

ABSTRACT
Genome-wide analysis of adeno-associated virus (AAV) type 2 integration in HeLa cells has shown that wild-type AAV integrates at numerous genomic sites, including AAVS1 on chromosome 19q13.42. Multiple GAGY/C repeats, resembling consensus AAV Rep-binding sites are preferred, whereas rep-deficient AAV vectors (rAAV) regularly show a random integration profile. This study is the first study to analyze wild-type AAV integration in diploid human fibroblasts. Applying high-throughput third-generation PacBio-based DNA sequencing, integration profiles of wild-type AAV and rAAV are compared side by side. Bioinformatic analysis reveals that both wild-type AAV and rAAV prefer open chromatin regions. Although genomic features of AAV integration largely reproduce previous findings, the pattern of integration hot spots differs from that described in HeLa cells before. DNase-Seq data for human fibroblasts and for HeLa cells reveal variant chromatin accessibility at preferred AAV integration hot spots that correlates with variant hot spot preferences. DNase-Seq patterns of these sites in human tissues, including liver, muscle, heart, brain, skin, and embryonic stem cells further underline variant chromatin accessibility. In summary, AAV integration is dependent on cell-type-specific, variant chromatin accessibility leading to random integration profiles for rAAV, whereas wild-type AAV integration sites cluster near GAGY/C repeats.
IMPORTANCE Adeno-associated virus type 2 (AAV) is assumed to establish latency by chromosomal integration of its DNA. This is the first genome-wide analysis of wild-type AAV2 integration in diploid human cells and the first to compare wild-type to recombinant AAV vector integration side by side under identical experimental conditions. Major determinants of wild-type AAV integration represent open chromatin regions with accessible consensus AAV Rep-binding sites. The variant chromatin accessibility of different human tissues or cell types will have impact on vector targeting to be considered during gene therapy.
INTRODUCTION
Adeno-associated viruses (AAV) represent defective, helper-dependent viruses that need to establish latency to ensure persistence in their primate hosts (1). The mechanisms leading to genomic integration were characterized for prototype AAV type 2 (AAV2) that preferentially integrates in the vicinity of a site on human chromosome 19q13.42, called AAVS1 (2). The specificity of wild-type (wt) AAV2 integration into AAVS1 is mediated by the large regulatory proteins, Rep78/68 (3). During helper-dependent productive AAV2 replication in the presence of either adenovirus or herpesvirus, Rep78/68 is required for AAV gene expression and DNA replication initiated at the 145-bp inverted terminal repeats (ITRs). These serve as AAV origins of DNA replication and flank the 4.7-kb single-stranded AAV genome at either end. Rep78 and/or Rep68 expressed from the p5 promoter were shown to bind to a Rep-binding site (RBS) within the AAV-ITRs (4). Rep unwinds the ITR and introduces a single-strand nick at the adjacent terminal resolution site (trs) (5). The AAV-ITRs also serve as cis elements for chromosomal integration (3). DNA sequences homologous to the RBS and a nearby trs element were also found in AAVS1 (6, 7) and, in vitro, ternary complex formation of Rep68 with the AAV-ITR and AAVS1 was shown (8). A 33-bp sequence of AAVS1 spanning the RBS and the trs element was sufficient to mediate AAV integration in vivo (3, 9). Upon AAV2 infection at high multiplicities of infection (MOIs), integration into AAVS1 was detected in up to 20% of infected HeLa cells within the first 4 to 8 h (10, 11). In AAV-infected and subsequently single cell-selected clonal cell lines, up to 80% of AAVS1-specific integration was described previously (12).
The preferred integration of AAV2wt in chromosome 19q13.42 was long considered unique and was viewed as a specifically evolved virus-encoded targeting mechanism. AAV vectors lack the integration-promoting rep gene and therefore only rarely and randomly integrate into the human genome (13, 14). Multiple studies have attempted to exploit the Rep-mediated targeting specificity for chromosome 19q13.42 aiming to direct AAV vectors to AAVS1, an assumed safe harbor for the integration of therapeutic genes (15–18). However, chromosome 19q13.42 is not the only genomic target region for AAV2wt. The first genome-wide survey of AAVwt integration sites revealed that these were distributed over the entire human genome (19). Bioinformatic analysis of AAVwt integration sites compared to those of rep-deficient recombinant AAV vectors (rAAV) demonstrated a highly significant overrepresentation of AAV chromosome junctions in the vicinity of consensus RBS displaying repeated GAGY/C motifs, including the one described for AAVS1. Expression of rep in the presence of rep-deficient rAAV vectors shifted targeting preferences from random integration back to hot spots in the neighborhood of consensus RBS in genomic open chromatin regions (19). Recent high-throughput DNA sequence analysis of AAVwt integration retrieved several orders of magnitude higher numbers of AAV2 integration sites, confirming and further extending our findings (20).
Until now, all AAVwt integration studies were conducted with HeLa cells, a highly proliferative carcinoma cell line. The HeLa cell karyotype is hypertriploid and aneuploid with infinite genomic rearrangements accumulated over the decades in culture, as documented by karyotype analysis (21) and by whole-genome DNA sequence analysis (22, 23). We present here integration profiles in diploid human fibroblasts infected side by side with AAV2wt and rAAV2 vectors, analyzed by third-generation, PacBio-based genomic DNA sequencing.
MATERIALS AND METHODS
Plasmids.
Plasmid pTAV2-0 (24) covers the AAV2 wild-type genome (GenBank accession number AF043303). Plasmid pTR-UF11ΔPvuII was derived from pTR-UF11, which represents a plasmid for a recombinant AAV vector, from which PvuII sites were removed by PflMI and PvuII digestion and religation. This led to disruption of the gfpneo cassette.
Cells.
MRC-5 cells (human fetal lung fibroblasts) obtained from the American Type Culture Collection (ATCC) and HEK 293 cells were grown in Dulbecco modified Eagle medium with GlutaMAX, 4.5 g/liter glucose (MRC-5) or 1 g/liter glucose (HEK 293), and sodium pyruvate (Gibco), supplemented with 10% fetal calf serum, penicillin (100 U/ml), and streptomycin (100 μg/ml).
AAV production, purification, and quantification.
Stocks of AAVwt or rep/cap-deficient rAAV used as a control were produced in HEK 293 cells by cotransfection of pDG and either pTAV2-0 or pTRUF11ΔPvuII, respectively, as described previously (10). AAV-containing freeze-thaw cell supernatants were treated with benzonase (Merck) to remove plasmid DNA originating from the transfection. AAVwt or rAAV were purified on iodixanol discontinuous density gradients, followed by heparin affinity chromatography, as described previously (25). Alternatively, freeze-thaw supernatants were purified by high-pressure liquid chromatography on an ÄKTA purifier using one-step AVB Sepharose affinity chromatography on prepacked HiTrap columns (GE Healthcare), as described previously (26). Highly purified AAV preparations were quantified as genomic particles (gp) by LightCycler-based quantitative PCR (qPCR) as described previously (25). For PCR amplification of rAAV genomes primers specific for the bovine growth hormone-derived poly(A) site in the vector genome were used as described previously (26). Infectious titers of AAVwt were determined by endpoint dilution on adenovirus type 2-infected cells as described previously (27).
Cell infection.
For the analysis of AAV integration, MRC-5 cells at passages 10 to 15 were seeded overnight on 10-cm-diameter dishes and infected with AAVwt or rAAV at 6,000 gp/cell. Cells were incubated for 6 days to allow limited cell proliferation and viral integration.
Isolation and fragmentation of genomic DNA.
High-molecular-weight DNA was extracted by sodium dodecyl sulfate/proteinase K digestion, followed by repeated phenol-chloroform extractions and ethanol precipitation. Genomic DNA equivalent to roughly 105 cells (0.6 μg) was digested with EcoRV, DraI, or PvuII. These enzymes produce blunt ends ready for linker/adapter ligation. Digestion with MfeI, Bsu36I, or NsiI produced sticky ends that were polished with T4 DNA polymerase. Fragmented genomic DNA was purified by extraction with phenol-chloroform and precipitation with ethanol.
Linker-selection-mediated (LSM) PCR.
In order to amplify virus-chromosome junctions a linker-based strategy was applied as described previously (19). Briefly, partially double-stranded linkers were prepared and ligated to the DNA fragments. Virus-chromosome junctions derived from six restriction enzyme digests were amplified from either end of AAVwt or rAAV resulting in 12 PCRs for each virus. One biotin-labeled primer specific for the linker and a second primer either specific for the left end of AAVwt (AAV2p5) or the right end of AAVwt (CAPgsp1) were used. For rAAV the left-end-specific primer bound the cytomegalovirus (CMV)/β-actin hybrid promoter (5′-ACCGTAAGTTATGTAACGCGGAACTCCA) and the right-end-specific primer was located in the bovine growth hormone (BGH)-derived poly(A) site (5′-CAGGACAGCAAGGGGGAGGATTG). One-third of the linker-ligation reaction was used as a template for PCR. Linker-specific products were captured with paramagnetic streptavidin-labeled Dynabeads M280 (Invitrogen) to deplete “AAV-only” amplification products. The nested PCRs were performed with primer pairs incorporating a 10-bp barcode at their 5′ ends, allowing unambiguous identification of the endonuclease used initially to fragment genomic DNA. “P linker nested” and “AAV2p5nested” or “CAPgsp2” were used for AAVwt, as described previously (19). “CMVnested” (5′-TGGGCTATGAACTAATGACCCCGT) or “BGHnested” (5′-AATAGCAGGCATGCTGGGGAGAG) were used for rAAV amplification. The 5′-end barcodes were as follows: PvuII, TCCTAATCCT; EcoRV, TGGTAATGGT; DraI, TCCTGGTAAT; MfeI, TAATCCTCCT; Bsu36I, TGGTCCTAAT; and NsiI, TAATGGTGGT.
Purification of LSM PCR products.
The products of 12 nested PCRs were pooled and then separated on agarose gels, and the size ranges between 300 to 450 bp and 450 to 1,300 bp were excised. Gel slices were equilibrated in 0.3 M sodium acetate (pH 7)–1 mM EDTA for 30 min and frozen at −80°C for 10 min. Samples were centrifuged through a glass wool filter, precipitated with ethanol, and column purified using a DNA agarose extraction kit (EURx [Poland] or Qiagen [Germany]). The DNA concentration of samples was quantified using a Qubit fluorometer (Life Technologies). The fragment size distribution was assayed by Bioanalyzer (Agilent).
Library preparation and single molecule sequencing.
Nested PCR and size fractionation were repeated and suitable samples were pooled to obtain sufficient material. After AMPure bead purification (Agencourt; Beckman Coulter) of the sample pools sequencing libraries were prepared using the PacBio standard 250-bp template prep protocol and C2 chemistry (250 bp to 3 kb) DNA preparation kit (Pacific Biosciences, Menlo Park, CA) according to the manufacturer's guidelines. Briefly, end repair, A-tailing, and ligation of universal hairpin adapters were performed. The library quality was verified using Qubit and Bioanalyzer. The PacBio sequencing primer was annealed and polymerase was added to bind to this complex. The first data collection on the PacBio RS platform was done using C2 chemistry and C2 polymerase on v2 single-molecule real-time (SMRT) cells with 2×45 min movies. One SMRT cell was used for each library. With the sequencer upgrade to PacBio RSII the libraries were resequenced with 1×180 min collecting time on V3-SMRT cells using P4 polymerase.
Determination of integration sites.
From the raw sequencing reads we derived circular consensus sequences (CCS) using PacBio standard software. The following analysis steps were performed both on the raw and CCS reads in order to determine the AAV integration sites. First, we searched the reads for the virus-specific primer sequences allowing up to three errors (insertions, deletions, or mismatches) using the Biostrings R package (28). For reads comprising a valid barcode directly upstream of the primer, we locally aligned the sequence downstream of the primer against the human reference genome (GRCh37/hg19) using Bowtie2 (29). For each read uniquely mapped to a single genomic location (Phred score of ≥20), we estimated the position of the exact integration site by computing an optimal “jumping” alignment of S against V and H, where S was the part of the read flanked by the virus-specific primer on one side and by the part of the read mapped to the human genome by Bowtie2 on the other side; V was the part of the virus genome downstream of the virus-specific primer; H was the part of the human reference genome upstream of the Bowtie2 hit. We split S into two parts—S1S2 = S—and computed the minimum edit distance between S1 and prefixes of V and the minimum edit distance between S2 and suffixes of H using the Needleman-Wunsch algorithm (30). From all splittings of S for which the sum of these two edit distances was minimal, we selected the one with the longest S1. The integration site was then defined to be the start position of the suffix of G with minimum edit distance to the S2 of the selected splitting. By aligning S1 to prefixes of V, we could also derive how many bases were lost during the virus integration. In order to retain only high-quality hits, we discarded all reads mapped with <90% sequence identity or to <20 bp of the human genome. To avoid false-positive sites due to wrong priming, we also required the read to contain at least 10 bp of the virus genome in addition to the virus-specific primer sequence. Note that the same integration event could be detected several times by different reads or by the raw and CCS versions of the same read. Moreover, both the integration position and the virus loss length could be slightly inaccurate because of sequencing errors. For these reasons, we considered all reads to belong to the same integration event, if the differences between their integration positions and their virus loss lengths both were ≤4 bp.
Statistical analysis of integration site distributions.
For each data set we generated a set of random control sites, which account for any bias due to the used restriction enzymes and mappability. For each integration site we determined the distance to the closest restriction site of the respective enzyme and randomly selected 400 genomic positions with the same distance to restriction sites in the genome. For integration sites found by using several restriction enzymes the random sites were generated proportionally. We then generated artificial reads at the selected positions, which emulate the mapping length of the corresponding sequencing reads. The artificial reads were mapped to the human genome with the same criteria as were used for the real sequencing reads, keeping only the uniquely mapped artificial reads. From the obtained sites we randomly picked 100,000 forming the set of random control sites. These sets were used to estimate the expected number of integration sites falling into genomic features. Statistical significances were determined using the Fisher exact test. We used Bowtie (31) to find the location of all consensus Rep-binding sites (RBS) in the human genome. For the calculations presented in Fig. 6C, a set of 5 × 107 random control sites was generated.
FIG 6.

Integration in the vicinity of consensus Rep-binding sites (RBS). (A) AAVwt integration site distribution with reverse (cap-rep) or forward (rep-cap) orientation of the AAVwt genome, in relation to the orientation of GAGC repeats in 1-kb bins around a genomic consensus RBS defined as (GAGC)4, allowing two mismatches. (B) Percentage of AAV integration sites in the vicinity of (GAGY/C)n. Distances from the RBS in intervals from 1 kb up to 50 kb are indicated. (C) Enrichment of AAVwt over random control sites in the vicinity of (GAGC)n. Values are reported as the proportion of AAVwt integration events divided by the proportion of integration events of random controls. Distances from RBS are given in intervals up to 1, 2, 5, 10, 20, and 50 kb, as indicated by black triangles.
Analysis of chromatin states.
We downloaded read alignment data (.bam files) of ChIP-Seq and DNase-Seq studies from the ENCODE Data Coordination Center (https://genome.ucsc.edu/ENCODE). Read densities were measured by RPKM (i.e., reads per kilobase per million mapped reads) values, which we computed by counting the number of sequencing reads falling into a given region and dividing it by the length of the region (in kb) and the total number of 106 mapped reads.
RESULTS
AAV infection of human diploid fibroblasts.
For third-generation PacBio-based, genome-wide AAV integration analysis in human diploid fibroblasts, we aimed to identify the initial AAV integration targets in the absence of cell selection. We have previously shown in HeLa cells that AAVwt integration into AAVS1 on chromosome 19 becomes detectable within a few hours postinfection (p.i.), reaching the peak of integration frequencies at 96 h p.i. (10). These conditions were used in our previous genome-wide analysis of AAV2wt integration sites in HeLa cells (19). Limited cell proliferation was expected to minimize the chances for selection of particular AAV integration sites. Human diploid fibroblasts proliferate more slowly. Therefore, the kinetics of AAV2wt integration into chromosome 19q13.42 (AAVS1) was determined in diploid human fibroblasts (MRC-5) using the AAVS1-specific quantitative PCR, as described previously (10). With high AAV MOIs (6,000 gp/cell), AAVwt integration into AAVS1 reached its peak with 1.700 copies/μg genomic DNA at 6 days p.i. (data not shown). To study the AAVwt integration pattern in human diploid fibroblasts and analyze the contribution of AAV Rep in target site selection, cells were infected in parallel with AAVwt, or with rAAV (6,000 gp/cell, each) and processed as outlined in Fig. 1A.
FIG 1.

Library preparation for third-generation PacBio sequencing of AAV integration sites. (A) Genomic DNA of AAVwt- or rAAV-infected human fibroblast (MRC-5) was digested by endonucleases, as indicated by vertical arrows. Integrated AAV genomes are represented as white and gray-shaded boxes. A linker-selection-mediated (LSM) PCR strategy was used for amplification of AAV-chromosomal junctions, followed by the generation of circular libraries for single molecule sequencing by PacBio third-generation sequencing. (B) Theoretical genome coverage upon genome fragmentation by parallel use of six different endonucleases. The percentage of total genome coverage is represented, assuming AAV integration at a maximal distance of 500 bp or 1 kb from these sites. (C) Total numbers of genomic AAV integration sites retrieved per endonuclease and per viral end.
PacBio DNA sequence analysis.
To allow sufficiently long DNA sequence reads for unambiguous assignment of exact AAV-chromosome junctions, third-generation PacBio-based single-molecule DNA sequencing was applied. The decision for the analysis of integration early after AAV infection had to put up with the retrieval of considerable numbers of free AAV episomes, which likely obscure the analysis. To amplify AAV-chromosome junctions the human genome was fragmented by parallel digests with six different endonucleases, as described previously (19) (Fig. 1A). All endonucleases represented noncutting enzymes for the amplified AAVwt or rAAV parts of the junctions. Assuming a maximal distance of 500 bp between any potential AAV insertion site and the nearest endonuclease restriction site, which represents a rather conservative estimation, a theoretical genome coverage of 91% was achieved (Fig. 1B). To both ends of the amplified PCR products hairpin adapters were ligated (Fig. 1A) since PacBio single-molecule real-time (SMRT) technology relies on continuous replication of single-stranded circular DNA templates, leading to repeated reads of single molecules. Sequence reads displayed AAV integrations in forward and reverse orientations. An unbiased distribution of barcodes was seen that allowed assignment of each endonuclease used initially to fragment the genome (Fig. 1C). The PacBio long DNA read lengths are an advantage for the analysis of viral integration sites. In contrast to second-generation, Illumina sequencing-based short reads, where only the termini of PCR fragments are determined, PacBio reads allow the sequencing through the entire viral and chromosomal parts of the junction. This facilitates the exact determination of the breakpoints and thus leads to high mapping quality on the human genome. PacBio-derived DNA sequence data were collected and analyzed under identical experimental conditions, as outlined in Materials and Methods. In total, about 450,000 reads were retrieved for each data set. Bioinformatic analysis eventually led to 1,457 AAVwt and 1,163 rAAV junctions that fulfilled the criteria for unambiguous mapping to specific chromosomal sites (Fig. 2A).
FIG 2.
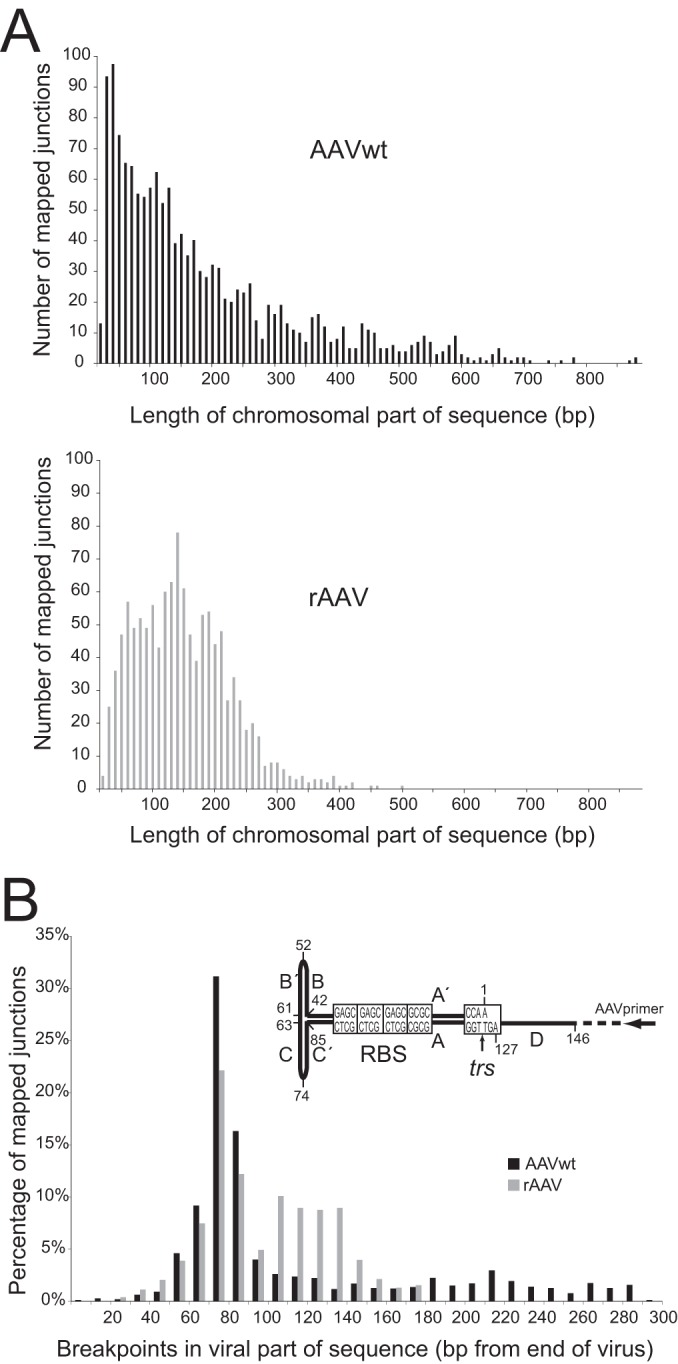
Third-generation PacBio-based DNA sequencing results. (A) Number of reads per given lengths of the chromosomal parts of sequenced junctions, displayed for AAVwt or rAAV, as indicated. (B) Percentage of junctions with breakpoints at indicated distances (in bp) from the end of the AAV genome. Numbers indicate the lengths of the viral genome sequences (in bp) lost during integration. The hairpin structure of one ITR flanking the AAV genome is depicted. Displayed are the nucleotide numbers of the AAV2-ITR, the orientation of the PCR primer (black arrow), the Rep-binding-site (RBS), and the terminal resolution site (trs).
AAV breakpoints.
The primer design for PCR-based retrieval of AAV-chromosome junctions allowed analysis of up to 300 bp at either end of the AAV genome, including ITRs (Fig. 1). Junctions with breakpoints within internal AAV sequences escaped detection. The AAV parts of the junctions revealed that half of the accumulated DNA sequencing reads displayed deletions of the first 60 to 90 bp of the AAV-ITR (Fig. 2B). In both AAVwt and rAAV data sets, the deletions peaked within the AAV-ITR at a distance of 70 to 80 bp from either AAV end. This cluster of AAV breakpoints is located within the first hairpin of the ITR (C/C′ repeat) downstream of the primer-binding site (Fig. 2). The observation is consistent with published data on AAVwt integration in HeLa cells (10, 19, 32). Our results show that the distribution of breakpoints within the ITR is largely identical for AAVwt and rAAV.
Distribution of chromosomal integration sites.
AAVwt and rAAV integration sites were distributed over the entire human genome with integration events in every chromosome (Fig. 3A). In addition, 1% of the AAVwt integration sites and 0.4% of the rAAV integration sites were detected in rRNA. For AAVwt, a series of integration hot spots was detected (Fig. 3A), defined as genomic regions of up to 100 kb length with at least five independent integrations. The list of hot spots included AAVS1 on chromosome 19q13.42 (Table 1). rAAV integration sites displayed a random integration pattern without hot spots, as expected from previous studies (13, 19).
FIG 3.

Chromosomal distribution of AAV integration sites and associated genomic features. (A) Chromosomal distribution of mapped AAVwt or rAAV integration sites represented as ideogram. Integration sites for AAVwt are indicated by black triangles above the chromosomes, and rAAV integration are indicated sites by gray triangles below the chromosomes. Each triangle represents one site. Hot spots, defined as ≥5 independent AAV integration sites within 100 kb, are summed up and displayed by larger triangles, with the total number of sites indicated. Sites located in repetitive rRNA genes could not be assigned to distinct chromosomes and were displayed separately. (B) Enrichment of AAVwt or rAAV integrations compared to random sites in the vicinity of specific genomic features, as depicted. Values are given as the proportion of integration events divided by the proportion of random events. Gene bodies are analyzed without the first 3 kb, and promoters are analyzed from 5 kb upstream to 3 kb downstream of transcription start sites.
TABLE 1.
Hotspots of wild-type AAV2 integration in diploid human fibroblasts (MRC-5)
| Rank | Chromosome | Band | UCSC gene | RBS motifa | No. of sitesb | Range (bp) |
|---|---|---|---|---|---|---|
| 1 | 19 | q13.42 | PPP1R12C | CAGC (GAGC)3 | 37‡ | 11,646 |
| 2 | 1 | q25.3 | RGL1 | (GAGC)3 | 37‡ | 5,916 |
| 3 | 3 | p21.31 | PTH1R | (GAGC)6 | 13‡ | 7,375 |
| 4 | 7 | q32.3 | LOC646329 | –* | 8 | 27,480 |
| 5 | 14 | q23.2 | (GAGC)2 | 7 | 4,568 | |
| 6 | 5 | q31.2 | (GAGC)2 | 6 | 5,744 | |
| 7 | X | q13.1 | GDPD2 | –† | 6 | 362 |
| 8 | 1 | p36.22 | (GAGC)5(CAGC)2 | 5‡ | 1,763 | |
| 9 | 2 | p22.3 | LINC00486 | –† | 5 | 342 |
| 10 | 6 | q12 | EYS | –† | 5 | 59 |
| 11 | 12 | q21.2 | SYT1 | –† | 5 | 27 |
| 12 | 13 | q34 | COL4A1/2 | (GAGC)2 | 5 | 34,859 |
*, No consensus RBS motif could be identified in the 27.4-kb interval of the identified integration sites; †, no consensus RBS motif could be identified in ±10-kb intervals of identified integration sites.
‡, Region corresponding to one of the previously described AAVwt integration hotspots in HeLa cells (20).
Comparison of the integration patterns previously described in aneuploid HeLa cells to those of diploid human fibroblasts showed that preferential AAVwt integration near AAVS1 on chromosome 19q13.42 was maintained but reduced frequencies (2.5%), and another equally frequent hot spot was identified in diploid human fibroblasts on chromosome 1q25.3 (Table 1). The latter had rarely been detected in HeLa cells before (19, 20). In addition, a series of novel hot spots was detected, e.g., on chromosome 7q32.3 and on chromosome 5q31.2 (Table 1), only some of which have been described before at reduced frequencies in AAVwt-infected HeLa cells (20). A number of hot spots from the previous HeLa data sets were not detected in human diploid fibroblasts. The top hot spots in HeLa cells on chromosome 5p13.3 (AAVS2) and on chromosome 3p25.1 (AAVS3) (19, 20) displayed only three and one AAVwt integrations, respectively, in human fibroblasts, despite over 10-fold-higher total junction numbers.
Of all the hot spots found in fibroblasts and in HeLa cells (19), only AAVS1 displays a conserved trs element (5′-GTTGG-3′) for Rep-induced nicking at a defined distance (10 to 30 bp) from the RBS. The expected mean occurrence of the trs motif is frequent (around every 500 bp), and we did not find candidate trs sites near any of the hot spot RBS. In vivo, Rep78/68 was shown to induce DNA damage and single-strand nicking (33) and to bind to key chromatin constituents (34). These interactions may explain in vivo AAV targeting in the absence of an adjacent trs element, as discussed before (19).
The association of AAVwt and rAAV integration sites with specific genomic features was evaluated using sets of 100,000 random controls generated computationally. The data displayed in Fig. 3B show that the patterns of AAVwt and rAAV integration were similar to those described before in HeLa cells (19, 20). Compared to rAAV, the integration frequencies of AAVwt were significantly increased in 10-kb intervals surrounding transcriptional start sites or CpG islands (Fig. 3B).
Association with epigenetic modifications and chromatin status.
The divergent distribution of AAV integration sites in fetal lung-derived diploid human fibroblasts (MRC-5) compared to HeLa cells prompted us to further analyze AAV target site selection. ChIP-Seq and DNase-Seq data for different human fetal lung fibroblast cell lines (AG04450, NHLF, and IMR90) from ENCODE were used to compare chromatin accessibility and transcriptional activity (35). DNase I-hypersensitive sites (HS), H3K4me3 and H3K27ac, each associated with active transcription and open chromatin (36, 37), showed significant enrichment compared to random controls for either AAVwt or rAAV integrations (P < 0.001) (Fig. 4A). AAV integrations were less frequent near marks for posttranslational histone modifications H3K9me3 and H3K27me3, both associated with heterochromatin (Fig. 4A). The data show that both AAVwt and rAAV favor open chromatin regions for integration.
FIG 4.
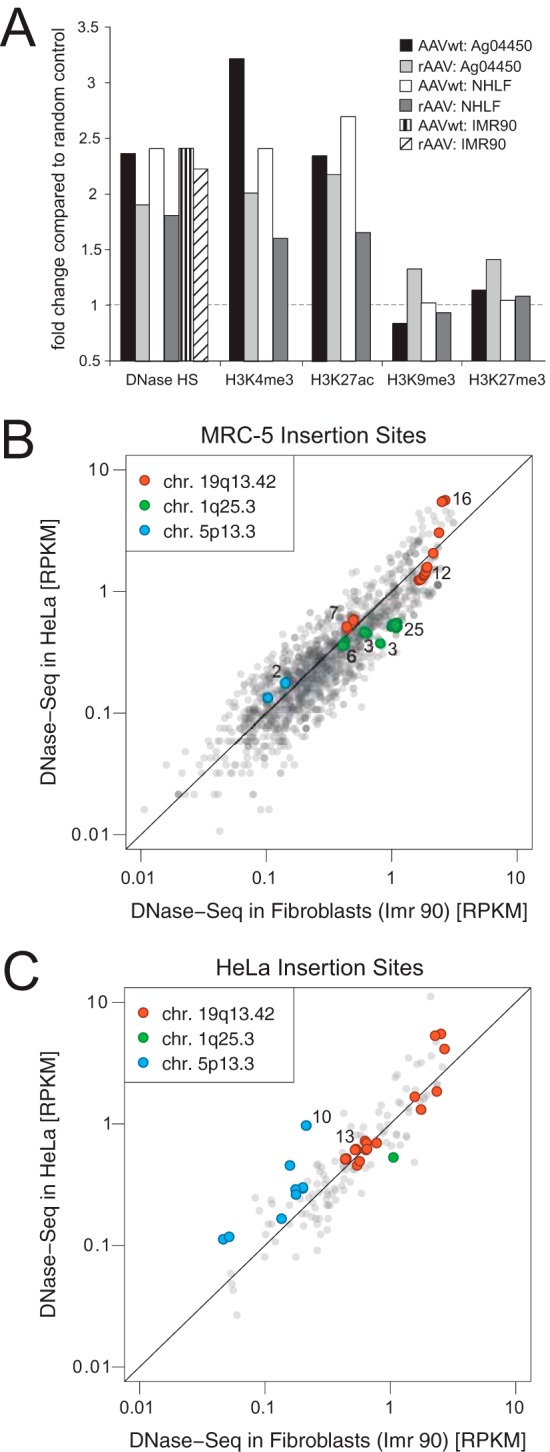
Association of AAV integration near DNase I sites and markers for epigenetic modifications. (A) Enrichment of AAV integration near DNase I cleavage sites and markers for epigenetic modifications associated with open or closed chromatin in human fibroblast cell lines, as indicated. The respective associations with integration sites are compared to random control sites. (B) AAVwt integration sites from human diploid fibroblasts (MRC-5) correlated with DNase-Seq data from HeLa-S3 and from IMR90 fibroblasts. The DNase-Seq read densities (RPKM) are measured in 5-kb windows centered around the AAVwt integration sites. (C) Published AAVwt integration sites from HeLa cells (19) were correlated with DNase-Seq data from HeLa-S3 and from IMR90 fibroblasts. Hot spot integration sites, chr.19q13.42 (AAVS1), and chr.1q25.3 described in fibroblasts (see Table 1) and chr.5p13.3 (AAVS2) described in HeLa cells previously (19) are differentiated by colors, as indicated. The numbers represent the counts of multiple overlapping integration sites.
The distribution of open or closed chromatin regions varies among different cell types. We therefore hypothesized that the divergent AAVwt integration profiles in human diploid fibroblasts and in HeLa cells might be explained by cell-type-specific differences in the accessibility of particular chromatin regions. To analyze this assumption, DNase-Seq data in the surroundings of the preferred AAVwt insertion sites were compared between diploid human fibroblasts and the HeLa cell data set, analyzed previously (19). Indeed, higher read densities for the region around chromosome 5p13.3 were found in HeLa cells compared to human fibroblasts (Fig. 4B and C), an observation consistent with a higher chromatin accessibility of this region in HeLa cells. Conversely, higher read densities for the hot spot regions on chromosomes 1q25.3 were found in fibroblasts, which is consistent with the higher chromatin accessibility of the latter. Scores for sites around the AAVS1 region on chromosome 19q13.42 spread over a broader range but were very comparable in HeLa cells and in fibroblasts, an observation consistent with a high chromatin accessibility in either cell type (Fig. 4B and C).
To further evaluate DNaseSeq data in primary human tissues, specific cells derived thereof, and in human embryonic stem cells, the chromatin accessibilities of the prime hot spot region in HeLa cells and human diploid fibroblasts were compared. As displayed in Fig. 5, chr.19q13.42 (AAVS1) represents a particularly accessible chromosome region in all human tissues and cell types, including embryonic stem cells. The chromatin accessibility of the top hot spots in fibroblasts on chromosome 1q25.3 and 3p21.31 (Table 1) is higher in human diploid cells than in HeLa. It is remarkable that human embryonic stem cells display particularly accessible chromatin at chromosome 3p21.31. Conversely, chromatin accessibilities at the top hot spots in HeLa cells at chr.5p13.3 (AAVS2) and chr3p25.1 (AAVS3) are higher in HeLa cells than in human diploid cells (Fig. 5). Taken together, there are considerable cell-type-dependent variations in chromatin accessibilities at the AAV integration hot spots.
FIG 5.
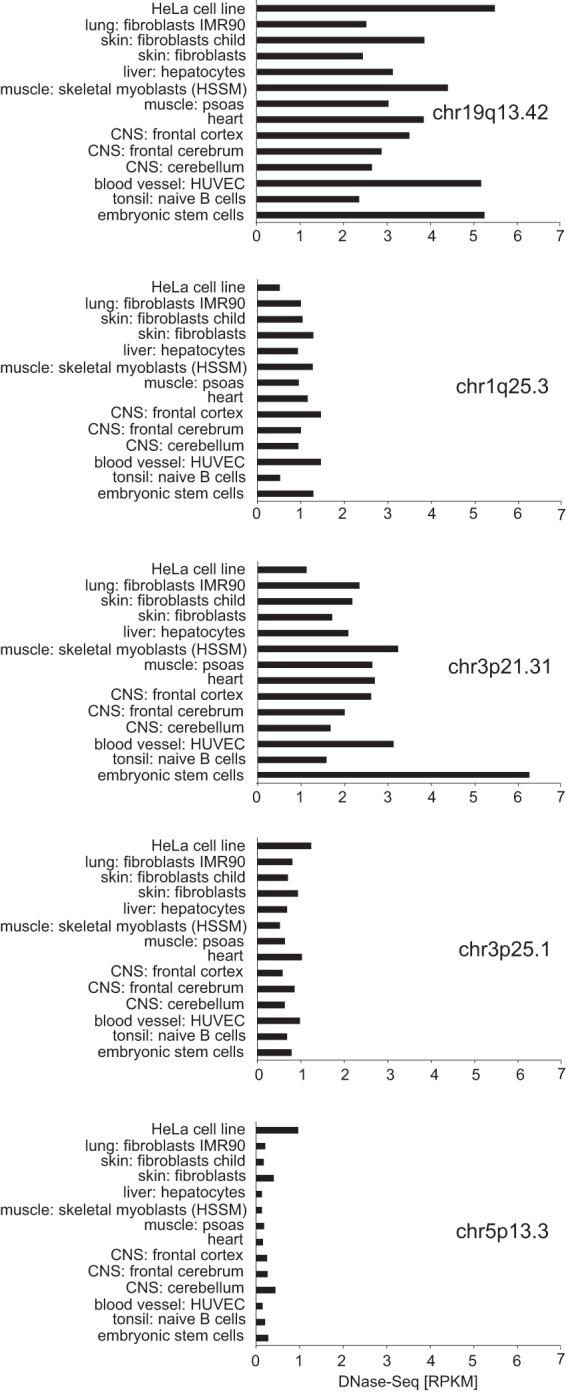
DNase-Seq data of AAV integration hot spots from primary human tissues, isolated cell types, and embryonic stem cells. DNase-Seq data for HeLa cells, diploid human fibroblasts, and primary human tissues and cell types are displayed for the major AAVwt integration hot spots, as indicated. The genomic region of the indicated chromosome band is analyzed in 5-kb intervals. The higher the RPKM score for a particular tissue or cell type, the more accessible is the chromatin. Chromosome designations are detailed in Fig. 4. chr.3p25.1 (AAVS3) represents the updated chr.3p24.3 band described previously (19). chr.3p21.31 represents fibroblast hot spot 3 in Table 1.
Integration of AAVwt in the vicinity of genomic RBS motifs.
In the previous genome-wide analysis of AAVwt integration in HeLa cells, we identified integration at genomic hot spots, allowing the definition of minimal (GAGC)2 and preferred GAGY(GAGC)2 genomic consensus RBS in the vicinity of which AAVwt integrates with a high preference (19). In human diploid fibroblast high-throughput DNA, sequence analysis detects two prime hot spots for AAVwt integration (Table 1). The hot spot on chromosome 19q13.42 (AAVS1) clusters within 12 kb around the described optimal genomic RBS (GAGC)3. The second hot spot lies on chromosome 1q25.3 and clusters within 6 kb, also around an optimal RBS (GAGC)3. The majority of hot spots in human fibroblasts are associated with a nearby consensus RBS (Table 1). By use of DNA mobility shift assays (electrophoretic mobility shift assays), the detected RBS sequences were previously shown to specifically bind to Rep78/68 (19, 38, 39). The integration sites lie preferably upstream and in the immediate vicinity of the RBS, as described previously in HeLa cells (19, 20). To examine the distribution of the entire set of AAVwt integrations in fibroblasts, we analyzed the surroundings of any genomic consensus RBS (GAGC)4 allowing two mismatches. The three prime AAVwt hot spots in human fibroblasts fall in this category and rAAV integrations were rarely found. The enrichment of AAVwt over rAAV integrations ranged between 14- and 44-fold. In addition, AAVwt integration was highly enriched upstream of the RBS and in reverse (cap-rep) orientation, whereas downstream of the RBS, AAV integrated less frequently and predominantly in forward (rep-cap) orientation (Fig. 6A). These findings are in agreement with our previous analysis of the integration site distribution at AAVS1 (11) and with the recent Illumina-based study (20), both in HeLa cells.
To analyze the association of AAV integration to variations of defined minimal (GAGC)2 or optimal (GAGY)2GAGC repeats in human fibroblasts, calculations for additional combinations of GAGC and GAGT motifs were performed. The data set for AAVwt infection was calculated against the data set for rAAV infection and also against a set of random control sites. AAVwt integration sites were significantly enriched in the surrounding of the (GAGC)2 motif (Fig. 6B). (GAGY)2GAGC with at most one mismatch showed the highest Rep-specific integration rate calculated as AAVwt minus rAAV integration rates. The rAAV integration frequency near (GAGC)2 is only marginally enriched over random controls. In the vicinity of (GAGY)2GAGC the integration frequency is undistinguishable from random controls. These data reproduce our findings in HeLa cells (19). Also, in fibroblasts the (GAGC)2 motif represents the minimal sequence requirement for Rep-dependent targeted AAV integration, and (GAGY)2GAGC was found to be the optimal, though less frequent, AAVwt targeting sequence. AAVwt integration rates are further enriched over those of rAAV or of random controls. Additional extensions of the optimal (GAGY)2GAGC motifs were evaluated by probing the vicinity of (GAGC)3+n motifs, as displayed in Fig. 6C. The results demonstrate that the probability of AAVwt integration increases with the number of GAGC repeats and decreases with increasing distance from the respective RBS. The motif (GAGC)5 showed the highest enrichment for AAVwt integration. In a 5-kb interval we found over 500 times more AAVwt integrations (16 sites) than were statistically expected from the random control set (P < 10−37). In summary, AAVwt clearly prefers integration near “(GAGC)” multimers: the more GAGC repeats, the higher the probability of Rep-dependent AAV integration.
DISCUSSION
This study represents the first genome-wide analysis of AAVwt integration in diploid human cells and the first using third-generation, PacBio-based, single-molecule real-time sequencing to study integration of any virus. Unambiguous assignment of numerous AAV-chromosome junctions was achieved. Moreover, side-by-side analysis of human diploid fibroblasts infected with AAVwt, or with rAAV under identical conditions allowed comparative bioinformatic analysis of integration patterns. The results demonstrate that AAVwt integration sites at genomic hot spots in human diploid fibroblasts differed from those described before in aneuploid HeLa cells, correlating preferred hot spots with cell-type-specific variations of chromatin accessibility.
AAV breakpoints.
Comparative analysis of breakpoints within the ITRs of AAVwt and rAAV showed consistent clustering within the first hairpin (C/C′) downstream of the primer-binding site. In a recent study it was speculated that Rep, by dual binding to the RBS and the RBE′ site at the tip of the hairpin, might be responsible for the clustering of AAV breakpoints (32). Here the parallel analysis of rep-negative rAAV and rep-positive AAVwt integration lead to identical breakpoint clusters. Our findings are in line with previous data on AAVwt or rAAV breakpoint clusters in human cells and tissues (10, 13, 14, 19). Therefore, Rep cannot be responsible for the breakpoint clustering within the C/C′ stem-loop of the AAV-ITR. The observed clustering more likely reflects insufficient DNA polymerase reads through complete ITRs or deletions due to recombination. In fact, AAV-chromosome junctions with longer, intact ITRs were detected but were under-represented, as described previously (10, 13, 19, 32).
Fibroblast-specific AAV integration profiles.
In human diploid fibroblasts AAVwt integrations cluster in the surroundings of RBS-like sequence elements. These consist of minimal (GAGC)2 or preferred (GAGY)2GAGC repeats, whereas the integration profile of rAAV vectors is random with respect to consensus RBS motifs. This is in perfect agreement with our previous results in HeLa cells, where the genome-wide association of AAVwt integration in the vicinity of these motifs was first described (19).
In diploid human fibroblasts the AAVS1 hot spot on chromosome 19q13.42 represents one of two prime integration hot spots. However, only 2.5% of all AAVwt integrations target this region, whereas in HeLa cells up to 45% of all integrations were detected there (19, 20). The second, equally frequent hot spot in diploid human fibroblasts lies on chromosome 1q25.3 and a third on chromosome 3p21.31. In HeLa cells these hot spot are underrepresented (20). On the other hand, the previously described prime hot spots in HeLa cells on chromosome 5p13.3 (AAVS2) and on chromosome 3p24.3 (AAVS3) (19) only display single integrations in fibroblasts. Most other hot spots of fibroblasts have not been found in HeLa cells (19, 20). The choice of HeLa cells for analysis has historic reasons. Preferential AAV type 2 integration on chromosome 19 q13.42 was initially described there (2). Unfortunately, many variants of the original HeLa cell line exist, and the karyotype is aneuploid and highly unstable (21). The recent release of the HeLa cell genome disclosed that six copies of the chromosome 5p13.3 region exist, the highest copy number of all regions in the HeLa genome (22, 23). As shown here, the chromatin of this region in HeLa is more accessible than in fibroblasts. Combined with the efficiently Rep-binding consensus RBS described before (19), it is very plausible that the site represents a prime hot spot in HeLa cells (19, 20).
Chromatin status directs AAV integration.
Global analysis of AAV integration site-associated genomic features, DNase-hypersensitive sites, and histone marks for active chromatin (H3K4me3) in human fibroblasts mimic the associations described in HeLa cells before (19, 20). Detailed chromatin analysis of single AAV integration hot spots in either cell type by DNase-Seq revealed that the chromosome 19q13.42 hot spot displays highly accessible chromatin in either cell type. The hot spot encompasses a site immediately upstream of the RBS, where previous in vivo DNase footprint studies had identified open chromatin and active transcription in HeLa cells before (40). These findings served as an early explanation for the preferred chromosome 19q13.42 targeting of AAVwt at the AAVS1 site. In diploid human fibroblasts and in most primary human tissues and cell types isolated therefrom, chromatin accessibility is very similar but is reduced compared to HeLa cells. The high chromatin accessibility at chromosome 19q13.42 in human embryonic stem cells acknowledges previous results that highlighted AAVS1 as a particularly suitable site for gene targeting (41). Conversely, the novel hot spots in diploid human fibroblasts on chromosomes 1q25.3 and 3p21.31 display higher chromatin accessibility in primary human cells than in HeLa cells, likely explaining the preferential AAVwt targeting shown here.
In summary, our findings demonstrate that AAV integration preferences depend on variant chromatin accessibilities of different cell types. Whereas rAAV targets accessible sites in a random fashion, the probability of AAVwt integration increases dramatically with the presence and the copy numbers of RBS-like GAGY/C repeats. As a result, genomic hot spot patterns of AAVwt integration emerge that vary depending on the cell type under study. It is anticipated that these findings have impact on safety assessments during clinical trials, where the rAAV integration profiles may have to be evaluated for each target organ separately.
ACKNOWLEDGMENTS
We thank Stefan Weger and Mario Mietzsch (Charité–Universitätsmedizin Berlin) for helpful discussions and critical readings of the manuscript and Eva Guhl, Melanie Hessler (Charité–Universitätsmedizin Berlin), and Claudia Quedenau (Max-Delbrück-Centrum für Molekulare Medizin) for expert technical assistance.
As part of the Berlin Institute for Medical Systems Biology at the MDC, the research group of W.C. is funded by the Federal Ministry for Education and Research (BMBF) and the Senate of Berlin, Berlin, Germany (BIMSB 0315362A and 0315362C).
Footnotes
Published ahead of print 16 July 2014
REFERENCES
- 1.Muzyczka N, Berns KI. 2001. Parvoviridae: the viruses and their replication, p 2327–2359 In Knipe DM, Howley PM. (ed), Fields virology, vol 2 Lippincott, Philadelphia, PA [Google Scholar]
- 2.Kotin RM, Siniscalco M, Samulski RJ, Zhu XD, Hunter L, Laughlin CA, McLaughlin S, Muzyczka N, Rocchi M, Berns KI. 1990. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. U. S. A. 87:2211–2215. 10.1073/pnas.87.6.2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linden RM, Winocour E, Berns KI. 1996. The recombination signals for adeno-associated virus site-specific integration. Proc. Natl. Acad. Sci. U. S. A. 93:7966–7972. 10.1073/pnas.93.15.7966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snyder RO, Im D-S, Ni T, Xiao X, Samulski RJ, Muzyczka N. 1993. Features of the adeno-associated virus origin involved in substrate recognition by the viral Rep protein. J. Virol. 67:6096–6104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Im D-S, Muzyczka N. 1990. The AAV origin-binding protein Rep68 is an ATP-dependent site-specific endonuclease with helicase activity. Cell 61:447–457. 10.1016/0092-8674(90)90526-K [DOI] [PubMed] [Google Scholar]
- 6.Samulski RJ, Zhu X, Xiao X, Brook JD, Housman DE, Epstein N, Hunter LA. 1991. Targeted integration of adeno-associated virus (AAV) into human chromosome 19. EMBO J. 10:3941–3950 (Erratum, 11:1228, 1992.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kotin RM, Linden RM, Berns KI. 1992. Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. EMBO J. 11:5071–5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weitzman MD, Kyöstiö SRM, Kotin RM, Owens RA. 1994. Adeno-associated virus (AAV) Rep. proteins mediate complex formation between AAV DNA and its integration site in human DNA. Proc. Natl. Acad. Sci. U. S. A. 91:5808–5812. 10.1073/pnas.91.13.5808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meneses P, Berns KI, Winocour E. 2000. DNA sequence motifs which direct adeno-associated virus site-specific integration in a model system. J. Virol. 74:6213–6216. 10.1128/JVI.74.13.6213-6216.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hüser D, Weger S, Heilbronn R. 2002. Kinetics and frequency of adeno-associated virus site-specific integration into human chromosome 19 monitored by quantitative real-time PCR. J. Virol. 76:7554–7559. 10.1128/JVI.76.15.7554-7559.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hüser D, Heilbronn R. 2003. Adeno-associated virus integrates site-specifically into human chromosome 19 in either orientation and with equal kinetics and frequency. J. Gen. Virol. 84:133–137. 10.1099/vir.0.18726-0 [DOI] [PubMed] [Google Scholar]
- 12.McCarty DM, Young SM, Jr, Samulski RJ. 2004. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu. Rev. Genet. 38:819–845. 10.1146/annurev.genet.37.110801.143717 [DOI] [PubMed] [Google Scholar]
- 13.Miller DG, Trobridge GD, Petek LM, Jacobs MA, Kaul R, Russell DW. 2005. Large-scale analysis of adeno-associated virus vector integration sites in normal human cells. J. Virol. 79:11434–11442. 10.1128/JVI.79.17.11434-11442.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaeppel C, Beattie SG, Fronza R, van Logtenstein R, Salmon F, Schmidt S, Wolf S, Nowrouzi A, Glimm H, von Kalle C, Petry H, Gaudet D, Schmidt M. 2013. A largely random AAV integration profile after LPLD gene therapy. Nat. Med. 19:889–891. 10.1038/nm.3230 [DOI] [PubMed] [Google Scholar]
- 15.Recchia A, Perani L, Sartori D, Olgiati C, Mavilio F. 2004. Site-specific integration of functional transgenes into the human genome by adeno/AAV hybrid vectors. Mol. Ther. 10:660–670. 10.1016/j.ymthe.2004.07.003 [DOI] [PubMed] [Google Scholar]
- 16.Zhang C, Cortez NG, Berns KI. 2007. Characterization of a bipartite recombinant adeno-associated viral vector for site-specific integration. Hum. Gene Ther. 18:787–797. 10.1089/hum.2007.056 [DOI] [PubMed] [Google Scholar]
- 17.Wang H, Lieber A. 2006. A helper-dependent capsid-modified adenovirus vector expressing adeno-associated virus rep78 mediates site-specific integration of a 27-kilobase transgene cassette. J. Virol. 80:11699–11709. 10.1128/JVI.00779-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howden SE, Voullaire L, Wardan H, Williamson R, Vadolas J. 2008. Site-specific, Rep.-mediated integration of the intact beta-globin locus in the human erythroleukaemic cell line K562. Gene Ther. 15:1372–1383. 10.1038/gt.2008.84 [DOI] [PubMed] [Google Scholar]
- 19.Hüser D, Gogol-Döring A, Lutter T, Weger S, Winter K, Hammer EM, Cathomen T, Reinert K, Heilbronn R. 2010. Integration preferences of wild-type AAV-2 for consensus rep-binding sites at numerous loci in the human genome. PLoS Pathog. 6:e1000985. 10.1371/journal.ppat.1000985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janovitz T, Klein IA, Oliveira T, Mukherjee P, Nussenzweig MC, Sadelain M, Falck-Pedersen E. 2013. High-throughput sequencing reveals principles of adeno-associated virus serotype 2 integration. J. Virol. 87:8559–8568. 10.1128/JVI.01135-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Macville M, Schrock E, Padilla-Nash H, Keck C, Ghadimi BM, Zimonjic D, Popescu N, Ried T. 1999. Comprehensive and definitive molecular cytogenetic characterization of HeLa cells by spectral karyotyping. Cancer Res. 59:141–150 [PubMed] [Google Scholar]
- 22.Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, Qiu R, Lee C, Shendure J. 2013. The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line. Nature 500:207–211. 10.1038/nature12064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landry JJ, Pyl PT, Rausch T, Zichner T, Tekkedil MM, Stutz AM, Jauch A, Aiyar RS, Pau G, Delhomme N, Gagneur J, Korbel JO, Huber W, Steinmetz LM. 2013. The genomic and transcriptomic landscape of a HeLa cell line. G3 3:1213–1224. 10.1534/g3.113.005777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heilbronn R, Bürkle A, Stephan S, zur Hausen H. 1990. The adeno-associated virus rep gene suppresses herpes simplex virus-induced DNA-amplification. J. Virol. 64:3012–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hüser D, Weger S, Heilbronn R. 2003. Packaging of human chromosome 19-specific adeno-associated virus (AAV) integration sites in AAV virions during AAV wild-type and recombinant AAV vector production. J. Virol. 77:4881–4887. 10.1128/JVI.77.8.4881-4887.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mietzsch M, Grasse S, Zurawski C, Weger S, Bennett A, Agbandje-McKenna M, Muzyczka N, Zolotukhin S, Heilbronn R. 2014. OneBac: platform for scalable and high-titer production of adeno-associated virus serotype 1–12 vectors for gene therapy. Hum. Gene Ther. 25:212–222. 10.1089/hum.2013.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weindler FW, Heilbronn R. 1991. A subset of herpes simplex virus replication genes provides helper functions for productive adeno-associated virus replication. J. Virol. 65:2476–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pages H, Aboyoun P, Gentleman R, DebRoy S. 2007. Biostrings: string objects representing biological sequences, and matching algorithms. http://bioconductor.wustl.edu/bioc/html/Biostrings.html [Google Scholar]
- 29.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Needleman SB, Wunsch CD. 1970. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48:443–453. 10.1016/0022-2836(70)90057-4 [DOI] [PubMed] [Google Scholar]
- 31.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janovitz T, Sadelain M, Falck-Pedersen E. 2014. Adeno-associated virus type 2 preferentially integrates single genome copies with defined breakpoints. Virol. J. 11:15. 10.1186/1743-422X-11-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berthet C, Raj K, Saudan P, Beard P. 2005. How adeno-associated virus Rep78 protein arrests cells completely in S phase. Proc. Natl. Acad. Sci. U. S. A. 102:13634–13639. 10.1073/pnas.0504583102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Costello E, Saudan P, Winocour E, Pizer L, Beard P. 1997. High mobility group chromosomal protein 1 binds to the adeno-associated virus replication protein (Rep) and promotes Rep-mediated site-specific cleavage of DNA, ATPase activity, and transcriptional repression. EMBO J. 16:5943–5954. 10.1093/emboj/16.19.5943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Consortium EP, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129:823–837. 10.1016/j.cell.2007.05.009 [DOI] [PubMed] [Google Scholar]
- 37.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. 2008. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 40:897–903. 10.1038/ng.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCarty DM, Pereira DJ, Zolotukhin I, Zhou X, Ryan JH, Muzyczka N. 1994. Identification of linear DNA sequences that specifically bind the adeno-associated virus Rep protein. J. Virol. 68:4988–4997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiorini JA, Yang L, Safer B, Kotin RM. 1995. Determination of adeno-associated virus Rep68 and Rep78 binging sites by random sequence oligonucleotide selection. J. Virol. 69:7334–7338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamartina S, Sporeno E, Fattori E, Toniatti C. 2000. Characteristics of the adeno-associated virus preintegration site in human chromosome 19: open chromatin conformation and transcription-competent environment. J. Virol. 74:7671–7677. 10.1128/JVI.74.16.7671-7677.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, Meng X, Miller JC, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jaenisch R. 2009. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 27:851–857. 10.1038/nbt.1562 [DOI] [PMC free article] [PubMed] [Google Scholar]