

ABSTRACT
Resting CD4+ T lymphocytes resist human immunodeficiency virus (HIV) infection. Here, we provide evidence that exosomes from HIV-1-infected cells render resting human primary CD4+ T lymphocytes permissive to HIV-1 replication. These results were obtained with transwell cocultures of HIV-1-infected cells with quiescent CD4+ T lymphocytes in the presence of inhibitors of exosome release and were confirmed using exosomes purified from supernatants of HIV-1-infected primary CD4+ T lymphocytes. We found that the expression of HIV-1 Nef in exosome-producing cells is both necessary and sufficient for cell activation as well as HIV-1 replication in target CD4+ T lymphocytes. We also identified a Nef domain important for the effects we observed, i.e., the 62EEEE65 acidic cluster domain. In addition, we observed that ADAM17, i.e., a disintegrin and metalloprotease converting pro-tumor necrosis factor alpha (TNF-α) in its mature form, associates with exosomes from HIV-1-infected cells, and plays a key role in the HIV-1 replication in quiescent CD4+ T lymphocytes. Treatment with an inhibitor of ADAM17 abolished both activation and HIV-1 replication in resting CD4+ T lymphocytes. TNF-α is the downstream effector of ADAM17 since the treatment of resting lymphocytes with anti-TNF-α antibodies blocked the HIV-1 replication. The data presented here are consistent with a model where Nef induces intercellular communication through exosomes to activate bystander quiescent CD4+ T lymphocytes, thus stimulating viral spread.
IMPORTANCE Overall, our findings support the idea that HIV evolved to usurp the exosome-based intercellular communication network to favor its spread in infected hosts.
INTRODUCTION
Cells infected by human immunodeficiency virus type 1 (HIV-1) release nanovesicles in the forms of viral particles and nonviral particles termed exosomes. The latter are lipid bilayer vesicles of 50 to 100 nm which form intracellularly upon inward invagination of endosome membranes (1). These intraluminal vesicles become part of multivesicular bodies and either undergo lysosomal degradation or are released into extracellular space upon fusion of multivesicular bodies with plasma membrane. Nanovesicles similar to exosomes can be released also through direct extrusion of plasma membrane (2). Current protocols of purification and marker analysis cannot distinguish between endosome-produced nanovesicles and vesicles with similar size but extruding from cell membranes. For the sake of clarity, these nanovesicles are here defined as exosomes regardless of their biogenesis.
Exosomes are part of the intercellular communication network (3). They incorporate messenger RNAs, microRNAs, and proteins which can be functional in target cells (4). Exosomes from HIV-1-infected cells incorporate Gag (5) and Nef HIV-1 proteins (6, 7). The latter is incorporated in exosomes upon anchoring into lipid raft microdomains through its N-terminal myristoylation and a stretch of basic amino acids residing in its alpha-helix 1.
The treatment with exosomes from Nef-expressing cells increases the expression of the activation marker CD69 in quiescent CD4+ T lymphocytes (6) and the release of tumor necrosis factor alpha (TNF-α) from peripheral blood mononuclear cells (PBMCs) (8). TNF-α release requires the activity of ADAM17. This protease needs to be activated at the plasma membrane in juxtaposition to TNF-α but can also be transferred/provided by exosomes (8). ADAM17 belongs to the family of ADAM (a disintegrin and metalloprotease) enzymes (9). It is a multidomain, transmembrane, Zn2+-dependent proteinase whose inactive form is cleaved by furin in the trans-Golgi network. The most studied function of ADAM17 is the processing of pro-TNF-α to its active form. For this reason, ADAM17 is also known as TNF-α-converting enzyme (TACE). In Nef-expressing cells, activated ADAM17 is shuttled into exosomes upon activation by a complex formed by the integrin effector paxillin, the polycomb protein Eed, and Nef. PBMCs ingesting these exosomes release TNF-α (8). However, whether similar mechanisms apply to exosomes from HIV-1-infected cells, and whether quiescent CD4+ T lymphocytes targeted by these exosomes become competent for HIV-1 replication, is still unknown. Here, we provide evidence that HIV-1-infected primary CD4+ T lymphocytes release exosomes able to activate quiescent human primary CD4+ T lymphocytes which thereby replicate HIV-1. Nef, ADAM17, and TNF-α are part of the underlying mechanism.
MATERIALS AND METHODS
Cell culture, isolation, transfections, and HIV-1 infections.
CD4+ T lymphocytes were isolated from PBMCs of healthy donors by negative selection using an immunomagnetic-based kit (Miltenyi) and cultivated in RPMI medium plus 10% heat-inactivated fetal calf serum (FCS). The cell cultures were checked for their purity through fluorescence-activated cell sorting (FACS) analysis for CD4, CD8, and CD14 markers. Preparations having >2% of CD8+ cells and/or detectable CD14+ cells were discarded. For activation, 2 μg/ml of phytohemagglutinin (PHA) was added to CD4+ T lymphocyte cultures. Rev-CEM cells (10) were grown in RPMI medium plus 10% FCS. Human embryonic kidney 293T cells were grown in Dulbecco's modified Eagle's medium plus 10% FCS.
HIV-1 preparations were obtained from the supernatants of 293T cells 48 h after transfection with the pNL4-3 HIV-1 molecular clone. To produce HIV-1 pseudotyped with the G protein from vesicular stomatitis virus (VSV-G), a cytomegalovirus (CMV) immediate early-promoted VSV-G-expressing vector was cotransfected in a molar ratio of 1:5 with vectors expressing either wild-type (wt), Δnef (11), or Nef4EA HIV-1. The latter molecular clone was obtained by amplifying the pcDNA3/Nef4EA vector (12) with primers carrying the MluI (forward) and ClaI (reverse) restriction sites. The amplification product was then inserted in the respective restriction sites of a pNL4-3 clone where MluI and ClaI sites were created at the 5′ and 3′ ends of the nef gene (13). The nef sequence of the resulting HIV-1 molecular clones was finally checked for the presence of nucleotide substitutions.
Transfections were performed using Lipofectamine 2000 (Invitrogen). Supernatants were clarified and concentrated by ultracentrifugation as previously described (14). Virus preparations were titrated in terms of HIV-1 CAp24 content using quantitative enzyme-linked immunosorbent assay (ELISA; Innogenetic). Infections with HIV-1 were carried out by spinoculation at 400 × g for 30 min at room temperature (RT). For 106 cells, 500 CAp24 equivalents of HIV-1 or 50 ng of VSV-G HIV-1 was used. The infectivity of HIV-1 in supernatants of activated CD4+ T lymphocytes was evaluated by infecting the indicator Rev-CEM cells. A total of 105 cells were spinoculated in microwells with scaled dilutions of the supernatants, and 48 h later the HIV-1 infectious units were calculated in terms of the percentages of green fluorescent protein (GFP)-positive cells as evaluated by FACS analysis.
For the production of exosomes from 293T-transfected cells, IE-CMV-promoted expression vectors expressing either ADAM17 (8), wt Nef (15), or Nef4EA (12) were used.
Azidothymidine (AZT) was obtained from the NIH AIDS Research and Reference Reagent Program. TAPI-2 was purchased from Santa Cruz Biotechnology. For anti-TNF-α neutralization experiments, either anti-TNF-α neutralizing antibodies (polyclonal rabbit antibodies; Fitzgerald Industries) or normal rabbit IgGs were added to CD4+ T lymphocyte cultures immediately after exosome challenge. The antibodies were then readded after HIV-1 challenge and every 24 h of culture.
Transwell cocultures.
Transwell cocultures were carried out in 12-well plates using a Falcon cell culture insert membrane (25-mm diameter, 0.4-μm pore size; Becton Dickinson). CD4+ T lymphocytes were activated with 2 μg/ml of PHA for 48 h and then infected with VSV-G HIV-1. After an additional 48 h, transwell cocultures were set up by putting 106 infected CD4+ T lymphocytes in the upper chamber, while 2 × 106 quiescent, isogenic CD4+ T lymphocytes were seeded in the bottom chamber. The cocultures were then run for 3 days in complete medium without cell growth/activator factors and in the presence or not of either 10 μM AZT, 1 μM the inhibitors of exosome release GW4869 (Sigma) and spiroepoxide (Santa Cruz), or 1 μM TAPI-2. Finally, HIV-1 contents in supernatants were measured by CAp24 ELISA, and cells from both chambers were analyzed by FACS for the HIV-1 expression through the detection of intracytoplasmic HIV-1 CAp24.
Nanovesicle purification and challenge.
Cell culture supernatants were processed following already described methods for exosome purification. In detail, supernatants were centrifuged at 500 × g for 10 min and filtered with a 0.22-μM-pore-size filter. Then, the supernatants underwent differential centrifugations consisting of a first ultracentrifugation at 10,000 × g for 30 min. Supernatants were then harvested and ultracentrifuged at 70,000 × g for l h. The pelleted vesicles were resuspended in 1× phosphate-buffered saline (PBS) and ultracentrifuged again at 70,000 × g for 1 h. Afterward, the pellet was resuspended in 200 to 400 μl of 1× PBS and, in some cases, subjected to discontinuous iodixanol (Axis-Shield) gradient. It was performed essentially as described previously (16). Briefly, concentrated vesicles were ultracentrifuged at 200,000 × g for 1.5 h at 4°C in an SW41 Ti rotor (Beckman) through a 6 to 18% iodixanol density gradient formed by layering iodixanol in 1.2% increments. Then, 0.7-ml fractions were collected starting from the top. In some instances, half of each fraction was diluted with 2 volumes of 0.9% sodium chloride and ultracentrifuged for 30 min at 95,000 rpm in a TL-100 tabletop ultracentrifuge. Finally, the pellet was resuspended in 50 μl of 10 mM Tris-HCl (pH 7.4), 100 mM NaCl, 1 mM EDTA, and 0.1% Triton X-100. Challenges of cells with exosomes were performed by spinoculation as described for HIV-1 infection.
FACS analysis of cells and nanovesicles.
For the detection of intracytoplasmic HIV-1 CAp24, cells were treated with trypsin for 15 min at 37°C. Then, they were labeled using the KC57-RD anti-CAp24 monoclonal antibody (Coulter) upon permeabilization with Cytofix/Cytoperm solutions (BD Pharmingen) as previously described (17).
CD63 staining of nanovesicles was performed by incubating them with 0.5 μl of surfactant-free white aldehyde/sulfate latex beads (Invitrogen Molecular Probes) for 2 h at RT on a rotating plate. Afterwards, nanovesicle-bead complexes were washed and incubated at 4°C for 2 h with fluorescein isothiocyanate (FITC)-conjugated anti-CD63 monoclonal antibody (BD Pharmingen). Finally, the beads were washed, resuspended in 1× PBS-2% (vol/vol) formaldehyde, and FACS analyzed.
Acetylcholinesterase and ADAM17 activity assays.
The nanovesicle-associated acetylcholinesterase (AchE) activity was evaluated through the Amplex Red kit (Molecular Probes) by following the manufacturer's recommendations. The AchE activity was measured as mU/ml, where 1 mU is defined as the amount of enzyme which hydrolyzes 1 pmol of acetylcholine to choline and acetate per minute at pH 8.0 at 37°C. The exosome preparations were quantified throughout in terms of units of AchE activity.
The amounts of active ADAM17 associated with exosomes were assayed through the Innozyme TACE activity kit (Calbiochem). The assay was carried out on 1 mU equivalent of AchE activity of each exosome preparation by following the manufacturer's recommendations.
Western blot assay.
Western blot analysis on cell lysates was performed by washing cells twice with 1× PBS (pH 7.4) and lysing them for 20 min on ice with lysis buffer (20 mM HEPES [pH 7.9], 50 mM NaCl, 10 mM EDTA, 2 mM EGTA, 0.5% nonionic detergent IGEPAL CA-630, 0.5 mM dithiothreitol, 20 mM sodium molybdate, 10 mM sodium orthovanadate, 100 mM sodium fluoride, 10 μg/ml leupeptin, 0.5 mM phenylmethylsulfonyl fluoride). Whole-cell lysates were centrifuged at 6,000 × g for 10 min at 4°C. The protein concentration of cell extracts was determined by the Lowry protein quantitation assay. Aliquots of cell extracts containing 30 to 50 μg of total proteins were resolved by 8 to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred by electroblotting on a 0.45-μM-pore-size nitrocellulose membrane (Amersham) overnight using a Bio-Rad Trans-Blot. For Western blot analysis of exosomes, they were lysed and analyzed as described for cell lysates. For immunoassays, membranes were blocked with 5% nonfat dry milk in PBS containing 0.1% Triton X-100 for 1 h at room temperature and then incubated overnight at 4°C with specific antibodies diluted in PBS containing 0.1% Triton X-100. The antibodies used in immunoblots were sheep polyclonal anti-Nef ARP444 (a generous gift from Mark Harris), rabbit polyclonal anti-ADAM17 from Cell Signaling, anti-ICAM-1 monoclonal antibody 15.2 from Santa Cruz Biotech, and anti-β-actin monoclonal antibody AC-74 (Sigma). Immune complexes were detected with horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse antibodies (GE Healthcare) and enhanced chemiluminescence reaction (Euroclone).
TNF-α and interleukin 2 (IL-2) detection.
The measurement of TNF-α in the cell supernatants was performed by using an ELISA kit from Immunological Sciences, by following the manufacturer's recommendations.
The release of IL-2 was detected by enzyme-linked immunosorbent spot (ELISPOT) assay. In detail, cells were treated with exosomes and then cultivated for 48 to 72 h in ELISPOT microwells (Millipore) previously coated with a monoclonal antibody against human IL-2 (Mabtech). Afterwards, the cells were removed, and a biotinylated antibody against human IL-2 was added, followed by the addition of a streptavidin-alkaline phosphatase. The plate was finally developed using 5-bromo-4-chloro-3-indolylphosphate (BCIP)/nitroblue tetrazolium (NBT) substrate (Sigma). Spot-forming cells were analyzed and counted using an ELISPOT reader (Amplimedical Bioline A-EL-VIS GmbH).
Statistical analysis.
When appropriate, data are presented as means + standard deviations (SD). In some instances, the paired Student t test was used and confirmed using the nonparametric Wilcoxon rank sum test. P values of <0.05 were considered significant.
RESULTS
Release of exosomes from HIV-1-infected lymphocytes associates with HIV-1 replication in cocultured quiescent CD4+ T lymphocytes.
We asked whether exosomes released by HIV-1-infected cells would impact the susceptibility to HIV-1 infection of quiescent CD4+ T lymphocytes. To this aim, we set up cocultures of activated lymphocytes infected by wt HIV-1 with resting CD4+ T lymphocytes. The cultures of resting CD4+ T lymphocytes consisted of cells isolated from healthy donors and cultivated in complete medium in the absence of activation/growth factors. The contribution of exosomes was assessed using the specific inhibitors of exosome release, GW4869 and spiroepoxide, i.e., two structurally unrelated inhibitors of neutral sphingomyelinase (18–23). The effectiveness of these compounds was proven in advance in cultures of HIV-1-infected lymphocytes, whose supernatants were assayed for the exosome contents by measuring the AchE activity, i.e., a classical exosome marker (Fig. 1A). To set up the cocultures, CD4+ T lymphocytes were activated by PHA and, 2 days later, infected with VSV-G-pseudotyped wt HIV-1. After 2 additional days, the cells were put in the upper chamber of a 0.4 μM transwell plate, where quiescent CD4+ T lymphocytes from the same donor were seeded in the bottom chamber. The cocultures were run in the presence or not of either 10 μM AZT or 1 μM of both GW4869 and spiroepoxide. After 3 days, HIV-1 replication in quiescent cells was evaluated by FACS analysis for the presence of intracytoplasmic HIV-1 Gag-related products. At this time, the majority of donor CD4+ T lymphocytes expressed HIV-1 (Fig. 1B), and the treatment with the exosome inhibitors affected neither the number of HIV-1 expressing cells (Fig. 1B) nor the amounts (Fig. 1C) and infectivity (Fig. 1D) of HIV-1 in supernatants. We observed that quiescent CD4+ T lymphocytes replicated HIV-1 when cocultured with cells infected by wt HIV-1. The viral replication was significantly reduced in the presence of the exosome inhibitors. As expected, AZT blocked the HIV-1 replication (Fig. 1E).
FIG 1.

HIV-1 replicates in quiescent CD4+ T lymphocytes cocultured in transwell plates with activated CD4+ T lymphocytes infected by HIV-1. (A) Inhibition of exosome release from HIV-1-infected cells treated with GW4869 and spiroepoxide. A total of 3 ×106 CD4+ T lymphocytes were activated with 2 μg/ml of PHA and, 2 days later, infected with VSV-G wt HIV-1. After an additional 2 days, the cells were treated with GW4869 and spiroepoxide for 16 h. Finally, exosomes in the supernatants were isolated and quantified in terms of mU of AchE. Values are the means + SD of results for duplicates from three independent experiments. *, P < 0.05. (B) FACS analysis for the HIV-1 CAp24 expression in activated CD4+ T lymphocytes from cocultures with resting lymphocytes. CD4+ T lymphocytes were activated and infected as described for panel A. Two days after infection, the cells were put in the upper chambers of transwell plates, and quiescent CD4+ T lymphocytes were seeded in the bottom chambers. The cocultures were run in the presence or not of either AZT or inhibitors of exosome release. Three days later, supernatants and cells from both the upper and bottom chambers were analyzed. The FACS analysis has been carried out in activated CD4+ T lymphocytes uninfected or infected with VSV-G wt HIV-1 in the presence or not of either AZT or the inhibitors of exosome release, GW4869 and spiroepoxide. The results are representative of data from three independent experiments. (C) HIV-1 CAp24 levels in supernatants of cocultures. nd, not detectable. Values are the means + SD of results for triplicates from one experiment representative of three independent experiments. (D) Infectivity of HIV-1 released by activated CD4+ T lymphocytes in the presence of GW4869 and spiroepoxide as measured on Rev-CEM indicator cells. Values are the means + SD of results for triplicates from one experiment representative of two independent experiments. (E) HIV-1 replication in resting CD4+ T lymphocytes. The cells were washed, treated with trypsin, and then scored for the expression of HIV-1 by means of intracellular FACS analysis. Shown are the mean percentages of HIV-1-positive cells as calculated from duplicate conditions using resting CD4+ T lymphocytes from three healthy donors. The interdonor mean percentages + SD are also shown. *, P < 0.05.
These results suggested that the release of exosomes from HIV-1-infected CD4+ T lymphocytes renders quiescent CD4+ T lymphocytes susceptible to HIV-1 infection.
Characterization of exosomes from primary human CD4+ T lymphocytes infected by HIV-1.
To strengthen our data, next we looked at the effects of exosomes purified from the supernatants of HIV-1-infected CD4+ T lymphocytes. Exosomes from HIV-1-infected cultures were isolated through iodixanol gradients of nanovesicles concentrated from culture supernatants (24). In detail, CD4+ T lymphocytes were activated for 2 days with PHA and then infected with 200 ng of CAp24 equivalent/106 cells of a VSV-G-pseudotyped HIV-1 T-tropic strain. At the time of peak replication, as evaluated by the percentage of infected cells, the supernatants were harvested, and nanovesicles were isolated through differential centrifugations. Finally, nanovesicles were loaded on a 6 to 18% discontinuous gradient. The fractions recovered after ultracentrifugation were analyzed in terms of both AchE activity and HIV-1 CAp24 contents. As a control, the same procedure was applied on supernatants of uninfected, activated CD4+ T lymphocytes. As previously described (24), iodixanol gradients efficiently separated exosomes, floating in fractions 5 to 8, from viral particles, which accumulated in denser fractions (Fig. 2A).
FIG 2.

Characterization of exosomes from supernatants of activated CD4+ T lymphocytes either uninfected or HIV-1 infected. (A) AchE activity and, for HIV-1-infected cells only, HIV-1 Gag CAp24 contents were measured in fractions from 6 to 18% iodixanol gradients loaded with vesicles obtained by differential centrifugations of supernatants from activated CD4+ T lymphocytes either uninfected or infected with wt HIV-1. The results are the mean values from duplicate conditions and are representative of results from two independent experiments. (B) Detection by FACS of CD63 on pools of AchE-positive fractions from iodixanol gradients loaded with vesicles from uninfected or HIV-1-infected cells. Vesicles were bound to aldehyde/sulfate latex beads and then labeled with FITC-conjugated anti-CD63 monoclonal antibody or, as a control, FITC-conjugated isotype IgGs. On the left, a representative forward and side scatter dot plot of the exosome-bead complexes is shown. The results are representative of results from three independent experiments carried out on vesicles from two iodixanol gradient preparations.
Nanovesicles from iodixanol gradients were formally identified as exosomes by FACS analysis by the presence of CD63 (Fig. 2B).
The treatment with exosomes from HIV-1-infected cells renders quiescent CD4+ T lymphocytes susceptible to HIV-1 replication.
We were interested in assessing whether the treatment of resting CD4+ T lymphocytes with exosomes from HIV-1-infected cells associates with HIV-1 replication. To this aim, purified exosomes were used to challenge quiescent CD4+ T lymphocytes. As a control, cells were treated with exosomes derived from uninfected cells. In view of the previously published evidence that resting CD4+ T lymphocytes replicate HIV-1 more efficiently when the stimulus preceded the viral challenge (25), we treated the cells with exosomes 6 h before infection with a T-tropic NL4-3 HIV-1 strain. The amount of exosomes challenging 105 cells was equalized at 120 μU of AchE activity, i.e., the quantity of exosomes which we recovered from the supernatant of approximately 60,000 HIV-1-infected CD4+ T lymphocytes. The HIV-1 replication in cell cultures was monitored by anti-HIV-1 Gag intracytoplasmic FACS analysis starting from 48 h postinfection. In cell cultures not treated with exosomes, we measured 0.2 to 0.8% of HIV-1 CAp24-positive cells. The strongest increase in the number of HIV-1 Gag-expressing cells within the cultures treated with exosomes from HIV-1-infected cells was observed 3 days postchallenge. Conversely, no increase was detectable in cells either treated with exosomes from uninfected cells or left untreated (Fig. 3A). The extents of HIV-1 replication in CD4+ T lymphocytes correlated with the exosome input (Fig. 3B). Notably, the treatment of cultures with 10 μM AZT led to a strong reduction of HIV-1 CAp24-expressing CD4+ T lymphocytes (Fig. 3C). This result formally excluded that what we detected in FACS analysis derived from carryover of exosome- and/or virus-associated CAp24.
FIG 3.
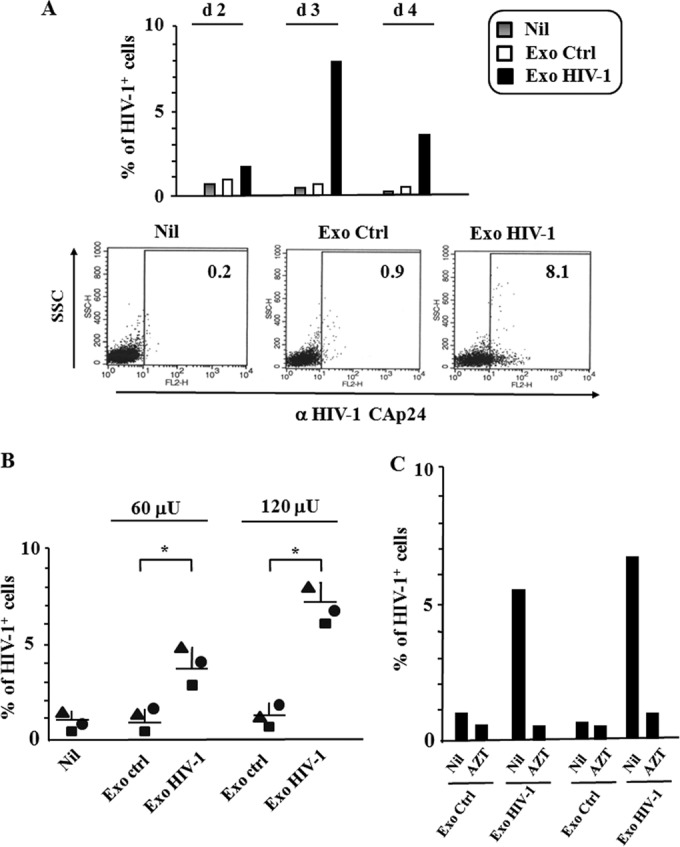
HIV-1 replication in quiescent CD4+ T lymphocytes treated with exosomes from HIV-1-infected CD4+ T lymphocytes. (A) Kinetic of HIV-1 infection in resting CD4+ T lymphocytes. A total of 105 quiescent CD4+ T lymphocytes were challenged with 120 μU of exosomes from either uninfected (Ctrl) or HIV-1-infected CD4+ T lymphocytes and then infected with 50 ng of a T-tropic HIV-1 strain. As a control, quiescent cells were challenged with HIV-1 alone (Nil). From 2 to 4 days later, the cells were analyzed for HIV-1 expression by FACS analysis. Shown are the mean percentages of infected cells from duplicate conditions representative of results from two independent experiments carried out with cells from two healthy donors. On the bottom, shown are the dot plots of the FACS analysis from a representative experiment at day 3 postchallenge. (B) Dose-response effect of exosomes from HIV-1-infected cells on HIV-1 susceptibility of resting CD4+ T lymphocytes. A total of 105 quiescent CD4+ T lymphocytes were challenged with 60 or 120 μU of exosomes from either uninfected (Ctrl) or HIV-1-infected cells and then infected with 50 ng of a T-tropic HIV-1 strain. As a control, quiescent cells were challenged with HIV-1 alone (Nil). Three days later, the cells were analyzed for HIV-1 expression. Shown are the mean percentages of HIV-1-positive cells as calculated from duplicate conditions using resting CD4+ T lymphocytes from three healthy donors. The interdonor mean percentages + SD are also shown. *, P < 0.05. (C) Effects of AZT on HIV-1 replication in resting CD4+ T cells. A total of 105 quiescent CD4+ T lymphocytes were challenged with 120 μU of exosomes from either uninfected (Ctrl) or HIV-1-infected cells and then infected with 50 ng of a T-tropic HIV-1 strain. Cell cultures were run in the absence (Nil) or in the presence of 10 μM AZT. Three days later, the cells were analyzed for HIV-1 expression. Shown are the mean percentages of HIV-1-positive cells as calculated from duplicate conditions using resting CD4+ T lymphocytes from two (I, II) healthy donors.
These data enforce the conclusions drawn through coculture experiments, hence supporting the idea that exosomes from HIV-1-infected cells render resting CD4+ T lymphocytes susceptible to HIV-1 infection.
Quiescent CD4+ T lymphocytes replicate HIV-1 when targeted by exosomes released by Nef-expressing cells.
In order to explore the mechanism underlying the exosome-dependent induction of HIV-1 susceptibility in quiescent CD4+ T lymphocytes, we next investigated the role of Nef. We focused on Nef since it was already demonstrated that resting CD4+ T lymphocytes can be activated upon ingestion of exosomes from Nef-expressing cells (6). 293T cells were transiently transfected with vectors expressing wt Nef or a number of Nef mutants. Exosomes were purified, and 120 μU of them was used to challenge 105 quiescent CD4+ T lymphocytes, which were then infected by a T-tropic HIV-1 strain. For a control, cells were treated with exosomes derived from cells transfected with empty vector. After 3 days, the lymphocytes were analyzed for HIV-1 expression. We observed a significant increase of HIV-1 replication in cells treated with exosomes from wt Nef-expressing cells compared to that in untreated cells and cells challenged with control exosomes (Fig. 4A). When the cells were treated with exosomes derived from cells expressing the Nef4EA mutant, where the acidic cluster domain at amino acids (aa) 62 to 65 was inactivated by the substitution with four alanines, the percentage of HIV-1-expressing cells was significantly reduced compared to that in cells challenged with wt Nef exosomes. We analyzed by Western blotting the presence of Nef4EA in both transfected cells and exosomes released by them (Fig. 4B). The results we obtained allowed us to exclude the possibility that the lack of effects on resting lymphocytes of exosomes from Nef4EA-expressing cells depended on the intrinsic instability of the Nef mutant or a defect in its incorporation in exosomes. Of note, and as previously reported (12), in SDS-PAGE analysis, Nef4EA migrates faster than the wt counterpart.
FIG 4.
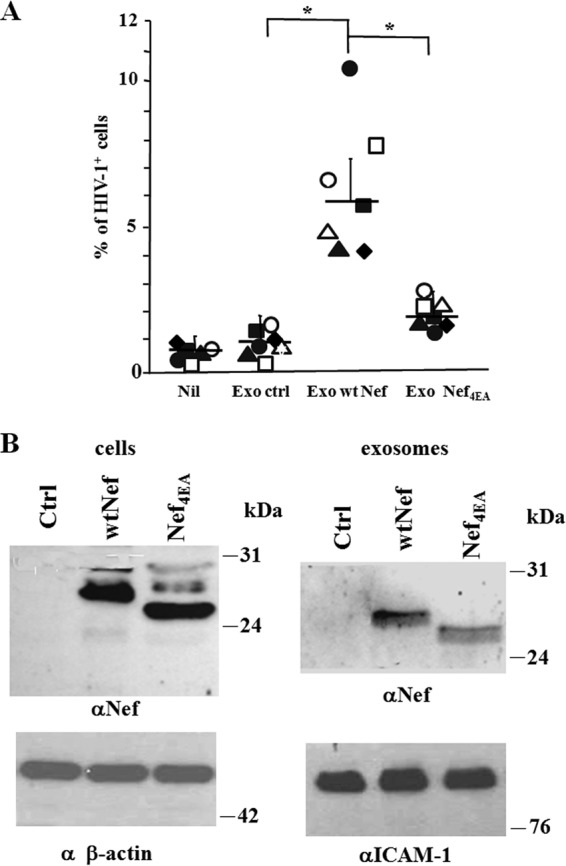
HIV-1 replicates in quiescent CD4+ T lymphocytes treated with exosomes from Nef-expressing cells. (A) HIV-1 replication in resting CD4+ T lymphocytes treated with exosomes from cells expressing either wt Nef or Nef4EA. A total of 105 quiescent CD4+ T lymphocytes were challenged with 120 μU of exosomes from 293T cells transfected with wt Nef- or Nef4EA-expressing vector, or the empty vector (Ctrl), and then infected with 50 ng of a T-tropic HIV-1 strain. As a control, quiescent cells were challenged with HIV-1 alone (Nil). Three days later, the cells were analyzed for HIV-1 expression. Shown are the mean percentages of HIV-1 positive cells as calculated from duplicate conditions using resting CD4+ T lymphocytes from seven healthy donors. The interdonor mean percentages + SD are also shown. *, P < 0.05. (B) Detection of Nef in cells transfected with either wt Nef or Nef4EA and in exosomes purified from the respective supernatants. Shown is the anti-Nef Western blot analysis of both cells and exosomes from 293T cells transiently transfected with vectors expressing wt Nef, Nef4EA, or the empty vector (Ctrl). Signals from cellular Nef were normalized with β-actin detection, while anti-ICAM-1 analysis served to normalize exosome signals. The molecular markers are given in kDa. Results are representative of results from two independent experiments.
These results indicate that the expression of Nef in exosome-producing cells is sufficient to render quiescent CD4+ T lymphocytes susceptible to HIV-1 infection. In addition, we newly identified a Nef domain important for the HIV-1 replication in resting lymphocytes.
Treatment with exosomes from cells infected by the Δnef HIV-1 strain or the Nef4EA HIV-1 strain does not affect the susceptibility of resting CD4+ T lymphocytes to HIV-1.
To verify the results we obtained with exosomes from transfected cells in a more physiological context, i.e., using exosomes purified from primary lymphocytes, activated CD4+ T lymphocytes were infected with the VSV-G Δnef HIV-1 strain or the VSV-G Nef4EA HIV-1 strain. By reproducing the above-described procedures, purified exosomes were recovered and analyzed in terms of both AchE activity and HIV-1 Gag CAp24 contents (Fig. 5A), as well as for CD63 expression (Fig. 5B). A total of 120 μU of these purified exosomes was used to challenge 105 quiescent CD4+ T lymphocytes. As a control, cells were challenged with the same amount of exosomes from uninfected cells or cells infected with wt HIV-1. After 6 h, the lymphocytes were infected by HIV-1 and 3 days later analyzed for HIV-1 expression. We observed that the challenge with exosomes from cells infected with either Δnef HIV-1 or Nef4EA HIV-1 appeared ineffective. Conversely, and as expected, the percentage of infected lymphocytes increased compared to results under control conditions when exosomes from wt HIV-1-infected cells were used (Fig. 5C). These data were consistent with those we obtained using exosomes from Nef-transfected cells, meanwhile strengthening the importance of the expression of Nef in HIV-1-infected cells for the exosome-mediated induction of HIV-1 susceptibility in resting CD4+ T lymphocytes.
FIG 5.

HIV-1 does not replicate in resting CD4+ T lymphocytes challenged with exosomes from cells infected by either the Δnef HIV-1 strain or the Nef4EA HIV-1 strain. (A) Characterization of exosomes from supernatants of activated CD4+ T lymphocytes infected by either the Δnef HIV-1 strain or the Nef4EA HIV-1 strain. AchE activity and HIV-1 Gag CAp24 contents were measured in fractions from 6 to 18% iodixanol gradients loaded with vesicles obtained by differential centrifugations of supernatants from activated CD4+ T lymphocytes infected by the Δnef HIV-1 strain or the Nef4EA HIV-1 strain. The results are the mean values for duplicate conditions and are representative of results from two independent experiments. (B) Detection by FACS of CD63 on pools of AchE-positive fractions from the iodixanol gradients described for panel A. (C) HIV-1 replication in resting CD4+ T lymphocytes treated with exosomes from cells infected by the wt, Δnef, or Nef4EA HIV-1 strain. A total of 105 quiescent CD4+ T lymphocytes were challenged with 120 μU of exosomes from the indicated infected cell populations, as well as from uninfected cells (Ctrl). As a control, cells were challenged with HIV-1 alone (Nil). Three days later, the cells were analyzed for HIV-1 expression. Shown are the mean percentages of HIV-1-positive cells as calculated from duplicate conditions using resting CD4+ T lymphocytes from two (I, II) healthy donors.
Primary quiescent CD4+ T lymphocytes release both TNF-α and IL-2 in response to the treatment with exosomes from HIV-1-infected cells.
CD4+ T lymphocytes replicate HIV upon cell activation. Thus, it was expected that the HIV-1 replication we observed in resting CD4+ T lymphocytes treated with exosomes was a consequence of the induction of cell activation. To support this hypothesis, we investigated the release of TNF-α and IL-2 from exosome-treated quiescent CD4+ T lymphocytes. For TNF-α, 105 cells were challenged with 120 μU of exosomes from uninfected or HIV-1-infected lymphocytes. The release of TNF-α was monitored over time within a 48-h interval. We noticed that the TNF-α release peaked at 6 h, afterwards remaining sustained through the observation time (Fig. 6A). The exosome-dependent TNF-α release appeared dose dependent (Fig. 6B), and resting lymphocytes challenged with exosomes from cells infected by either the Δnef HIV-1 strain or the Nef4EA HIV-1 strain did not increase the TNF-α release (Fig. 6C). No TNF-α was detected in the exosome preparations we used for the challenges (not shown).
FIG 6.

Resting CD4+ T lymphocytes release TNF-α and IL-2 when treated with exosomes from HIV-1-infected cells. (A) Kinetic of TNF-α release. A total of 105 quiescent CD4+ T lymphocytes were challenged with 120 μU of exosomes from HIV-1-infected or uninfected (Ctrl) cells. After extensive washes, the cells were seeded in complete medium, and supernatants were harvested at the indicated time points. Shown are the mean concentrations of TNF-α as calculated from duplicate conditions using resting CD4+ T lymphocytes from a representative of two healthy donors. (B) Detection of TNF-α on supernatants of 105 quiescent CD4+ T lymphocytes challenged with either increasing amounts (i.e., from 30 to 120 μU) of exosomes from HIV-1-infected cells or 120 μU of exosomes from uninfected cells (Ctrl). As a control, cells were treated with 2 μg/ml of PHA or left untreated (Nil). After extensive washes, the cells were seeded in complete medium, and supernatants were harvested 6 h later. Shown are the mean concentrations of TNF-α as calculated from duplicate conditions using resting CD4+ T lymphocytes from two (I, II) healthy donors. (C) TNF-α detection in supernatants from resting CD4+ T lymphocytes challenged with exosomes from cells infected by the Δnef HIV-1 strain or the Nef4EA HIV-1 strain. A total of 105 quiescent CD4+ T lymphocytes were challenged with 120 μU of exosomes from cells infected with the wt, Δnef HIV-1, or Nef4EA HIV-1 strain. As a control, cells were challenged with equal amounts of exosomes from uninfected cells (Ctrl), treated with 2 μg/ml of PHA, or left untreated (Nil). Supernatants were harvested 6 h later, and TNF-α contents were determined by ELISA. Shown are the mean concentrations of TNF-α as calculated from duplicate conditions using resting CD4+ T lymphocytes from two (I, II) healthy donors. (D) Detection of IL-2-producing cells. A total of 105 resting CD4+ T lymphocytes were challenged with 120 μU of exosomes from cells infected with the wt, Δnef, or Nef4EA HIV-1 strain. As a control, cells were challenged with equal amounts of exosomes from uninfected cells (Ctrl), treated with 2 μg/ml of PHA, or left untreated (Nil). Afterwards, the cells were washed and seeded in ELISPOT microwells previously coated with an anti-IL-2 monoclonal antibody. The spots were counted after both 48 and 72 h. Shown are the mean number of spots + SD as calculated from duplicate conditions evaluated in triplicate wells using resting CD4+ T lymphocytes from two (I, II) healthy donors. nd, no spots found.
We evaluated the exosome-dependent activation in resting CD4+ T lymphocytes also in terms of IL-2 release by ELISPOT assay from 24 to 72 h after exosome challenge. A total of 105 cells were challenged with 120 μU of exosomes from uninfected cells as well as from cells infected by the wt, Δnef, or Nef4EA HIV-1 strain. Whereas no IL-2 production was detectable at the 24-h time point (not shown), starting from 48 h postchallenge we observed a well-detectable number of cells producing IL-2 under the conditions treated with exosomes from cells infected with wt HIV-1 but not from those infected with HIV-1 mutants (Fig. 6D).
Together, these data imply that the treatment of resting CD4+ T lymphocytes with exosomes from HIV-1-infected cells leads to cell activation which relies on the expression of a functional Nef in exosome-producing cells.
ADAM17 is involved in both activation and HIV-1 replication in quiescent CD4+ T lymphocytes treated with exosomes from HIV-1-infected cells.
The expression of HIV-1 Nef leads to incorporation of activated ADAM17 into exosomes (8). Upon exosome ingestion, PBMCs release TNF-α as a consequence of the cleavage of pro-TNF-α driven by exosome-associated ADAM17 (8). We investigated whether this mechanism was on the basis of CD4+ T lymphocyte activation and HIV-1 replication we describe here. To this aim, we first assayed the presence of activated ADAM17 in exosomes from HIV-1-infected cells. The ADAM17-specific Western blot analysis revealed an increase of active ADAM17 associating with a decrease of the inactive form in cells infected by wt HIV-1 compared to cells either uninfected or infected with the Δnef HIV-1 strain or the Nef4EA HIV-1 strain (Fig. 7A). Consistently, we found a strong ADAM17 activity associated with exosomes from lymphocytes expressing wt HIV-1, which was significantly reduced in exosomes from cells either uninfected or infected by the Δnef HIV-1 strain or the Nef4EA HIV-1 strain (Fig. 7B).
FIG 7.

Active ADAM17 associates with exosomes from wt HIV-1-infected cells and is important for both TNF-α release and induction of HIV-1 susceptibility in resting CD4+ T lymphocytes. (A) Western blot analysis for the expression of ADAM17 in cells infected with the wt, Δnef, or Nef4EA HIV-1 strain or mock infected (Ctrl). On the left of blots for ADAM17, arrows identify both inactive and active ADAM17 forms. Signals from cellular ADAM17 were normalized with both β-actin and Nef signals. On the right of each panel, molecular mass markers are given in kDa. The results are representative of results from two independent experiments. (B) ADAM17 activity detected in 1 mU of exosomes purified from the supernatants of lymphocytes either uninfected (Ctrl) or infected with the wt, Δnef, or Nef4EA HIV-1 strain. Mean values + SD of ng of active ADAM17 detected in duplicate samples from three exosome preparations are shown. *, P < 0.05. (C) Effect of TAPI-2 on TNF-α release. A total of 105 quiescent CD4+ T lymphocytes were challenged with 120 μU of exosomes from HIV-1-infected cells in the presence of increasing concentrations of TAPI-2. As a control, quiescent cells were challenged with HIV-1 alone (Nil) or in the presence of 2 μg/ml of PHA. The cells were left in culture for 6 h, and then TNF-α contents in supernatants were determined. Shown are the mean concentrations of TNF-α as calculated from duplicate conditions using resting CD4+ T lymphocytes from two (I, II) healthy donors. (D) Effects of TAPI-2 on HIV-1 replication. A total of 105 quiescent CD4+ T lymphocytes were challenged with 120 μU of exosomes from HIV-1-infected cells in the presence of increasing concentrations of TAPI-2. Alternatively, the cells were challenged with the same amounts of exosomes from wt Nef-expressing 293T cells in the presence of the highest TAPI-2 concentration. As a control, quiescent cells were challenged with HIV-1 alone (Nil). Cells were then infected with HIV-1 and 3 days later were analyzed for HIV-1 expression. Shown are the mean percentages of HIV-1-positive cells as calculated from duplicate conditions using resting CD4+ T lymphocytes from a representative of two healthy donors analyzed. (E) Transwell cocultures of resting CD4+ T lymphocytes with HIV-1-infected cells in the presence of AZT or TAPI-2. CD4+ T lymphocytes were activated with 2 μg/ml of PHA and 2 days later infected with VSV-G wt HIV-1. After an additional 2 days, the cells were put in the upper chamber of transwell plates, and quiescent CD4+ T lymphocytes were seeded in the bottom chamber. The cocultures were run in the presence or not of either AZT or 1 μM TAPI-2. Three days later, resting cells were harvested and analyzed for the percentage of HIV-1-infected cells by FACS. Shown are the mean percentages of HIV-1-positive cells as calculated from duplicate conditions using cells from a representative of two healthy donors analyzed. In the inset, shown is the infectivity of HIV-1 released by activated CD4+ T lymphocytes in the presence or not of TAPI-2 as measured in Rev-CEM cells. Shown are the mean values + SD of the infectious units as calculated from triplicate conditions using the supernatants from a representative of two independent experiments. (F) Effects of exosomes from ADAM17-transfected cells. A total of 120 μU of exosomes from 293T cells cotransfected with vectors expressing wt Nef and ADAM17 was used to challenge quiescent CD4+ T lymphocytes, which were then infected by HIV-1. As a control, cells were left untreated (Nil), activated with 2 μg/ml of PHA, or treated with exosomes from cells transfected with either an empty vector (Ctrl) or a wt Nef-expressing vector. Three days later, resting cells were analyzed for the percentage of HIV-1-infected cells by FACS. Shown are the mean percentages of HIV-1-positive cells as calculated from duplicate conditions using cells from a representative of two healthy donors analyzed.
To test the role of ADAM17 shuttled by exosomes, we treated CD4+ T lymphocytes with TAPI-2, i.e., a potent ADAM17 inhibitor (26), at the time of exosome challenge. We observed that TAPI-2 inhibited the release of TNF-α from CD4+ T lymphocytes challenged with exosomes from HIV-1-infected cells in a dose-dependent manner (Fig. 7C). Consistently, TAPI-2 also inhibited the viral replication in quiescent CD4+ T lymphocytes challenged with exosomes either from HIV-1-infected cells or from wt Nef-expressing 293T cells and then infected with HIV-1 (Fig. 7D). In a similar fashion, TAPI-2 abrogated the HIV-1 replication in resting CD4+ T lymphocytes cocultivated in transwell plates with HIV-1-infected lymphocytes (Fig. 7E), in the apparent absence of relevant effects on HIV-1 infectivity (Fig. 7E, inset).
Next, we sought to further enforce the concept that the association of ADAM17 to exosomes is critical for the induction of HIV-1 susceptibility in resting lymphocytes regardless of the cell source of exosomes and the mechanism of ADAM17 uploading. To this end, we tested exosomes associating active ADAM17 as a consequence of its overexpression in exosome producer cells as previously described (8). In detail, 293T cells were cotransfected with wt Nef- and ADAM17-expressing vectors, and exosomes were recovered from supernatants harvested 2 days later. The coexpression of Nef ensured the optimal uploading of active ADAM17 in exosomes (8). Afterwards, 105 resting CD4+ T lymphocytes were challenged with 120 μU of the exosomes and then with HIV-1. As a control, equal amounts of exosomes from cells transfected with empty or wt Nef-expressing vectors were used. Three days later, we observed that the levels of HIV-1 replication in resting lymphocytes treated with ADAM17 exosomes were larger than those in cells treated with exosomes from Nef-expressing cells (Fig. 7F).
Taken together, these data highlight the key role of exosome-associated ADAM17 in the activation and HIV-1 replication observed in CD4+ T lymphocytes treated with exosomes from HIV-1-infected cells.
The neutralization of TNF-α inhibits the HIV-1 replication in primary quiescent CD4+ T lymphocytes treated with exosomes from HIV-1-infected cells.
TAPI-2 can inhibit different ADAMs and metalloproteinases. To verify that the TAPI-2-dependent inhibition we observed was a result of the specific block of ADAM17 activity, we next investigated the role of the major ADAM17 substrate, i.e., TNF-α, in our experimental setting. Quiescent CD4+ T lymphocytes were challenged with exosomes and HIV-1 and then cultured for 3 days in the presence of either increasing amounts of anti-TNF-α neutralizing antibodies or, as a control, isotype-matched IgGs. We observed that anti-TNF-α antibodies abolished HIV-1 replication in CD4+ T lymphocytes treated with exosomes from HIV-1-infected cells in a dose-dependent way (Fig. 8). Consistent results were obtained when cells were challenged with exosomes from wt Nef-expressing 293T cells (Fig. 8).
FIG 8.
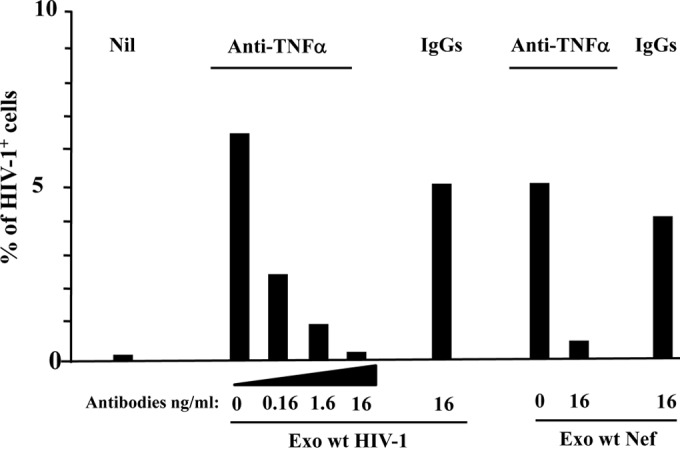
TNF-α neutralization blocks the HIV-1 replication in quiescent CD4+ T lymphocytes treated with exosomes from HIV-1-infected cells. A total of 105 quiescent CD4+ T lymphocytes were challenged with 120 μU of exosomes from HIV-1-infected cells. Then, the cells were incubated for 6 h in the presence of the indicated amounts of anti-TNF-α neutralizing antibodies or unrelated, isotype-specific IgGs. Alternatively, the cells were treated with equal amounts of exosomes from 293T cells and then incubated with the highest dose of antibodies. Afterwards, the cells were infected with HIV-1 and then washed and reseeded in the presence of the antibodies. As a control, resting CD4+ T cells were treated with HIV-1 alone (Nil). Three days after challenges, the cells were analyzed for HIV-1 expression by FACS. Shown are the mean percentages of HIV-1 positive cells as calculated from duplicate conditions using cells from a representative of two healthy donors analyzed.
These results are consistent with the idea that TNF-α is the main activation stimulus in exosome-treated resting CD4+ T lymphocytes and a downstream effector of exosome-associated ADAM17.
DISCUSSION
HIV-1-infected cells release exosomes associating with viral proteins (5–7) and RNA (27, 28). Recently, it has been reported that exosomes from cells expressing Nef upload activated ADAM17 and induce release of TNF-α from unstimulated PBMCs (8). We investigated whether a similar mechanism occurs in resting CD4+ T lymphocytes challenged with exosomes from primary CD4+ T lymphocytes and the possible consequences in terms of HIV-1 replication.
We found that resting CD4+ T lymphocytes challenged by exosomes from HIV-1-infected cells release both TNF-α and IL-2. The release of TNF-α peaked at 6 h after exosome challenge, thereby remaining sustained within the 48 h of observation, whereas we noticed the highest number of IL-2-producing cells at the 48-h time point. These data suggested that the IL-2 release was a consequence of cellular activation induced by the autocrine/paracrine stimulus of TNF-α.
Treatment with TNF-α has been found to activate 5 to 7% of resting lymphocytes (29). In addition, the activation of downstream effectors of TNF-α supports HIV-1 transcription by targeting long terminal repeats (LTRs) and activating TAK, i.e., a kinase required for Tat-dependent trans-activation (29). On the other hand, activation of resting lymphocytes is required for HIV replication, since both retrotranscription and integration are heavily compromised in quiescent CD4+ T lymphocytes (30, 31). On the basis of the literature data, we hypothesize that the HIV-1 replication we observed in quiescent lymphocytes may be a consequence of the TNF-α-dependent transcription of already integrated HIV-1 genomes and/or the result of the increase in retrotranscription/integration efficiency, as previously reported in resting lymphocytes stimulated with anti-CD3/anti-CD28 antibodies (25).
Although the HIV-1 preparations we used to challenge CD4+ T lymphocytes were obtained by an ultracentrifugation-based procedure excluding the coprecipitation of exosomes (32), the presence of residual contaminating exosomes in HIV-1 preparations cannot be formally ruled out. However, the lack of HIV-1 infection we observed when HIV-1 was given to resting CD4+ T lymphocytes alone indicated that the presence of residual exosomes contaminating our HIV-1 preparations was negligible.
HIV-1 replication was observed in quiescent CD4+ T lymphocytes also when they were challenged with exosomes from cells expressing wt Nef alone. Hence, the expression of wt Nef in exosome-producer cells is sufficient to render exosome-treated CD4+ T lymphocytes permissive to HIV-1 replication. In addition, our data originally identified the Nef acidic cluster as a domain important for HIV replication in quiescent CD4+ T lymphocytes. This region was originally characterized for the binding with phosphofurin acidic cluster sorting protein 1 (PACS-1) (33), i.e., a protein controlling the trafficking of furin between trans-Golgi and endosomes (34). Furin is the protease involved in the processing of pro-ADAM17 to its active form (9). It is tempting to speculate that Nef–PACS-1 interaction contributes to the increased ADAM17 activation we observed in HIV-1-infected cells and exosomes released by them. The Nef4E4A mutant could be a great tool to deeply investigate the interactions among Nef, PACS-1, ADAM17, and exosomes.
The results from transwell cocultures well recapitulated those we obtained with purified exosomes; however, in a more physiological context. The transwell coculture setting likely mirrors events occurring in tissues of infected patients where HIV actively replicates, e.g., gut mucosa and lymph nodes. Meanwhile, it was useful to avoid misinterpretations due to possible effects induced by cell-to-cell contact. The data we obtained with transwell cocultures confirmed that the expression of a functional Nef in HIV-1-infected donor cells is required for virus replication in bystander, resting CD4+ T lymphocytes.
We found activated ADAM17 in HIV-1-infected cells and exosomes released by them but not in cells either uninfected of expressing an HIV-1 lacking a functional Nef. The association of activated ADAM17 with exosomes from Nef-expressing cells was recently described (8). Here, we extend this observation to exosomes from HIV-1-infected cells, hence adding relevance to the previous finding. We observed an impressive increase of HIV-1 replication in resting CD4+ T lymphocytes treated with exosomes from 293T cells overexpressing ADAM17. These data support the idea that the association of ADAM17 to exosomes is the key event for the induction of viral replication in quiescent cells, which was observed regardless of the source of exosomes, i.e., primary cells or cell lines.
Our results allow us to propose a model where HIV-1-infected cells release ADAM17-loaded exosomes which, when ingested by bystander quiescent CD4+ T lymphocytes, can induce the cleavage of prestored pro-TNF-α. The mature cytokine may act in both autocrine and paracrine ways by activating intracellular signals supporting the progression of the life cycle of incoming HIV-1. The very early production of Nef, as documented in HIV-1-infected resting CD4+ T lymphocytes (35), could induce additional release of ADAM17-containing exosomes fostering viral spread to quiescent bystander CD4+ T lymphocytes. Therefore, exosomes, ADAM17, and TNF-α could be of paramount importance for HIV replication in the infected host.
On the basis of the results presented here, the role of Nef in AIDS pathogenesis could be, at least in part, reconsidered. Our data imply a model where Nef expression is necessary to arm exosomes with active ADAM17 in order to render resting bystander lymphocytes permissive for HIV replication. This mechanism would be particularly relevant in primary infection and would explain the rapid spread of viral infection in, for example, the gut. In HIV reservoirs, the expression of Nef would lead to the release of exosomes providing a microenvironment highly favorable for virus replication, in particular for viral mutants escaping the antiretroviral treatments. In these as-yet-theoretical scenarios, Nef, ADAM17, and TNF-α could represent targets of new therapies aimed at containing virus spread, hampering primary infection, and limiting the emergence of drug-resistant HIV-1 quasispecies.
ACKNOWLEDGMENTS
This work was supported by grants from AIDS National and “Ricerca Finalizzata” projects, both from the Ministry of Health, Italy.
Rev-CEM cells and AZT were obtained from the NIH AIDS Research and Reference Reagent Program. We are indebted to Pietro Arciero for excellent technical support.
Footnotes
Published ahead of print 23 July 2014
REFERENCES
- 1.Gyorgy B, Szabo TG, Pasztoi M, Pal Z, Misjak P, Aradi B, Laszlo V, Pallinger E, Pap E, Kittel A, Nagy G, Falus A, Buzas EI. 2011. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell. Mol. Life Sci. 68:2667–2688. 10.1007/s00018-011-0689-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Booth AM, Fang Y, Fallon JK, Yang JM, Hildreth JEK, Gould SJ. 2006. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J. Cell Biol. 172:923–935. 10.1083/jcb.200508014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathivanan S, Ji H, Simpson RJ. 2010. Exosomes: extracellular organelles important in intercellular communication. J. Proteomics 73:1907–1920. 10.1016/j.jprot.2010.06.006 [DOI] [PubMed] [Google Scholar]
- 4.Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT, Carter BS, Krichevsky AM, Breakefield XO. 2008. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 10:1470–1476. 10.1038/ncb1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fang Y, Wu N, Gan X, Yan WH, Morrell JC, Gould SJ. 2007. Higher-order oligomerization targets plasma membrane proteins and HIV Gag to exosomes. PLoS Biol. 5:1267–1283. 10.1371/journal.pbio.0050158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lenassi M, Cagney G, Liao MF, Vaupotic T, Bartholomeeusen K, Cheng YF, Krogan NJ, Plemenitas A, Peterlin BM. 2010. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4(+) T cells. Traffic 11:110–122. 10.1111/j.1600-0854.2009.01006.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muratori C, Cavallin LE, Kratzel K, Tinari A, De Milito A, Fais S, D'Aloja P, Federico M, Vullo V, Fomina A, Mesri EA, Superti F, Baur AS. 2009. Massive secretion by T cells is caused by HIV Nef in infected cells and by Nef transfer to bystander cells. Cell Host Microbe 6:218–230. 10.1016/j.chom.2009.06.009 [DOI] [PubMed] [Google Scholar]
- 8.Lee JH, Wittki S, Brau T, Dreyer FS, Kratzel K, Dindorf J, Johnston ICD, Gross S, Kremmer E, Zeidler R, Schlotzer-Schrehardt U, Lichtenheld M, Saksela K, Harrer T, Schuler G, Federico M, Baur AS. 2013. HIV Nef, paxillin, and Pak1/2 regulate activation and secretion of TACE/ADAM10 proteases. Mol. Cell 49:668–679. 10.1016/j.molcel.2012.12.004 [DOI] [PubMed] [Google Scholar]
- 9.Gooz M. 2010. ADAM-17: the enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 45:146–169. 10.3109/10409231003628015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu Y, Beddall MH, Marsh JW. 2007. Rev-dependent indicator T cell line. Curr. HIV Res. 5:394–402. 10.2174/157016207781024018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chowers MY, Spina CA, Kwoh TJ, Fitch NJS, Richman DD, Guatelli JC. 1994. Optimal infectivity in-vitro of human immunodeficiency virus type 1 requires an intact nef gene. J. Virol. 68:2906–2914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mangino G, Percario ZA, Fiorucci G, Vaccari G, Acconcia F, Chiarabelli C, Leone S, Noto A, Horenkamp FA, Manrique S, Romeo G, Polticelli F, Geyer M, Affabris E. 2011. HIV-1 Nef induces proinflammatory state in macrophages through its acidic cluster domain: involvement of TNF alpha receptor associated factor 2. PLoS One 6(8):e22982. 10.1371/journal.pone.0022982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olivetta E, Federico M. 2006. HIV-1 Nef protects human-monocyte-derived macrophages from HIV-1-induced apoptosis. Exp. Cell Res. 312:890–900. 10.1016/j.yexcr.2005.12.003 [DOI] [PubMed] [Google Scholar]
- 14.Federico M, Percario Z, Olivetta E, Fiorucci G, Muratori C, Micheli A, Romeo G, Affabris E. 2001. HIV-1 Nef activates STAT1 in human monocytes/macrophages through the release of soluble factors. Blood 98:2752–2761. 10.1182/blood.V98.9.2752 [DOI] [PubMed] [Google Scholar]
- 15.D'Aloja P, Olivetta E, Bona R, Nappi F, Pedacchia D, Pugliese K, Ferrari G, Verani P, Federico M. 1998. gag, vif and nef genes contribute to the homologous viral interference induced by a nonproducer human immunodeficiency virus type 1 (HIV-1) variant: identification of novel HIV-1-inhibiting viral protein mutants. J. Virol. 72:4308–4319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dettenhofer M, Yu XF. 1999. Highly purified human immunodeficiency virus type 1 reveals a virtual absence of vif in virions. J. Virol. 73:1460–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Muratori C, Sistigu A, Ruggiero E, Falchi M, Bacigalupo I, Palladino C, Toschi E, Federico M. 2007. Macrophages transmit human immunodeficiency virus type 1 products to CD4-negative cells: involvement of matrix metalloproteinase 9. J. Virol. 81:9078–9087. 10.1128/JVI.00675-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chairoungdua A, Smith DL, Pochard P, Hull M, Caplan MJ. 2010. Exosome release of beta-catenin: a novel mechanism that antagonizes Wnt signaling. J. Cell Biol. 190:1079–1091. 10.1083/jcb.201002049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kogure T, Lin W-L, Yan IK, Braconi C, Patel T. 2011. Intercellular nanovesicle-mediated microRNA transfer: a mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology 54:1237–1248. 10.1002/hep.24504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kosaka N, Iguchi H, Yoshioka Y, Takeshita F, Matsuki Y, Ochiya T. 2010. Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 285:17442–17452. 10.1074/jbc.M110.107821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kosaka N, Iguchi H, Yoshioka Y, Hagiwara K, Takeshita F, Ochiya T. 2012. Competitive interactions of cancer cells and normal cells via secretory microRNAs. J. Biol. Chem. 287:1397–1405. 10.1074/jbc.M111.288662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Bruegger B, Simons M. 2008. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319:1244–1247. 10.1126/science.1153124 [DOI] [PubMed] [Google Scholar]
- 23.Yuyama K, Sun H, Mitsutake S, Igarashi Y. 2012. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. J. Biol. Chem. 287:10977–10989. 10.1074/jbc.M111.324616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cantin R, Diou J, Belanger D, Tremblay AM, Gilbert C. 2008. Discrimination between exosomes and HIV-1: purification of both vesicles from cell-free supernatants. J. Immunol. Methods 338:21–30. 10.1016/j.jim.2008.07.007 [DOI] [PubMed] [Google Scholar]
- 25.Vatakis DN, Bristol G, Wilkinson TA, Chow SA, Zack JA. 2007. Immediate activation fails to rescue efficient human immunodeficiency virus replication in quiescent CD4(+) T cells. J. Virol. 81:3574–3582. 10.1128/JVI.02569-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moss ML, Rasmussen FH. 2007. Fluorescent substrates for the proteinases ADAM17, ADAM10, ADAM8, and ADAM12 useful for high-throughput inhibitor screening. Anal. Biochem. 366:144–148. 10.1016/j.ab.2007.04.043 [DOI] [PubMed] [Google Scholar]
- 27.Columba Cabezas S, Federico M. 2013. Sequences within RNA coding for HIV-1 Gag p17 are efficiently targeted to exosomes. Cell. Microbiol. 15:412–429. 10.1111/cmi.12046 [DOI] [PubMed] [Google Scholar]
- 28.Narayanan A, Iordanskiy S, Das R, Van Duyne R, Santos S, Jaworski E, Guendel I, Sampey G, Dalby E, Iglesias-Ussel M, Popratiloff A, Hakami R, Kehn-Hall K, Young M, Subra C, Gilbert C, Bailey C, Romerio F, Kashanchi F. 2013. Exosomes derived from HIV-1-infected cells contain trans-activation response element RNA. J. Biol. Chem. 288:20014–20033. 10.1074/jbc.M112.438895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ghose R, Liou LY, Herrmann CH, Rice AP. 2001. Induction of TAK (cyclin T1/P-TEFb) in purified resting CD4(+) T lymphocytes by combination of cytokines. J. Virol. 75:11336–11343. 10.1128/JVI.75.23.11336-11343.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, Chen ISY. 1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61:213–222. 10.1016/0092-8674(90)90802-L [DOI] [PubMed] [Google Scholar]
- 31.Zack JA, Haislip AM, Krogstad P, Chen ISY. 1992. Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. J. Virol. 66:1717–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park I-W, He JJ. 2010. HIV-1 is budded from CD4+ T lymphocytes independently of exosomes. Virol. J. 7:234. 10.1186/1743-422X-7-234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piguet V, Wan L, Borel C, Mangasarian A, Demaurex N, Thomas G, Trono D. 2000. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat. Cell Biol. 2:163–167. 10.1038/35004038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wan L, Molloy SS, Thomas L, Liu G, Xiang Y, Rybak SL, Thomas G. 1998. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell 94:205–216. 10.1016/S0092-8674(00)81420-8 [DOI] [PubMed] [Google Scholar]
- 35.Wu YT, Marsh JW. 2001. Selective transcription and modulation of resting T cell activity by preintegration HIV DNA. Science 293:1503–1506. 10.1126/science.1061548 [DOI] [PubMed] [Google Scholar]