

ABSTRACT
The effects of heightened microbial translocation on B cells during HIV infection are unknown. We examined the in vitro effects of HIV and lipopolysaccharide (LPS) on apoptosis of CD27+ IgD− memory B (mB) cells from healthy controls. In vivo analysis was conducted on a cohort of 82 HIV+ donors and 60 healthy controls. In vitro exposure of peripheral blood mononuclear cells (PBMCs) to LPS and HIV led to mB cell death via the Fas/Fas ligand (FasL) pathway. Plasmacytoid dendritic cells (pDCs) produced FasL in response to HIV via binding to CD4 and chemokine coreceptors. HIV and LPS increased Fas expression on mB cells in PBMCs, which was dependent on the presence of pDCs and monocytes. Furthermore, mB cells purified from PBMCs and pretreated with both HIV and LPS were more sensitive to apoptosis when cocultured with HIV-treated pDCs. Blocking the interferon receptor (IFNR) prevented HIV-stimulated FasL production in pDCs, HIV-plus-LPS-induced Fas expression, and apoptosis of mB cells. In vivo or ex vivo, HIV+ donors have higher levels of plasma LPS, Fas expression on mB cells, and mB cell apoptosis than controls. Correspondingly, in HIV+ donors, but not in controls, a positive correlation was found between plasma FasL and HIV RNA levels and between Fas expression on mB cells and plasma LPS levels. This work reveals a novel mechanism of mB cell apoptosis mediated by LPS and HIV through the Fas/FasL pathway, with key involvement of pDCs and type I IFN, suggesting a role for microbial translocation in HIV pathogenesis.
IMPORTANCE This study demonstrates that lipopolysaccharide (LPS) and type I interferon (IFN) play an important role in memory B cell apoptosis in HIV infection. It reveals a previously unrecognized role of microbial translocation in HIV pathogenesis.
INTRODUCTION
Perturbations of B lymphocytes in human immunodeficiency virus (HIV) disease include memory B (mB) cell depletion, polyclonal B cell activation, and impaired antibody (Ab) responses after vaccination (1–3). Although some of these deficiencies stem from a lack of CD4+ T cell help, intrinsic B cell dysfunction also appears to be present in HIV disease (4).
Spontaneous B cell apoptosis ex vivo has been observed in both acute and chronic HIV infection (5, 6). Memory B cell depletion may stem from the increased susceptibility of these cells to apoptosis in HIV disease. The tumor necrosis factor alpha (TNF-α)/tumor necrosis factor receptor (TNFR), TRAIL/DR5, Fas/Fas ligand (FasL), and Foxo3a cell death signaling pathways have been reported to play a role in HIV pathogenesis (7–10). Plasmacytoid dendritic cells (pDCs) have been reported to produce TRAIL in response to HIV (mediated through type I interferon [IFN]) and play a role in T cell depletion in HIV infection (11). Additionally, there is evidence of a role for the Fas/FasL signaling pathway in B cell apoptosis in HIV disease (8). Naive B (nB) cells express low levels of Fas, whereas activated mB cells express high levels of Fas (12). However, FasL induction is much more restricted. Previous studies showed that opsonized zymosan, CD4 cross-linking, or HIV in vitro could induce FasL on monocytes or macrophages (13–15). More importantly, inhibition of the Fas/FasL pathway by an anti-FasL Ab (RNOK203) resulted in decreased B cell apoptosis and increased Ab production against viral proteins in simian immunodeficiency virus (SIV)-infected macaques in vivo (16). Furthermore, it was reported that Fas surface expression on B cells from HIV+ donors was related to exogenous FasL-induced B cell apoptosis in vitro (8), suggesting that the Fas/FasL signaling pathway is critical for mB cell apoptosis in HIV infection.
Our recent studies indicated that increased microbial translocation from the damaged gut in chronically HIV-infected patients is at least partially responsible for the chronic immune dysregulation observed in HIV-infected patients (17, 18). Toll-like receptors (TLRs), which recognize a wide variety of microbe-associated molecular patterns (MAMPs), play an important role in B cell homeostasis. Microbial products, such as TLR ligands, can maintain mB cell numbers and recall Ab titers in the absence of protein antigens (Ags) in healthy individuals (19). Although TLR ligands released from the gut have long-term effects on the humoral system, they do not appear to maintain mB cell numbers and functions in HIV-infected subjects as they do in healthy subjects (1, 2, 20). However, B cells from HIV-infected subjects are still polyclonally activated and are able to produce auto-Abs during chronic infection (20). Therefore, B cell dysfunction does not result solely from repeated stimulation by microbial products and subsequent desensitization. We considered the possibility that increased MT in the context of HIV infection might have deleterious effects on B cell function and survival. Given the lack of a direct association between the increased concentrations of microbial products in serum and impaired B cell responses in other chronic diseases associated with heightened MT (e.g., inflammatory bowel disease or chronic hepatitis infection) (21–24), we asked if the concurrent exposure of B cells to microbial products and HIV might contribute to B cell dysfunction during chronic HIV infection. We found that indeed, lipopolysaccharide (LPS) and HIV synergistically induced mB cell apoptosis in a manner that was dependent on pDCs through the Fas/FasL signaling pathway.
MATERIALS AND METHODS
Study subjects.
In the present study, 60 healthy controls, 39 HIV+ antiretroviral therapy (ART)-naïve (ART−) patients, and 43 HIV+ ART-treated (ART+) subjects were studied. In order to investigate the effects of HIV and LPS on memory B cell apoptosis, 21% of the ART-treated subjects were viremic. The median CD4 T cell counts and plasma levels of HIV RNA in the ART-treated subjects were 386 cells/μl (interquartile range [IQR], 132 to 607 cells/μl) and 455 cells/μl (IQR, 323 to 565 cells/μl), respectively; in the ART-naive subjects, the median CD4 T cell counts and plasma levels of HIV RNA were 48 copies/ml (IQR, 48 to 38,425 copies/ml) and 29,886 cells/μl (IQR, 9,215 to 79,701 copies/ml), respectively.
Ethics statement.
These studies were approved by the Institutional Review Boards for Human Research (IRBs) at the Medical University of South Carolina, Case Western Reserve University, and University Hospitals Case Medical Center of Cleveland. All subjects were adults and provided written informed consent.
Reagents.
HIV (CL.4/SUPT1, lot number P4509, MN, X4 tropic; CL.30/SUPT1, lot number P4101, ADA-M, R5 tropic) was provided by NCI, NIH. HIV-1 gp120 (MN, X4 tropic) was obtained through the Reference Reagent Program, Division of AIDS, NIAID, NIH. LPS was purchased from Sigma (St. Louis, MO), and the CXCR4 inhibitor (CXCR4inh) (AMD3100) was obtained from Sigma. A neutralizing Ab against Fas was obtained from Millipore (Billerica, MA), a neutralizing Ab against FasL was obtained from MBL (Woburn, MA), a neutralizing Ab against IFN receptor was obtained from PBL (Piscataway, NJ), and neutralizing Abs against TNF-α and TRAIL were obtained from R&D Systems (Minneapolis, MN). The IgG1 isotype Ab was purchased from BD Pharmingen (San Jose, CA). KFL9 and Jurkat cells were purchased from the ATCC (Manassas, VA). The IFN receptor inhibitor was purchased from PBL (Piscataway, NJ), soluble CD4 (sCD4) was obtained from Progenics Pharmaceuticals (New York, NY), and the CCR5 inhibitor (CCR5inh) maraviroc was obtained from Selzentry (Middlesex, United Kingdom).
Cells.
Blood was collected in heparin-coated tubes, and peripheral blood mononuclear cells (PBMCs) were isolated over a Ficoll-Hypaque cushion. The isolated PBMCs were cultured for 30 h in 96-well plates (0.4 × 106 cells/200 μl) in fresh complete medium in the presence of bacterial LPS (20 ng/ml; Escherichia coli O111:B4; Sigma), HIV (150 ng/ml of p24), or both. Fas expression and B cell apoptosis were evaluated by flow cytometry. In some assays, cells were treated with neutralizing Abs (5 μg/ml) against TNF-α; TRAIL; or Fas, FasL, or both or with the isotype Ab 3 h before adding HIV and LPS. mB cells were tested for annexin V binding, or the cells were treated with soluble CD4 (10 μg/ml), inhibitors of CXCR4 (10 μg/ml), and the IFN receptor inhibitor (20 μg/ml) or the isotype Ab (20 μg/ml) 3 h before adding HIV (X4 or R5 tropic). pDCs were tested for FasL expression after 20 h by flow cytometry.
Purified B cells were obtained by negative selection (purity, >97% for B cells; Miltenyi Biotec, Bergisch Gladbach, Germany). pDCs were isolated by negative selection and then positive selection (Diamond pDC isolation kit; purity, >90%; Miltenyi Biotec). Monocyte-depleted, pDC-depleted, or myeloid dendritic cell (MDC)-depleted PBMCs were obtained using CD14 microbeads, BDCA4 microbeads, or BDCA1 and BDCA3 microbeads, respectively (negative selection; depletion purity, >98%; Miltenyi Biotec).
In coculture assays, purified pDCs were cultured with or without HIV (X4; 150 ng/ml of p24) for 20 h, and PBMCs from the same donor were cultured with medium, LPS, HIV, or both for 20 h. B cells were purified from 20-h-cultured PBMCs under different conditions by positive selection using CD19 microbeads (purity, >90%; Miltenyi Biotec). pDCs and B cells were washed with complete medium and cocultured at a ratio of 1:1 for 3 h. In the transwell system, pDCs were added to the top well, and B cells were added to the bottom well. Annexin V binding among mB cells was measured by flow cytometry.
Cell surface and intracellular staining.
For surface staining, Abs were incubated with blood or PBMCs at room temperature for 10 min. After surface staining, the red cells were lysed or the PBMCs were washed and stained intracellularly using Perm/fix reagents (BD) according to the manufacturer's protocol. The cells were constantly cultured at 37°C after cell permeabilization for FasL intracellular staining. After intracellular staining, the cells were immediately analyzed by flow cytometry.
Flow cytometry.
The fluorochrome-labeled monoclonal Abs used in this study included Abs against CD95-phycoerytherin (PE) (BD Pharmingen), IgD-PE-Cy7 (Biolegend, San Diego, CA), CD20-peridinin chlorophyll protein (PerCP) (Miltenyi Biotec), CD19-Pacific Blue, CD27-allophycocyanin (APC) (BD Pharmingen), CD38-fluorescein isothiocyanate (FITC) (BD Pharmingen), FasL-PE (BD Pharmingen), CD123-PerCP-Cy5.5 (Miltenyi Biotec), BDCA2-FITC (Miltenyi Biotec), BDCA1-APC (Miltenyi Biotec), BDCA3-APC (Miltenyi Biotec), CD14-Pacific Blue (BD Pharmingen), CD11c-PE-Cy7 (Biolegend), 7-amino-actinomycin D (7-AAD) (BD), annexin V-FITC (BD Pharmingen), and isotype control Abs (BD Pharmingen). The cells were identified by their forward and side scatter characteristics and were analyzed by flow cytometry on a MACSQuant flow cytometer (Miltenyi Biotec).
Plasma LPS and FasL levels.
Plasma samples were collected into tubes containing EDTA and stored at −80°C until they were thawed once for analysis of LPS and FasL. For LPS analysis, the plasma samples were diluted to 10% with endotoxin-free water, and LPS was quantified using a commercially available Limulus amebocyte assay kit (Lonza Inc., Allendale, NJ) according to the manufacturer's protocol. Plasma FasL levels were quantified using a commercial kit according to the manufacturer's protocol (R&D).
Statistical analysis.
Conventional measurements of central location and dispersion were used to describe the data, and the differences in continuous measurements between the groups were compared by the Mann-Whitney U test (unpaired) or Wilcoxon matched-paired signed-rank test (paired). To explore associations between pairs of continuous variables, Spearman's rank correlation was used. Comparison analysis was performed using SPSS software (version 16.01). All tests were 2 sided, and a P value of ≤0.05 was considered to indicate statistical significance.
RESULTS
HIV and LPS synergistically induce CD27+ IgD− mB cell apoptosis in PBMCs in vitro.
To evaluate the impact of LPS on mB cell apoptosis in HIV infection, PBMCs were pretreated with medium alone or medium supplemented with LPS, HIV (R5 or X4), and HIV plus LPS. Cell apoptosis was assessed by annexin V and 7-AAD staining in fluorescence-activated cell sorter (FACS)-gated memory (CD19+ CD27+ IgD−) and naive (CD19+ CD27− IgD+) B cells (Fig. 1A). Viable cells (dual negative), early apoptotic cells (annexin V positive), and end-stage apoptotic cells and dead cells (dual positive) are shown in Fig. 1A (25). Because annexin V binding is an indicator of both early- and late-stage apoptosis, only the annexin V staining was used to define apoptotic cells in subsequent assays. These data suggest that costimulation with HIV and LPS induces B cell, and especially mB cell, apoptosis. In contrast, cell-derived control microvesicles (150 ng/ml of protein) did not cooperate with LPS to induce CD27+ memory B cell death (data not shown). The control microvesicles were isolated from the supernatants of uninfected cell cultures in a manner identical to that used for virus preparation from infected cells. Moreover, the effect of LPS on mB cell death was dose dependent in vitro. A concentration of LPS (200 pg/ml) comparable to what has been measured in HIV-infected viremic patients (17) was found to have a modest effect on mB cell death. Increasing the LPS concentration to 20 ng/ml had a significant effect on mB cell death; therefore, we used 20 ng/ml to investigate the impact of LPS on mB apoptosis in subsequent assays.
FIG 1.

mB cell apoptosis in PBMCs is induced by HIV and LPS in vitro. PBMCs were cultured with medium alone (med) or medium supplemented with LPS, HIV (R5 or X4), or both HIV and LPS for 30 h. B cell apoptosis was defined by annexin V binding and 7-AAD among nB (CD27− IgD+) and mB (CD27+ IgD−) cells by flow cytometry. (A) Representative dot plots revealing the gating strategy used to assess the frequencies of apoptotic nB and mB cells and the frequencies of early apoptosis (annexin V+ 7-AAD−) and late apoptosis (annexin V+ 7-AAD+) of nB and mB cells from one representative donor. The numbers represent the frequencies of cells in early apoptosis or late apoptosis among nB or mB cells. FSC, forward scatter; SSC, side scatter. (B to E) B cell subset apoptosis in the presence of LPS and HIV (R5) in different culture systems. (F to I) B cell subset apoptosis in the presence of LPS and HIV (X4) in different culture systems. (B and F) Frequencies of nB cell apoptosis induced by various treatments in PBMCs. (C and G) Frequencies of mB cell apoptosis induced by various treatments in PBMCs. (D and H) Frequencies of plasma cell (CD19+ CD27+ CD38+) apoptosis induced by various treatments in PBMCs. (E and I) Frequencies of mB cell apoptosis induced by different treatments in purified B cells. The data are presented as medians. Sample sizes (N) and P values are shown.
To further evaluate the effects of HIV virions and LPS on CD27+ IgD− mB cell apoptosis, PBMCs were cultured in medium, LPS (20 ng/ml), HIV (X4 or R5; 150 ng/ml of p24), or both HIV and LPS for 30 h. A combination of HIV (R5) and LPS caused significant apoptosis of both nB and mB cells, but not plasma cells (Fig. 1B to D). Since mB cell depletion has been extensively observed in HIV infection (26), to clarify the impacts of HIV and LPS on mB cell apoptosis, we purified B cells from PBMCs. Interestingly, mB cell apoptosis induced by HIV plus LPS was observed only in whole PBMCs and not in purified B cells (Fig. 1E). A similar effect on mB cell apoptosis in PBMCs was observed in the combination of HIV (X4) and LPS, but not in purified B cells (Fig. 1F to I), indicating that mB cell apoptosis requires the presence of other non-B cells in PBMCs.
mB cell apoptosis in response to HIV and LPS requires pDCs acting through the Fas/FasL pathway.
pDCs have been implicated in B cell growth and differentiation (27). To investigate whether pDCs are involved in mB cell apoptosis induced by LPS and HIV, pDCs were depleted from PBMCs in the apoptosis assay. Intriguingly, depletion of pDCs abrogated the synergistic effect of HIV (X4) and LPS on mB cell apoptosis (Fig. 2A), indicating that pDCs participated in the regulation of mB cell apoptosis. This point was corroborated by the restoration of mB cell apoptosis after coculturing HIV-plus-LPS-treated B cells with HIV-treated pDCs (Fig. 2B). Cell-to-cell contact is required for this effect (Fig. 2B and C). Furthermore, apoptosis was induced in Fas-expressing Jurkat cells by HIV-treated pDCs (Fig. 2D), raising the possibility of pDC involvement through the Fas/FasL pathway. Indeed, the introduction of a soluble-FasL inhibitor or Fas inhibitor significantly reduced mB cell apoptosis in response to HIV plus LPS (Fig. 2E). Blocking TNF-α and TRAIL with neutralizing Abs also had a significant effect (Fig. 2E). In the current study, we focused on Fas/FasL, the main cell death pathway mediating HIV- and LPS-induced mB cell apoptosis.
FIG 2.

mB cell apoptosis induced by HIV and LPS is mediated by pDCs and the Fas/FasL signaling pathway. (A) Frequencies of mB cell apoptosis induced by treatment with medium alone or medium supplemented with LPS, HIV (X4), or LPS plus HIV in pDC-depleted PBMCs. n = 6. P > 0.05 for a comparison between any two treatments. (B) mB cell apoptosis in a coculture of pDCs and B cells. Purified pDCs were incubated with or without HIV (X4) for 20 h. PBMCs were stimulated with medium alone or medium supplemented with LPS, HIV (X4), or LPS plus HIV for 20 h. Total B cells isolated from PBMCs were cocultured with pDCs at a ratio of 1:1. mB cell apoptosis was examined 3 h after coculture. n = 5. (C) mB cell apoptosis in a transwell system with separate culture of pDCs and B cells. Purified pDCs were incubated with or without HIV (X4) for 20 h. PBMCs were stimulated with medium alone or medium supplemented with LPS, HIV (X4), or LPS plus HIV for 20 h. Total B cells (bottom wells) isolated from PBMCs were separately cocultured with pDCs (top wells) at a ratio of 1:1 in a transwell system to prevent direct cell-to-cell contacts. mB cell apoptosis was examined 3 h after coculture. n = 7. P > 0.05 for comparisons between any two conditions. (D) Cytotoxicity of pDCs on Fas-expressing Jurkat cells. PBMCs were cultured with medium or HIV (X4) for 20 h, and then stimuli were removed. pDCs were isolated and cocultured with Fas-expressing Jurkat cells for 3 h, and the percentage of apoptosis in Jurkat cells (CD4+) was tested by flow cytometry. n = 5. (E) PBMCs were incubated with neutralizing Abs against TNF-α, TRAIL, Fas, FasL, both Fas and FasL, or a control isotype IgG1 for 3 h before adding HIV (X4) and LPS. The frequency of mB cell apoptosis was measured after 20 h of incubation. n = 9. The data are presented as medians.
Next, to verify the contribution of Fas expression to mB apoptosis in response to HIV plus LPS, PBMCs were cultured with medium alone or medium supplemented with LPS, HIV (X4), and HIV plus LPS for 24 h, as shown in Fig. 3A. Fas expression on mB cells was induced by HIV plus LPS compared to medium, HIV, or LPS alone. Moreover, HIV-plus-LPS-induced Fas expression on mB cells was directly associated with HIV-plus-LPS-induced mB apoptosis (Fig. 3B). Indeed, HIV-plus-LPS-activated B cells were more susceptible to apoptosis induced by FasL-expressing KFL9 cells (Fig. 3C). Human monocytes express TLR4 (28) and are supposed to be involved in mB apoptosis via direct interaction between TLR4 and LPS. Depletion of pDCs and monocytes induced a remarkable inhibition of Fas induction on HIV-plus-LPS-treated mB cells (Fig. 3D). Additionally, a neutralizing Ab against interferon receptor (IFNR) prevented Fas induction on mB cells by HIV and LPS (Fig. 3E), and a neutralizing Ab against TNF-α reduced Fas induction on mB cells by HIV and LPS (Fig. 3F), suggesting that HIV-plus-LPS-induced Fas expression on mB cells is driven by multiple pathways and that type I IFN is one of the most important mediators. Furthermore, inhibition of type I IFNR reduced mB cell apoptosis by HIV and LPS (Fig. 3G), confirming that type I IFN is critical for mB cell apoptosis in response to HIV and LPS through the Fas/FasL pathway.
FIG 3.

Fas expression on mB cells is induced by HIV plus LPS via pDCs and monocytes. (A) (Left) Representative dot plots displaying the gating strategy used to assess the percentage of surface Fas expression on mB cells from one representative donor. (Right) Median percentages of Fas expression on mB cells after 24 h of treatment with medium alone or medium supplemented with LPS, HIV (X4), or both HIV and LPS. n = 20. (B) PBMCs from control donors were treated with medium or HIV (X4) plus LPS. The correlation between apoptotic mB cell induction (% annexin V+) and Fas-positive mB cell induction (% Fas+) by treatment with HIV (X4) plus LPS was analyzed after the subtraction of control medium values. n = 15. (C) PBMCs were cultured with medium or HIV (X4) plus LPS for 20 h, cells were washed in medium to remove the stimuli and cocultured with FasL-expressing KFL9 cells, and the percentage of mB cell apoptosis was evaluated. n = 4. (D) pDCs or monocytes were depleted from PBMCs and then treated with medium alone or medium supplemented with LPS, HIV (X4), or both HIV and LPS for 20 h. The percentage of Fas-positive induction on mB cells was analyzed after the subtraction of control medium values. n = 7. (E and F) Impact of type I IFN (n = 8) (E) and TNF-α (n = 7) (F) on Fas induction in mB cells by HIV (X4) plus LPS. PBMCs were first cultured with TLR4 inhibitor (10 μg/ml), IFNR inhibitor (20 μg/ml), TNF-α neutralizing Ab (10 μg/ml), an isotype IgG2a antibody (20 μg/ml), or IgG1 (10 μg/ml) for 3 h and then incubated with medium or HIV (X4; 150 ng/ml) plus LPS (20 ng/ml) for another 20 h. The percentage of Fas-positive mB cells was analyzed by flow cytometry. (G) mB cell apoptosis by HIV (R5) and LPS through type I IFN (n = 6). PBMCs were first cultured with isotype control IgG2a antibody (20 μg/ml) or IFNR inhibitor (20 μg/ml) for 3 h and then incubated with medium, HIV (R5; 150 ng/ml), LPS (20 ng/ml), or HIV plus LPS. The data are presented as medians.
To further address the role of pDCs in Fas/FasL-mediated mB cell apoptosis, we first assessed soluble FasL in the PBMC culture supernatants with medium, LPS, HIV (X4), or HIV plus LPS and did not detect any significant changes in soluble-FasL expression in the supernatants from PBMCs cultured under various conditions (Fig. 4A). To further confirm the pDC effect, whole PBMCs were cultured with HIV (X4 or R5 tropic) and HIV gp120 protein (MN, X4 tropic) with or without sCD4, the CXCR4 or CCR5 inhibitors, and the neutralizing Ab against IFNR. FasL intracellular expression in pDCs was examined after 20 h of cultivation. The data showed that HIV (X4 and R5; 150 ng/ml of p24) and HIV gp120 protein (X4; 150 ng/ml) induced intracellular FasL expression in pDCs. sCD4 and CXCR4inh or CCR5inh significantly decreased FasL induction by pDCs in response to HIV (Fig. 4B and C). Furthermore, neutralizing Abs against IFNR inhibited the induction of FasL on pDCs by HIV (Fig. 4B and C). In contrast, there was no significant change in intracellular FasL expression in monocytes or MDCs in response to HIV (data not shown). Direct ex vivo analysis of pDCs, MDCs, and monocytes revealed that HIV-infected subjects displayed increased frequencies of intracellular FasL expression compared to healthy donors only in pDCs (and not in monocytes or MDCs) (Fig. 4D). Moreover, the percentage of FasL+ pDCs was directly related to plasma HIV RNA levels (Fig. 4E). These results suggest that Fas/FasL pathway-mediated mB cell apoptosis is dependent on pDCs and IFN.
FIG 4.

HIV activates pDCs to produce FasL. (A) PBMCs were cultured with medium, LPS, HIV (X4), or LPS plus HIV for 24 h. FasL production in the supernatants was measured by enzyme-linked immunosorbent assay (ELISA) in vitro. n = 5. P > 0.05 for a comparison between any two treatments. (B) Representative dot plots displaying the gating strategy used to assess the percentage of FasL+ in pDCs from one representative donor. PBMCs were first treated with sCD4 (10 μg/ml), IFNR inhibitor (20 μg/ml), CXCR4 inhibitor (AMD3100; 10 μg/ml), or CCR5 inhibitor (10 μg/ml) for 3 h and then incubated with medium, HIV (X4, 150 ng/ml, or R5, 150 ng/ml), or gp120 (150 ng/ml) for an additional 20 h. The intracellular levels of FasL in pDCs (CD123+ BDCA2+) were analyzed by flow cytometry. (C) Median frequencies of FasL+ pDCs and P values between pairs of conditions. (D) The frequencies of intracellular FasL expression were examined in monocytes (CD14+), MDCs (BDCA1/3+ CD11c+), and pDCs (CD123+ BDCA2+) from 4 controls and 5 viremic ART-naive HIV+ donors in fresh peripheral blood samples ex vivo. (E) Correlation between the percentages of FasL+ pDCs and plasma HIV RNA levels. The data are presented as medians.
Collectively, these data suggest that pDCs are essential for the synergistic effect of HIV and LPS on mB cell apoptosis. CD4, the HIV coreceptor, and type I IFN are responsible for the induction of FasL in pDCs.
HIV infection is associated with increases in Fas expression on mB cells, plasma LPS, and apoptosis of mB cells.
To address whether the synergistic effect of HIV and LPS contributes to mB cell apoptosis in vivo, we next examined levels of LPS, Fas/FasL expression, and mB cell apoptosis in plasma of healthy controls and HIV+ donors. High levels of plasma LPS were detected in HIV+ donors, particularly in ART-naive HIV+ donors (Fig. 5A). Accordingly, increased frequencies of mB cell apoptosis in freshly isolated PBMCs were observed in ART-naive HIV+ donors (Fig. 5B). Moreover, Fas expression on mB cells in HIV+ donors, particularly ART-naive HIV+ donors, was higher than that on control cells (Fig. 5C and D). No significant difference in plasma FasL among healthy controls and two groups of HIV+ donors was found (Fig. 5E). These data indicate that HIV infection is associated with increased plasma LPS levels, higher Fas expression on mB cells, and increased mB cell apoptosis.
FIG 5.

High levels of Fas expression on mB cells, plasma LPS, and mB cell apoptosis were found in HIV+ donors in vivo or ex vivo. The data are shown as the median values for three groups: HIV− donors and ART-naive and ART-treated HIV+ donors. (A) Plasma LPS levels (nHIV− = 53, nHIV+ ART+ = 24, and nHIV+ ART− = 39). (B) mB cell apoptosis in freshly isolated PBMCs (nHIV− = 60, nHIV+ ART+ = 43, and nHIV+ ART− = 36). (C) Geometric means of Fas expression on mB cells (nHIV− = 53, nHIV+ ART+ = 24, and nHIV+ ART− = 39). (D) Frequencies of Fas+ mB cells (nHIV− = 53, nHIV+ ART+ = 24, and nHIV+ ART− = 39). (E) Plasma FasL levels (nHIV− = 48, nHIV+ ART− = 32, and nHIV+ ART+ = 32).
Fas expression on mB cells positively correlates with plasma LPS levels, and plasma FasL levels are highly associated with plasma HIV RNA levels among HIV+ donors.
To further investigate the interactions between LPS, plasma HIV RNA, and mB cell apoptosis in vivo, we quantified each of these parameters in healthy controls and HIV+ donors, followed by correlation tests. The results revealed that there was a significant correlation between the plasma LPS levels and Fas expression on mB cells (r = 0.38; P = 0.003) (Fig. 6A) in HIV+ donors but not in HIV− controls (r = −0.24; P = 0.13) (Fig. 6B). There was no positive relationship between plasma LPS levels and plasma FasL levels in HIV− and HIV+ donors (Fig. 6C and D). In contrast, a significant correlation was obtained between plasma HIV RNA levels and plasma soluble-FasL levels in HIV+ donors (r = 0.56; P < 0.0001) (Fig. 6E). A correlation between Fas expression on mB cells and plasma HIV RNA levels was noted (r = 0.33; P = 0.01) (Fig. 6F). These results suggest that in HIV+ donors, Fas surface expression on mB cells might be partially induced by heightened levels of LPS and HIV replication, whereas FasL production is mediated by HIV infection per se.
FIG 6.

Fas/FasL expression was associated with plasma LPS and HIV RNA levels in HIV+ donors in vivo or ex vivo. (A and B) Correlation analysis between plasma LPS levels and Fas expression on mB cells in HIV+ donors (nHIV+ ART− = 38 and nHIV+ ART+ = 22) (A) and HIV− donors (nHIV− = 53) (B). (C and D) Correlation analysis between plasma LPS levels and FasL in HIV− donors (nHIV− = 38) (C) and HIV+ donors (nHIV+ ART− = 27 and nHIV+ ART+ = 14) (D). (E and F) Correlation analysis between plasma HIV RNA levels and plasma FasL levels (nHIV+ ART− = 30 and nHIV+ ART+ = 16) (E) or between plasma levels of HIV RNA and Fas expression on mB cells in HIV+ donors (nHIV+ ART− = 41 and nHIV+ ART+ = 23 (F). The open circles represent ART-naive HIV+ donors, and the solid circles represent ART-treated HIV+ donors.
DISCUSSION
Several mechanisms have been proposed to contribute to mB cell depletion and dysfunction in HIV disease, including impaired CD4 T cell help, direct viral binding to B cells through CD21 (29) or through integrin α4β7 (30), polyclonal B cell activation (3), perturbations in B cell trafficking and differentiation (31), and impaired function of T follicular helper cells (32, 33). B cell dysfunction can lead to loss of the ability to control microbial translocation at mucosal sites due to inadequate levels of mucosal IgA (34). In the present study, we found that HIV and LPS exert a synergistic effect in inducing the apoptosis of CD27+ IgD− mB cells, suggesting that MT could play an important role in mB cell apoptosis during chronic HIV infection.
Microbial TLR agonists are important factors in maintaining normal immune function (19). Sterile conditions cause immunodeficiency in mice (35). However, long-term exposure to microbial TLR ligands (likely also including HIV-derived components serving as TLR7/8 ligands) may induce persistent immune activation and perturbation of B cell function, manifested as hyperimmunoglobulinemia or a reduced ability to produce Ag-specific Abs in HIV disease. However, it is clear that patients with other diseases also characterized by heightened MT can exhibit polyclonal B cell activation in the absence of increasing B cell apoptosis and depletion. Chronic hepatitis B infection is such an example (22, 36). Thus, exposure to TLR ligands alone is not sufficient to result in B cell apoptosis and humoral immunodeficiency, arguing for additional mechanisms of B cell dysfunction in HIV infection. Moreover, B cell depletion and functional impairment have been found in pathogenic but not in nonpathogenic SIV-infected models (37–40). Nonpathogenic animal models also do not exhibit a “leaky” gut or heightened levels of MT (17, 41) during chronic SIV infection. Consistent with these findings, a high level of plasma LPS was found in HIV+ ART− and HIV+ ART+ donors in this study, indicating that mB cells in these patients are subjected to long-term LPS exposure and may be affected through direct or indirect LPS-mediated signaling. Importantly, our study revealed that HIV exposure did not directly cause mB cell apoptosis unless PBMCs were concomitantly exposed to LPS in vitro. This finding demonstrates that mB cell apoptosis and dysfunction are caused by the synergistic interaction between HIV and LPS and indicates the roles of HIV and LPS in modulating non-B cells, most notably pDCs.
It has been demonstrated that pDCs, one major subset of DCs, substantially contribute to immune defense against viral infection and microbial pathogens in vivo. The persistent secretion of type I IFN and TRAIL by pDCs has been implicated in CD4 T cell decline and HIV pathogenesis (11, 42). Although chronic HIV infection is associated with reduced numbers and dysfunction of pDCs (43–45), pDCs are at least partially responsible for the increased production of TRAIL and type I IFN in HIV infection (11, 46, 47). In the current study, soluble CD4 and a coreceptor inhibitor reduced HIV-1 virions or gp120-mediated FasL production in pDCs, indicating a direct correlation between pDCs and mB cell apoptosis during HIV infection. Consistently, HIV infection is associated with increased levels of FasL in pDCs ex vivo (Fig. 4D). The suppression of FasL in pDCs and decreased Fas expression on mB cells caused by an IFN inhibitor in vitro suggest a cascade of mB cell apoptosis via interferon secretion and FasL production from pDCs in HIV infection. Moreover, we did not find that soluble FasL by pDCs could induce mB cell apoptosis in a transwell system (Fig. 2D), implying that direct cell-to-cell communication between pDCs and mB cells is required for mB cell apoptosis. HIV gp120 contains binding regions for CD4 and the HIV coreceptor and has been shown to induce significant FasL expression via cellular receptors (48). Consistent with this information, gp120 induced more FasL expression in pDCs than HIV (Fig. 4B and C). Nevertheless, there was no difference in the plasma FasL levels in controls and patients (Fig. 5E), perhaps because pDCs are a relatively rare cell population. We speculate that CD27+ IgD− mB cells are killed by pDCs in lymphoid tissues rather than in the blood, as lymphoid tissues exhibit heightened levels of HIV and LPS, facilitating a closer interaction between pDCs and B cells in HIV disease. However, we cannot eliminate the possibility that the other cells with elevated FasL expression (e.g., NK cells) in HIV infection mediate mB cell apoptosis through the Fas/FasL pathway in vivo. The concentrations of HIV and LPS used in this study were higher than physiological doses, calling into question this effect in vivo; however, lymph nodes from HIV-infected patients exhibit heightened levels of inflammation and virus replication compared to the periphery (49), and the amounts of virus and LPS in contact with pDCs and mB cells in lymph nodes may be much greater than their concentrations in plasma, suggesting that this effect may contribute to mB cell apoptosis in vivo.
Our results suggest a model to explain humoral immune dysregulation mediated by HIV infection and microbial products (Fig. 7). HIV infection results in the increased apoptosis of gut epithelial cells and the depletion of Th17 cells, which permits MT from the damaged gut (50, 51). As a consequence of MT, TLR ligands stimulate B cells and induce polyclonal activation, as reflected in the increased levels of auto-Abs in HIV infection (52). Our data show that TLR ligands also increase the sensitivity of mB cells to apoptosis through pDCs and IFN. However, HIV infection plays a central role in this model by virtue of its ability to induce apoptotic signals via the persistent production of inflammatory factors, such as IFN and FasL.
FIG 7.
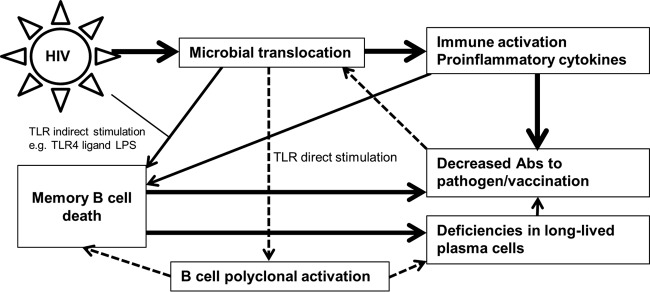
Effects of MT on B cell function in HIV infection. It was hypothesized that microbial TLR ligands (e.g., bacterial DNA) can directly stimulate B cells and induce polyclonal activation, as reflected by increased plasma auto-Ab levels in HIV infection. In addition, TLR ligands play a major role in activating and enhancing mB cell susceptibility to apoptosis. However, HIV is the key inducer of apoptosis-triggering molecules, such as FasL. As a consequence of mB cell death and a lack of CD4 T cell help, B cell Ag-specific Ab production in response to pathogens and vaccination is impaired in chronic HIV infection. Reduced antimicrobial IgA levels in the mucosal sites might also contribute to the “leaky” gut, thereby permitting greater translocation of microbial products into the systemic circulation. Therefore, microbial TLR ligands (e.g., LPS) cooperate with HIV to induce mB cell apoptosis. Released bacterial products lead to a vicious circle of mB cell death and poor mB cell responses in HIV disease. The solid lines represent known directions, and the dashed lines represent predicted directions.
In summary, we have uncovered a novel mechanism for mB cell apoptosis in HIV attributable to the synergistic activities of viral infection and LPS. This synergistic effect was at least in part mediated by the Fas/FasL apoptotic pathway via pDCs. Our findings provide new insight into mechanisms of B cell dysfunction during chronic HIV infection and suggest that blocking MT in HIV-infected patients might be a useful therapeutic strategy to inhibit the perturbation of humoral immunity.
ACKNOWLEDGMENTS
We thank the Bad Boys of Cleveland/Cleveland Immunopathogenesis Consortium (AI076174) for helpful comments and discussions related to this project.
This work was supported by NIH grants AI91526, AI034343, and AI36219; by a STERIS grant; and by the National 12th Five-Year Plan in China (2012ZX10001-003 and 2012ZX10001-006), the Beijing Municipal Science and Technology Commission (D09050703590901), and the Beijing Key Laboratory (BZ0089). This study utilized facilities and resources of the Center for Oral Health Research (COHR) at Medical University of South Carolina, which is supported by National Institute of General Medicine grant P30GM103331.
Footnotes
Published ahead of print 23 July 2014
REFERENCES
- 1.Malaspina A, Moir S, Orsega SM, Vasquez J, Miller NJ, Donoghue ET, Kottilil S, Gezmu M, Follmann D, Vodeiko GM, Levandowski RA, Mican JM, Fauci AS. 2005. Compromised B cell responses to influenza vaccination in HIV-infected individuals. J. Infect. Dis. 191:1442–1450. 10.1086/429298 [DOI] [PubMed] [Google Scholar]
- 2.Titanji K, De Milito A, Cagigi A, Thorstensson R, Grutzmeier S, Atlas A, Hejdeman B, Kroon FP, Lopalco L, Nilsson A, Chiodi F. 2006. Loss of memory B cells impairs maintenance of long-term serologic memory during HIV-1 infection. Blood 108:1580–1587. 10.1182/blood-2005-11-013383 [DOI] [PubMed] [Google Scholar]
- 3.Levesque MC, Moody MA, Hwang KK, Marshall DJ, Whitesides JF, Amos JD, Gurley TC, Allgood S, Haynes BB, Vandergrift NA, Plonk S, Parker DC, Cohen MS, Tomaras GD, Goepfert PA, Shaw GM, Schmitz JE, Eron JJ, Shaheen NJ, Hicks CB, Liao HX, Markowitz M, Kelsoe G, Margolis DM, Haynes BF. 2009. Polyclonal B cell differentiation and loss of gastrointestinal tract germinal centers in the earliest stages of HIV-1 infection. PLoS Med. 6:e1000107. 10.1371/journal.pmed.1000107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malaspina A, Moir S, Kottilil S, Hallahan CW, Ehler LA, Liu S, Planta MA, Chun TW, Fauci AS. 2003. Deleterious effect of HIV-1 plasma viremia on B cell costimulatory function. J. Immunol. 170:5965–5972. 10.4049/jimmunol.170.12.5965 [DOI] [PubMed] [Google Scholar]
- 5.Samuelsson A, Brostrom C, van Dijk N, Sonnerborg A, Chiodi F. 1997. Apoptosis of CD4+ and CD19+ cells during human immunodeficiency virus type 1 infection—correlation with clinical progression, viral load, and loss of humoral immunity. Virology 238:180–188. 10.1006/viro.1997.8790 [DOI] [PubMed] [Google Scholar]
- 6.Titanji K, Chiodi F, Bellocco R, Schepis D, Osorio L, Tassandin C, Tambussi G, Grutzmeier S, Lopalco L, De Milito A. 2005. Primary HIV-1 infection sets the stage for important B lymphocyte dysfunctions. AIDS 19:1947–1955. 10.1097/01.aids.0000191231.54170.89 [DOI] [PubMed] [Google Scholar]
- 7.de Oliveira Pinto LM, Garcia S, Lecoeur H, Rapp C, Gougeon ML. 2002. Increased sensitivity of T lymphocytes to tumor necrosis factor receptor 1 (TNFR1)- and TNFR2-mediated apoptosis in HIV infection: relation to expression of Bcl-2 and active caspase-8 and caspase-3. Blood 99:1666–1675. 10.1182/blood.V99.5.1666 [DOI] [PubMed] [Google Scholar]
- 8.Moir S, Malaspina A, Pickeral OK, Donoghue ET, Vasquez J, Miller NJ, Krishnan SR, Planta MA, Turney JF, Justement JS, Kottilil S, Dybul M, Mican JM, Kovacs C, Chun TW, Birse CE, Fauci AS. 2004. Decreased survival of B cells of HIV-viremic patients mediated by altered expression of receptors of the TNF superfamily. J. Exp. Med. 200:587–599. 10.1084/jem.20032236 [DOI] [PubMed] [Google Scholar]
- 9.Mueller YM, De Rosa SC, Hutton JA, Witek J, Roederer M, Altman JD, Katsikis PD. 2001. Increased CD95/Fas-induced apoptosis of HIV-specific CD8(+) T cells. Immunity 15:871–882. 10.1016/S1074-7613(01)00246-1 [DOI] [PubMed] [Google Scholar]
- 10.van Grevenynghe J, Cubas RA, Noto A, DaFonseca S, He Z, Peretz Y, Filali-Mouhim A, Dupuy FP, Procopio FA, Chomont N, Balderas RS, Said EA, Boulassel MR, Tremblay CL, Routy JP, Sekaly RP, Haddad EK. 2011. Loss of memory B cells during chronic HIV infection is driven by Foxo3a- and TRAIL-mediated apoptosis. J. Clin. Invest. 121:3877–3888. 10.1172/JCI59211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barblu L, Machmach K, Gras C, Delfraissy JF, Boufassa F, Leal M, Ruiz-Mateos E, Lambotte O, Herbeuval JP, ANRS EP36 HIV Controllers Study Group 2012. Plasmacytoid dendritic cells (pDCs) from HIV controllers produce interferon-alpha and differentiate into functional killer pDCs under HIV activation. J. Infect. Dis. 206:790–801. 10.1093/infdis/jis384 [DOI] [PubMed] [Google Scholar]
- 12.Miyawaki T, Uehara T, Nibu R, Tsuji T, Yachie A, Yonehara S, Taniguchi N. 1992. Differential expression of apoptosis-related Fas antigen on lymphocyte subpopulations in human peripheral blood. J. Immunol. 149:3753–3758 [PubMed] [Google Scholar]
- 13.Badley AD, McElhinny JA, Leibson PJ, Lynch DH, Alderson MR, Paya CV. 1996. Upregulation of Fas ligand expression by human immunodeficiency virus in human macrophages mediates apoptosis of uninfected T lymphocytes. J. Virol. 70:199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown SB, Savill J. 1999. Phagocytosis triggers macrophage release of Fas ligand and induces apoptosis of bystander leukocytes. J. Immunol. 162:480–485 [PubMed] [Google Scholar]
- 15.Oyaizu N, Adachi Y, Hashimoto F, McCloskey TW, Hosaka N, Kayagaki N, Yagita H, Pahwa S. 1997. Monocytes express Fas ligand upon CD4 cross-linking and induce CD4+ T cells apoptosis: a possible mechanism of bystander cell death in HIV infection. J. Immunol. 158:2456–2463 [PubMed] [Google Scholar]
- 16.Salvato MS, Yin CC, Yagita H, Maeda T, Okumura K, Tikhonov I, Pauza CD. 2007. Attenuated disease in SIV-infected macaques treated with a monoclonal antibody against FasL. Clin. Dev. Immunol. 2007:93462. 10.1155/2007/93462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, Kazzaz Z, Bornstein E, Lambotte O, Altmann D, Blazar BR, Rodriguez B, Teixeira-Johnson L, Landay A, Martin JN, Hecht FM, Picker LJ, Lederman MM, Deeks SG, Douek DC. 2006. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12:1365–1371. 10.1038/nm1511 [DOI] [PubMed] [Google Scholar]
- 18.Jiang W, Lederman MM, Hunt P, Sieg SF, Haley K, Rodriguez B, Landay A, Martin J, Sinclair E, Asher AI, Deeks SG, Douek DC, Brenchley JM. 2009. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J. Infect. Dis. 199:1177–1185. 10.1086/597476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bernasconi NL, Traggiai E, Lanzavecchia A. 2002. Maintenance of serological memory by polyclonal activation of human memory B cells. Science 298:2199–2202. 10.1126/science.1076071 [DOI] [PubMed] [Google Scholar]
- 20.Atlas A, Thanh Ha TT, Lindstrom A, Nilsson A, Alaeus A, Chiodi F, De Milito A. 2004. Effects of potent antiretroviral therapy on the immune activation marker soluble CD27 in patients infected with HIV-1 subtypes A-D. J. Med. Virol. 72:345–351. 10.1002/jmv.20006 [DOI] [PubMed] [Google Scholar]
- 21.Melmed GY, Agarwal N, Frenck RW, Ippoliti AF, Ibanez P, Papadakis KA, Simpson P, Barolet-Garcia C, Ward J, Targan SR, Vasiliauskas EA. 2010. Immunosuppression impairs response to pneumococcal polysaccharide vaccination in patients with inflammatory bowel disease. Am. J. Gastroenterol. 105:148–154. 10.1038/ajg.2009.523 [DOI] [PubMed] [Google Scholar]
- 22.Sandler NG, Koh C, Roque A, Eccleston JL, Siegel RB, Demino M, Kleiner DE, Deeks SG, Liang TJ, Heller T, Douek DC. 2011. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology 141:1220–1230. 10.1053/j.gastro.2011.06.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sayyad B, Alavian SM, Najafi F, Mokhtari Azad T, Ari Tabarestani MH, Shirvani M, Behnava B, Afshrian M, Vaziri S, Janbakhsh AR, Mansouri F, Kaviani S. 2012. Efficacy of influenza vaccination in patients with cirrhosis and inactive carriers of hepatitis B virus infection. Iran. Red Crescent Med. J. 14:623–630 [PMC free article] [PubMed] [Google Scholar]
- 24.Salim SY, Soderholm JD. 2011. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm. Bowel Dis. 17:362–381. 10.1002/ibd.21403 [DOI] [PubMed] [Google Scholar]
- 25.Reif RD, Martinez MM, Wang K, Pappas D. 2009. Simultaneous cell capture and induction of apoptosis using an anti-CD95 affinity microdevice. Anal. Bioanal. Chem. 395:787–795. 10.1007/s00216-009-3024-1 [DOI] [PubMed] [Google Scholar]
- 26.De Milito A, Morch C, Sonnerborg A, Chiodi F. 2001. Loss of memory (CD27) B lymphocytes in HIV-1 infection. AIDS 15:957–964. 10.1097/00002030-200105250-00003 [DOI] [PubMed] [Google Scholar]
- 27.Poeck H, Wagner M, Battiany J, Rothenfusser S, Wellisch D, Hornung V, Jahrsdorfer B, Giese T, Endres S, Hartmann G. 2004. Plasmacytoid dendritic cells, antigen, and CpG-C license human B cells for plasma cell differentiation and immunoglobulin production in the absence of T-cell help. Blood 103:3058–3064. 10.1182/blood-2003-08-2972 [DOI] [PubMed] [Google Scholar]
- 28.Lore K, Betts MR, Brenchley JM, Kuruppu J, Khojasteh S, Perfetto S, Roederer M, Seder RA, Koup RA. 2003. Toll-like receptor ligands modulate dendritic cells to augment cytomegalovirus- and HIV-1-specific T cell responses. J. Immunol. 171:4320–4328. 10.4049/jimmunol.171.8.4320 [DOI] [PubMed] [Google Scholar]
- 29.Moir S, Malaspina A, Li Y, Chun TW, Lowe T, Adelsberger J, Baseler M, Ehler LA, Liu S, Davey RT, Jr, Mican JA, Fauci AS. 2000. B cells of HIV-1-infected patients bind virions through CD21-complement interactions and transmit infectious virus to activated T cells. J. Exp. Med. 192:637–646. 10.1084/jem.192.5.637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jelicic K, Cimbro R, Nawaz F, Huang DA, Zheng WX, Yang J, Lempicki RA, Pascuccio M, Van Ryk D, Schwing C, Hiatt J, Okwara N, Wei D, Roby G, David A, Hwang IY, Kehrl JH, Arthos J, Cicala C, Fauci AS. 2013. The HIV-1 envelope protein gp120 impairs B cell proliferation by inducing TGF-beta1 production and FcRL4 expression. Nat. Immunol. 14:1256–1265. 10.1038/ni.2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peruchon S, Chaoul N, Burelout C, Delache B, Brochard P, Laurent P, Cognasse F, Prevot S, Garraud O, Le Grand R, Richard Y. 2009. Tissue-specific B-cell dysfunction and generalized memory B-cell loss during acute SIV infection. PLoS One 4:e5966. 10.1371/journal.pone.0005966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindqvist M, van Lunzen J, Soghoian DZ, Kuhl BD, Ranasinghe S, Kranias G, Flanders MD, Cutler S, Yudanin N, Muller MI, Davis I, Farber D, Hartjen P, Haag F, Alter G, Schulze zur Wiesch J, Streeck H. 2012. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J. Clin. Invest. 122:3271–3280. 10.1172/JCI64314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cubas RA, Mudd JC, Savoye AL, Perreau M, van Grevenynghe J, Metcalf T, Connick E, Meditz A, Freeman GJ, Abesada-Terk G, Jr, Jacobson JM, Brooks AD, Crotty S, Estes JD, Pantaleo G, Lederman MM, Haddad EK. 2013. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat. Med. 19:494–499. 10.1038/nm.3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaoul N, Burelout C, Peruchon S, van Buu BN, Laurent P, Proust A, Raphael M, Garraud O, Le Grand R, Prevot S, Richard Y. 2012. Default in plasma and intestinal IgA responses during acute infection by simian immunodeficiency virus. Retrovirology 9:43. 10.1186/1742-4690-9-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szeri I, Anderlik P, Banos Z, Radnai B. 1976. Decreased cellular immune response of germ-free mice. Acta Microbiol. Acad. Sci. Hung. 23:231–234 [PubMed] [Google Scholar]
- 36.Manns MP, Rambusch EG. 1999. Autoimmunity and extrahepatic manifestations in hepatitis C virus infection. J. Hepatol. 31(Suppl 1):S39–S42 [DOI] [PubMed] [Google Scholar]
- 37.Dykhuizen M, Mitchen JL, Montefiori DC, Thomson J, Acker L, Lardy H, Pauza CD. 1998. Determinants of disease in the simian immunodeficiency virus-infected rhesus macaque: characterizing animals with low antibody responses and rapid progression. J. Gen. Virol. 79:2461–2467 [DOI] [PubMed] [Google Scholar]
- 38.Holznagel E, Norley S, Holzammer S, Coulibaly C, Kurth R. 2002. Immunological changes in simian immunodeficiency virus (SIV(agm))-infected African green monkeys (AGM): expanded cytotoxic T lymphocyte, natural killer and B cell subsets in the natural host of SIV(agm). J. Gen. Virol. 83:631–640 [DOI] [PubMed] [Google Scholar]
- 39.Miller CJ, Genesca M, Abel K, Montefiori D, Forthal D, Bost K, Li J, Favre D, McCune JM. 2007. Antiviral antibodies are necessary for control of simian immunodeficiency virus replication. J. Virol. 81:5024–5035. 10.1128/JVI.02444-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mao H, Lafont BA, Igarashi T, Nishimura Y, Brown C, Hirsch V, Buckler-White A, Sadjadpour R, Martin MA. 2005. CD8+ and CD20+ lymphocytes cooperate to control acute simian immunodeficiency virus/human immunodeficiency virus chimeric virus infections in rhesus monkeys: modulation by major histocompatibility complex genotype. J. Virol. 79:14887–14898. 10.1128/JVI.79.23.14887-14898.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pandrea IV, Gautam R, Ribeiro RM, Brenchley JM, Butler IF, Pattison M, Rasmussen T, Marx PA, Silvestri G, Lackner AA, Perelson AS, Douek DC, Veazey RS, Apetrei C. 2007. Acute loss of intestinal CD4+ T cells is not predictive of simian immunodeficiency virus virulence. J. Immunol. 179:3035–3046. 10.4049/jimmunol.179.5.3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herbeuval JP, Nilsson J, Boasso A, Hardy AW, Kruhlak MJ, Anderson SA, Dolan MJ, Dy M, Andersson J, Shearer GM. 2006. Differential expression of IFN-alpha and TRAIL/DR5 in lymphoid tissue of progressor versus nonprogressor HIV-1-infected patients. Proc. Natl. Acad. Sci. U. S. A. 103:7000–7005. 10.1073/pnas.0600363103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyers JH, Justement JS, Hallahan CW, Blair ET, Sun YA, O'Shea MA, Roby G, Kottilil S, Moir S, Kovacs CM, Chun TW, Fauci AS. 2007. Impact of HIV on cell survival and antiviral activity of plasmacytoid dendritic cells. PLoS One 2:e458. 10.1371/journal.pone.0000458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Desai S, Chaparro A, Liu H, Haslett P, Arheart K, Scott G, Pahwa R, Pahwa S. 2007. Impaired CCR7 expression on plasmacytoid dendritic cells of HIV-infected children and adolescents with immunologic and virologic failure. J. Acquir. Immune Defic. Syndr. 45:501–507. 10.1097/QAI.0b013e3180654811 [DOI] [PubMed] [Google Scholar]
- 45.Zhang L, Jiang Q, Li G, Jeffrey J, Kovalev GI, Su L. 2011. Efficient infection, activation, and impairment of pDCs in the BM and peripheral lymphoid organs during early HIV-1 infection in humanized rag2(-)/(-)gamma C(-)/(-) mice in vivo. Blood 117:6184–6192. 10.1182/blood-2011-01-331173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stary G, Klein I, Kohlhofer S, Koszik F, Scherzer T, Mullauer L, Quendler H, Kohrgruber N, Stingl G. 2009. Plasmacytoid dendritic cells express TRAIL and induce CD4+ T-cell apoptosis in HIV-1 viremic patients. Blood 114:3854–3863. 10.1182/blood-2009-04-217927 [DOI] [PubMed] [Google Scholar]
- 47.O'Brien M, Manches O, Sabado RL, Baranda SJ, Wang Y, Marie I, Rolnitzky L, Markowitz M, Margolis DM, Levy D, Bhardwaj N. 2011. Spatiotemporal trafficking of HIV in human plasmacytoid dendritic cells defines a persistently IFN-alpha-producing and partially matured phenotype. J. Clin. Invest. 121:1088–1101. 10.1172/JCI44960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anand AR, Ganju RK. 2006. HIV-1 gp120-mediated apoptosis of T cells is regulated by the membrane tyrosine phosphatase CD45. J. Biol. Chem. 281:12289–12299. 10.1074/jbc.M511786200 [DOI] [PubMed] [Google Scholar]
- 49.Josefsson L, Palmer S, Faria NR, Lemey P, Casazza J, Ambrozak D, Kearney M, Shao W, Kottilil S, Sneller M, Mellors J, Coffin JM, Maldarelli F. 2013. Single cell analysis of lymph node tissue from HIV-1 infected patients reveals that the majority of CD4+ T-cells contain one HIV-1 DNA molecule. PLoS Pathog. 9:e1003432. 10.1371/journal.ppat.1003432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cecchinato V, Trindade CJ, Laurence A, Heraud JM, Brenchley JM, Ferrari MG, Zaffiri L, Tryniszewska E, Tsai WP, Vaccari M, Parks RW, Venzon D, Douek DC, O'Shea JJ, Franchini G. 2008. Altered balance between Th17 and Th1 cells at mucosal sites predicts AIDS progression in simian immunodeficiency virus-infected macaques. Mucosal Immunol. 1:279–288. 10.1038/mi.2008.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Epple HJ, Schneider T, Troeger H, Kunkel D, Allers K, Moos V, Amasheh M, Loddenkemper C, Fromm M, Zeitz M, Schulzke JD. 2009. Impairment of the intestinal barrier is evident in untreated but absent in suppressively treated HIV-infected patients. Gut 58:220–227. 10.1136/gut.2008.150425 [DOI] [PubMed] [Google Scholar]
- 52.Kuwata T, Nishimura Y, Whitted S, Ourmanov I, Brown CR, Dang Q, Buckler-White A, Iyengar R, Brenchley JM, Hirsch VM. 2009. Association of progressive CD4(+) T cell decline in SIV infection with the induction of autoreactive antibodies. PLoS Pathog. 5:e1000372. 10.1371/journal.ppat.1000372 [DOI] [PMC free article] [PubMed] [Google Scholar]