

ABSTRACT
Eriophyid mite-transmitted, multipartite, negative-sense RNA plant viruses with membrane-bound spherical virions are classified in the genus Emaravirus. We report here that the eriophyid mite-transmitted Wheat mosaic virus (WMoV), an Emaravirus, contains eight genomic RNA segments, the most in a known negative-sense RNA plant virus. Remarkably, two RNA 3 consensus sequences, encoding the nucleocapsid protein, were found with 12.5% sequence divergence, while no heterogeneity was observed in the consensus sequences of additional genomic RNA segments. The RNA-dependent RNA polymerase, glycoprotein precursor, nucleocapsid, and P4 proteins of WMoV exhibited limited sequence homology with the orthologous proteins of other emaraviruses, while proteins encoded by additional genomic RNA segments displayed no significant homology with proteins reported in GenBank, suggesting that the genus Emaravirus evolved further with a divergent octapartite genome. Phylogenetic analyses revealed that WMoV formed an evolutionary link between members of the Emaravirus genus and the family Bunyaviridae. Furthermore, genomic-length virus- and virus-complementary (vc)-sense strands of all WMoV genomic RNAs accumulated asymmetrically in infected wheat, with 10- to 20-fold more virus-sense genomic RNAs than vc-sense RNAs. These data further confirm the octapartite negative-sense polarity of the WMoV genome. In WMoV-infected wheat, subgenomic-length mRNAs of vc sense were detected for genomic RNAs 3, 4, 7, and 8 but not for other RNA species, suggesting that the open reading frames present in the complementary sense of genomic RNAs are expressed through subgenomic- or near-genomic-length vc-sense mRNAs.
IMPORTANCE Wheat mosaic virus (WMoV), an Emaravirus, is the causal agent of High Plains disease of wheat and maize. In this study, we demonstrated that the genome of WMoV comprises eight negative-sense RNA segments with an unusual sequence polymorphism in an RNA encoding the nucleocapsid protein but not in the additional genomic RNA segments. WMoV proteins displayed weak or no homology with reported emaraviruses, suggesting that the genus Emaravirus further evolved with a divergent octapartite genome. The current study also examined the profile of WMoV RNA accumulation in wheat and provided evidence for the synthesis of subgenomic-length mRNAs of virus complementary sense. This is the first report to demonstrate that emaraviruses produce subgenomic-length mRNAs that are most likely utilized for genome expression. Importantly, this study facilitates the examination of gene functions and virus diversity and the development of effective diagnostic methods and management strategies for an economically important but poorly understood virus.
INTRODUCTION
Eriophyid mite-transmitted, multipartite single-stranded RNA plant viruses with negative polarity recently were classified in the genus Emaravirus (1). The Emaravirus virions are membrane-bound particles of 80 to 200 nm in diameter, resembling those of Tospovirus. Members of the genus Emaravirus possess four to six negative (−)-sense genomic RNA segments, with each segment encoding one open reading frame (ORF) in its complementary (+) sense. The RNA-dependent RNA polymerase (RdRp), glycoprotein precursor (GP), nucleocapsid (NC), and putative movement protein encoded by RNAs 1, 2, 3, and 4, respectively, exhibit relatively low but detectable sequence homology among Emaravirus species. In contrast, little or no homology was found among emaraviruses for the protein encoded by RNA 5 (1). Although emaravirus genomes are multipartite in nature, individual members differ substantially in the number of genomic RNAs they possess. European mountain ash ringspot-associated virus (EMARaV), the type species of the Emaravirus genus (2, 3), and Rose rosette virus (RRV) (4) are quadripartite; Pigeonpea sterility mosaic virus (PPSMV) (5, 6) and Raspberry leaf blotch virus (RLBV) (7) are pentapartite; and Fig mosaic virus (FMV) (8–10) is a hexapartite virus. The diverse number of genomic RNA segments suggests that emaraviruses evolve by acquiring additional genomic RNAs in order to facilitate precise virus-host and virus-vector interactions for virus survival.
Viruses employ efficient genome expression strategies for the successful invasion of hosts by overcoming the hostile host environment (11). However, the genome expression strategy of emaraviruses is poorly understood. The accumulation profile of genomic-length virus- and virus-complementary (vc)-sense RNAs and synthesis of messenger RNAs (mRNAs) in infected plants have not been examined for emaraviruses. Moreover, it is not clearly established whether ORFs present in the complementary strand of genomic RNAs directly translate from the genome-length vc-sense RNAs or subgenomic-length mRNAs of virus-complementary sense.
High Plains (HP) disease, an economically important disease of wheat (Triticum aestivum L.) and maize (Zea mays L.), was first found in the Great Plains region of the United States in 1993 and 1994 (12). Since then, HP disease has been reported from different regions in the United States, Australia, and New Zealand (13–18). The causal agent of HP disease is transmitted by the wheat curl mite (Aceria tosichella Keifer) (19, 20) and was identified as High Plains virus based on double-membrane virus-like particles and a 32-kDa NC protein from partially purified virion preparations (12, 21). However, subsequent studies renamed the causal agent of HP disease Wheat mosaic virus (WMoV), based on International Committee on Taxonomy of Viruses conventions for naming taxa, and suggested it as a tentative member of the Emaravirus genus (1, 22). WMoV is a poorly studied virus because attempts to characterize its genome have not been successful, mainly due to the inability to transmit the virus mechanically and difficulty in long-term maintenance of live virus due to complex virus-mite transmission interactions and the annual nature of wheat and maize. Additionally, most of the field-collected samples are often coinfected with eriophyid mite-transmitted Wheat streak mosaic virus (WSMV) and/or Triticum mosaic virus (TriMV). Except for the partial sequence of RNA 3 encoding the NC protein, the genome sequence of WMoV is not available (15, 17, 22, 23). The unusually low (18 to 31%) amino acid identity of the NC protein with other reported emaraviruses prompted further examination of the phylogenetic relationships of WMoV.
In this study, we examined how the eriophyid mite-transmitted negative-sense RNA plant viruses, with a diverse number of divergent RNA segments, further evolved by determining the genome composition of WMoV. The combination of relatively pure virion RNA and high-throughput RNA sequencing technology allowed us to determine that WMoV contains eight genomic RNA segments, the most found in any known negative-strand RNA plant virus. Interestingly, significant sequence polymorphism was found in an RNA encoding the nucleocapsid protein but not in additional genomic RNA segments. Phylogenetic analysis of NC proteins suggested that WMoV formed an evolutionary link between emaraviruses and members of the family Bunyaviridae. Additionally, the accumulation profile of WMoV-specific RNAs in wheat demonstrated that WMoV produces virus- and vc-sense strands of all genomic RNAs plus subgenomic- or near-genomic-length mRNAs of vc sense for its genome expression.
MATERIALS AND METHODS
Maintenance of WMoV-viruliferous wheat curl mite colony and WMoV-infected wheat tissue.
The WMoV-viruliferous wheat curl mite colony was established by collecting wheat tillers with virus-like symptoms from several wheat fields in three western Nebraska counties (Box Butte, Scottsbluff, and Chase) during the 2011 spring season. These tillers were placed in contact with 14-day-old wheat (cv. Millennium) plants in 4-cm-diameter Cone-tainers (Stuewe and Sons Inc.) to enable wheat curl mites to transfer to the new plants. All plants were covered with cylindrical plastic cages with screened vent holes to prevent possible contamination and were maintained in a growth chamber at 25 to 27°C with a 14-h light and 10-h dark photoperiod. Three weeks after infestation, wheat plants were assayed for WMoV infection by reverse transcription-PCR (RT-PCR), using RNA 3-specific oligonucleotides (22) (see Table S1 in the supplemental material). Subsequently, wheat curl mites from WMoV-positive plants were used in a series of single-mite transfers to eliminate possible contamination with WSMV and/or TriMV, followed by mite-virus propagation as described in McMechan et al. (24).
Nucleocapsid purification and isolation of viral RNA.
Symptomatic leaves from wheat infested with WMoV-viruliferous mites were used for partial purification of nucleocapsids as described in Lane (25), with slight modifications (26). Viral RNA was isolated from partially purified nucleocapsids as described in Tatineni et al. (26) and analyzed on a 1.2% formamide-formaldehyde agarose gel (27).
Illumina sequencing library construction.
TruSeq RNA library construction of partially purified virion RNA was performed at the Interdisciplinary Center for Biotechnology Research Gene Expression Core Facility, University of Florida, Gainesville, FL, using an Illumina TruSeq RNA sample preparation kit (Illumina, San Diego, CA). Briefly, 1.5 μg of WMoV RNA was fragmented using a divalent cation solution and incubated at 94°C. This step was followed by first-strand cDNA synthesis using avian myeloblastosis virus (AMV) reverse transcriptase (Roche, Indianapolis, IN) and random primers (Promega, Madison, WI). Synthesis of double-stranded cDNA was performed using a second-strand master mix provided with the kit, followed by end repair, dA-tailing, and Illumina adaptor ligation. Finally, the library was enriched by 12 cycles of amplification and purified by Agencourt AMPure beads (Beckman Coulter, Brea, CA). The library size and mass were assessed by analysis in a Bioanalyzer (Agilent Technologies, Santa Clara, CA). Typically, a 200- to 2,000-bp-broad library peak was observed, with the highest peak at ∼500 bp. Quantitative PCR was used to validate the library's functionality, using a Kapa library quantification kit (Kapa Biosystems, Wilmington, MA) with monitoring on an ABI 7900HT real-time PCR system.
Illumina MiSeq sequencing.
Sequencing was performed using the reagents provided in the Illumina 500-cycle MiSeq, version 2, sequencing kit. Ten microliters of library was mixed with 10 μl of 0.1 N NaOH for 5 min. Then the library was diluted to 20 pM in the HT1 buffer provided with the kit. A final dilution to 13 pM was performed with HT1 buffer for a final volume of 1 ml. A volume of 600 μl was loaded onto the reagent cartridge for sequencing. Denatured, diluted libraries were sequenced on an Illumina MiSeq benchtop sequencer with the sequencing-by-synthesis technology. Runs were set for “generate FASTQ only” workflow in Illumina Experiment Manager. Reagent cartridges, 500-cycle MiSeq, version 2 (Illumina), were used to sequence libraries with paired-end indexed runs of 251 cycles per read (two 251-cycle reads).
Bioinformatics.
The filtered paired-end reads were mapped onto the wheat genome sequence (28) using Bowtie (version 2.1.0) with the “very-sensitive” option (29). To identify the possible adapter contamination, the filtered paired-end reads were mapped onto the UniVec database. The Trinity platform (30) was used for de novo assembly of contigs from reads that were subtracted from the wheat genome. The gene expression levels (the number of reads in a contig) were determined using Bowtie (version 2.1.0) (29). The numerical values of gene expression were measured by reads per kilobase per million mapped reads (RPKM) to normalize for the number of sequencing reads and total read length (31).
Determination of the 5′ end of genomic RNAs.
The exact 5′ end sequence of WMoV RNAs was determined using partially purified virion RNA as a template for a 5′ rapid amplification of cDNA ends (RACE) system (Life Technologies, Carlsbad, CA). The first-strand cDNA was synthesized using respective minus-sense gene-specific primer 1 of RNAs 1 to 8 (see Table S1 in the supplemental material), followed by column purification, C-tailing of the 3′ end of first-strand cDNA, and PCR amplification with minus-sense gene-specific primer 2 of RNAs 1 to 8 (see Table S1) and abridged anchor primer, essentially as described in the 5′ RACE kit. The PCR products were ligated into pGEM-T Easy vector (Promega), and the inserts were sequenced from 15 to 20 clones per genomic RNA.
Sequence analyses.
Pairwise sequence comparisons of WMoV proteins and other reported emaraviruses and selected members of the Bunyaviridae were performed using the ALIGN program from a set of online analysis tools (http://molbiol-tools.ca). Multiple protein sequence alignments were performed with the ClustalW program (32). Phylogenetic analyses were performed with the MEGA, version 6.0, analysis package (33) using the neighbor-joining (NJ) method with the JTT matrix and pairwise gap deletion, with 1,000 bootstrap replicates as the test of phylogeny.
Northern blot hybridization.
Total RNA was extracted from 400 mg of WMoV-infected and healthy wheat leaves as described in Tatineni et al. (34). Total RNA was separated through 1.2% agarose gels containing formaldehyde, followed by electrotransfer to nylon membranes (Roche). Nylon membranes were probed with digoxigenin (DIG)-labeled virus- or vc-sense RNA-specific riboprobes of WMoV RNAs 1 to 8. Prehybridization and hybridization were carried out in a hybridization buffer containing 50% formamide, 5× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 2% blocking solution (Roche), 0.02% SDS, and 0.1% N-lauroylsarcosine at 65°C for 2 to 3 h and overnight, respectively. The nylon membranes were immunologically developed using an anti-DIG-alkaline phosphatase (ALP) conjugate (Roche), essentially as described by the supplier. The WMoV-specific RNA bands were captured and quantified using a Molecular Imager ChemiDoc XRS+ with an Image Lab software system (Bio-Rad).
Nucleotide sequence accession numbers.
The genome sequence of WMoV has been submitted to GenBank under accession numbers KJ939623 RNA 1), KJ939624 (RNA 2), KJ939625 (RNA 3A), KJ939626 (RNA 3B), and KJ939627 to KJ939631 (RNAs 4 to 8).
RESULTS
Purification of nucleocapsids and RNA isolation.
Nucleocapsids were purified from symptomatic wheat leaves at 21 days postinfestation with WMoV-viruliferous mites and from healthy wheat leaves (negative control). A major protein band of 32 kDa was detected from the nucleocapsid preparation of WMoV-infected leaves but not from healthy tissue (Fig. 1A), suggesting that the nucleocapsid preparation was relatively pure, except for a small amount of wheat proteins.
FIG 1.
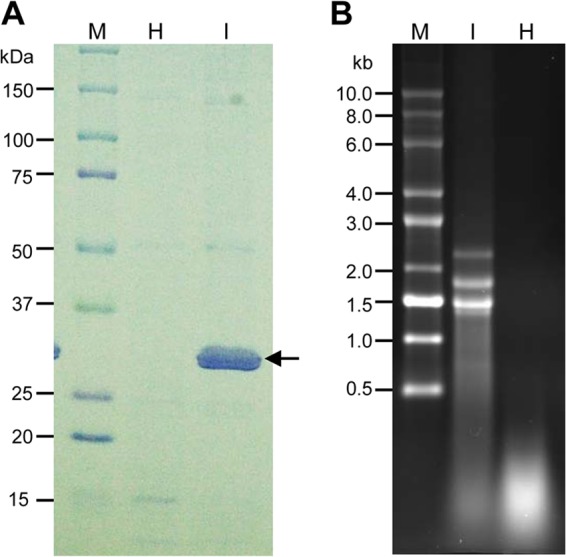
Analyses of nucleocapsids and virion RNA of WMoV. (A) SDS-PAGE gel showing partially purified nucleocapsids from WMoV-infected wheat. Lane M, protein markers; lane H, mock purification from healthy wheat leaves; lane I, nucleocapsids purified from WMoV-infected symptomatic wheat leaves. The position of nucleocapsids is indicated with an arrow. (B) Formaldehyde-agarose gel (1.2%) electrophoresis of partially purified virion RNA of WMoV (I) and RNA isolated from similarly purified preparation from healthy wheat leaves (H) as a negative control. Lane M, RNA size ladder.
RNA isolated from the healthy nucleocapsid (mock) preparation contained no discrete RNA bands but did produce a smear toward the bottom of the gel (Fig. 1B). In contrast, four RNA bands with sizes of 2,300 nucleotides (nt), 1,750 nt, 1,450 nt, and 1,350 nt were found in RNA isolated from the nucleocapsid preparation of WMoV-infected tissue (Fig. 1B). A faint RNA band of ∼7,000 nt was found when an excess amount of virion RNA was loaded (data not shown). These data suggested that the RNA isolated from partially purified nucleocapsids from WMoV-infected tissue was relatively free of host RNAs, and it was used for Illumina MiSeq sequencing. Additionally, protein and RNA profiles from partially purified nucleocapsids were similar to those observed by Skare et al. (22), further confirming that the RNA isolated from the nucleocapsid preparation was indeed WMoV genomic RNA.
High-throughput sequencing of the WMoV genome.
A total of 14.8 million reads obtained from the Illumina MiSeq run (2 times 250 sequencing cycles), with a mean length of 198 nt, were filtered by removing reads with nucleotide Ns and reads that did not have the minimum quality score of 30 (Q30) per base across the whole read length, resulting in 14.7 million reads. These paired-end reads were further filtered by removing 70.4% of the reads that were matched to the wheat genome (28). The remaining 4.4 million reads were used for de novo assembly of contigs using the Trinity platform (30). Twenty-one contigs were obtained, with sizes ranging from 563 to 7,611 nt. As shown in Table 1, some of the contigs corresponded to fewer reads of 12 to 2,926, with low RPKM expression values of 1.68 to 88.18 (Table 1); hence, no further analyses of these sequences were performed.
TABLE 1.
RNA contigs found in high-throughput sequencing of WMoV RNA
| Contig no.a | Sequence length (nt) | No. of reads in contig | RPKMb |
|---|---|---|---|
| 12 | 1,339 | 190,309 | 32,589.61 |
| 13 | 6,850 | 185 | 6.19 |
| 14 | 1,386 | 55 | 9.10 |
| 16 | 1,080 | 33 | 7.01 |
| 17 | 752 | 38 | 11.59 |
| 18 | 2,204 | 490,538 | 51,034.21 |
| 19 | 1,746 | 498,711 | 65,494.54 |
| 19-1 | 6,981 | 468,852 | 15,399.92 |
| 20-1 | 7,611 | 2,926 | 88.15 |
| 20-2 | 2,830 | 904 | 73.25 |
| 23 | 1,636 | 12 | 1.68 |
| 27-1 | 1,439 | 341,883 | 54,477.49 |
| 27-2 | 1,441 | 245,797 | 39,112.27 |
| 28 | 1,715 | 312,065 | 41,723.56 |
| 29 | 1,434 | 170,401 | 27,247.29 |
| 30 | 1,671 | 100,258 | 13,757.61 |
| 44 | 1,346 | 73 | 12.44 |
| 47 | 563 | 27 | 11.00 |
| 51 | 1,001 | 39 | 8.93 |
| 54 | 1,880 | 52 | 6.34 |
| 60 | 4,266 | 83 | 4.46 |
Contigs with excessive coverage of reads are indicated in bold.
Reads per kilobase per million mapped reads.
In contrast, nine contigs were found with an abundance of coverage at 100,258 to 498,711 reads with RPKM values of 13,758 to 65,495 (Table 1), suggesting that these contigs might represent the genomic RNA segments of WMoV. Since all previously reported emaraviruses share a 12-nt conserved motif at both ends of their genomic RNA segments (1), the presence of these conserved nucleotides was examined in overexpressed contigs, and a search was performed using blastx of the BLAST program. WMoV genomic RNAs 1 to 5 were named based on their sequence homology to orthologous proteins of other reported emaraviruses, while RNAs 6 to 8 were named in order of decreasing size.
The 6,981-nt contig 19-1 contained the 12-nt conserved motif at both ends and displayed a weak homology with the RdRp-encoding RNA 1 of other emaraviruses; it was therefore designated RNA 1 of WMoV (Table 1). The 2,204-nt contig 18 contained the 12-nt conserved motif at its 3′ end and 5 of the 12 conserved nucleotides at the 5′ end and was predicted to encode a protein homologous to GP of other emaraviruses (Table 1). Therefore, this contig was designated RNA 2 of WMoV. Contigs 27-1 and 27-2 comprised 1,439 and 1,441 nt RNAs, respectively, with coverage of 341,883 and 245,797 reads (Table 1). These two contigs contained the 12-nt conserved motif at both ends with a significant homology to the WMoV NC protein (22, 23), suggesting that two consensus sequences of WMoV RNA 3 were present in partially purified nucleocapsid preparation. The 1,671-nt contig 30 contained an emaraviral conserved 12-nt motif at the 3′ end but not at the 5′ end and was predicted to encode a protein that displayed a weak homology with emaraviral P4, suggesting that this contig represents RNA 4 of WMoV. The 1,715-nt contig 28 comprised the 12-nt conserved motif at both ends and possessed a weak homology with emaraviral P5 and was therefore designated RNA 5 of WMoV.
The remaining three contigs of 1,746 nt (contig 19), 1,434 nt (contig 29), and 1,339 nt (contig 12) RNAs were found with a coverage of 498,711, 170,401, and 190,309 reads, respectively (Table 1). Contig 19 contained the 12-nt conserved motif at the 3′ end but was missing 6 nt at the 5′ end, while contigs 29 and 12 comprised the 12-nt conserved motif at both ends, suggesting that RNAs of these three contigs might belong to the WMoV genome. The RNA of contig 19 displayed a weak homology with the RLBV P5, while no significant homology was found for RNAs of contigs 29 and 12 with reported GenBank sequences in a BLAST search. Altogether, high-throughput RNA sequencing of partially purified virion RNA found nine RNA species with abundant reads, and these RNAs likely represent eight genomic RNA species of WMoV with two RNA 3 variants. After the discovery of two consensus sequences for RNA 3, we reexamined the high-throughput sequence reads for possible variants in the consensus sequences of other genomic RNAs of WMoV, but none was found.
Authentication of contigs 19, 29, and 12 as WMoV-specific genomic RNAs.
Although contigs 19, 29, and 12 were covered with a large number of reads, it is not clear whether the RNA species represented by these contigs belong to the genomic RNAs of WMoV as no significant homology (except a weak homology for contig 19 RNA) was found with reported emaraviral proteins. Moreover, so far only four to six RNA species have been reported in the genomes of other emaraviruses (1). Authenticity of the RNA sequences of these three contigs was examined by performing RT-PCR with two sets of primers, using total RNA extracted from two WMoV-infected wheat plants.
Authenticity of contig 19 was examined with the primer pairs H-9/H-10 and H-15/H-16 (see Table S1 in the supplemental material). As expected, 600- and 500-bp products were obtained from total RNA from the infected plants but not from the healthy sample (Fig. 2), suggesting that contig 19 belongs to the WMoV genome. Contig 19 was designated genomic RNA 6. Contig 29 was validated with PCR primer pairs H-1/H-4 and H-3/H-2 (see Table S1); 750- and 500-bp products were obtained, as expected, for primer positions from total RNA of the WMoV-infected sample, but no product was obtained from the healthy sample (Fig. 2). These results indicate that contig 29 is a WMoV-specific RNA; it was tentatively designated genomic RNA 7. Two pairs of primers, H-5/H-8 and H-7/H-6, corresponding to contig 12 were used for its validation (see Table S1). As expected for the primer positions in contig 12, RT-PCR products with sizes of 550 and 400 bp were obtained from WMoV-infected samples (Fig. 2), suggesting that contig 12 is specific to WMoV. This contig was named genomic RNA 8. As a positive control, primers corresponding to RNA 3 of WMoV were used for RT-PCR, obtaining a 400-bp-sized product from infected samples (Fig. 2). These data revealed that RNAs representing contigs 19, 29, and 12 are genomic RNAs 6, 7, and 8, respectively, of WMoV.
FIG 2.

Authentication of contigs 19, 29, and 12. Agarose gel (1.2%) electrophoresis of RT-PCR products from total RNA extracted from WMoV-infected wheat plants using two pairs of primers for each contig. Primers specific to RNA 3 were used as a positive control. Lane M, DNA 1.0-kbp ladder; lanes 1 and 2, total RNA from two WMoV-infected wheat plants; lane 3, RNA from healthy wheat; and lane 4, water control.
WMoV is an octapartite virus.
Sequence analyses revealed that WMoV contains eight distinct RNA species in its genome, the most genomic RNA species reported for any known emaravirus. Each genomic RNA encodes an ORF in the vc strand with various sizes of 5′ and 3′ nontranslated regions (NTRs) (Fig. 3). WMoV RNAs 1 to 5 correspond to those found in other emaraviruses, albeit with limited sequence homology, while RNAs 6 to 8 are unique to WMoV, with no significant sequence homology with other emaraviruses. Although six RNA species were reported in the FMV genome (8, 10), none of the WMoV genomic RNAs 6 to 8 possesses significant homology with FMV RNA 6.
FIG 3.

Genome organization of WMoV. The presented schematic representations are genomic RNA segments with an encoded ORF (open rectangles) in each of the genomic RNAs. The genomic RNAs are numbered from the 5′ to 3′ end. The columns at the right show the length of the 5′ nontranslated region (NTR), coding region of an ORF, and 3′ NTR. Genomic RNAs 1 to 5 were named based on sequence homology with orthologous proteins of reported emaraviruses, and RNAs 6 to 8 were designated in order of decreasing RNA size. aa, amino acids.
The 5′ ends of all eight genomic RNAs were verified by the 5′ RACE system. The first 14 nt of all genomic RNAs of WMoV were conserved (5′-AGU AGU GAU CUC CC…) and are complementary with the 3′ end, with the exception of 2 nt, as observed in members of the Emaravirus and Tenuivirus genera and the Bunyaviridae and Arenaviridae families (1, 35, 36). The extreme 3′ end conserved sequences were found in the contigs of all RNA species obtained by high-throughput sequencing. The 13 nt at the 5′ and 3′ ends of all emaravirus and orthobunyavirus genomic RNAs are conserved, with two mismatches (1).
RNA 1 is 6,981 nt long and contains a single large ORF between nt 6913 and 98 with 94- and 68-nt-long 5′ and 3′ NTRs, respectively (Fig. 3). This ORF encodes a 266-kDa protein (P1) of 2,272 amino acids and has a sequence identity of only 33 to 42% with other emaraviral RdRp proteins (Table 2). The P1 of WMoV contains the following Bunyaviridae RdRp signature motifs: motif A (DXKWS1114–1118), motif B (QGXXXXXSS1200–1208), motif C (SDD1241–1243), motif D (KK1284–1285), and motif E (EFLST1294–1298), similar to those found in RdRps of other emaraviruses (4, 7). RNA 2 is 2,211 nt long with a single ORF between nt 2132 and 132 that encodes a putative GP protein (P2). This RNA contains 128- and 79-nt NTRs at the 5′ and 3′ ends, respectively (Fig. 3). WMoV P2 possesses low (24 to 35%) amino acid identity with other emaraviral GP proteins (Table 2). A potential cleavage site was predicted between Ala224 and Asp225, which would release 25.7-kDa (GP1) and 50.9-kDa (GP2) proteins as predicted for RRV (4).
TABLE 2.
Percent amino acid identities of RdRp and GP proteins (above the diagonal) and NC and P4 proteins (below the diagonal) between members of the Emaravirus genus
| Virusa | % Amino acid identities of RdRp and GP proteins (above the diagonal) or NC and P4 proteins (below the diagonal)b |
|||||
|---|---|---|---|---|---|---|
| EMARaV | PPSMV | FMV | RRV | RLBV | WMoV | |
| EMARaV | 100 | 48.1 and 38.7 | 49.3 and 37.6 | 48.8 and 36.8 | 35.2 and 26.4 | 33.7 and 24.7 |
| PPSMV | 33.3 and 15.8 | 100 | 52.7 and 43.4 | 53.3 and 43.2 | 34.6 and 28.0 | 32.8 and 24.4 |
| FMV | 38.7 and 15.7 | 40.9 and 39.6 | 100 | 68.6 and 50.0 | 34.1 and 25.5 | 33.5 and 25.6 |
| RRV | 31.8 and 15.2 | 40.9 and 38.4 | 59.7 and 59.3 | 100 | 33.7 and 26.4 | 33.4 and 28.0 |
| RLBV | 23.8 and 14.1 | 27.7 and 24.1 | 22.2 and 20.3 | 24.8 and 23.7 | 100 | 42.2 and 35.1 |
| WMoVc | 19.1 (18.4) and 13.8 | 20.3 (19.9) and 20.1 | 21.8 (20.3) and 22.7 | 20.4 (19.4) and 22.3 | 31.0 (29.6) and 44.2 | 100 |
EMARaV, European mountain ash ringspot-associated virus (GenBank accession number NC_013105-08); PPSMV, Pigeon pea sterility mosaic virus (HF568801-04); FMV, Fig mosaic virus (HQ703343 to HQ703346); RRV, Rose rosette virus (HQ871942 to HQ871945); RLBV, Raspberry leaf blotch virus (FR823299 to FR823302), WMoV: Wheat mosaic virus (this study).
RdRP, RNA-dependent RNA polymerase encoded by RNA 1; GP, glycoprotein precursor protein encoded by RNA 2; NC, nucleocapsid protein encoded by RNA 3. P4 protein was encoded by RNA 4.
Identities of WMoV P3-A and P3-B proteins encoded by two RNA 3 sequences with the P3s of reported emaraviruses are shown outside and inside the parentheses, respectively. The P3-A and P3-B proteins of WMoV are 88.9% identical to each other.
RNA 3 contains two variant sequences of 1,439 nt (RNA 3A) and 1,441 nt (RNA 3B) and encodes the 33-kDa NC protein (P3) of 286 (P3-A) or 289 (P3-B) amino acids (Fig. 3 and 4). RNA 3A and 3B, respectively, contain a 351- and 352-nt 5′ NTR and a 224- and 219-nt 3′ NTR (Fig. 3). It is unusual to have two divergent sequences encoding the NC protein in a purified virion RNA preparation. The P3-A and P3-B proteins possess amino acid identities of 82.6% and 89.0% with a Texas isolate (22) and of 86.4% and 99.0% with a Kansas isolate (23), respectively, of WMoV. The two NC proteins of WMoV displayed 18 to 31% and 13 to 19% amino acid identity with reported NC proteins of other emaraviruses (Table 2) and tospoviruses (Table 3), respectively.
FIG 4.

Alignment of nucleocapsid protein sequences encoded by WMoV RNAs 3A and 3B. The two P3 sequences (P3-A and P3-B) differ from each other by 11.1% at the amino acid identity level. RNAs 3A and 3B of WMoV are 1,439 and 1,441 nt in length, encoding 286 and 289 amino acids, respectively.
TABLE 3.
Amino acid identity of Wheat mosaic virus proteins with those of selective members of the Bunyaviridae family
| Genus | Virusa | % amino acid identity |
|||
|---|---|---|---|---|---|
| RdRP (P1)b | GC precursor (P2)c | Nucleocapsid (P3)d |
|||
| P3A | P3B | ||||
| Tospovirus | TSWV | 16.6 | 14.9 | 16.1 | 13.3 |
| INSV | 16.8 | 14.4 | 17.6 | 18.5 | |
| PBNV | 17.5 | 14.2 | 13.7 | 13.9 | |
| Orthobunyavirus | LACV | 17.9 | 11.5 | 14.0 | 15.0 |
| BUNV | 18.1 | 12.0 | 16.0 | 12.8 | |
| Nairovirus | CCHFV | 13.9 | 9.9 | 12.9 | 11.0 |
| DUGV | 13.4 | 10.9 | 12.8 | 13.0 | |
| Hantavirus | HTNV | 14.6 | 13.6 | 12.4 | 12.6 |
TSWV, Tomato spotted wilt virus (GenBank accession numbers D10066, S48091, and D00645); INSV, Impatiens necrotic spot virus (X93218, M74904, and X66972); PBNV, Peanut bud necrosis virus (AF025538, U42555, and U27809); LACV, La Crosse virus (AF528165 to AF528167); BUNV, Bunyamwera virus (X14383, M11852, and D00353); CCHFV, Crimean-Congo hemorrhagic fever virus (AY389361, AF467768, and U88410); DUGV, Dugbe virus (U15018, M94133, and AF434161); HTNV, Hantaan virus (X55901, M14627, and M14626).
RNA-dependent RNA polymerase encoded by RNA 1 or large RNA.
Glycoprotein precursor protein encoded by RNA 2 or medium RNA.
Nucleocapsid protein encoded by RNA 3 or small RNA.
WMoV RNA 4 is 1,682 nt in length, encoding an ORF (nt 1570 to 479) of a 42-kDa protein of 364 amino acids (P4). RNA 4 contains 475- and 112-nt-long 5′ and 3′ NTRs, respectively (Fig. 3). The P4 possesses 44% amino acid identity with RLBV P4 and only 14 to 23% identity with the P4 proteins of other emaraviruses (Table 2).
RNA 5 is 1,715 nt long, with a single ORF (nt 1595 to 162) of a 56-kDa protein (P5). RNA 5 contains 158 and 120 nt as the 5′ and 3′ NTRs, respectively (Fig. 3). The P5 protein of WMoV possesses 18 to 24% amino acid sequence identity with the P5 proteins of FMV, RLBV, and PPSMV, and the P5 protein has not been reported for EMARaV and RRV. WMoV RNA 6 is 1,752 nt long with an ORF encoding a 58-kDa protein (P6) between nt 1634 and 159 (Fig. 3). This RNA contains 5′ and 3′ NTRs of 155 and 118 nt, respectively, lengths similar to those in RNA 5 of WMoV (Fig. 3). A BLAST search revealed that the WMoV P6 possesses 23% amino acid identity with 72% coverage with the P5 of RLBV but no significant homology with other GenBank sequences. It is interesting that both the P5 and P6 proteins of WMoV possess similar levels of homology with the RLBV P5 protein.
RNA 7 is 1,434 nt long and comprises an ORF (nt 1317 to 403) encoding a 36-kDa protein (P7) of 305 amino acids. It possesses no sequence homology with proteins in GenBank. This RNA contains 399- and 117-nt-long NTRs at the 5′ and 3′ ends, respectively (Fig. 3). RNA 8 is 1,339 nt long with a single ORF, and 715- and 93-nt-long NTRs at the 5′ and 3′ ends, respectively (Fig. 3). The ORF (nt 1246 to 719) encodes a 21-kDa protein of 176 amino acids that has no sequence homology with any other reported proteins in GenBank.
Heterogeneity in RNA 3 sequence but not in other genomic RNAs.
Assembly of WMoV-specific reads into contigs revealed one consensus sequence for each genomic RNA except for RNA 3, for which two consensus sequences were found (Table 1). The sequences of both RNA 3 contigs were covered with an abundance of reads and differed significantly from each other by 12.5% and 11.1% at the nucleotide and amino acid levels, respectively. We further examined RNA 3 divergence by amplifying a nearly complete RNA 3 using a conserved forward primer corresponding to nt 15 to 40 plus an XhoI site and a reverse primer complementary to nt 1419 to 1395 plus a BamHI site. The RT-PCR product from virion RNA was ligated into pGEM-7Zf(+) between XhoI and BamHI restriction sites, and 60 independent clones were sequenced from both ends.
The length of RNA 3 (including the 5′ and 3′ end sequences that were not present in the RT-PCR product) from the sequenced clones was 1,439 nt and 1,441 nt in 47 and 10 clones, respectively. However, a few additions or deletions in the U-rich region of the 5′ NTR resulted in 1,438 nt (1 clone) and 1,440 nt (2 clones) of RNA 3. Sixty sequences aligned by ClustalW that clustered into 50 RNA 3A sequences and 10 RNA 3B sequences further confirm the presence of two divergent RNA 3 sequences at a 5 to 1 ratio. The NC proteins of RNA 3A and 3B differed from each other by 11.1%, with a 3-amino-acid insertion in P3-B at positions 24 and 25 and at the C terminus of the protein (Fig. 4). These data demonstrate that the purified virion RNA preparation of WMoV contains two divergent RNA 3 sequences.
To determine whether the heterogeneity observed in RNA 3 also exists in other WMoV genomic RNAs, RNAs 4, 5, and 7 were selected as representative RNA segments and amplified by RT-PCR from virion RNA using RNA segment-specific forward (with an XhoI site) and reverse (with a BamHI site) primers positioned downstream and upstream of the 5′ and 3′ conserved sequences, respectively. The RT-PCR products were ligated into pGEM-7Zf(+), and 30 clones per RNA segment were sequenced in both directions. No clone with >1% sequence divergence from the RNA 4, 5, and 7 consensus sequences was found. The nucleotide and amino acid differences in the total sequence of 30 clones were estimated to be, respectively, 0.07% and 0.04% for RNA 4, 0.02% and 0.0% for RNA 5, and 0.09% and 0.07% for RNA 7. As observed within the RNA 3A and 3B sequences, a few additions or deletions were observed in the U-rich regions of 5′ NTRs, and a few random nucleotide substitutions were observed in the coding regions without forming specific sequence groups. These data, together with the absence of divergent consensus sequences for RNAs 1, 2, and 4 to 8 from high-throughput sequencing, suggest that these genomic RNA sequences are nearly homogeneous.
WMoV is a distinct emaravirus.
Phylogenetic analysis was performed with the MEGA, version 6.0, analysis package using the neighbor-joining method with RdRp, GP, and NC proteins of all known emaraviruses and representative members of the family Bunyaviridae and the genus Tenuivirus (Fig. 5). Phylogenetic trees with RdRp, GP, and NC proteins resulted in similar topologies, with all emaraviruses clustered into two distinct clades (Fig. 5). WMoV and RLBV formed as sister taxa in a separate clade from other emaraviruses, suggesting that emaraviruses evolved into two distinct lineages. These two emaraviral clades share a most recent common ancestor with members of the genera Orthobunyavirus and Tospovirus (for RdRp and GP) and Tospovirus, Nairovirus, and Orthobunyavirus (for NC protein) (Fig. 5). Though WMoV and RLBV formed as sister taxa in a clade, these two viruses are distinct from each other, with only 42%, 35%, and 31% amino acid identity between the RdRp, GP, and NC proteins, respectively (Table 2). The two NC proteins of WMoV formed into two separate branches with a strong bootstrap support value in a clade along with RLBV (Fig. 5C), further confirming the presence of two divergent copies of NC protein. These data suggest that WMoV is a distinct virus, which evolved with eight divergent genomic RNA segments with significant polymorphism in RNA 3 encoding the NC protein.
FIG 5.

Phylogenetic analyses of emaraviruses, representative members of Bunyaviridae, and Tenuivirus. Unrooted bootstrap consensus phylogenetic trees were generated from the amino acid sequences of RdRp (A), glycoprotein precursor protein (B), and nucleocapsid protein (C). Phylogenetic trees were constructed by the neighbor-joining method using the JTT matrix and pairwise gap deletion with 1,000 bootstrap replicates; bootstrap support is indicated at branch points. The bar represents the number of amino acid replacements per site. Note that WMoV formed a separate clade with RLBV from other members of the genus Emaravirus. GenBank accession numbers of proteins and the names of viruses used for phylogenetic analyses are given in Tables 2 and 3.
Accumulation of WMoV-specific RNAs in wheat.
The accumulation profile of WMoV-specific RNAs in wheat was examined by Northern blot hybridization using strand-specific riboprobes of genomic RNAs 1 to 8. PCR products of 459 to 971 bp of genomic RNAs 1 to 8 were amplified using RNA segment-specific primers containing the SP6 and T7 RNA polymerase promoter sequences in the forward and reverse primers, respectively (see Table S1 in the supplemental material). Gel-eluted PCR products of RNAs 1 to 8 were used for in vitro transcription to synthesize the virus- or vc-sense DIG-labeled riboprobes with SP6 or T7 RNA polymerase, respectively. The strength and specificity of virus- and vc-sense riboprobes for each genomic RNA were examined by including 5 ng of in vitro transcripts (without DIG-labeled nucleotide mix) of each probe in Northern blot hybridization and probing with the virus- or vc-sense riboprobes of respective RNAs. All riboprobes were hybridized with the opposite sense transcript, and both the virus- and vc-sense riboprobes hybridized approximately with the same intensities with in vitro transcripts of the opposite polarity (Fig. 6B). These data suggest that the riboprobes are highly specific and that the virus- and vc-sense riboprobes of each genomic RNA segment are approximately equal in strength.
FIG 6.

Accumulation of WMoV-specific RNAs in wheat. (A) Northern blot hybridization of total RNA from healthy (H) and WMoV-infected (I) wheat. The virus- and virus complementary-sense RNA-specific riboprobes were generated for portions of ORFs (459 to 971 nt) encoded by RNAs 1 to 8. Note that virus (v)-sense RNA strands accumulated in large amounts compared to virus complementary (vc)-sense RNA strands. RNAs 3, 4, 7, and 8 produced shorter-than-genomic vc-strand RNAs, which are most likely subgenomic-length mRNAs. Subgenomic-length mRNAs were not detected for RNAs 1, 2, 5, and 6, which most likely express through near-genomic-length mRNAs that are difficult to separate from the respective genomic-length vc-strand RNAs. Solid and open arrowheads indicate the positions of genomic-length virus- and vc-sense RNAs, respectively. Subgenomic-length mRNAs are indicated with asterisks. (B) The specificity and strength of DIG-labeled riboprobes were examined by hybridizing each probe with the positive- and negative-sense transcripts of the corresponding probe. Riboprobes for RNA 6 are presented as representative of the probes of all other genomic RNAs.
Total RNA (200 ng per lane) from WMoV-infected and healthy wheat leaves was used for Northern blot hybridization. The virus- and vc-sense-strand RNA-specific riboprobes of RNAs 1 to 8 readily detected the virus- and vc-sense strands of respective genomic RNAs (Fig. 6A). Accumulation of virus-sense genomic RNA strands was 10- to 20-fold greater than that of genomic complementary-strand RNAs (Fig. 6A). Additionally, accumulation of shorter-than-genomic RNAs of vc sense was detected with probes specific to genomic RNAs 3, 4, 7, and 8 (Fig. 6A). Shorter-than-genomic vc-strand RNAs most likely represent subgenomic-length mRNAs for the expression of ORFs present in the vc-sense genomic RNA strands. However, subgenomic-length mRNAs were not detected for the genomic RNAs 1, 2, 5, and 6 (Fig. 6A), and this is most likely due to shorter 5′ NTRs and lack of a putative transcription termination signal containing U-rich regions in these RNAs (37, 38). Our data suggest that each WMoV genomic RNA species produces genomic-length virus- and vc-sense RNAs and subgenomic- or near-genomic-length mRNAs of vc sense in wheat. Taken together, detection of virus- and vc-sense strands of eight genomic RNA segments and of subgenomic-length mRNAs of vc strand for genomic RNAs 3, 4, 7, and 8 in wheat further confirmed that WMoV is an octapartite negative-strand RNA virus that employs subgenomic- or near-genomic-length mRNAs of vc sense for genome expression.
DISCUSSION
The use of relatively pure virion RNA for high-throughput RNA sequencing enabled us to determine that the eriophyid mite-transmitted WMoV comprises eight genomic RNAs, the largest number of genomic RNAs found in any known negative-strand RNA plant virus. Interestingly, we found two significantly divergent RNA 3 consensus sequences, but the additional genomic RNAs were homogeneous in nature. The octapartite nature of the WMoV genome was further confirmed by detecting asymmetric accumulation of virus- and vc-sense strands of all genomic RNAs in infected wheat. Moreover, vc-sense subgenomic-length mRNAs were detected for genomic RNAs 3, 4, 7, and 8.
Proteins encoded by WMoV RNAs 1 to 4 displayed weak homology with the respective homologous proteins of known emaraviruses. The P4 proteins of FMV and RLBV have been demonstrated to be involved in cell-to-cell movement (39, 40). Based on sequence homology with other emaraviral P4 proteins, it is likely that the P4 of WMoV might also be involved in cell-to-cell movement. Although the P5 and P6 proteins of WMoV are unrelated, both of these proteins displayed 23 to 24% amino acid identity with the RLBV P5 protein in a BLAST search. Additionally, the P5, but not the P6, of WMoV possesses weak homology with the P5 proteins of FMV and PPSMV. It is unusual that two WMoV-encoded proteins show sequence homology, though weak, with one protein of RLBV. Perhaps the P5 and P6 of WMoV complement each other's functions in the virus life cycle. The possibility of genomic RNAs 6, 7, and 8 being defective RNAs was excluded because these RNAs do not have sequence homology with any other genomic RNAs of WMoV which likely acts as helper RNAs for defective RNAs. Moreover, these RNAs encode an ORF in vc strands similar to other genomic RNAs, and genomic-length vc-sense RNA strands and subgenomic-length mRNAs have been detected (for RNAs 7 and 8) (see below). It is possible that the P5 to P8 proteins of WMoV are involved in the virus life cycle through roles such as virus transport, determination of host range and pathogenicity, suppression of host RNA silencing, and transmission by wheat curl mites.
Members of the genus Emaravirus are unusually divergent, with a variable number of genomic RNA segments (1). The RdRp, GP, and NC proteins possess weak to moderate identities of 33 to 67%, 24 to 50%, and 19 to 60%, respectively, among members of the genus Emaravirus (Table 2). Although WMoV genome organization is similar, except for the octapartite nature, to that of other emaraviruses, WMoV-encoded RdRp, GP, and NC proteins are divergent from other emaraviruses, with homologies comparable to those between members of different genera in a family (41). Based on lack of significant homology of WMoV RNAs 6 to 8 with GenBank sequences, it is possible that WMoV might have evolved by acquiring some or all of these genomic RNAs as virus-specific genes, as shown for Citrus tristeza virus (42). Although WMoV is distinct from other emaraviruses, it should be maintained as a definitive species in the genus Emaravirus based on similar genome organization, conserved 5′ and 3′ end sequences, limited sequence homology, and eriophyid mite transmission.
Phylogenetic analyses revealed that WMoV formed a separate clade with RLBV from those of other emaraviruses sharing common ancestors with members of the genera Orthobunyavirus, Tospovirus, and Nairovirus. Additionally, the NC protein of WMoV displayed 19% amino acid identity with that of EMARaV, the type species of Emaravirus, as well as with Impatiens necrotic spot virus, a Tospovirus (Tables 2 and 3), suggesting that WMoV formed an evolutionary link between emaraviruses and members of the genus Tospovirus of the family Bunyaviridae.
We found two distinct RNA 3 sequences in virion RNA with 87.5% nucleotide homology. The sequence difference found between these two RNA 3 variants is similar to what is often observed among strains of various other viruses (41). The presence of sequence heterogeneity in only one of the eight genomic RNA segments indicates that the RNA 3 heterogeneity might not be due to mixed infection of wheat with two strains of WMoV. RNA 3 is the least expected genomic RNA to have heterogeneity because it encodes the NC protein, a structural protein that encapsidates genomic RNAs. It is possible that WMoV might tolerate variations in the NC protein but not in other proteins, and it is not clear why an RNA encoding a structural protein is more flexible than the other genomic RNA segments. What is the advantage for a virus to have two distinct RNA 3 sequences? What is the functional role of the two divergent RNA 3 sequences in virus biology? It is possible that WMoV might have acquired RNA 3A and 3B as the result of functional speciation and that the duplicated RNA 3 species might be involved in closely related but distinct functions. Moreover, the presence of RNA 3A and 3B at a 5-to-1 ratio may reflect their differential functional requirements in virus biology. Perhaps the major (P3-A) and minor (P3-B) components of the NC protein might be required for encapsidation of WMoV in wheat and wheat curl mites, respectively, although it remains to be known whether WMoV replicates in its vector. It has been shown that EMARaV is located internally in the pear leaf blister mite (43) and that Tomato spotted wilt tospovirus replicates in thrips (44). However, exploring the biological significance of RNA 3 heterogeneity is beyond the scope of the present work. Future studies should shed light on the nature and biological significance of the two RNA 3 variants found in virion RNA. Though we found nine genomic RNA segments with two copies of RNA 3 in a partially purified nucleocapsid preparation, additional experiments are needed to unequivocally claim WMoV as a nine-segment virus.
WMoV replication in wheat might have resulted in 10- to 20-fold more accumulation of virus-sense genomic RNAs than genomic-length vc-sense strands. Furthermore, detection of virus- and vc-sense strands of all genomic RNAs in wheat confirmed the presence of eight RNA segments in the WMoV genome. Detection of virus- and vc-sense strands of RNAs 2 and 3 in WMoV-infected wheat by Skare et al. (22) further supported the synthesis of genomic-length virus- and vc-sense RNAs of all genomic RNAs in infected wheat. Accumulation of virus-sense genomic RNAs in abundant amounts, as is the case for RNA viruses compared to ORF-containing vc-strand RNAs, further confirmed the negative-sense polarity of genomic RNAs. Additionally, WMoV genomic RNAs 3, 4, 7, and 8 produced shorter-than-genomic complementary-sense RNAs in wheat, which probably represent subgenomic-length mRNAs for the expression of ORFs present in vc-sense genomic RNA strands. In contrast, subgenomic-length mRNAs were not detected for genomic RNAs 1, 2, 5, and 6, possibly due to a small size difference between the genomic-length vc-sense RNAs and subgenomic-length mRNAs. WMoV genomic RNAs 1, 2, 5, and 6 possess short 5′ NTRs without a U-rich region, which may have potential transcription termination signals (37, 38). Our data suggest that asymmetric accumulation of genomic-length virus- and vc-sense RNA strands and production of vc-sense subgenomic-length mRNAs demonstrated replication of all genomic RNAs in wheat and expression of ORFs, possibly through subgenomic-length (for RNAs 3, 4, 6, and 7) or near-genomic-length (for RNAs 1, 2, 5, and 6) mRNAs of vc sense. Previously, it has been reported that mRNAs of FMV RNAs 2 and 3 were generated by a cap-snatching mechanism (45) and that genomic-length virus- and vc-sense RNAs, but not subgenomic-length mRNAs, were detected for genomic RNAs 3, 5, and 6 (10). We demonstrated for the first time that emaraviruses produce subgenomic-length mRNAs that are most likely utilized for genome expression. Additionally, detection of subgenomic-length mRNAs in WMoV-infected wheat could provide a unique opportunity to examine the promotion and termination mechanisms of mRNAs of emaraviruses using WMoV as a model system.
Availability of the genome sequence of WMoV will have practical implications for the management of High Plains disease of wheat and maize. The highly variable nature of the WMoV NC protein (22, 23; also this study) may explain why antibodies produced against one strain of WMoV failed to detect some field isolates (15). The significantly variable nature of RNA 3 suggests that NC protein-based diagnostic methods are unreliable and that serology- or PCR-based diagnostic methods based on other proteins or genomic RNAs would facilitate broad-spectrum and reliable detection of WMoV isolates. Additionally, heterogeneity in RNA 3 suggests that RNA 3-based disease management strategies may have serious consequences on the development of transgenic resistant plants and that WMoV isolates may easily overcome such resistance due to the highly variable nature of RNA 3. Moreover, availability of the genome sequence will facilitate the management of High Plains disease through the development of transgenic plants resistant to WMoV using RNA interference technology.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jonathan Horrell and Melissa Bartels for their technical assistance. We thank David M. Amador and Yanping Zhang for MiSeq RNA sequencing and William G. Farmerie for sequence data analyses at the Interdisciplinary Center for Biotechnology Research, University of Florida, Gainesville, FL, and Jean-Jack M. Riethoven and Seong-il Eyun, Bioinformatics Core Research Facility, University of Nebraska—Lincoln, Lincoln, NE, for high-throughput sequence data analyses.
Funding for this work was partially provided by the Agriculture and Food Research Initiative Competitive Grants Program, grant number 2013-68004-20358, from the National Institute of Food and Agriculture.
Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Footnotes
Published ahead of print 6 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01901-14.
REFERENCES
- 1. Mielke-Ehret N, Mühlbach HP. 2012. Emaravirus: A novel genus of multipartite, negative strand RNA plant viruses. Viruses 4:1515–1536. 10.3390/v4091515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mühlbach HP, Mielke-Ehret N. 2011. Emaravirus, p 767–770 In King AMQ, Adams MJ, Carstens EB, Lefkovitz EJ. (ed), Virus taxonomy: classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses. Academic Press, London, United Kingdom [Google Scholar]
- 3. Mielke N, Muehlbach HP. 2007. A novel, multipartite, negative-strand RNA virus is associated with the ringspot disease of European mountain ash (Sorbus aucuparia L.). J. Gen. Virol. 88:1337–1346. 10.1099/vir.0.82715-0 [DOI] [PubMed] [Google Scholar]
- 4. Laney AG, Keller KE, Martin RR, Tzanetakis IE. 2011. A discovery 70 years in the making: characterization of the rose rosette virus. J. Gen. Virol. 92:1727–1732. 10.1099/vir.0.031146-0 [DOI] [PubMed] [Google Scholar]
- 5. Kumar PL, Jones AT, Reddy DVR. 2003. A novel mite-transmitted virus with a divided RNA genome closely associated with pigeonpea sterility mosaic disease. Phytopathology 93:71–81. 10.1094/PHYTO.2003.93.1.71 [DOI] [PubMed] [Google Scholar]
- 6. Elbeaino T, Digiaro M, Uppala M, Sudini H. 2014. Deep sequencing of Pigeonpea sterility mosaic virus discloses five RNA segments related to emaraviruses. Virus Res. 188:27–31. 10.1016/j.virusres.2014.03.022 [DOI] [PubMed] [Google Scholar]
- 7. McGavin WJ, Mitchell C, Cock PJA, Wright KM, MacFarlane SA. 2012. Raspberry leaf blotch virus, a putative new member of the genus Emaravirus, encodes a novel genomic RNA. J. Gen. Virol. 93:430–437. 10.1099/vir.0.037937-0 [DOI] [PubMed] [Google Scholar]
- 8. Elbeaino T, Digiaro M, Martelli GP. 2009. Complete nucleotide sequence of four RNA segments of fig mosaic virus. Arch. Virol. 154:1719–1727. 10.1007/s00705-009-0509-3 [DOI] [PubMed] [Google Scholar]
- 9. Walia JJ, Salem NM, Falk BW. 2009. Partial sequence and survey analysis identify a multipartite, negative-sense RNA-virus associated with fig mosaic. Plant Dis. 93:4–10. 10.1094/PDIS-93-1-0004 [DOI] [PubMed] [Google Scholar]
- 10. Ishikawa K, Maejima K, Komatsu K, Kitazawa Y, Hashimoto M, Takata D, Yamaji Y, Namba S. 2012. Identification and characterization of two novel genomic RNA segments of fig mosaic virus, RNA5 and RNA6. J. Gen. Virol. 93:1612–1619. 10.1099/vir.0.042663-0 [DOI] [PubMed] [Google Scholar]
- 11. Hull R. 2002. Matthews' plant virology, 4th ed. Academic Press, New York, NY [Google Scholar]
- 12. Jensen SG, Lane LC, Seifers DL. 1996. A new disease of maize and wheat in the high plains. Plant Dis. 80:1387–1390. 10.1094/PD-80-1387 [DOI] [Google Scholar]
- 13. Lebas BSM, Ochoa-Corona FM, Elliott DR, Tang Z, Alexander BJR. 2005. Development of an RT-PCR for High Plains virus indexing scheme in New Zealand post-entry quarantine. Plant Dis. 89:1103–1108. 10.1094/PD-89-1103 [DOI] [PubMed] [Google Scholar]
- 14. Burrows M, Franc G, Rush C, Blunt T, Ito D, Kinzer K, Olson J, O'Mara J, Price J, Tande C, Ziems A, Stack J. 2009. Occurrence of viruses in wheat in the Great Plains region 2008. Plant Health Progress. 10.1094/PHP-2009-0706-01-RS [DOI] [Google Scholar]
- 15. Seifers DL, Martin TJ, Harvey TL, Haber S, Krokhin O, Spicer V, Ying S, Standing KG. 2009. Identification of variants of the High Plains virus infecting wheat in Kansas. Plant Dis. 93:1265–1274. 10.1094/PDIS-93-12-1265 [DOI] [PubMed] [Google Scholar]
- 16. Byamukama E, Seifers DL, Hein GL, De Wolf E, Tisserat NA, Langham MAC, Osborne LE, Timmerman A, Wegulo SN. 2013. Occurrence and distribution of Triticum mosaic virus in the central Great Plains. Plant Dis. 97:21–29. 10.1094/PDIS-06-12-0535-RE [DOI] [PubMed] [Google Scholar]
- 17. Stewart LR, Paul PA, Qu F, Redinbaugh MG, Miao H, Todd J, Jones M. 2013. Wheat mosaic virus (WMoV), the causal agent of High Plains disease, is present in Ohio wheat fields. Plant Dis. 97:1125. 10.1094/PDIS-03-13-0243-PDN [DOI] [PubMed] [Google Scholar]
- 18. Coutts BA, Cox BA, Thomas GJ, Jones RAC. 2014. First report of Wheat mosaic virus infecting wheat in Western Australia. Plant Dis. 98:285. 10.1094/PDIS-03-13-0288-PDN [DOI] [PubMed] [Google Scholar]
- 19. Seifers DL, Harvey TL, Martin TJ, Jensen SG. 1997. Identification of the wheat curl mite as the vector of the High Plains virus of corn and wheat. Plant Dis. 81:1161–1166. 10.1094/PDIS.1997.81.10.1161 [DOI] [Google Scholar]
- 20. Skare JM, Wijkamp I, Rezende JAM, Michels GJ, Rush CM, Scholthof KBG, Scholthof HB. 2003. Colony establishment and maintenance of the eriophyid wheat curl mite Aceria tosichella for controlled transmission studies on a new virus-like pathogen. J. Virol. Methods 108:133–137. 10.1016/S0166-0934(02)00257-4 [DOI] [PubMed] [Google Scholar]
- 21. Louie R, Seifers DL, Bradfute OE. 2006. Isolation, transmission and purification of the High Plains virus. J. Virol. Methods 135:214–222. 10.1016/j.jviromet.2006.03.023 [DOI] [PubMed] [Google Scholar]
- 22. Skare JM, Wijkamp I, Denham I, Rezende JAM, Kitajima EW, Park JW, Desvoyes B, Rush CM, Michels G, Scholthof KBG, Scholthof HB. 2006. A new eriophyid mite-borne membrane-enveloped virus-like complex isolated from plants. Virology 347:343–353. 10.1016/j.virol.2005.11.030 [DOI] [PubMed] [Google Scholar]
- 23. She Y-M, Seifers DL, Haber S, Ens W, Standing KG. 2004. Characterization of the agent of “High Plains disease.” J. Biol. Chem. 279:488–494. 10.1074/jbc.M308506200 [DOI] [PubMed] [Google Scholar]
- 24. McMechan AJ, Tatineni S, French R, Hein GL. 2014. Differential transmission of Triticum mosaic virus by wheat curl mite populations collected in the Great Plains. Plant Dis. 98:806–810. 10.1094/PDIS-06-13-0582-RE [DOI] [PubMed] [Google Scholar]
- 25. Lane LC. 1986. Propagation and purification of RNA plant viruses. Methods Enzymol. 118C:687–696 [Google Scholar]
- 26. Tatineni S, Ziems AD, Wegulo SN, French R. 2009. Triticum mosaic virus: a distinct member of the family Potyviridae with an unusually long leader sequence. Phytopathology 99:943–950. 10.1094/PHYTO-99-8-0943 [DOI] [PubMed] [Google Scholar]
- 27. Sambrook J, Russell D. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 28. Brenchley R, Spannagl M, Pfeifer M, Barker GLA, D'Amore R, Allen AM, McKenzie N, Kramer M, Kerhornou A, Bolser D, Kay S, Waite D, Trick M, Bancroft I, Gu Y, Huo N, Luo MC, Sehgal S, Gill B, Kianian S, Anderson O, Kersey P, Dvorak J, McCombie WR, Hall A, Mayer KFX, Edwards KJ, Bevan MW, Hall N. 2012. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 491:705–710. 10.1038/nature11650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A. 2013. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8:1494–1512. 10.1038/nprot.2013.084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat. Methods 5:621–628. 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 32. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins D. 1997. The Clustal_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25:4876–4882. 10.1093/nar/25.24.4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30:2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tatineni S, Graybosch RA, Hein GL, Wegulo SN, French R. 2010. Wheat cultivar-specific disease synergism and alteration of virus accumulation during co-infection with Wheat streak mosaic virus and Triticum mosaic virus. Phytopathology 100:230–238. 10.1094/PHYTO-100-3-0230 [DOI] [PubMed] [Google Scholar]
- 35. Falk BW, Tsai JH. 1998. Biology and molecular biology of viruses in the genus Tenuivirus. Annu. Rev. Phytopathol. 36:139–163. 10.1146/annurev.phyto.36.1.139 [DOI] [PubMed] [Google Scholar]
- 36. Walter TC, Barr JN. 2011. Recent advances in the molecular and cellular biology of bunyaviruses. J. Gen. Virol. 92:2467–2484. 10.1099/vir.0.035105-0 [DOI] [PubMed] [Google Scholar]
- 37. Hutchinson KL, Peters CJ, Nichol ST. 1996. Sin Nombre virus mRNA synthesis. Virology 224:139–149. 10.1006/viro.1996.0515 [DOI] [PubMed] [Google Scholar]
- 38. van Knippenberg I, Lamine M, Goldbach R, Kormelink R. 2005. Tomato spotted wilt virus transcriptase in vitro displays a preference for cap donors with multiple base complementarity to the viral template. Virology 335:122–130. 10.1016/j.virol.2005.01.041 [DOI] [PubMed] [Google Scholar]
- 39. Ishikawa K, Maejima K, Komatsu K, Netsu O, Keima T, Shiraishi T, Okano Y, Hashimoto M, Yamaji Y, Namba S. 2013. Fig mosaic emaravirus p4 protein is involved in cell-to-cell movement. J. Gen. Virol. 94:682–686. 10.1099/vir.0.047860-0 [DOI] [PubMed] [Google Scholar]
- 40. Yu C, Karlin DG, Lu Y, Wright K, Chen J, MacFarlane S. 2013. Experimental and bioinformatic evidence that raspberry leaf blotch emaravirus P4 is a movement protein of the 30K superfamily. J. Gen. Virol. 94:2117–2128. 10.1099/vir.0.053256-0 [DOI] [PubMed] [Google Scholar]
- 41. King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. (ed). 2011. Virus taxonomy: classification and nomenclature of viruses. Ninth report of the International Committee on Taxonomy of Viruses. Academic Press, London, United Kingdom [Google Scholar]
- 42. Tatineni S, Robertson CJ, Garnsey SM, Dawson WO. 2011. A plant virus evolved by acquiring multiple nonconserved genes to extend its host range. Proc. Natl. Acad. Sci. U. S. A. 108:17366–17371. 10.1073/pnas.1113227108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mielke-Ehret N, Thoma J, Schlatermund N, Mühlbach HP. 2010. Detection of European mountain ash ringspot-associated virus-specific RNA and protein P3 in the pear leaf blister mite Phytoptus pyri (Eriophyidae). Arch. Virol. 155:987–991. 10.1007/s00705-010-0667-3 [DOI] [PubMed] [Google Scholar]
- 44. Wijkamp I, Lent JV, Kormelink R, Goldbach R, Peters D. 1993. Multiplication of Tomato spotted wilt virus in its insect vector, Frankliniella occidentalis. J. Gen. Virol. 74:341–349. 10.1099/0022-1317-74-3-341 [DOI] [PubMed] [Google Scholar]
- 45. Walia JJ, Falk BW. 2012. Fig mosaic virus mRNAs show generation by cap-snatching. Virology 426:162–166. 10.1016/j.virol.2012.01.035 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.