

ABSTRACT
Vaccinia virus (VACV) L1 is an important target for viral neutralization and has been included in multicomponent DNA or protein vaccines against orthopoxviruses. To further understand the protective mechanism of the anti-L1 antibodies, we generated five murine anti-L1 monoclonal antibodies (MAbs), which clustered into 3 distinct epitope groups. While two groups of anti-L1 failed to neutralize, one group of 3 MAbs potently neutralized VACV in an isotype- and complement-independent manner. This is in contrast to neutralizing antibodies against major VACV envelope proteins, such as H3, D8, or A27, which failed to completely neutralize VACV unless the antibodies are of complement-fixing isotypes and complement is present. Compared to nonneutralizing anti-L1 MAbs, the neutralization antibodies bound to the recombinant L1 protein with a significantly higher affinity and also could bind to virions. By using a variety of techniques, including the isolation of neutralization escape mutants, hydrogen/deuterium exchange mass spectrometry, and X-ray crystallography, the epitope of the neutralizing antibodies was mapped to a conformational epitope with Asp35 as the key residue. This epitope is similar to the epitope of 7D11, a previously described potent VACV neutralizing antibody. The epitope was recognized mainly by CDR1 and CDR2 of the heavy chain, which are highly conserved among antibodies recognizing the epitope. These antibodies, however, had divergent light-chain and heavy-chain CDR3 sequences. Our study demonstrates that the conformational L1 epitope with Asp35 is a common site of vulnerability for potent neutralization by a divergent group of antibodies.
IMPORTANCE Vaccinia virus, the live vaccine for smallpox, is one of the most successful vaccines in human history, but it presents a level of risk that has become unacceptable for the current population. Studying the immune protection mechanism of smallpox vaccine is important for understanding the basic principle of successful vaccines and the development of next-generation, safer vaccines for highly pathogenic orthopoxviruses. We studied antibody targets in smallpox vaccine by developing potent neutralizing antibodies against vaccinia virus and comprehensively characterizing their epitopes. We found a site in vaccinia virus L1 protein as the target of a group of highly potent murine neutralizing antibodies. The analysis of antibody-antigen complex structure and the sequences of the antibody genes shed light on how these potent neutralizing antibodies are elicited from immunized mice.
INTRODUCTION
Variola virus and monkeypox virus are orthopoxviruses that are highly pathogenic to humans, are considered to be potential bioterrorism agents (1), and are emerging pathogens (2). A related orthopoxvirus, vaccinia virus (VACV), serves as the vaccine against these pathogens. Live VACV immunization is capable of eliciting neutralizing antibodies against a variety of targets on two antigenically distinct forms of virions, the intracellular mature virions (MV) and the extracellular enveloped virions (EV) (3, 4). Vaccinia vaccine is arguably the most successful vaccine in human history, having led to the eradication of smallpox (5). However, it was also associated with a relatively high rate of adverse events (6). Consequently, safer multicomponent DNA or protein vaccines that include a subset of MV and EV antigens (Ag) have been developed, and they showed protection against orthopoxvirus challenges in mice and nonhuman primates (7–10). While many MV antigens have been shown to be neutralization targets (11, 12), the MV antigen that is invariably included in these subunit vaccines is L1. L1 is an immunodominant neutralizing antibody target in mice, although it is a less common target in humans (13). It is a 250-amino-acid myristoylated protein with a C-terminal transmembrane domain that spans residues 186 to 204 (14, 15). L1 associates with the virus-encoded multiprotein entry-fusion complex (EFC) and plays an essential role in viral entry (16).
Despite the importance of L1 as a neutralizing target and subunit vaccine component, relatively little is known about its neutralizing epitopes and the corresponding paratopes. A conformational epitope with Asp35 as the key residue is recognized by several murine monoclonal antibodies (MAbs) (17), which potently neutralize MV. The sequence of one of the MAbs, 7D11, has been reported, and a structure of the Fab domain of 7D11 bound to L1 has been determined (18). A linear epitope (residues 118 to 128) of L1 is recognized by several antibodies, which neutralized MV with reduced potency compared to 7D11 (19). In an effort to gain a more comprehensive understanding of neutralizing epitopes on L1 and the neutralizing mechanism of anti-L1 antibodies, we developed additional MAbs against L1, examined their neutralizing abilities in vitro and in vivo, and determined their epitopes and the corresponding paratopes.
MATERIALS AND METHODS
Viruses and antibodies.
VACVWR stocks were grown on HeLa cells in T175 flasks, infecting them at a multiplicity of infection of 0.5. Cells were harvested at 60 h, and virus was isolated by rapidly freeze-thawing the cell pellet three times in a volume of 2.3 ml RPMI plus 1% fetal calf serum (FCS). Cell debris was removed by centrifugation. Clarified supernatant was frozen at −80°C as virus stock. Titers of VACVWR stocks were determined on Vero cells (∼2 × 108 PFU/ml). VACVACAM2000 was obtained from the CDC. Strain Mos-3-P2 (Moscow strain) of ectromelia virus, as described in Chen et al. (20), was used for ectromelia in vivo studies. Monoclonal antibodies used in the study are the following: anti-B5, B126 (21); anti-A10, BG3.1 (22); anti-H3, JH4 (22) and 41 (23); anti-A27, 1G6 and 12G2 (unpublished data); anti-D8, JE10 (22) and LA5 (24).
Hybridoma generation and characterization.
The generation and characterization of the hybridomas were performed as described previously (22), except for some changes in the immunization procedure described below. BALB/c mice initially were infected intranasally with 5 × 103 PFU of VACVWR. Subsequently, the mice were boosted twice with intraperitoneal (i.p.) injection of 100 μg of recombinant L1 proteins together with incomplete Freund's adjuvant. Recombinant L1 proteins were either a glutathione S-transferase (GST) fusion of L1(1-185) or His-tagged L1(1-185). Both forms of recombinant L1 proteins were expressed in Escherichia coli, and the His-tagged L1(1-185) protein was refolded as described below. The hybridomas were screened with enzyme-linked immunosorbent assays (ELISAs) against recombinant L1 proteins and with an immunofluorescence assay of VACVWR-infected cells as described previously (22).
L1 expression and purification.
The VACV-L1 (residues 1 to 185) expression construct was kindly provided by David Garboczi, and expression and refolding was carried out as reported previously (15). Refolded protein was purified by nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography (5-ml HisTrap FF column; GE Healthcare) by elution with 0.2 M imidazole, 20 mM Tris, pH 8.0, 0.3 M NaCl. The eluted protein then was further purified by gel filtration chromatography using a Superdex 200 size-exclusion column (GE Healthcare). Proper protein folding was verified by ELISA using three different anti-L1 antibodies.
Cross-blocking ELISA.
Refolded L1(1-185) was prepared at 0.5 μg/ml and used to coat Nunc polysorbent flat-bottom 96-well plates with 100 μl per well. Plates were incubated overnight at 4°C and washed four times with phosphate-buffered saline (PBS) plus 0.05% Tween 20. One hundred μl of blocking buffer (PBS plus 10% fetal bovine serum [FBS]) was added to the plate and incubated for 90 min at room temperature. Blocking buffer was discarded, and 100 μl of the antibodies of interest was added to the plate at 10 μg/ml and incubated for 90 min. Horseradish peroxidase (HRP)-conjugated antibodies of interest (Innova Biosciences lightning-link HRP conjugation kit) were prepared at 0.5 μg/ml and added to the plates for 20 min. The plates were developed using o-phenylenediamine (OPD), and optical density (OD) at 490 nm was read on a SpectraMax 250 (Molecular Devices).
Flow cytometry-based in vitro neutralization.
Vero E6 cells (1 × 105 cells/well) were seeded in 96-well Costar plates (Corning Inc., Corning, NY) and incubated for 5 h to adhere. Subsequently, cells were infected with 12.5 μl purified VACV-green fluorescent protein (GFP) at 1 × 106 PFU/ml (final PFU of 1.25 × 104) and 12.5 μl MAbs at 80 μg/ml (final concentration of 20 μg/ml) for 12 h at 37°C and 5% CO2 in a total volume of 50 μl in the presence (2% final concentration) or absence of sterile baby rabbit complement (Cedarlane). Samples were prepared in duplicates. Cells subsequently were tested using flow cytometry as described previously (21).
Plaque reduction neutralization tests (PRNT).
Vero E6 cells were seeded into 6-well Costar plates (Corning Inc., Corning, NY) and used within 2 days of reaching confluence. Purified antibodies were prepared in duplicates at 20 μg/ml, and 5-fold serial dilutions were performed in PBS. Ten μl of sonicated VACVWR at 1 × 105 PFU/ml was added to 970 μl for every titrated antibody sample, resulting in a final concentration of 1 × 103 PFU/sample. For samples to be tested in the presence of complement, 20 μl of complement was added, resulting in a final concentration of 2%. PBS was used instead for other samples. Samples were incubated for 1 h at 4°C with shaking. Medium was aspirated from 6-well plates, and 1 ml of sample mixtures was added to each well to adsorb for 60 min at 37°C with periodic swirling. The mix was aspirated, 2 ml of Dulbecco's modified Eagle medium (DMEM) with 1% FBS plus 1% penicillin-streptomycin was added, and the plates were incubated for 2.5 days for the plaques to develop. The medium was aspirated, the cells were fixed and stained in one step with 0.1% crystal violet in 20% ethanol, and the plaques were quantified over white-light transillumination. The data were plotted, and a curve of best fit was applied by eye. The neutralization ability was investigated as a comparison of the samples, including antibody to the mean number of plaques of controls with no antibody.
Vaccinia intranasal infections and protection studies.
Female BALB/c mice were used at an age of 7 to 8 weeks. Animal husbandry and experimental procedures were approved by the Department of Laboratory Animal Care and the Animal Care Committee of the La Jolla Institute. To infect the mice, a Pipetman was used to place 10 μl of VACVWR on each nare of an isoflurane-anesthetized mouse (total volume, 20 μl), and the liquid was rapidly inhaled by the mouse. Mice were weighed daily to assess disease progression. After intranasal infection with VACVWR, mice develop a systemic infection and exhibit severe weight loss. Mice were euthanized if and when 25% weight loss occurred. A dose of 1 × 105 PFU of VACVWR was the standard lethal dose given to 7-week-old BALB/c females. For L1 MAb protection studies, mice were treated by i.p. injection with 100 μg of antibodies 1 day before infection. Control mice received anti-A10 BG3.1 antibody, which is known to provide no protection. An additional group received anti-B5 B126 as a positive control.
Vaccinia intravenous infection protection studies.
To infect mice, 1 × 105 PFU of VACVACAM2000 was injected retro-orbitally. Mice were weighed daily to assess disease progression. The clinical score, a composite score of the pox lesion abundance on the four paws plus the tail, was evaluated as described previously (23). For L1 MAb protection studies, mice were inoculated i.p. with 100 μg of antibodies 1 day before infection. Control mice received anti-A10 BG3.1 antibody. An additional group received anti-H3 number 41 as a positive control.
Ectromelia infection and protection studies.
Four- to 6-week-old female C57BL/6 mice were obtained from Harlan Laboratories (Indianapolis, IN), housed in filter-top microisolator cages, and fed commercial mouse chow and water ad libitum. The mice were housed in an animal biosafety level 3 containment area. Animal husbandry and experimental procedures were approved by the Institutional Animal Care and Use Committee of Saint Louis University. The day before challenge, mice were treated i.p. with 100 μg of the specified antibody. Immediately before challenge, mice were anesthetized with 0.1 ml/10 g body weight of ketamine HCl (6 mg/ml) and xylazine (0.5 mg/ml) by i.p. injections. One thousand PFU of ECTV (Moscow strain) in PBS without Ca2+ and Mg2+ was slowly loaded into nares (5 μl/nare). Mice subsequently were left in situ for 2 to 3 min before being returned to their cages (25, 26). Mice were monitored daily for mortality and morbidity, as measured by weight change.
Epitope mapping by ELISA.
Overlapping 20-mer peptides for the L1 antigen were synthesized (AnaSpec) and tested for MAb binding using an ELISA. Flat-bottom 96-well microtiter plates were coated with 100 μl of neutravidin biotin-binding protein (1 mg/ml) diluted in PBS overnight at 4°C (ThermoScientific Pierce). Coated plates were washed with washing buffer (PBS, pH 7.2, 0.05% Tween 20) and blocked with blocking buffer (PBS, pH 7.2, 1% bovine serum albumin [BSA], 0.1% Tween 20) for 2 h at room temperature (RT). Plates were incubated with 100 μl of overlapping linear biotinylated peptides (200 ng/ml) in blocking buffer for 90 min at RT. Plates were washed and incubated with purified MAb at 10 μg/ml for 90 min at RT. Plates were washed, and the bound MAb was detected by adding a streptavidin-HRP-conjugated secondary antibody to mouse immunoglobulin G (Invitrogen) and incubated for 60 min at RT, followed by incubation in OPD substrate (Sigma-Aldrich).
Peptide truncation and alanine scan.
Variant peptides with N- or C-terminal truncations and/or alanine substitutions were tested for their ability to block binding to the parent 20-mer peptides in ELISA. Ninety-six-well plates were coated with 100 μl neutravidin per well at a concentration of 0.5 μg/ml. Plates were incubated overnight at 4°C and washed 4 times with PBS plus 0.05% Tween 20. One hundred μl of blocking buffer (PBS plus 10% FBS) was added to the plates and incubated for 90 min at 4°C. Blocking buffer was discarded, and 100 μl of biotinylated 20-mer peptides was added to the plate at 200 ng/ml and incubated for 90 min at 4°C. Simultaneously, selected antibodies were incubated with variant peptides. We used 30 μl/well of MAb at 600 ng/ml and incubated it with 30 μl/well of alanine-modified peptides at 100 μg/ml for 90 min at 4°C. After washing the plates, 50 μl of the antibody/alanine peptide mix was added to plate-bound peptides and incubated for 20 min at 4°C. Plates were washed, and 100 μl/well of secondary goat anti-mouse anti-IgGγ HRP (1:1,000 diluted in blocking buffer) was added and incubated for 90 min at 4°C. A final wash step was performed, plates were developed using o-phenylenediamine, and the OD at 490 nm was read on a SpectraMax 250 (Molecular Devices).
Epitope mapping by deuterium exchange mass spectrometry.
To maximize peptide sequence coverage, the optimized quench and proteolysis conditions were determined prior to deuterium exchange mass spectrometry (DXMS) experiments (27). For each sample, 0.4 μl of stock solution of L1 at 6.2 mg/ml was diluted with 7.6 μl of H2O buffer (8.3 mM Tris-HCl, 150 mM NaCl, in H2O, pH 7.2) at 0°C and then quenched with 12 μl of a quench solution containing 0.8% formic acid, 16% glycerol, 1.4 M guanidine HCl, and 100 mM tris(2-carboxyethyl)phosphine (TCEP). The quenched samples were incubated on ice for 5 min, frozen on dry ice, and stored at −80°C. Procedures for pepsin digestion from DXMS have been described elsewhere (28). In addition, nondeuterated samples (incubated in the H2O buffer mentioned above) and equilibrium-deuterated back-exchange control samples (incubated in D2O buffer containing 0.5% formic acid overnight at 25°C) were prepared as described elsewhere (29). The centroids of the isotopic envelopes of nondeuterated, functionally deuterated, and fully deuterated peptides were measured using HDExaminer and then converted to corresponding deuteration levels with corrections for back exchange (30).
Isolation of neutralization escape mutants.
A crude stock of mutant VACV was prepared from cells that were infected with VACVWR in the presence of ethyl methanesulfonate (Sigma-Aldrich). Ethyl methanesulfonate was present in the culture medium at 500 μg/ml from 2 h to 6 h postinfection. The mutant viral stock was mixed with M12B9 antibody at a final concentration of 100 μg/ml for 1 h, and the mixture was applied to fresh cells. After 2 h of incubation, the mixture was removed from the cells and crude viruses subsequently were prepared. This process was repeated 3 to 4 times with a constant antibody concentration but with increasingly less crude viruses than the previous round. Clones of viruses were plaque purified from the final viral stock, and individual clones were tested for neutralization by M12B9. Clones that resisted M12B9 neutralization were selected, and their L1 gene was sequenced.
Fab digestion and purification.
Purified M12B9 and 8C8 MAbs (mouse IgG2a and IgG2b, respectively) at ∼1 mg/ml were incubated with 2% (wt/wt) activated papain for 4 h at 37°C in digestion buffer (50 mM sodium acetate [NaOAc], pH 5.5). Papain was activated by incubating 24.4 μl papain solution (Sigma stock solution at 20.5 mg/ml) with 100 μl 10× papain buffer (1 M sodium acetate [NaOAc] [pH 5.5], 12 mM EDTA) and 100 μl cysteine (12.2 mg/ml) for 15 min at 37°C. The papain digestion was stopped by adding 13 mM iodoacetamide (IAM). Samples were dialyzed overnight against PBS. Subsequent protein A purification was performed to remove any uncleaved IgG and Fc fragments. The protein A flowthrough containing Fab was concentrated and purified by size-exclusion chromatography (SEC) on a Superdex 200 column using 20 mM Tris, pH 8.0, 200 mM NaCl.
Fab-Ag complex preparation.
L1 and M12B9-Fab were mixed together at a L1:Fab stoichiometric ratio of 1.5:1 and at a low concentration (<1 mg/ml). The L1/Fab complex then was concentrated to a volume of 1.2 ml using centrifugal filtration devices (Amicon Ultra 15; 10-kDa-molecular-weight cutoff) and subjected to SEC purification (Superdex 200 16/60) to separate the L1/Fab complex from any excess of L1. Before crystallization, the nature of the complex was verified by nonreducing SDS-PAGE. Fractions corresponding to the complex were pooled and concentrated to 6.2 and 9.5 mg/ml in SEC buffer using centrifugal filtration devices.
Crystallization and structure determination.
Initial crystallization experiments were carried out by sitting-drop vapor diffusion in a 96-well format, using a Phoenix liquid-handling robot (Art Robbins Instruments), with a panel of commercial sparse-matrix screens (PEG/Ion 1 and 2 from Hampton Research, Wizard 2 from Emerald Biosciences, JCSG+ Suite from Qiagen, and JBScreen 6 from Jena BioScience). Quality diffracting crystals of L1/M12B9-Fab complex were obtained manually at RT by mixing 0.5 μl of protein solution at 9.5 mg/ml with 0.5 μl of precipitant [100 mM Tris, pH 7.0, 20% (wt/vol) polyethylene glycol (PEG) 3000, and 200 mM Ca(OAc)2] and seeding with initial crystals obtained at 6.5 mg/ml. Crystals were flash-frozen at 100 K in mother liquor containing 20% glycerol. Diffraction data were collected at the Stanford synchrotron radiation light source (SSRL), beamline 11.1 (366 images, 0.5° oscillation, 5.0-s exposure, 2.95-Å edge resolution) and processed with iMosflm software with space group p3121 (31). Scaling up to 3.1 Å was performed with Scala (32), and crystal structure was determined by molecular replacement (MR) with Phaser MR, allowing alternative space groups of the common point group p321 as possible solutions; both are part of the CCP4i suite (32, 33). Starting MR templates were unbound L1 (PDB code 1YPY) and OKT3-Fab (extracted from PDB code 1SY6), individualized as two components (Fv and constant domains). OKT3 was chosen as a template because it presents the highest sequence identity to M12B9-Fab heavy chain (HC) (34). The model was refined by performing multiple runs of model building in Coot (35, 36), followed by maximum-likelihood restrained refinement with Refmac 5 (36). The final model (R/Rfree = 21.17/26.39%) ranks in the 100th percentile for clashes and geometry compared to structures of similar resolution (37). Figures were generated using Pymol (http://www.pymol.org). Buried surface areas were reported as the output from the PISA server (38), and shape correlation (Sc) values were calculated with CCP4i.
Antibody sequencing.
Total RNA from 300 μl hybridoma cells in solution was isolated using the NucleoSpin RNA II kit according to the manufacturer's instructions (Macherey-Nagel). cDNA was amplified using the OneStep reverse transcription-PCR kit (Qiagen). The reverse transcription-PCR was performed using primers 5′MsVHE and 3′Cy1 (for isotype IgG1 antibodies M2E9 and M7B6), 3′Cy2c outer (for isotype IgG2a antibodies M12B9 and 39D4) or 3′Cy2b outer (for isotype IgG2b antibody 8C8) for the heavy chains, and primers 5′mVκ and 3′mCκ for the kappa light chains (LC) (39). The cycling profile was slightly modified from the manufacturer's recommendations and was set up as 1 cycle of 30 min at 50°C and 15 min at 95°C; 40 cycles of 30 s at 94°C, 45 s at 60°C (for heavy chains)/58°C (for light chains), and 55 s at 72°C; followed by 1 cycle of 10 min at 72°C and a 12°C cool down. PCR products were verified by gel electrophoresis with an ∼500-bp product for heavy chains and ∼450-bp product for light chains. Afterwards, PCR products were purified using the QIAquick PCR purification kit (Qiagen) and subsequently sequenced by Invitrogen (provided with the respective 5′ primer for heavy and light chains). Sequences include V-D-J regions for heavy chains and V-J regions for light chains. Finally, antibody germ lines were determined using IMGT's V-Quest service (40).
Alternatively, an Illumina MiSeq library preparation was performed by standard Illumina methods. Briefly, the Nextera XT DNA sample preparation kit uses an engineered transposome to fragment and tag (“tagment”) input DNA simultaneously, adding unique adapter sequences in the process. A limited-cycle PCR using those adapter sequences was performed to amplify the inserted DNA. The PCR also adds index sequences on both ends of the DNA, enabling dual-indexed sequencing of pooled libraries on the Illumina MiSeq instrument to generate approximately 100,000 to 300,000 paired-end reads per sample. Data then were analyzed via IMGT/HighV-QUEST.
L1 site-directed mutagenesis.
Three individual L1 mutants, N27A, Q31A, and D35A, were generated by site-directed mutagenesis of the L1 wild-type expression vector using the QuikChange II mutagenesis kit (Agilent Technologies) and verified by sequencing. The protein was produced as reported above and used for subsequent binding studies.
Real-time binding assay using BLI.
To test the effect of the single-alanine mutations on antibody binding, biolayer interferometry (BLI)-based assays using the OctetRED96 instrument (ForteBio, Inc.) were performed. As L1 nonspecifically bound to mouse Fc antibody capture chips, all assays were performed by immobilizing L1 on Ni-NTA biosensors via its C-terminal hexahistidine tag. There was no measurable dissociation of L1 from the Ni-NTA tips. Binding kinetics of WT-L1 and mutants to M12B9-Fab, 8C8-Fab, and 39D4-IgG were determined using Octet Data Analysis 7.1 software (Forte Bio, Inc.) with curve-fitting statistics. Ni-NTA biosensors were loaded with 20 nM L1 protein in 1× kinetics buffer (PBS, pH 7.4, 0.002% Tween 20, 0.01% BSA) over 5 min. L1-loaded tips were tested against 2-fold serial dilutions of Fab and IgG analytes with a 50 nM starting concentration of M12B9-Fab and 8C8-Fab and 400 nM starting concentration of 39D4-IgG. The association steps were performed over 10 min, and dissociation steps were performed over 15 min. The buffer control was subtracted from raw data, and curves were aligned to the baseline. The dissociation constant (KD), on rate (kon), and off rate (koff) were determined by global fitting of association and dissociation steps for all dilutions using a 1:1 binding model.
Fluorescence microscopy.
BHK cells grown on coverslips were infected with WT-GFP+, a strain of VACVWR that expresses GFP (41), at 0.5 PFU/cell. At 8 h postinfection, the cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.5% Saponin (Sigma-Aldrich) for 5 min, blocked with 10% FBS for 60 min, and stained with 1 μg/ml of anti-L1 MAb for 1 h and goat anti-mouse antibody conjugated with Cy3 for one additional hour. The DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). For virion staining, sucrose gradient-purified VACVWR virions were absorbed to glass coverslips coated with fibronectin (BD Biosciences) and fixed with 4% paraformaldehyde. The virions then were stained with anti-L1 MAb and DAPI as described above.
Accession numbers.
The GenBank accession numbers for the antibody sequences determined in the course of this work were deposited in GenBank under accession numbers KM116467, KM116468, KM116469, KM116470, KM116471, KM116472, KM116473, and KM116474.
RESULTS
Generation of anti-L1 MAbs.
In our previous screens of B cell hybridomas derived from mice immunized with live VACV (22), no anti-L1 hybridomas were found. To increase the chance of getting anti-L1 MAbs, we modified the immunization strategy by performing additional boosting with recombinant L1 protein after immunizing the mice with live VACV. Initially, a GST fusion of L1(1-185) was used in the boosting, and 2 anti-L1 MAbs (8C8 and 39D4) were obtained. Subsequently, a His-tagged L1 (1-185) protein that had undergone refolding was used, and 3 additional anti-L1 MAbs (M12B9, M2E9, and M7B6) were obtained (Table 1). All 5 L1 antibodies bound refolded L1 protein in ELISA (shown later) and immunoprecipitated L1 protein from VACV-infected cells (data not shown). Cross-blocking ELISA was performed, and the MAbs clustered into three distinct groups (Fig. 1). 8C8 and 39D4 each have a unique epitope, whereas M2E9, M7B6, and M12B9 share a mutual binding site.
TABLE 1.
Summary of immunization scheme and characteristics of anti-L1 MAbs
| MAb group and clone | Immunization scheme | Isotype | Epitope |
|---|---|---|---|
| I | |||
| M2E9 | Live VACV plus refolded L1(1-185) | IgG1 | Conformational |
| M7B6 | Live VACV plus refolded L1(1-185) | IgG1 | Conformational |
| M12B9 | Live VACV plus refolded L1(1-185) | IgG2a | Conformational |
| II | |||
| 8C8 | Live VACV plus GST-L1(1-185) | IgG2b | Conformational |
| III | |||
| 39D4 | Live VACV plus GST-L1(1-185) | IgG2a | Linear (121-140) |
FIG 1.
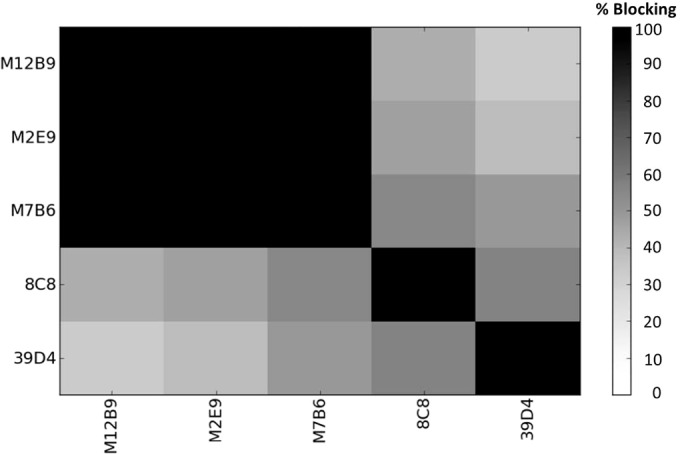
Cross-blocking results for five L1 MAbs. Anti-L1 antibodies were tested for cross-blocking ability. 8C8 and 39D4 were found to exclusively bind sites 2 and 3, respectively. M12B9, M2E9, and M7B6 share a mutual binding site, 1.
Group I anti-L1 MAbs potently neutralized MV.
We tested the ability of the anti-L1 MAbs to neutralize VACV MV in two different neutralization assays (Fig. 2). First, Vero E6 cells were incubated overnight with purified VACVWR MV expressing a green fluorescent protein (GFP) in the presence or absence of antibody and complement. Samples were evaluated the next day using a flow cytometer. Group I MAbs (M12B9, M2E9, and M7B6) neutralized more than 70% of the viruses at 20 μg/ml (Fig. 2A). In contrast, 8C8 did not neutralize the virus, while 39D4 neutralized less than 20%. The level of neutralization achieved by the group I MAbs was significantly higher than that by any other in-house MV neutralizing antibodies at similar concentrations. In the absence of complement, MAbs against other major MV antigens, like A27, H3, and D8, neutralized a maximum of 15% MV. They neutralized closer to 70% only in the presence of complement. In contrast, complement did not further enhance the neutralization by 39D4, although 39D4 was of the complement-fixing isotype.
FIG 2.

In vitro neutralization assay with anti-L1 MAbs. Anti-L1 antibodies have been tested for their ability to neutralize in vitro. (A) FACS-based neutralization assay featuring anti-L1 MAbs M12B9, M2E9, M7B6, 39D4, and 8C8. Anti-H3 MAbs (41 and JH4), anti-A27 MAbs (1G6 and 12G2), and anti-D8 MAbs (JE10 and LA5) were used as controls. All antibodies were used at a final concentration of 20 μg/ml. Anti-L1 MAbs 39D4 and 8C8 did not neutralize the virus in the absence or presence of complement. Anti-L1 group I MAbs (M12B9, M7B6, and M2E9) are capable of neutralizing in the absence of complement, whereas control MAbs required the presence of complement to provide neutralization. (B, top) Neutralization levels for two L1 MAbs (M12B9 and M2E9) and one control MAb for A27, H3, and D8 each in the absence of complement. (Bottom) Results in the presence of complement. A 5-fold titration of antibodies was done for all antibodies, starting with a concentration of 20 μg/ml (green bars). Consistent with the FACS results, the 2 anti-L1 MAbs were able to neutralize in a complement-independent manner, whereas anti-H3 MAb 41 was not able to neutralize in the absence of complement. Error bars are based on four data points per sample (averages from duplicates in two independent experiments, normalized).
We then tested the ability of the group I anti-L1 MAbs to neutralize VACVWR MV with a plaque reduction assay to confirm our initial results. Purified VACVWR MV was incubated in the presence or absence of antibody and complement for 1 h and then tested for infectivity in a plaque assay. L1 antibodies reduced plaque numbers dramatically in the absence and presence of complement, showing strong neutralizing ability (Fig. 2B). Anti-H3 MAb 41 did not neutralize the virus in the absence of complement (Fig. 2B, top) but provided excellent neutralization in the presence thereof (Fig. 2B, bottom). Anti-A27 MAb 1G6 showed only a small effect in this particular assay. However, a neutralization-enhancing effect was detectable in the presence of complement. Anti-D8 MAb JE10 showed similar results. Based on those results, we conclude that group I anti-L1 MAbs can potently neutralize MV in a largely complement-independent manner. Although complement is not required for potent neutralization of VACV by the group I MAbs, it did enhance the neutralization by the MAb of the complement-fixing isotype (M12B9) at low antibody concentration.
Group I anti-L1 MAbs protected against vaccinia virus infection.
Anti-L1 MAbs were tested in two in vivo vaccinia virus protection systems. First, they were assessed for their ability to protect BALB/c mice against a lethal intranasal dose (105 PFU) of VACVWR. Anti-B5 MAb B126 provided outstanding protection against weight loss at day 7 (P < 0.0001) and death (P = 0.005) after lethal VACVWR infection (Fig. 3A and B). As expected, anti-A10 MAb BG3.1 had no effect, and neither did anti-L1 MAb 39D4 (P = 0.27 for weight loss, P = 0.60 for death). 8C8 had very modest efficacy (P = 0.0088 for weight loss, P = 0.0005 for death). Anti-L1 MAbs M12B9, M2E9, and M7B6 all provided significant protection against death (50%, 100%, and 80%, respectively) but poor protection against weight loss (P = 0.062, P = 0.0075, and P = 0.011, respectively). This indicates that EV is the primary virion form important in lung infection of BALB/c mice, and there are limited opportunities for anti-L1 Abs to neutralize virus in the lung. Anti-L1 MAbs then were tested in a second in vivo protection model, SCID mice infected with 105 PFU ACAM2000 retro-orbitally (Fig. 3C to E). This model is much more MV dependent than intranasal infection with VACVWR (12, 23, 42, 43). Anti-A10 MAb BG3.1 was used as a negative control, and anti-H3 #41 was used as a positive control. In this model, M12B9, M2E9, and M7B6 each provided outstanding protection against weight loss (P < 0.001) (Fig. 3C), death (P = 0.0002) (Fig. 3D), and pox lesions (clinical score) at day 46 (23) (Fig. 3E). 8C8 again had modest efficacy (P = 0.042 for weight loss, P = 0.068 for death, and P = 0.0026 for pox at day 21). 39D4 had some measurable activity but was the least effective of the MAbs, as measured by protection against weight loss (P = 0.098) or pox development at day 21 (P = 0.22). From these two studies, we conclude that anti-L1 MAbs M12B9, M2E9, and M7B6 are highly effective in vivo at neutralizing MV but not EV and provide excellent protection in proportion to the involvement of MV in disease pathogenesis.
FIG 3.

In vivo protection assays with anti-L1 MAbs. (A and B) Protection of BALB/c mice against lethal intranasal VACVWR. (A) Body weights. (B) Survival. (C to E) Protection of SCID mice against VACV ACAM2000. Also shown are body weight (C), survival (D), and clinical scores (E) over time. There were 6 to 8 mice per group. Significance ranges were the following: *, P = 0.05 to 0.01; **, P = 0.01 to 0.005; ***, P = 0.005 to 0.0001; ****, P < 0.0001; ns, not significant.
Group I anti-L1 MAbs failed to protect against ectromelia virus infection.
One goal of researching immunity induced by smallpox vaccination is to be able to develop vaccination or treatment strategies that work across different poxviruses in order to be able to quickly deploy such strategies in the event of a different poxvirus emerging as a human health threat. Given that the anti-L1 antibodies in group I were highly protective against vaccinia virus challenge, we asked if they also could protect from infection with ectromelia virus (ECTV). We chose M12B9 as a representative for our group I antibodies.
C57BL/6 mice were intranasally challenged with 1,000 PFU of ECTV on day zero (D0) (∼10× 50% lethal dose [LD50]). Monoclonal antibodies (100 μg) were administered by intraperitoneal injection 1 day prior to challenge (D−1). As shown in Fig. 4, B126 provided protection against a lethal challenge with ECTV (P = 0.002); however, M12B9 did not protect against death (P = 0.13), as all animals were dead by D12. The reason why M12B9 did not protect against ectromelia virus will become clear later.
FIG 4.
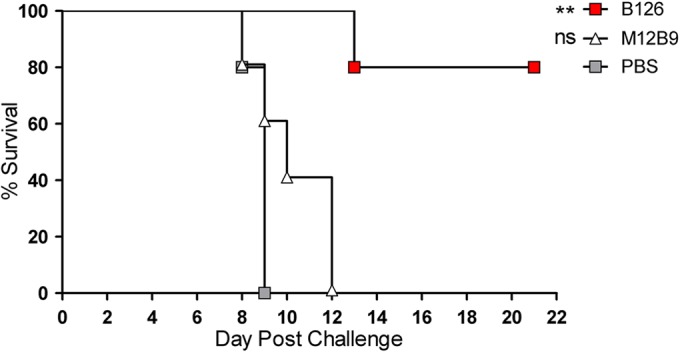
Survival of C57BL/6 mice treated with M12B9 and B126 following a lethal ectromelia virus challenge. Survival of C57BL/6 mice treated with anti-L1 M12B9 MAb at D−1, followed by a 1,000-PFU ECTV challenge at D0. By day 12 of the experiment, all mice of group M12B9 were dead, but B126-treated mice were protected. There were 5 mice per group.
Epitope mapping of group III anti-L1 MAb.
To understand why the anti-L1 MAbs have such a big difference in neutralization abilities, we continued characterization of those MAbs by mapping their epitopes. ELISAs were performed with a series of 20-mer peptides spanning L1, overlapping by 10 residues. All anti-L1 MAbs recognized L1 protein (1-185). However, except for 39D4, no other MAbs recognized any L1 peptides, indicating that they bind conformational epitopes. 39D4 recognized a linear peptide spanning residues 121 to 140 (Fig. 5A). Truncation analysis of the peptide showed that the binding site was contained in residues 123 to 132 (Fig. 5B and D). Alanine substitutions of peptide 123-132 identified four residues (N124, K125, I128, and I132) as critical for binding by 39D4 (Fig. 5C and E).
FIG 5.

Linear epitope determination of 39D4 MAb. (A) Summary of epitope mapping of VACV anti-L1 MAbs by linear peptide ELISA. All 5 MAbs bind to the full-length L1 protein, but only 39D4 binds an individual peptide (121-140). (B and D) Truncation assay discovered the epitope to be amino acids 123 to 132 of peptide 121-140. A low optical density (OD) indicates that the peptide fragment preincubated with the antibody fully occupies the antibody's episome and prevents it from binding to plate-bound full-length peptide. (C and E) Alanine scan of the linear (lin.) epitope 123-132 revealed N124, K125, I128, and I132 to be key residues for antibody binding. A lowered OD indicates the decreased ability of the antibody to bind to that particular peptide; thus, the alanine-substituted residue in the original peptide has a large effect on the binding ability. Dashed lines indicate a cutoff for positive results (OD of 1.0). w/o, without.
Epitope mapping of group I anti-L1 MAb.
As group I antibodies potently neutralize MV, we next focused on mapping the epitopes of group I MAbs. Hydrogen/deuterium exchange mass spectrometry experiments were performed with M12B9. L1 (1-185) alone and in complex with M12B9 was incubated with deuterium for 10, 30, 100, and 1,000 s. Following pepsin digestion, L1 peptide fragments were detected by mass spectrometry. The rate of deuterium exchange is highest in solvent-exposed amino acids. Regions 25-34 and 113-131 of L1 have the most slowing of deuterium exchange in the presence of M12B9 (Fig. 6), suggesting that they are part of the epitope.
FIG 6.

Epitope mapping of anti-L1 M12B9 MAb using DXMS. Deuterium exchange data show differences in deuteration levels in the presence compared to the absence of MAb binding at four time points (10 s, 30 s, 100 s, and 1,000 s). Slower deuterium exchange is marked in blue, and faster exchange is marked in red. Residues 25 to 34 and 113 to 131 (red boxes) show the most marked slowing, indicating regions likely incorporating epitope binding sites.
To identify key epitope residues of group I MAbs, VACV mutants that resisted the neutralization by M12B9 were selected. Cells infected with VACVWR were incubated in the presence of ethyl methanesulfonate (EMS) to induce transition mutations in viral DNA. M12B9 then was used to neutralize the mutant viruses. The titer of the initial viral stock was greatly reduced by M12B9. However, after passaging the viruses three times following M12B9 neutralization, the passaged viral stock was no longer significantly neutralized by M12B9. Clones of the mutants were plaque purified, and two of the clones showed complete resistance to neutralization by M12B9. The L1 coding sequences of the mutants were determined and found to have a single-amino-acid substitution mutation at residue D35. One mutant had a D35N substitution, while the other one had a D35Y substitution (Fig. 7). Both escape mutants also were resistant to neutralization by the other two group I MAbs (M2E9 and M7B6) (Fig. 7B), indicating that D35 is essential for the binding of all group I MAbs. Previously, D35N substitution in L1 was found to be responsible for escaping the neutralization by a highly potent anti-L1 MAb, 2D5 (17). Therefore, the group I MAbs and 2D5 target a similar epitope that includes D35 as a critical residue. Amino acid comparison of L1 and its ectromelia ortholog (EVM072) showed four differences (Fig. 7C), one of which is residue 35 (D35 in vaccinia versus N35 in ectromelia). This difference could explain the lack of in vivo protection against ectromelia virus by M12B9.
FIG 7.

Identification of Asp35 as a key epitope residue for group I MAbs. (A) Part of the L1 sequence of two VACV mutants that are resistant to neutralization by M12B9. The two mutants were plaque purified from VACV that had been mutagenized with ethyl methanesulfonate and escaped the neutralization by M12B9. The L1 coding sequence of the mutants was determined. Shown in the dashed box is a single-nucleotide substitution compared to the wild-type sequence. (B) VACV mutants with D35N or D35Y substitution in L1 were resistant to neutralization by all group I anti-L1 MAbs. The abilities of the anti-L1 MAbs to neutralize wild-type or mutant VACV were determined with the plaque reduction assay. The labels below the x axis indicate the viruses that were used for neutralization. (C) Amino acid comparison of L1 and its ectromelia ortholog (EVM072) shows four differences, one of which is residue 35 (D35 in vaccinia versus N35 in ectromelia).
Crystal structure of M12B9/L1 complex.
We also determined the structure of L1 in complex with the Fab domain of M12B9 at a resolution of 3.1 Å and compared its mode of L1 binding with that of the previously determined 7D11 antibody (18) (Table 2). Four copies of the Fab/L1 complex were present in the asymmetric unit. M12B9 Fab binds to four long loops that connect the 4-helix bundle at the tip of L1, presumably away from the viral membrane, in an orientation that is very similar to that of the antibody 7D11 (Fig. 8A).
TABLE 2.
Data collection and refinement statistics
| Parameter | Value(s) for L1/M12B9-Fab |
|---|---|
| Data collection statistics | |
| Space group | p3221 |
| Cell dimensions | |
| a, b, c (Å) | 102.83,102.83, 238.35 |
| α, β, γ (°) | 90, 90, 120 |
| Resolution range (Å) | 47.67-3.10 |
| Outer shella | 3.27-3.10 |
| No. of reflections | 27,308 |
| Rmergea (%) | 19.4 (89.4) |
| Rpima (%) | 6.7 (30.5) |
| Multiplicitya | 9.1 (9.4) |
| Avg I/σIa | 11.2 (2.8) |
| Completenessa (%) | 99.9 (100.0) |
| Refinement statistics | |
| No. of atoms | |
| Total | 9,196 |
| Protein | 9,196 |
| Ramachandran plot (%) | |
| Favored | 95.29 |
| Allowed | 99.83 |
| RMSD | |
| Bonds (Å) | 0.004 |
| Angles (°) | 0.865 |
| B factor (Å2) | |
| Protein | 64.14 |
| R factor (%) | 21.17 |
| Rfree (%) | 26.39 |
Values in parentheses refer to the highest-resolution shell.
FIG 8.

M12B9-MAb targets the same epitope as 7D11-MAb. (A) Superimposed structures of L1 bound to M12B9 or 7D11-Fab domains. Models were superimposed on L1 using the secondary-structure matching algorithm (SSM) within Coot 0.7 (root mean square deviations [RMSD] of 0.53 Å for L1 using PDB code 2I9L). For simplicity, only L1 of the M12B9 complex is shown in gray throughout. (B) M12B9 footprint on L1 surface colored by contacting CDR, with H1, H2, and H3 shown in green, cyan, and blue, respectively. D35 occupies a central position in the epitope. The associated table lists contact residues. Boldface black labels highlight residues chosen for site-directed mutagenesis. (C, D, E, and F) Detailed contacts of M12B9/L1 and comparison to 7D11/L1. CDRs of M12B9 are shown in orange superimposed on 7D11 (gray), while L1 is gray with side-chain orientation drawn as thin lines. Detailed MAb/L1 interactions are shown for H1 (C), H2 (D), and H3 (E). (F) Framework residue R74 forms a salt bridge with D62 of L1. Yellow dashed lines indicate hydrogen bonds or salt bridges (for L1/M12B9-Fab). Note how H2 in the L1/7D11 interface forms almost identical contacts with L1 residues.
L1 residues E25, N27, A28, S29, Q31, T32, K33, D35, S58, A59, D60, A61, D62, K125, K127, and S153 are bound directly by the M12B9 heavy chain (Fig. 8B) (residues in boldface elicit salt bridges). The heavy chain forms a total of 11 hydrogen bonds (H-bonds), five salt bridges, and 99 Van der Waals (VdW) interactions. The HC-L1 interface is characterized by a buried surface area of 1,366 Å2 and a shape complementarity (Sc) of 0.70 versus 0.65 for the 7D11 heavy chain (44). As expected from previous Ag-Fab structures, the heavy chain dominates Ag-MAb interactions (45).
The LC-L1 interface has a much lower buried surface area (219 Å2), and it has an Sc value of 0.62; that of the 7D11/L1 complex is 0.26, suggesting the light chain, despite forming fewer contacts, also interacts specifically with L1. In total, the M12B9 light chain forms one potential salt bridge between complementarity-determining region (CDR) L3 residue D98 and L1 residue K33 and one hydrogen bond between L1 residue Q31 and N34 of CDR L1 (Fig. 8B). In the 7D11/L1 structure, there were no interactions between the light chain and L1 within a 4-Å distance (18).
The L1/7D11 and L1/M12B9 complex structures superimpose closely with a low root mean square deviation (RMSD) over all Cα atoms (L1, 0.53 Å; HC, 1.00 Å; LC, 1.51 Å), which correlates well with the sequence identity, especially within the CDRs of the heavy chain that binds L1 (Fig. 8). Despite the relatively high structural homology of the Fab domains, Fv light chains present a relatively low sequence identity (67.6%), consistent with the fact that it barely participates in L1 binding. The heavy chains display a high sequence identity even for CDRs H1 and H2 (83.2%), along with a low RMSD (1.00 Å), with the exception of H3 residues that are not conserved (Fig. 8). Therefore, we compared how these subtle sequence differences affect binding to the same epitope. In the M12B9-Fab/L1 complex, H3 elicits a few extra VdW contacts compared to L1/7D11; while 7D11 uses H3:D102 to form an H bond with L1 residue K33, M12B9 instead uses H3:Q101 to hydrogen bond to the backbone oxygen of N27 of L1 (distance [d], 3.2 Å). Also, we observed increased VdW contacts relative to CDR H3 in our model, elicited by H3 residues 101 to 104 and L1 residues 27, 28, 29, 31, 32, and 33. This contrasts with the fewer interactions found in the 7D11 complex (VdW interactions between L1 residues Q31 and T32 and H3 residues D102 and Y104).
However, the bulk of specific interactions (H bonds and salt bridges) are formed by CDRs H1 and H2 in a manner very similar to that reported for the 7D11 complex, while CDR H3 adopts an orientation similar to that of 7D11 but almost exclusively contacts L1 through nondirected VdW interactions (Fig. 8E). H1 elicits five hydrogen bonds and 36 VdW contacts the L1 antigen, while H2 forms four hydrogen bonds, one salt bridge (E59 with L1 residue K127), and 36 VdW contacts. A detailed Ag-Fab interface contact comparison of M12B9/L1 and 7D11/L1 complexes is shown in Fig. 8B to F. L1 residue D35 was suggested to be a critical residue for viral escape (18) (Fig. 7). Both antibodies 7D11 and M12B9 interact with the D35 carboxyl group via two hydrogen bonds (H1:W33 side chain Nε and H2:Y50 hydroxyl). Therefore, those interactions appear to crucial for antibody binding. Interestingly, an additional salt bridge is formed by non-CDR heavy-chain residue R74 with L1 residue D62 (d, 2.80 Å), which appears not to be formed in the 7D11 structure (Fig. 8F).
Sequence characteristics of highly potent neutralizing MAbs targeting L1.
Our group I MAbs and the previously described L1 MAb 7D11 are highly potent MV neutralization antibodies, and they target a similar epitope. To gain some insight on antibody sequences that determine the recognition of the epitope, the sequences of the three group I MAbs were determined and compared to that of 7D11. The heavy chains also were compared to the germ line IGHV1S26*01, as all four MAbs originate from this germ line. CDR3 regions of the heavy chains are quite different, but CDR1 and CDR2 regions of the heavy chains are highly conserved between the four MAbs (Fig. 9A). The light-chain sequences of the four L1 MAbs were quite divergent (Fig. 9B). According to 7D11/L1 and M12B9/L1 structures, the heavy chain dominates the binding with L1. 7D11 has 12 heavy-chain residues contacting L1 in the crystal structure (T30, R31, W33, Y50, N52, S54, T55, G56, Y57 T58, D102, and Y104), while M12B9 has four additional heavy-chain residues contacting L1 in the structure (F32, E59, Q101, and N103). Among these residues, the CDR1 and CDR2 residues are nearly identical between the four MAbs except for a G56D substitution for two of the MAbs (M7B6 and M2E9), a conservative Y32F substitution for M12B9, and a conservative Y57F substitution for M2E9. In contrast, while one of the CDR3 residues that contact L1 in the crystal structure is conserved (Y104 or W104), the rest (residues 101, 102, and 103) are not conserved between the four MAbs, suggesting that the CDR3 residues play a limited role in binding L1. Many of the critical CDR1 and CDR2 residues (30, 32, 50, 52, 54, 55, 57, and 58) also are conserved in the germ line. Only residues 31, 33, and 59 have nonconservative substitutions compared to the germ line sequence.
FIG 9.

Sequence alignment of 4 anti-L1 MAbs. (A) Heavy-chain sequence alignment of germ line IGHV1S26*01 versus 7D11, M12B9, M7B6, and M2E9. Identical amino acids are marked by a dot, green boxes are marked CDR1 through CDR3, and dashes indicate where the germ line sequence ends. Residues in CDR1 and CDR2 are highly conserved between all 4 MAbs and the germ line, whereas residues in CDR3 show high diversity. 7D11 has 12 heavy-chain residues contacting L1 in the crystal structure (T30, R31, W33, Y50, N52, S54, T55, G56, Y57 T58, D102, and Y104), while M12B9 has four additional heavy-chain residues contacting L1 in the structure (F32, E59, Q101, and N103), highlighted in red. Many of the critical CDR1 and CDR2 residues (30, 32, 50, 52, 54, 55, 57, and 58) are either conserved in the germ line or have conservative substitutions (black arrows). Only residues 31, 33, and 59 have nonconservative substitutions compared to the germ line sequence (red arrows). (B) Light-chain sequence alignment of 7D11 versus M12B9, M7B6, and M2E9. Identical amino acids are marked by a dot, green boxes mark CDR1 through CDR3, and gaps (insertions) are marked by a tilde symbol. Whereas M12B9 and M2E9 show quite similar sequences, overall all 4 MAbs show a highly diverse CDR sequence. Light chains originate from different germ lines; thus, no comparison to a mutual germ line was possible.
Binding of anti-L1 MAbs with L1 protein and virion.
To assess whether differences in binding affinity could account for differences in neutralization among the 3 groups of anti-L1 MAbs, we measured binding affinities of the MAbs with recombinant L1 protein by biolayer interferometry (BLI). Refolded L1(1-185) protein was immobilized on an Ni-NTA tip, and its binding to different MAbs was monitored in real time with BLI (Fig. 10). For M12B9 (group I) and 8C8 (group II), purified Fab fragments generated by papain digestion of IgG were used for the binding study. The binding affinity was approximately 200-fold higher for M12B9 (KD, 0.09 nM) than for 8C8 (KD, 17 nM), predominantly due to the difference in off rate (Table 3). For 39D4 (group III), its Fab fragment had a very low binding signal due to a much lower binding affinity. Therefore, undigested 39D4 IgG was used for the binding study, and it had a modest affinity to L1 (KD, 32 nM).
FIG 10.

Real-time binding curves using biolayer interferometry. Curves show binding of analytes M12B9-Fab, 8C8-Fab, and 39D4-IgG in solution to wild-type antigen L1 (residues 1 to 184) and indicated mutants immobilized on Ni-NTA biosensors. Association (600 s) and dissociation (900 s) steps are represented. Curves are colored according to their specific analyte (Fab or IgG) concentration (middle right, 1.56, 3.12, 6.25, 12.5, 25, 50, 100, and 200 nM). The affinity constant (KD) is reported as an average from duplicate assays with standard errors.
TABLE 3.
Binding kinetics of WT-L1 and mutants to M12B9-Fab, 8C8-Fab, and 39D4-IgG
| Specificity group and L1 variant | KD (nM) | Kon (104 M−1 s−1) | Koff (10−3 s−1) | R2 | X2 |
|---|---|---|---|---|---|
| Group I (M12B9-Fab) | |||||
| Wild type | 0.09 ± 0.04 | 51.25 ± 2.25 | 0.048 ± 0.023 | 0.99 ± 0.001 | 0.87 ± 0.20 |
| N27A | 0.05 ± 0.04 | 55.90 ± 3.40 | 0.064 ± 0.009 | 0.99 ± 0.001 | 0.81 ± 0.10 |
| Q31A | 0.11 ± 0.08 | 49.00 ± 1.60 | 0.11 ± 0.01 | 0.99 ± 0.001 | 0.85 ± 0.02 |
| D35A | No binding | NAa | NA | NA | NA |
| Group II (8C8-Fab) | |||||
| Wild type | 17.4 ± 2.6 | 11.2 ± 1.7 | 1.91 ± 0.01 | 0.99 ± 0.001 | 0.13 ± 0.02 |
| N27A | 14.5 ± 0.2 | 10.5 ± 0.9 | 1.53 ± 0.10 | 0.99 ± 0.01 | 0.38 ± 0.11 |
| D35A | 18.1 ± 1.1 | 8.43 ± 0.9 | 1.51 ± 0.07 | 0.97 ± 0.01 | 0.50 ± 0.05 |
| Group III (39D4-IgG) | |||||
| Wild type | 32.0 ± 2.1 | 2.00 ± 1.13 | 0.629 ± 0.371 | 0.99 ± 0.01 | 0.49 ± 0.09 |
| N27A | 18.5 ± 2.7 | 1.11 ± 0.33 | 0.195 ± 0.030 | 0.99 ± 0.001 | 0.77 ± 0.19 |
NA, not applicable.
To assess the contribution of predicted epitope residues to the binding of the antibodies, we performed similar BLI studies with recombinant L1 proteins containing single-alanine substitution (Fig. 10). Residues N27, Q31, and D35 were chosen for substitution based on the M12B9/L1 crystal structure. Remarkably, D35A substitution completely disrupted binding with M12B9, while it had no effect on binding with 8C8, illustrating the specific importance of D35 in binding with the group I antibodies. N27A or Q31A, however, had no significant effect on the binding with any of the L1 MAbs, suggesting that those residues are not crucial binding hot spots for any of the tested antibodies (Table 3).
The binding of the MAbs with native L1 protein expressed by VACV was assessed by immunofluorescence analysis. The group I MAb M12B9 stained viral factories (site of virion assembly) and many virion-size particles in infected cells (Fig. 11). It also stained purified virions, indicating that the MAb binds to native L1 and its epitope is well exposed on the MV surface. In contrast, group II MAb 8C8 stained viral factories but not virion-size particles in infected cells. It also failed to stain purified virions, indicating that while 8C8 binds to native L1, its epitope is either not exposed on the surface of the MV or the binding affinity is very low. The staining of infected cells by group III MAb 39D4 was barely above the level of background staining in uninfected cells, indicating that 39D4 has a very low binding affinity to native L1. These results are consistent with the different binding affinities of the antibodies to the recombinant L1 proteins shown in the BLI assay.
FIG 11.

Immunofluorescence analysis with anti-L1 MAbs. BHK cells were infected with a GFP-expressing VACV at an MOI of 0.5 PFU/cell for 8 h. The cells were fixed, permeabilized, and stained with the indicated anti-L1 MAb, followed by DAPI and goat anti-mouse IgG coupled to Cy3. The DAPI and antibody staining are shown separately in the left and middle panels, respectively. They are shown together with GFP fluorescence in the right panel. F, viral factory. For virion images, sucrose gradient-purified VACV virions were absorbed to glass coverslips coated with fibronectin and fixed with paraformaldehyde. The virions then were stained with DAPI and the indicated anti-L1 MAb. DAPI and antibody staining are shown separately in the left and middle panels, respectively. They are shown together in the right panel. The white arrows point to individual virion particles.
DISCUSSION
In this study, we developed and comprehensively characterized five anti-L1 MAbs. Group I of three MAbs (M12B9, M2E9, and M7B6) potently neutralized VACV in a largely complement-independent manner, while group II and III MAbs failed to neutralize VACV in the presence or absence of complement. By using a variety of techniques, the epitope of the group I MAbs was mapped to a conformational epitope with Asp35 as the key residue. The crystal structure of L1-M12B9 complex showed that the epitope is surface exposed and distal to the transmembrane domain. Indeed, M12B9 was able to bind to purified virion in an immunofluorescence assay. Thus, the potent neutralization by the group I MAbs could be explained by their high-affinity binding to a critical region of L1 that is well exposed on the virion surface. The epitope of group III MAb (39D4) was mapped by peptide ELISA to residues 123 to 132, which are largely surface exposed and adjacent to the group I epitope in the L1 structure. The 39D4 epitope overlaps extensively with the linear epitope (residues 118 to 128) of several previously described, potent VACV neutralizing antibodies (19). However, 39D4 binds to L1 with low affinity. Only the intact 39D4 IgG showed some binding to the recombinant L1 protein, while the Fab fragment did not bind at all. 39D4 also failed to bind significantly to the native L1 protein expressed by VACV in the cells or displayed on virions in an immunofluorescence assay. Therefore, we conclude that 39D4 fails to neutralize MV due to its low affinity to L1 on the virions. We have not mapped the conformational epitope recognized by the group II MAb 8C8. While 8C8 binds to recombinant L1 as well as native L1 protein expressed by VACV, its affinity to recombinant L1 is at least 200-fold less than that of the group I MAbs, and it does not bind to virions in an immunofluorescence assay. Therefore, we propose that 8C8 fails to neutralize MV mostly because its epitope is not well exposed on the virion.
An initial goal of our study was to expand the knowledge of immune epitopes in L1, an important neutralizing target in smallpox vaccine. Quite unexpectedly, we found that all three of our anti-L1 neutralizing MAbs recognized the same conformational epitope that was known to be the target of previously described murine anti-L1 MAbs, 2D5, 7D11, and 10F5 (17, 46). Our anti-L1 MAbs appear to have a lower virus neutralizing potency than that reported for 7D11 (50% inhibitory concentration of ∼400 ng/ml for M12B9 versus 16 ng/ml for 7D11 [19]), which could explain why the group I MAbs appear to be less protective than 7D11 in mouse models (47). Since antibodies targeting this common L1 epitope represent the most potent MV neutralizing antibodies ever reported in the literature and L1 is an important component of experimental subunit vaccines against orthopoxviruses, our study is important in that it provides a better understanding of the molecular basis of epitope recognition by these antibodies and how these antibodies could be elicited through immunization. Previously, only the 7D11 sequence had been reported, and its molecular basis for epitope recognition had been revealed by the crystal structure of 7D11 and L1 complex (18). In this study, we determined the sequences of three additional antibodies targeting this epitope and solved the structure of one of those antibodies in complex with L1. Sequence comparison between the 4 MAbs showed that the light chain and CDR3 of the heavy chain are quite different, but CDR1 and CDR2 of the heavy chain are highly conserved. The structures of 7D11/L1 and M12B9/L1 also showed that CDR1 and CDR2 of the heavy chain are used predominantly for binding the epitope. Together, these studies show that the binding of this neutralizing L1 epitope requires antibodies that have the appropriate heavy-chain CDR1 and CDR2 and can tolerate divergent light-chain and heavy-chain CDR3. Since heavy-chain CDR1 and CDR2 of these anti-L1 antibodies are highly homologous to a mouse germ line gene, IGHV1S26*01, only a very limited number (3 nonconservative substitutions) of somatic mutations would be required for the germ line gene to evolve into antibodies that recognize this L1 epitope. In contrast, the closest matching human germ line gene (IGHV1-8*02) would require 6 nonconservative substitutions. This may be one reason why L1 is an immunodominant neutralizing antibody target in mice but a less common target in humans (13). This also highlights the need for caution in extrapolating results of vaccine studies in mice to human vaccine design.
While antibodies targeting this L1 neutralizing epitope are among the most potent MV neutralizing antibodies, they are also vulnerable to neutralization escape due to their heavy reliance on a single L1 residue for binding. Previously, VACV with an L1 D35N substitution was found to escape neutralization by 2D5 (17). We discovered in this study that VACV with either L1 D35N or D35Y substitution completely escaped the neutralization by three anti-L1 neutralizing MAbs. While M12B9 binds with L1 protein with very high affinity, a single D35A substitution in L1 was able to completely abolish the binding. These results demonstrate that while D35 side chain is not critical for L1 function during viral replication, it is essential for the binding of a group of potent neutralizing antibodies. While alkylating agents were used to induce the escape mutants in previous and current studies, similar mutants may emerge naturally and be selected by neutralizing antibody response. L1 D35N substitution resulted from a transition mutation of guanine to adenine, which is the type of mutation that is expected to be caused by alkylating agents. In contrast, L1 D35Y substitution resulted from a guanine-to-thymine mutation, which is unlikely to be caused by alkylating agents and may have emerged spontaneously. Even more interestingly, although L1 orthologs in different orthopoxviruses have almost identical sequences, all ectromelia virus strains have Asn instead of Asp at residue 35 of the L1 orthologs. This explains why passive administration of M12B9 failed to protect mice from ectromelia virus challenge. As discussed above, it may be relatively easy to elicit in mice neutralizing antibodies targeting the L1 epitope with D35. Since mouse is the natural host for ectromelia virus, it is tempting to speculate that the D35N substitution in the L1 ortholog is an adaptation for ectromelia virus to escape this particular neutralizing antibody response.
At least 8 MV proteins (D8, H3, A26, A27, A13, L1, A28, and A17) are known to be the targets of neutralizing antibodies. However, for practical and technical reasons, vaccine formulation can only include a few selected immunogens. Therefore, it is important to know whether all MV targets are equally valuable or if some are much more important than others as targets for protective immunity. Anti-L1 MAbs are known to be very potent neutralizing antibodies, but they have rarely been compared directly with MAbs against other MV targets for their neutralization potency. We have recently developed a large panel of MAbs against VACV, including MV neutralizing antibodies targeting H3, D8, A13, and A27 (12, 22). This allows us to directly compare anti-L1 and many other anti-MV antibodies in terms of their neutralization potency. We find that the anti-L1 MAbs are distinct from other MV neutralizing antibodies in that they can completely neutralize MV independent of complement. In contrast, MAbs against H3, D8, A13, and A27 can achieve a high level of neutralization only if the antibodies are of the complement-fixing isotypes and complement is present in the experiment. This is despite the fact that there are more binding sites available on the surface of MV for these antibodies than for anti-L1 antibodies, as D8, H3, A13, and A27 are major MV proteins and are present on the MV at a molar ratio of at least 10:1 with respect to the L1 protein (48). The requirement of complement does not appear to be an unusual requirement for antibodies targeting a particular epitope, as multiple anti-D8 MAbs targeting four different epitopes all require complement for potent MV neutralization (data not shown). The reason for this difference in complement requirement may be that anti-L1 antibodies use a different mechanism than other antibodies in neutralizing MV. L1 plays an essential role in viral entry, so antibodies binding to a critical functional region of L1 should be sufficient to prevent viral entry. In contrast, A13 plays no direct role in viral entry, while D8 (49), H3 (50), and A27 (51) play a redundant role in viral attachment to cell surface receptors; thus, the occupation of all functional sites of these proteins by antibodies is not sufficient to prevent viral attachment or entry. Antibodies against these major MV proteins may have to rely on the binding of the complement to increase their footprints on the viral surface, so there will be sufficient steric hindrance to block the functions of other envelope proteins that are essential for viral binding or entry. The entry of VACV into cells requires virus-encoded multiprotein entry-fusion complex (EFC), which consists of at least 11 proteins (52). It is possible that antibodies against EFC members L1 or A28 could neutralize MV directly in a complement-independent manner, while antibodies against non-EFC membrane proteins (D8, H3, A13, A17, A26, and A27) neutralize MV by a different, complement-enhanced mechanism.
ACKNOWLEDGMENTS
We thank David Garboczi (NIH-NIAID) for supplying the L1 expression construct. We also thank the SSRL beamline 11.1 staff for their kind support.
Portions of this research were carried out at the SSRL, a Directorate of the SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393), and the National Center for Research Resources (P41RR001209).
This project has been funded in whole or in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract no. HHSN272200900048C and HHSN272201000021I.
Footnotes
Published ahead of print 16 July 2014
REFERENCES
- 1.Henderson DA, Inglesby TV, Bartlett JG, Ascher MS, Eitzen E, Jahrling PB, Hauer J, Layton M, McDade J, Osterholm MT, O'Toole T, Parker G, Perl T, Russell PK, Tonat K. 1999. Smallpox as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA 281:2127–2137 [DOI] [PubMed] [Google Scholar]
- 2.Parker S, Nuara A, Buller RM, Schultz DA. 2007. Human monkeypox: an emerging zoonotic disease. Future Microbiol. 2:17–34. 10.2217/17460913.2.1.17 [DOI] [PubMed] [Google Scholar]
- 3.Davies DH, Liang X, Hernandez JE, Randall A, Hirst S, Mu Y, Romero KM, Nguyen TT, Kalantari-Dehaghi M, Crotty S, Baldi P, Villarreal LP, Felgner PL. 2005. Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc. Natl. Acad. Sci. U. S. A. 102:547–552. 10.1073/pnas.0408782102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies DH, Molina DM, Wrammert J, Miller J, Hirst S, Mu Y, Pablo J, Unal B, Nakajima-Sasaki R, Liang X, Crotty S, Karem KL, Damon IK, Ahmed R, Villarreal L, Felgner PL. 2007. Proteome-wide analysis of the serological response to vaccinia and smallpox. Proteomics 7:1678–1686. 10.1002/pmic.200600926 [DOI] [PubMed] [Google Scholar]
- 5.Henderson DA. 2011. The eradication of smallpox–an overview of the past, present, and future. Vaccine 29(Suppl 4):D7–D9. 10.1016/j.vaccine.2011.06.080 [DOI] [PubMed] [Google Scholar]
- 6.Fulginiti VA, Papier A, Lane JM, Neff JM, Henderson DA. 2003. Smallpox vaccination: a review, part II. Adverse events. Clin. Infect. Dis. 37:251–271. 10.1086/375825 [DOI] [PubMed] [Google Scholar]
- 7.Buchman GW, Cohen ME, Xiao Y, Richardson-Harman N, Silvera P, DeTolla LJ, Davis HL, Eisenberg RJ, Cohen GH, Isaacs SN. 2010. A protein-based smallpox vaccine protects non-human primates from a lethal monkeypox virus challenge. Vaccine 28:6627–6636. 10.1016/j.vaccine.2010.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fogg CN, Americo JL, Lustig S, Huggins JW, Smith SK, Damon I, Resch W, Earl PL, Klinman DM, Moss B. 2007. Adjuvant-enhanced antibody responses to recombinant proteins correlates with protection of mice and monkeys to orthopoxvirus challenges. Vaccine 25:2787–2799. 10.1016/j.vaccine.2006.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hooper JW, Thompson E, Wilhelmsen C, Zimmerman M, Ichou MA, Steffen SE, Schmaljohn CS, Schmaljohn AL, Jahrling PB. 2004. Smallpox DNA vaccine protects nonhuman primates against lethal monkeypox. J. Virol. 78:4433–4443. 10.1128/JVI.78.9.4433-4443.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao Y, Aldaz-Carroll L, Ortiz AM, Whitbeck JC, Alexander E, Lou H, Davis HL, Braciale TJ, Eisenberg RJ, Cohen GH, Isaacs SN. 2007. A protein-based smallpox vaccine protects mice from vaccinia and ectromelia virus challenges when given as a prime and single boost. Vaccine 25:1214–1224. 10.1016/j.vaccine.2006.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moss B. 2011. Smallpox vaccines: targets of protective immunity. Immunol. Rev. 239:8–26. 10.1111/j.1600-065X.2010.00975.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu C, Meng X, Yan B, Crotty S, Deng J, Xiang Y. 2011. An epitope conserved in orthopoxvirus A13 envelope protein is the target of neutralizing and protective antibodies. Virology 418:67–73. 10.1016/j.virol.2011.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benhnia MR, McCausland MM, Su HP, Singh K, Hoffmann J, Davies DH, Felgner PL, Head S, Sette A, Garboczi DN, Crotty S. 2008. Redundancy and plasticity of neutralizing antibody responses are cornerstone attributes of the human immune response to the smallpox vaccine. J. Virol. 82:3751–3768. 10.1128/JVI.02244-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franke CA, Wilson EM, Hruby DE. 1990. Use of a cell-free system to identify the vaccinia virus L1R gene product as the major late myristylated virion protein M25. J. Virol. 64:5988–5996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su HP, Garman SC, Allison TJ, Fogg C, Moss B, Garboczi DN. 2005. The 1.51-Angstrom structure of the poxvirus L1 protein, a target of potent neutralizing antibodies. Proc. Natl. Acad. Sci. U. S. A. 102:4240–4245. 10.1073/pnas.0501103102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bisht H, Weisberg AS, Moss B. 2008. Vaccinia virus l1 protein is required for cell entry and membrane fusion. J. Virol. 82:8687–8694. 10.1128/JVI.00852-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ichihashi Y, Oie M. 1996. Neutralizing epitope on penetration protein of vaccinia virus. Virology 220:491–494. 10.1006/viro.1996.0337 [DOI] [PubMed] [Google Scholar]
- 18.Su HP, Golden JW, Gittis AG, Hooper JW, Garboczi DN. 2007. Structural basis for the binding of the neutralizing antibody, 7D11, to the poxvirus L1 protein. Virology 368:331–341. 10.1016/j.virol.2007.06.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aldaz-Carroll L, Whitbeck JC, Ponce de Leon M, Lou H, Pannell LK, Lebowitz J, Fogg C, White CL, Moss B, Cohen GH, Eisenberg RJ. 2005. Physical and immunological characterization of a recombinant secreted form of the membrane protein encoded by the vaccinia virus L1R gene. Virology 341:59–71. 10.1016/j.virol.2005.07.006 [DOI] [PubMed] [Google Scholar]
- 20.Chen W, Drillien R, Spehner D, Buller RM. 1992. Restricted replication of ectromelia virus in cell culture correlates with mutations in virus-encoded host range gene. Virology 187:433–442. 10.1016/0042-6822(92)90445-U [DOI] [PubMed] [Google Scholar]
- 21.Benhnia MR, McCausland MM, Laudenslager J, Granger SW, Rickert S, Koriazova L, Tahara T, Kubo RT, Kato S, Crotty S. 2009. Heavily isotype-dependent protective activities of human antibodies against vaccinia virus extracellular virion antigen B5. J. Virol. 83:12355–12367. 10.1128/JVI.01593-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng X, Zhong Y, Embry A, Yan B, Lu S, Zhong G, Xiang Y. 2011. Generation and characterization of a large panel of murine monoclonal antibodies against vaccinia virus. Virology 409:271–279. 10.1016/j.virol.2010.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCausland MM, Benhnia MR, Crickard L, Laudenslager J, Granger SW, Tahara T, Kubo R, Koriazova L, Kato S, Crotty S. 2010. Combination therapy of vaccinia virus infection with human anti-H3 and anti-B5 monoclonal antibodies in a small animal model. Antiviral Ther. 15:661–675. 10.3851/IMP1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matho MH, Maybeno M, Benhnia MR, Becker D, Meng X, Xiang Y, Crotty S, Peters B, Zajonc DM. 2012. Structural and biochemical characterization of the vaccinia virus envelope protein D8 and its recognition by the antibody LA5. J. Virol. 86:8050–8058. 10.1128/JVI.00836-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Esteban D, Parker S, Schriewer J, Hartzler H, Buller RM. 2012. Mousepox, a small animal model of smallpox. Methods Mol. Biol. 890:177–198. 10.1007/978-1-61779-876-4_11 [DOI] [PubMed] [Google Scholar]
- 26.Parker S, Chen NG, Foster S, Hartzler H, Hembrador E, Hruby D, Jordan R, Lanier R, Painter G, Painter W, Sagartz JE, Schriewer J, Mark Buller R. 2012. Evaluation of disease and viral biomarkers as triggers for therapeutic intervention in respiratory mousepox–an animal model of smallpox. Antiviral Res. 94:44–53. 10.1016/j.antiviral.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marsh JJ, Guan HS, Li S, Chiles PG, Tran D, Morris TA. 2013. Structural insights into fibrinogen dynamics using amide hydrogen/deuterium exchange mass spectrometry. Biochemistry 52:5491–5502. 10.1021/bi4007995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burns-Hamuro LL, Hamuro Y, Kim JS, Sigala P, Fayos R, Stranz DD, Jennings PA, Taylor SS, Woods VL., Jr 2005. Distinct interaction modes of an AKAP bound to two regulatory subunit isoforms of protein kinase A revealed by amide hydrogen/deuterium exchange. Protein Sci. 14:2982–2992. 10.1110/ps.051687305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamuro Y, Anand GS, Kim JS, Juliano C, Stranz DD, Taylor SS, Woods VL., Jr 2004. Mapping intersubunit interactions of the regulatory subunit (RIalpha) in the type I holoenzyme of protein kinase A by amide hydrogen/deuterium exchange mass spectrometry (DXMS). J. Mol. Biol. 340:1185–1196. 10.1016/j.jmb.2004.05.042 [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z, Smith DL. 1993. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2:522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. 2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67:271–281. 10.1107/S0907444910048675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collaborative Computational Project N. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 33.McCoy AJ. 2007. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63:32–41. 10.1107/S0907444906045975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. 2008. NCBI BLAST: a better web interface. Nucleic Acids Res. 36:W5–W9. 10.1093/nar/gkn201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132. 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 36.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240–255. 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 37.Chen VB, Arendall WB, III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66:12–21. 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372:774–797. 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 39.Tiller T, Busse CE, Wardemann H. 2009. Cloning and expression of murine Ig genes from single B cells. J. Immunol. Methods 350:183–193. 10.1016/j.jim.2009.08.009 [DOI] [PubMed] [Google Scholar]
- 40.Brochet X, Lefranc MP, Giudicelli V. 2008. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 36:W503–W508. 10.1093/nar/gkn316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meng X, Jiang C, Arsenio J, Dick K, Cao J, Xiang Y. 2009. Vaccinia virus K1L and C7L inhibit antiviral activities induced by type I interferons. J. Virol. 83:10627–10636. 10.1128/JVI.01260-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fogg CN, Americo JL, Earl PL, Resch W, Aldaz-Carroll L, Eisenberg RJ, Cohen GH, Moss B. 2008. Disparity between levels of in vitro neutralization of vaccinia virus by antibody to the A27 protein and protection of mice against intranasal challenge. J. Virol. 82:8022–8029. 10.1128/JVI.00568-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaufman DR, Goudsmit J, Holterman L, Ewald BA, Denholtz M, Devoy C, Giri A, Grandpre LE, Heraud JM, Franchini G, Seaman MS, Havenga MJ, Barouch DH. 2008. Differential antigen requirements for protection against systemic and intranasal vaccinia virus challenges in mice. J. Virol. 82:6829–6837. 10.1128/JVI.00353-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawrence MC, Colman PM. 1993. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234:946–950. 10.1006/jmbi.1993.1648 [DOI] [PubMed] [Google Scholar]
- 45.Davies DR, Padlan EA, Sheriff S. 1990. Antibody-antigen complexes. Annu. Rev. Biochem. 59:439–473. 10.1146/annurev.bi.59.070190.002255 [DOI] [PubMed] [Google Scholar]
- 46.Wolffe EJ, Vijaya S, Moss B. 1995. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology 211:53–63. 10.1006/viro.1995.1378 [DOI] [PubMed] [Google Scholar]
- 47.Lustig S, Fogg C, Whitbeck JC, Eisenberg RJ, Cohen GH, Moss B. 2005. Combinations of polyclonal or monoclonal antibodies to proteins of the outer membranes of the two infectious forms of vaccinia virus protect mice against a lethal respiratory challenge. J. Virol. 79:13454–13462. 10.1128/JVI.79.21.13454-13462.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chung CS, Chen CH, Ho MY, Huang CY, Liao CL, Chang W. 2006. Vaccinia virus proteome: identification of proteins in vaccinia virus intracellular mature virion particles. J. Virol. 80:2127–2140. 10.1128/JVI.80.5.2127-2140.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsiao JC, Chung CS, Chang W. 1999. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 73:8750–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin CL, Chung CS, Heine HG, Chang W. 2000. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol. 74:3353–3365. 10.1128/JVI.74.7.3353-3365.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chung CS, Hsiao JC, Chang YS, Chang W. 1998. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 72:1577–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Senkevich TG, Ojeda S, Townsley A, Nelson GE, Moss B. 2005. Poxvirus multiprotein entry-fusion complex. Proc. Natl. Acad. Sci. U. S. A. 102:18572–18577. 10.1073/pnas.0509239102 [DOI] [PMC free article] [PubMed] [Google Scholar]