

ABSTRACT
Autophagy is an intracellular degradation pathway that provides a host defense mechanism against intracellular pathogens. However, many viruses exploit this mechanism to promote their replication. This study shows that lytic induction of Epstein-Barr virus (EBV) increases the membrane-bound form of LC3 (LC3-II) and LC3-containing punctate structures in EBV-positive cells. Transfecting 293T cells with a plasmid that expresses Rta also induces autophagy, revealing that Rta is responsible for autophagic activation. The activation involves Atg5, a key component of autophagy, but not the mTOR pathway. The expression of Rta also activates the transcription of the genes that participate in the formation of autophagosomes, including LC3A, LC3B, and ATG9B genes, as well as those that are involved in the regulation of autophagy, including the genes TNF, IRGM, and TRAIL. Additionally, treatment with U0126 inhibits the Rta-induced autophagy and the expression of autophagy genes, indicating that the autophagic activation is caused by the activation of extracellular signal-regulated kinase (ERK) signaling by Rta. Finally, the inhibition of autophagic activity by an autophagy inhibitor, 3-methyladenine, or Atg5 small interfering RNA, reduces the expression of EBV lytic proteins and the production of viral particles, revealing that autophagy is critical to EBV lytic progression. This investigation reveals how an EBV-encoded transcription factor promotes autophagy to affect viral lytic development.
IMPORTANCE
Autophagy is a cellular process that degrades and recycles nutrients under stress conditions to promote cell survival. Although autophagy commonly serves as a defense mechanism against viral infection, many viruses exploit this mechanism to promote their replication. This study finds that a transcription factor that is encoded by Epstein-Barr virus (EBV), Rta, activates autophagy, and the inhibition of autophagy reduces the ability of the virus to express viral lytic proteins and to generate progeny. Unlike other virus-encoded proteins that modulate autophagy by interacting with proteins that are involved in the autophagic pathway, Rta activates the transcription of the autophagy-related genes via the ERK pathway. The results of this study reveal how the virus manipulates autophagy to promote its lytic development.
INTRODUCTION
Epstein-Barr virus (EBV) is the etiological agent of infectious mononucleosis and is associated with Burkitt's lymphoma, nasopharyngeal carcinoma, Hodgkin's lymphoma, and gastric cancer (1–4). Following infection, EBV is usually maintained under latent conditions. However, EBV enters a lytic cycle to generate progeny after cells that have been latently infected by EBV are exposed to 12-O-tetradecanoylphorbol-13-acetate (TPA), sodium butyrate (SB), calcium ionophores, transforming growth factor β1 (TGF-β1), or anti-IgG (5–7). The lytic activation of EBV is closely related to the expression of two EBV-encoded transcription factors, Rta and Zta (8, 9), which act either alone or synergistically to transcribe lytic genes on which viral lytic progression depends (10–12).
Rta often binds to Rta response elements (RRE) to activate the transcription of EBV and cellular genes, including BALF2, BMRF1, BMLF1, and cellular Decoy receptor 3 (13–16). However, transactivation by Rta also involves mechanisms that are independent of RRE binding. For example, Rta forms a complex with Sp1 through an adaptor protein, MCAF1, to autoregulate the transcription of its own gene (17, 18). It also activates the transcription of the Zta gene, BZLF1, by activating phosphatidylinositol 3-kinase and mitogen-activated protein kinase pathways (19–21). Additionally, Rta induces cell cycle arrest in the G1 phase and initiates a cellular senescence program in epithelial cells (22).
Autophagy, which is an evolutionarily conserved cellular degradation process, is responsible for the degradation of long-lived proteins and damaged organelles. Autophagy proceeds at a basal level in cells under normal conditions but is stimulated by starvation, oxidative stress, and the accumulation of misfolded proteins (23). Upon the induction of autophagy, two-layered membrane structures, called autophagosomes, which contain cytoplasmic materials and damaged organelles, are formed. The autophagosomes then mature by fusion with lysosomes to form autolysosomes to degrade their content, liberating amino acids and fatty acids that can be metabolized or recycled to improve cell survival (24, 25). This intracellular degradation system is tightly controlled by the coordinated action of proteins that are encoded by autophagy-related genes (Atg) (26).
Autophagy is a cellular defense mechanism by which invading pathogens, including viruses, are removed by encapsulation and degradation by autophagic vesicles (27–29). Autophagy also eliminates viruses by promoting the presentation of viral antigens that are processed in autolysosomes that are loaded onto major histocompatibility complex class II (MHC-II) antigen and presented to CD4+ T cells (30). Autophagy has a deleterious effect on the pathogenesis of the neurotropic Sindbis virus and limits the replication of the tobacco mosaic virus (31, 32). However, various viruses exploit the autophagic machinery of the host for their own survival and replication. For example, the stimulation of autophagy increases the yields of poliovirus, hepatitis C virus (HCV), Dengue virus, and coxsackie B virus (33–35). Additionally, HIV activates autophagy, which promotes the process of Gag and prevents the degradation of virions in the autolysosomes, increasing virion yield (36). Several herpesviruses are known to regulate autophagy negatively. The herpes simplex virus type 1 (HSV-1) protein ICP34.5 suppresses autophagy via dephosphorylation of the α-subunit of eukaryotic initiation factor 2 (eIF2α) and the binding of ICP34.5 to Beclin-1 (37, 38). Kaposi's sarcoma-associated herpesvirus (KSHV) encodes vFLIP, a homolog of cellular FLICE-like inhibitor protein (cFLIP), and binds to Atg3, preventing it from binding to LC3 (39). On the other hand, autophagy is essential to the lytic reactivation of KSHV, since inhibition of autophagy reduces RTA-mediated lytic gene expression and KSHV replication (40). In EBV, latent membrane protein 1 (LMP1) activates the autophagic machinery to regulate its own turnover (41). Furthermore, autophagy affects MHC class II-EBNA1 presentation in EBV-immortalized lymphoblastoid cells by delivering EBNA1 to the lysosomal compartment (30, 42). However, the interplay between autophagy and the lytic production cycle of EBV is unknown. This investigation reveals that the lytic cycle of EBV activates autophagic activity. The activation is associated with EBV-encoded transcription factor Rta via the extracellular signal-regulated kinase (ERK) signaling pathway and affects viral lytic development.
MATERIALS AND METHODS
Cell cultures and reagents.
293T cells, a human embryonic kidney cell line, were maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS). P3HR1, Akata, and Raji cells were cultured in RPMI 1640 containing 10% FBS. 293EBV(2089) cells, which are stably transfected with Maxi-EBV plasmid p2089 (43), were maintained in DMEM containing 100 μg/ml hygromycin. EBV in P3HR1 cells was lytically activated by treating the cells with 20 ng/ml 12-O-tetradecanoylphorbol-13-acetate (TPA; Sigma-Aldrich) and 3 mM sodium butyrate (SB; Sigma-Aldrich). Akata cells were activated by treating cells with anti-IgG (MP Biomedicals). 3-Methyladenine (3-MA), bafilomycin, rapamycin, and hydroxycholoquine (CQ) were purchased from Sigma-Aldrich. U0126 was purchased from Cell Signaling.
Plasmids.
Plasmids pCMV-Rta and pCMV-Zta were used to express Rta and Zta as described elsewhere (14). Plasmids pR550 and pRtaF600/605A carry Rta mutated genes for R550 and RtaF600/605A (44). Plasmid pGFP-LC3 has been described elsewhere (45).
Transfection and electroporation.
293T and 293EBV(2089) cells were transfected using Lipofectamine 2000 (Invitrogen), following the instructions of the manufacturer. P3HR1 cells were transfected by electroporation using an Amaxa Nucleofector. A total of 5 × 106 cells were electroporated with 10 μg DNA in Ingenio electroporation solution (Mirus) using the program C-009. Transfection efficiency was estimated to be 62% by flow cytometry of the red fluorescence of P3HR1 cells that had been transfected with 10 μg mRFP DNA for 48 h.
Immunofluorescence staining.
Cells were fixed using 4% paraformaldehyde and processed for immunofluorescence staining with a primary antibody against Rta (Argene), gp350/220 (72A1 clone; ATCC HB-168), or LC3B (Sigma). Following incubation with fluorescence-labeled secondary antibodies (Invitrogen), cells were observed under a Nikon E600 or a Leica SP5 fluorescence microscope.
Immunoblot analysis.
Cells were lysed in PBS that contained 1% Triton X-100 and protease inhibitor cocktail (Sigma). The lysates were then centrifuged at 12,000 × g at 4°C for 15 min. The proteins in the lysate were separated by SDS-PAGE, transferred to a polyvinylidene difluoride membrane and detected by immunoblotting. Anti-Rta and anti-Zta antibodies were purchased from Argene; anti-EA-D antibody was obtained from Millipore; anti-tubulin and anti-Atg5 antibodies came from Sigma-Aldrich; anti-mTOR, anti-pS2448-mTOR, anti-p70S6K, anti-pT371-p70S6K, anti-4EBP1, anti-pT37/46–4EBP1, anti-p44/42 ERK1/2, and anti-ERK1/2 antibodies were obtained from Cell Signaling; and anti-LC3 antibodies came from MBL. Anti-BBLF1 antibody was generated in rabbits by our laboratory.
siRNA and shRNA knockdown.
Double-stranded small interfering RNA (siRNA) against Atg5 was purchased from Santa Cruz Biotechnology. 293T cells were transfected with 200 to 400 pmol siRNAs by using RNAiMax (Invitrogen). Lentiviral vectors that expressed Atg5 shRNA and control shRNA (TRCN0000330394 and TRCN0000072224) were purchased from the National RNAi Core Facility, Academia Sinica, Taiwan. Recombinant lentiviruses were generated by cotransfecting 293T cells with plasmids pLKO.1 Atg5 shRNA or pLKO.1 lacZ shRNA, pCMVDR8.2, and pMD.G, using Lipofectamine 2000. Culture medium was collected at 48 and 72 h posttransfection. P3HR1 cells were infected with lentiviruses by mixing cells with the culture supernatant in the presence of 8 μg/ml Polybrene and then centrifuging the mixture at 450 × g for 2 h. The cells that were infected by lentiviruses were selected in the medium that contained 0.5 μg/ml puromycin for 5 to 7 days.
Reverse transcription-quantitative PCR (RT-qPCR).
Total RNA was isolated using an RNAeasy minikit (Qiagen), according to the method that was recommended by the manufacturer. Reverse transcription was performed using the SuperScript III first-strand synthesis supermix (Invitrogen). An equal amount of cDNA product was used in PCR that was performed using a Bio-Rad CFX apparatus. PCR amplification was conducted using the following primers; MAP1LC3A F, 5′-CGCTACAAGGGTGAGAAGCA; MAP1LC3A R, 5′-AGAAGCCGAAGGTTTCCTGG; MAP1LC3B F, 5-GCGAGTCACCTGACCAGGCTG; MAP1LC3B R, 5-GCGAGTCACCTGACCAGGCTG; ATG9B F, 5′-GGACTCTCCTGGGCTGCGGGTAG; ATG9B R, 5′-GCAGGCAAAGCCATTCCGCTGGTGG; TNF F, 5′-GGCAGGCGCCACCACGCTCTTC; TNF R, 5′-GCATTGGCCCGGCGGTTCAGC; IRGM F, 5′-GCAGATGGGAACTTGCCAGA; IRGM R, 5′-AGGCCTTACCCTCATGTCCT; TNFSF10 F, 5′-TTGGGACCCCAATGACGAAG; TNFSF10 R, 5′-TGGTCCCAGTTATGTGAGCTG; ACTB F, 5′-GGACTTCGAGCAAGAGATGG; ACTB R, 5′-AGCACTGTGTTGGCGTACAG.
Enumeration of virus particles.
The amount of encapsidated viral DNA was determined following a method that was described elsewhere (46). Following lytic induction for 4 days, cells were collected by centrifugation. The supernatant fraction contained viral particles that were released into the medium. The viral particles within the cells were also released from the cell pellets by three rounds of freezing and thawing. DNA from broken cells was removed by treatment with DNase I. Next, SDS and proteinase K were added to remove the viral envelope and capsid. EBV DNA was extracted using phenol-chloroform, precipitated with isopropanol, and then recovered by centrifugation. The DNA pellet was washed with 70% ethanol and suspended in Tris-EDTA buffer. The amount of EBV DNA was analyzed by real-time PCR using an iCycler iQ multicolor real-time PCR detection system (Bio-Rad) with primers and a probe that were specific to BKRF1 (47).
Infection of cells by EBV.
Culture supernatant was collected from 293EBV(2089) cells 4 days after transfection. Raji cells were then infected by the virus in the culture supernatant. Cells were then treated with TPA (20 mg/ml) and butyrate (3 mM) at day 2 postinfection to enhance expression of the green fluorescent protein (GFP) gene. The expression of GFP from EBV(2098) was observed 3 days postinfection. The percentage of cells that expressed GFP was determined by flow cytometry.
Fluorescence-activated cell sorting (FACS).
At 48 h after lytic induction, P3HR1 cells were washed in phosphate-buffered saline (PBS) and incubated with anti-gp350/220 antibody for 1 h at 4°C and then incubated with fluorescein isothiocyanate (FITC)-labeled secondary antibody. Cells that were labeled with FITC fluorescence were separated from unlabeled cells by using a FACSAria cell sorter (BD Biosciences). Labeled and unlabeled cells were collected and analyzed by immunoblotting.
TEM analysis.
293T cells that had been transfected with pCMV3 or pCMV-Rta for 48 h were prepared for transmission electron microscopic (TEM) analysis, as described elsewhere (48). Briefly, cells were fixed in a solution that contained 2% paraformaldehyde and 2.5% glutaradehyde for 30 min at 4°C. Cells were washed and postfixed in 1% osmium tetroxide for 15 min and then stained with 1% uranyl acetate for 1 h at room temperature. Samples were dehydrated using increasing concentrations of ethanol from 50 to 100% and then embedded in Spurr resin. Embedded samples were sliced into thin sections and stained with uranyl acetate and lead citrate. Images of the samples were obtained using a JEOL JEM-1200 transmission electron microscope.
Statistics.
Data are presented as means ± standard deviations (SD). Student's t test was performed on these means; a P value less than 0.05 was considered significant.
RESULTS
EBV lytic activation and formation of autophagosomes.
P3HR1 cells were transfected with pCMV-Zta to activate the lytic cycle of EBV and to study how the activation affected autophagy. The induction of autophagy was monitored by observing the translocation of the autophagosome protein LC3 from the cytosol to the membrane of the newly formed autophagosomes. The translocation converts the diffused cytosolically distributed LC3, LC3-I, to membrane-bound LC3, LC3-II, which appears as cytoplasmic puncta (49). As expected, the virus expressed EBV lytic proteins, including Rta, Zta, EA-D, and BBLF1, after lytic induction (Fig. 1A, lanes 2 and 3). The expression of LC3-II was also detected by immunoblotting (Fig. 1A, lanes 2 and 3). A parallel experiment showed that transfecting the cells with an empty vector did not result in the expression of LC3-II (Fig. 1A, lane 1), suggesting that activation of the EBV lytic cycle by Zta promotes the synthesis of autophagosomes. We also sorted P3HR1 cells by FACS, based on the presence of an EBV lytic marker, gp350/220, after lytic induction using TPA and sodium butyrate. Immunoblot analysis revealed that P3HR1 cells with gp350/220 on their surfaces expressed Zta, Rta, EA-D, BBLF1, and LC3-II (Fig. 1B, lane 2). However, the population that did not express gp350/220 expressed LC3-II at a level lower than in cells expressing gp350/220 (Fig. 1B, lane 1), showing a correlation between EBV lytic activation and autophagy. Similar results were also detected in Akata and 293EBV(2089) cells (Fig. 1C and D). Notably, the amount of LC3-II that was expressed after lytic induction by TPA and sodium butyrate (Fig. 1B, lane 2) exceeded that expressed after transfection using pCMV-Zta (Fig. 1A, lanes 2 and 3), which is likely attributable to the fact that TPA itself activates autophagy (50) and to the low transfection efficiency of B cells. Additionally, confocal laser scanning microscopy revealed that after P3HR1 cells were transfected with pGFP-LC3 and pCMV3, the cells exhibited a weak GFP-LC3 signal in the cytosol (Fig. 1E). However, after they were transfected with pCMV-Zta, those cells that expressed Rta or gp350/220 also exhibited punctate GFP-LC3 fluorescence (Fig. 1E), confirming that EBV lytic activation promotes the formation of autophagosomes.
FIG 1.

EBV lytic cycle and autophagy activation. (A) P3HR1 cells were transfected with control plasmid pCMV3 (Crt) or pCMV-Zta (Zta) to activate the lytic cycle. Cell lysates were prepared 24 h (lane 2) and 48 h (lane 1 and lane 3) after transfection and analyzed by immunoblotting using antibodies against the indicated proteins. (B) P3HR1 cells that were treated with TPA and SB for 48 h were incubated with anti-gp350/220 antibody and then labeled with fluorescence-conjugated secondary antibody. The cells that expressed gp350/220 (lane 2) were separated from those did not (lane 1) by using a cell sorter. Cell lysates were prepared and analyzed by immunoblotting. (C) Akata cells were treated with anti-IgG for 24 and 48 h. Cell lysates were prepared and analyzed by immunoblotting using antibodies against the indicated proteins. (D) 293EBV(2089) cells were transfected with control plasmid pCMV3 (Crt; lanes 1 and 3) or pCMV-Zta (Z; lanes 2 and 4). Cell lysates were prepared at 48 and 72 h after transfection and analyzed by immunoblotting. GAPDH, glyceraldehyde 3-phosphate dehydrogenase. (E) P3HR1 cells were cotransfected with pGFP-LC3 and pCMV3 or pCMV-Zta to induce the lytic cycle. Cells were fixed and adhered to poly-l-lysine-coated coverslips and then stained using anti-Rta or anti-gp350/220 antibodies. 4′,6-Diamidino-2-phenyldole staining revealed the nucleus. The images of fluorescence were captured under a Leica SP5 confocal laser scanning microscope. z-stacks with 20 planes were acquired in each experiment. Compressed images of multiple z-stacks are shown. Bar, 5 μm.
Activation of autophagy by Rta.
Since EBV lytic activation induces autophagy, this study further examined whether the induction is associated with the expression of Zta and Rta. Our immunoblot study revealed that transfecting 293T cells with control plasmid or pCMV-Zta did not increase the expression of LC3-II (Fig. 2A, lanes 1 and 2). However, transfecting the cells with pCMV-Rta increased LC3-II expression (Fig. 2A, lane 3), whereas Beclin 1 remained at the same level of expression. Measuring the band intensity revealed that the amount of LC3-II expressed by the cells that were transfected with pCMV-Rta was about 2.6 times higher than that expressed by the cells transfected with pCMV3 or pCMV-Zta (Fig. 2A, lanes 1 to 3, and B), indicating that Rta increases the formation of autophagosomes. Since the increase in the amount of LC3-II could be interpreted as being caused by an increase in the amount of generated autophagosomes or an inhibition of autophagosome maturation, cells were further treated with bafilomycin A1, which is an inhibitor of autophagosomal maturation. As expected, the treatment increased intracellular levels of LC3-II (Fig. 2A, lanes 4 to 6). However, the amount of LC3-II in 293T(pCMV-Rta) cells (Fig. 2A, lane 6) remained higher than that in 293T(pCMV-Zta) and 293(pCMV3) cells (Fig. 2A, lanes 4 and 5), indicating that the increase in the amount of LC3-II after the expression of Rta is attributable to an increase in the formation of autophagosomes rather than an inhibition of autophagosome maturation. To verify this finding, the formation of autophagosomes was studied under a fluorescence microscope. We cotransfected 293T cells with plasmids that expressed LC3-GFP and also Rta or Zta. LC3-GFP punctate structures were observed in the cells that expressed Rta but not in those that expressed Zta (Fig. 2C), confirming that Rta induces the formation of autophagosomes. In addition, we examined the formation of autophagosomes under starvation conditions and found that when cells were cultured in Earle's balanced salt solution (EBSS) that contained hydroxycholoquine (CQ), endogenous LC3B, an isoform of LC3, was present as punctate structures in cells with or without Rta expression (Fig. 2D, panels d, e and f). However, when the cells were cultured in a nutrient-rich medium, DMEM, LC3B punctate structures were observed only in the cells that expressed Rta (Fig. 2C, arrow, panels a, b, and c), suggesting that Rta induces autophagic activation under nutrient-rich conditions. Under an electron microscope, vesicular structures were observed in the cells that had been transfected by pCMV-Rta (Fig. 2E). At a magnification of ×35,000, double-layered membranes that contained undigested cytoplasmic contents, which are typical of autophagosomes (49), were observed (Fig. 2E, panel a). Autolysosomes, which have a single-membrane cytoplasmic structure at various stages of the degradation process, were also observed in Rta-transfected cells (Fig. 2E, panels a and b). These results revealed that Rta induces autophagy, including the formation of autophagosomes and their maturation into autolysosomes. After 24 h of transfection and lytic induction, 293T(pCMV-Rta) (lane 1) was found to have produced less Rta than did the P3HR1 cells following lytic induction (lane 2), indicating that the induction of autophagy in 293T cells is not attributable to the expression of excessive Rta.
FIG 2.

Expression of Rta and autophagic activation. (A) 293T cells were transfected with an empty vector pCMV3 (Ctr), pCMV-Zta (Z), and pCMV-Rta (R). Cells were treated with 100 nM bafilomycin A1 for 2 h, and lysates were prepared at 48 h after transfection. Proteins were separated by SDS-PAGE and analyzed by immunoblot analysis using antibodies against the indicated proteins. (B) The intensity of the LC3-II band from the cells that were transfected with pCMV-Zta or pCMV-Rta was compared to that from cells that were transfected with an empty vector. The intensity of LC3-II was measured using ImageJ software and normalized to the band intensity of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) based on three blots. (C) 293T cells were cotransfected with both pLC3-GFP and pCMV-Zta or pCMV-Rta. (D) 293T cells were transfected with pCMV-Rta and cultured in various media as indicated. Cells were fixed at 48 h after transfection. Indirect immunofluorescence staining was performed using anti-Zta or anti-Rta (C) and anti-LC3B and anti-Rta (D). 4′,6-Diamidino-2-phenylindole staining revealed the nucleus. z-stacks of 20 planes were acquired in each experiment. Compressed images of multiple z-stacks are presented. Bar, 10 μm; white arrow, Rta-expressing cell. (E) Electron micrographs of 293T cells transfected with pCMV3 and pCMV-Rta. Two boxed regions in the image were enlarged (a and b). The black arrow indicates an autophagosome; the blue arrow indicates an autolysosome. (F) Lysates were prepared from P3HR1 cells that had been treated with TPA and sodium butyrate and from 293T cells that had been transfected with pCMV-R. Rta and tubulin (Tub) in the lysates were analyzed by immunoblotting.
Involvement of Atg5 in Rta-induced autophagy.
Atg5 participates in the formation of autophagosomes and is conjugated to an ubiquitin-like protein, Atg12 (51). Therefore, we reduced the expression of Atg5 by siRNA knockdown to examine whether the reduction influenced Rta-induced autophagy. Immunoblotting revealed that Atg5 siRNA, but not control siRNA (scramble [scr]), reduced the level of Atg5-Atg12 in 293T cells (Fig. 3A, lanes 3 and 4). Meanwhile, Atg5 siRNA reduced the amount of LC3-II that was induced by Rta (Fig. 3A, lanes 2 and 4), indicating that Atg5 is involved in Rta-induced autophagic activity. Furthermore, autophagosomes were examined by immunofluorescence using anti-LC3B antibody. The results showed that among 400 cells examined, about 30% of the cell population that expressed Rta (shown in red) exhibited autophagic vesicles (green dots) (Fig. 3B and C). However, after introducing Atg5 siRNA, the percentage dropped to 12% (Fig. 3B and C). These results revealed that Atg5 is involved in Rta-induced autophagy.
FIG 3.

Involvement of Atg5 in Rta-induced autophagy activation. (A) 293T cells were cotransfected with control siRNA (scr) or Atg5 siRNA (100 pmol) and pCMV3 (Crt) or pCMV-Rta (R), as indicated. The lysates were prepared at 48 h following transfection and analyzed by immunoblotting using antibodies against the indicated proteins. (B) 293T cells were cotransfected with control siRNA (scr) or Atg5 siRNA (100 pmol), and pCMV-Rta, and examined by indirect immunofluorescence using anti-Rta and anti-LC3B antibodies. 4′,6-Diamidino-2-phenylindole staining revealed the nucleus. Bar, 10 μm. (C) The percentages of Rta expressing cells (red) containing autophagic punctate vesicles (green dots) was determined by counting the numbers of cells that exhibited autophagic vesicles (green dots) in approximately 400 Rta-expressing cells (red).
Rta-induced autophagy and mTOR signaling.
mTOR is a serine-threonine protein kinase that regulates autophagy in response to nutrient starvation (23). To determine whether Rta induces autophagy through the mTOR pathway, the activity of mTOR and its downstream targets, p70S6K and 4E-BP1, was studied by immunoblot analysis with phospho-specific antibodies. Rapamycin, an inhibitor of mTOR, inhibits the activity of mTOR and phosphorylation of its downstream targets, p70S6K and 4E-BP1, which leads to activation of autophagy, as identified by the increased amount of LC3-II (Fig. 4, lanes 1 and 2). In contrast, the expression of Rta did not change the amounts of phospho-mTOR, phospho-p70S6K, and phospho-4E-BP1 (Fig. 4, lanes 3 and 4), suggesting that Rta-induced autophagy is independent of mTOR signaling.
FIG 4.
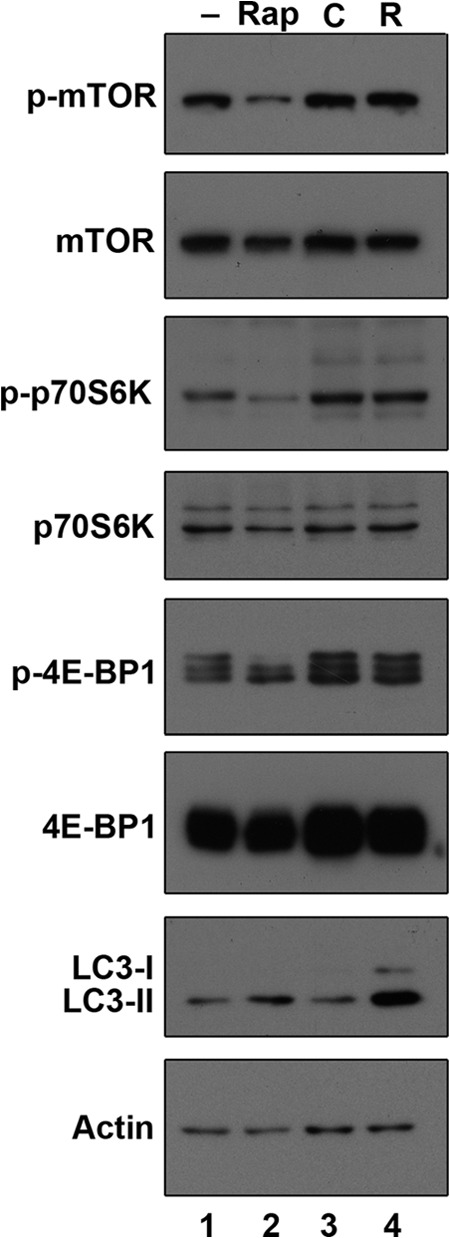
Rta-induced autophagy is independent of the mTOR signaling pathway. 293T cells were cultured for 24 h, and lysates were prepared after 1 h treatment with dimethyl sulfoxide (lane 1) or rapamycin (Rap; 100 mM) (lane 2). 293T cells were transfected with control plasmid (C, lane 3) or pCMV-Rta (R, lane 4) and lysates were prepared at 48 h after transfection. Lysates from cells treated with rapamycin or transfected with plasmids were analyzed by immunoblotting using antibodies against the indicated proteins.
Correlation between autopahgic and transactivation functions of Rta.
To determine whether autophagic activation depends on the transactivational activity of Rta, two mutants, R600/605A and R550 (Fig. 5A), were used to study how Rta affects autophagy. Mutant R550 contains a deletion in the transactivation domain, which eliminates Rta's transactivation activity; mutant F600/605A, while binding strongly to RRE, exhibits lower transactivation activity than does the wild-type Rta (44). The ability of Rta and its mutants to activate the BZLF1 promoter was confirmed in a transient-reporter assay (Fig. 5B). An immunoblot study revealed that the amount of LC3-II that was expressed by the cells correlated with Rta's transactivation activity, as the expression of R550 in 293T cells did not increase the level of LC3-II whereas that of F600/605A increased the amount of LC3-II, although to a level lower than that achieved using Rta (Fig. 5C and D). LC3B punctate structures that were induced by Rta and its mutants were also studied by indirect immunofluorescence (Fig. 5E). The percentage of cells with autophagic punctate vesicles that expressed Rta or its mutants was determined (Fig. 5E and F): 38% of the Rta-expressing cells contained LC3B punctate structures, of which hardly any were detected in those cells that expressed R550. The corresponding fraction of cells that expressed F600/605A was 11% (Fig. 5E and F). These results indicate that autophagic activation by Rta correlates with the ability of the transactivation activity of Rta.
FIG 5.

Mutations in Rta and autophagy activation. (A) Schematic diagram of the structure of Rta and its mutant derivatives. Dimerization, DNA binding, transactivation domain, and mutations in Rta are indicated. (B) The reporter plasmid pZpluc was cotransfected with pCMV3 (Crt), pCMV-Rta (Rta), pCMV-R550, or pCMV-Rta F600/605. Luciferase activity was measured at 24 h after transfection. (C) 293T cells were transfected with pCMV3, a control plasmid (Crt), pCMV-Rta (Rta), or plasmids that expressed Rta mutants, as indicated. The lysates were prepared 48 h after transfection and analyzed by immunoblotting using antibodies against the indicated proteins. (D) Quantification of LC3-II levels shown in panel C. The amount of LC3-II was measured using ImageJ software and normalized using that of the internal control, tubulin. (E) 293T cells were transfected with pCMV-Rta (Rta) or plasmids that expressed Rta mutants. At 48 h after transfection, cells were fixed and stained with anti-Rta and anti-LC3B antibodies. 4′,6-Diamidino-2-phenylindole reveals the nucleus. Bar, 20 μm. (F) The percentage of the cells in the population that had autophagic punctate vesicles in panel E was determined by counting the numbers of cells that exhibited autophagic vesicles (red dot) in approximately total 400 Rta-expressing cells (green).
Dependence of Rta-induced autophagic activity on ERK activity.
Since ERK signaling crucially influences autophagy (52) and Rta is known to transactivate the transcription of the Zta gene through ERK signaling (21), this study examined whether Rta affects autophagy through the ERK pathway. As expected, immunoblotting with phospho-specific ERK antibody revealed that the expression of Rta activated ERK activity and the phosphorylation of ERK induced by Rta was prevented by the presence of U0126, an inhibitor of MEK (Fig. 6A). Additionally, U0126 treatment reduced the amount of LC3-II (Fig. 6A, lanes 2 and 3) and of Rta-induced LC3B punctate structures (Fig. 6B). Counting the autophagic punctate vesicles in Rta-expressing cells revealed that adding U0126 reduced the fraction of the cells that contained autophagic vesicles from 48% to 15% (Fig. 6C). Not only was the percentage of the cell population that exhibited autophagic punctate vesicles reduced, but also the average number and size of the autophagic punctate structures within each cell decreased (Fig. 6B), indicating that Rta affects autophagy via ERK signaling.
FIG 6.
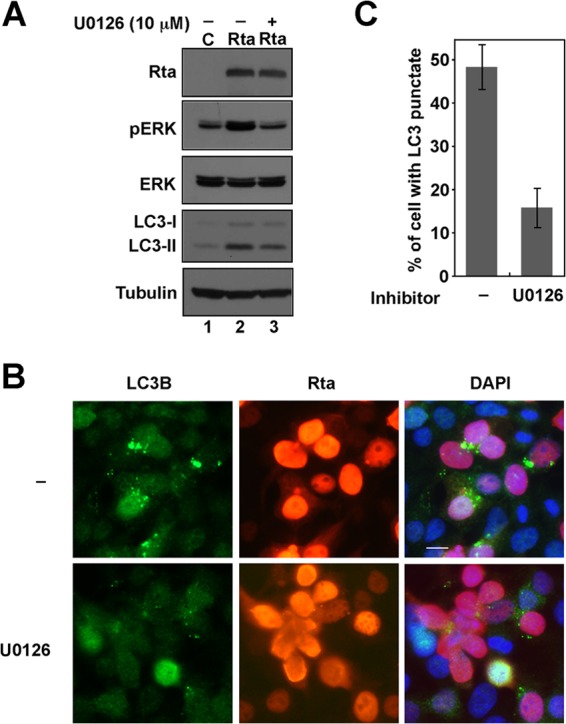
Involvement of ERK activity in Rta-induced autophagy activation. (A) 293T cells that had been transfected with pCMV-Rta (R) and cells were treated with U0126 (10 μM) after 6 h transfection. The lysates were harvested after 48 h of transfection and analyzed by immunoblotting using antibodies against the indicated proteins. (B) Cells were fixed and stained with anti-Rta and anti-LC3B antibodies. 4′,6-Diamidino-2-phenylindole stain reveals the nucleus. Bar, 10 μm. (C) The number of cells that had autophagic punctate vesicles (green) in a total population of about 400 Rta-expressing cells (red) was counted and the percentages were calculated.
Activation of genes that are involved in autophagosomal formation by Rta.
An autophagy RT-qPCR array (SABiosciences) was utilized to determine whether Rta transcriptionally activates the expression of autophagy-related genes (data not shown). The autophagy-related genes whose transcription was activated by Rta were selected, and their transcription was verified further by RT-qPCR. A transient-transfection study revealed that the expression of Rta, but not that of R550, increased the transcription of genes that are involved in the formation of autophagosomes, including LC3A, LC3B, and ATG9B, by factors of 16, 2.5, and 27, respectively (Fig. 7A, B, and C). The expression of Rta, but not that of R550, also increased the transcription of the genes that are involved in the regulation of autophagosome formation, including TNF, IRGM, and TNFSF10 (TRAIL), by factors of 3.8, 22.5, and 3.5, respectively (Fig. 7D, E, and F). U0126 also inhibited the transcriptional activity (Fig. 7A to F). These results revealed that Rta induces autophagy mediated by transcriptional activation of the genes that are involved in the formation or regulation of autophagosomal formation and influences the auotphagy process.
FIG 7.

Rta activates transcription of autophagy-related genes. The expression levels of LC3A (A), LC3B (B), ATG9B (C), TNF (D), IRGM (E) and TNFSF10 (F) in control (C) and Rta- (R) and R550-expressing cells that were treated (+) or were not treated (−) with U0126 were determined by RT-qPCR. The control (C) or Rta-expressing cells were treated with U0126 (10 μM) at 6 h after transfection. The relative expression of each gene was normalized to the amount of actin and expressed as the fold change relative to that in control cells. *, P < 0.05; **, P < 0.01 (determined with Student's t test).
Autophagy and EBV lytic progression.
P3HR1 cells were treated with 3-MA, an autophagy inhibitor, after lytic induction to study how autophagy affected the lytic cycle of EBV. The treatment decreased the expression of LC3-II and the expression of Zta, Rta, EA-D, and BBLF1 (Fig. 8A, lanes 2 and 3), indicating that 3-MA inhibits the lytic cycle of EBV. The treatment also reduced the numbers of virus particles released into culture medium by about 90% (Fig. 8B). The viral particles retained within cells were also reduced by 60% (Fig. 8B), suggesting that autophagy may affect EBV maturation (Fig. 8B). We observed that TPA and sodium butyrate treatment was toxic to P3HR1 cells; however, adding 3-MA did not increase cell death further (Fig. 8C), suggesting that the effect of 3-MA on the lytic cycle of EBV is probably not associated with its toxicity. To elucidate further the effects of autophagy on the lytic cycle of EBV, the autophagy pathway was attenuated by using Atg5 shRNA in P3HR1 cells by lentiviral infection. The expression of Atg5 was significantly reduced after infection, and this lowering was accompanied by a reduction in the level of LC3-II following lytic induction (Fig. 8D, lanes 2, 4, and 6). Meanwhile, expression of Rta, Zta, EA-D, and BBLF1 decreased in Atg5 knockdown cells compared with the control cells (Fig. 8D, lanes 3 to 6); the production of EBV virions was also reduced by 80% (Fig. 8E). The knockdown of Atg5 did not seem to influence cell viability in the latent phase (Fig. 8F). The knockdown of Atg5 in 293EBV(2098) cells reduced the expression of EBV lytic proteins EA-D and BBLF1 (Fig. 8G) and reduced the production of viral particles by 60% (Fig. 8H). Since 293EBV(2089) cells harbor recombinant EBV carrying a GFP gene, the relative virion infectivity could be measured by infecting Raji cells and determining the percentage of cells that were green by flow cytometry. Although the Atg5 siRNA treatment of cells reduced the production of virus particles by 60%, viral infectivity was 9 times that of the cells that were transfected with control siRNA (Fig. 8I). These results suggest that autophagy activation promotes lytic reactivation but reduces viral infectivity.
FIG 8.

Relationship between autophagy and EBV lytic development. (A) P3HR1 cells were pretreated with 10 mM 3-MA for 1 h and then treated with TPA and SB to activate the EBV lytic cycle. Lysates were prepared at 48 or 24 h (for LC3) after lytic induction and analyzed by immunoblotting using antibodies to the indicated proteins. (B) Viral particles in the cell (intracellular) or in the culture medium (extracellular) were analyzed by qPCR after 4 days of lytic induction. (C) The numbers for P3HR1 cells that had been treated with 3-MA or/and TPA and SB were determined at different times. (D) P3HR1 cells that were infected with lentiviruses expressing β-galactosidase (lacZ) or Atg5 shRNA and were treated with TPA and SB. The lysates were prepared 48 h after infection and analyzed by immunoblotting using antibodies against the indicated proteins. (E) The production of viral particles after 4 days of lytic induction was analyzed by qPCR. (F) The numbers of latent P3HR1 cells that were infected with lentiviruses that expressed lacZ or Atg5 shRNA were determined at different times. (G) 293EBV(2089) cells were first transfected with control siRNA (scr) (lanes 1 and 2) or Atg5 siRNA (lanes 3 and 4) and then transfected with control plasmid (Crt) or pCMV-Zta (Z) a day later. Proteins in cell lysates were harvested 48 h after transfection and were analyzed by immunblotting using antibodies against the indicated proteins. (H) The production of viral particles after 4 days of lytic induction was analyzed by qPCR. The numbers of virus particles as percentages of those detected from cells treated with control siRNA are presented. (I) Raji cells were infected with virus particles that were harvested from cells treated with control siRNA or Atg5 siRNA following lytic induction. After 3 days, the percentage of Raji cells that were GFP positive was determined by FACS analysis. The relative infectivity of EBV virions is presented as the fold change of virion infectivity from 293EBV(2089) treated with control siRNA after normalization for virus particles.
DISCUSSION
Autophagy is a cell defense mechanism against viral infections (27). To combat this antiviral response, viruses have developed ways to subvert or manipulate this cellular pathway to facilitate viral propagation (53). This study demonstrates that EBV induces autophagy during lytic reactivation and that the EBV-encoded transcription factor, Rta, participates in autophagic activation via mechanisms that involve transcriptional activation and ERK signaling. The activation of autophagy promotes lytic progression but reduces viral infectivity.
EBV is typically maintained under latent conditions following infection. However, the virus must enter the lytic cycle to generate viral progeny. During EBV latency, LMP1 activates autophagy via the unfolded protein response to regulate LMP1's own synthesis and degradation, modulating LMP1-mediated signaling (41). In this study, EBV induced the formation of autophagosomes in P3HR1, Akata, and 293EBV(2098) cells (Fig. 1) after the virus entered the lytic cycle, suggesting that EBV lytic reactivation induces autophagy. KSHV also activates autophagy after lytic reactivation (40), revealing that autophagic activation in the lytic cycle may be common among gammaherperviruses. Autophagic activation is also observed in cells that are lytically infected by murine gammaherpesvirus 68 (γMV68) (54), varicella-zoster virus (55), and HSV-1 and human cytomegalovirus (HCMV) (56). After infection, HSV-1 activates double-stranded RNA-activated protein kinase (PKR), which induces autophagy (57). Although the role of PKR in autophagy activation during the lytic cycle in gammaherpesviruses is unknown, EBV and KHSV are known to encode viral proteins to activate autophagy (40).
Like KSHV (40), EBV encodes the transcription factor Rta, which participates in autophagic activation (Fig. 2). The present study demonstrates that autophagic induction by Rta depends on Atg5 in 293T cells (Fig. 3). Rta is known to activate the transcription of EBV lytic genes and cellular genes (13–16, 18, 19). In this study, Rta and its mutants were utilized to demonstrate that the capacity of Rta to transactivate Zp correlates with its ability to induce autophagy (Fig. 5). Rta is known to active Zp indirectly by activating mitogen-activated kinases, resulting in the activation of ATF2, which targets the ZII element of Zp (19, 58). This study revealed that Rta increases the expression of autophagy-related genes involves ERK-mediated transcriptional activation (Fig. 5, 6, and 7), suggesting that Rta may regulate autophagy at the transcriptional level. Although autophagy is regarded as a cellular process that is regulated largely at the posttranscriptional level (23, 59), several studies have revealed that autophagic activation is also regulated at the transcriptional level (59). For instance, transcription factors such as FoxO3, HIF-1, TFEB, and ZKSAN3 activate autophagy by the transcriptional activation of autophagy-related genes in response to stresses (60–63). Our RT-qPCR array and transient-transfection studies indicated that Rta transactivates Map1lc3a, Map1LC3b, Atg9B, TNF, TNFSF10, and IRGM, which are crucially involved in the formation of autophagosomes (26, 64) and the induction of autophagy (65, 66). Therefore, this study suggests that the Rta-activated expression of genes that are essential to autophagy is important to the induction of autophagy activation.
Although an increasing number of studies suggest that ERK is involved in modulating autophagy, the roles of ERK in autophagy are diverse (67). ERK signaling regulates the expression of autophagy and lysosomal genes by regulating the nuclear localization of transcription factor EB (TFEB) (63). ERK8 has been shown to stimulate autophagy by interacting with LC3 (68). Exactly how ERK affects Rta-induced autophagic activation remains unknown. Whether the activation of downstream targets of ERK signaling by Rta, such as ATF2 (19), activates the transcription of the genes that are associated with autophagy is yet to be confirmed. A recent study revealed that a noncanonical AMPK-MEK/ERK-TSC signaling pathway regulates autophagic activation by increasing the expression of Beclin-1 (52). However, an increase in the amount of Beclin-1 in cells that express Rta was not observed (Fig. 2), suggesting that Rta activates autophagy independently of this pathway.
The mTOR pathway is a key regulator of autophagy in response to nutrients. Abundant nutrients, including growth factors, glucose, and amino acids, activate mTOR and suppress autophagy, whereas nutrient deprivation suppresses mTOR, leading to the activation of autophagy (23, 24). Rta does not influence autophagic activation under nutrient starvation conditions (Fig. 2D). Additionally, the expression of Rta does not appear to affect the mTOR signaling pathway (Fig. 4), suggesting that Rta induces autophagy by a mechanism that is independent of mTOR.
Autophagic activation in the lytic cycle of herpesviruses is common, but its effects on viral growth and pathogenesis vary among the viruses. In alphaherpesvirus, HSV-1 encodes ICP34.5 to suppress PKR-mediated autophagy activation during lytic infection, increasing its replication and pathogenesis (37, 57). Other herpesviruses also encode proteins that suppress autophagy, including TRS1 of HCMV (69), a viral homolog of Bcl-2 that is encoded by KSHV and γMV68 (70), and vFLIP of KSHV (39), suggesting that autophagy maybe negatively affects these herpesviruses. In contrast, the present study and others (40) revealed that EBV and KSHV each encode a lytic transcription factor, Rta and RTA, respectively, that activates autophagy. The activation of autophagy appears to favor viral progression, since the inhibition of autophagy reduces lytic gene expression and viral replication following the lytic activation of EBV and KSHV. This phenomenon is unique to gammaherpesviruses, which are typically maintained under latency after infection. We hypothesize that autophagy is important and beneficial during the lytic switch from latency but is harmful after the lytic program has begun. Viruses may encode proteins, such as vFLIP and vBcL-2, to suppress autophagy and thereby counteract this cellular defense pathway. Accordingly, the functions of autophagy in herpesviruses are complex and depend on the stage of the lytic cycle. How autophagy influences the expression of lytic genes following reactivation in EBV and KSHV remains unclear. The autophagy machinery probably degrades cellular or viral factors that repress EBV lytic reactivation. Additionally, the induction of the EBV lytic cycle causes endoplasmic reticulum and oxidative stresses (71), and autophagic induction may prevent stress-induced cell death, eventually favoring lytic development. This study also found that 3-MA, an inhibitor of autophagy, led to viral particles being retained within cells (Fig. 8B), suggesting that autophagy may also influence viral egress, although the possibility that 3-MA inhibits protein trafficking events and thereby affects viral egress indirectly cannot be excluded. Interestingly, although the production of viral particles is lowered in autophagic-defective cells after EBV lytic reactivation, increased infectivity of the virions was observed, suggesting that autophagy reduces EBV infectivity. This study reveals that autophagy critically affects EBV lytic development.
ACKNOWLEDGMENTS
We thank the Chang Gung Medicine Research Center (CMRPD680023, CMRPD6C0011, and CMRPD1B0371) for financially supporting this research.
Chia-Shu Liu's (National Cheng Kung University, Taiwan) provision of the plasmid GFP-LC3 is greatly appreciated.
Footnotes
Published ahead of print 13 August 2014
REFERENCES
- 1. Diehl V, Henle G, Henle W, Kohn G. 1968. Demonstration of a herpes group virus in cultures of peripheral leukocytes from patients with infectious mononucleosis. J. Virol. 2:663–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Epstein MA, Barr YM. 1964. Cultivation in vitro of human lymphoblasts from Burkitt's malignant lymphoma. Lancet i:252–253 [DOI] [PubMed] [Google Scholar]
- 3. Hoshikawa Y, Satoh Y, Murakami M, Maeta M, Kaibara N, Ito H, Kurata T, Sairenji T. 2002. Evidence of lytic infection of Epstein-Barr virus (EBV) in EBV-positive gastric carcinoma. J. Med. Virol. 66:351–359. 10.1002/jmv.2152 [DOI] [PubMed] [Google Scholar]
- 4. Klein G, Geering G, Old LJ, Henle G, Henle W, Clifford P. 1970. Comparison of the anti-EBV titer and the EBV-associated membrane reactive and precipitating antibody levels in the sera of Burkitt lymphoma and nasopharyngeal carcinoma patients and controls. Int. J. Cancer 5:185–194. 10.1002/ijc.2910050204 [DOI] [PubMed] [Google Scholar]
- 5. Daibata M, Humphreys RE, Takada K, Sairenji T. 1990. Activation of latent EBV via anti-IgG-triggered, second messenger pathways in the Burkitt's lymphoma cell line Akata. J. Immunol. 144:4788–4793 [PubMed] [Google Scholar]
- 6. Faggioni A, Zompetta C, Grimaldi S, Barile G, Frati L, Lazdins J. 1986. Calcium modulation activates Epstein-Barr virus genome in latently infected cells. Science 232:1554–1556. 10.1126/science.3012779 [DOI] [PubMed] [Google Scholar]
- 7. zur Hausen H, O'Neill FJ, Freese UK, Hecker E. 1978. Persisting oncogenic herpesvirus induced by the tumour promotor TPA. Nature 272:373–375. 10.1038/272373a0 [DOI] [PubMed] [Google Scholar]
- 8. Chevallier-Greco A, Manet E, Chavrier P, Mosnier C, Daillie J, Sergeant A. 1986. Both Epstein-Barr virus (EBV)-encoded trans-acting factors, EB1 and EB2, are required to activate transcription from an EBV early promoter. EMBO J. 5:3243–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feederle R, Kost M, Baumann M, Janz A, Drouet E, Hammerschmidt W, Delecluse HJ. 2000. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 19:3080–3089. 10.1093/emboj/19.12.3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carey M, Kolman J, Katz DA, Gradoville L, Barberis L, Miller G. 1992. Transcriptional synergy by the Epstein-Barr virus transactivator ZEBRA. J. Virol. 66:4803–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang LK, Chuang JY, Nakao M, Liu ST. 2010. MCAF1 and synergistic activation of the transcription of Epstein-Barr virus lytic genes by Rta and Zta. Nucleic Acids Res. 38:4687–4700. 10.1093/nar/gkq243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ragoczy T, Heston L, Miller G. 1998. The Epstein-Barr virus Rta protein activates lytic cycle genes and can disrupt latency in B lymphocytes. J. Virol. 72:7978–7984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ho CH, Hsu CF, Fong PF, Tai SK, Hsieh SL, Chen CJ. 2007. Epstein-Barr virus transcription activator Rta upregulates decoy receptor 3 expression by binding to its promoter. J. Virol. 81:4837–4847. 10.1128/JVI.02448-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hung CH, Liu ST. 1999. Characterization of the Epstein-Barr virus BALF2 promoter. J. Gen. Virol. 80:2747–2750 [DOI] [PubMed] [Google Scholar]
- 15. Kenney S, Holley-Guthrie E, Mar EC, Smith M. 1989. The Epstein-Barr virus BMLF1 promoter contains an enhancer element that is responsive to the BZLF1 and BRLF1 transactivators. J. Virol. 63:3878–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Quinlivan EB, Holley-Guthrie EA, Norris M, Gutsch D, Bachenheimer SL, Kenney SC. 1993. Direct BRLF1 binding is required for cooperative BZLF1/BRLF1 activation of the Epstein-Barr virus early promoter, BMRF1. Nucleic Acids Res. 21:1999–2007. 10.1093/nar/21.8.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chang LK, Chung JY, Hong YR, Ichimura T, Nakao M, Liu ST. 2005. Activation of Sp1-mediated transcription by Rta of Epstein-Barr virus via an interaction with MCAF1. Nucleic Acids Res. 33:6528–6539. 10.1093/nar/gki956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ragoczy T, Miller G. 2001. Autostimulation of the Epstein-Barr virus BRLF1 promoter is mediated through consensus Sp1 and Sp3 binding sites. J. Virol. 75:5240–5251. 10.1128/JVI.75.11.5240-5251.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Adamson AL, Darr D, Holley-Guthrie E, Johnson RA, Mauser A, Swenson J, Kenney S. 2000. Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 activate the ATF2 transcription factor by increasing the levels of phosphorylated p38 and c-Jun N-terminal kinases. J. Virol. 74:1224–1233. 10.1128/JVI.74.3.1224-1233.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Darr CD, Mauser A, Kenney S. 2001. Epstein-Barr virus immediate-early protein BRLF1 induces the lytic form of viral replication through a mechanism involving phosphatidylinositol-3 kinase activation. J. Virol. 75:6135–6142. 10.1128/JVI.75.13.6135-6142.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee YH, Chiu YF, Wang WH, Chang LK, Liu ST. 2008. Activation of the ERK signal transduction pathway by Epstein-Barr virus immediate-early protein Rta. J. Gen. Virol. 89:2437–2446. 10.1099/vir.0.2008/003897-0 [DOI] [PubMed] [Google Scholar]
- 22. Chen YL, Chen YJ, Tsai WH, Ko YC, Chen JY, Lin SF. 2009. The Epstein-Barr virus replication and transcription activator, Rta/BRLF1, induces cellular senescence in epithelial cells. Cell Cycle. 8:58–65. 10.4161/cc.8.1.7411 [DOI] [PubMed] [Google Scholar]
- 23. Kroemer G, Marino G, Levine B. 2010. Autophagy and the integrated stress response. Mol. Cell 40:280–293. 10.1016/j.molcel.2010.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mizushima N. 2007. Autophagy: process and function. Genes Dev. 21:2861–2873. 10.1101/gad.1599207 [DOI] [PubMed] [Google Scholar]
- 25. Shintani T, Klionsky DJ. 2004. Autophagy in health and disease: a double-edged sword. Science 306:990–995. 10.1126/science.1099993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xie Z, Klionsky DJ. 2007. Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9:1102–1109. 10.1038/ncb1007-1102 [DOI] [PubMed] [Google Scholar]
- 27. Deretic V, Levine B. 2009. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5:527–549. 10.1016/j.chom.2009.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Orvedahl A, Levine B. 2009. Eating the enemy within: autophagy in infectious diseases. Cell Death Differ. 16:57–69. 10.1038/cdd.2008.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saitoh T, Akira S. 2010. Regulation of innate immune responses by autophagy-related proteins. J. Cell Biol. 189:925–935. 10.1083/jcb.201002021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Munz C. 2005. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307:593–596. 10.1126/science.1104904 [DOI] [PubMed] [Google Scholar]
- 31. Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. 1998. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 72:8586–8596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Y, Schiff M, Czymmek K, Talloczy Z, Levine B, Dinesh-Kumar SP. 2005. Autophagy regulates programmed cell death during the plant innate immune response. Cell 121:567–577. 10.1016/j.cell.2005.03.007 [DOI] [PubMed] [Google Scholar]
- 33. Dreux M, Gastaminza P, Wieland SF, Chisari FV. 2009. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. U. S. A. 106:14046–14051. 10.1073/pnas.0907344106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jackson WT, Giddings TH, Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. 2005. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156. 10.1371/journal.pbio.0030156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sir D, Liang C, Chen WL, Jung JU, Ou JH. 2008. Perturbation of autophagic pathway by hepatitis C virus. Autophagy 4:830–831. 10.4161/auto.6566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A, Federico M, Panganiban A, Vergne I, Deretic V. 2009. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 186:255–268. 10.1083/jcb.200903070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B. 2007. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35. 10.1016/j.chom.2006.12.001 [DOI] [PubMed] [Google Scholar]
- 38. Talloczy Z, Jiang W, Virgin HWt Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. 2002. Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 99:190–195. 10.1073/pnas.012485299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, Jung JU. 2009. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 11:1355–1362. 10.1038/ncb1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wen HJ, Yang Z, Zhou Y, Wood C. 2010. Enhancement of autophagy during lytic replication by the Kaposi's sarcoma-associated herpesvirus replication and transcription activator. J. Virol. 84:7448–7458. 10.1128/JVI.00024-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee DY, Sugden B. 2008. The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 27:2833–2842. 10.1038/sj.onc.1210946 [DOI] [PubMed] [Google Scholar]
- 42. Taylor GS, Long HM, Haigh TA, Larsen M, Brooks J, Rickinson AB. 2006. A role for intercellular antigen transfer in the recognition of EBV-transformed B cell lines by EBV nuclear antigen-specific CD4+ T cells. J. Immunol. 177:3746–3756. 10.4049/jimmunol.177.6.3746 [DOI] [PubMed] [Google Scholar]
- 43. Delecluse HJ, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. U. S. A. 95:8245–8250. 10.1073/pnas.95.14.8245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen LW, Raghavan V, Chang PJ, Shedd D, Heston L, Delecluse HJ, Miller G. 2009. Two phenylalanines in the C-terminus of Epstein-Barr virus Rta protein reciprocally modulate its DNA binding and transactivation function. Virology 386:448–461. 10.1016/j.virol.2009.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–5728. 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chiu YF, Tung CP, Lee YH, Wang WH, Li C, Hung JY, Wang CY, Kawaguchi Y, Liu ST. 2007. A comprehensive library of mutations of Epstein Barr virus. J. Gen. Virol. 88:2463–2472. 10.1099/vir.0.82881-0 [DOI] [PubMed] [Google Scholar]
- 47. Ryan JL, Fan H, Swinnen LJ, Schichman SA, Raab-Traub N, Covington M, Elmore S, Gulley ML. 2004. Epstein-Barr Virus (EBV) DNA in plasma is not encapsidated in patients with EBV-related malignancies. Diagn. Mol. Pathol. 13:61–68. 10.1097/00019606-200406000-00001 [DOI] [PubMed] [Google Scholar]
- 48. Wang WH, Chang LK, Liu ST. 2011. Molecular interactions of Epstein-Barr virus capsid proteins. J. Virol. 85:1615–1624. 10.1128/JVI.01565-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mizushima N, Yoshimori T, Levine B. 2010. Methods in mammalian autophagy research. Cell 140:313–326. 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mishima Y, Terui Y, Mishima Y, Taniyama A, Kuniyoshi R, Takizawa T, Kimura S, Ozawa K, Hatake K. 2008. Autophagy and autophagic cell death are next targets for elimination of the resistance to tyrosine kinase inhibitors. Cancer Sci. 99:2200–2208. 10.1111/j.1349-7006.2008.00932.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. 1998. A protein conjugation system essential for autophagy. Nature 395:395–398. 10.1038/26506 [DOI] [PubMed] [Google Scholar]
- 52. Wang J, Whiteman MW, Lian H, Wang G, Singh A, Huang D, Denmark T. 2009. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J. Biol. Chem. 284:21412–21424. 10.1074/jbc.M109.026013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sir D, Ou JH. 2010. Autophagy in viral replication and pathogenesis. Mol. Cells 29:1–7. 10.1007/s10059-010-0014-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Suarez AL, Kong R, George T, He L, Yue Z, van Dyk LF. 2011. Gammaherpesvirus 68 infection of endothelial cells requires both host autophagy genes and viral oncogenes for optimal survival and persistence. J. Virol. 85:6293–6308. 10.1128/JVI.00001-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Carpenter JE, Jackson W, Benetti L, Grose C. 2011. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. J. Virol. 85:9414–9424. 10.1128/JVI.00281-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McFarlane S, Aitken J, Sutherland JS, Nicholl MJ, Preston VG, Preston CM. 2011. Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. J. Virol. 85:4212–4221. 10.1128/JVI.02435-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Talloczy Z, Virgin HWt Levine B. 2006. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2:24–29. 10.4161/auto.2176 [DOI] [PubMed] [Google Scholar]
- 58. Morton S, Davis RJ, Cohen P. 2004. Signalling pathways involved in multisite phosphorylation of the transcription factor ATF-2. FEBS Lett. 572:177–183. 10.1016/j.febslet.2004.07.031 [DOI] [PubMed] [Google Scholar]
- 59. He C, Klionsky DJ. 2009. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43:67–93. 10.1146/annurev-genet-102808-114910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM. 2009. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 29:2570–2581. 10.1128/MCB.00166-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chauhan S, Goodwin JG, Manyam G, Wang J, Kamat AM, Boyd DD. 2013. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol. Cell 50:16–28. 10.1016/j.molcel.2013.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. 2007. FoxO3 controls autophagy in skeletal muscle in vivo. Cell. Metab. 6:458–471. 10.1016/j.cmet.2007.11.001 [DOI] [PubMed] [Google Scholar]
- 63. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. 2011. TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433. 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, Ichikawa R, Kinjo M, Ohsumi Y. 2012. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J. Cell Biol. 198:219–233. 10.1083/jcb.201202061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Keller CW, Fokken C, Turville SG, Lunemann A, Schmidt J, Munz C, Lunemann JD. 2011. TNF-alpha induces macroautophagy and regulates MHC class II expression in human skeletal muscle cells. J. Biol. Chem. 286:3970–3980. 10.1074/jbc.M110.159392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mills KR, Reginato M, Debnath J, Queenan B, Brugge JS. 2004. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc. Natl. Acad. Sci. U. S. A. 101:3438–3443. 10.1073/pnas.0400443101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sridharan S, Jain K, Basu A. 2011. Regulation of autophagy by kinases. Cancers 3:2630–2654. 10.3390/cancers3022630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Colecchia D, Strambi A, Sanzone S, Iavarone C, Rossi M, Dall'Armi C, Piccioni F, Verrotti di Pianella A, Chiariello M. 2012. MAPK15/ERK8 stimulates autophagy by interacting with LC3 and GABARAP proteins. Autophagy 8:1724–1740. 10.4161/auto.21857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chaumorcel M, Lussignol M, Mouna L, Cavignac Y, Fahie K, Cotte-Laffitte J, Geballe A, Brune W, Beau I, Codogno P, Esclatine A. 2012. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J. Virol. 86:2571–2584. 10.1128/JVI.05746-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liang C, E X, Jung JU. 2008. Downregulation of autophagy by herpesvirus Bcl-2 homologs. Autophagy 4:268–272. 10.4161/auto.5210 [DOI] [PubMed] [Google Scholar]
- 71. Gargouri B, Van Pelt J, El Feki Ael F, Attia H, Lassoued S. 2009. Induction of Epstein-Barr virus (EBV) lytic cycle in vitro causes oxidative stress in lymphoblastoid B cell lines. Mol. Cell. Biochem. 324:55–63. 10.1007/s11010-008-9984-1 [DOI] [PubMed] [Google Scholar]