

ABSTRACT
Human immunodeficiency virus type 1 (HIV-1) infection is chronic and presently still incurable. Antiretroviral drugs effectively suppress replication; however, persistent activation of inflammatory pathways remains a key cause of morbidity. Recent studies proposed that purinergic signaling is required for HIV-1 infection. Purinergic receptors are distributed throughout a wide variety of tissue types and detect extracellular ATP as a danger signal released from dying cells. We have explored how these pathways are involved in the transmission of HIV-1 from cell to cell through virological synapses. Infection of CD4+ T lymphocytes with HIV-1 in the presence of an inhibitor of P2X receptors effectively inhibited HIV-1 infection through both cell-free and cell-to-cell contact in a dose-dependent manner. Inhibition of direct cell-to-cell infection did not affect the formation of virological synapses or the subsequent cell-to-cell transfer of HIV-1. During both cell-free and cell-to-cell CD4+ T lymphocyte infection, purinergic antagonists blocked infection at the level of viral membrane fusion. During cell-to-cell transmission, we observed CXCR4 colocalization with the newly internalized virus particles within target lymphocytes and found that the purinergic antagonists did not impair the recruitment of the coreceptor CXCR4 to the site of Gag internalization in the target cell. In a screen of a library of purinergic antagonists, we found that the most potent inhibitors of HIV-1 fusion were those that target P2X receptors, while P2Y-selective receptor antagonists or adenosine receptor antagonists were ineffective. Our results suggest that P2X receptors may provide a therapeutic target and that purinergic antagonists may have potent activity against viral infection of CD4+ T lymphocytes by both cell-free and cell-to-cell transmission.
IMPORTANCE This study identifies purinergic antagonists to be potent inhibitors of HIV-1 cell-free and cell-to-cell-mediated infection and provides a stepwise determination of when these compounds inhibit HIV-1 infection. These data provide a rationale for the development of novel antiretroviral therapies that have a dual role in both direct antiviral activity and the reduction of HIV-associated inflammation. Purinergic antagonists are shown here to have equivalent efficacy in inhibiting HIV infection via cell-free and cell-to-cell infection, and it is shown that purinergic receptors could provide an attractive therapeutic anti-HIV target that might avoid resistance by targeting a host signaling pathway that potently regulates HIV infection. The high-throughput screen of HIV-1 fusion inhibitors further defines P2X-selective compounds among the purinergic compounds as being the most potent HIV entry inhibitors. Clinical studies on these drugs for other inflammatory indications suggest that they are safe, and thus, if developed for use as anti-HIV agents, they could reduce both HIV replication and HIV-related inflammation.
INTRODUCTION
Effective treatment of human immunodeficiency virus type 1 (HIV-1) infection can inhibit CD4+ cell decline and acquired immunodeficiency, yet the infection remains a major cause of morbidity and mortality as the population living with the virus ages. Patients on antiretroviral therapy now routinely survive long enough to develop diseases associated with aging and chronic disease. HIV-1 infection has been associated with premature aging and an increased risk for heart disease, cancer, bone disease, and cognitive decline (1–4). These sequelae are proposed to relate to the chronic inflammation that occurs despite antiretroviral therapy. In recent years, extracellular ATP (eATP) has been recognized as a signaling molecule important in chronic inflammation that signals through purinergic receptors on the cell membrane (5–11).
Recent studies suggest a requirement for eATP and purinergic receptor signaling in HIV-1 infection (12), and these signaling molecules appear to localize at the interface between an infected cell and a target cell, known as the virological synapse (VS) (13–15). Most studies regarding the pathogenesis of HIV-1 transmission have focused on cell-free viral infection. The direct spread of HIV-1 from T cell to T cell that occurs through VS is initiated when the viral envelope (Env) on the surface of an infected donor cell interacts with CD4+ on the surface of an uninfected target cell. The internalization of HIV-1 following cell-to-cell contact is more efficient than internalization by cell-free exposure, and HIV-1 can resist antibody neutralization when it is transmitted by this route (14, 16, 17). Cell-to-cell infection can result in a high multiplicity of infection that can reduce the efficiency of blocking of infection by some antiretroviral drugs compared to the efficiency of blocking of infection via cell-free virus (18–20). The signaling events that occur during VS formation have not been clearly delineated. In the study described here, we studied the role that purinergic signaling plays during HIV-1 entry and early infection through the VS. Recent studies suggest that HIV-1 Env interactions with the surface of CD4+ T lymphocytes can induce the release of ATP to the extracellular milieu (12). A study by Séror et al. found that inhibition of P2Y2 receptors that detect ATP can block HIV-1 infection by inhibiting viral entry into CD4+ T lymphocytes (12). Another study by Hazleton et al. found that P2X1 antagonists can block HIV-1 infection of macrophages (21). A third study by Orellana et al. described the ATP channel pannexin1, which is triggered in response to HIV-1 envelope binding to CD4+ and coreceptor, and indicated that triggering of this channel stimulates viral internalization (22).
Here, we examined the impact of purinergic antagonists on the efficiency of cell-to-cell transmission and found that HIV-1 requires purinergic signaling for viral membrane fusion following transfer across the VS. Our results support a model whereby P2X-mediated purinergic receptor signaling regulates a pathway following coreceptor recruitment to the site of viral entry.
MATERIALS AND METHODS
Viral constructs.
HIV-1 NL-GI contains green fluorescent (GFP) in place of nef, and nef expression is directed by a downstream internal ribosome entry site (IRES) (23). Primary CD4+ T cells were infected with NL-GI, which contains the NL4-3 envelope (X4 tropic), or NL-GI-RHPA, which was constructed by insertion of the R5-tropic B-clade primary envelope from pRHPA4259 clone 7 (SVPB14) into NL-GI (24). The gene for the RHPA clone was obtained through the AIDS Reagent Program (ARP), Division of AIDS, NIAID, NIH, from B. H. Hahn and J. F. Salazar-Gonzalez. HIV Gag-iCre, a virus that packages the Cre enzyme, was generated by cloning the cre recombinase in place of GFP in the molecular clone HIV-1 Gag-iGFP, a GFP-tagged HIV-1 containing the NL4-3 (an X4-tropic virus) envelope (25). The resulting virus, called HIV Gag-iCre, carries an insertion of cre into Gag between the MA and CA domains, and native protease cleavage sites were reproduced at either end of the Cre enzyme.
Cells and cell culture.
Cells of the human cell lines Jurkat E6-1 and MT4 (provided by Arthur Weiss and Douglass Richman, respectively, ARP) were maintained in RPMI 1640 medium containing 10% fetal bovine serum (FBS), 100 U/ml penicillin, 10 U/ml streptomycin, and 2 mM glutamine (complete RPMI). The CCR5-expressing T cell line, MT4-R5, was kindly provided by J. Robinson. Primary CD4+ T lymphocytes were purified from peripheral blood mononuclear cells (PBMCs) obtained from deidentified HIV-negative blood donors (New York Blood Center) using a Miltenyi Biosciences CD4+ T cell isolation kit (to purify untouched T cells) according to the manufacturer's instructions and stored in liquid nitrogen prior to use. Primary CD4+ T lymphocytes were thawed in RPMI containing 10% fetal calf serum, 5% human serum (HS), interleukin-2 (IL-2; 50 IU), and phytohemagglutinin (PHA) at 4 μg/ml.
Antibodies and inhibitors.
A panel of inhibitors was tested for the ability to block cell-free and cell-to-cell infection using 5-fold serial dilutions beginning at the concentration given below, unless otherwise stated. These included 100 μM pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid tetrasodium salt (PPADS; Sigma), a nonselective P2 antagonist, 10 μM the reverse transcriptase inhibitor azidothymidine (AZT; Sigma), 10 μM AMD3100 (a CXCR4 coreceptor antagonist; Sigma), and the library of 71 purinergic antagonists and agonists (Enzo BML-2820, v4.3). The CXCR4 antibody 44708.111 (catalog number 4084) was obtained through ARP.
Flow cytometry.
An LSR II flow cytometer (BD Biosciences) was used to detect infection and discriminate donor and target cell populations. All cells were initially discriminated by side scatter (SSC) area versus forward scatter (FSC) area (SSC-A/FSC-A); doublets were excluded using FSC width (FSC-W) versus FSC height (FSC-H). GFP was detected using the fluorescein isothiocyanate channel, dsRed-Express was detected using the phycoerythrin-Texas Red channel, CellTracker Blue was detected using the Pacific Blue channel, and CellTrace Far Red was detected using the allophycocyanin channel. All cells within a single experiment were detected using the same voltage settings.
Cell-free infection assay.
Cell-free virus particles were produced in HEK293T cells by calcium phosphate transfection. Viral supernatants were quantified by a p24 enzyme-linked immunosorbent assay. Target cells were either MT4 cells or PBMCs activated with PHA (4 μg/ml) and IL-2 (50 IU) for 3 days, as previously described (26), and were infected in 96-well plates with 75 ng per well HIV NL-GI to obtain up to 10% infection after 48 h in the absence of inhibitors. Virus supernatant and MT4 cells were preincubated separately with equal volumes of inhibitors for 30 min at 37°C before mixing. After 18 h, the culture medium was replaced with complete RPMI containing 10 μM AZT. At 48 h after mixing, cells were treated with trypsin and fixed in 2% paraformaldehyde.
Cell-to-cell infection assay.
Jurkat (donor) cells were transfected by nucleofection (nucleofected; Lonza) with 5 μg HIV NL-GI DNA, cultured overnight in antibiotic-free medium, and purified by Ficoll-Hypaque density gradient centrifugation. MT4 (target) cells were dye labeled with 5 μM eFluor 450 (eBioscience) for 10 min at 37°C. Donor or target cells (0.125 × 106) were preincubated separately with inhibitors for 30 min at 37°C before mixing at a ratio of approximately 1:1, cocultured at 37°C for 36 h with a medium change after 18 h, treated with trypsin, fixed, and analyzed by flow cytometry.
Cell-to-cell transfer assay.
Jurkat (donor) cells were transfected by nucleofection (Amaxa Biosystems) with 5 μg HIV-1 Gag-iGFP or HIV-1 Gag-iCherry DNA, cultured overnight in antibiotic-free medium, and purified by Ficoll-Hypaque density gradient centrifugation. Primary CD4+ T lymphocytes or MT4 (target) cells were dye labeled with 5 μM eFluor 450 (eBioscience) or Far Red 9-H-(1,3-dichloro-9,9-dimethylacridin-2-one-7-yl) succinimidyl ester (DDAO-SE; CellTrace; Life Sciences) for 10 min at 37°C. Donor or target cells (0.125 × 106) were preincubated separately with inhibitors for 30 min at 37°C before mixing at a ratio of approximately 1:1, cocultured at 37°C for 4 h, treated with trypsin to remove surface-adsorbed virus, and fixed in phosphate-buffered saline (PBS) containing 2% paraformaldehyde.
Cell-free and cell-to-cell viral fusion assay.
A stable cell line of Jurkat cells called Jurkat floxRG (where RG indicates red to green) was generated by retroviral transduction with the pMSCV-loxP-dsRed-loxP-eGFP-Puro-WPRE vector (plasmid 32702; Addgene), which expresses a dsRed reporter flanked by loxP sites followed by a Cre-activated enhanced GFP (eGFP) gene (27). Jurkat (donor) cells were transfected by nucleofection (Amaxa Biosystems) with 5 μg HIV-1 Gag-iCre DNA, cultured overnight in antibiotic-free medium, and purified by Ficoll-Hypaque density gradient centrifugation. Donor or target cells (1.25 × 105) were preincubated separately with inhibitors for 30 min at 37°C before mixing at a ratio of approximately 1:1, cocultured at 37°C for 48 h, treated with trypsin, and fixed.
Vpr-BlaM fusion assay.
Viral membrane fusion directed by cell-free virions was measured as described previously (28). Briefly, HIV-1 was produced by cotransfecting HEK293T cells with wild-type proviral DNA (pNL4-3) and a plasmid (pMM310) that encodes a Vpr–beta-lactamase fusion protein (Vpr-BlaM). The resulting viral supernatant, which contained 30 ng p24 antigen, was added to 2 × 105 HEK293T cells in a volume of 200 μl. After coculture, cells were incubated in CO2-independent medium for 12 h to allow substrate cleavage at 37°C. Cells were washed in PBS, fixed in 3.7% formaldehyde, and read on an LSR II flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Cleavage of coumarin cephalosporin fluorescein acetoxymethyl ester (CCF2-AM) was determined by flow cytometry using a 405-nm excitation and acquiring emissions at 450 nm (±50 nm) and 525 nm (±50 nm). Flow cytometry data were exported and analyzed using FlowJo software (Tree Star, Ashland, OR). Gates were set using cocultures of untransfected cells. The small percentage of positive cells present in the untransfected controls were subtracted as background for all samples. In each experiment, the results for control conditions were normalized to 100%, and the results for the experimental conditions were expressed as a percentage of the results for the control.
Confocal microscopy/imaging flow cytometry.
Colocalization studies were performed on mixed donor and target cells. Jurkat (donor) cells were transfected by nucleofection (Amaxa Biosystems) with 5 μg HIV-1 Gag-iGFP or HIV-1 Gag-iCherry DNA, as described above for cell-to-cell transfer. Primary CD4+ T lymphocytes or MT4 (target) cells were incubated with monoclonal mouse anti-CXCR4 (catalog number 4084; ARP) (1:100) for 2 h and then washed and incubated with goat anti-mouse secondary Alexa Fluor 488 IgG (1:200) for 1 h. Target cells were washed and dye labeled with 5 μM Alexa Fluor 450 (eBioscience) for 10 min at 37°C. Donor or target cells (1.25 × 105) were preincubated separately with inhibitors for 30 min at 37°C before mixing at a ratio of approximately 1:1, cocultured at 37°C for 4 h, and then transferred to a coverslip coated with 0.01% poly-l-lysine for confocal microscopy or a 96-well plate for imaging flow cytometry. Cells were fixed and imaged on a Leica DM-5 laser scanning confocal microscope. Images were acquired using a ×63 objective and analyzed using Volocity (PerkinElmer) or ImageJ (NIH) software. Quantification of cell number and puncta was performed using Metamorph software (Molecular Devices). For imaging flow cytometry, data were acquired using Inspire acquisition software and a 488-nm solid-state laser with appropriate compensation controls and settings. Data from a minimum of 50,000 cells were collected for each sample and analyzed using IDEAS software. Single in-focus cells were identified using data from the bright-field images.
RESULTS
Inhibition of purinergic receptors results in reduction of productive infection.
Several studies have implicated purinergic receptor signaling as being important for HIV-1 infection in CD4+ T lymphocytes and macrophages (21, 29). Immunofluorescence imaging studies have revealed that key purinergic signaling components localize to the donor cell-target cell interface. This observation could indicate that purinergic receptor signaling is particularly important for cell-to-cell transmission of HIV-1 (12). Studies examining antiretroviral therapies have revealed that cell-to-cell infection can exhibit sensitivities different from those of cell-free infection (18, 19). We therefore examined the relative sensitivity of cell-free versus cell-associated HIV-1 to the purinergic antagonist pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS). The assay utilizes Jurkat cells nucleofected with a GFP-expressing HIV strain, HIV NL-GI (23), mixed with target cells in the presence or absence of drug. Measurement of infection of target cells by flow cytometry revealed strong inhibition of cell-free and cell-to-cell infection by PPADS (Fig. 1A and B). A titration experiment demonstrated the dose-dependent inhibition of HIV-1 infection under both cell-to-cell and cell-free infection conditions, with the 50% inhibitory concentrations (IC50s) being nearly identical (Fig. 1C).
FIG 1.

PPADS treatment results in dose-dependent inhibition of cell-to-cell and cell-free productive infection. (A) Cell-free infection is inhibited by PPADS. Representative fluorescence-activated cell sorter plots are shown for uninfected cells (top) or HIV NL-GI-infected Alexa Fluor 450-labeled MT4 cells (cell-free infection) in the absence (middle) or presence (bottom) of 100 μM PPADS. (B) Cell-cell infection is inhibited by PPADS. Representative fluorescence-activated cell sorter plots are shown for HIV NL-GI-nucleofected Jurkat (donor) cells mixed with Alexa Fluor 450-labeled MT4 (target) cells. MT4 cells mixed with uninfected Jurkat cells (uninfected, top), infected cells (middle), or infected cells in the presence of 100 μM PPADS (bottom). (C) Dose-response curves for both NL-GI cell-free and cell-cell infections by concentration of PPADS. Nonlinear regression curve fits for cell-to-cell and cell-free infection are shown. (D) Dose-response curves of NL-GI (X4) and NL-GI-RHPA (a primary R5-tropic virus cloned into the NL-GI backbone) cell-free infections, using MT4 or MT4-R5 target cells, by concentration of PPADS. Infections were conducted in the presence of serial 5-fold dilutions of PPADS from 1 mM, and samples were incubated for 36 h and then fixed and analyzed by flow cytometry. (E) PPADS inhibition of NL-GI infection of activated PBMCs after 48 h. PPADS inhibited productive infection by 82%. Results are the means ± SEMs of three independent experiments. *, P < 0.05.
We next asked whether inhibition of productive infection was coreceptor specific. We tested this by infecting CD4+ MT4 cells stably expressing CCR5 (MT4-R5) with an R5-tropic virus that carries a primary isolate Env gene cloned into the NL-GI backbone (NL-GI-RHPA). Cell-free infection with the R5-tropic virus was blocked by PPADS with a 50% inhibitory concentration (IC50) similar to that seen with the infection of MT4 cells with an X4-tropic virus (Fig. 1D). Treatment of the cells with PPADS did not affect cell viability, as determined by trypan blue staining at 36 h. These findings suggest an important role of purinergic receptor activation in both cell-to-cell and cell-free HIV-1 infection. To test the effect of PPADS on the infection of primary cells, we infected PHA-activated peripheral blood mononuclear cells with GFP-expressing virus NL-GI in the presence or absence of PPADS (Fig. 1E). After 48 h, PPADS inhibited productive infection by 82%.
Inhibition of purinergic receptors does not affect cell-to-cell transfer or stable donor-target associations.
With confirmation that PPADS potently blocks productive infection in both cell-to-cell and cell-free systems, we next examined the ability of the inhibitor to block virological synapse initiation and/or the transfer of viral materials from cell to cell. During cell-to-cell-mediated entry, the contact of the infected donor with the target cell membrane through interaction between CD4+ and Env leads to the internalization of HIV-1 into a trypsin-resistant compartment, followed by viral maturation, fusion, and budding of nascent virions (26). To study the transfer of virus across virological synapses, we employed the fluorescent virus particles produced by HIV-1 Gag-iGFP, a GFP-tagged infectious clone of HIV-1, to examine the transfer of viral antigen from cell to cell that occurs in a CD4-dependent manner (14). Jurkat T cells transfected with HIV-1 Gag-iGFP, which produce highly fluorescent virus particles, were mixed with uninfected Far Red-labeled primary CD4+ T lymphocytes. The efficiency of virological synapse formation and transfer of virus from one cell to the next was assessed by measuring the uptake of GFP-labeled virus into target cells labeled with an inert dye (CellTracker) to distinguish target cells from donor cells. As assessed by the percentage of acceptor CD4+ T lymphocytes with a GFP signal, the cell-to-cell transfer of HIV-1 was efficient and nearly achieved a steady state at 4 h. Representative flow cytometry plots (Fig. 2A) indicate that cell-to-cell transfer was insensitive to purinergic inhibition by PPADS. Quantification of transfer (Fig. 2B) did not show any significant inhibition of viral transfer by 100 μM PPADS, and no dose-response effect was observed. We also observed by confocal microscopy that cell-to-cell associations were still readily observed (Fig. 2C).
FIG 2.

PPADS does not inhibit cell-to-cell transfer of HIV. Jurkat cells were transfected with HIV-1 Gag-iGFP. Target primary CD4+ T cells were stained, and both donor and target cells were preincubated with inhibitor for 30 min prior to mixing. Cells were coincubated for 4 h and then fixed and analyzed by flow cytometry. (A) Representative fluorescence-activated cell sorter plots of uninfected target cells (top), cells infected with Gag-iGFP (middle), and cells infected with Gag-iGFP in the presence of 100 μM PPADS (bottom). (B) Quantification of transfer efficiency from flow cytometry of uninfected target cells, cells infected with Gag-iGFP, and cells infected in the presence of 100 μM PPADS. Results are the means ± SEMs of three independent experiments. NS, nonsignificant. (C) Confocal micrographs of infected Jurkat cells associated with target primary CD4+ T cells. Nuclei are stained with DAPI (4′,6-diamidino-2-phenylindole), and HIV (mCherry-HIV) is in red. Bar, 10 μm. (D, E) Flow cytometric imaging was performed on cells infected with Gag-iGFP (D) or infected in the presence of 100 μM PPADS (E) after incubation for 4 h. (Top) Representative plots of the cells are shown (results are the means ± SEMs of three independent experiments); (bottom) examples of cells identified by flow cytometry imaging are shown below for each condition. Stable associations of donor and target cells were identified as follows: cells were sorted for doublets in which a single cell stained positive for CellTracker (target cells) and the second cell stained positive for iCherry (donor). Bars, 10 μm. DIC, differential interference contrast.
Using an Amnis flow cytometric imaging system, cells can be sorted by flow cytometry and images of cells with specific fluorescence profiles can be visualized. This is particularly helpful in capturing and measuring relatively rare events and identifying related structures. Figure 2D demonstrates the use of flow cytometric imaging of stable associations between infected donor cells and CellTracker-labeled target cells (2 to 4% transfer). While there is a small reduction in the transfer efficiency with PPADS, this was not found to be statistically significant (P > 0.5). These results suggest that the step of HIV-1 replication inhibited by PPADS occurs after the formation of the virological synapse and transfer of virus. Because cell-to-cell transfer of HIV across the VS is dependent on CD4-Env interactions, these results suggest that the purinergic antagonism does not interfere with the engagement of Env with CD4 during VS formation.
Purinergic inhibition blocks viral membrane fusion.
We next examined whether viral fusion may be blocked in target cell endocytic compartments. We employed a novel cre recombinase-activated assay to test viral fusion (A. M. Esposito, P. Cheung, T. H. Swartz, D. P. Felsenfeld, and B. K. Chen, unpublished data). As target cells, we employed CD4+ Jurkat cells that express a cre-activated GFP expression cassette (Jurkat-floxRG). These cells are stably transduced with a murine stem cell virus (MSCV) vector expressing dsRed, a red fluorescent protein (RFP) that is flanked by two loxP sites and followed by a GFP gene (27). When this target cell is infected with a virus that packages the Cre enzyme, HIV Gag-iCre, Cre-mediated recombination deletes the floxed dsRed and induces the expression of GFP. GFP expression indicates that virus-cell fusion has occurred (Fig. 3A).
FIG 3.
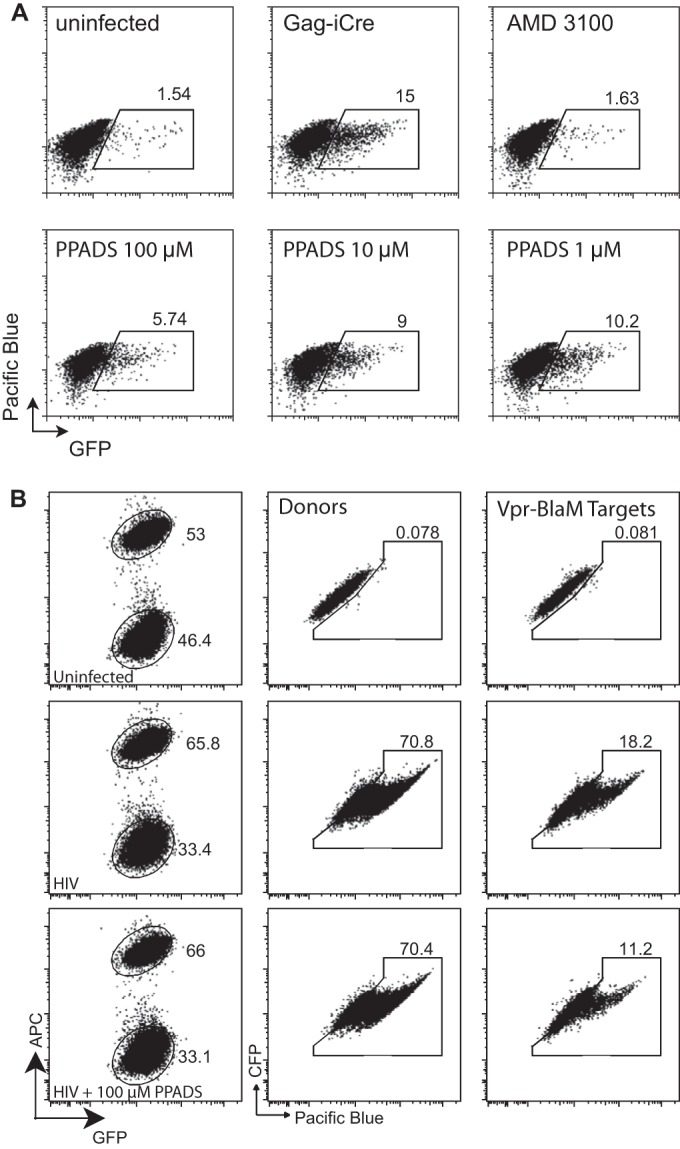
HIV-membrane fusion following cell-to-cell infection is inhibited by PPADS. (A) Jurkat-floxRG cells were mixed with Jurkat cells nucleofected with HIV-1 Gag-iCre, and the inhibitors AMD3100 and PPADS were tested for their ability to block viral membrane fusion. Cell-to-cell infection was observed, and PPADS inhibition was noted for concentrations of PPADS between 1 and 100 μM in the representative fluorescence-activated cell sorter plots. (B) PPADS inhibited viral membrane fusion during cell-to-cell infection, as measured by a Vpr-BlaM assay. Representative fluorescent-activated cell sorting plots of uninfected target cells (top), cells infected with HIV Vpr-BlaM (middle), and cells infected with HIV Vpr-BlaM in the presence of 100 μM PPADS (bottom) are shown. APC, allophycocyanin.
When Jurkat-floxRG cells were exposed to cell-free HIV Gag-iCre (carrying the strain NL4-3 Env), viral membrane fusion was sensitive to AMD3100, an inhibitor of HIV-1 fusion that works by antagonizing CXCR4, as indicated by GFP expression (30), but was insensitive to the reverse transcriptase inhibitor azidothymidine (AZT) (not shown). We also examined the ability of PPADS to block viral membrane fusion after cell-cell coculture using the Vpr–beta-lactamase assay, where donor cells expressing a Vpr–beta-lactamase fusion are mixed with target cells loaded with the beta-lactamase substrate CCF2-AM. We observed that the level of inhibition of viral membrane fusion was comparable to that observed in the Gag-iCre assay (Fig. 3B). Treatment of the cells with PPADS during cell-to-cell infection with HIV-1 Gag-iCre resulted in inhibition, indicating that the purinergic signaling acts at or prior to the induction of viral membrane fusion (Fig. 3). On the basis of the findings presented above, the stage of inhibition can be specified to an event that follows cell-cell adhesion, synapse formation, and transfer of virus into an endocytic compartment, but prior to viral membrane fusion.
PPADS has no effect on colocalization of coreceptor CXCR4 with HIV-1 Gag in infected target cells.
We next probed how purinergic inhibition might block HIV-1 fusion by testing the effect of PPADS on CXCR4 colocalization with HIV Gag in target cells. As a coreceptor is known to be required for HIV-1 fusion but is not required for cell-to-cell transfer of HIV-1 across the VS (16), we examined whether CXCR4 localization to the endocytic compartment following cell-to-cell entry may be abrogated in the presence of PPADS. Figure 4A shows confocal microscopy images in which donor Jurkat cells were nucleofected with HIV-Gag-iCherry and cocultured with primary CD4+ T lymphocytes isolated from human PBMCs. The target cells were discriminated by labeling with Far Red DDAO-SE (CellTrace; Life Sciences) and prestained with fluorescein isothiocyanate (FITC)-conjugated anti-CXCR4. Cells not treated with PPADS (Fig. 4A) showed a colocalization of CXCR4 with HIV-1 Gag. Target cells were evaluated for their expression of CXCR4 by flow cytometry and confocal microscopy in the presence or absence of 100 μM PPADS. MT4 cells were labeled with FITC-conjugated anti-CXCR4 and incubated with or without 100 μM PPADS for 3 h. Figure 4B shows a histogram, obtained by flow cytometry, which showed no difference in CXCR4 surface expression on PPADS-treated target cells. Measurement of CXCR4 puncta from three independent experiments with both uninfected and infected target cells in the presence or absence of 100 μM PPADS also did not reveal any significant changes among the groups in the number of CXCR4-positive puncta internalized into target cells by a t test of pooled data from three separate experiments (Fig. 4C).
FIG 4.

Colocalization of CXCR4 with HIV-1 Gag in infected target cells. (A) Confocal microscopy images demonstrate an HIV-1 Gag-iCherry-infected Jurkat cell attached to three primary CD4 T cells. CXCR4 (green) is shown associated with Gag-iCherry (red). Bar, 10 μm. (B) PPADS does not affect CXCR4 staining of MT4 target cells, as determined by flow cytometric analysis of target cells labeled with anti-CXCR4. (C) Quantification of CXCR4 puncta on the basis of confocal microscopy demonstrates no difference in the number of CXCR4 puncta in the presence or absence of PPADS (results are the means ± SEMs of three independent experiments; P > 0.5, t test). (D) Amnis flow cytometry imaging was performed on infected cells, and a colocalization percentage was calculated on the basis of the proportion of HIV-positive target cells positive for CXCR4 in the presence or absence of 100 μm PPADS (results are the means ± SEMs of three independent experiments; P = 0.8, t test; NS, nonsignificant). (E, F) Amnis flow cytometry imaging illustrating gating of infected target cells. The x axis is Gag-iCherry (infected) cells, and the y axis is eFluor 450 (positive targets). The red rectangular gates in the right upper quadrants indicate HIV Gag-iCherry-positive target cells in untreated cocultures (E) or in cocultures treated with 100 μM PPADS (F). Representative images of colocalization of CXCR4 with HIV-1 Gag-iCherry on infected target cells without inhibitor treatment (E, right) or treatment with 100 μM PPADS (F, right). Bar, 10 µm.
Using flow cytometric imaging, we analyzed samples of >100,000 cells to identify those target cells with recently transferred virus (HIV Gag-iCherry), which accounted for 5% of the total population. We then identified HIV Gag-iCherry-positive target cells and determined the extent of CXCR4 with Gag colocalization in this cell population (Fig. 4D). Using this flow cytometric imaging analysis, we observed the comparable colocalization of CXCR4 with Gag in the presence or absence of PPADS, with no statistically significant difference among the groups being detected by a t test of pooled data from three separate experiments. Gating for these cells is shown for those untreated (Fig. 4E) or treated with 100 μM PPADS (Fig. 4F). Representative images of colocalization in the absence or presence of PPADS are shown (Fig. 4E and F, respectively).
Chemical profiling of purinergic inhibitors of HIV-1 fusion following cell-to-cell infection identifies P2X antagonists to be potent HIV-1 entry inhibitors.
The purinergic antagonist PPADS is thought to inhibit several different receptors, with the greatest potency being against the P2X1, P2X7, and P2Y1 receptors. To better define the pharmacological profile of related purinergic compounds that can antagonize HIV-1 fusion, we examined a library of 71 purinergic compounds, including endogenous neurotransmitters, agonists, antagonists, and marketed drugs (Enzo BML-2820, v4.3). Using the cell-to-cell Cre-mediated viral fusion assay, the inhibitory activity of each member of the entire library was tested at 100 μM, and the results were compared with those for the untreated control. The assay was performed twice, and inhibition values were averaged. At a 100 μM concentration, 5 of the compounds showed significant (greater than 30%) inhibition of HIV-1 fusion (Fig. 5A), while they did not significantly alter cell viability, as measured by changes in forward and side scatter (not shown). Five of the compounds caused a decrease in viability of greater than 30% compared to that for the untreated control (Fig. 5B). The remaining compounds did not have a significant effect on viability or viral fusion.
FIG 5.

Screen of a purinergic library reveals that compounds selective for P2X receptors inhibit viral membrane fusion. (A) Seventy-one purinergic compounds listed on the basis of their level of inhibition of HIV-1 membrane fusion, which was based on the averages of two independent determinations with the compounds at 100 μM. (B) The compounds were categorized on the basis of their toxicity and inhibitory activity. Five compounds had inhibitory activity without toxicity, while five compounds had inhibitory activity at a toxic level. The remaining 61 compounds had no effect. (C) Dot plot of HIV fusion-inhibitory activity of all compounds grouped on the basis of their reported receptor specificity. Compounds reported to have nonselective inhibition against either P2X or P2Y receptors are placed in the P2X/P2Y category, compounds reported to have only P2Y activity are placed in the P2Y category, and the remaining compounds have some reported activity on the adenosine receptors and are placed in the A1/A2 category. *, P < 0.0005. (D) Titrations of the four most potent inhibitors were conducted using the Cre-Lox assay to measure viral fusion during cell-free and cell-to-cell infection. IC50s were calculated using nonlinear regression analysis of a log concentration (agonist) versus the normalized response with a variable slope. The structure of each compound is listed below the plot.
While these compounds exhibited various degrees of cross-reactivity, they can be grouped into several broad categories: those that exhibited selectivity for the P2X class of receptors, those that exhibited selectivity for the P2Y class of receptors, those that exhibited selectivity for adenosine receptors or are involved in adenosine metabolism, and lastly, a number that are not well characterized with respect to purinergic receptors. When sorted by viral fusion-inhibitory activity, compounds with higher levels of inhibition showed a strong bias toward being selective toward the P2X class of receptors. These were NF279, suramin, NF023, pyridoxal-5′-phosphate-6-(2′-naphthylazo-6′-nitro-4′,8′-disulfonate) (PPNDS), and PPADS (Fig. 5C).
A total of five compounds that inhibit HIV-1 fusion by greater than 50% were identified (Table 1). The broad-spectrum P2 inhibitor suramin and its structural analogue, NF279, showed the greatest level of inhibition of viral fusion (95%). NF279 and NF023 are structurally related and have selectivity toward the P2X1 receptor. They showed a high level of inhibition (95% and 68%, respectively). The broad-spectrum P2 inhibitor PPADS showed partial inhibition of viral fusion (38%), and PPNDS, the PPADS analogue with increased selectivity for P2X1, strongly inhibited fusion (75%) and has not previously been shown to inhibit HIV-1 replication. Table 2 lists the IC50s for cell-free and cell-to-cell viral fusion using the Cre recombinase assay. We observed no significant difference in inhibition of the cell-free and cell-to-cell entry assays. Notably, the compounds that target adenosine and adenosine receptors showed no inhibition of HIV-1 fusion. Our studies provide a chemical profile of the purinergic antagonists that mediate strong inhibition of HIV-1 cell-to-cell infection. While previous studies that implicate P2Y2 as an important target in CD4+ T lymphocytes (12), the findings of the inhibitor studies presented here are consistent with an important role for P2X receptors in CD4+ T lymphocytes, with a preference for P2X1 selectivity. An earlier study in primary human macrophages has reported that P2X1 signaling influences HIV-1 infection (21).
TABLE 1.
Compounds that inhibit HIV-1 fusion by greater than 50%
| Compound | % inhibition at 100 μM | Published mechanism | References |
|---|---|---|---|
| NF279 | 95 | Suramin analogue with high selectivity for P2X over P2Y receptors; selective for P2X1 and P2X7 over other P2X receptors | 21, 54–56 |
| Suramin | 95 | Broad-spectrum P2 inhibitor; also blocks calmodulin binding to recognition sites and G protein coupling to G-protein-coupled receptors | 37, 57–59 |
| PPNDS | 75 | PPADS analogue with enhanced selectivity toward P2X1 | 60–63 |
| NF023 | 68 | Suramin analogue with enhanced selectivity for P2X over P2Y; selectivity toward P2X1 | 59, 64, 65 |
| PPADS | 38 | Broad-spectrum P2 Inhibitor | 12, 66, 67 |
TABLE 2.
IC50s for cell-free and cell-to-cell viral fusion
| Compound | Cell-free viral fusion |
Cell-to-cell viral fusion |
||
|---|---|---|---|---|
| IC50 (μM) | 95% CIa | IC50 (μM) | 95% CI | |
| NF279 | 13.4 | 11.4–15.7 | 13.3 | 11.4–15.7 |
| Suramin | 19.4 | 16.4–22.9 | 19.3 | 15.6–23.8 |
| NF023 | 34.5 | 16.9–70.2 | 30.3 | 11.3–81.4 |
| PPNDS | 34.2 | 27.4–42.6 | 30.9 | 26.1–36.6 |
CI, confidence interval.
DISCUSSION
Here we demonstrate that PPADS, a nonselective purinergic antagonist, has potent inhibitory effects on cell-to-cell and cell-free productive HIV-1 infection and that this occurs in both X4- and R5-tropic virus infections. We demonstrate that inhibition of purinergic signaling inhibits HIV-1 membrane fusion. The fact that the IC50 is similar for cell-to-cell and cell-free productive infection raises a plausible role for these compounds as inhibitors of both modalities of HIV-1 infection, particularly as literature suggests that certain classes of antiretroviral inhibitors have reduced efficacy during cell-cell infection (18, 19). Because these inhibitors target a common cellular pathway that blocks entry through both cell-free and cell-to-cell infection, these agents could be useful in diminishing the residual replication that is driven by cell-to-cell infection and would inhibit both modes of infection with equal efficacy.
An emerging literature demonstrates the importance of extracellular ATP purinergic signaling in viral infections, specifically, those caused by HIV-1 (21, 22, 31–33). While limited data on other enveloped viruses exist, it is reported that internalization of pseudotyped HIV was not reduced by purinergic inhibitors (12). Our data and those of others support a role for purinergic signaling in promoting viral membrane fusion in HIV-1 infection (11, 12, 21). Here we report the first systematic screen of purinergic compounds and their effects on inhibition of HIV-1 fusion. Our purinergic library screen revealed a high level of inhibition of viral fusion by inhibitors specific for the P2X receptors. Interestingly, PPADS, a nonselective P2X/P2Y inhibitor, appeared to inhibit viral fusion less potently than the more selective P2X inhibitors, such as suramin, NF279, and NF023 (Fig. 5A). This was unexpected, on the basis of the observation that PPADS inhibited productive infection with a high potency (Fig. 1C). This may indicate that inhibition of productive infection by PPADS represents inhibition at more than one point in the infection cycle. The receptors that are the most highly targeted by these compounds and that are expressed in lymphocytes include the P2X1 and P2X7 receptors, which are ATP-gated receptors permeable to calcium. The results of our screen further suggest that inhibitors of P2Y receptors or adenosine receptors alone do not have an effect on HIV-1 fusion. Purinergic targets in a number of inflammatory and pain conditions, including rheumatoid arthritis and neuropathic pain, are in development (34, 35). Phase II and III studies have demonstrated the safety of some P2X inhibitors and have shown that they have minimal toxicity.
It is important to note that while titration of PPADS resulted in 100% inhibition of productive infection (Fig. 1), it did not fully inhibit viral fusion, as measured by the HIV Gag-iCre entry assay (Fig. 3). This may indicate that the inhibition of PPADS may occur at more than one stage of infection. PPADS has in the past been described to be a putative reverse transcriptase inhibitor (36, 37). The other compounds identified in the screen, i.e., suramin, NF279, and NF023, did inhibit fusion at a higher level (Fig. 5) and may represent more selective viral fusion inhibitors.
Additionally, we show the application of flow cytometry imaging for identifying populations of cells with specific subset labeling. Using this technology, it is possible to identify stable associations of donor and target cells and identify cofactors at the interface. We show with a large population of cells that the coreceptor and Gag appear to colocalize in infected target cells (Fig. 4D to F). Figure 4D indicates a comparable level of HIV-1 Gag and CXCR4 colocalization. We conclude that CXCR4 colocalization does not likely explain the effect of PPADS on viral fusion.
The model illustrated in Fig. 6 describes the role of purinergic inhibition of HIV-1 infection via cell-free or cell-to-cell entry where a donor cell infected with HIV-1 can transmit virus to a target cell. As depicted on the upper portion of Fig. 6, virus maturation and budding take place, resulting in a cell-free virion that can attach and undergo viral membrane fusion. As depicted on the bottom of Fig. 6, virus maturation occurs within the target cell after transfer of immature virions across the virological synapse. In both cell-free and cell-cell infection, the recruitment of coreceptors, CXCR4 or CCR5, is not disrupted; and fusion can be blocked by P2X inhibitors independently of coreceptor recruitment.
FIG 6.
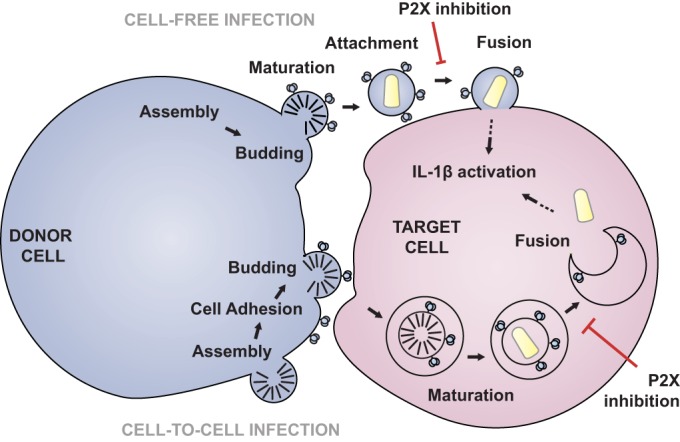
Model of HIV-1 infection via purinergic signaling. Two cells, a donor cell (left) and a target cell (right), are shown. The scheme at the top indicates cell-free infection, and that at the bottom shows cell-to-cell infection. In the cell-free model, HIV-1 assembles and then buds off the cell. Mature cell-free virions then attach to a target cell and fusion occurs. This step is inhibited by purinergic inhibition. In the cell-to-cell mode, HIV-1 Env triggers cell adhesion, virus (Gag) is recruited to the VS, and budding is coordinated with uptake into the target cell. Virus is transferred across the virological synapse into a target cell compartment, where it undergoes fusion, which is also inhibited by P2X antagonists. The arrows with a dotted tail indicate that in both cell-free and cell-to-cell infections this process may result in inflammasome activation, as indicated by the activation of IL-1β.
Our data support a role for purinergic inhibition at the stage between coreceptor recruitment and viral membrane fusion. The mechanism of this inhibition still remains an important subject of investigation. Given that the inhibitors do not block synapse formation, it is unlikely that the inhibitors bind to CD4 in a manner that would block attachment. Purinergic receptors are known to be upstream of several inflammatory pathways. It is interesting to speculate that, in addition to facilitating viral entry, the activation of P2X receptors by HIV could cause downstream inflammatory activation. There are downstream signaling pathways that may account for the immediate and long-term inflammatory effects after HIV-1 infection. These include the activation of the NLRP3 inflammasome and pyroptosis, which are known downstream effectors of purinergic signaling (38–41). NLRP3, a multimeric protein complex that activates the maturation and release of proinflammatory cytokines, is the best-characterized inflammasome. Inflammasome activation is known to occur downstream of purinergic signaling, most notably, P2X7 signaling (42–45). Inflammasome activation also mediates the release of IL-1β triggered by caspase-1 cleavage. With specific external triggers, lymphocytes are able to undergo pyroptosis, which is a proinflammatory caspase-1-specific cell death that is marked by inflammasome activation and IL-1β release. Recently, it has been proposed that pyroptosis may account for a significant proportion of T cell death in HIV-1-infected lymphoid tissue. Furthermore, this cell death can be blocked by caspase-1 inhibition (46, 47). It is plausible that purinergic antagonists may also inhibit these HIV-triggered inflammatory responses. The potential adverse events of blocking purinergic signaling could be immunosuppressive effects; however, these could be offset by a reduction in lymphocyte cell death, which may be a desired effect of an HIV anti-infective.
Our data indicate that purinergic signaling represents an essential pathway that HIV-1 utilizes during viral entry. The purinergic antagonists are being actively studied for their therapeutic potential and have been demonstrated to be safe in clinical trials testing P2X7 antagonists in the treatment of rheumatoid arthritis (including trials AZD9056, CE-224,535, and GSK1482160) (48–52). We postulate that the development of targeted therapies that block these signaling pathways may provide simultaneous treatment of HIV-1 infection, the chronic inflammation associated with HIV-1 infection, and, possibly, the associated long-term sequelae, including neurotoxic effects (53). Because the activation of purinergic signaling during HIV-1 infection may trigger inflammatory mediators that are involved in HIV-1-associated inflammation, purinergic receptors are an attractive target for future antiretroviral therapeutic development.
ACKNOWLEDGMENTS
We thank the members of the B. K. Chen laboratory for meaningful discussion and the Mount Sinai Core Microscopy Facility and Flow Cytometry Facility for services.
T. H. Swartz was supported in part by NIH grant T32AI007605 in immunology, NIH grant T32DK007792 in investigative gastroenterology, and a grant through Conduits, which is supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences of the National Institutes of Health through grant UL1TR000067. A. M. Esposito was supported by NIH grant T32 5T32AI007647 (Training Program in Mechanisms of Virus-Host Interactions). This work was supported by a grant to B. K. Chen from NIH, NIAID (R01AI074420), NIDA Avant Garde grant DP1DA028866, and the Burroughs Wellcome Investigators in the Pathogenesis of Infectious Disease Fund.
We have no conflicting financial interests.
T. H. Swartz, A. M. Esposito, and N. D. Durham performed the experiments. T. H. Swartz and N. D. Durham carried out the cell-free and cell-to-cell infection and transfer assays. T. H. Swartz carried out all the imaging and flow cytometry imaging. N. D. Durham carried out the Vpr-BlaM assay. A. M. Esposito developed the stable cell line of floxRG cells and carried out experiments using HIV-1 Gag-iCre. T. H. Swartz and A. M. Esposito wrote the paper. B. Hartmann carried out the Amnis flow cytometric imaging. T. H. Swartz, A. M. Esposito, and B. K. Chen designed the experiments. B. K. Chen conceived the approach.
Footnotes
Published ahead of print 16 July 2014
REFERENCES
- 1.Guaraldi G, Orlando G, Zona S, Menozzi M, Carli F, Garlassi E, Berti A, Rossi E, Roverato A, Palella F. 2011. Premature age-related comorbidities among HIV-infected persons compared with the general population. Clin. Infect. Dis. 53:1120–1126. 10.1093/cid/cir627 [DOI] [PubMed] [Google Scholar]
- 2.Ofotokun I, McIntosh E, Weitzmann MN. 2012. HIV: inflammation and bone. Curr. HIV/AIDS Rep. 9:16–25. 10.1007/s11904-011-0099-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sigel K, Dubrow R, Silverberg M, Crothers K, Braithwaite S, Justice A. 2011. Cancer screening in patients infected with HIV. Curr. HIV/AIDS Rep. 8:142–152. 10.1007/s11904-011-0085-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hunt PW, Sinclair E, Rodriguez B, Shive C, Clagett B, Funderburg N, Robinson J, Huang Y, Epling L, Martin JN, Deeks SG, Meinert CL, Van Natta ML, Jabs DA, Lederman MM. 21 April 2014. Gut epithelial barrier dysfunction and innate immune activation predict mortality in treated HIV infection. J. Infect. Dis. 10.1093/infdis/jiu238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belete HA, Hubmayr RD, Wang S, Singh RD. 2011. The role of purinergic signaling on deformation induced injury and repair responses of alveolar epithelial cells. PLoS One 6:e27469. 10.1371/journal.pone.0027469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Busillo JM, Azzam KM, Cidlowski JA. 2011. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J. Biol. Chem. 286:38703–38713. 10.1074/jbc.M111.275370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deli T, Csernoch L. 2008. Extracellular ATP and cancer: an overview with special reference to P2 purinergic receptors. Pathol. Oncol. Res. 14:219–231. 10.1007/s12253-008-9071-7 [DOI] [PubMed] [Google Scholar]
- 8.McIlvain HB, Ma L, Ludwig B, Manners MT, Martone RL, Dunlop J, Kaftan EJ, Kennedy JD, Whiteside GT. 2010. Purinergic receptor-mediated morphological changes in microglia are transient and independent from inflammatory cytokine release. Eur. J. Pharmacol. 643:202–210. 10.1016/j.ejphar.2010.06.046 [DOI] [PubMed] [Google Scholar]
- 9.Pillai S, Bikle DD. 1992. Adenosine triphosphate stimulates phosphoinositide metabolism, mobilizes intracellular calcium, and inhibits terminal differentiation of human epidermal keratinocytes. J. Clin. Invest. 90:42–51. 10.1172/JCI115854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reigada D, Lu W, Zhang M, Mitchell CH. 2008. Elevated pressure triggers a physiological release of ATP from the retina: possible role for pannexin hemichannels. Neuroscience 157:396–404. 10.1016/j.neuroscience.2008.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paoletti A, Raza SQ, Voisin L, Law F, Pipoli da Fonseca J, Caillet M, Kroemer G, Perfettini JL. 2012. Multifaceted roles of purinergic receptors in viral infection. Microbes Infect. 14:1278–1283. 10.1016/j.micinf.2012.05.010 [DOI] [PubMed] [Google Scholar]
- 12.Séror C, Melki MT, Subra F, Raza SQ, Bras M, Saidi H, Nardacci R, Voisin L, Paoletti A, Law F, Martins I, Amendola A, Abdul-Sater AA, Ciccosanti F, Delelis O, Niedergang F, Thierry S, Said-Sadier N, Lamaze C, Metivier D, Estaquier J, Fimia GM, Falasca L, Casetti R, Modjtahedi N, Kanellopoulos J, Mouscadet JF, Ojcius DM, Piacentini M, Gougeon ML, Kroemer G, Perfettini JL. 2011. Extracellular ATP acts on P2Y2 purinergic receptors to facilitate HIV-1 infection. J. Exp. Med. 208:1823–1834. 10.1084/jem.20101805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanco J, Bosch B, Fernandez-Figueras MT, Barretina J, Clotet B, Este JA. 2004. High level of coreceptor-independent HIV transfer induced by contacts between primary CD4 T cells. J. Biol. Chem. 279:51305–51314. 10.1074/jbc.M408547200 [DOI] [PubMed] [Google Scholar]
- 14.Chen P, Hubner W, Spinelli MA, Chen BK. 2007. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J. Virol. 81:12582–12595. 10.1128/JVI.00381-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. 2004. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199:283–293. 10.1084/jem.20030648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hubner W, McNerney GP, Chen P, Dale BM, Gordon RE, Chuang FY, Li XD, Asmuth DM, Huser T, Chen BK. 2009. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 323:1743–1747. 10.1126/science.1167525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Durham ND, Yewdall AW, Chen P, Lee R, Zony C, Robinson JE, Chen BK. 2012. Neutralization resistance of virological synapse-mediated HIV-1 infection is regulated by the gp41 cytoplasmic tail. J. Virol. 86:7484–7495. 10.1128/JVI.00230-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, Baltimore D. 2011. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 477:95–98. 10.1038/nature10347 [DOI] [PubMed] [Google Scholar]
- 19.Agosto LM, Zhong P, Munro J, Mothes W. 2014. Highly active antiretroviral therapies are effective against HIV-1 cell-to-cell transmission. PLoS Pathog. 10:e1003982. 10.1371/journal.ppat.1003982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jolly C, Mitar I, Sattentau QJ. 2007. Adhesion molecule interactions facilitate human immunodeficiency virus type 1-induced virological synapse formation between T cells. J. Virol. 81:13916–13921. 10.1128/JVI.01585-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hazleton JE, Berman JW, Eugenin EA. 2012. Purinergic receptors are required for HIV-1 infection of primary human macrophages. J. Immunol. 188:4488–4495. 10.4049/jimmunol.1102482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orellana JA, Velasquez S, Williams DW, Saez JC, Berman JW, Eugenin EA. 2013. Pannexin1 hemichannels are critical for HIV infection of human primary CD4+ T lymphocytes. J. Leukoc. Biol. 94:399–407. 10.1189/jlb.0512249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, Baltimore D. 1999. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10:661–671. 10.1016/S1074-7613(00)80065-5 [DOI] [PubMed] [Google Scholar]
- 24.Li M, Gao F, Mascola JR, Stamatatos L, Polonis VR, Koutsoukos M, Voss G, Goepfert P, Gilbert P, Greene KM, Bilska M, Kothe DL, Salazar-Gonzalez JF, Wei X, Decker JM, Hahn BH, Montefiori DC. 2005. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 79:10108–10125. 10.1128/JVI.79.16.10108-10125.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dale BM, McNerney GP, Thompson DL, Hubner W, de Los Reyes K, Chuang FY, Huser T, Chen BK. 2011. Cell-to-cell transfer of HIV-1 via virological synapses leads to endosomal virion maturation that activates viral membrane fusion. Cell Host Microbe 10:551–562. 10.1016/j.chom.2011.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koo BK, Stange DE, Sato T, Karthaus W, Farin HF, Huch M, van Es JH, Clevers H. 2012. Controlled gene expression in primary Lgr5 organoid cultures. Nat. Methods 9:81–83. 10.1038/nchembio.1138 [DOI] [PubMed] [Google Scholar]
- 28.Cavrois M, De Noronha C, Greene WC. 2002. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 20:1151–1154. 10.1038/nbt745 [DOI] [PubMed] [Google Scholar]
- 29.Barat C, Gilbert C, Imbeault M, Tremblay MJ. 2008. Extracellular ATP reduces HIV-1 transfer from immature dendritic cells to CD4+ T lymphocytes. Retrovirology 5:30. 10.1186/1742-4690-5-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blanco J, Barretina J, Henson G, Bridger G, De Clercq E, Clotet B, Este JA. 2000. The CXCR4 antagonist AMD3100 efficiently inhibits cell-surface-expressed human immunodeficiency virus type 1 envelope-induced apoptosis. Antimicrob. Agents Chemother. 44:51–56. 10.1128/AAC.44.1.51-56.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tovar-y-Romo LB, Kolson DL, Bandaru VV, Drewes JL, Graham DR, Haughey NJ. 2013. Adenosine triphosphate released from HIV-infected macrophages regulates glutamatergic tone and dendritic spine density on neurons. J. Neuroimmune Pharmacol. 8:998–1009. 10.1007/s11481-013-9471-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eugenin EA. 2014. Role of connexin/pannexin containing channels in infectious diseases. FEBS Lett. 588:1389–1395. 10.1016/j.febslet.2014.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Velasquez S, Eugenin EA. 2014. Role of pannexin-1 hemichannels and purinergic receptors in the pathogenesis of human diseases. Front. Physiol. 5:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gunosewoyo H, Kassiou M. 2010. P2X purinergic receptor ligands: recently patented compounds. Expert Opin. Ther. Patents 20:625–646. 10.1517/13543771003702424 [DOI] [PubMed] [Google Scholar]
- 35.Gum RJ, Wakefield B, Jarvis MF. 2012. P2X receptor antagonists for pain management: examination of binding and physicochemical properties. Purinergic Signal. 8:41–56. 10.1007/s11302-011-9272-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Clercq E. 1979. Suramin: a potent inhibitor of the reverse transcriptase of RNA tumor viruses. Cancer Lett. 8:9–22. 10.1016/0304-3835(79)90017-X [DOI] [PubMed] [Google Scholar]
- 37.De Clercq E. 1987. Suramin in the treatment of AIDS: mechanism of action. Antiviral Res. 7:1–10. 10.1016/0166-3542(87)90034-9 [DOI] [PubMed] [Google Scholar]
- 38.Riteau N, Baron L, Villeret B, Guillou N, Savigny F, Ryffel B, Rassendren F, Le Bert M, Gombault A, Couillin I. 2012. ATP release and purinergic signaling: a common pathway for particle-mediated inflammasome activation. Cell Death Dis. 3:e403. 10.1038/cddis.2012.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L, Kanellopoulos J, Quesniaux VF, Marchand-Adam S, Crestani B, Ryffel B, Couillin I. 2010. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 182:774–783. 10.1164/rccm.201003-0359OC [DOI] [PubMed] [Google Scholar]
- 40.Wang Q, Imamura R, Motani K, Kushiyama H, Nagata S, Suda T. 2013. Pyroptotic cells externalize eat-me and release find-me signals and are efficiently engulfed by macrophages. Int. Immunol. 25:363–372. 10.1093/intimm/dxs161 [DOI] [PubMed] [Google Scholar]
- 41.Gombault A, Baron L, Couillin I. 2012. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front. Immunol. 3:414. 10.3389/fimmu.2012.00414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eltom S, Stevenson CS, Rastrick J, Dale N, Raemdonck K, Wong S, Catley MC, Belvisi MG, Birrell MA. 2011. P2X7 receptor and caspase 1 activation are central to airway inflammation observed after exposure to tobacco smoke. PLoS One 6:e24097. 10.1371/journal.pone.0024097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franchi L, Kanneganti TD, Dubyak GR, Nunez G. 2007. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J. Biol. Chem. 282:18810–18818. 10.1074/jbc.M610762200 [DOI] [PubMed] [Google Scholar]
- 44.Harder J, Franchi L, Munoz-Planillo R, Park JH, Reimer T, Nunez G. 2009. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-kappa B activation but proceeds independently of TLR signaling and P2X7 receptor. J. Immunol. 183:5823–5829. 10.4049/jimmunol.0900444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jorgensen NR, Husted LB, Skarratt KK, Stokes L, Tofteng CL, Kvist T, Jensen JE, Eiken P, Brixen K, Fuller S, Clifton-Bligh R, Gartland A, Schwarz P, Langdahl BL, Wiley JS. 2012. Single-nucleotide polymorphisms in the P2X7 receptor gene are associated with post-menopausal bone loss and vertebral fractures. Eur. J. Hum. Genet. 20:675–681. 10.1038/ejhg.2011.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, Hebbeler AM, Greene WC. 2010. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 143:789-801. 10.1016/j.cell.2010.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Munoz-Arias I, Greene WC. 2014. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505:509-514. 10.1038/nature12940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ali Z, Laurijssens B, Ostenfeld T, McHugh S, Stylianou A, Scott-Stevens P, Hosking L, Dewit O, Richardson JC, Chen C. 2013. Pharmacokinetic and pharmacodynamic profiling of a P2X7 receptor allosteric modulator GSK1482160 in healthy human subjects. Br. J. Clin. Pharmacol. 75:197–207. 10.1111/j.1365-2125.2012.04320.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stock TC, Bloom BJ, Wei N, Ishaq S, Park W, Wang X, Gupta P, Mebus CA. 2012. Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J. Rheumatol. 39:720–727. 10.3899/jrheum.110874 [DOI] [PubMed] [Google Scholar]
- 50.Elsby R, Fox L, Stresser D, Layton M, Butters C, Sharma P, Smith V, Surry D. 2011. In vitro risk assessment of AZD9056 perpetrating a transporter-mediated drug-drug interaction with methotrexate. Eur. J. Pharm. Sci. 43:41–49. 10.1016/j.ejps.2011.03.006 [DOI] [PubMed] [Google Scholar]
- 51.Keystone EC, Wang MM, Layton M, Hollis S, McInnes IB, D1520C00001 Study Team 2012. Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann. Rheum. Dis. 71:1630–1635. 10.1136/annrheumdis-2011-143578 [DOI] [PubMed] [Google Scholar]
- 52.Wagner MC. 2011. The therapeutic potential of adenosine triphosphate as an immune modulator in the treatment of HIV/AIDS: a combination approach with HAART. Curr. HIV Res. 9:209–222. 10.2174/157016211796320289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sorrell ME, Hauser KF. 2014. Ligand-gated purinergic receptors regulate HIV-1 Tat and morphine related neurotoxicity in primary mouse striatal neuron-glia co-cultures. J. Neuroimmune Pharmacol. 9:233–244. 10.1007/s11481-013-9507-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Damer S, Niebel B, Czeche S, Nickel P, Ardanuy U, Schmalzing G, Rettinger J, Mutschler E, Lambrecht G. 1998. NF279: a novel potent and selective antagonist of P2X receptor-mediated responses. Eur. J. Pharmacol. 350:R5–R6. 10.1016/S0014-2999(98)00316-1 [DOI] [PubMed] [Google Scholar]
- 55.Klapperstuck M, Buttner C, Nickel P, Schmalzing G, Lambrecht G, Markwardt F. 2000. Antagonism by the suramin analogue NF279 on human P2X(1) and P2X(7) receptors. Eur. J. Pharmacol. 387:245–252. 10.1016/S0014-2999(99)00826-2 [DOI] [PubMed] [Google Scholar]
- 56.Rettinger J, Schmalzing G, Damer S, Muller G, Nickel P, Lambrecht G. 2000. The suramin analogue NF279 is a novel and potent antagonist selective for the P2X(1) receptor. Neuropharmacology 39:2044–2053. 10.1016/S0028-3908(00)00022-8 [DOI] [PubMed] [Google Scholar]
- 57.Busch W, Brodt R, Ganser A, Helm EB, Stille W. 1985. Suramin treatment for AIDS. Lancet ii:1247. [DOI] [PubMed] [Google Scholar]
- 58.Kaplan LD, Wolfe PR, Volberding PA, Feorino P, Levy JA, Abrams DI, Kiprov D, Wong R, Kaufman L, Gottlieb MS. 1987. Lack of response to suramin in patients with AIDS and AIDS-related complex. Am. J. Med. 82:615–620. 10.1016/0002-9343(87)90108-2 [DOI] [PubMed] [Google Scholar]
- 59.Hui EK, Nayak DP. 2002. Role of G protein and protein kinase signalling in influenza virus budding in MDCK cells. J. Gen. Virol. 83:3055–3066 [DOI] [PubMed] [Google Scholar]
- 60.Croci R, Tarantino D, Milani M, Pezzullo M, Rohayem J, Bolognesi M, Mastrangelo E. 2014. PPNDS inhibits murine norovirus RNA-dependent RNA-polymerase mimicking two RNA stacking bases. FEBS Lett. 588:1720–1725. 10.1016/j.febslet.2014.03.021 [DOI] [PubMed] [Google Scholar]
- 61.Lambrecht G, Rettinger J, Baumert HG, Czeche S, Damer S, Ganso M, Hildebrandt C, Niebel B, Spatz-Kumbel G, Schmalzing G, Mutschler E. 2000. The novel pyridoxal-5′-phosphate derivative PPNDS potently antagonizes activation of P2X(1) receptors. Eur. J. Pharmacol. 387:R19–R21. 10.1016/S0014-2999(99)00834-1 [DOI] [PubMed] [Google Scholar]
- 62.Suzuki E, Kessler M, Montgomery K, Arai AC. 2004. Divergent effects of the purinoceptor antagonists suramin and pyridoxal-5′-phosphate-6-(2′-naphthylazo-6′-nitro-4′,8′-disulfonate) (PPNDS) on alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. Mol. Pharmacol. 66:1738–1747. 10.1124/mol.104.003038 [DOI] [PubMed] [Google Scholar]
- 63.Wood CR, Hennessey TM. 2003. PPNDS is an agonist, not an antagonist, for the ATP receptor of Paramecium. J. Exp. Biol. 206:627–636. 10.1242/jeb.00105 [DOI] [PubMed] [Google Scholar]
- 64.Soto F, Lambrecht G, Nickel P, Stuhmer W, Busch AE. 1999. Antagonistic properties of the suramin analogue NF023 at heterologously expressed P2X receptors. Neuropharmacology 38:141–149. 10.1016/S0028-3908(98)00158-0 [DOI] [PubMed] [Google Scholar]
- 65.Ziyal R, Ziganshin AU, Nickel P, Ardanuy U, Mutschler E, Lambrecht G, Burnstock G. 1997. Vasoconstrictor responses via P2X-receptors are selectively antagonized by NF023 in rabbit isolated aorta and saphenous artery. Br. J. Pharmacol. 120:954–960. 10.1038/sj.bjp.0700984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Borsani E, Albertini R, Colleoni M, Sacerdote P, Trovato AE, Lonati C, Labanca M, Panerai AE, Rezzani R, Rodella LF. 2008. PPADS, a purinergic antagonist reduces Fos expression at spinal cord level in a mouse model of mononeuropathy. Brain Res. 1199:74–81. 10.1016/j.brainres.2007.12.066 [DOI] [PubMed] [Google Scholar]
- 67.Martucci C, Trovato AE, Costa B, Borsani E, Franchi S, Magnaghi V, Panerai AE, Rodella LF, Valsecchi AE, Sacerdote P, Colleoni M. 2008. The purinergic antagonist PPADS reduces pain related behaviours and interleukin-1 beta, interleukin-6, iNOS and nNOS overproduction in central and peripheral nervous system after peripheral neuropathy in mice. Pain 137:81–95. 10.1016/j.pain.2007.08.017 [DOI] [PubMed] [Google Scholar]