

ABSTRACT
Nuclear targeting of capsid proteins (VPs) is important for genome delivery and precedes assembly in the replication cycle of porcine parvovirus (PPV). Clusters of basic amino acids, corresponding to potential nuclear localization signals (NLS), were found only in the unique region of VP1 (VP1up, for VP1 unique part). Of the five identified basic regions (BR), three were important for nuclear localization of VP1up: BR1 was a classic Pat7 NLS, and the combination of BR4 and BR5 was a classic bipartite NLS. These NLS were essential for viral replication. VP2, the major capsid protein, lacked these NLS and contained no region with more than two basic amino acids in proximity. However, three regions of basic clusters were identified in the folded protein, assembled into a trimeric structure. Mutagenesis experiments showed that only one of these three regions was involved in VP2 transport to the nucleus. This structural NLS, termed the nuclear localization motif (NLM), is located inside the assembled capsid and thus can be used to transport trimers to the nucleus in late steps of infection but not for virions in initial infection steps. The two NLS of VP1up are located in the N-terminal part of the protein, externalized from the capsid during endosomal transit, exposing them for nuclear targeting during early steps of infection. Globally, the determinants of nuclear transport of structural proteins of PPV were different from those of closely related parvoviruses.
IMPORTANCE Most DNA viruses use the nucleus for their replication cycle. Thus, structural proteins need to be targeted to this cellular compartment at two distinct steps of the infection: in early steps to deliver viral genomes to the nucleus and in late steps to assemble new viruses. Nuclear targeting of proteins depends on the recognition of a stretch of basic amino acids by cellular transport proteins. This study reports the identification of two classic nuclear localization signals in the minor capsid protein (VP1) of porcine parvovirus. The major protein (VP2) nuclear localization was shown to depend on a complex structural motif. This motif can be used as a strategy by the virus to avoid transport of incorrectly folded proteins and to selectively import assembled trimers into the nucleus. Structural nuclear localization motifs can also be important for nuclear proteins without a classic basic amino acid stretch, including multimeric cellular proteins.
INTRODUCTION
Porcine parvovirus (PPV, newly reclassified as ungulate protoparvovirus) is a member of the Protoparvovirus genus of the Parvovirinae subfamily of the Parvoviridae family (1). It is an important cause of reproductive failure in swine (2, 3). Infection of sows hardly results in clinical signs, but some strains can breach the placental barrier and reach the fetus (3). Infection can then result in death of the fetus, depending on the strain and time of gestation at which infection occurred (4–6). Despite the fact that vaccines are widely used and efficiently prevent reproductive problems, the virus can still replicate and be shed from infected sows (7). This results in the constant presence of PPV in herds, enabling the occurrence of mutated field strains. This virus has a single-stranded DNA (ssDNA) genome, known to have a high mutation rate (8). Monitoring the emergence of new strains and understanding cell-virus interactions are therefore important to gain a better knowledge of the life cycle of this pathogen.
The limited coding capacity (5 kb) of parvoviruses suggests that they require elaborate and efficient interactions with infected cells, and possibly many cellular proteins are required for proper infection. Likewise, early PPV infection begins with at least two entry mechanisms inside the cells: clathrin-coated endocytosis and macropinocytosis (9). While virions are in the endosomal pathway, acidification and translocation to late endosomes are required (9). This environment triggers conformational changes in the virion, leading to externalization of the N terminus of capsid proteins (10). PPV capsids consist of two proteins. VP2, the major protein, is cleaved into VP3 in the endosomes (11–14), which is believed to create the necessary space for VP1 N terminus externalization. The VP1 sequence comprises the VP2 sequence with a 150-amino-acid N-terminal extension, called VP1up, for VP1 unique part (15). VP1up externalization in the endosomes is absolutely required for PPV infection since it contains the phospholipase A2 (PLA2) activity, essential to breach the endosomal membrane barrier (10, 16, 17).
Viral replication of parvoviruses occurs in the nucleus and requires specific transport of capsid proteins toward the nucleus at two distinct steps of the replication cycle. In early steps, incoming capsids need to either enter the nucleus or at least dock to the nuclear membrane to inject the viral genome into the nucleus (18–21). In late steps of infection, newly synthesized capsid proteins rapidly assemble into trimers before nuclear transport (22, 23). In the nucleus, trimers assemble into capsids prior to DNA packaging and export of complete viruses from the cell (24). The ratio of VP1 and VP2 proteins in the capsid varies between 1:5 and 1:10, and therefore trimers contain either three copies of VP2 or two copies of VP2 with one copy of VP1 (23, 25).
Nuclear transport of a small protein (<50 kDa) can be achieved by slow, passive diffusion through the nuclear pore complex (26). Larger proteins require active transport. Classic nuclear transport mechanisms proceed after recognition of a nuclear localization signal (NLS) by the transport machinery (usually importin-α or importin-β) (26). Several NLSs have been identified, and they are composed of a cluster of basic amino acids in close proximity in the protein sequence. Two common motifs are used in bioinformatics to identify potential NLSs within a protein sequence. The Pat4 motif consists of four continuous basic amino acids, and the Pat7 motif consists of a proline followed by three out of four basic residues after an interruption of 1 to 3 amino acids [P-X(1-3)-(3-4K/R)] (26). In addition to these “monopartite” NLSs, there are also “bipartite” NLSs if the transport depends on two basic regions separated by a short amino acid linker. A key point to ensure transport is accessibility of the motif to interact with cellular transport proteins. Specific accessibility can help coordinate complex transport, such as required for ribosome formation. The ribosomal subunits, synthesized in the cytoplasm, display their NLSs to migrate to the nucleus for final assembly and then return to the cytoplasm to exercise their functions. Assembly of the ribosomes enables the switch from exposed NLSs to exposed nuclear export sequences (NES) (27). This demonstrates that structural dynamics can alter the signals presented by the protein to the nuclear transport machinery. Numerous viruses use classic NLSs for nuclear targeting of viral proteins (28–30).
Several parvovirus structural proteins were shown to contain basic regions (BRs) that can act as nuclear localization signal (NLSs). For some viruses, these regions can be found in all structural proteins (for example, adeno-associated virus type 2 [AAV-2]) (31), while for others, such as minute virus of mice (MVM) (32) and canine parvovirus (CPV) (33), active NLSs are found only in the VP1 minor protein. Sequence analysis of PPV showed that there are five BRs that could potentially act as NLSs; all of them are located in VP1up (Fig. 1). It was also demonstrated for the B19 human parvovirus that an unconventional NLS (KLGPRKATGRW) containing few basic amino acids was responsible for VP2 protein transport to the nucleus (34). This region is not conserved in PPV (TAKMRSSMNW). Furthermore, another unconventional signal was found in the common region of VP1/VP2 for MVM (35). This region contained five basic amino acids (K and R; underlined and in boldface) in an 11-amino-acid sequence (528-KGKLTMRAKLR-538; GenBank accession number J02275.1). This region is located in a beta-sheet, enabling close proximity of basic amino acids when the protein is folded and assembled into a trimer. This region is essential for VP2 nuclear localization and is called the nuclear localization motif (NLM) since it is dependent on the correct folding of the protein. This region is well conserved with the closely related CPV (530-KGKLVFKAKLR-540; GenBank accession number NC_001539.1). However, this region is poorly conserved for PPV. The corresponding beta-sheet in PPV contains only three basic amino acids (525-KGTLTFTAKMR-535; GenBank accession number NC_001718.1), suggesting that nuclear localization of PPV VP2 depends on a different NLM. Moreover, VP2 does not contain any potential classic NLSs; there is no region with more than two basic amino acids in close proximity in the primary sequence of the protein.
FIG 1.

Basic region (BR) identification and protein construction. (A) Basic regions corresponding to potential nuclear localization signals (NLSs) were identified as clusters of more than two basic amino acids in close proximity. Alignment of VP1up proteins from PPV and MVM showed that one basic region was unique to PPV (BR3) and that four were conserved in MVM (BR1, BR2, BR4, and BR5). Critical amino acids of the catalytic site of PLA2 (HD) and its associated calcium-binding site (GPG) are also shown. (B) The fusion protein used for mutagenesis and localization experiments corresponded to the first 412 amino acids of VP1 (VP1-412), fused with mCherry fluorescent protein. This protein was big enough to prevent entry in the nucleus by passive diffusion and lacked VP sequences that enable trimer interactions (C). aa, amino acids.
In this study, evaluation of the five potential NLSs in VP1up by localization of different mutants of a fluorescent fusion protein demonstrated that active NLSs were BR1 and BR45 (a combination of BR4 and BR5), two classic NLSs, i.e., a Pat7 NLS and a bipartite NLS, respectively. Mutation of both of these NLSs in an infectious clone resulted in replication-incompetent virus, demonstrating the importance of VP1 NLSs for viral replication. Nuclear transport of VP2 protein depended on a structural motif (NLM) that differed from the NLM already identified for MVM, including basic amino acids outside the beta-sheet (35). This NLM was also required for PPV infection.
MATERIALS AND METHODS
Cell lines and viral strains.
Porcine fibroblast cells (porcine testis [PT]), a clone derived from ST cells (ATCC CRL-1746), and Cos-7 cells (ATCC CRL-1651) were grown at 37°C in Dulbecco's modified Eagle's medium containing d-glucose and l-glutamine and supplemented with 7% heat-inactivated fetal bovine serum and antibiotics (penicillin-streptomycin), all from Wisent. Viral infections were conducted with the NADL-2 strain of PPV (ATCC VR-742). Viral stocks were obtained by collection of supernatants from infected, lysed cells and used directly in experiments after a brief centrifugation to remove cellular debris.
Virus titration.
Viral titers were obtained as described previously (9) by immunofluorescence (IF) in 96-well plates at 20 h postinfection (p.i.) with a monoclonal mouse capsid-specific antibody (3C9-D11-H11), together with anti-mouse Alexa Fluor 488 as a secondary antibody (Life Technologies). Capsid-positive, fluorescent nuclei were scored, and virus titers were expressed in fluorescent focus-forming units/ml (FFU/ml).
Cloning and mutagenesis of potential NLSs.
First, the DNA fragment corresponding to amino acids 1 to 412 of VP1 (VP1-412) protein (start codon to EcoRI site) was cloned into the pBlueScript KS+ vector for mutagenesis (nucleotides 2287 to 3595 according the numbering of GenBank NC_001718). The sequence contained a mutation to inactivate the viral PLA2 (41-HD/AN-42) to avoid cellular toxicity when the protein was expressed. Basic amino acids of the different basic regions (BRs) were mutated into neutral hydrophilic polar amino acids (N, T, or Q) to conserve global polarity of the protein. Each BR was inactivated individually and in all possible combinations with the others BRs. Mutagenesis was carried out according to a QuikChange protocol (Stratagene). Final amino acid sequences including the different mutations are presented in Table 1. All resulting proteins (including the wild-type [wt)]) contained the mutation to inactivate the viral PLA2 (data not shown). Mutated sequences were cloned into pcDNA3.1+ vector (Life Technologies) containing the mCherry fluorescent protein sequence. This vector enabled expression of a fluorescent fusion protein to assess the localization of VP1-412 inside the cells. All clones were verified by sequencing, and positive clones were used for transfection experiments.
TABLE 1.
Inactivation mutations of putative NLSs in VP1upa

Amino acid positions of the BRs are indicated at the top, with double slashes indicating breaks in the sequence. Basic residues are in boldface, and substitutions with neutral hydrophilic polar amino acids are shaded.
Cloning of the basic regions with EGFP.
To confirm NLS activity of the BRs, they were cloned into pEGFP (where EGFP is enhanced green fluorescent protein) from Clontech. BR1, BR2, BR3, and BR5 were cloned by oligonucleotide annealing between BspEI and BamHI restriction sites. BR23 (a combination of BR2 and BR3) and BR45 were cloned by PCR cloning. A simian virus 40 (SV40) NLS (PKKKRKV) was used as a positive control and was also cloned by oligonucleotide annealing.
Cloning NLS mutants into the infectious clone.
Selected mutations were cloned into the infectious clone of NADL-2 (in pSmart vector) in two steps. First, the native PLA2 sequence was restored by mutagenesis, and then mutations were transferred into the infectious clone using existing cloning sites. All clones were sequenced, and positive clones were used in transfection experiments. Clones that displayed active viral replication were amplified in a small viral stock (no more than two passages on cells), and DNA from viral stock was amplified by PCR and sequenced to ensure that no reversion or compensatory mutations arose.
Mutagenesis and cloning of potential NLMs.
The genome fragment between the natural EcoRI site and the stop codon of the VPs was cloned into pBlueScript KS+ vector for mutagenesis (nucleotides 3590 to 4549 according to the numbering of GenBank NC_001718). The nucleotides around the stop codon were modified from ACTAGA to ACTAGT to include an SpeI restriction site, which enabled restoration of the natural viral sequence when XbaI was used for cloning into the infectious clone. Mutagenesis was used to replace basic amino acids of a potential NLM with glycines to give flexibility to the mutated protein. After sequence confirmation, mutations were transferred into the infectious clone of NADL-2 and sequenced again before transfection experiments. Mutagenesis of conserved amino acids of VP2 to alanines was done by site-directed mutagenesis, and the mutations were also transferred into the infectious clone for transfection experiments to assess localization of VP2.
Transfection into PT cells.
VP1-412-mCherry constructs (wt and BR mutants), pEGFP-BR constructs, and NLM (or VP2) mutated infectious clones were transiently expressed in PT cells. Cells were plated in 24-well plates with glass coverslips 1 day prior to experiments to achieve 80% confluence by the next day (5 × 104 cells/well). Transfections were carried out with Lipofectamine 2000 (Life Technologies), according to the manufacturer's instructions (1 μg of DNA per transfection). Incubation was then carried out at 37°C in the dark for 24 h (30 or 48 h for infectious clones). Cells were then fixed in 3% formaldehyde in IF buffer (see the immunofluorescence section) for 30 min. For infectious clones, immunofluorescence was performed as described in the next section, and DNA was stained with Hoechst 33258 after transfection with mCherry or EGFP clones as described in the immunofluorescence section. Coverslips were then mounted on glass slides and kept at 4°C in the dark until analysis. Localization of the fusion proteins was determined using fluorescence microscopy for mCherry or EGFP fluorescence. Results for VP1-412-mCherry localization are expressed as the percentages of cells where proteins were present either in the nucleus (N > C, where N represents the nucleus and C is the cytoplasm), diffuse throughout the cell (N = C), or mostly in the cytoplasm (C > N). For each protein construct, at least 300 cells were scored from at least three independent transfections experiments.
Immunofluorescence and confocal microscopy.
Cells were fixed with 3% formaldehyde in IF buffer (1× phosphate-buffered saline [PBS], 0.02% sodium azide, 0.1% Tween, 0.1% bovine serum albumin [BSA]) for 30 min and washed three times with 1× PBS. Cells were permeabilized with 3% Triton X-100 in IF buffer for 30 min and washed three times with 1× PBS. PPV capsids were detected with monoclonal mouse anti-capsid primary antibody (3C9-D11-H11, ATCC CRL-1745) (staining both empty and DNA containing capsids; diluted 1:2,000 in IF buffer) or polyclonal rabbit anti-VP2 (staining subunits and DNA-containing capsids [36]; diluted 1:1,000 in IF buffer) for 1 h. Cells were washed with 1× PBS and incubated with secondary antibodies, including goat anti-mouse or goat anti-rabbit conjugated with Alexa Fluor 488 or 568 as indicated in the figure legends (diluted 1:2,000 in IF buffer; Life Technologies) for 1 h. Finally, DNA was stained with Hoechst 33258 (2 μg/ml) for 30 min, and coverslips were fixed on slides, using Fluoroshield (Sigma), that were kept at 4°C in the dark until fluorescence was read. Confocal microscopy images were collected on a Zeiss LSM780 system equipped with a 30-mW 405-nm diode laser, 25-mW 458/488/514-nm argon multiline laser, 20-mW DPSS 561-nm laser, and a 5-mW HeNe 633-nm laser mounted on a Zeiss Axio Observer Z1 and operated with Zen 2011 software (Zeiss). We used a Plan-Apochromat 63× oil differential interference contrast (DIC) objective (numerical aperture [NA], 1.4) for our observations. All images were minimally and equally processed to ensure good contrast.
qPCR.
In fitness experiments, viral DNA was measured by quantitative PCR (qPCR) from supernatant samples at 24 h postinfection. qPCR for an infectious clone with mutations of BR1, BR4, and BR5 (IC-ΔBR145) was performed after immunoprecipitation (IP) as described below. After samples were washed, DNA was released by boiling them in 50 μl of 50 mM Tris (pH 7.5)–0.1% NP-40 (final wash solution) for 5 min. Samples were diluted 1:10 after elution for the purpose of PCR. qPCRs were performed as described previously (9), using SYBR mix from either ABScience or Life Technologies as per the manufacturer's recommendations. Primer specificity was verified using BLAST analysis and melting curve analysis from 60°C to 95°C. Viral DNA was expressed in genome copy equivalents (GCE) per ml (fitness) or per sample (IP).
Immunoprecipitation and Western blotting.
Immunoprecipitation of PPV capsids after transfection with infectious clones was performed as described previously (9). Briefly, cell lysates (pooled from four transfection wells) were first precleared with 20 μl of protein G-agarose beads for 2 h on ice. After centrifugation, supernatants were transferred into new tubes, and IP was done with monoclonal anti-capsid antibody (3C9-D11-H11, diluted 1:10; 20 μl) and 50 μl of protein G-agarose beads overnight at 4°C. Then, samples were washed and eluted as described previously (9). Samples were loaded on a 10% acrylamide gel and migrated at 200 V for 50 min. Proteins were electrotransferred to a nitrocellulose membrane at 300 mA for 60 min. Signals were enhanced using a Pierce Western Blot Signal Enhancer kit according to the manufacturer's protocol. After nonspecific sites were blocked with 1.5% nonfat dry milk in TBS-T (Tris-buffered saline plus Tween), membranes were incubated with primary antibody (polyclonal anti-VP2 at 1:1,000 in TBS-T) for 1 h and washed three times with TBS-T. Incubation with the secondary antibody, goat anti-rabbit conjugated to alkaline phosphatase ([AP] Bio-Rad) (1:1,000 in TBS-T), and washes were performed as for the primary antibody. Detection was performed using nitroblue tetrazolium/5-bromo-4-chloro-3-indolylphosphate (NBT/BCIP), as suggested by the manufacturer (Roche). Western blot images were minimally processed to ensure good contrast.
Evaluation of trimer formation.
Cos-7 cells were plated and transfected as explained in the section on transfection. At 30 h posttransfection, cells were washed three times with 1× PBS. Cells were then treated with the cross-linker dimethyl suberimidate (DMS; Thermo Scientific/Pierce) at a concentration of 10 mM in HNEM buffer (50 mM HEPES, pH 8.0, 150 mM NaCl, 2 mM MgCl2, 1 mM EDTA) for 30 min. The reaction was quenched by the addition of 10 mM Tris, pH 7.5, for 10 min. Cells were lysed as explained in the immunoprecipitation section. Proteins were separated in 10% acrylamide gels (SDS-PAGE) and electrotransferred to nitrocellulose membrane as described in the Western blotting section. Western blotting was performed with rabbit anti-VP2 primary antibody.
RESULTS
Identification of potential NLSs of VP1up and mutagenesis.
Basic regions (BRs) possibly involved in nuclear localization of structural proteins of PPV were identified as regions containing at least three basic amino acids in close proximity. These putative nuclear localization signals (NLSs) are highlighted in Fig. 1A. All BRs were located in the VP1 unique part (VP1up). Therefore, no classic NLSs were expected in the VP2 protein. BR1 was a potential Pat7 classic motif (26), and the combination of BR4 and BR5 was a potential classic bipartite NLS (26). Comparison with the MVM VP1up sequence showed that BR1, BR2, BR4, and BR5 are conserved between both viruses while BR3 is unique to PPV (32).
To identify active NLSs present in VP1 protein, the 1,236 first nucleotides corresponding to the first 412 amino acids of VP1 (VP1-412) were cloned into pBlueScript KS+ vector for mutagenesis. This part of the protein was chosen since the complete protein would assemble in trimers after synthesis in the cells. These trimers would be expected to contain a nuclear localization motif (NLM) responsible for the transport of VP2 trimers into the nucleus for viral assembly (35). Therefore, as shown in Fig. 1C, working with VP1-412 prevented trimer formation. Moreover, important basic amino acids identified for VP2 nuclear localization started at amino acid K272 (see Fig. 5), corresponding to K422 in VP1. Constructs from only VP1up alone might have yielded protein that appeared to be too small to prevent passive diffusion to the nucleus (about 45 kDa) (unpublished observations). Thus, using a longer fragment of VP1 naturally circumvented the passive diffusion without requiring fusion to several fluorescent proteins that could potentially result in a structure masking some of the VP1 features that were being investigated (72 kDa for VP1-412-mCherry).
FIG 5.

NLM identification for VP2 protein. Three basic clusters were identified using the trimer structure information (Protein Data Bank accession number 1K3V) (basic amino acids are in white). (A) One external basic cluster is located outside the capsid and includes three basic amino acids (green). (B) A central basic cluster is located inside the capsid at the center of the trimer and includes two basic amino acids for each protein (a total of 6 amino acids; red). The image also shows basic amino acids in the beta-sheet corresponding to MVM NLM (orange). (C) Another internal cluster is located inside the capsid and is partly conserved with the NLM of MVM and includes six basic amino acids (orange). Basic amino acids were replaced by glycines in the infectious clone of PPV (IC). VP2 localization and capsid formation were investigated in transfection experiments. Immunofluorescence experiments were performed with rabbit anti-VP2 antibody together with anti-rabbit Alexa Fluor 488 antibody and mouse anti-capsid antibody together with anti-mouse Alexa Fluor 568 antibody. (D) Mutations in the external basic cluster or central basic cluster did not significantly change global VP2 localization. (E) Mutation of some of the basic amino acids of the internal cluster resulted in a VP2 located either in a diffuse pattern between the nucleus and the cytoplasm or mostly in the cytoplasm. All images are representative fields for each clone from at least three transfections. For each clone at least 300 positive cells were analyzed. The percentage of cells with capsid formation is also indicated.
Next, site-directed mutagenesis was performed to inactivate each BR alone and in all possible combinations. Basic amino acids (K and R) were replaced by polar uncharged amino acids (N, Q, and T) to conserve global polarity of the protein. Protein sequences of all BR mutants are presented in Table 1. These constructs also contained the viral PLA2 inactivation mutation (41-HD/AN-42) since expression of large amounts of VP1 protein in cells could be toxic. Finally, mutated DNA was transferred in the pcDNA3.1+ vector for transient expression experiments. This vector also contained the mCherry sequence to produce fluorescent fusion proteins enabling easy localization of the VP1-412-mCherry products (Fig. 1B).
Roles of basic regions for VP1up nuclear localization.
All different VP1-412-mCherry constructs were transfected into porcine PT cells. At 24 h posttransfection (p.t.), cells were fixed, and DNA was stained. Then, intracellular localization of VP1 fusion proteins was determined by the fluorescence of mCherry. Typical localizations are shown in Fig. 2. Three possible patterns were distinguished: either nuclear (N > C), diffuse (N = C), or cytoplasmic (C > N). Wild type (wt) VP1-412-mCherry was located exclusively in the nucleus of almost all cells (90%). On the other hand, mCherry expressed alone resulted in a diffuse pattern, indicating that the protein was present in both the cytoplasm and the nucleus (data not shown). The mCherry protein alone contains no NLS activity but is small enough (25 kDa) to passively diffuse between the two cell compartments.
FIG 2.
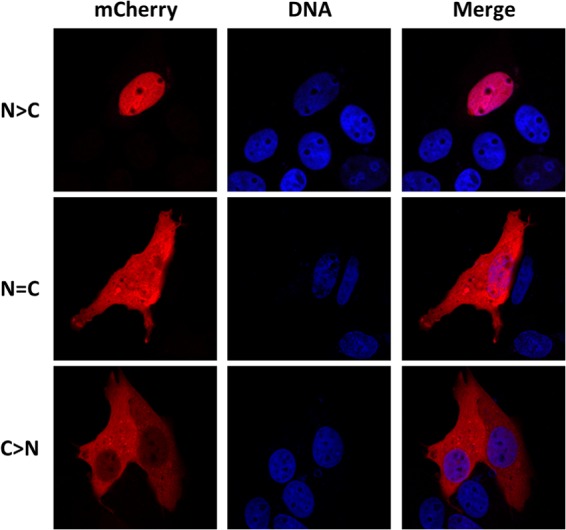
Localization patterns of VP1-412-mCherry mutants. The fusion protein was detected through mCherry fluorescence, and nuclei were stained with Hoechst DNA marker. wt protein was present exclusively in the nucleus (N > C). Proteins from some constructs were equally present in the nucleus and the cytoplasm (N = C), and some were almost exclusively in the cytoplasm (C > N).
Relative proportions of the three localization patterns for each VP1-412-mCherry construct were scored from at least 300 cells (Fig. 3A to D). As demonstrated, no single BR inactivation led to efficient inhibition of nuclear localization of VP1-412-mCherry protein. However, mutations in both BR1 and either half of the putative bipartite NLS (BR4 or BR5) resulted in 80% of the protein being diffused in both the cytoplasm and nucleus, while the protein was exclusively cytoplasmic in the remainder of cells. Further mutations (BR1 plus BR4 plus BR5) resulted in 35% of cells with VP1-412-mCherry in the cytoplasm, whereas localization in the rest of the cells was diffuse. BR2 and BR3 mutations did not significantly affect protein nuclear localization alone; however, the mutation of all BRs resulted in a greater proportion of cells with the protein only in the cytoplasm (65%). From all BRs investigated, BR1 and the bipartite BR4-BR5 were shown to display strong NLS activity.
FIG 3.

Cellular localization of the VP1-412-mCherry mutants and EGFP fusion proteins. (A) For each mutant, the percentage of cells displaying the fusion protein localized in the nucleus (N > C), diffused (N = C), or localized in the cytoplasm (C>N) was scored for at least 300 cells in at least three independent transfections. No single mutation resulted in an alteration of protein localization. (B) Double mutants containing mutation of BR1 together with BR4 or BR5 resulted in a diffuse localization of the protein. Mutation of three (C) or more (D) BRs confirmed the importance of BR1, BR4, and BR5 for nuclear localization of the fusion protein. (E) Confirmation of NLS activity of BRs. Indicated BRs were cloned in fusion with EGFP and expressed by transfection of PT cells. Localization of the proteins was evaluated by confocal microscopy through EGFP fluorescence in comparison with DNA staining with Hoechst reagent. Images for each clone are representative of three independent transfection experiments.
Confirmation of NLS activity.
To confirm the classic NLS activity of BR1 and BR45, we cloned these BRs with EGFP. Transfection and confocal microscopy showed that EGFP alone was present in both the nucleus and the cytoplasm, due to its small size (26 kDa) (Fig. 3E). EGFP-BR1 and EGFP-BR45 were completely located in the nucleus in almost all cells, confirming NLS activity. In contrast, EGFP-BR2 localization was not different from that of EGFP alone. EGFP-BR3 and EGFP-BR23 were slightly enriched in the nucleus, confirming weak NLS activity. Cloning only half of the bipartite NLS (EGFP-BR5) resulted in a partial nuclear accumulation. BR4 alone was not cloned with EGFP since it contained only two basic amino acids. Cloning of the classic SV40 NLS to EGFP was used as a positive control of nuclear transport. All images in Fig. 3E represent the localization that was observed in >90% of transfected cells for each construct.
Roles of VP1up NLSs in infection cycle.
The effects of these mutations in the context of the whole protein during the course of infection were then investigated. Mutated BR23, BR1, BR45, and BR145 were chosen for further experiments. The mutated sequences were cloned into the PPV infectious clone (IC) with an active PLA2 and transfected into PT cells. Supernatants were titrated at 48 h posttransfection. As demonstrated in Fig. 4A, IC-ΔBR1, IC-ΔBR45, and IC-ΔBR23 all produced infectious virions after transfection. However, mutant IC-ΔBR145 was completely nonproductive as no infectious virus was detected. These results demonstrated that VP1up NLSs are essential for PPV infection. Since results greatly depended on the transfection efficiency of the infectious clones, which is quite low (<5% efficiency), small viral stocks of the mutants were amplified for regular infection of cells at a multiplicity of infection (MOI) of 1. DNA from viral stocks obtained of the various mutants was amplified by PCR and sequenced to ensure that no mutation occurred to restore infectivity (data not shown). Results shown in Fig. 4B demonstrate that all infections resulted in an equivalent amplification of viral DNA, suggesting equivalent efficiencies for the delivery of the DNA to the nucleus in early infection. Infection with IC-ΔBR23 and IC-ΔBR45 produced slightly less infectious virus (as expressed in FFU) (Fig. 4C), and infection with IC-ΔBR1 resulted in a 4-fold-lower FFU production. The GCE/FFU ratios of the mutants were thus higher (Fig. 4D), suggesting that late steps of infection could be less efficient or that capsid is less stable. However, differences observed were all relatively small (<1 log), suggesting that mutations had only a minor effect on viral reproduction.
FIG 4.

Infectious clones with BR mutations. Selected mutations were cloned in the infectious clone of PPV. (A) Transfection and reinfection experiments showed that mutation of one NLS (BR1 or BR45) did not prevent viral amplification. However, mutation of both NLSs (BR145) completely abrogated viral amplification. Small stocks of replicating mutant infectious clones were made to evaluate the impact of the mutations on viral fitness. All mutants produced equivalent amounts of viral DNA (B) but fewer infectious particles (C), leading to higher GCE/FFU ratios (D). (E) Transfection of the infectious clones and immunofluorescence using 3C9 anti-capsid antibody together with goat anti-mouse Alexa Fluor 488 antibody demonstrated capsid formation for all mutants. These capsids were immunoprecipitated and contained viral DNA (F) as shown by qPCR and VP1 (G) as shown by Western blotting using anti-VP2 antibody (reacting with both structural proteins). This indicated that transfection of the mutant of regions BR145 produced complete capsids (*, P < 0,05; **, P < 0,005; ***, P < 0,001).
Mutant IC-ΔBR145.
To rule out the possibility that the mutation affected virus assembly following transfection, immunofluorescence and confocal microscopy were carried out at 30 h p.t. using anti-capsid antibody. Figure 4E shows that all mutants resulted in capsid formation that was similar to that of the wild-type infectious clone (pN2). Then, the amount of encapsidated viral DNA was examined by qPCR after immunoprecipitation of the capsid at 30 h p.t. The clone with both NLSs mutated successfully produced full capsids (containing DNA) (Fig. 4F). A control qPCR using primers from the infectious clone vector (pSmart) was performed to assess the presence of contaminant transfection DNA in the sample. Results revealed that there was no transfection DNA present in the immunoprecipitation-eluted capsids (data not shown). Finally, VP1 incorporation into the capsids was assessed by immunoprecipitation of the capsid at 30 h p.t. and Western blotting (Fig. 4G) demonstrating that capsids included both DNA and VP1. Transfection with this mutant produced complete virions that were, however, unable to proceed to a second round of infection. Transfection experiments with infectious clones were inefficient (low percentage of cells with virus formation), and this technical limitation prevented further characterization and evaluation of the PLA2 activity, also an essential part of VP1up. Insufficient virus production following transfection of this double NLS mutant clone also precluded immunofluorescence experiments.
Targeting VP2 to the nucleus: NLM.
Sequence analysis of the VP2 protein showed that there was no region containing more than two basic amino acids in a cluster. Therefore, it was postulated that protein folding and assembly into trimers would generate regions rich in basic amino acids, as previously demonstrated for MVM parvovirus. The MVM nuclear localization motif (NLM) consists of a cluster of five basic amino acids (underlined and in boldface) located in a beta-sheet of 11 amino acids (KGKLTMRAKLR) (35). This motif is highly conserved in the VP2 protein of the canine parvovirus (CPV) but poorly conserved in PPV VP2 (KGTLTFTAKMR), which contains only three basic amino acids. Moreover, mutagenesis of the MVM NLM resulting in a sequence equivalent to the PPV sequence (three remaining basic amino acids: KGNLTMTAKLR) almost completely prevented nuclear localization of MVM VP2 (35). This suggested that the NLM for PPV might be different from the MVM NLM. We used the data from the crystal structure of PPV and PyMol software to find basic amino acid clusters that could be potential NLMs for PPV. As shown in Fig. 5A, an external basic cluster, comprising amino acids R374, R393, and R565, was identified on the exterior face of the assembled capsid. A central basic cluster (K475 and R477), was identified in the center of the interior side of the trimer (Fig. 5B) and contained six basic amino acids in the assembled trimer. Some amino acids of the NLM of MVM were also conserved, such as K525, which is too far from K533 and R535 to act as an NLM as for MVM VP2. However, further analysis of this region showed that K272, K275, K487, and R576 are located in close proximity to K533 and R535, and this region was named the internal basic cluster and is also located in the interior face of the capsid (Fig. 5C).
Site-directed mutagenesis was used to replace basic amino acids of potential NLM with glycines to ensure flexibility. After mutagenesis, these mutants were cloned directly into the infectious clone containing or lacking both NLS mutations (BR1 and BR45) to prevent nuclear transport caused by VP1. Transfection experiments and confocal microscopy were used to localize VP2 and observe capsid formation for each mutant. Results shown in Fig. 5D and 6A demonstrated that neither the external basic cluster nor the central basic cluster mutations efficiently prevented VP2 transport to the nucleus. In sharp contrast, two single mutations of the internal basic cluster were sufficient to prevent most of VP2 transport to the nucleus (K272G and K533G), and VP2 K275G was present equally in the cytoplasm and nucleus (Fig. 5E and 6B). Since the VP1 NLS could be responsible for some VP2 transport in the nucleus, by their association in trimer, infectious clones were created containing both the NLM and NLS mutations (BR145). Results showed no major difference in VP2 localization with or without the presence of the NLS of VP1 (Fig. 6).
FIG 6.

NLS and NLM mutation combinations. Infectious clones containing each NLM mutation together with the mutation of BR145 were transfected in porcine PT cells. Immunofluorescence experiments at 30 h posttransfection were performed with rabbit anti-VP2 antibody together with anti-rabbit Alexa Fluor 488 antibody and mouse anti-capsid antibody together with anti-mouse Alexa Fluor 568 antibody. Results showed that the localization of VP2 was not significantly different from that of the mutants containing only the NLM mutations (Fig. 5). VP2 distribution was slightly more diffused or more cytoplasmic for some mutants. External and central basic clusters were not implicated in the efficient VP2 transport to the nucleus (A), but several single amino acid mutations of the internal basic cluster resulted in a deficient VP2 nuclear transport (B). All images are representative fields for each clone from at least three representative transfections. For each clone at least 300 positive cells were analyzed. The percentage of cells with capsid formation is also indicated.
For the mutants with nuclear localization of VP2 and capsid formation, infectious virus production after transfection was assessed with the transfection supernatants (Fig. 7A). Despite relatively poor VP2 transport to the nucleus, viral amplification for mutant IC-K272G was comparable to that of the wt infectious clone. Mutant IC-R477G also produced large amounts of infectious particles. In contrast, there was no infectious virus production for mutants IC-R374G, IC-R393G, IC-R565G, and IC-K487G. Fitness experiments were performed with replicative infectious clones as explained for VP1up mutants. Figure 7B to D show that both mutant K272G and R477G produced less infectious virus, but the global fitness of the viruses was only statistically different for mutant K272G, for which nuclear transport of VP2 trimers was impaired. This result suggested that mutation of a single amino acid (K272) from the NLM was not sufficient to completely block VP2 transport to the nucleus. It also suggests that virus production is efficient as soon as there is some VP2 in the nucleus. The location of R477G at the 5-fold axis probably impaired genome packaging and consequently resulted in the production of fewer infectious particles.
FIG 7.

Viral amplification of NLM mutants. The ability to produce infectious virus was investigated for mutants with capsid assembly. (A) Only two mutants produced infectious viruses: K272G and R477G. Fitness experiments were performed for these mutants. Infection of cells at an MOI of 1 resulted in less viral DNA amplification for both mutants (B), and less infectious virus was released from the cells 24 h p.i. (C). (D) Mutant K272G displayed a statistically lower global fitness, having a higher GCE/FFU ratio (*, P < 0,05; **, P < 0,005; ***, P < 0,001). Trimer formation was investigated for mutants that could not efficiently assemble into capsids. Cos-7 cells were transfected with the infectious clones containing mutations that prevented nuclear localization or capsid formation. At 30 h p.t., cell lysate was either directly used for Western blotting (E) or proteins were cross-linked with DMS before lysis and Western blotting using rabbit anti-VP2 antibody (F), enabling identification of trimer formation.
Mutant IC-R374G assembled into capsids in only 30% of transfected cells, suggesting that assembly problems may have prevented viral amplification. Mutants IC-R393G and IC-R565G were also not infectious, and their localization on the exterior of the capsid suggested that viral binding to the cell could be affected for those mutants. Using transfection supernatants of these clones, the amounts of viral DNA before and after incubation with fresh cells for 2 h were compared by qPCR. There was no difference in the binding/entry of wt virus compared to that of mutants R393G and R565G, suggesting that the defect is not at this step of infection (data not shown). Mutants IC-K275G, IC-K533G, and IC-R535G were not able to assemble into capsid, suggesting either assembly problems or too few VP2 trimers in the nucleus (35).
Evaluation of VP2 trimer formation.
Trimer formation is essential for nuclear transport of VP2 proteins. Protein association into trimers was investigated for mutants in which VP2 transport to the nucleus was prevented and for mutants with poor or no capsid formation. Transfection of porcine cells was not efficient enough to get significant signal in Western blot experiments. It was recently shown that transfection of nonpermissive cells was an efficient system to produce parvoviral proteins. Some nonpermissive cells, including Cos-7 cells, transfected with the infectious clone of canine minute virus were efficient to amplify viral DNA and produced infectious virus (37). Transfection of Cos-7 cells with the infectious clone of PPV also enabled production of viral protein and production of infectious virus in the supernatant (data not shown). These cells were transfected much more efficiently than the porcine cells and could produce enough VP proteins to allow investigation of trimer formation (Fig. 7E). Protein cross-linking before cell lysis showed that all mutants could associate in trimers as did the wt VP proteins (Fig. 7F, top bands). Two mutants also showed an intermediate band that could represent dimer or misformed trimer (IC-R374G and IC-R535C). This result suggested that for most of NLM mutations, nuclear transport impairment was not due to the absence of trimer formation.
DISCUSSION
Identification of active NLS in VP1up.
Several parvoviruses are known to contain active NLSs in the unique region of VP1 (32, 33). Sequence analysis of PPV VP1up revealed five potential NLSs. Mutagenesis experiments with fluorescent fusion proteins showed that BR1 was an active classic Pat7 NLS and that the combination BR4-BR5 (BR45) was an active classic bipartite NLS (Fig. 3). Moreover, these NLSs were essential to complete the PPV infectious cycle (Fig. 4). These results showed that PPV minor capsid protein contained one more active NLS than the VP1up of the closely related parvoviruses MVM and CPV (32, 33). Transfection of different mutant fusion proteins always resulted in some cells where the protein was present in both the cytoplasm and the nucleus (Fig. 3A to D). The proportion of cells with diffuse protein was less important for the mutants of all basic regions than for mutant BR145, suggesting that BR23 may provide weak NLS activity. A small nuclear accumulation of EGFP-BR3 and EGFP-BR23 (Fig. 3E) was also observed, supporting this supposition. However, these basic regions were not essential or sufficient for proper nuclear transport of VP1up. Finally, it is possible that a subset of the small proportion of cells with diffused protein is caused by cellular division after protein expression and before cell fixation, leading to the inclusion of some protein in the newly formed nucleus.
VP1up NLSs were located in the flexible N-terminal part of VP1, which becomes externalized in the endosomal pathway. Their location suggested that they can be used either to transport the capsid to the nucleus in the early steps of infection or to transport newly synthesized trimers at the late steps of infection. However, since VP1 is the minor protein, the NLM was expected to be the major determinant for the nuclear targeting of the trimers in late infection. Moreover, since the NLM identified for PPV (Fig. 5) is located inside assembled capsids, the two active NLSs of VP1up are the only exposed nuclear targeting sequences in the early steps of infection. Our experiments showed that transfection of an infectious clone containing the mutated BR1, BR4, and BR5 resulted in the formation of complete capsids (Fig. 4). These capsids were unable to complete the infectious cycle after infection of fresh cells using virus from the transfection supernatants, strongly suggesting that the NLSs are important to target the capsid to the nucleus in the early phase of infection.
Identification of a complex motif responsible for VP2 nuclear transport.
The linear VP2 sequence does not contain regions with more than two basic amino acids in close proximity, which could be potential classic NLSs. However, as much as 40% of cellular nuclear protein sequences do not contain such classic NLSs (38). Some nonclassic NLSs were identified in the literature, and others remain to be found. In our hypothesis, VP2 folding and assembly into trimers were necessary to form a motif containing enough basic amino acids to be recognized and transported by the cellular import machinery. It was shown that MVM VP2 nuclear import depends on a beta-sheet containing five basic amino acids in close proximity in the tertiary structure of the protein after correct protein folding (35). This region is also well conserved in the canine parvovirus major capsid protein but not in VP2 from PPV. However, structural analysis of basic amino acids in that region showed that basic residues outside the beta-sheet were also in close proximity in the correctly folded trimer. Two basic amino acids conserved with MVM NLM (K533 and R535) were also close to K272, K275, K487, and R576 in PPV trimers (Fig. 5). Mutagenesis of single amino acids from that region was enough to prevent efficient VP2 nuclear localization (K272G, K275G, and K533G), strongly suggesting that this region was the NLM for PPV VP2, responsible for transport of trimers to the nucleus. Since most trimers do not contain VP1 and its NLS, the NLM is essential for trimers containing only VP2, and these trimers are essential for proper capsid assembly (32); thus, this NLM is essential for PPV infection. Combinations of two amino acid mutations resulted in localization patterns similar to single mutations (data not shown). Further mutations were expected to result in protein assembly difficulties and were not attempted.
Interestingly, the localization of VP2 mutants was much more constant from cell to cell than for VP1 mutants. The representative immunofluorescence images in Fig. 5 correspond to VP2 localization patterns that were observed in most transfected cells (>80%), and it was therefore unnecessary to evaluate the localization in percentages of cells as for VP1up (Fig. 3). It is possible that the fusion of VP1 with mCherry resulted in a protein for which folding was not stable or consistent, resulting in some cells displaying more or less accessible NLSs.
The transfection experiments also showed that capsid formation was sensitive to single amino acid mutations. Indeed, several mutants accumulated high concentrations of VP2 in the nucleus but without capsid assembly (IC-K475G, IC-K275G, IC-R535G, and IC-R576G). Several possibilities can explain this observation. First, it was shown for MVM that nuclear import solely by the NLS of VP1up results in aggregate formations without capsids, suggesting that the ratio of VP2 to VP1 protein is important and that an excess of VP1 can prevent proper capsid assembly (35). This is the most likely explanation for the IC-K533G clone with a particularly low accumulation of VP2 in the nucleus. It is also possible that some mutations caused distortions of the trimers, preventing the strong contacts required for capsid assembly or antibody recognition in IF experiments. Analysis of several single amino acid mutations at 22 VP2 amino acid positions, conserved among the Parvovirinae subfamily, indicated that all but three (T170A, T258A, and G259A) prevented capsid formation, supporting our hypothesis that capsid formation is sensitive to mutations (E. Akl and P. Tijssen, unpublished observations).
Trimer association for parvovirus capsid protein is believed to be spontaneous and involve multiple strong contacts between the proteins. It was demonstrated for MVM that trimer association and transport to the nucleus could not be prevented by single amino acid mutations (39). The amino acids chosen in that study were those displaying strong intratrimeric interactions. In the present work, several single amino acid mutations of the identified NLM prevented nuclear accumulation of the protein. However, protein cross-linking experiments showed that most mutants could associate in trimer as efficiently as wt proteins. The only mutants with different protein migration patterns were IC-R374G and IC-R535G. These mutants showed the same trimeric band as the other mutants and wt clone but also a band of lower molecular mass that could represent a dimer or malformed trimer. This experiment demonstrated that mutants IC-K272G, IC-K275G, and IC-K533G could assemble into trimer, but these trimers were incompetent for nuclear transport. This supports our hypothesis that this region is the NLM for PPV VP proteins.
There was a subset of mutants for which capsid formation was successful but viral amplification failed: IC-R374G, IC-R393G, IC-R565G, and IC-K487G. Our initial hypothesis for the mutations located outside the capsid was that virus binding could be impaired such that these viruses could not enter cells. However, a qPCR experiment showed that these mutants could enter cells as well as the wt virus. Since macropinocytosis is an important entry mode for PPV (9) and since this entry mode does not require specific receptor binding, it is possible that uptake of the mutants was mostly achieved by that mechanism. It is also possible that mutations located on the outer surface of the capsid prevented other interactions with cellular proteins, proper capsid disassembly, or genome delivery to the nucleus. A more efficient system for transfection and production of mutant virus will be required to increase the production of mutant virus to pursue the analysis of nonreplicative mutants.
In summary, we demonstrated that capsid protein targeting to the nucleus for porcine parvovirus depended on both classic nuclear localization signals and a novel structural motif, requiring proper folding of structural proteins and assembly into trimers. PPV was also shown to be different from the other closely related protoparvoviruses CPV and MVM, containing one more active NLSs, and a more complex NLM, including basic residues outside the beta-sheet. Structural motifs like the one described here are likely to be responsible for nuclear transport of other proteins, both viral and cellular, for which no classic NLSs were identified by bioinformatics pattern recognition software.
ACKNOWLEDGMENTS
This research was funded by a grant from the Natural Sciences and Engineering Research Council of Canada (NSERCC) to P.T., an NSERCC scholarship to M.B., and Fondation Armand-Frappier scholarships to M.B., V.B.-L., and S.F.
We thank Jessy Tremblay for technical support with the confocal microscopy imaging, Angela Pearson for the mCherry plasmid, and Jean-François Laliberté for the EGFP plasmid.
Footnotes
Published ahead of print 30 July 2014
REFERENCES
- 1. Tattersall P. 2006. The evolution of parvovirus taxonomy, p 5–14 In Kerr JR, Cotmore SF, Bloom ME, Linden MR, Parrish CR. (ed), Parvoviruses. Hodder Arnold, London, United Kingdom [Google Scholar]
- 2. Dunne HW, Gobble JL, Hokanson JF, Kradel DC, Bubash GR. 1965. Porcine reproductive failure associated with a newly identified “SMEDI” group of picorna viruses. Am. J. Vet. Res. 26:1284–1297 [PubMed] [Google Scholar]
- 3. Mengeling WL, Cutlip RC. 1976. Reproductive disease experimentally induced by exposing pregnant gilts to porcine parvovirus. Am. J. Vet. Res. 37:1393–1400 [PubMed] [Google Scholar]
- 4. Mengeling WL, Pejsak Z, Paul PS. 1984. Biological assay of attenuated strain NADL-2 and virulent strain NADL-8 of porcine parvovirus. Am. J. Vet. Res. 45:2403–2407 [PubMed] [Google Scholar]
- 5. Cutlip RC, Mengeling WL. 1975. Pathogenesis of in utero infection: experimental infection of eight- and ten-week-old porcine fetuses with porcine parvovirus. Am. J. Vet. Res. 36:1751–1754 [PubMed] [Google Scholar]
- 6. Choi CS, Molitor TW, Joo HS, Gunther R. 1987. Pathogenicity of a skin isolate of porcine parvovirus in swine fetuses. Vet. Microbiol. 15:19–29. 10.1016/0378-1135(87)90125-8 [DOI] [PubMed] [Google Scholar]
- 7. Jozwik A, Manteufel J, Selbitz HJ, Truyen U. 2009. Vaccination against porcine parvovirus protects against disease, but does not prevent infection and virus shedding after challenge infection with a heterologous virus strain. J. Gen. Virol. 90:2437–2441. 10.1099/vir.0.012054-0 [DOI] [PubMed] [Google Scholar]
- 8. Streck AF, Bonatto SL, Homeier T, Souza CK, Goncalves KR, Gava D, Canal CW, Truyen U. 2011. High rate of viral evolution in the capsid protein of porcine parvovirus. J. Gen. Virol. 92:2628–2636. 10.1099/vir.0.033662-0 [DOI] [PubMed] [Google Scholar]
- 9. Boisvert M, Fernandes S, Tijssen P. 2010. Multiple pathways involved in porcine parvovirus cellular entry and trafficking toward the nucleus. J. Virol. 84:7782–7792. 10.1128/JVI.00479-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Venkatakrishnan B, Yarbrough J, Domsic J, Bennett A, Bothner B, Kozyreva OG, Samulski RJ, Muzyczka N, McKenna R, Agbandje-McKenna M. 2013. Structure and dynamics of adeno-associated virus serotype 1 VP1-unique N-terminal domain and its role in capsid trafficking. J. Virol. 87:4974–4984. 10.1128/JVI.02524-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Farr GA, Cotmore SF, Tattersall P. 2006. VP2 cleavage and the leucine ring at the base of the fivefold cylinder control pH-dependent externalization of both the VP1 N terminus and the genome of minute virus of mice. J. Virol. 80:161–171. 10.1128/JVI.80.1.161-171.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sanchez-Martinez C, Grueso E, Carroll M, Rommelaere J, Almendral JM. 2012. Essential role of the unordered VP2 N-terminal domain of the parvovirus MVM capsid in nuclear assembly and endosomal enlargement of the virion fivefold channel for cell entry. Virology 432:45–56. 10.1016/j.virol.2012.05.025 [DOI] [PubMed] [Google Scholar]
- 13. Ros C, Burckhardt CJ, Kempf C. 2002. Cytoplasmic trafficking of minute virus of mice: low-pH requirement, routing to late endosomes, and proteasome interaction. J. Virol. 76:12634–12645. 10.1128/JVI.76.24.12634-12645.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ros C, Gerber M, Kempf C. 2006. Conformational changes in the VP1-unique region of native human parvovirus B19 lead to exposure of internal sequences that play a role in virus neutralization and infectivity. J. Virol. 80:12017–12024. 10.1128/JVI.01435-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bergeron J, Menezes J, Tijssen P. 1993. Genomic organization and mapping of transcription and translation products of the NADL-2 strain of porcine parvovirus. Virology 197:86–98. 10.1006/viro.1993.1569 [DOI] [PubMed] [Google Scholar]
- 16. Zadori Z, Szelei J, Lacoste MC, Li Y, Gariepy S, Raymond P, Allaire M, Nabi IR, Tijssen P. 2001. A viral phospholipase A2 is required for parvovirus infectivity. Dev. Cell 1:291–302. 10.1016/S1534-5807(01)00031-4 [DOI] [PubMed] [Google Scholar]
- 17. Canaan S, Zadori Z, Ghomashchi F, Bollinger J, Sadilek M, Moreau ME, Tijssen P, Gelb MH. 2004. Interfacial enzymology of parvovirus phospholipases A2. J. Biol. Chem. 279:14502–14508. 10.1074/jbc.M312630200 [DOI] [PubMed] [Google Scholar]
- 18. Lux K, Goerlitz N, Schlemminger S, Perabo L, Goldnau D, Endell J, Leike K, Kofler DM, Finke S, Hallek M, Buning H. 2005. Green fluorescent protein-tagged adeno-associated virus particles allow the study of cytosolic and nuclear trafficking. J. Virol. 79:11776–11787. 10.1128/JVI.79.18.11776-11787.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cohen S, Pante N. 2005. Pushing the envelope: microinjection of Minute virus of mice into Xenopus oocytes causes damage to the nuclear envelope. J. Gen. Virol. 86:3243–3252. 10.1099/vir.0.80967-0 [DOI] [PubMed] [Google Scholar]
- 20. Cohen S, Marr AK, Garcin P, Pante N. 2011. Nuclear envelope disruption involving host caspases plays a role in the parvovirus replication cycle. J. Virol. 85:4863–4874. 10.1128/JVI.01999-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Porwal M, Cohen S, Snoussi K, Popa-Wagner R, Anderson F, Dugot-Senant N, Wodrich H, Dinsart C, Kleinschmidt JA, Pante N, Kann M. 2013. Parvoviruses cause nuclear envelope breakdown by activating key enzymes of mitosis. PLoS Pathog. 9:e1003671. 10.1371/journal.ppat.1003671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu H, Rossmann MG. 1993. The canine parvovirus empty capsid structure. J. Mol. Biol. 233:231–244. 10.1006/jmbi.1993.1502 [DOI] [PubMed] [Google Scholar]
- 23. Riolobos L, Reguera J, Mateu MG, Almendral JM. 2006. Nuclear transport of trimeric assembly intermediates exerts a morphogenetic control on the icosahedral parvovirus capsid. J. Mol. Biol. 357:1026–1038. 10.1016/j.jmb.2006.01.019 [DOI] [PubMed] [Google Scholar]
- 24. Cotmore SF, Tattersall P. 2005. Encapsidation of minute virus of mice DNA: aspects of the translocation mechanism revealed by the structure of partially packaged genomes. Virology 336:100–112. 10.1016/j.virol.2005.03.007 [DOI] [PubMed] [Google Scholar]
- 25. Tullis GE, Burger LR, Pintel DJ. 1993. The minor capsid protein VP1 of the autonomous parvovirus minute virus of mice is dispensable for encapsidation of progeny single-stranded DNA but is required for infectivity. J. Virol. 67:131–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Macara IG. 2001. Transport into and out of the nucleus. Microbiol. Mol. Biol. Rev. 65:570–594. 10.1128/MMBR.65.4.570-594.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tschochner H, Hurt E. 2003. Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends Cell Biol. 13:255–263. 10.1016/S0962-8924(03)00054-0 [DOI] [PubMed] [Google Scholar]
- 28. Krauer K, Buck M, Flanagan J, Belzer D, Sculley T. 2004. Identification of the nuclear localization signals within the Epstein-Barr virus EBNA-6 protein. J. Gen. Virol. 85:165–172. 10.1099/vir.0.19549-0 [DOI] [PubMed] [Google Scholar]
- 29. Li M, Wang S, Cai M, Zheng C. 2011. Identification of nuclear and nucleolar localization signals of pseudorabies virus (PRV) early protein UL54 reveals that its nuclear targeting is required for efficient production of PRV. J. Virol. 85:10239–10251. 10.1128/JVI.05223-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mears WE, Lam V, Rice SA. 1995. Identification of nuclear and nucleolar localization signals in the herpes simplex virus regulatory protein ICP27. J. Virol. 69:935–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grieger JC, Snowdy S, Samulski RJ. 2006. Separate basic region motifs within the adeno-associated virus capsid proteins are essential for infectivity and assembly. J. Virol. 80:5199–5210. 10.1128/JVI.02723-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lombardo E, Ramirez JC, Garcia J, Almendral JM. 2002. Complementary roles of multiple nuclear targeting signals in the capsid proteins of the parvovirus minute virus of mice during assembly and onset of infection. J. Virol. 76:7049–7059. 10.1128/JVI.76.14.7049-7059.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vihinen-Ranta M, Kakkola L, Kalela A, Vilja P, Vuento M. 1997. Characterization of a nuclear localization signal of canine parvovirus capsid proteins. Eur. J. Biochem. 250:389–394. 10.1111/j.1432-1033.1997.0389a.x [DOI] [PubMed] [Google Scholar]
- 34. Pillet S, Annan Z, Fichelson S, Morinet F. 2003. Identification of a nonconventional motif necessary for the nuclear import of the human parvovirus B19 major capsid protein (VP2). Virology 306:25–32. 10.1016/S0042-6822(02)00047-8 [DOI] [PubMed] [Google Scholar]
- 35. Lombardo E, Ramirez JC, Agbandje-McKenna M, Almendral JM. 2000. A beta-stranded motif drives capsid protein oligomers of the parvovirus minute virus of mice into the nucleus for viral assembly. J. Virol. 74:3804–3814. 10.1128/JVI.74.8.3804-3814.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zadori Z, Szelei J, Tijssen P. 2005. SAT: a late NS protein of porcine parvovirus. J. Virol. 79:13129–13138. 10.1128/JVI.79.20.13129-13138.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li F, Zhang Q, Yao Q, Chen L, Li J, Qiu J, Sun Y. 2014. The DNA replication, virogenesis and infection of canine minute virus in non-permissive and permissive cells. Virus Res. 179:147–152. 10.1016/j.virusres.2013.10.019 [DOI] [PubMed] [Google Scholar]
- 38. Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, Corbett AH. 2007. Classical nuclear localization signals: definition, function, and interaction with importin alpha. J. Biol. Chem. 282:5101–5105. 10.1074/jbc.R600026200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Perez R, Castellanos M, Rodriguez-Huete A, Mateu MG. 2011. Molecular determinants of self-association and rearrangement of a trimeric intermediate during the assembly of a parvovirus capsid. J. Mol. Biol. 413:32–40. 10.1016/j.jmb.2011.08.020 [DOI] [PubMed] [Google Scholar]