

ABSTRACT
Paramyxoviruses are enveloped negative-strand RNA viruses that are significant human and animal pathogens. Most paramyxoviruses infect host cells via the concerted action of a tetrameric attachment protein (variously called HN, H, or G) that binds either sialic acid or protein receptors on target cells and a trimeric fusion protein (F) that merges the viral envelope with the plasma membrane at neutral pH. F initially folds to a metastable prefusion conformation that becomes activated via a cleavage event during cellular trafficking. Upon receptor binding, the attachment protein, which consists of a globular head anchored to the membrane via a helical tetrameric stalk, triggers a major conformation change in F which results in fusion of virus and host cell membranes. We recently proposed a model for F activation in which the attachment protein head domains move following receptor binding to expose HN stalk residues critical for triggering F. To test the model in the context of wild-type viral glycoproteins, we used a restricted-diversity combinatorial Fab library and phage display to rapidly generate synthetic antibodies (sAbs) against multiple domains of the paramyxovirus parainfluenza 5 (PIV5) pre- and postfusion F and HN. As predicted by the model, sAbs that bind to the critical F-triggering region of the HN stalk do not disrupt receptor binding or neuraminidase (NA) activity but are potent inhibitors of fusion. An inhibitory prefusion F-specific sAb recognized a quaternary antigenic site and may inhibit fusion by preventing F refolding or by blocking the F-HN interaction.
IMPORTANCE The paramyxovirus family of negative-strand RNA viruses cause significant disease in humans and animals. The viruses bind to cells via their receptor binding protein and then enter cells by fusion of their envelope with the host cell plasma membrane, a process mediated by a metastable viral fusion (F) protein. To understand the steps in viral membrane fusion, a library of synthetic antibodies to F protein and the receptor binding protein was generated in bacteriophage. These antibodies bound to different regions of the F protein and the receptor binding protein, and the location of antibody binding affected different processes in viral entry into cells.
INTRODUCTION
Paramyxoviruses are enveloped, nonsegmented, negative-stranded RNA viruses that infect host cells by fusing their membranes with the cells' plasma membranes at neutral pH (1). The family Paramyxoviridae includes many major clinically and economically important pathogens of humans and animals, including parainfluenza viruses 1 to 5 (PIV1 to PIV5), mumps virus (MuV), Newcastle disease virus (NDV), Sendai virus, measles virus (MeV), canine distemper virus (CDV), Nipah virus (NiV), Hendra virus (HeV), respiratory syncytial virus (RSV), and human metapneumovirus (hMPV).
Paramyxoviruses mediate membrane fusion and cell entry by the concerted action of two viral glycoproteins: the attachment protein (HN, H, or G) and the fusion protein (F). The attachment protein binds cellular surface receptors and interacts with F. This interaction triggers a conformational change in F to induce membrane fusion, thereby releasing the viral ribonucleoprotein complex into the host cell cytoplasm.
Atomic structures of the attachment proteins (HN, H, or G) reveal a globular head harboring a typical sialidase domain created by a six-bladed β-propeller fold (2–11). PIV1 to PIV5, MuV, and NDV have HN-type receptor binding proteins possessing both hemagglutinating and neuraminidase (NA) activities, and HN binds sialic acid as receptor through a central binding site within the β-propeller fold. In contrast, H proteins of MeV and CDV and G proteins of HeV and NiV bind cell surface-expressed protein receptors through specific sites on the globular head.
The attachment proteins exist as dimers of dimers, with dimerization occurring through covalent and noncovalent interactions primarily within a stalk domain that connects the globular heads to the transmembrane domain (8, 12–17). Recently obtained atomic structures of HN stalk domains from NDV HN (12) and PIV5 HN (18) showed the stalks to be four-helix bundles (4HB). A large body of data suggests that F interacts with the attachment protein through the stalk domains (19–30).
Paramyxovirus F protein is a type I viral fusion protein with mechanistic features common to the fusion proteins of several other viruses, including human immunodeficiency virus (HIV) Env, influenza virus hemagglutinin (HA), and Ebola virus glycoprotein (GP) (31). F initially folds to a metastable trimeric precursor (F0) that is proteolytically cleaved into the covalently associated F1 and F2 subunits. Atomic structures of F trimers in the prefusion form have been determined for PIV5 and RSV (32–34). An interesting structure of prefusion MPV F monomers bound to an inhibitory antibody (Ab) has also been reported (35). Prefusion F has a short C-terminal cytoplasmic tail, a single transmembrane domain, a helical stalk, and a globular head domain. Atomic structures of NDV, human PIV3 (hPIV3), and RSV F in the postfusion form reveal that a large refolding event occurs to convert prefusion F to postfusion F in which part of the globular head domain rearranges to form a six-helix bundle (36–39). These structures, along with peptide inhibitory data, suggest a model for F-mediated membrane fusion where, upon activation, F1/F2 rearranges to insert a hydrophobic fusion peptide from the N terminus of F1 into the target cell membrane, forming a prehairpin intermediate (40). This relatively extended structure tethers the virus to the cell membrane and collapses to form the stable six-helix bundle of the postfusion structure. The transition from the metastable prefusion form, to the prehairpin intermediate, to the postfusion conformation proceeds down an energy gradient, with the postfusion form representing the most stable state, and the energy released during F refolding is coupled with membrane fusion.
HN, H, or G receptor binding is linked to F activation to ensure the correct timing of the F fusion peptide insertion into the target cell membrane. Upon binding receptor, the attachment protein globular heads are thought to initiate a rearrangement, and this movement is believed to be responsible for F activation during the F-HN/H/G interaction (1, 41–44).
The mechanistic details of how attachment proteins trigger F have been elusive. Monoclonal antibodies (MAbs) have proven to be powerful tools for revealing such details, including virus entry into cells, in many systems (45, 46). For PIV5, two previously described MAbs, F1a and 6-7, have been used with success in several studies (47, 48). MAb 6-7 is specific for postfusion F, while F1a recognizes cleaved F in the prefusion conformation better than uncleaved F (cleavage specific). Whereas F1a binds cleaved prefusion F more readily than cleaved postfusion F, it is not completely prefusion specific. Therefore, it would be useful to have PIV5 prefusion F-specific Abs as well as Abs that recognize additional unique antigenic sites on both F and HN. However, MAb discovery can be time-consuming and expensive. Furthermore, the MAb screening process offers limited control over screening conditions (e.g., pH, halide concentrations, etc.), including the ability to counterscreen, which can be especially important for antigens with closely related subtypes or for antigens that adopt multiple conformation states.
Antibody engineering using combinatorial libraries and phage display selection strategies can overcome many of the limitations associated with traditional MAb discovery. Phage display is an extraordinarily high-throughput and versatile screening process that isolates target molecule binders from highly diverse libraries (often >1010 unique members). However, phage display libraries that encode all 20 amino acids at each position can only comprehensively cover the sequence diversity resulting from randomizing a limited number of residues (full randomization of eight residues results in 2.5 × 1010 possible peptide sequences). Importantly, structural and bioinformatics studies have revealed significant bias for a subset of amino acids in protein interfaces (reviewed in reference 49). Combinatorial libraries with minimal sequence diversity (e.g., restricted to Tyr or Ser) based on various scaffolds have demonstrated that conformational diversity is more important than sequence diversity for generating specific and high-affinity protein interfaces (50, 51). Fellouse and coworkers (52) described previously a high-diversity phage library displaying engineered antibodies comprised of two disulfide-linked Fab fragments with extensive conformational diversity but minimal chemical diversity in their complementarity-determining regions (CDRs). They screened this library against a wide variety of antigenic targets and successfully produced high-affinity synthetic Fabs (synthetic antibodies, sAbs) against all of them (52). Recently, Koellhoffer and coworkers (53) used an advanced variant of this library, library F, to produce Fabs that bind and neutralize proteolytically cleaved and uncleaved forms of the Ebola virus envelope glycoprotein (53).
Here, we used synthetic antibody technology to generate novel sAbs, which were used to test a model for paramyxovirus F-HN/H/G interaction. In total, 53 unique sAb sequences were identified and shown to be specific for pre- and postfusion PIV5 F-GCNt (32) and HN. Surprisingly, despite strong affinity, only a single anti-prefusion F sAb inhibited fusion, whereas none of the anti-postfusion F-specific sAbs inhibited fusion. The inhibitory prefusion-specific sAb maps to a novel antigenic site in contrast to the well-characterized neutralizing sites on RSV F. Conversely, each of the anti-HN sAbs inhibited fusion; however, we show that the inhibitory mechanisms differ depending on the location of the antigenic site. sAbs that bind to the HN stalk likely inhibit by preventing association with F, and we discuss this inhibitory mechanism in the context of a recently proposed model for paramyxovirus F triggering by HN/H/G.
MATERIALS AND METHODS
Cells, plasmids, and antibodies.
CV-1, MDBK, and 293T cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). CHO-K1 cells were maintained in DMEM/F12 medium containing 10% GlutaMax (Gibco), 10% FBS, and 1% Pen-Strep (Invitrogen). Hi5 insect cells were maintained in Express 5 serum-free medium (Gibco) supplemented with 10% GlutaMax. S2 cells were maintained in Schneider's Drosophila medium containing 10% heat-inactivated FBS and 1% Pen-Strep.
pCAGGS-F and pCAGGS-HN are mammalian expression vectors expressing PIV5 F and HN proteins, respectively. pCAGGS-MCS (where MCS is multiple cloning site) is a negative control lacking insert. pT7 luciferase (Promega) and T7 RNA polymerase plasmids express firefly luciferase under T7 polymerase control and T7 RNA polymerase, respectively.
Antibodies specific for F included polyclonal antibodies (pAbs) 245 and R9716, raised in rabbits against purified F-GCNt. Antibodies specific for HN include anti-SDS-HN and R471, raised in rabbits against purified HN ectodomain. F-GCNt and HN ectodomain constructs (residues 56 to 565) were expressed by a recombinant baculovirus in insect cells.
Target protein expression, purification, and Western blot verification.
Hi5 insect cells were infected (multiplicity of infection [MOI] of 2) with a recombinant baculovirus stock containing a construct consisting of residues 37 to 565 of PIV5 HN (PIV5-HN37–565), and the supernatant was harvested at 65 h postinfection (8). Protein was purified by affinity chromatography using Ni-nitrilotriacetic acid (NTA)-agarose (Qiagen) as described previously (18). Protein was then concentrated and further purified by size exclusion chromatography (SEC) on a Superdex 200 column using 50 mM sodium phosphate, pH 7.4, and 150 mM NaCl as the running buffer.
F-GCNt was expressed from Drosophila S2 cells, and supernatant was dialyzed into column loading buffer (50 mM phosphate, pH 7.0, 300 mM NaCl, 1 mM imidazole) and purified using Ni-NTA as described previously (18). The sample was further purified by SEC as described above. Following purification, both HN37–565 and F-GCNt protein samples were >90% pure by SDS-PAGE and silver staining analysis. A portion of the F-GCNt protein was heated to 60°C for 10 min to convert it to the postfusion form as previously described (48).
Western blotting was performed using the anti-F 245 and anti-SDS-HN (bleed 10) pAbs at dilutions of 1:1,000 as the primary Abs for F-GCNt and HN37–565 detection, respectively. Goat anti-rabbit IRDye 6800RD (Li-Cor) at a 1:10,000 dilution was used as the secondary Ab. Blots were imaged with an Odyssey Infrared Imaging System (Li-Cor).
Target protein biotinylation.
Pre- and postfusion PIV5 F-GCNt and HN37–565 targets were biotinylated using NHS-PEO-biotin (N-hydroxysuccinimide ester, polyethylene glycol, biotin) as described previously (52, 54–56). The extent of biotinylation was quantified using an EZ-link biotin quantification kit (Pierce) according to the manufacturer's protocol. Biotinylation reactions were optimized such that each target molecule contained 2 to 3 biotins/molecule on average.
Phage library, screening, and phage enzyme-linked immunosorbent assay (ELISA).
A high-diversity M13 phage library expressing synthetic Fab fragments (sAbs) as p3 phage coat protein fusions was used for screening. Library generation methods have been described previously (52, 54–56). The library used employs limited amino acid diversity (Tyr or Ser) at most positions in CDRs H1 and H2, while H3 and L1 are enriched for Tyr, Ser, Gly, and Trp and have variable lengths.
Phage display selection and phage ELISA (including soluble-target competition ELISA) validation of selected clones was performed as described previously (54).
sAb reformatting, expression, and purification.
ELISA-validated phage clones were reformatted for sAb protein expression using Kunkel mutagenesis to insert a stop codon between the sAb heavy chain and gene III. Details of this method and sAb expression and purification have been described previously (54).
Antigenic site binning assay.
sAbs at 100 nM in HBS (HEPES-buffered saline) buffer were labeled with 1 mM Cy3-NHS or Cy5-NHS for 1 h and quenched with an equal volume of 100 mM Tris, pH 8.0, for 1 h. HisSorb 96-well plates (Qiagen) were blocked for 30 min with Tris-buffered saline (100 mM Tris, pH 8.0, 150 mM NaCl) plus 0.5% bovine serum albumin (TBS-BSA), and wells were coated with 50 μl of 30 nM His-tagged pre- or postfusion F-GCNt or HN37–565 diluted in TBS for 60 min. Wells were washed five times with TBS–0.05% Tween 20 (TBST), and 50 μl of Cy3-labeled sAbs was added to appropriate wells (1 sAb per column). After 30 min, 50 μl of 50 nM Cy5-labeled sAbs was added (1 sAb per row), and the plate was sealed and incubated in the dark at 4°C for 24 h. Plates were then washed five times with TBST, and 100 μl of TBS was added to each well. Fluorescence was quantified on a Tecan plate reader using excitation and emission wavelengths for Cy3 signal detection of 540 to 550 nm and 565 to 600 nm, respectively, and of 640 to 650 nm and 665 to 700 nm, respectively, for Cy5.
Surface plasmon resonance (SPR).
Binding kinetics were determined using a BIAcore 2000 with His-tagged pre- or postfusion F-GCNt or HN37–565 immobilized on a Ni-NTA chip (GE Healthcare). The purified sAbs were dialyzed into HBS running buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% Tween 20), and serial dilutions were injected (50 μl each) at a flow rate of 25 μl/min. Binding responses were corrected by subtraction of the response on a blank flow cell. A 1:1 Langmuir model using global fitting of kinetic parameters was used, and KD (equilibrium dissociation constant) values were determined from the kon and koff values.
Immunoprecipitation (IP) and SDS-PAGE.
293T cell monolayers in six-well plates transfected with 1 μg of pCAGGS-MCS, 1 μg of pCAGGS-F, or 2 μg of pCAGGS-HN were starved in DMEM deficient in cysteine and methionine for 30 min, followed by labeling with 67 μCi of 35S label (Promix, GE Healthcare) in the same medium for 90 min. The cells were subsequently lysed in cold DH buffer (50 mM HEPES, pH 7.4, 10 mM lauryl maltoside, 150 mM NaCl). The lysate was then clarified by centrifugation at 16,000 relative centrifugal force (RCF) for 10 min at 4°C. Clarified lysates were incubated with a suitable antibody for 1 h at 4°C, following which protein A-Sepharose beads were added, and the samples were further incubated for 30 min at 4°C. Antibody-antigen complexes were washed twice with DH buffer and once with 50 mM HEPES (pH 7.4)–150 mM NaCl. The proteins were eluted from the beads by boiling for 3 min in protein lysis buffer (containing 100 mM dithiothreitol and 1% SDS) and separated on a 15% acrylamide gel. Radioactivity was detected using a Fuji FLA-5100 image reader with Multi Gauge, version 3.0, software (Fuji Medical Systems, Stamford, CT).
EM.
Solutions of F-GCNt or HN37–565 alone or with sAbs at a total protein concentration of approximately 5 mg/ml were absorbed onto 300-mesh copper grids covered with a carbon film that had been freshly glow discharged. Grids were stained with a 1% aqueous solution of freshly prepared and filtered uranyl formate. Grids were observed in a JEOL 1230 electron microscope (EM) operated at 100 kV, and images were acquired with a Gatan 831 charge-coupled-device (CCD) camera at the Biological Imaging Facility, Northwestern University, Evanston, IL.
Viral inhibition assay.
Serial dilutions of sAbs were prepared in medium containing recombinant PIV5 virus expressing a green fluorescent protein (GFP) reporter gene. Virus and sAb were incubated for 30 min at 4°C before the addition of 100 μl to CV1-E cell monolayers in black-walled 96-well plates (Costar) at an MOI of 1.3. Following 12 to 16 h of incubation at 37°C, cells were washed, and 100 μl of phosphate-buffered saline (PBS) was added. GFP signal was quantified via a SpectraMax M5 plate reader (Molecular Devices) using excitation and emission wavelengths of 488 nm and 510 nm, respectively.
Luciferase reporter cell-cell fusion inhibition assay.
CHO-K1 effector cell monolayers in 96-well plates were transfected with 0.1 μg each of pCAGGS-F, pCAGGS-HN, and pT7-luciferase plasmids. CHO-K1 target cell monolayers in six-well plates were similarly transfected with 2 μg of the plasmid carrying T7 RNA polymerase. After 2 h, effector cells were washed with PBS, and 50 μl of a 1:1 solution of PBS and Opti-Mem containing sAb at 2× the final concentration was added to the cells. Target cells were washed with 530 μM EDTA in PBS and incubated with 150 μl of EDTA in PBS for 10 min at room temperature with shaking. Opti-MEM was then added at 1.6 ml per well, and 50 μl of suspended target cells was overlaid on the effector cells. Following a 12- to 16-h incubation at 37°C, cells were lysed by incubation with 50 μl of lysis buffer (Promega) per well for 10 min at room temperature (RT) with vigorous shaking. Subsequently, 50 μl of luciferase assay substrate (Promega) was mixed with the cell lysate. Ninety microliters of the lysate/substrate solution was transferred to a white opaque 96-well plate, and luciferase activity was determined using a SpectraMax M5 plate reader (Molecular Devices).
NA inhibition assay.
Serial dilutions of sAbs at 4× final concentrations were prepared in 2× final concentrations of neuraminidase (NA) activity assay buffer (200 mM Tris, pH 7.0, 10 mM CaCl2) and added to an equal volume of 40 nM (4× stock) purified PIV5 HN37–565 in 2× NA activity assay buffer in a black-walled 96-well plate. Following a 30-min incubation at RT, an equal volume of 2 mM methylumbelliferyl N-acetylneuraminic acid (MUNANA) in H2O was added, and the plate was incubated for 30 min at 37°C. The reaction was quenched by the addition of 20 mM sodium carbonate, pH 10.4, to a final concentration of 37.5%. Fluorescence was quantified via a SpectraMax M5 plate reader (Molecular Devices) using excitation and emission wavelengths of 356 nm and 450 nm, respectively.
HA activity inhibition assay.
Serial dilutions of sAb at 4×the final concentration were prepared in PBS and added to an equal volume of PIV5 virus diluted 1:8 (4× the final concentration) in PBS in a 96-well plate. Following a 30-min incubation at 4°C, an equal volume of 2% fresh chicken red blood cells (RBCs) in Alsever's solution was added to each well and incubated for 60 min at 4°C, and plates were photographed. The highest dilution of virus for which HA activity was observable in the absence of inhibitor was 1:128 (4-fold more dilute than the virus dilution used in this assay).
Selection for resistance mutations.
Resistance mutations to selected inhibitory sAbs and MAbs F1a and 4b were generated by serial passage of PIV5-infected MDBK cells in the presence of increasing concentrations of inhibitor. An amount of virus equivalent to an MOI of 0.2 was preincubated for 30 min at 4°C in the presence of inhibitor at a concentration that inhibited ∼75% of the virus (IC75) in DMEM containing 1% BSA. The solution was then transferred to washed 90% confluent MDBK cell monolayers in a T25 flask and incubated at 4°C for 60 min. Four milliliters of DMEM containing 2% FBS and the same inhibitor concentration as above was then added, and the infected cells were incubated for 3 to 4 days at 37°C. HA activity assays were performed on cell supernatant on days 3 and 4 to assess the viral titer (HA titer had been previously correlated with plaque assay titer for wild-type [wt] virus). Based on the HA titer, inhibitor concentrations were adjusted in order to increase selective pressure while maintaining sufficient viral titer. The HA titer was also used to determine how much supernatant was required to infect a fresh monolayer at an MOI of 0.2. After five passages, viral titers were adequate in the presence of inhibitor at ≥10× the original IC75 concentration. Supernatants potentially containing a pool of multiple viral clones resistant to each inhibitor were harvested.
Isolation of resistant virus clones and verification of resistance.
Individual sAb- and MAb-resistant viral clones were isolated from supernatant containing resistant virus by standard plaque assay. BHK-21F cells were infected with dilutions of passaged MDBK cell supernatants for 1 h at 4°C. Following infection, monolayers were washed with PBS and overlaid with 1% low-melting-point agarose in DMEM containing 4% FBS and 2% tryptose phosphate broth. Upon solidification, plates were incubated for 96 h at 37°C, and a second overlay was added (same as above but with 0.01% neutral red). After an additional 16 to 24 h of incubation at 37°C, plaques were picked using a sterile Pasteur pipette. The agar plug was transferred to 1 ml of DMEM containing 1% BSA and used to infect MDBK cell monolayers as described above. In some cases, this procedure was repeated using the amplified supernatant from the original plaque to ensure a clonal population of virus.
Plaque reduction assays were performed on viral clones to verify resistance. Following infection of BHK-21F cells in the presence of inhibitor, these assays were performed as described above, except that agarose overlays were replaced with a 1:1 mix of 2.4% Avicel and DMEM containing 8% FBS and 4% tryptose phosphate broth and 2× the final concentration of inhibitor; cells were incubated for 72 h following Avicel overlay, and cells were stained with naphthol blue-black for 60 min at RT.
Viral sequencing.
Viral RNA was isolated from the cell supernatant using a QIAamp Viral RNA minikit (Qiagen). Viral RNA was reverse transcribed and amplified using a SuperScript III One-Step PCR System (Invitrogen) using F- or HN-specific primers. Resulting amplicons were sequenced using an Applied Biosystems 3100-Avant automated DNA sequencer.
RESULTS AND DISCUSSION
Target preparation and phage display.
A prefusion stabilized version of the PIV5 F ectodomain (F-GCNt) and the HN ectodomain (residues 37 to 565; HN37–565) were expressed using recombinant baculovirus-infected insect cells (8, 32). The proteins were purified by Ni-NTA affinity chromatography, and reducing SDS-PAGE and silver staining indicated that the purity of each protein was >90%. Western blotting using PIV5 F- and HN-specific antibodies confirmed the identity of each protein (Fig. 1A and B). Subsequent size exclusion chromatography (SEC) indicated that F-GCNt and HN37–565 were trimeric and tetrameric, respectively. SEC also removed a significant amount of aggregates, especially from the F-GCNt sample (Fig. 1C to E). A portion of the purified F-GCNt was heated to convert it to the postfusion form as previously described (48), and portions of the pre- and postfusion F-GCNt and HN37–565 were lightly biotinylated to allow subsequent capture on streptavidin (SA)-coated beads during phage display. Electron microscopy (EM) was used to estimate the homogeneity of the pre- and postfusion F-GCNt samples. The prefusion F-GCNt sample was an ∼70/30 mixture of the pre- and postfusion conformations, respectively, whereas the postfusion F sample was nearly homogenous (data not shown). The EM data also confirmed that biotinylation did not cause gross misfolding or aggregation of the purified proteins.
FIG 1.
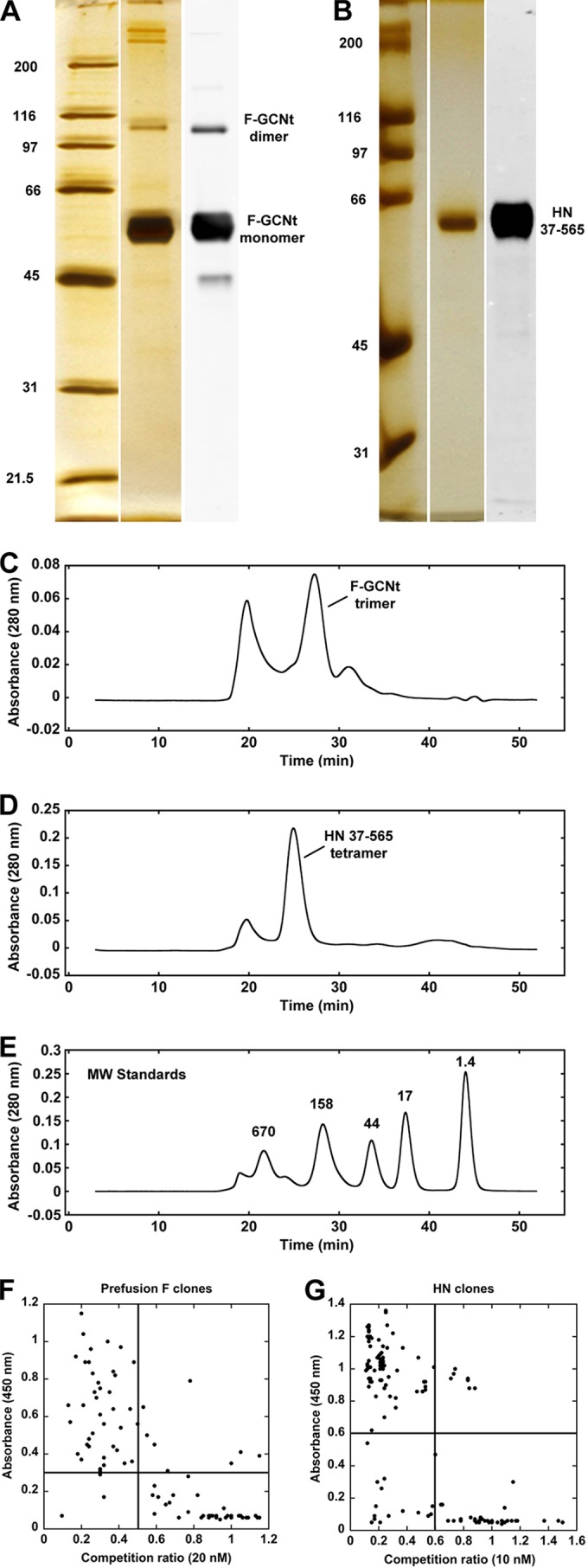
Target preparation and phage ELISA results. (A) Silver-stained SDS-PAGE analysis of Ni-NTA-purified F-GCNt used for phage display. A corresponding Western blot with anti-PIV5 F Ab confirms protein identity. (B) A similar analysis as in panel A of the HN37–565 protein used for phage display. (C) SEC verified that the F-GCNt protein was trimeric and removed aggregates and/or high-molecular-weight contaminants. (D) SEC of HN37–565 tetramers. (E) Profile of molecular weight (MW) markers (in thousands) under the same conditions as for F-GCNt and HN37–565. Aggregates in each sample eluted in the void volume peak at ∼20 min. (F) Phage ELISA results of individual clones selected against the biotin-labeled prefusion form of F-GCNt. The y axis shows the magnitude of the phage ELISA signal while the x axis shows the ratio of the ELISA signal in the presence (numerator) and absence (denominator) of nonbiotinylated F-GCNt used as a soluble competitor at 20 nM. Clones in the upper left quadrant represent the highest-affinity clones with target specificity. Arbitrary cutoffs of 0.3 (A450) and 0.5 (competition ratio) were used to select phage for subsequent analysis. (G) Similar plot as in panel F of the ELISA and solution competition ELISA results for HN37–565. Nonbiotinylated HN37–565 was used at 10 nM in the competition ELISA, and an arbitrary cutoff of 0.6 was used for both parameters to select clones for subsequent analysis.
Phage display was performed essentially as described by Paduch and coworkers (54). M13 phage bound to biotinylated targets were captured from solution using magnetic streptavidin-coated beads; the beads were washed, and the specifically bound phage were eluted and used in subsequent rounds of panning to enrich for phage with the highest target affinity. In addition to simple target screens, counterscreens were employed. For example, nonbiotinylated postfusion F-GCNt was added to the nonhomogenous biotinylated prefusion F-GCNt sample to prevent capture of anti-postfusion phage. Following the selections, clones were amplified and a phage ELISA was performed to verify target binding. The ELISA was repeated in the presence of soluble competitor (nonbiotinylated version of the same target) for initial specificity assessment. The tightest binding (highest ELISA signal) and most specific (lowest competition ratio) phage (Fig. 1F and G, upper left quadrant) were then sequenced to identify unique clones (see Table S1 in the supplemental material). Altogether, 24 unique anti-prefusion F, 17 unique anti-postfusion F (one sAb recognized both pre- and postfusion F), and 12 unique anti-HN sAbs were generated.
Antigenic site binning and affinity and specificity measurements of sAbs.
To characterize the sAbs outside the context of phage, DNA from each clone was reformatted for protein expression in Escherichia coli by the insertion of a stop codon after the last residue of the heavy chain (before gene III of the phage). sAbs were then expressed and purified in a 96-well microscale format as previously described (54).
To sort sAbs into bins recognizing distinct antigenic sites, we performed a two-color fluorescence competition assay. sAbs were labeled with Cy3-NHS or Cy5-NHS ester, and a limiting amount of the relevant His-tagged target was captured onto Ni-NTA plates. Each Cy3-labeled sAb competed with the Cy5-labeled version of itself or each of the other sAbs (at equal concentrations) for target binding (and vice versa). Assays were normalized to each sAb signal in the absence of competition. Each sAb competed with itself, resulting in ∼50% signal from each color (Fig. 2, e.g., 3C:3E). sAbs that did not compete for the same antigenic site gave ∼100% signal for each color for each sAb (Fig. 2, e.g., 3E:3C/3C:3E). sAbs that competed for the same antigenic site either gave ∼50% signal for each color (if they had similar affinities), or the signal from the higher-affinity sAb dominated (Fig. 2, e.g., 3D:3C/3C:3D). We determined that the anti-prefusion F sAbs bound to five distinct antigenic sites and that all but one of the anti-postfusion F sAbs bound to the same antigenic site. Additionally, with one exception, all of the anti-HN sAbs bound to a single antigenic site.
FIG 2.
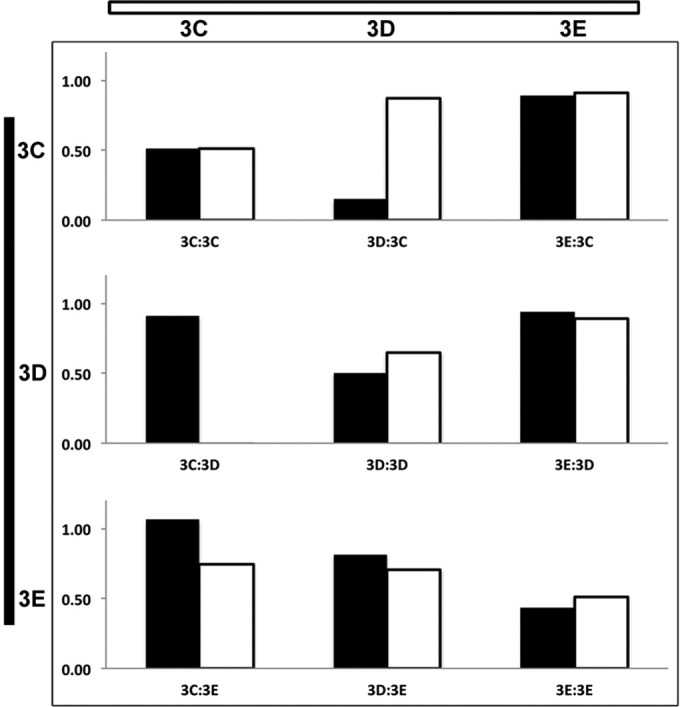
Binning of sAbs by unique antigenic site. (A) Representative data from a two-color solution competition ELISA is shown. Each sAb was labeled with Cy3 and Cy5 fluorescent dye, and competitive binding to immobilized target was assessed for pairwise combinations. sAbs competing with themselves resulted in half-maximal binding for each color label compared to the signal in the absence of competition (3C:3C). Different sAbs that competed for the same antigenic site that had the same affinity also resulted in half-maximal binding. However, the higher-affinity sAb dominated when sAbs with different affinities competed for binding to the same antigenic site (e.g., 3D:3C/3C:3D). When sAbs do not compete for binding, both sAbs bind at similar levels as in the absence of competition (3E:3C/3C:3E and 3E:3D/3D:3E).
Surface plasmon resonance (SPR) was performed to assess the kinetics and affinity of each sAb. His-tagged pre- and postfusion F-GCNt and HN37–565 targets were captured onto a four-channel Ni-NTA chip (one channel was used as a reference surface), and purified sAbs were individually flowed over each surface. This experimental design also served to validate target specificity. Because the prefusion F-GCNt target was somewhat contaminated with the postfusion form, postfusion-specific sAbs bound to that surface but with significantly reduced responses. Binding of sAb 3D was unique in that it gave a significant signal for both pre- and postfusion F-GCNt surfaces. All of the sAbs to F and HN bound with affinities ranging from ∼125 nM to subnanomolar range (the vast majority have a KD in the low-nanomolar to picomolar range). The highest-affinity sAbs recognizing unique antigenic sites were chosen for further characterization based on these results (Fig. 3; Table 1).
FIG 3.

Surface plasmon resonance (SPR) of various sAbs. SPR sensorgrams of representative sAbs binding to targets are shown. His-tagged pre- and postfusion forms of F-GCNt and HN37–565 were captured onto separate channels of a four-channel Ni-NTA surface, and sAbs were flowed over each surface at multiple concentrations. sAb 3D uniquely bound significantly to both conformations of F-GCNt (right). Raw data (black lines) and curve fits (red lines) are shown. Approximate KD values are indicated.
TABLE 1.
sAb properties
| Target and Ab | Affinity (∼KD [nM]) | ∼IC50 (nM) for: |
Antigenic site | Resistance mutation(s) | |||
|---|---|---|---|---|---|---|---|
| Virus neutralization | Syncytium formation | NA activity | HA activity | ||||
| Anti-prefusion F | |||||||
| 5D | <1 | 160 | 60 | NAc | NA | Side of head | N34D, E85G, F372S |
| 3E | <1 | >10,000 | >10,000 | NA | NA | Top of head | NT |
| 1B | 5 | >10,000 | >10,000 | NA | NA | Upper stalk | NT |
| 3C | <1 | >10,000 | >10,000 | NA | NA | Middle stalk | NT |
| 3Da | 10 | >10,000 | >10,000 | NA | NA | Middle stalk | NT |
| 1G | 30 | >10,000 | >10,000 | NA | NA | Lower stalk | NT |
| MAb F1a | NT | 1:220 dilutiond | 1:200 dilution | NA | NA | Head | E132G |
| Anti-postfusion F | |||||||
| 1D | 2 | >10,000 | >10,000 | NA | NA | Top of crutch | NT |
| 3Da | 30 | >10,000 | >10,000 | NA | NA | Bottom of crutch | NT |
| Anti-HN | |||||||
| 1H | <1 | 30 | <10 | >1,000 | >1,000 | Stalk | A82T, A82V |
| 4E | <1 | 25 | <10 | >1,000 | >1,000 | Stalk | Q86K |
| F4 | 10 | 300 | 100 | 100 | 100 | Top of head | F457S |
| MAb 4b | NTb | NT | <1:100 dilution | >1:100 dilution | >1:100 dilution | Head | R248K |
Pre- and postfusion F specific.
NT, not tested.
NA (body of table), not applicable.
Dilution of MAb containing ascites fluid or culture medium with specified effect.
We then demonstrated the ability of selected sAbs to bind purified targets by immunoprecipitating soluble pre- or postfusion F-GCNt or HN56–565 using protein A-coated beads (Fig. 4A and B). Anti-prefusion F-GCNt sAbs were unable to immunoprecipitate postfusion F-GCNt (Fig. 4A) and vice versa (data not shown). sAb 03D was uniquely able to immunoprecipitate pre- and postfusion F-GCNt. Furthermore, selected sAbs were able to immunoprecipitate wild-type F or HN from transfected cells (Fig. 4C and D). These data demonstrate our success in producing conformation-specific sAbs that bind to PIV5 F and HN and that serve as novel tools for studying PIV5 membrane fusion.
FIG 4.
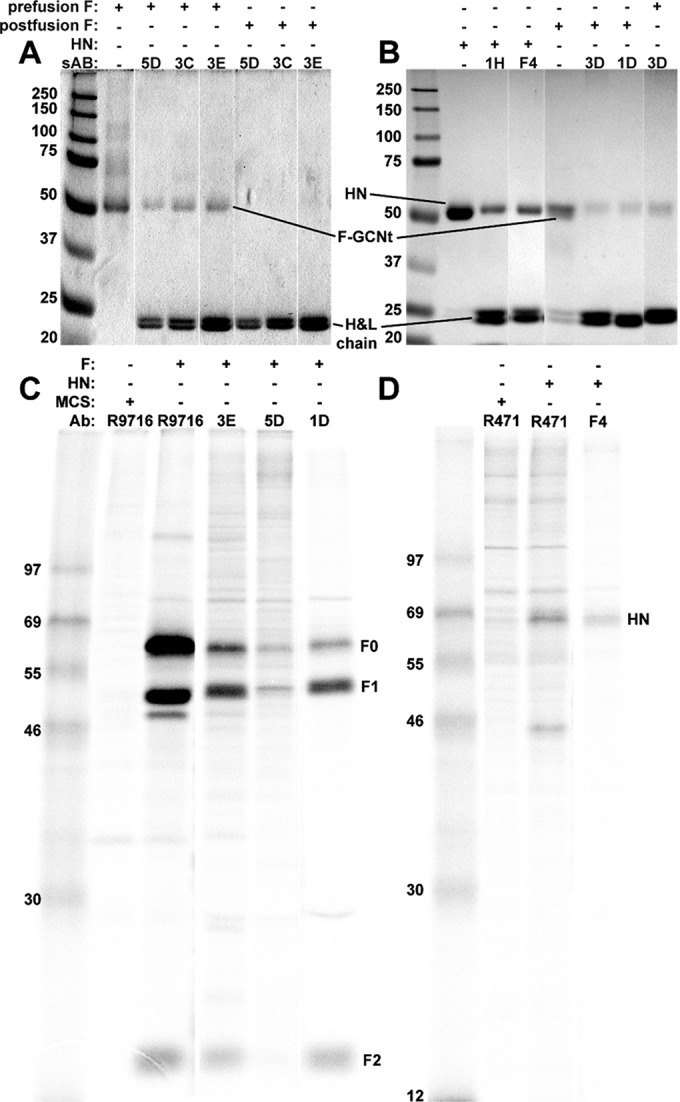
Immunoprecipitation (IP) of various sAbs. (A) IP followed by reducing SDS-PAGE and Coomassie staining of the purified prefusion forms of F-GCNt using prefusion conformation-specific sAbs (third to fifth lanes). These sAbs did not immunoprecipitate F-GCNt in the postfusion conformation (sixth to eighth lanes). (B) IP followed by reducing SDS-PAGE and Coomassie staining of HN56–565 using anti-HN-specific sAbs (third and fourth lanes) and IP of the postfusion form of F-GCNt using anti-postfusion-specific sAbs (sixth and seventh lanes). 3D was able to immunoprecipitate both forms of F-GCNt (sixth and eighth lanes). (C) IP and SDS-PAGE of radiolabeled PIV5 F from transfected cells using an anti-F pAb (lane 3), anti-prefusion-specific sAbs (fourth and fifth lanes), and an anti-postfusion-specific sAb (sixth lane). (D) IP and SDS-PAGE of radiolabeled PIV5 HN from transfected cells using an anti-HN-specific pAb (third lane) and an anti-HN-specific sAb (fourth lane). H&L chain, heavy and light chains. Values to left of panels indicate molecular masses (in kilodaltons).
Functional assays.
The ability of sAbs representing each unique antigenic site on F and HN to neutralize PIV5 entry and replication was tested (Fig. 5A and Table 1). Surprisingly, only a single anti-prefusion F sAb, 5D, blocked virus replication or syncytium formation despite the high affinity (KD of ≤10 nM) of each of these prefusion-specific sAbs. 5D likely inhibits fusion either by stabilizing F in the prefusion form, thus preventing it from refolding to the postfusion conformation, or, alternatively, by preventing the interaction with HN that triggers F to refold. None of the postfusion F-specific sAbs inhibited membrane fusion (data not shown), and this may explain why many neutralizing antibodies are directed to the attachment protein.
FIG 5.

Virus-cell fusion neutralization, cell-cell fusion inhibition, and neuraminidase (NA) inhibition data for representative sAbs. (A) Virus-cell fusion neutralization data. sAbs were preincubated for 30 min with replication-competent, GFP-labeled virus prior to infection at an MOI of 1.3. The anti-prefusion-specific sAb, 5D, was uniquely inhibitory among the anti-F-GCNt sAbs (dashed lines). All of the anti-HN sAbs (solid black lines) were inhibitory. (B) NA inhibition data generated from incubating soluble purified HN56–565 and anti-HN sAbs with MUNANA substrate. Only sAb F4 inhibited NA enzymatic activity. (C) Cell-cell fusion inhibition. F and HN were expressed in CHO-K1 cells and subsequently with CHO-K1 cells expressing T7 RNA polymerase in the presence and absence of inhibitors. Fusion was quantified using a luciferase reporter assay. AU, arbitrary units. (D) BHK cells expressing F and HN or F and HN1–117 stalk (41) at 15 h posttransfection were incubated with 200 nM sAb 1H for 4 h and photographed.
In contrast to the anti-F sAbs, each of the anti-HN sAbs was inhibitory, albeit F4, the anti-HN sAb recognizing an antigenic site distinct from the others, was markedly less potent in neutralization (Fig. 5A). The ability of anti-HN sAbs to inhibit NA (Fig. 5B and Table 1) and HA (Table 1) activity was also assessed to determine their inhibitory mechanism(s). Interestingly, F4 uniquely inhibited HA and NA activity and at concentrations similar to its IC50 in neutralization and inhibition assays. We discuss these results below in light of antigenic site location and a recently proposed model for F activation by HN.
Inhibition of syncytium formation by the sAbs mirrored the ability of the antibodies to neutralize infectivity, with 5D, 1H, 4E, and F4 all inhibiting fusion activity (Fig. 5A and C). sAb 1H, as an example of an HN stalk-specific antibody, was tested for its ability to block activation of F syncytium formation by binding to the HN stalk region consisting of residues 1 to 117 (HN1–117) (41). As shown in Fig. 5D, sAb 1H addition to cells expressing either F and HN or F and HN1–117 stalk blocked syncytium formation.
Antigenic site mapping by EM and mutagenesis.
We visualized sAb/target complexes by single-molecule EM and negative staining to determine the approximate location of each unique antigenic site (Table 2). Figure 6 shows at least one target-bound sAb representing each unique antigenic site identified by the two-color competition assay for each of the targets. F4, the unique sAb that inhibited both NA and HA activity, bound to the HN NA domain (Fig. 6, top row, right). Interestingly, the sAbs that did not inhibit HA and NA function, represented by 1H and 4E, but potently neutralized viral entry/replication and inhibited syncytium formation clearly bound to the HN stalk (Fig. 6, top row, left). Cartoons of the EM images are shown in Fig. 6 to help orientate the reader, with F or HN in black and the sAb in blue.
TABLE 2.
Location of sAb antigenic sites as determined by competition ELISA and electron microscopy
| Target (site no.) | sAb(s)a |
|---|---|
| Prefusion F head | |
| Side (1) | 5D, 5E, H12, G12 |
| Top (2) | 3E |
| Prefusion F stalkb | |
| Upper (3) | 1B |
| Middle (4) | 1A, 2D, 2G, 3A, 3C, 3Dc, 3F, 4J |
| Lower (5) | 1G, 3H |
| Postfusion F | |
| Top of crutch (6) | 1C, 1D, 1F, 4C, 4H, 6D, 6G, 7F, 8B, 8F, 9C, 9D, 9E, 9F, 10A, 10B |
| Bottom of crutch (7) | 3Dc |
| HN | |
| Head (8) | F4 |
| Stalk (9) | 1H, 3B, 3H, 4E, 4G, 6D, 6E, 6F, 7B, 7E, B6, C8 |
The epitopes for 3G and 4D were not characterized. Boldface type indicates sAb found in Fig. 6.
sAbs 2A, 5A, 5H, 6A, 6B, and 6C bind to the prefusion F stalk, but epitopes were not further delineated.
Dual specificity.
FIG 6.

Single-molecule electron microscopy of pre- and postfusion forms of F-GCNt and HN37–565 alone and with representative sAbs from each unique antigenic site bound. These images reveal the approximate locations of the different antigenic sites on the target molecules (i.e., stalk versus NA domain for HN and stalk, side of head, and top of head for F). sAbs have been artificially colored to make them more visible. First row, anti-HN stalk sAbs and anti-HN head (NA domain) sAb; second row, anti-prefusion F stalk sAbs; third row, anti-prefusion F head sAbs and anti-postfusion F sAbs; fourth row, unbound HN, prefusion F (Fpre), postfusion F (Fpost), and sAb alone. Cartoons of the EM images are shown to help orientate the reader, with F or HN in black and the sAb in blue. α, anti.
It is surprising that the stalk antigenic site of HN was dominant in our selection, given that the antigenic response to viral particles in vivo is mostly against the HN NA domain (57–60). The unique HN NA domain binder, F4, was identified in an alternate phage display selection in which biotinylated tris-nitrilotriacetic acid (BT-NTA) was used to noncovalently attach biotin to the N-terminal His tag of HN37–565 versus random biotinylation of exposed Lys residues (the biotin labeling strategy otherwise used). Steric constraints may have precluded phage bound to the NA domain from being captured onto the SA surface with random Lys biotinylation, but this would not be expected when biotin is located at the N terminus of HN′s stalk domain. Alternatively, the dense packing of F and HN on the surface of a virion could sterically obscure the stalk domain antigenic site when Abs are produced against whole virions. Additionally, the variance could be attributed to differences in how the immune system “selects” antigenic sites in vivo compared to phage display selection.
sAbs representing three of the five different anti-prefusion F antigenic sites identified appeared to bind to the F stalk (Fig. 6, row 2). While it is difficult to distinguish these antigenic sites based on EM, 1B appeared to bind to the top of the stalk just below the globular head, 1G appeared to bind to the bottom of the stalk (possibly the GCNt trimerization domain), and 3C appeared to bind to the middle region of the stalk. sAb 3D competed with 3C for binding to prefusion F-GCNt (Fig. 2), and both of these sAbs appeared to bind in similar locations in the middle stalk region, as observed by EM, indicating that they have distinct but overlapping antigenic sites. The two categories of anti-prefusion F head-binding sAbs also appeared distinct by EM. 3E appeared to bind an antigenic site near the top of the globular head while 5D-like sAbs bound to an antigenic site on the side of the head (Fig. 6, row 3, left).
Each of the postfusion-specific sAbs, represented by 1D, recognized the same antigenic site near the widest part of the golf-tee-shaped postfusion F protein. Conversely, 3D recognized an antigenic site at the narrow end (Fig. 6, row 3, right). Given the location of the 3D antigenic sites in pre- and postfusion F-GCNt, it is likely that 3D recognizes a single antigenic site in heptad repeat B (HRB), with conserved secondary structure between the two forms of F.
In addition to mapping antigenic sites by EM, we selected for resistance mutations of representative inhibitory sAbs to determine more precisely their antigenic sites on F and HN. Selected sAbs included 1H and 4E (anti-HN stalk), F4 (anti-HN NA domain), and 5D (anti-prefusion F, side of head). Neutralizing MAbs 4b (anti-HN) and F1a (anti-F) (61) were also included in the resistance selections for comparison. To select for mutations, PIV5-infected MDCK cells were passaged with increasing concentrations of inhibitory antibody, and viral titers were monitored at each passage via HA titer assay. The concentration of inhibitor was adjusted as seemed appropriate based on the HA titers at each passage. After five rounds of passaging, resistant virus grew in the presence of inhibitor at a concentration at least 10-fold above the initial ∼IC75. Resistant strains from cell supernatants were plaque purified and amplified, and the F and HN gene nucleotide sequences were obtained to identify resistance mutations (Table 1). Plaque reduction assays were also performed on clonal resistant virus stocks to ensure resistance compared to wild-type virus (data not shown).
Subtle structural changes at the cleavage site occur in prefusion F upon protease cleavage of PIV5 F0 into F1/F2 (33), and MAb F1a reactivity is sensitive to these changes. In accordance with this observation, the identified F1a-resistant mutation, E132G, is a surface-exposed residue in the heptad repeat A (HRA) helix adjacent to the cleavage site in the PIV5 cleaved and uncleaved prefusion atomic structures. Also, in the postfusion F atomic structure of hPIV3, a closely related paramyxovirus, HRA is an extended helix (36), and this conservation of secondary structure for the F1a antigenic site likely explains why F1a retains some reactivity for PIV5 postfusion F (Fig. 7A and B).
FIG 7.
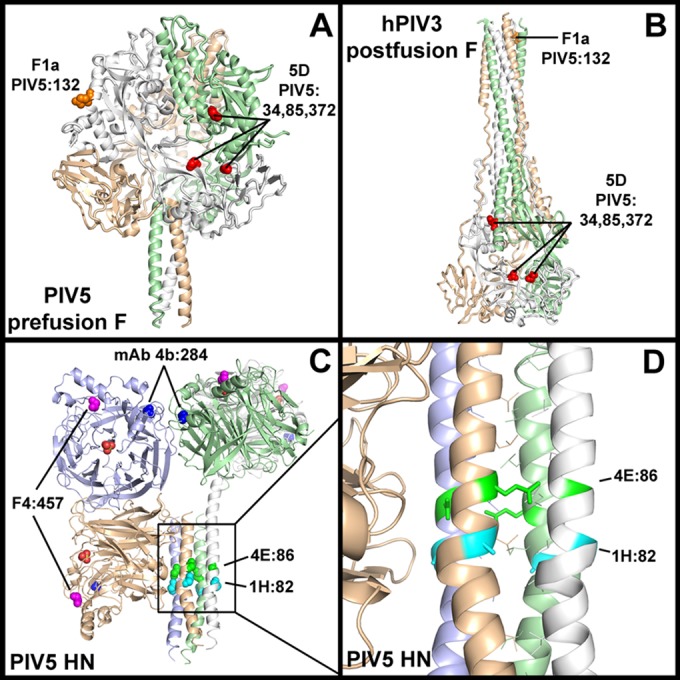
Ab resistance mutations in F and HN. (A) Cartoon representation of the prefusion cleaved F-GCNt crystal structure (Protein Data Bank [PDB] entry 4GIP). Anti-PIV5 F sAb 5D and MAb F1a resistance mutations are shown as red and orange spheres, respectively, and mutated PIV5 residue numbers are indicated. (B) Cartoon representation of the postfusion hPIV3 F crystal structure (PDB 1ZTM) showing the location of the same antigenic sites (based on primary sequence alignment). For clarity, only a single antigenic site is shown for each Ab within the F trimers in panels A and B. (C) Cartoon representation of the PIV5 HN56–565 ectodomain two-heads-up/two-heads-down structure (PDB 4JF7), with resistance mutations of anti-PIV5 HN sAbs 1H, 4E, F4, and MAb 4b (61) shown as cyan, green, magenta, and blue spheres, respectively. The PIV5 residue numbers corresponding to the resistance mutations are indicated. A sulfate ion in the active site of each NA domain is shown as orange and red spheres. The resistance mutations in all four monomers are shown. (D) A zoomed-in view of the stalk region of HN reveals that the identified stalk-binding sAb resistance mutations (shown as sticks) are surface exposed.
Multiple resistance mutations, N34D, E85G, and F372S, were found in separate clones from the anti-F inhibitory sAb 5D resistance selection. These residues map to a noncontiguous antigenic site comprised of two adjacent protomers that is distinct from that of F1a or the PIV5 equivalent antigenic sites (based on multiple-sequence alignment) of well-characterized antigenic sites in RSV (34). The movement of protomers relative to each other upon transition from the pre- to postfusion conformation disrupts the 5D antigenic site, as shown by mapping the identified residues onto the PIV5 prefusion and hPIV3 postfusion structures (Fig. 7A and B).
F-HN interaction and fusion-triggering mechanism.
We recently proposed a simple and potentially general model of paramyxovirus F protein activation in which the head domains of the attachment protein move from a down position to an up position following receptor binding (Fig. 8). This movement exposes critical residues in the stalk domain that interact with and trigger F to initiate membrane fusion (41, 62, 63). This model is based on the following observations: (i) mutations in HN that affect only fusion activity map to the stalk region of the attachment protein of several paramyxoviruses (12, 18, 42, 43, 64, 65); (ii) headless PIV5 and NDV HN, MeV H, and NiV G are sufficient to trigger fusion (41, 63, 66, 67); (iii) the head domains of HN have been observed by EM to be in various orientations relative to the stalk for NDV (12) and PIV5 (41); (iv) a crystal structure of the tetrameric NDV HN ectodomain reveals the heads in the down position, forming an interface with the stalk that overlaps with the critical fusion promoting region (“four-heads-down” conformation) (12); (v) different crystal structures of PIV5 HN tetramers have revealed the heads can adopt a four-heads-up conformation or a hybrid two-heads-up/two-heads-down conformation (8, 30); (vi) there is an energetic requirement for the movement of the heads, likely indicating that receptor binding is the trigger (41).
FIG 8.
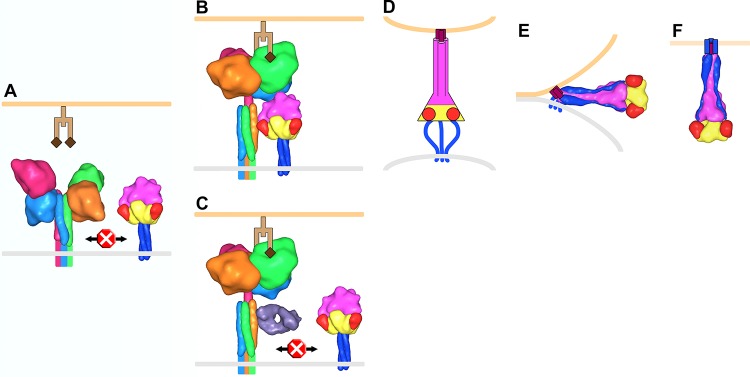
Model of PIV5 membrane fusion. (A) F trimers (yellow, red, magenta, and blue corresponding to domains I, II, III, and HRB, respectively) and HN tetramers (monomers shown as light blue, hot pink, orange, and green) reside on the surface of the virus membrane (gray). Structural data are lacking for the membrane-proximal portion of the HN stalk, and this region, the transmembrane domain, and the cytoplasmic tails are depicted as colored lines. Prior to receptor (light and dark brown representing a glycan chain terminating with sialic acid) engagement on a host cell membrane (light orange), HN is in a four-heads-down conformation in which the heads sterically block interaction between the membrane-distal portion of the HN stalk and the head domain of F. (B) HN head domains transition to a four-heads-up position upon receptor engagement allowing the F/HN interaction. HN is rotated ∼45° clockwise in panels B and C compared to the orientation in panel A. (C) Antibodies (Fab, violet) that bind to the membrane-distal portion of the HN stalk inhibit membrane fusion by blocking the interaction with F. (D) The F-HN interaction results in a major conformation change in F such that domain III refolds into an extended trimeric coiled coil (HRA) and inserts a hydrophobic fusion peptide (purple) into the target cell membrane (for simplicity HN and receptor are not shown in panels D to F). Structural data are lacking for this “prehairpin intermediate” conformation; therefore, it is depicted exclusively as a cartoon. (E) HRB interacts with HRA, forming a thermostable six-helix bundle domain in the postfusion conformation, which juxtaposes the virus and target cell membranes leading to membrane fusion (F). To make this figure, Sculptor (version 2.1.1_r1 [http://sculptor.biomachina.org]) (69) was used to morph high-resolution structures of HN, F, and the humanized anti-HER2 Fab, 4D5 (which the sAb phage library was based on), to 15-Å surface representations. Protein Data Bank (PDB) entries 3T1E and 3TSI were overlaid and used for the HN four-heads-down model. Similarly, PDB 1Z4X, 3TSI, and 4JF7 were used for the HN four-heads-up model. PDB entries 2B9B and 1ZTM were used for the pre- and postfusion representations of F, respectively. The GCNt trimerization domain was used as a surrogate to represent the TM and CT domains of prefusion F. PDB 1FVD was used for the Fab.
A key tenant of this model (Fig. 8) is that when the heads are in the down position, access to the fusion-promoting region of the attachment protein stalk is sterically blocked, but that when the heads move to the up position (or are absent), the block surrounding this critical region is removed, allowing access of this region to F. The model predicts that antibodies (or other inhibitors) that bind to the fusion-promoting region of attachment protein stalks would inhibit fusion. sAb 1H, an example of an HN stalk-specific antibody, inhibits headless HN1–117 stalk from activating F and blocks syncytium formation (Fig. 5D). By EM, sAbs 1H and 4E bind to the middle region of the HN stalk, and their resistant mutations map to surface-exposed residues A82 and Q86, respectively, which comprise a portion of the head-stalk interface as observed in the two-heads-up/two-heads-down crystal structure (Fig. 7C and D). Interestingly, we recently showed that these residues could tolerate a variety of mutations while maintaining the ability to trigger F but that directly adjacent hydrophobic residues V81 and L85, critical for triggering PIV5 F, were much less tolerant of substitution (63). This may partially explain why A82 and Q86 were identified when we were selecting for viable sAb resistance mutations. Additionally, as the anti-HN stalk-binding sAbs do not disrupt NA or HA activity, they further establish the fusion-promoting region of the HN stalk. The generality of this inhibitory mechanism is supported by previously identified neutralizing MAbs against the hPIV2 HN stalk (68).
The model (Fig. 8) also predicts that Abs binding to the HN interaction site on F would prevent fusion by blocking interaction with the HN stalk. 5D recognizes a quaternary antigenic site and may neutralize by stabilizing F trimers in the prefusion form or by blocking an association with HN. Interestingly, one of the 5D resistance mutations, F372, is directly adjacent to one of the PIV5 equivalent residues (D373, based on sequence alignment) identified in MeV F as being important for interaction with MeV H. However, we recently identified a role for several hydrophobic residues in the immunoglobulin-like domain within F that mediate the PIV5 F-HN interaction, and these residues are not close to the 5D antigenic site (60).
Conclusions.
We used synthetic antibody technology and phage display to rapidly generate a large number of unique sAbs to various antigenic sites on pre- and postfusion F and HN, including conformation-specific sAbs. Our results show the feasibility of using synthetic antibody technology and phage display to generate a wide variety of specific and custom antibodies that can be used to test specific models. The inhibitory activities of the anti-HN stalk sAbs further establish the emerging model for paramyxovirus F-HN interaction and fusion promotion independently of attachment protein mutations (including chimeric and headless constructs) and high-resolution structural information.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported in part by National Institutes of Health research grants R01 AI-23173 (to R.A.L.) and U01GM094588 (to A.A.K.). B.D.W. was an Associate and R.A.L. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Published ahead of print 13 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01707-14.
REFERENCES
- 1. Lamb RA, Parks GD. 2013. Paramyxoviridae: The viruses and their replication, p 957–995 Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Rancaniello VR, Roizman B. (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Bowden TA, Aricescu AR, Gilbert RJ, Grimes JM, Jones EY, Stuart DI. 2008. Structural basis of Nipah and Hendra virus attachment to their cell-surface receptor ephrin-B2. Nat. Struct. Mol. Biol. 15:567–572. 10.1038/nsmb.1435 [DOI] [PubMed] [Google Scholar]
- 3. Colf LA, Juo ZS, Garcia KC. 2007. Structure of the measles virus hemagglutinin. Nat. Struct. Mol. Biol. 14:1227–1228. 10.1038/nsmb1342 [DOI] [PubMed] [Google Scholar]
- 4. Crennell S, Takimoto T, Portner A, Taylor G. 2000. Crystal structure of the multifunctional paramyxovirus hemagglutinin-neuraminidase. Nat. Struct. Biol. 7:1068–1074. 10.1038/81002 [DOI] [PubMed] [Google Scholar]
- 5. Hashiguchi T, Kajikawa M, Maita N, Takeda M, Kuroki K, Sasaki K, Kohda D, Yanagi Y, Maenaka K. 2007. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc. Natl. Acad. Sci. U. S. A. 104:19535–19540. 10.1073/pnas.0707830104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lawrence MC, Borg NA, Streltsov VA, Pilling PA, Epa VC, Varghese JN, McKimm-Breschkin JL, Colman PM. 2004. Structure of the haemagglutinin-neuraminidase from human parainfluenza virus type III. J. Mol. Biol. 335:1343–1357. 10.1016/j.jmb.2003.11.032 [DOI] [PubMed] [Google Scholar]
- 7. Xu K, Rajashankar KR, Chan YP, Himanen JP, Broder CC, Nikolov DB. 2008. Host cell recognition by the henipaviruses: crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc. Natl. Acad. Sci. U. S. A. 105:9953–9958. 10.1073/pnas.0804797105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yuan P, Thompson T, Wurzburg BA, Paterson RG, Lamb RA, Jardetzky TS. 2005. Structural studies of the parainfluenza virus 5 hemagglutinin-neuraminidase tetramer in complex with its receptor, sialyllactose. Structure 13:803–815. 10.1016/j.str.2005.02.019 [DOI] [PubMed] [Google Scholar]
- 9. Bowden TA, Crispin M, Harvey DJ, Jones EY, Stuart DI. 2010. Dimeric architecture of the Hendra virus attachment glycoprotein: evidence for a conserved mode of assembly. J. Virol. 84:6208–6217. 10.1128/JVI.00317-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuan P, Paterson RG, Leser GP, Lamb RA, Jardetzky TS. 2012. Structure of the Ulster strain Newcastle disease virus hemagglutinin-neuraminidase reveals auto-inhibitory interactions associated with low virulence. PLoS Pathog. 8:e1002855. 10.1371/journal.ppat.1002855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Santiago C, Celma ML, Stehle T, Casasnovas JM. 2010. Structure of the measles virus hemagglutinin bound to the CD46 receptor. Nat. Struct. Mol. Biol. 17:124–129. 10.1038/nsmb.1726 [DOI] [PubMed] [Google Scholar]
- 12. Yuan P, Swanson KA, Leser GP, Paterson RG, Lamb RA, Jardetzky TS. 2011. Structure of the Newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc. Natl. Acad. Sci. U. S. A. 108:14920–14925. 10.1073/pnas.1111691108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bowden TA, Crispin M, Harvey DJ, Aricescu AR, Grimes JM, Jones EY, Stuart DI. 2008. Crystal structure and carbohydrate analysis of Nipah virus attachment glycoprotein: a template for antiviral and vaccine design. J. Virol. 82:11628–11636. 10.1128/JVI.01344-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yuan P, Leser GP, Demeler B, Lamb RA, Jardetzky TS. 2008. Domain architecture and oligomerization properties of the paramyxovirus PIV 5 hemagglutinin-neuraminidase (HN) protein. Virology 378:282–291. 10.1016/j.virol.2008.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ng DT, Hiebert SW, Lamb RA. 1990. Different roles of individual N-linked oligosaccharide chains in folding, assembly, and transport of the simian virus 5 hemagglutinin-neuraminidase. Mol. Cell. Biol. 10:1989–2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bossart KN, Crameri G, Dimitrov AS, Mungall BA, Feng YR, Patch JR, Choudhary A, Wang LF, Eaton BT, Broder CC. 2005. Receptor binding, fusion inhibition, and induction of cross-reactive neutralizing antibodies by a soluble G glycoprotein of Hendra virus. J. Virol. 79:6690–6702. 10.1128/JVI.79.11.6690-6702.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brindley MA, Plemper RK. 2010. Blue native PAGE and biomolecular complementation reveal a tetrameric or higher-order oligomer organization of the physiological measles virus attachment protein H. J. Virol. 84:12174–12184. 10.1128/JVI.01222-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bose S, Welch BD, Kors CA, Yuan P, Jardetzky TS, Lamb RA. 2011. Structure and mutagenesis of the parainfluenza virus 5 hemagglutinin-neuraminidase stalk domain reveals a four-helix bundle and the role of the stalk in fusion promotion. J. Virol. 85:12855–12866. 10.1128/JVI.06350-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bishop KA, Hickey AC, Khetawat D, Patch JR, Bossart KN, Zhu Z, Wang LF, Dimitrov DS, Broder CC. 2008. Residues in the stalk domain of the Hendra virus G glycoprotein modulate conformational changes associated with receptor binding. J. Virol. 82:11398–11409. 10.1128/JVI.02654-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deng R, Wang Z, Mirza AM, Iorio RM. 1995. Localization of a domain on the paramyxovirus attachment protein required for the promotion of cellular fusion by its homologous fusion protein spike. Virology 209:457–469. 10.1006/viro.1995.1278 [DOI] [PubMed] [Google Scholar]
- 21. Deng R, Wang Z, Mahon PJ, Marinello M, Mirza A, Iorio RM. 1999. Mutations in the Newcastle disease virus hemagglutinin-neuraminidase protein that interfere with its ability to interact with the homologous F protein in the promotion of fusion. Virology 253:43–54. 10.1006/viro.1998.9501 [DOI] [PubMed] [Google Scholar]
- 22. Melanson VR, Iorio RM. 2004. Amino acid substitutions in the F-specific domain in the stalk of the Newcastle disease virus HN protein modulate fusion and interfere with its interaction with the F protein. J. Virol. 78:13053–13061. 10.1128/JVI.78.23.13053-13061.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee JK, Prussia A, Paal T, White LK, Snyder JP, Plemper RK. 2008. Functional interaction between paramyxovirus fusion and attachment proteins. J. Biol. Chem. 283:16561–16572. 10.1074/jbc.M801018200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paal T, Brindley MA, St Clair C, Prussia A, Gaus D, Krumm SA, Snyder JP, Plemper RK. 2009. Probing the spatial organization of measles virus fusion complexes. J. Virol. 83:10480–10493. 10.1128/JVI.01195-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stone-Hulslander J, Morrison TG. 1999. Mutational analysis of heptad repeats in the membrane-proximal region of Newcastle disease virus HN protein. J. Virol. 73:3630–3637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Takimoto T, Taylor GL, Connaris HC, Crennell SJ, Portner A. 2002. Role of the hemagglutinin-neuraminidase protein in the mechanism of paramyxovirus-cell membrane fusion. J. Virol. 76:13028–13033. 10.1128/JVI.76.24.13028-13033.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yao Q, Hu X, Compans RW. 1997. Association of the parainfluenza virus fusion and hemagglutinin-neuraminidase glycoproteins on cell surfaces. J. Virol. 71:650–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Plemper RK, Hammond AL, Gerlier D, Fielding AK, Cattaneo R. 2002. Strength of envelope protein interaction modulates cytopathicity of measles virus. J. Virol. 76:5051–5061. 10.1128/JVI.76.10.5051-5061.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aguilar HC, Ataman ZA, Aspericueta V, Fang AQ, Stroud M, Negrete OA, Kammerer RA, Lee B. 2009. A novel receptor-induced activation site in the Nipah virus attachment glycoprotein (G) involved in triggering the fusion glycoprotein (F). J. Biol. Chem. 284:1628–1635. 10.1074/jbc.M807469200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Welch BD, Yuan P, Bose S, Kors CA, Lamb RA, Jardetzky TS. 2013. Structure of the parainfluenza virus 5 (PIV5) hemagglutinin-neuraminidase (HN) ectodomain. PLoS Pathog. 9:e1003534. 10.1371/journal.ppat.1003534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698. 10.1038/nsmb.1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44. 10.1038/nature04322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Welch BD, Liu Y, Kors CA, Leser GP, Jardetzky TS, Lamb RA. 2012. Structure of the cleavage-activated prefusion form of the parainfluenza virus 5 fusion protein. Proc. Natl. Acad. Sci. U. S. A. 109:16672–16677. 10.1073/pnas.1213802109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, Zhou T, Baxa U, Yasuda E, Beaumont T, Kumar A, Modjarrad K, Zheng Z, Zhao M, Xia N, Kwong PD, Graham BS. 2013. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340:1113–1117. 10.1126/science.1234914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wen X, Krause JC, Leser GP, Cox RG, Lamb RA, Williams JV, Crowe JE, Jr, Jardetzky TS. 2012. Structure of the human metapneumovirus fusion protein with neutralizing antibody identifies a pneumovirus antigenic site. Nat. Struct. Mol. Biol. 19:461–463. 10.1038/nsmb.2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yin H-S, Paterson RG, Wen X, Lamb RA, Jardetzky TS. 2005. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. U. S. A. 102:9288–9293. 10.1073/pnas.0503989102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McLellan JS, Yang Y, Graham BS, Kwong PD. 2011. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing antigenic sites. J. Virol. 85:7788–7796. 10.1128/JVI.00555-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Swanson K, Wen X, Leser GP, Paterson RG, Lamb RA, Jardetzky TS. 2010. Structure of the Newcastle disease virus F protein in the post-fusion conformation. Virology 402:372–379. 10.1016/j.virol.2010.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Swanson KA, Settembre EC, Shaw CA, Dey AK, Rappuoli R, Mandl CW, Dormitzer PR, Carfi A. 2011. Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers. Proc. Natl. Acad. Sci. U. S. A. 108:9619–9624. 10.1073/pnas.1106536108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim YH, Donald JE, Grigoryan G, Leser GP, Fadeev AY, Lamb RA, DeGrado WF. 2011. Capture and imaging of a prehairpin fusion intermediate of the paramyxovirus PIV5. Proc. Natl. Acad. Sci. U. S. A. 108:20992–20997. 10.1073/pnas.1116034108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bose S, Zokarkar A, Welch BD, Leser GP, Jardetzky TS, Lamb RA. 2012. Fusion activation by a headless parainfluenza virus 5 hemagglutinin-neuraminidase stalk suggests a modular mechanism for triggering. Proc. Natl. Acad. Sci. U. S. A. 109:E2625–E2634. 10.1073/pnas.1213813109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ader N, Brindley MA, Avila M, Origgi FC, Langedijk J, Orvell C, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2012. Structural rearrangements of the central region of the morbillivirus attachment protein stalk domain trigger F protein refolding for membrane fusion. J. Biol. Chem. 287:16324–16334. 10.1074/jbc.M112.342493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Navaratnarajah CK, Negi S, Braun W, Cattaneo R. 2012. Membrane fusion triggering: three modules with different structure and function in the upper half of the measles virus attachment protein stalk. J. Biol. Chem. 287:38543–38551. 10.1074/jbc.M112.410563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Navaratnarajah CK, Oezguen N, Rupp L, Kay L, Leonard VH, Braun W, Cattaneo R. 2011. The heads of the measles virus attachment protein move to transmit the fusion-triggering signal. Nat. Struct. Mol. Biol. 18:128–134. 10.1038/nsmb.1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sugrue RJ, Bahadur G, Zambon MC, Hall-Smith M, Douglas AR, Hay AJ. 1990. Specific structural alteration of the influenza haemagglutinin by amantadine. EMBO J. 9:3469–3476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sanchez-San Martin C, Sosa H, Kielian M. 2008. A stable prefusion intermediate of the alphavirus fusion protein reveals critical features of class II membrane fusion. Cell Host Microbe 4:600–608. 10.1016/j.chom.2008.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Waning DL, Russell CJ, Jardetzky TS, Lamb RA. 2004. Activation of a paramyxovirus fusion protein is modulated by inside-out signaling from the cytoplasmic tail. Proc. Natl. Acad. Sci. U. S. A. 101:9217–9222. 10.1073/pnas.0403339101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Connolly SA, Leser GP, Yin HS, Jardetzky TS, Lamb RA. 2006. Refolding of a paramyxovirus F protein from prefusion to postfusion conformations observed by liposome binding and electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 103:17903–17908. 10.1073/pnas.0608678103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Koide S, Sidhu SS. 2009. The importance of being tyrosine: lessons in molecular recognition from minimalist synthetic binding proteins. ACS Chem. Biol. 4:325–334. 10.1021/cb800314v [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Koide A, Gilbreth RN, Esaki K, Tereshko V, Koide S. 2007. High-affinity single-domain binding proteins with a binary-code interface. Proc. Natl. Acad. Sci. U. S. A. 104:6632–6637. 10.1073/pnas.0700149104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fellouse FA, Barthelemy PA, Kelley RF, Sidhu SS. 2006. Tyrosine plays a dominant functional role in the paratope of a synthetic antibody derived from a four amino acid code. J. Mol. Biol. 357:100–114. 10.1016/j.jmb.2005.11.092 [DOI] [PubMed] [Google Scholar]
- 52. Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, Koide S, Sidhu SS. 2007. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J. Mol. Biol. 373:924–940. 10.1016/j.jmb.2007.08.005 [DOI] [PubMed] [Google Scholar]
- 53. Koellhoffer JF, Chen G, Sandesara RG, Bale S, Saphire EO, Chandran K, Sidhu SS, Lai JR. 2012. Two synthetic antibodies that recognize and neutralize distinct proteolytic forms of the Ebola virus envelope glycoprotein. Chembiochem. 13:2549–2557. 10.1002/cbic.201200493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Paduch M, Koide A, Uysal S, Rizk SS, Koide S, Kossiakoff AA. 2013. Generating conformation-specific synthetic antibodies to trap proteins in selected functional states. Methods 60:3–14. 10.1016/j.ymeth.2012.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Miller KR, Koide A, Leung B, Fitzsimmons J, Yoder B, Yuan H, Jay M, Sidhu SS, Koide S, Collins EJ. 2012. T cell receptor-like recognition of tumor in vivo by synthetic antibody fragment. PLoS One 7:e43746. 10.1371/journal.pone.0043746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tonikian R, Zhang Y, Boone C, Sidhu SS. 2007. Identifying specificity profiles for peptide recognition modules from phage-displayed peptide libraries. Nat. Protoc. 2:1368–1386. 10.1038/nprot.2007.151 [DOI] [PubMed] [Google Scholar]
- 57. Iorio RM, Bratt MA. 1983. Monoclonal antibodies to Newcastle disease virus: delineation of four antigenic sites on the HN glycoprotein. J. Virol. 48:440–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Iorio RM, Bratt MA. 1984. Monoclonal antibodies as functional probes of the HN glycoprotein of Newcastle disease virus: antigenic separation of the hemagglutinating and neuraminidase sites. J. Immunol. 133:2215–2219 [PubMed] [Google Scholar]
- 59. Iorio RM, Bratt MA. 1985. Selection of unique antigenic variants of Newcastle disease virus with neutralizing monoclonal antibodies and anti-immunoglobulin. Proc. Natl. Acad. Sci. U. S. A. 82:7106–7110. 10.1073/pnas.82.20.7106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Iorio RM, Lawton KA, Nicholson PM, Bratt MA. 1984. Monoclonal antibodies identify a strain-specific antigenic site on the HN glycoprotein of Newcastle disease virus strain Australia-Victoria. Virus Res. 1:513–525. 10.1016/0168-1702(84)90009-1 [DOI] [PubMed] [Google Scholar]
- 61. Randall RE, Young DF, Goswami KKA, Russell WC. 1987. Isolation and characterization of monoclonal antibodies to simian virus 5 and their use in revealing antigenic differences between human, canine and simian isolates. J. Gen. Virol. 68:2769–2780. 10.1099/0022-1317-68-11-2769 [DOI] [PubMed] [Google Scholar]
- 62. Bose S, Heath CM, Shah PA, Alayyoubi M, Jardetzky TS, Lamb RA. 2013. Mutations in the parainfluenza virus 5 fusion (F) protein reveal domains important for fusion triggering and metastability. J. Virol. 87:13520–13531. 10.1128/JVI.02123-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bose S, Song AS, Jardetzky TS, Lamb RA. 2014. Fusion activation through attachment protein stalk domains indicates a conserved core mechanism of paramyxovirus entry into cells. J. Virol. 88:3925–3941. 10.1128/JVI.03741-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Iorio RM, Mahon PJ. 2008. Paramyxoviruses: different receptors—different mechanisms of fusion. Trends Microbiol. 16:135–137. 10.1016/j.tim.2008.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Brindley MA, Takeda M, Plattet P, Plemper RK. 2012. Triggering the measles virus membrane fusion machinery. Proc. Natl. Acad. Sci. U. S. A. 109:E3018–E3027. 10.1073/pnas.1210925109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Liu Q, Stone JA, Bradel-Tretheway B, Dabundo J, Benavides-Montano JA, Santos-Montanez J, Biering SB, Nicola AV, Iorio RM, Lu X, Aguilar HC. 2013. Unraveling a three-step spatiotemporal mechanism of triggering of receptor-induced Nipah virus fusion and cell entry. PLoS Pathog. 9:e1003770. 10.1371/journal.ppat.1003770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Brindley MA, Suter R, Schestak I, Kiss G, Wright ER, Plemper RK. 2013. A stabilized headless measles virus attachment protein stalk efficiently triggers membrane fusion. J. Virol. 87:11693–11703. 10.1128/JVI.01945-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yuasa T, Kawano M, Tabata N, Nishio M, Kusagawa S, Komada H, Matsumura H, Ito Y, Tsurudome M. 1995. A cell fusion-inhibiting monoclonal antibody binds to the presumed stalk domain of the human parainfluenza type 2 virus hemagglutinin-neuraminidase protein. Virology 206:1117–1125. 10.1006/viro.1995.1035 [DOI] [PubMed] [Google Scholar]
- 69. Bimanns S, Rusu M, Wriggers W. 2011. Using Sculptor and Situs for simultaneous assembly of atomic components into low-resolution shapes. J. Struct. Biol. 173:428–435. 10.1016/j.jsb.2010.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.