

ABSTRACT
Previous reports showed that raltegravir, a recently approved antiviral compound that targets HIV integrase, can inhibit the nuclease function of human cytomegalovirus (HCMV terminase) in vitro. In this study, subtoxic levels of raltegravir were shown to inhibit the replication of four different herpesviruses, herpes simplex virus 1 (HSV-1), HSV-2, HCMV, and mouse cytomegalovirus, by 30- to 700-fold, depending on the dose and the virus tested. Southern blotting and quantitative PCR revealed that raltegravir inhibits DNA replication of HSV-1 rather than cleavage of viral DNA. A raltegravir-resistant HSV-1 mutant was generated by repeated passage in the presence of 200 μM raltegravir. The genomic sequence of the resistant virus, designated clone 7, contained mutations in 16 open reading frames. Of these, the mutations F198S in unique long region 15 (UL15; encoding the large terminase subunit), A374V in UL32 (required for DNA cleavage and packaging), V296I in UL42 (encoding the DNA polymerase accessory factor), and A224S in UL54 (encoding ICP27, an important transcriptional regulator) were introduced independently into the wild-type HSV-1(F) genome, and the recombinant viruses were tested for raltegravir resistance. Viruses bearing both the UL15 and UL32 mutations inserted within the genome of the UL42 mutant were also tested. While the UL15, UL32, and UL54 mutant viruses were fully susceptible to raltegravir, any virus bearing the UL42 mutation was as resistant to raltegravir as clone 7. Overall, these results suggest that raltegravir may be a valuable therapeutic agent against herpesviruses and the antiviral activity targets the DNA polymerase accessory factor rather than the nuclease activity of the terminase.
IMPORTANCE This paper shows that raltegravir, the antiretrovirus drug targeting integrase, is effective against various herpesviruses. Drug resistance mapped to the herpesvirus DNA polymerase accessory factor, which was an unexpected finding.
INTRODUCTION
Herpesviruses cause a number of important diseases in animals and humans, including recurrent skin lesions, blindness, birth defects, transplant rejection, encephalitis, and cancers of the skin and lymphoid tissue. Moreover, herpesvirus infection commonly worsens the clinical course of human immunodeficiency virus (HIV) infection, and HIV type 1 (HIV-1) has frequently been recovered from genital herpes lesions in coinfected individuals (1).
All herpesviruses encode a highly conserved terminase comprising three subunits (1–3). The terminase cleaves concatameric DNA that has accumulated within the nuclei of infected cells into unit-length genomes, docks with the portal vertex of the capsid, and pumps the DNA into the capsid through the hydrolysis of ATP (4, 5). Given its high conservation and importance to replication, the terminase represents an important antiviral drug target (2, 6). The three-dimensional structures of the nuclease domains of terminase subunits encoded by unique long region 89 (UL89) of human cytomegalovirus (HCMV; or human herpesvirus 5) and UL15 of herpes simplex virus 1 (HSV-1) are highly similar and reveal an RNase H/integrase-like fold (7, 8).
Raltegravir (MK-0518; Isentress; Merck & Co. Inc., Whitehouse Station, NJ) is a member of a novel class of antiretroviral drugs that blocks the integration of HIV-1 cDNA into the cellular genome (9, 10). This compound binds an active site within an RNase H-like fold of integrase to prevent the initiation of strand transfer (11–13). Importantly for the hypotheses initiating this study, the nuclease activity of the UL89 protein (pUL89) could be inactivated by raltegravir in vitro (8). As a follow-up to these observations, the current studies were undertaken to determine whether raltegravir could inhibit herpesvirus replication, with the expectation that raltegravir would inhibit HSV and HCMV replication by blocking the nuclease activity of the terminase. Although we did note substantial antiviral activity against herpesviruses, we were surprised to find that raltegravir inhibited DNA replication rather than terminase-mediated viral DNA cleavage and packaging. Moreover, drug resistance to raltegravir mapped to the DNA polymerase accessory factor encoded by HSV-1 UL42. These data suggest a novel therapeutic avenue against dual HIV and herpesvirus infections but suggest that long-term raltegravir treatment may favor the emergence of drug-resistant viruses.
MATERIALS AND METHODS
Cells, viruses, and plasmids.
The cell lines CV1 and FS-2 were purchased from ATCC, and cells of these lines were propagated in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% newborn calf serum, 100 U/ml of penicillin, and 100 μg/ml streptomycin. The cell line M210B4 was purchased from ATCC, and M210B4 cells were propagated in RPMI 1640 medium supplemented with 10% newborn calf serum, 100 U/ml of penicillin, and 100 μg/ml streptomycin. To minimize genetic changes, all viral stocks were maintained as mother pools that were used to seed working stocks. The herpes simplex virus 1 F strain [HSV-1(F)], referred to as the wild type in this study, was from Bernard Roizman (14). Herpes simplex virus 2 strain G was also a gift from Bernard Roizman (14). The mouse cytomegalovirus (MCMV) Smith strain was a gift from Brian D. Rudd (Department of Microbiology and Immunology, Cornell University), and the HCMV Towne strain was a gift from Greg Pari, University of Nevada, Reno, School of Medicine. The HSV-1(F) bacterial artificial chromosome (BAC) was obtained from Y. Kawaguchi, University of Tokyo (15), and plasmid pEP-kan-S, used in BAC mutagenesis, was obtained from Klaus Osterrieder, University of Berlin. The GS1783 bacterial Escherichia coli host strain was obtained from Greg Smith, Northwestern University. Plasmid pCAGGS-nlsCre, expressing Cre recombinase, was from Michael Kotlikoff, Cornell University.
Toxicity testing.
Raltegravir cytotoxicity was determined by a cell proliferation assay according to the manufacturer's protocol (CellTiter 96 AQueous One Solution cell proliferation assay; Promega). Briefly, cells of the CV1, FS-2, and M210B4 cell lines were grown in 24-well plates. At 80% confluence, medium only or medium containing either dimethyl sulfoxide (DMSO) or raltegravir at various concentrations was added. After incubation for 24 h at 37°C, 40 μl of the manufacturer's reagent was added to the cells. The plates were then incubated for 2 h at 37°C, and the absorbance at 490 nm was read with a plate reader (ELX800; Bio-Tex).
Raltegravir treatment.
Four separate experiments were performed to test for the antiherpesvirus activity of raltegravir. First, 1.2 × 106 CV1 cells were seeded into 6-well plates and infected with HSV-1(F) or HSV-2 at a multiplicity of infection (MOI) of 0.01. After virus adsorption and washing, the cells were maintained in medium containing raltegravir at various concentrations (10, 100, 200, or 400 μM) or an equivalent volume of the DMSO carrier. At 24 h postinfection (hpi), the virus within the cells was harvested by three cycles of freezing and thawing, and standard plaque assays were performed on CV1 cells to quantify viral infectivity. Second, 1.2 × 106 FS-2 cells were seeded into 6-well plates and infected with HCMV at an MOI of 0.01. After virus adsorption and washing, raltegravir at various concentrations (10, 100, 200, or 400 μM) or an equivalent volume of DMSO was added. Drug or carrier levels were maintained by replacing the medium every 24 h. At 72 hpi, the virus within the cells was harvested by freezing and thawing three times, and to quantify infectivity a plaque assay was performed as described above but with some modifications. In brief, 1.2 × 106 FS-2 cells were seeded into 6-well plates, infected with 10-fold serially diluted HCMV, and then treated with various concentrations of raltegravir. At 5 days postinfection (dpi), the plaques were counted to quantify the viral infectivity. Third, 1.2 × 106 M210B4 cells were seeded into each well of 6-well plates and infected with MCMV at an MOI of 0.01. After virus adsorption and washing, the cells were maintained in medium containing various concentrations (10, 100, 200, or 400 μM) of raltegravir or the amount of DMSO that served as the carrier. Medium was replaced every 24 h with fresh medium containing similar levels of drug or carrier. At 72 hpi, the virus within the cells was harvested by freezing-thawing three times, and the amount of infectivity was quantified by plaque assay on M210B4 cells as described above but with some modifications. In brief, 1.2 × 106 M210B4 cells were seeded into 6-well plates and infected with 10-fold serially diluted MCMV from cells treated with various concentrations of raltegravir, as indicated in Results. At 5 dpi, the viral infectivity was determined by plaque assay on fresh monolayers of the same cells.
Southern blot assays.
Three 60-mm plates of CV1 cells were infected with HSV-1(F) at an MOI of 5. After virus adsorption and washing, infected cells were maintained in the presence of 200 μM raltegravir or the DMSO carrier. Uninfected cells served as an additional control. At 18 hpi, the medium was removed and the cells were washed in phosphate-buffered saline (PBS), removed by scraping, and pelleted by centrifugation at 1,000 × g for 5 min. The cells were lysed, and total cellular DNA was extracted as described previously (16). Briefly, nuclei were lysed in SDS, followed by treatment with proteinase K and phenol-chloroform extraction. Approximately 10 μg of total DNA was digested with BamHI and electrophoretically separated on 0.8% agarose gels. The DNA fragments were denatured, neutralized, and transferred to a nylon membrane as described previously (17). The bound DNA was UV cross-linked to the nylon membranes and hybridized with the [32P]dCTP-labeled BamHI P fragment of HSV-1(F) DNA. The bound probe was visualized by exposure to X-ray film at −80°C in the presence of an intensifying screen.
qPCR.
Viral genome replication was measured by real-time quantitative PCR (qPCR) as described with some modifications (18). Target primers for UL42 and reference primers for 18S rRNA (listed in Table 1) were used to measure DNA replication. qPCR was carried out with SYBR green (Thermo Scientific, Pittsburgh, PA) according to the manufacturer's directions. Briefly, amplification reactions were performed in a volume of 25 μl with 5 pmol of each primer, 100 ng DNA, and 12.5 μl 2 × SYBR green mix. The thermal cycling conditions included an initial denaturation for 10 min at 95°C and 40 cycles consisting of a denaturation step at 95°C for 15 s, an annealing step at 60°C for 30 s, and an extension step for 30 s at 72°C. Each sample was analyzed in triplicate, and average threshold cycle (CT) values were used for further analysis. Relative amounts of DNA were determined by the 2−ΔΔCT method, which uses the difference of the CT value of the target gene subtracted from the value of 18S rRNA in the raltegravir-treated sample minus this value in the DMSO-treated sample (18).
TABLE 1.
Primers used for qPCR
| Primer name | Primer sequence (orientation 5′ → 3′) |
|---|---|
| 18S forward | CCA GTA AGT GCG GGT CAT AAG C |
| 18S reverse | GCC TCA CTA AAC CAT CCA ATC GG |
| UL42 forward | GCG GTA TCG GCG GTA TTT |
| UL42 reverse | CCC GTC TTA GGT TTC TTT AGG G |
Generation of drug-resistant virus.
HSV-1-infected CV1 cells were maintained in medium containing 200 μM raltegravir. Fresh medium containing the drug of the same concentration was exchanged every 24 h, and progeny virus was harvested after a cytopathic effect (CPE) was evident. These F1 progeny were passaged under the same conditions, and F2 progeny were harvested after the onset of a CPE. This process of infection and collection of progeny in the presence of raltegravir was repeated a total of 10 times. The F10 progeny viruses were plaque purified, grown to a high titer, and tested for raltegravir resistance by plaque assay. Viruses were plaque purified in the presence of raltegravir, and a virus with a high level of resistance was designated clone 7. This virus was passaged five more times in the presence of 200 μM raltegravir, after which its genome was sequenced. The level of drug resistance was then determined by plaque assay in the presence and absence of 200 μM raltegravir.
Pure genomic DNA preparation.
Genomic DNA was extracted from capsids purified from cells infected with the clone 7 mutant as described previously (19). Viral DNA was purified by phenol-chloroform extraction and ethanol precipitation, resuspended in deionized water, and stored at −20°C.
Genome sequencing and alignment of mutant virus.
Genome sequencing was performed by the Cornell University DNA Sequencing and Genotyping Core Facility. Illumina libraries were constructed using 1-μg purified DNA samples according to the manufacturer's protocol. Libraries were sequenced using single-end cluster generation kits and 100-cycle sequencing kits (Illumina) on an Illumina HiSeq2000 sequencing system following the manufacturer's instructions. Base calling and initial data processing were performed using the standard Illumina pipeline. Reads that passed Illumina quality control were aligned to the HSV-1(F) reference genome sequence (GenBank accession number GU734771) using the Burrows-Wheeler aligner (BWA). Unique mapping reads with no more than 2 mismatches were used for single nucleotide polymorphism (SNP) calling. SNPs were called by use of the SAMtools Mpileup command with default options (20, 21).
Virus growth kinetics analysis.
CV1 cells were infected with HSV-1(F) or clone 7 at an MOI of 0.01. At different time points, cell cultures were collected, cell-associated virus was released by 3 freeze-thaw cycles, and virus titers were determined by plaque assay.
Genetic rescue and generation of recombinant viruses.
Mutations in UL15 (F198S), UL32 (A374V), UL42 (V296I), and UL54 (A224S) matching the changes in clone 7 viral DNA were introduced into the HSV-1(F) viral genome by en passant mutagenesis (22). Infectious virus was generated by transfection of BAC DNA into CV1 cells as described previously (15, 23). The primers used were as follows (underlining indicates the mutated codon): 5′ GGG CGG TAC CGC GAC GAT TAT ATC ATC TTT GCC CTG GAG CAC TCT TTT CTC CGC GCG CTC ACG GGC TAG GGA TAA CAG GGT AAT CGA TTT 3′ (forward) and 5′ GGC GAT GTC GGC GGG GGC CGA GCC CGT GAG CGC GCG GAG AAA AGA GTG CTC CAG GGC AAA GAT GAT GCC AGT GTT ACA ACC AAT TAA CC 3′ (reverse) for the UL15 mutation, 5′ CGC AAA GCG CGG AGC CAC GTC GCG CGT GCG TGC CCC GCG ATG CAC TTC CCA GGA CTG GCG GAC CGT TAG GGA TAA CAG GGT AAT CGA TTT 3′ (forward) and 5′ GCC GCG GAG GCC CGT CGC GCC ACG GTC CGC CAG TCC TGG GAA GTG CAT CGC GGG GCA CGC ACG CGC GCC AGT GTT ACA ACC AAT TAA CC 3′ (reverse) for the UL32 mutation, 5′ CAG GTC GCC GGG GGC ACC CTC AAG TTC TTC CTC ACG ACC CCC ATC CCC AGT CTG TGC GTC ACC GCC TAG GGA TAA CAG GGT AAT CGA TTT 3′ (forward) and 5′ CGA TAC CGC GTT GGG ACC GGT GGC GGT GAC GCA CAG ACT GGG GAT GGG GGT CGT GAG GAA GAA CTT GCC AGT GTT ACA ACC AAT TAA CC 3′ (reverse) for the UL42 mutation, and 5′ GGC GTG CGC CAA GCA CCC CCC CCG CTA ATG ACG CTG GCG ATT TCC CCC CCG CCC GCG GAC CCC CGC TAG GGA TAA CAG GGT AAT CGA TTT 3′ (forward) and 5′ CTT TCG CTC CGG GGC CGG GGC GCG GGG GTC CGC GGG CGG GGG GGA AAT CGC CAG CGT CAT TAG CGG GCC AGT GTT ACA ACC AAT TAA CC 3′ (reverse) for the UL54 mutation. Viruses with double mutations in UL15/UL42 and UL32/UL42 were derived by using the primers for UL15 and UL32 to insert the respective mutations into UL42 mutant BAC DNA.
The resulting BAC DNAs were cotransfected separately into CV1 cells with a Cre recombinase expression plasmid, and the reconstituted viruses were plaque purified. The genotypes of the mutant viruses were confirmed by restriction fragment length polymorphism analysis and DNA sequencing of PCR amplicons of the relevant regions (data not shown).
Statistical analysis.
Student's t test was used to compare the amounts of DNA purified from pairs of treated or untreated groups of cells. All statistical analyses and calculations were done using GraphPad Prism (version 5) software (GraphPad Software Inc., La Jolla, CA).
Nucleotide sequence accession numbers.
The sequences determined in this study have been deposited in GenBank under accession numbers KM259925 to KM259928.
RESULTS
To ensure that experiments were conducted using nontoxic doses of raltegravir, the cell lines CV1, FS-2, and M210B4 were treated with 0 to 1,600 μM raltegravir for 24 h. In the presence of phenazine methosulfate (PMS), 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) is converted by cellular NAD(P)H reduction in living cells into a formazan product with a maximum absorbance at 490 to 500 nm. PMS and MTS were therefore added to raltegravir-treated cells, the cells were incubated for a further 2 h, and the optical density at 490 nm was then measured. As shown in Fig. 1A, up to 400 μM raltegravir did not reduce the viability of FS-2 and M210B4 cells, while CV1 cell viability was reduced only slightly by treatment with 100 to 200 μM raltegravir. Once the concentration of raltegravir was increased to 800 or 1,600 μM, the viability of all three cell lines was significantly impaired, with a 40% to 70% decline in the absorbance at 490 nm.
FIG 1.

Antiviral activities of raltegravir. (A) Cytotoxic effect of raltegravir. CV1, FS-2, and M210B4 cells at 80% confluence in 24-well plates were treated with various concentrations of raltegravir for 24 h. After treatment, a cell proliferation reagent (Promega) was added to each well, and 2 h later, the absorbance at 490 nm was recorded. (B and C) Specific anti-HSV-1 (B) and anti-HSV-2 (C) activity of raltegravir. Subconfluent CV1 cells in 6-well plates were infected with HSV-1(F) or HSV-2(G) at an MOI of 0.01 PFU per cell. After 60 min of incubation at 37°C, the inocula were removed, residual extracellular infectivity associated with the cells was reduced by treatment with a low-pH citrate buffer, and growth media containing the indicated concentrations of raltegravir were added. At 24 h after virus infection, the amount of infectious virus was determined by plaque assay in CV1 cells. (D) Specific anti-HCMV activity of raltegravir. Subconfluent FS-2 cells in 6-well plates were infected with HCMV at an MOI of 0.01 PFU per cell. After 60 min of incubation at 37°C, the inocula were removed, residual extracellular infectivity associated with the cells was reduced by treatment with a low-pH citrate buffer, and growth media containing the indicated concentrations of raltegravir were added. At 72 h after virus infection, the amount of infectious virus was determined by plaque assay in FS-2 cells. (E) Specific anti-MCMV activity of raltegravir. Subconfluent M210B4 cells in 6-well plates were infected with MCMV at an MOI of 0.01 PFU per cell. After 60 min of incubation at 37°C, the inocula were removed, residual extracellular infectivity associated with the cells was reduced by treatment with a low-pH citrate buffer, and growth media containing the indicated concentrations of raltegravir were added. At 72 h after virus infection, the amount of infectious virus was determined by plaque assay in M210B4 cells. In all panels, the data represent the means and standard errors of three replicates.
To test for the effects of raltegravir on HSV replication, CV1 cells were infected with 0.01 PFU/cell HSV-1 or HSV-2 and then treated with various concentrations of raltegravir, starting at 1 h after infection. At 24 hpi, the cells were lysed by three cycles of freeze-thawing, and the amount of infectious virus in CV1 cells was determined by plaque assay. As shown in Fig. 1B, the virus titer did not significantly decrease at drug concentrations up to 10 μM, indicating that HSV-1 replication was not inhibited. At 100 μM, virus infectivity was reduced 14-fold, and at 200 μM the virus titer was reduced by approximately 100-fold. HSV-2 titers (Fig. 1C) were reduced 4- to 321-fold over the range of 100 μM to 400 μM. On the basis of these data, the estimated effective inhibitory concentrations required to reduce infectivity by 50% (EIC50s) were 67.7 ± 8.0 μM and 85.8 ± 10.0 μM for HSV-1 and HSV-2, respectively.
To determine whether raltegravir was effective against HCMV or MCMV, FS-2 or M210B4 cells were infected with 0.01 PFU/cell HCMV or MCMV and then treated with the same concentrations of raltegravir 1 h after infection. At 72 hpi, the cells were lysed by three cycles of freeze-thawing, and the amount of infectious virus was determined by plaque assay. As shown in Fig. 1D and E, the viral titers of HCMV were reduced 80-fold by treatment with 400 μM raltegravir, whereas the viral titers of MCMV were reduced more than 700-fold by treatment with 400 μM raltegravir. The EIC50s estimated on the basis of these data were 20.0 ± 6.5 μM and 2.1 ± 0.6 μM for HCMV and MCMV, respectively. We conclude that raltegravir has antiviral activity against both alphaherpesviruses (HSV-1 and HSV-2) and betaherpesviruses (HCMV and MCMV).
The next series of experiments was designed to determine the stage of viral replication affected by raltegravir treatment. Because we suspected that DNA cleavage might be affected, we used an established Southern blot assay to assess this parameter (24). Briefly, CV1 cells were infected with 5 PFU per cell of HSV-1(F). After virus adsorption and washing, cells were maintained in medium supplemented with either the DMSO carrier or 200 μM raltegravir. Uninfected cells were used as a negative control. Cells were harvested at 18 hpi, and total cellular DNA was purified, digested with BamHI, subjected to electrophoresis in a 0.8% agarose gel, denatured, and transferred to a Zeta-Probe blotting membrane. DNA on the membrane was then reacted with a denatured 32P-labeled BamHI P fragment, which is specific for both genomic ends and the junction between the unique long and unique short components of the HSV-1 genome.
The results are presented in Fig. 2A. In wild-type HSV-1(F) DNA from untreated cells, the probe recognized the S-P junction fragment and the smaller P fragment, which is specific for the end of the short component in linear genomes. Remarkably, raltegravir did not eliminate production of the P fragment, as would be expected if DNA cleavage were inhibited. Instead, the intensities of both bands were reduced in the drug-treated sample compared to those of the bands from DMSO-treated cells. Moreover, a decreased intensity of the BamHI viral DNA ladder in ethidium bromide-stained gels was noted in raltegravir-treated samples compared with that of the viral DNA ladder in DMSO-treated cells (Fig. 2B). These data indicate that raltegravir inhibits DNA replication directly or indirectly. Indirect mechanisms might include lower levels of expression of DNA replication enzymes or the initiation of infection.
FIG 2.

Raltegravir can inhibit DNA replication of HSV-1(F). CV1 cells were infected with virus at an MOI of 5 PFU per cell. After virus adsorption and washing, cells were maintained in medium containing DMSO alone or with 200 μM raltegravir. (A) The cells were lysed, and total cellular DNA was extracted, digested with BamHI, electrophoretically separated on 0.8% agarose gels, and transferred to nitrocellulose. (B) The gels were then probed with radiolabeled BamHI P fragment, which is specific for sequences at genomic ends (P fragments), and internal junction fragments (S-P fragment). (C) Comparison of the DNA copies of the viral genome of HSV-1(F) with DMSO treatment and raltegravir treatment using real-time quantitative PCR. Target primers for UL42 and reference primers for 18S rRNA were used to quantify viral DNA. The relative amount of amplicon DNA was calculated by subtraction of the CT value of target genes and 18S rRNA (control) gene in the raltegravir-treated samples. This difference was then substracted from the same calculation derived from the DMSO-treated samples by the 2−ΔΔCT method. qPCR was performed in triplicate, and data are shown as the mean ± SD of three independent experiments. ***, P < 0.001.
To confirm the inhibition of HSV-1 DNA replication by raltegravir, viral DNA in infected cells was measured by qPCR. As shown in Fig. 2C, there was an approximately 77% decrease in the amount of UL42 target sequences in cells treated with raltegravir compared with that in DMSO-treated samples (P < 0.001). Experiments were also performed with several other primer sets. Depending on the locus analyzed, the amount of DNA in drug-treated cells was decreased from 75 to 83% compared with that in the DMSO-treated cells examined in parallel (data not shown). These data indicate that raltegravir inhibits HSV DNA replication.
The absence of an apparent effect of raltegravir on DNA cleavage was unexpected. To determine the target of inhibition, a series of experiments was undertaken to generate raltegravir-resistant HSV isolates. CV1 cells infected with HSV-1 were maintained in DMEM containing 200 μM raltegravir. Fresh medium containing the same concentration of raltegravir was added at 24-h intervals. Progeny virus was harvested after a cytopathic effect was evident. This virus stock was subjected to the same conditions for nine additional cycles of infection in the presence of drug, followed by collection of progeny virus. These progeny were then plaque purified, grown to a high titer, and tested for raltegravir resistance by plaque assay. Of 12 isolates from this 10th passage, 2 that exhibited the greatest resistance to raltegravir were chosen for further study (data not shown). One of these was passaged five more times in the presence of 200 μM raltegravir and was designated clone 7. As shown in Fig. 3A, plaque assay results show that the titer of virus clone 7 decreased by only 3-fold in the presence of raltegravir, while that of wild-type virus was reduced by more than 50-fold in the presence of the drug at the same concentration.
FIG 3.
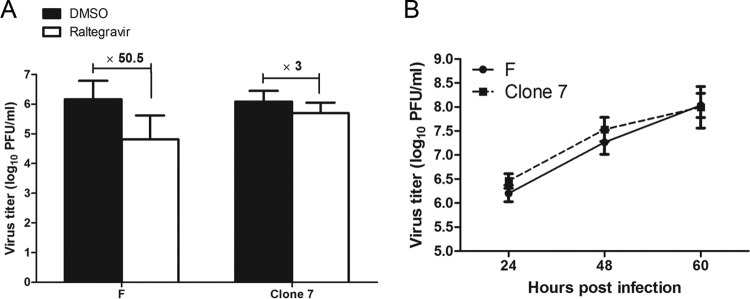
Generation of mutant virus with raltegravir-resistant activity under continuous raltegravir treatment. (A) Plaque-purified virus with high resistance was designated clone 7 and was passaged five more times in the presence of 200 μM raltegravir. Subconfluent CV1 cells in 6-well plates were infected with HSV-1(F) or clone 7 at an MOI of 0.01 PFU per cell. After 60 min of incubation at 37°C, the inocula were removed, the residual extracellular infectivity associated with the cells was reduced by treatment with a low-pH citrate buffer, and growth media containing 200 μM raltegravir were added. At 24 hpi, the amount of infectious virus was determined by plaque assay in CV1 cells. (B) Growth kinetics of clone 7 virus and HSV-1(F). CV1 cells were infected with clone 7 or HSV-1(F) at an MOI of 0.01. At the indicated time points, cell cultures were collected and viral infectivity was measured by plaque assay in CV1 cells. Each point and error bar represents the mean ± SD of results from three individual experiments.
To ensure that the resistant virus was not impaired in growth by continuous passage in the presence of drug, CV1 cells were infected with clone 7 or HSV-1(F) at an MOI of 0.01. At 24, 48, and 60 hpi, the cultures were collected and virus titers were determined by plaque assay. As shown in Fig. 3B, the growth kinetics of clone 7 were similar to those of HSV-1(F).
A series of experiments was then undertaken to identify the mutation(s) responsible for raltegravir resistance. In the first experiment, viral DNA was extracted from capsids purified from cells infected with clone 7 and sequenced. Table 2 lists 16 mutations from the published sequence of HSV-1(F) (25). The mutations were distributed throughout the genome, with a single nucleotide change occurring in 16 different genes. All of the reads from these regions contained the mutations, suggesting that the derived sequence was from a single genome containing all 16 mutations. Of these mutations, 5 were silent, including a mutation in UL29 (26), encoding the single-strand DNA-binding protein (27). Amino acid-altering mutations were located in genes encoding virion structural proteins and proteins required for DNA replication and packaging, such as pUL15 (encoding the large terminase subunit) (28), pUL32 (a DNA cleavage/packaging protein) (29), and pUL42 (DNA polymerase accessory factor) (26, 30).
TABLE 2.
Nucleotide and amino acid changes in genome of clone 7 mutant virus
| Mutation no. | Change |
Gene | Function | |
|---|---|---|---|---|
| Nucleotide | Amino acid | |||
| 1 | A9604G | —a | UL1 | Envelope glycoprotein L |
| 2 | G11809A | R182W | UL4 | Nuclear protein UL4 |
| 3 | C12461T | — | UL5 | Helicase-primase helicase subunit |
| 4 | T15564C | — | UL6 | Capsid portal protein |
| 5 | C28512T | V109 M | UL14 | Tegument protein UL14 |
| 6 | T34226C | F198S | UL15 | DNA packaging terminase subunit 1 |
| 7 | A35711G | — | UL18 | Capsid triplex subunit 2 |
| 8 | G43838A | A821V | UL22 | Envelope glycoprotein H |
| 9 | C60590T | — | UL29 | Single-stranded DNA-binding protein |
| 10 | G67957A | A374V | UL32 | DNA packaging protein UL32 |
| 11 | T73069C | M2434V | UL36 | Large tegument protein |
| 12 | T84529C | C35R | UL38 | Capsid triplex subunit 1 |
| 13 | G93893A | V296I | UL42 | DNA polymerase accessory factor |
| 14 | T104340C | T212A | UL48 | trans-Activating tegument protein VP16 |
| 15 | C107306A | P134T | UL50 | Deoxyuridine triphosphatase |
| 16 | G114294T | A224S | UL54 | Multifunctional expression regulator |
—, no amino acid change.
Because the changes in sequence could have arisen at any time during viral passage, including within the original virus stock, we performed a second series of experiments to identify the target of raltegravir. Specifically, the mutations within UL15, UL32, UL42, and UL54 (encoding ICP27, which is an important regulatory protein) (31) were introduced into the wild type HSV-1(F) genome using two-step red-mediated recombination, as described by Tischer et al. (22). The recombinant viruses were designated Mu15, Mu32, Mu42, and Mu54, respectively. CV1 cells were infected with 0.01 PFU/cell of these mutant viruses or HSV-1(F) and after adsorption were treated with DMSO carrier or 200 μM raltegravir. At 24 hpi, the cells were lysed by three cycles of freeze-thawing, and the amount of infectious virus was determined by plaque assay in CV1 cells. As shown in Fig. 4A, the viral titers of three of the recombinant viruses (those with F198S in UL15, A374V in UL32, and A224S in UL54) showed that they were as susceptible to inhibition by raltegravir as HSV-1(F). In contrast, recombinant viruses bearing the V296I mutation in UL42 (Mu42, Mu15/42, and Mu32/42) were resistant to raltegravir, inasmuch as the drug reduced the infectious titers by only about 4-fold, similar to the effect of raltegravir on clone 7 replication. These data strongly suggest that the mutation in UL42 confers resistance to raltegravir.
FIG 4.
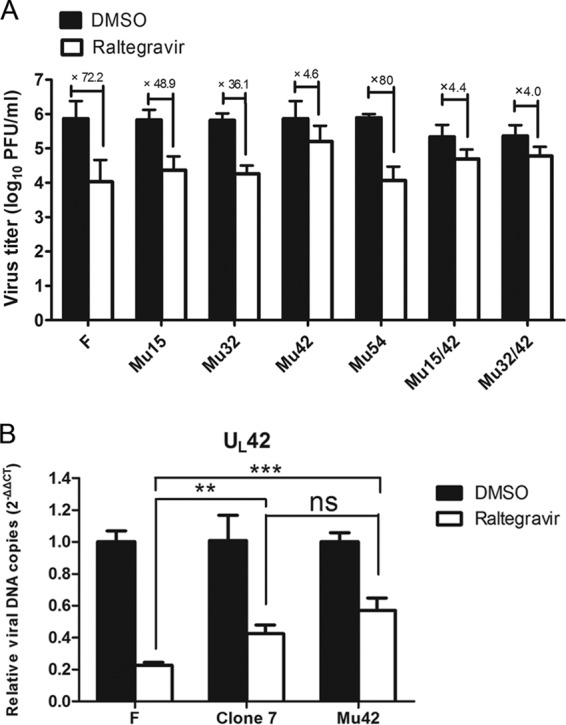
A single amino acid mutation in UL42 confers raltegravir resistance. (A) Identification of the virus titers of the recombinant viruses corresponds to different amino acid mutations. Subconfluent CV1 cells in 6-well plates were infected with recombinant viruses as well as HSV-1(F) at an MOI of 0.01 PFU per cell. After 60 min of incubation at 37°C, the inocula were removed, residual extracellular infectivity associated with the cells was reduced by treatment with a low-pH citrate buffer, and growth media containing 200 μM raltegravir were added. At 24 hpi, the amount of infectious virus was determined by plaque assay in CV1 cells. (B) Comparison of the viral DNA copies of HSV-1(F), clone 7, and Mu42 with DMSO treatment or raltegravir treatment using real-time qPCR. CV1 cells were infected with these three viruses at an MOI of 5 PFU per cell. After virus adsorption and washing, cells were maintained in medium containing 200 μM raltegravir or an equivalent of DMSO carrier. The cells were lysed, and total cellular DNA was extracted and used as the template for qPCR, as described previously (see Fig. 2C). qPCR was performed in triplicate, and data are shown as the mean ± SD of three independent experiments. ns, not significant (P > 0.05); **, P < 0.01; ***, P < 0.001.
To determine if the UL42 mutation restored DNA replication to HSV-1(F) in the presence of raltegravir, cells were infected with HSV-1(F), the clone 7 mutant, or the recombinant virus into which the UL42 mutation was inserted (Mu42). Total DNA was purified from the infected cells and subjected to qPCR using a pair of primers specific for the UL42 gene. As expected, the Mu42- and clone 7-infected cells both contained an increase in UL42 target sequences compared to the levels in cells infected with HSV-1(F) and treated with raltegravir. Additionally, there was no significant difference in the number of copies of UL42 between clone 7- and Mu42-infected cells (Fig. 4B). Similar observations were obtained using primer sets specific for UL44 and UL46 (data not shown). These data suggest that the UL42 mutation restores the replication of HSV-1 in the presence of raltegravir.
Figure 5A shows the structure of UL42 bound to a portion of the DNA polymerase, as previously published by Zuccola et al. (32). Modeling of the V296I mutation into this structure (Fig. 5B) suggests that while it is located near the polymerase-interacting region of pUL42, the mutation is unlikely to interfere with binding to the DNA polymerase.
FIG 5.
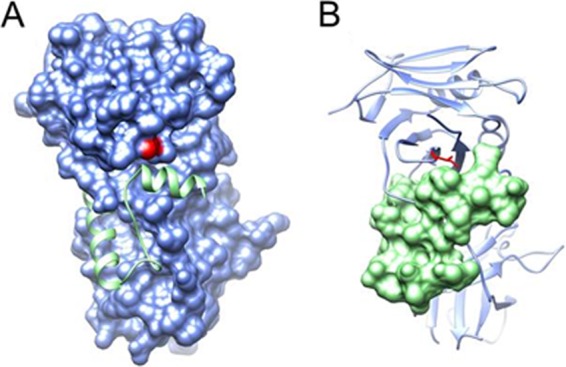
(A) pUL42 residue V296 (red) is located next to the interface of pUL42 (molecular surface in blue) and the bound C-terminal tail of the viral DNA polymerase catalytic subunit (ribbon diagram in green) (RCSB PDB accession code 1DML) (32). (B) V296 is mutated to Ile (stick model in red) in pUL42 (ribbon diagram in blue), showing no steric clash with binding of the DNA polymerase C-terminal tail (molecular surface in green). The two panels are in the same view.
DISCUSSION
A previous report showed that raltegravir can block the nuclease activity of terminase subunit pUL89 of HCMV expressed in E. coli (8). Given the highly conserved nature of this subunit in all herpesviruses and the requirement for its nuclease activity as a prerequisite for DNA packaging, we speculated that raltegravir would act against a variety of herpesviruses. In this study, we show that four herpesviruses from two subfamilies are sensitive to raltegravir and the drug reduced titers up to 700-fold, depending on the dose and the species of virus. Surprisingly, however, Southern blot and qPCR analyses indicated that raltegravir inhibited viral DNA replication rather than DNA cleavage (Fig. 2).
Passaging of HSV-1 in the presence of raltegravir over the course of 4 months caused an increase in resistance to the drug. By the 10th passage, the viral titer was reduced by 6-fold in the presence of drug (data not shown), while a further 5 passages produced a virus that replicated to titers reduced by only 3-fold (Fig. 3A). Sequencing of this highly passaged virus revealed a variety of mutations. Of the mutations tested, the V296I mutation in UL42 increased viral resistance to raltegravir to a level similar to that of the highly passaged virus, suggesting that UL42 was the most important target of the drug. UL42 also makes sense as the major target because it is necessary for optimal viral DNA synthesis (33). While a mutation (F198S) in the large terminase subunit encoded by UL15 was noted, this mutation did not contribute additively to drug resistance when paired with the UL42 mutation, nor did it confer substantial resistance to raltegravir by itself. Nevertheless, the data cannot exclude the possibility that one or more of the mutations other than UL42 V296I contribute to raltegravir resistance.
Structural modeling suggests that UL42 V296I is unlikely to interfere with the critical binding of the UL42 gene product to the HSV DNA polymerase (Fig. 5). This prediction is supported by the observation that the cadres of mutations, including UL42 V296I, did not impair viral growth in the absence of raltegravir (Fig. 3B). We hypothesize that the UL42 V296I mutation blocks drug binding to UL42 or to other components of the DNA replication complex which comprises UL42, the polymerase, and helicase and primase components.
The drug resistance of viruses is an important consideration for antiviral therapy. While raltegravir might be effective against HIV and herpesviruses, drug resistance arises due to the emergence of HIV variants (34, 35) that contain amino acid changes in the integrase. These are especially important in severely immunocompromised patients receiving prolonged antiviral therapy, such as transplant recipients and AIDS patients. The currently approved antiherpesvirus agents acyclovir (ACV), penciclovir (PCV), ganciclovir (GCV), cidofovir (CDV), and foscarnet (PFA) ultimately target the viral DNA polymerase. Molecular mechanisms of HSV resistance to ACV most often involve mutations within genes encoding either the viral thymidine kinase (UL23) or the DNA polymerase (UL30) (36, 37). In transplant recipients, the drug resistance of human cytomegalovirus is caused by mutations in either the viral kinase encoded by UL97 or the gene encoding viral DNA polymerase (UL54) (38). Following the development of genotypic testing (39, 40) and BAC technology (41), novel mutations in genes involved in viral DNA replication have been shown to confer drug resistance. Our finding that a mutation in HSV-1 UL42 can confer resistance to raltegravir therefore adds another mutation to a growing list. We also caution that raltegravir targets in HSV-2, MCMV, and HCMV may or may not map to the corresponding DNA polymerase accessory factors of these viruses; certainly, this possibility warrants further investigation.
Our results demonstrate that raltegravir treatment might comprise part of an effective strategy to control HSV and HIV coinfection. While the long-term use of raltegravir alone may cause the emergence of drug-resistant HIV and herpesviruses, its use as part of a combination therapy against these pathogens warrants evaluation.
ACKNOWLEDGMENTS
These studies were supported by NIH grant AI52341 to J.D.B.
We thank Yancheng Liu, Cornell University, for advice on bioinformatics and comparison of DNA sequences.
Footnotes
Published ahead of print 9 July 2014
REFERENCES
- 1.Conway JF, Homa FL. 2011. Nucleocapsid structure, assembly and DNA packaging of herpes simplex virus, p 175–193 In Weller SK. (ed), Alphaherpesviruses. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 2.Baines JD. 2011. Herpes simplex virus capsid assembly and DNA packaging: a present and future antiviral drug target. Trends Microbiol. 19:606–613. 10.1016/j.tim.2011.09.001 [DOI] [PubMed] [Google Scholar]
- 3.Beard PM, Taus NS, Baines JD. 2002. The DNA cleavage and packaging proteins encoded by genes UL28, UL15, and UL33 of herpes simplex virus 1 form a complex in infected cells. J. Virol. 76:4785–4791. 10.1128/JVI.76.10.4785-4791.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu D, Weller SK. 1998. Genetic analysis of the UL 15 gene locus for the putative terminase of herpes simplex virus type 1. Virology 243:32–44. 10.1006/viro.1998.9041 [DOI] [PubMed] [Google Scholar]
- 5.Yang K, Homa F, Baines JD. 2007. Putative terminase subunits of herpes simplex virus 1 form a complex in the cytoplasm and interact with portal protein in the nucleus. J. Virol. 81:6419–6433. 10.1128/JVI.00047-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldner T, Hewlett G, Ettischer N, Ruebsamen-Schaeff H, Zimmermann H, Lischka P. 2011. The novel anti-cytomegalovirus compound AIC246 inhibits HCMV replication through a specific antiviral mechanism that involves the viral terminase. J. Virol. 85:10884–10893. 10.1128/JVI.05265-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selvarajan Sigamani S, Zhao H, Kamau YN, Baines JD, Tang L. 2013. The structure of the herpes simplex virus DNA-packaging terminase pUL15 nuclease domain suggests an evolutionary lineage among eukaryotic and prokaryotic viruses. J. Virol. 87:7140–7148. 10.1128/JVI.00311-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nadal M, Mas PJ, Blanco AG, Arnan C, Solà M, Hart DJ, Coll M. 2010. Structure and inhibition of herpesvirus DNA packaging terminase nuclease domain. Proc. Natl. Acad. Sci. U. S. A. 107:16078–16083. 10.1073/pnas.1007144107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grinsztejn B, Nguyen BY, Katlama C, Gatell JM, Lazzarin A, Vittecoq D, Gonzalez CJ, Chen J, Harvey CM, Isaacs RD. 2007. Safety and efficacy of the HIV-1 integrase inhibitor raltegravir (MK-0518) in treatment-experienced patients with multidrug-resistant virus: a phase II randomised controlled trial. Lancet 369:1261–1269. 10.1016/S0140-6736(07)60597-2 [DOI] [PubMed] [Google Scholar]
- 10.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, Fiore F, Gardelli C, Gonzalez PO, Hazuda DJ, Jones P, Kinzel O, Laufer R, Monteagudo E, Muraglia E, Nizi E, Orvieto F, Pace P, Pescatore G, Scarpelli R, Stillmock K, Witmer MV, Rowley M. 2008. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J. Med. Chem. 51:5843–5855. 10.1021/jm800245z [DOI] [PubMed] [Google Scholar]
- 11.Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD. 2000. Inhibitors of strand transfer that prevent integration and inhibit HIV-1 replication in cells. Science 287:646–650. 10.1126/science.287.5453.646 [DOI] [PubMed] [Google Scholar]
- 12.Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P. 2010. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. U. S. A. 107:20057–20062. 10.1073/pnas.1010246107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. 2010. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 464:232–236. 10.1038/nature08784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behavior of infected cells. J. Gen. Virol. 2:357–364. 10.1099/0022-1317-2-3-357 [DOI] [PubMed] [Google Scholar]
- 15.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382–1391. 10.1128/JVI.77.2.1382-1391.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang K, Poon AP, Roizman B, Baines JD. 2008. Temperature-sensitive mutations in the putative herpes simplex virus type 1 terminase subunits pUL15 and pUL33 preclude viral DNA cleavage/packaging and interaction with pUL28 at the nonpermissive temperature. J. Virol. 82:487–494. 10.1128/JVI.01875-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Homa FL, Otal TM, Glorioso JC, Levine M. 1986. Transcriptional control signals of a herpes simplex virus type 1 late (g2) gene lie within bases −34 to +124 relative to the 5′ terminus of the mRNA. Mol. Cell. Biol. 6:3652–3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 19.Yang K, Baines JD. 2011. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc. Natl. Acad. Sci. U. S. A. 108:14276–14281. 10.1073/pnas.1108564108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ram JL, Karim AS, Sendler ED, Kato I. 2011. Strategy for microbiome analysis using 16S rRNA gene sequence analysis on the Illumina sequencing platform. Syst. Biol. Reprod. Med. 57:162–170. 10.3109/19396368.2011.555598 [DOI] [PubMed] [Google Scholar]
- 21.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat. Biotechnol. 29:24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step RED-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. 10.2144/000112096 [DOI] [PubMed] [Google Scholar]
- 23.Liang L, Tanaka M, Kawaguchi Y, Baines JD. 2004. Cell lines that support replication of a novel herpes simplex 1 UL31 deletion mutant can properly target UL34 protein to the nuclear rim in the absence of UL31. Virology 329:68–76. 10.1016/j.virol.2004.07.030 [DOI] [PubMed] [Google Scholar]
- 24.Jacobson JG, Yang K, Baines JD, Homa FL. 2006. Linker insertion mutations in the herpes simplex virus type 1 UL28 gene: effects on UL28 interaction with UL15 and UL33 and identification of a second-site mutation in the UL15 gene that suppresses a lethal UL28 mutation. J. Virol. 80:12312–12323. 10.1128/JVI.01766-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szpara ML, Parsons L, Enquist LW. 2010. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J. Virol. 84:5303–5313. 10.1128/JVI.00312-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hernandez TR, Lehman IR. 1990. Functional interaction between the herpes simplex-1 DNA polymerase and UL42 protein. J. Biol. Chem. 265:11227–11232 [PubMed] [Google Scholar]
- 27.Ruyechan WT, Weir AC. 1984. Interaction with nucleic acids and stimulation of the viral DNA polymerase by the herpes simplex virus type 1 major DNA-binding protein. J. Virol. 52:727–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davison AJ. 1992. Channel catfish virus: a new type of herpesvirus. Virology 186:9–14. 10.1016/0042-6822(92)90056-U [DOI] [PubMed] [Google Scholar]
- 29.Lamberti C, Weller SK. 1998. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J. Virol. 72:2463–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gottlieb J, Marcy AI, Coen DM, Challberg MD. 1990. The herpes simplex virus 1 UL42 gene product: a subunit of DNA polymerase that functions to increase processivity. J. Virol. 64:5976–5987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sacks WR, Greene CC, Aschman DP, Schaffer PA. 1985. Herpes simplex virus type 1 ICP27 is an essential regulatory protein. J. Virol. 55:796–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zuccola HJ, Filman DJ, Coen DM, Hogle JM. 2000. The crystal structure of an unusual processivity factor, herpes simplex virus UL42, bound to the C terminus of its cognate polymerase. Mol. Cell 5:267–278. 10.1016/S1097-2765(00)80422-0 [DOI] [PubMed] [Google Scholar]
- 33.Wu CA, Nelson NJ, McGeoch DJ, Challberg MD. 1988. Identification of herpes simplex virus type 1 genes required for origin-dependent DNA synthesis. J. Virol. 62:435–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malet I, Delelis O, Valantin MA, Montes B, Soulie C, Wirden M, Tchertanov L, Peytavin G, Reynes J, Mouscadet JF, Katlama C, Calvez V, Marcelin AG. 2008. Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob. Agents Chemother. 52:1351–1358. 10.1128/AAC.01228-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malet I, Gimferrer AL, Artese A, Costa G, Parrotta L, Alcaro S, Delelis O, Tmeizeh A, Katlama C, Valantin MA, Ceccherini-Silberstein F, Calvez V, Marcelin AG. 2014. New raltegravir resistance pathways induce broad cross-resistance to all currently used integrase inhibitors. J. Antimicrob. Chemother. 69:2118–2122. 10.1093/jac/dku095 [DOI] [PubMed] [Google Scholar]
- 36.Gilbert C, Bestman-Smith J, Boivin G. 2002. Resistance of herpesviruses to antiviral drugs: clinical impacts and molecular mechanisms. Drug Resist. Updat. 5:88–114. 10.1016/S1368-7646(02)00021-3 [DOI] [PubMed] [Google Scholar]
- 37.Burrel S, Deback C, Agut H, Boutolleau D. 2010. Genotypic characterization of UL23 thymidine kinase and UL30 DNA polymerase of clinical isolates of herpes simplex virus: natural polymorphism and mutations associated with resistance to antivirals. Antimicrob. Agents Chemother. 54:4833–4842. 10.1128/AAC.00669-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fischer L, Laib SK, Jahn G, Hamprecht K, Gohring K. 2013. Generation and characterization of a GCV resistant HCMV UL97-mutation and a drug sensitive UL54-mutation. Antiviral Res. 100:575–577. 10.1016/j.antiviral.2013.09.026 [DOI] [PubMed] [Google Scholar]
- 39.Bestman-Smith J, Schmit I, Papadopoulou B, Boivin G. 2001. Highly reliable heterologous system for evaluating resistance of clinical herpes simplex virus isolates to nucleoside analogues. J. Virol. 75:3105–3110. 10.1128/JVI.75.7.3105-3110.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bestman-Smith J, Boivin G. 2003. Drug resistance patterns of recombinant herpes simplex virus DNA polymerase mutants generated with a set of overlapping cosmids and plasmids. J. Virol. 77:7820–7829. 10.1128/JVI.77.14.7820-7829.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drouot E, Piret J, Boivin G. 2013. Novel method based on “en passant” mutagenesis coupled with a Gaussia luciferase reporter assay for studying the combined effects of human cytomegalovirus mutations. J. Clin. Microbiol. 51:3216–3224. 10.1128/JCM.01275-13 [DOI] [PMC free article] [PubMed] [Google Scholar]