C) Kinetics of GCs (identified as PNA+ Fas+ CD19+) in the medLN of WT and ICOS−/− mice following PR8 infection (n = 4 to 7 mice/group). nd, not determined. (D) At days 8 and 33 after PR8 infection, virus-specific IgM and IgG ASCs in medLN and BM from WT and ICOS−/− mice (n = 4 mice/group) were determined by ELISPOT. (E) WT and ICOS−/− mice were infected with PR8 or coinfected with PR8 and S. pneumoniae (PR8+Sp) (n = 5 to 10 mice/group), and PR8-specific IgG in serum was determined by ELISA. Significance was determined by Student's t test. ns, nonsignificant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Additional PR8 HA-specific CD4+ T cells do not alter the skewed GC/ASC response in bacterial coinfection.
To further examine how virus-specific T cells may regulate antiviral B cell responses in the context of S. pneumoniae coinfection, we analyzed antiviral B cell responses in mice with adoptively transferred PR8 HA-specific CD4+ T cells. After PR8 infection alone, the provision of additional HA-specific CD4+ T cells led to a higher number of PR8-specific IgG ASCs in the medLN at day 10 (Fig. 6A, left panel), while the numbers of total IgG ASCs were not altered (Fig. 6A, right panel). Interestingly, PR8 HA-specific CD4+ T cells did not affect the frequency of HA+ GC B cells in the medLN but increased the frequency of HA+ ASCs (Fig. 6B).
FIG 6.

Provision of additional PR8-HA-specific CD4+ T cells augments virus-specific Ab responses but does not alter the skewed GC/ASC response in bacterial coinfection. (A and B) MedLN from mice with or without adoptively transferred naive CD4+ T cells from TS1 mice were analyzed at day 10 after PR8 infection for PR8-specific and total IgG ASCs by ELISPOT assay (A) and the percentage of PR8 HA-specific GC B cells and CD138+ ASCs by cell surface staining with the PR8 HA probe (B). (C) Numbers of PR8 HA-specific GC B cells and PR8-specfic IgG ASCs in the medLN from indicated groups. (D) GC B cells and CD138+ ASCs in medLN from single PR8- and coinfected mice in the adoptive transfer model at indicated days after PR8 infection. (E and F) PR8-specific C12Id+ Ig (E) and IgM and IgG (F) Ab in serum of mice with or without adoptively transferred CD4+ T cells from TS1 mice (day 10 p.i. with PR8). (G) Total IgG serum Abs in indicated groups (days 34 and 35 p.i. with PR8). At least n = 5 mice/group. Significance was determined by Student's t test. ns, nonsignificant; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We next determined whether addition of HA-specific CD4+ T cells impacts the skewed GC/ASC differentiation that we have documented in the context of coinfection. As in mice without T cell transfers, Sp+PR8-coinfected mice had increased HA-specific GC B cell numbers compared to PR8 singly infected mice, whereas PR8+Sp-coinfected mice had a boosted virus-specific ASC response in the medLN at day 10 after PR8 infection (Fig. 6C). The increased availability of HA-specific T cell help did not alter overall GC numbers and ASCs compared to those in mice without transferred T cells (Fig. 6D). However, in the context of PR8+Sp coinfection, additional PR8 HA-specific CD4+ T cells further boosted C12Id+ serum Ab levels (Fig. 6E). This effect by HA-specific CD4+ T cells was also observed for the production of virus-specific IgM and IgG Ab levels (Fig. 6F). In contrast to virus-specific Abs, total IgG in the serum was not increased in the presence of additional virus-specific T cells and was even reduced in PR8+Sp-coinfected mice (Fig. 6G). Thus, provision of additional HA-specific T cell help enhances the generation of antiviral serum Abs, especially in the context of a secondary S. pneumoniae infection, without affecting total serum Ab levels. These results suggest that increasing the frequency of virus-specific T cell help could be beneficial in boosting antiviral Ab responses without increasing non-virus-specific IgG levels.
Impact of bacterial coinfection on viral clearance and inflammatory cytokines.
We considered the possibility that our observations are due to differences in viral antigen persistence and/or cytokine production in the coinfection models. We found that exposure to S. pneumoniae prior to influenza infection (Sp+PR8) did not alter initial viral infection and viral clearance in the lungs (Fig. 7A) (49). PR8+Sp-coinfected mice had increased viral titers by day 7.5, but by day 10, viral titers were indistinguishable from those of mice infected with PR8 alone, indicating that viral clearance is not impaired (Fig. 7A).
FIG 7.
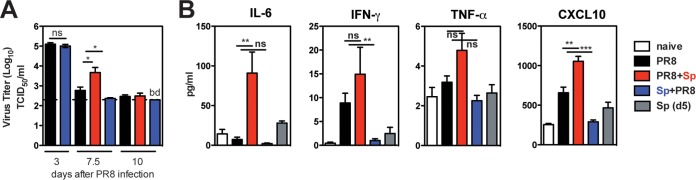
Impact of coinfection on viral clearance and systemic inflammatory mediators. (A) Lung titers of infectious virus at days 3, 7.5, and 10 after PR8 infection in indicated groups (n = 5 to 12 mice/group) were determined by MDCK titer assay. Significance was determined by Mann-Whitney test and two-part test. ns, nonsignificant; *, P < 0.05; bd, below detection. (B) Cytokines in sera from naive, infected, and coinfected mice (day 10 p.i. with PR8) (n = 3 to 9 mice) were analyzed by multiplex assay. Significance was determined by Student's t test. ns, nonsignificant; **, P < 0.01; ***, P < 0.001.
Independent of coinfection status, we detected similar bacterial loads in the noses of the majority of all mice up to 2 months after S. pneumoniae infection, although bacterial clearance from the lower RT was delayed in PR8+Sp-coinfected mice, consistent with other studies of secondary S. pneumoniae infections (6, 49, 55).
Exacerbated production of inflammatory cytokines (56), including IL-6 and IFN-γ, that can dysregulate B cell responses (32, 34, 36) often accompanies coinfections, especially those with increased pathogenesis. To test various cytokines, sera collected from infected and coinfected mice were analyzed by multiplex assay. IL-6 levels in PR8+Sp-coinfected mice were 10-fold increased compared to those in mice infected with PR8 alone, while IFN-γ and TNF-α were not significantly altered (Fig. 7B). In Sp+PR8-coinfected mice, IFN-γ levels were reduced compared to those in mice infected with PR8 alone, and the levels of all serum cytokines analyzed were indistinguishable from those in naive mice. CXCL10, which promotes cellular infiltration, including ASCs, into inflamed tissues (57), was significantly increased in sera from PR8-infected mice and further boosted in PR8+Sp-coinfected mice. In Sp+PR8-coinfected mice, levels of CXCL10 were comparable to those in naive mice. S. pneumoniae alone induced no systemic inflammatory cytokine/chemokine production, consistent with asymptomatic carriage.
DISCUSSION
Coinfections can subvert host immunity to unrelated pathogens and reduce efficacy of vaccines (reviewed in reference 9). IAV and S. pneumoniae are common participants in RT coinfections, yet, the impact on adaptive immunity, especially protective B cell responses, is almost completely unknown. B cells are critical players in the early defense against IAV infection and protect against reinfection through maintenance of Abs and memory B cells. Here, we investigated the impact of S. pneumoniae coinfection on the B cell response to IAV. Using a PR8 HA-specific probe to track PR8 HA-specific B cells, we showed that prior exposure to S. pneumoniae (Sp+PR8 coinfection) significantly increased the magnitude of HA-specific GC B cells in the medLN, whereas a secondary infection with S. pneumoniae (PR8+Sp coinfection) favored ASC responses within the same time frame. However, the initial GC response in Sp+PR8-coinfected mice was not maintained and correlated with lower serum antiviral Ab levels. In contrast, PR8+Sp-coinfected mice sustained increased virus-specific serum IgG, consistent with heightened GC persistence. Thus, this study reveals the novel finding that S. pneumoniae coinfection modulates both early IAV-specific B cell differentiation in the lung-draining medLN as well as the maintenance of antiviral humoral immunity and that the outcome differs, depending on the order of exposure to S. pneumoniae and IAV.
Studies with model antigens, such as hen egg lysozyme (HEL) and ovalbumin-conjugated to NP (NP-OVA), have revealed that increasing levels of antigen positively correlate with the magnitude and persistence of GC and TFH cells (58, 59). Interestingly, we detected elevated GC and TFH responses in Sp+PR8-coinfected mice 10 days after PR8 infection, although viral loads in the lungs were not significantly different between the coinfection groups and mice infected with PR8 alone. However, we cannot rule out that PR8+Sp coinfection may lead to increased persistence of viral antigen depots in secondary lymphoid organs and consequently account for the extended maintenance of GCs and TFHs at later times. Viral epitopes for CD4+ T cells have been detected by reverse transcription-PCR in the medLN for at least 10 to 14 days following IAV infection (60) and have been shown to promote T cell proliferation for at least 1 month later (60–62). It is unknown to what extent B cell epitopes persist in response to IAV infection. Studies are under way to determine if the maintenance and distribution of viral B and T cell epitopes are altered in bacterial coinfection.
There is strong evidence that cytokines can impact antigen-activated B cell differentiation. We detected prolonged and systemically increased levels of IL-6, which alone or together with type I IFNs induced by PR8 and S. pneumoniae (63, 64) could account for the exaggerated ASC response in the medLN of PR8+Sp-coinfected mice at day 10 p.i. This model is consistent with studies that demonstrated that IL-6, IFN-γ, and type I IFNs can enhance Ab responses (30, 65–67). Another candidate cytokine is IL-12, which is highly induced in cultures of human dendritic cells (DCs) treated with IAV and then subsequently exposed to S. pneumoniae (68). IL-12-producing DCs were shown to favor B cell differentiation to ASCs at the expense of GC formation (69). We speculate that the exaggerated ASC response in PR8+Sp coinfection could reflect a contribution of specialized cytokine-producing DCs in promoting nascent ASC differentiation at extrafollicular foci (EFF) (70, 71).
In the experiments using ICOS−/− mice, we demonstrated that bacterially derived signals in the context of PR8+Sp coinfection did not override the need for ICOS costimulation for the generation of antiviral Abs. To further examine the role of T cells, we hypothesized that the provision of additional virus-specific CD4+ T cell help would have distinct impacts on GC and ASC differentiation in the context of coinfection. We found that transferred PR8 HA-specific CD4+ T cells had no effect on HA-specific GC numbers but augmented antiviral ASC responses, including those directed against PR8 HA. On the other hand, the numbers of total IgG ASCs remained similar in mice with and mice without additional virus-specific CD4+ T cells, and consequently, antiviral IgG but not total IgG was increased in the serum. Importantly, in the context of coinfection with S. pneumoniae, our study showed that additional HA-specific CD4+ T cells did not alter the skewed GC/ASC response in the medLN early after infection. Together, these data suggest that the modulated GC/ASC response in IAV-S. pneumoniae coinfection is regulated through signals originating from cells other than virus-specific CD4+ T helper cells.
Although PR8+Sp coinfection leads to overall increased PR8-specific serum IgG, including HA-specific IgG, serum HI Abs were not increased. This could be due to a lack of sensitivity of the HI assay. Additional possibilities include that the HI activity could be affected by HI-interfering components and/or non-PR8-binding Abs that are increased in sera of the PR8+Sp group or that the affinity of overall anti-HA Abs produced in PR8+Sp coinfection compared to PR8 infection is reduced. It is intriguing to speculate that PR8+Sp coinfection causes a repertoire shift in the antiviral B cell response, such that representation of non-HI Abs or HA Abs of lower affinity are favored. The increase in early antiviral ASC responses (in the absence of altered GC responses) in PR8+Sp-coinfected mice suggests a preferential induction of Abs at EFF that have not undergone affinity maturation compared to Abs produced by post-GC-derived ASCs (71). Indeed, levels of C12Id+ Abs, a hallmark of early anti-PR8 HA EFF Ab responses (51), were significantly elevated in PR8+Sp-coinfected mice. However, C12Id+ serum Ab levels diminished in PR8+Sp-coinfected mice to the same extent as in mice infected with PR8 alone, arguing against the hypothesis that coinfection instructs short-lived ASCs, like C12-producing cells, to become long-lived.
Taken together, our results demonstrate that coinfection alters the antiviral B cell response and that the order of exposure to S. pneumoniae and IAV significantly changes the outcome. This is in contrast to antibacterial Ab responses that are not altered by IAV coinfection, indicating that S. pneumoniae modulates the immune response to influenza but not vice versa. Although we and others have demonstrated that colonization with S. pneumoniae or Staphylococcus aureus protects against acute influenza-induced morbidity (6, 8, 49), this study suggest that bacterial exposure may compromise long-term antiviral Ab-mediated immunity. Modulation of antiviral Ab levels as a consequence of the infection history with other microbes could have an impact on disease susceptibility and protection in humans.
ACKNOWLEDGMENTS
We thank Gary Nabel for HA probes and Michele Metzger for excellent technical assistance.
This work was supported by The Wistar Cancer Center Core grant P30 CA10815 and NIH grants U19AI083022 to J.E. and A.J.C., AI038446 to J.N.W., K22AI091651 to S.E.H., and T32CA09171 to A.I.W. and M.C.S.
A.I.W. and M.C.S designed and performed experiments, K.M., K.L.W., and S.E.H. helped with experiments, J.R.W., J.N.W., and A.J.C. provided essential reagents, J.E. designed experiments and provided overall direction, and A.I.W. and J.E. wrote the article.
The authors declare they have no commercial or financial conflicts of interest.
Footnotes
Published ahead of print 6 August 2014
REFERENCES
- 1. Morens DM, Taubenberger JK, Fauci AS. 2008. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J. Infect. Dis. 198:962–970. 10.1086/591708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mizgerd JP. 2008. Acute lower respiratory tract infection. N. Engl. J. Med. 358:716–727. 10.1056/NEJMra074111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McCullers JA. 2006. Insights into the interaction between influenza virus and pneumococcus. Clin. Microbiol. Rev. 19:571–582. 10.1128/CMR.00058-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Obaro SK, Monteil MA, Henderson DC. 1996. The pneumococcal problem. BMJ 312:1521–1525. 10.1136/bmj.312.7045.1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Austrian R. 1986. Some aspects of the pneumococcal carrier state. J. Antimicrob. Chemother. 18(Suppl A):35–45. 10.1093/jac/18.1.35 [DOI] [PubMed] [Google Scholar]
- 6. McCullers JA, Rehg JE. 2002. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J. Infect. Dis. 186:341–350. 10.1086/341462 [DOI] [PubMed] [Google Scholar]
- 7. McCullers JA. 2014. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Microbiol. 12:252–262. 10.1038/nrmicro3231 [DOI] [PubMed] [Google Scholar]
- 8. Wang J, Li F, Sun R, Gao X, Wei H, Li LJ, Tian Z. 2013. Bacterial colonization dampens influenza-mediated acute lung injury via induction of M2 alveolar macrophages. Nat. Comm. 4:2106. 10.1038/ncomms3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stelekati E, Wherry EJ. 2012. Chronic bystander infections and immunity to unrelated antigens. Cell Host Microbe 12:458–469. 10.1016/j.chom.2012.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gerhard W, Mozdzanowska K, Furchner M, Washko G, Maiese K. 1997. Role of the B-cell response in recovery of mice from primary influenza virus infection. Immunol. Rev. 159:95–103. 10.1111/j.1600-065X.1997.tb01009.x [DOI] [PubMed] [Google Scholar]
- 11. Gerhard W. 2001. The role of the antibody response in influenza virus infection. Curr. Top. Microbiol. Immunol. 260:171–190 [DOI] [PubMed] [Google Scholar]
- 12. Waffarn EE, Baumgarth N. 2011. Protective B cell responses to flu—no fluke! J. Immunol. 186:3823–3829. 10.4049/jimmunol.1002090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sealy R, Surman S, Hurwitz JL, Coleclough C. 2003. Antibody response to influenza infection of mice: different patterns for glycoprotein and nucleocapsid antigens. Immunology 108:431–439. 10.1046/j.1365-2567.2003.01615.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coro ES, Chang WLW, Baumgarth N. 2006. Type I IFN receptor signals directly stimulate local B cells early following influenza virus infection. J. Immunol. 176:4343–4351. 10.4049/jimmunol.176.7.4343 [DOI] [PubMed] [Google Scholar]
- 15. Sangster MY, Riberdy JM, Gonzalez M, Topham DJ, Baumgarth N, Doherty PC. 2003. An early CD4+ T cell-dependent immunoglobulin A response to influenza infection in the absence of key cognate T-B interactions. J. Exp. Med. 198:1011–1021. 10.1084/jem.20021745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boyden AW, Legge KL, Waldschmidt TJ. 2012. Pulmonary infection with influenza A virus induces site-specific germinal center and T follicular helper cell responses. PLoS One 7:e40733. 10.1371/journal.pone.0040733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jones PD, Ada GL. 1987. Persistence of influenza virus-specific antibody-secreting cells and B-cell memory after primary murine influenza virus infection. Cell. Immunol. 109:53–64. 10.1016/0008-8749(87)90291-7 [DOI] [PubMed] [Google Scholar]
- 18. Hyland L, Sangster M, Sealy R, Coleclough C. 1994. Respiratory virus infection of mice provokes a permanent humoral immune response. J. Virol. 68:6083–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wolf AI, Mozdzanowska K, Quinn WJ, III, Metzgar M, Williams KL, Caton AJ, Meffre E, Bram RJ, Erickson LD, Allman D, Cancro MP, Erikson J. 2011. Protective antiviral antibody responses in a mouse model of influenza virus infection require TACI. J. Clin. Invest. 121:3954–3964. 10.1172/JCI57362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Joo HM, He Y, Sangster MY. 2008. Broad dispersion and lung localization of virus-specific memory B cells induced by influenza pneumonia. Proc. Natl. Acad. Sci. U. S. A. 105:3485–3490. 10.1073/pnas.0800003105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Onodera T, Takahashi Y, Yokoi Y, Ato M, Kodama Y, Hachimura S, Kurosaki T, Kobayashi K. 2012. Memory B cells in the lung participate in protective humoral immune responses to pulmonary influenza virus reinfection. Proc. Natl. Acad. Sci. U. S. A. 109:2485–2490. 10.1073/pnas.1115369109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bertram EM, Tafuri A, Shahinian A, Chan VSF, Hunziker L, Recher M, Ohashi PS, Mak TW, Watts TH. 2002. Role of ICOS versus CD28 in antiviral immunity. Eur. J. Immunol. 32:3376–3385. 10.1002/1521-4141(200212)32:12<3376::AID-IMMU3376>3.0.CO;2-Y [DOI] [PubMed] [Google Scholar]
- 23. Lee BO, Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Makris M, Sprague F, Lund FE, Randall TD. 2005. CD4 T cell-independent antibody response promotes resolution of primary influenza infection and helps to prevent reinfection. J. Immunol. 175:5827–5838. 10.4049/jimmunol.175.9.5827 [DOI] [PubMed] [Google Scholar]
- 24. Choi Y, Baumgarth N. 2008. Dual role for B-1a cells in immunity to influenza virus infection. J. Exp. Med. 205:3053–3064. 10.1084/jem.20080979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Crotty S. 2011. Follicular helper CD4 T cells (T FH). Annu. Rev. Immunol. 29:621–663. 10.1146/annurev-immunol-031210-101400 [DOI] [PubMed] [Google Scholar]
- 26. McHeyzer-Williams M, Okitsu S, Wang N, McHeyzer-Williams L. 2012. Molecular programming of B cell memory. Nat. Rev. Immunol. 12:24–34. 10.1038/nri3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shlomchik MJ, Weisel F. 2012. Germinal center selection and the development of memory B and plasma cells. Immunol. Rev. 247:52–63. 10.1111/j.1600-065X.2012.01124.x [DOI] [PubMed] [Google Scholar]
- 28. Vinuesa CG, Tangye SG, Moser B, Mackay CR. 2005. Follicular B helper T cells in antibody responses and autoimmunity. Nat. Rev. Immunol. 5:853–865. 10.1038/nri1714 [DOI] [PubMed] [Google Scholar]
- 29. Karnowski A, Chevrier S, Belz GT, Mount A, Emslie D, D'Costa K, Tarlinton DM, Kallies A, Corcoran LM. 2012. B and T cells collaborate in antiviral responses via IL-6, IL-21, and transcriptional activator and coactivator, Oct2 and OBF-1. J. Exp. Med. 209:2049–2064. 10.1084/jem.20111504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dienz O, Eaton SM, Bond JP, Neveu W, Moquin D, Noubade R, Briso EM, Charland C, Leonard WJ, Ciliberto G, Teuscher C, Haynes L, Rincon M. 2009. The induction of antibody production by IL-6 is indirectly mediated by IL-21 produced by CD4+ T cells. J. Exp. Med. 206:69–78. 10.1084/jem.20081571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bessa J, Kopf M, Bachmann MF. 2010. Cutting edge: IL-21 and TLR signaling regulate germinal center responses in a B cell-intrinsic manner. J. Immunol. 184:4615–4619. 10.4049/jimmunol.0903949 [DOI] [PubMed] [Google Scholar]
- 32. Sweet RA, Lee SK, Vinuesa CG. 2012. Developing connections amongst key cytokines and dysregulated germinal centers in autoimmunity. Curr. Opin. Immunol. 24:658–664. 10.1016/j.coi.2012.10.003 [DOI] [PubMed] [Google Scholar]
- 33. Harker JA, Lewis GM, Mack L, Zuniga EI. 2011. Late interleukin-6 escalates T follicular helper cell responses and controls a chronic viral infection. Science 334:825–829. 10.1126/science.1208421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hunziker L, Recher M, Macpherson AJ, Ciurea A, Freigang S, Hengartner H, Zinkernagel RM. 2003. Hypergammaglobulinemia and autoantibody induction mechanisms in viral infections. Nat. Immunol. 4:343–349. 10.1038/ni911 [DOI] [PubMed] [Google Scholar]
- 35. Coutelier JP, van der Logt JT, Heessen FW, Vink A, van Snick J. 1988. Virally induced modulation of murine IgG antibody subclasses. J. Exp. Med. 168:2373–2378. 10.1084/jem.168.6.2373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Montes CL, Acosta-Rodríguez EV, Merino MC, Bermejo DA, Gruppi A. 2007. Polyclonal B cell activation in infections: infectious agents' devilry or defense mechanism of the host? J. Leukoc. Biol. 82:1027–1032. 10.1189/jlb.0407214 [DOI] [PubMed] [Google Scholar]
- 37. Monsalvo AC, Batalle JP, Lopez MF, Krause JC, Klemenc J, Hernandez JZ, Maskin B, Bugna J, Rubinstein C, Aguilar L, Dalurzo L, Libster R, Savy V, Baumeister E, Aguilar L, Cabral G, Font J, Solari L, Weller KP, Johnson J, Echavarria M, Edwards KM, Chappell JD, Crowe JE, Williams JV, Melendi GA, Polack FP. 2011. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat. Med. 17:195–199. 10.1038/nm.2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pone EJ, Zan H, Zhang J, Al-Qahtani A, Xu Z, Casali P. 2010. Toll-like receptors and B-cell receptors synergize to induce immunoglobulin class-switch DNA recombination: relevance to microbial antibody responses. Crit. Rev. Immunol. 30:1–29. 10.1615/CritRevImmunol.v30.i1.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kirberg J, Baron A, Jakob S, Rolink A, Karjalainen K, von Boehmer H. 1994. Thymic selection of CD8+ single positive cells with a class II major histocompatibility complex-restricted receptor. J. Exp. Med. 180:25–34. 10.1084/jem.180.1.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McAdam AJ, Greenwald RJ, Levin MA, Chernova T, Malenkovich N, Ling V, Freeman GJ, Sharpe AH. 2001. ICOS is critical for CD40-mediated antibody class switching. Nature 409:102–105. 10.1038/35051107 [DOI] [PubMed] [Google Scholar]
- 41. McCool TL, Weiser JN. 2004. Limited role of antibody in clearance of Streptococcus pneumoniae in a murine model of colonization. Infect. Immun. 72:5807–5813. 10.1128/IAI.72.10.5807-5813.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McCool TL, Cate TR, Moy G, Weiser JN. 2002. The immune response to pneumococcal proteins during experimental human carriage. J. Exp. Med. 195:359–365. 10.1084/jem.20011576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wolf AI, Buehler D, Hensley SE, Cavanagh LL, Wherry EJ, Kastner P, Chan S, Weninger W. 2009. Plasmacytoid dendritic cells are dispensable during primary influenza virus infection. J. Immunol. 182:871–879. 10.4049/jimmunol.182.2.871 [DOI] [PubMed] [Google Scholar]
- 44. Whittle JR, Wheatley AK, Wu L, Lingwood D, Kanekiyo M, Ma SS, Narpala SR, Yassine HM, Frank GM, Yewdell JW, Ledgerwood JE, Wei CJ, McDermott AB, Graham BS, Koup RA, Nabel GJ. 2014. Flow cytometry reveals that H5N1 vaccination elicits cross-reactive stem-directed antibodies from multiple Ig heavy chain lineages. J. Virol. 88:4047–4057. 10.1128/JVI.03422-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mozdzanowska K, Maiese K, Gerhard W. 2000. Th cell-deficient mice control influenza virus infection more effectively than Th- and B cell-deficient mice: evidence for a Th-independent contribution by B cells to virus clearance. J. Immunol. 164:2635–2643. 10.4049/jimmunol.164.5.2635 [DOI] [PubMed] [Google Scholar]
- 46. Kavaler J, Caton AJ, Staudt LM, Gerhard W. 1991. A B cell population that dominates the primary response to influenza virus hemagglutinin does not participate in the memory response. Eur. J. Immunol. 21:2687–2695. 10.1002/eji.1830211107 [DOI] [PubMed] [Google Scholar]
- 47. Scherle PA, Palladino G, Gerhard W. 1992. Mice can recover from pulmonary influenza virus infection in the absence of class I-restricted cytotoxic T cells. J. Immunol. 148:212–217 [PubMed] [Google Scholar]
- 48. Liang S, Mozdzanowska K, Palladino G, Gerhard W. 1994. Heterosubtypic immunity to influenza type A virus in mice. Effector mechanisms and their longevity. J. Immunol. 152:1653–1661 [PubMed] [Google Scholar]
- 49. Wolf AI, Strauman MC, Mozdzanowska K, Williams KL, Osborne LC, Shen H, Liu Q, Garlick D, Artis D, Hensley SE, Caton AJ, Weiser JN, Erikson J. 2014. Pneumolysin expression by Streptococcus pneumoniae protects colonized mice from influenza virus-induced disease. Virology 462-463C:254–265. 10.1016/j.virol.2014.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Marshall D, Sealy R, Sangster M, Coleclough C. 1999. TH cells primed during influenza virus infection provide help for qualitatively distinct antibody responses to subsequent immunization. J. Immunol. 163:4673–4682 [PubMed] [Google Scholar]
- 51. Rothaeusler K, Baumgarth N. 2010. B-cell fate decisions following influenza virus infection. Eur. J. Immunol. 40:366–377. 10.1002/eji.200939798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kavaler J, Caton AJ, Staudt LM, Schwartz D, Gerhard W. 1990. A set of closely related antibodies dominates the primary antibody response to the antigenic site CB of the A/PR/8/34 influenza virus hemagglutinin. J. Immunol. 145:2312–2321 [PubMed] [Google Scholar]
- 53. Mozdzanowska K, Feng J, Eid M, Zharikova D, Gerhard W. 2006. Enhancement of neutralizing activity of influenza virus-specific antibodies by serum components. Virology 352:418–426. 10.1016/j.virol.2006.05.008 [DOI] [PubMed] [Google Scholar]
- 54. Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, Flavell RA. 2001. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature 409:97–101. 10.1038/35051100 [DOI] [PubMed] [Google Scholar]
- 55. Ghoneim HE, Thomas PG, McCullers JA. 2013. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J. Immunol. 191:1250–1259. 10.4049/jimmunol.1300014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Smith MW, Schmidt JE, Rehg JE, Orihuela CJ, McCullers JA. 2007. Induction of pro- and anti-inflammatory molecules in a mouse model of pneumococcal pneumonia after influenza. Comp. Med. 57:82–89 [PMC free article] [PubMed] [Google Scholar]
- 57. Kunkel EJ, Butcher EC. 2003. Plasma-cell homing. Nat. Rev. Immunol. 3:822–829. 10.1038/nri1203 [DOI] [PubMed] [Google Scholar]
- 58. Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, Sallusto F. 2013. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity 38:596–605. 10.1016/j.immuni.2012.11.020 [DOI] [PubMed] [Google Scholar]
- 59. Paus D. 2006. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J. Exp. Med. 203:1081–1091. 10.1084/jem.20060087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim TS, Hufford MM, Sun J, Fu Y-X, Braciale TJ. 2010. Antigen persistence and the control of local T cell memory by migrant respiratory dendritic cells after acute virus infection. J. Exp. Med. 207:1161–1172. 10.1084/jem.20092017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jelley-Gibbs DM, Brown DM, Dibble JP, Haynes L, Eaton SM, Swain SL. 2005. Unexpected prolonged presentation of influenza antigens promotes CD4 T cell memory generation. J. Exp. Med. 202:697–706. 10.1084/jem.20050227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zammit DJ, Turner DL, Klonowski KD, Lefrançois L, Cauley LS. 2006. Residual antigen presentation after influenza virus infection affects CD8 T cell activation and migration. Immunity 24:439–449. 10.1016/j.immuni.2006.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nakamura S, Davis KM, Weiser JN. 2011. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J. Clin. Invest. 121:3657–3665. 10.1172/JCI57762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li W, Moltedo B, Moran TM. 2012. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of gammadelta T cells. J. Virol. 86:12304–12312. 10.1128/JVI.01269-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Köhler G. 1994. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 368:339–342. 10.1038/368339a0 [DOI] [PubMed] [Google Scholar]
- 66. Baumgarth N, Kelso A. 1996. In vivo blockade of gamma interferon affects the influenza virus-induced humoral and the local cellular immune response in lung tissue. J. Virol. 70:4411–4418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kopf M, Herren S, Wiles MV, Pepys MB, Kosco-Vilbois MH. 1998. Interleukin 6 influences germinal center development and antibody production via a contribution of C3 complement component. J. Exp. Med. 188:1895–1906. 10.1084/jem.188.10.1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kuri T, Sorensen AS, Thomas S, Hedestam GB, Normark S, Henriques-Normark B, McInerney GM, Plant L. 2013. Influenza A virus-mediated priming enhances cytokine secretion by human dendritic cells infected with Streptococcus pneumoniae. Cell. Microbiol. 15:1385–1400. 10.1111/cmi.12122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kim S, Caton M, Wang C, Khalil M, Zhou Z, Hardin J, Diamond B. 2008. Increased IL-12 inhibits B cells' differentiation to germinal center cells and promotes differentiation to short-lived plasmablasts. J. Exp. Med. 205:2437–2448. 10.1084/jem.20070731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. García de Vinuesa C, MacLennan IC, Holman M, Klaus GG. 1999. Anti-CD40 antibody enhances responses to polysaccharide without mimicking T cell help. Eur. J. Immunol. 29:3216–3224. 10.1002/(SICI)1521-4141(199910)29:10<3216::AID-IMMU3216>3.0.CO;2-X [DOI] [PubMed] [Google Scholar]
- 71. MacLennan ICM, Toellner K-M, Cunningham AF, Serre K, Sze DM-Y, Zúñiga E, Cook MC, Vinuesa CG. 2003. Extrafollicular antibody responses. Immunol. Rev. 194:8–18. 10.1034/j.1600-065X.2003.00058.x [DOI] [PubMed] [Google Scholar]