

ABSTRACT
Hepatitis C virus (HCV) nonstructural protein 5B (NS5B), an RNA-dependent RNA polymerase (RdRp), is the key enzyme for HCV RNA replication. We previously showed that HCV RdRp is phosphorylated by protein kinase C-related kinase 2 (PRK2). In the present study, we used biochemical and reverse-genetics approaches to demonstrate that HCV NS5B phosphorylation is crucial for viral RNA replication in cell culture. Two-dimensional phosphoamino acid analysis revealed that PRK2 phosphorylates NS5B exclusively at its serine residues in vitro and in vivo. Using in vitro kinase assays and mass spectrometry, we identified two phosphorylation sites, Ser29 and Ser42, in the Δ1 finger loop region that interacts with the thumb subdomain of NS5B. Colony-forming assays using drug-selectable HCV subgenomic RNA replicons revealed that preventing phosphorylation by Ala substitution at either Ser29 or Ser42 impairs HCV RNA replication. Furthermore, reverse-genetics studies using HCV infectious clones encoding phosphorylation-defective NS5B confirmed the crucial role of these PRK2 phosphorylation sites in viral RNA replication. Molecular-modeling studies predicted that the phosphorylation of NS5B stabilizes the interactions between its Δ1 loop and thumb subdomain, which are required for the formation of the closed conformation of NS5B known to be important for de novo RNA synthesis. Collectively, our results provide evidence that HCV NS5B phosphorylation has a positive regulatory role in HCV RNA replication.
IMPORTANCE While the role of RNA-dependent RNA polymerases (RdRps) in viral RNA replication is clear, little is known about their functional regulation by phosphorylation. In this study, we addressed several important questions about the function and structure of phosphorylated hepatitis C virus (HCV) nonstructural protein 5B (NS5B). Reverse-genetics studies with HCV replicons encoding phosphorylation-defective NS5B mutants and analysis of their RdRp activities revealed previously unidentified NS5B protein features related to HCV replication and NS5B phosphorylation. These attributes most likely reflect potential structural changes induced by phosphorylation in the Δ1 finger loop region of NS5B with two identified phosphate acceptor sites, Ser29 and Ser42, which may transiently affect the closed conformation of NS5B. Elucidating the effects of dynamic changes in NS5B phosphorylation status during viral replication and their impacts on RNA synthesis will improve our understanding of the molecular mechanisms of NS5B phosphorylation-mediated regulation of HCV replication.
INTRODUCTION
Posttranslational phosphorylation has important roles in regulating the structures and functions of proteins and modulating protein-protein interactions for the rapid regulation of phosphosignaling pathways (1). In virus-infected cells, the functions, stability, and subcellular localization of virus-encoded proteins can be altered by host kinase-mediated phosphorylation. Indeed, growing numbers of virus-encoded phosphoproteins implicated in viral pathogenesis, virion assembly, and genome replication have been identified recently (2–7). In plus-strand RNA viruses, including hepatitis C virus (HCV), the viral RNA genome is replicated by virus-encoded RNA-dependent RNA polymerases (RdRps) (7), and their phosphorylation has been suggested to be functionally linked to viral genome replication (6–13).
HCV is the major etiologic agent of non-A and -B hepatitis. The virus persistently infects approximately 170 million people worldwide and is responsible for most cases of severe chronic liver disease, including cirrhosis and hepatocellular carcinoma (14). HCV contains a 9.6-kb single-stranded, positive-polarity RNA genome consisting of a 5′ untranslated region (UTR), a large open reading frame encoding a single large precursor polyprotein, and a 3′ UTR (15). The viral polyprotein is processed by cellular and viral proteases into structural (C, E1, E2, and p7) and nonstructural (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins (15). The 68-kDa HCV NS5B protein is the viral RdRp, a key component of the RNA replicase complex formed with other NS (NS3 to NS5A) and cellular proteins (16, 17).
The HCV NS5B RdRp contains the typical finger, palm, and thumb structural subdomains common to all RdRps (18). The NS5B protein, which forms an RNA replicase complex through interactions with some of the viral NS proteins, is also able to form an oligomeric complex through its Δ1 loop and thumb subdomain to achieve the de novo RNA replication initiation-competent conformation (19). The RNA polymerase activity of NS5B was reported to be regulated by its interaction with the viral capsid protein (Core) (20). In addition to viral proteins, cellular proteins may also modulate the function of NS5B (21–24). With regard to HCV RNA replication, we previously demonstrated that protein kinase C-related kinase 2 (PRK2), a Ser/Thr kinase in the AGC kinase subfamily (25), binds to and phosphorylates HCV RdRp at its N-terminal finger subdomain (amino acids 1 to 187) (7). Silencing of PRK2 expression by small interfering RNA (siRNA), inhibiting PRK2 activity with its pharmacological inhibitors HA1077 and Y27632, or destabilizing the PRK2 upstream kinase phosphoinositide-dependent kinase 1 reduced HCV replication (7, 8, 26), demonstrating PRK2's regulatory role in the HCV life cycle.
In the present work, we identified phosphorylation sites on HCV NS5B and evaluated the role that NS5B phosphorylation plays in HCV RNA replication. We showed that PRK2 phosphorylates Ser29 and Ser42 residues at the Δ1 loop domain of HCV NS5B. Furthermore, reverse-genetics experiments with HCV subgenomic replicons and infectious HCV cDNA clones demonstrated that PRK2-mediated HCV NS5B phosphorylation, which can alter the NS5B conformation as predicted by molecular-modeling studies, has a positive regulatory role in HCV RNA replication.
MATERIALS AND METHODS
Plasmids, antibodies, and siRNAs.
For the expression of truncated NS5B proteins, pET-28a(+)-NS5B(80-591), pET-28a(+)-NS5B(54-371), and pET-28a(+)-NS5B(28-371) were constructed by cloning the cDNA encoding the amino acids indicated in parentheses. The partial HCV NS5B gene fragments were generated by conventional PCR using a full-length infectious HCV genotype 1b clone (27) as a template. The amplified PCR products were digested with NheI and EcoRI and subcloned into pET-28a(+) (Novagen, San Diego, CA, USA). To construct various HCV NS5B subgenomic replicons with mutations in the Ser29 and/or Ser42 residue, inserts for HCV NS5B point mutants were generated by two-step overlapping PCR using the pZS2 template (28), as previously described (29), and cloned into the XhoI-ClaI site of pZS2. Plasmid pTrcHisB-NS5BΔ21 expressing HCV NS5B lacking the C-terminal 21 hydrophobic amino acids (8) was used as a template to construct expression vectors for NS5B carrying S29A (mutation of Ser29 to Ala), S42A (mutation of Ser42 to Ala), or S29A S42A (mutations of both Ser29 and Ser42 to Ala). The two-step overlapping-PCR products carrying the above-mentioned mutations were cloned into the NheI- and EcoRI-digested pTrcHisB vector (Invitrogen, Carlsbad, CA, USA). Similarly, the GDD motif at the active site was mutated to GAA to produce an inactive form of NS5B (NS5B_GAA). Mammalian expression vectors for each of the above-described NS5B mutants were constructed by subcloning each of the corresponding genes into pcDNA3.1-Flag-NS5B (20) to obtain pcDNA3.1-Flag-NS5B_S29A, pcDNA3.1-Flag-NS5B_S42A, and pcDNA3.1-Flag-NS5B_S29AS42A. The JFH1 HCV genotype 2a infectious clone (30) was used to generate NS5B derivatives lacking phosphorylation sites by two-step overlapping PCR. The correct insertions of mutations were confirmed by sequencing the amplified genes. All the primer sequences used for cloning are available upon request.
The antibodies used in this study were obtained as follows: goat anti-NS5B antibody from Santa Cruz Biotechnology (Dallas, TX, USA), rabbit polyclonal anti-PRK2 antibody from Cell Signaling Technology (Danvers, MA, USA), mouse monoclonal anti-phosphoserine (pSer) antibody (clone PSR-45) from Sigma-Aldrich (St. Louis, MO, USA), anti-Flag antibody from Sigma-Aldrich, monoclonal anti-HCV Core (C7-50) antibody from Abcam (Cambridge, MA, USA), and anti-β-actin antibody from Santa Cruz Biotechnology. Affinity-purified NS5B antibody was prepared as described previously (8).
siRNAs were obtained from ST Pharm (Seoul, South Korea). The PRK2-targeting siRNA (siPRK2) and scrambled (SC) siRNA have the following sequences: siPRK2 passenger strand, 5′-UCAAAGAAGGAGCUGAAAAUU-3′; siPRK2 guide strand, 5′-UUUUCAGCUCCUUCUUUGAUU-3′; SC siRNA passenger strand, 5′-AUGAACGUGAAUUGCUCAAUU-3′; and SC siRNA guide strand, 5′-UUGAGCAAUUCACGUUCAUUU-3′.
Cell culture and transfection.
The human hepatocellular carcinoma cell line Huh7 was grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.1 mM nonessential amino acids under 5% CO2 at 37°C. The Huh7-derived cell line R-1, which harbors autonomously replicating HCV subgenomic RNA, was maintained in DMEM with 1 mg/ml G418 as previously described (7). Plasmids were transfected into cells using FuGene6 (Roche Diagnostics, Basel, Switzerland), and siRNAs were delivered into cells using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer's instructions.
Expression and purification of HCV NS5B protein.
The His6-tagged HCV NS5B lacking 21 C-terminal amino acids (Δ21NS5B) and other NS5B derivatives were expressed in Escherichia coli at 25°C for 12 h with 1 mM isopropyl-β-d-thiogalactopyranoside induction. Soluble NS5B proteins were purified using Ni-nitrilotriacetic acid–Sepharose resin (Qiagen, Hilden, Germany), followed by chromatography using heparin-Sepharose and SP Sepharose columns, if required, as previously described (7).
RdRp assay.
The in vitro RdRp assay was performed with 3 pmol recombinant HCV NS5BΔ21 or its derivatives in a total volume of 25 μl RdRp reaction buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 2.5 mM MgCl2, 1 mM dithiothreitol [DTT], and 20 U RNase inhibitor [Promega, Madison, WI, USA]) containing 1 μg poly(A) RNA template and 10 pmol oligo(U)20, 10 μM UTP, and 5 μCi [α-32P]UTP (3,000 Ci/mmol; Amersham Biosciences, NJ, USA). The RdRp reactions with the HCV 3′ UTR or 3′(−)341-nt (representing the 341-nucleotide [nt] 3′ end of the HCV genome minus strand) were carried out with 200 ng RNA template prepared by in vitro transcription, as described previously (31). The reaction products were processed and resolved on a denaturing 5% polyacrylamide gel containing 8 M urea. After electrophoresis, the gels were stained with ethidium bromide to locate template positions, photographed, dried, and then exposed to X-ray film for autoradiography. The amount of radiolabeled RNA was quantified using a phosphorimager.
In vitro PRK2 kinase assay.
An in vitro PRK2 kinase reaction was performed in a total volume of 20 μl kinase buffer (50 mM Tris-Cl, pH 7.5, 10 mM MgCl2, and 1 mM DTT) containing 10 μM purified NS5B, 0.1 mM ATP, 10 μCi [γ-32P]ATP, and 1 μM purified PRK2 (Invitrogen) for 30 min at 30°C. After the reaction was stopped by boiling at 95°C, protein samples were subjected to SDS-8% PAGE and analyzed by autoradiography. To identify the sites of phosphorylation on NS5B, the kinase assay was carried out in the presence of unlabeled ATP (10 mM).
Metabolic cell labeling, immunoprecipitation, and Western blot analysis.
Huh7 and R-1 cells transiently expressing Flag-tagged NS5B proteins were metabolically labeled with [32Pi]orthophosphate (PerkinElmer, Waltham, MA, USA) as previously described (7). When indicated, the transfected cells were treated with 100 nM siPRK2 or SC siRNA prior to metabolic labeling of the cells at 48 h posttransfection. The cells were washed, preincubated for 1 h in phosphate-free DMEM (Invitrogen) supplemented with 10% dialyzed FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin. After further incubation for 4 h in the presence of [32Pi]orthophosphate at a final concentration of 80 μCi/ml, the cells were washed with cold phosphate-buffered saline (PBS), harvested, and lysed in RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EGTA, 0.5 mM sodium deoxycholate, 1% Triton X-100, and 0.1% SDS) supplemented with an EDTA-free protease inhibitor mixture (Roche Diagnostics) and phosphatase inhibitors (10 mM NaF, 1 mM Na3VO4, 10 mM tetrasodium pyrophosphate, and 17.5 mM β-glycerophosphate). Labeled NS5B proteins were immunoprecipitated using affinity-purified NS5B antibody. Western blot analysis of the cell lysates for NS5B, PRK2, and β-actin was performed as previously described (28). Membrane-bound antibodies were detected with an enhanced-chemiluminescence (ECL) kit (GE Healthcare Life Sciences, Piscataway, NJ, USA).
Lambda phosphatase treatment.
Huh7 or R-1 cell lysates were prepared in a lysis buffer (20 mM Tris-HCl, pH 7.5, 1 mM EDTA, 1 mM DTT, 50 mM NaF, and 1 mM Na3VO4) supplemented with an EDTA-free protease inhibitor cocktail (Roche Diagnostics). After incubation for 10 min on ice, the cell debris was clarified by centrifugation at 21,000 × g for 20 min at 4°C to obtain cell lysates. The NS5B proteins in the cell lysates were immunoprecipitated as described above and incubated with or without 400 U λ-phosphatase (New England BioLabs, Ipswich, MA, USA) in 30 μl λ-phosphatase reaction buffer (50 mM Tris-HCl, pH 7.5, 2 mM MnCl2, 0.1 mM EDTA, 5 mM DTT, and 0.01% Brij 35) supplemented with a protease inhibitor cocktail (Roche Diagnostics) at 30°C for 30 min. Next, the reaction mixtures were analyzed by electrophoresis on an SDS-8% polyacrylamide gel, followed by Western blotting.
Two-dimensional phosphoamino acid analysis.
To analyze the phosphorylated amino acid residues on the NS5B protein, a two-dimensional (2D) phosphoamino acid assay was carried out as previously described (32). Briefly, radiolabeled NS5B protein was separated by SDS-8% PAGE and transferred to a Hybond-P polyvinylidene difluoride (PVDF) membrane (Amersham Biosciences). The transferred membrane was exposed to X-ray film, and the labeled NS5B protein band was detected by autoradiography. The NS5B protein band was cut from the membrane and hydrolyzed directly with 6 M HCl for 1 h at 110°C. The hydrolysates were lyophilized in a SpeedVac concentrator and subsequently analyzed by 2D electrophoresis at pH 1.9 (first dimension) and 3.5 (second dimension) on a thin-layer cellulose (TLC) plate (Merck, Whitehouse, NJ, USA) using the HTLE-7002 apparatus (CBS Scientific, Del Mar, CA, USA). The plate was sprayed with 0.25% (wt/vol) ninhydrin in acetone to detect the standard pSer, phosphothreonine (pThr), and phosphotyrosine (pTyr) (Sigma-Aldrich) and then analyzed by autoradiography.
Mass spectrometry.
Protein bands were excised from the gel, subjected to in-gel tryptic digestion, and analyzed by tandem mass spectrometry (MS-MS), as previously described (33). All MS-MS experiments for peptide identification were performed using a nano-liquid chromatography (LC)-MS system consisting of a Surveyor high-performance liquid chromatography (HPLC) system and a 7-Tesla Finningan LTQ-FT mass spectrometer (Thermo Electron, Bremen, Germany) equipped with a nano-electrospray ionization (ESI) source. Ten microliters of each sample was loaded by an autosampler onto a C18 trap column (inside diameter [i.d.], 300 μm; length, 5 mm; particle size, 5 μm; LC Packings) for desalting and concentration at a flow rate of 20 μl/min. Next, the trapped peptides were backflushed and separated on a homemade microcapillary column (length, 100 mm) packed with C18 (particle size, 5 μm) in 75-μm silica tubing (8-μm-i.d. orifice). The mass spectrometer was operated in the data-dependent mode to automatically switch between MS and MS-MS acquisition. Xcalibur software was enabled for each MS-MS spectrum. The general mass-spectrometric conditions were as follows: spray voltage, 2.2 kV; no sheath and auxiliary gas flow; ion transfer tube temperature, 220°C; collision gas pressure, 1.3 millitorrs; normalized collision energy using wide-band activation mode; and 35% for MS-MS. The ion selection thresholds were 500 counts for MS-MS. An activation q value of 0.25 and an activation time of 30 ms were applied in the MS-MS acquisitions.
Colony formation assay.
Wild-type and mutant subgenomic HCV RNAs prepared by in vitro transcription using the Megascript T7 kit (Ambion, Austin, TX, USA) were transfected into Huh7 cells by electroporation as previously described (26). At 24 h posttransfection, selection of G418-resistant colonies was initiated by adding 0.5 mg/ml G418. After 3 weeks, the resistant colonies were fixed with methanol and stained with crystal violet.
Real-time qRT-PCR.
Total RNA was extracted with TRIzol reagent (Invitrogen) and purified according to the manufacturer's instructions. HCV RNA levels were quantified by real-time quantitative reverse transcription (qRT)-PCR using a TaqMan probe targeting a region within the HCV 5′ UTR, as previously described (26).
Transient HCV replication assay.
The plasmid JFH1 and its derivatives with mutations at the NS5B phosphorylation site were used to prepare viral-RNA transcripts as previously reported (26). The viral RNAs were then electroporated into Huh7 cells as previously described (26) or using Lipofectamine 2000 (Invitrogen), and total RNA harvested from the transfected cells was analyzed by Northern blotting to detect HCV genomic RNA (34). Briefly, total RNA (10 μg) was separated by electrophoresis on a 0.7% denaturing agarose gel and transferred to a positively charged nylon membrane (Roche), which was then hybridized with DNA probe (HCV NS3 [3,446 nt]-NS5B [9,265 nt]) labeled with 32P using the Ready-To-Go DNA-labeling kit (GE Healthcare Life Sciences). The hybridized membrane was exposed for autoradiography, and the amount of radiolabeled RNA was quantified using a phosphorimager.
Immunostaining.
HCV-infected Huh7 cells grown in Lab-Tek four-well chamber coverslips (Nunc, Roskilde, Denmark) were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 15 min at 2 days postinfection. The fixed cells were permeabilized by incubation with 0.3% Triton X-100 and blocking solution (PBS containing 3% horse serum [Sigma-Aldrich]). After washing 3 times with PBS, HCV Core protein was immunostained with anti-HCV Core antibody, as described previously (20). Nuclei were visualized by staining with 1 μM 4′,6′-diamidino-2-phenylindole (DAPI). The immunostained images were analyzed by confocal laser scanning microscopy using a Zeiss LSM 510 (Carl Zeiss, Oberkochen, Germany). The HCV Core-positive cells among 1,000 total cells were counted using GSA Image Analyzer software (GSA, Rostock, Germany).
Molecular modeling of the phosphorylated NS5B polymerase three-dimensional structure.
In silico molecular models of phosphorylated NS5B polymerase and its substitution mutants were generated with the wild-type NS5B crystal structure (Protein Data Bank [PDB] ID 1GX5) (35) as a template using the UCSF Chimera package (36, 37). Side chain refinements of the structures were carried out with SCWRL (38), followed by the addition of hydrogen using MolProbity (39). Final structures were minimized with the AMBER force field (AMBER99SB) (40) and an MMTK module (41) in UCSF Chimera. During minimization, AMBER parameters were used for standard residues, and the Antechamber module was used to assign parameters for the nonstandard residues. One hundred steps of steepest minimization were performed to relieve unfavorable clashes, followed by 100 steps of conjugate gradient minimization. The size of the steepest descent and conjugate gradient minimization was 0.02 Å. The APBS (42) and Delphi (43) programs were used to calculate the electrostatic surface potentials of the final mutated and phosphorylated models. Molecular visualization was performed using UCSF Chimera and PyMOL (PyMOL Molecular Graphics System, version 1.2r3pre; Schrödinger, LLC). The hydrophobic pocket of the NS5B protein was analyzed using CASTp methods (44).
Statistical analysis.
Pairwise comparisons between groups were made using Student's t test. P values of less than 0.05 were considered statistically significant.
RESULTS
HCV NS5B is phosphorylated exclusively on Ser residues.
We previously demonstrated that HCV NS5B is a phosphoprotein by immunoprecipitating NS5B proteins from [32P]orthophosphate-labeled Huh7 cells harboring an HCV subgenomic replicon and by immunostaining with a phosphoserine-specific antibody (7). In the present work, we performed 2D phosphoamino acid analyses to provide clear evidence indicating that NS5B is indeed phosphorylated at its Ser residues. An in vitro kinase assay with purified NS5B and a catalytically active recombinant PRK2 detected a radiolabeled NS5B band representing NS5B phosphorylated by PRK2, along with radiolabeled autophosphorylated PRK2 (45) (Fig. 1A). The 32P-labeled NS5B was transferred to a PVDF membrane and subjected to acid hydrolysis. The resulting phosphoamino acids were separated by 2D electrophoresis on a TLC plate with standard phosphoamino acids to detect the labeled amino acids by autoradiography. Figure 1B shows that NS5B is phosphorylated exclusively on its Ser residues.
FIG 1.

HCV NS5B is phosphorylated at its Ser residues by PRK2. (A) Purified HCV NS5B protein was subjected to an in vitro kinase assay using an enzymatically active recombinant PRK2 protein. The reaction mixture, resolved by SDS-PAGE, was transferred to a PVDF membrane for autoradiography. Bands corresponding to phosphorylated NS5B (p-NS5B) and PRK2 (p-PRK2) are indicated by arrowheads. Molecular mass markers are shown on the left. (B) The band corresponding to radiolabeled NS5B was excised from the PVDF membrane, processed as described in Materials and Methods, and separated by 2D TLC. Standard phosphoamino acids detected by ninhydrin staining (a), radiolabeled phosphoamino acids detected by autoradiography (b), and the overlaid image (c) are shown. (C and D) R-1 cells harboring an HCV subgenomic replicon (a schematic representation of the replicon is shown in Fig. 5A) and Huh7 cells were labeled with [32P]orthophosphate. Radiolabeled NS5B proteins in cell lysates were immunoprecipitated with affinity-purified NS5B antibody, separated by SDS-PAGE, and transferred to a PVDF membrane for autoradiography. The same blot was also used for Western blot (WB) analysis of PRK2. Phosphoamino acid analysis was performed as for panel B. (E) The NS5B protein immunoprecipitated from R-1 or Huh7 cells was left untreated or was treated with λ-phosphatase (PPase) and subjected to SDS-PAGE for detection of p-NS5B and NS5B by WB analysis using pSer-specific antibody (top) and anti-NS5B antibody (bottom), respectively.
To investigate whether the NS5B in HCV-replicating cells is also phosphorylated at its Ser residues, we performed a similar phosphoamino acid analysis using NS5B proteins immunoprecipitated from the Huh7-derived cell line R-1, which harbors a genotype 1b HCV subgenomic replicon. In immunoprecipitates from R-1 cells metabolically labeled with [32P]orthophosphate, radiolabeled NS5B and an ∼140-kDa protein were detected; we identified the latter protein as PRK2 by Western blotting (Fig. 1C). Subsequent phosphoamino acid analysis of radiolabeled NS5B confirmed the in vitro kinase assay results shown in Fig. 1B (Fig. 1D). Furthermore, λ-phosphatase treatment of immunoprecipitated NS5B dramatically reduced phosphorylated NS5B levels, detectable by immunostaining using a phosphoserine-specific antibody (Fig. 1E, top, lane 4 versus lane 5), whereas similar levels of NS5B were present in both λ-phosphatase-treated and untreated immunoprecipitates (Fig. 1E, bottom). These results indicate that the NS5B in HCV-replicating cells is also phosphorylated primarily on its Ser residues.
Mapping of NS5B phosphorylation sites.
To determine which specific Ser residues are phosphorylated, in vitro phosphorylation assays were performed with serial deletion mutants of NS5B protein (Fig. 2A) and recombinant PRK2 in the presence of [γ-32P]ATP. The assay showed that NS5B(28-371), but not the NS5B mutant lacking the N-terminal 79 residues [NS5B(80-591)] or 53 residues [NS5B(54-591)], can be phosphorylated as effectively as the protein with the C-terminal 21 amino acids deleted [NS5B(1-570)] (Fig. 2B, top). These results suggest that the N-terminal region, spanning amino acids 28 to 53, which contain four Ser residues (Fig. 2C), is likely to possess potential phosphate acceptor sites and that the C-terminal hydrophobic 21 amino acids have no effect on PRK2-mediated NS5B phosphorylation.
FIG 2.
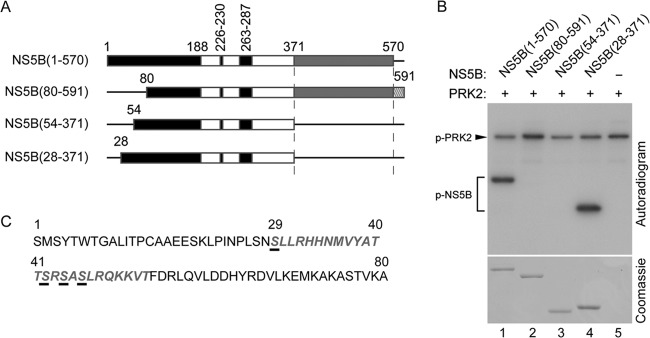
Deletion mapping of putative HCV NS5B phosphorylation sites. (A) Schematic of HCV NS5B derivatives. The fingers (black), palm (white), and thumb (gray) subdomains of NS5B and its C-terminal hydrophobic transmembrane domain (hatched) are shown. (B) In vitro kinase assays were performed with the indicated purified NS5B proteins and PRK2 in the presence of [γ-32P]ATP. Reaction mixtures were resolved by SDS-10% PAGE for autoradiography. The bottom gel shows equivalent amounts of NS5B used for the assays. (C) The PRK2 phosphorylation region mapped to the N-terminal part of NS5B is shown in gray italics. Potential phosphorylation sites are underlined.
The Ser29 and Ser42 residues are the two major phosphorylation sites on NS5B.
In experiments parallel to the NS5B phosphorylation assays described above, we sought to identify the phosphorylation site(s) with an MS-based approach. To this end, NS5B protein was phosphorylated with PRK2 in the presence of cold ATP, resolved by SDS-PAGE, and visualized by Coomassie staining. The NS5B protein was excised from the gel, digested in gel with trypsin, and subjected to MS analysis. We consistently identified two phosphorylated peptides in two independent experiments. As shown in Fig. 3A, the first phosphate acceptor Ser residue (Ser29) was on a peptide comprised of amino acids 21 to 32 (LPINALSNS29LLR). The second phosphorylation site was on a peptide comprised of amino acids 33 to 43 that included phospho-Ser42 (HHNLVYATTS42R) (Fig. 3B). These results were consistent with the phosphorylation site mapping results shown in Fig. 2B, in which the phosphorylation sites were mapped to the N-terminal region of the finger subdomain of NS5B spanning amino acids 28 to 53, where these two identified phosphorylation sites are located.
FIG 3.
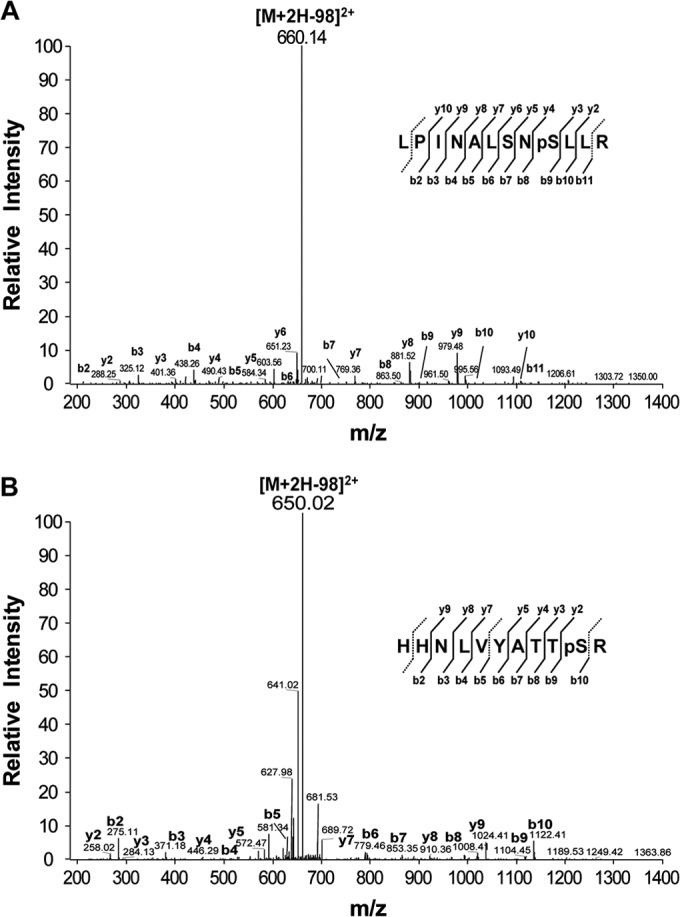
Identification of HCV NS5B protein phosphorylation sites. NS5B phosphorylated by PRK2 in vitro was resolved by SDS-PAGE, and the band corresponding to NS5B was excised and subjected to in-gel digestion using trypsin for tandem mass spectrometry. Spectra for the phosphopeptides in the digested HCV NS5B protein, LPINALSNpSLLR (A) and HHNLVYATTpSR (B), are shown.
To confirm the MS analysis results, in vitro kinase assays were performed with three mutant NS5B proteins, NS5B_S29A, NS5B_S42A, and NS5B_S29AS42A, in which Ser29 and Ser42, separately or together, were mutated to Ala to prevent phosphorylation (Fig. 4A). As shown in Fig. 4B, mutants NS5B_S29A and NS5B_S42A were partially labeled (24% ± 3.5% and 40% ± 1.5% of controls for NS5B_S29A and NS5B_S42A, respectively), indicating that both Ser residues serve as phosphate acceptors, with Ser29 preferentially phosphorylated. The NS5B_S29AS42A mutant was marginally phosphorylated (12% ± 3.5% of the control). This low, yet detectable, level of phosphorylation might be attributed to other potential phosphorylation sites not identified by MS analysis. Alternatively, it is possible that the mutations of these two Ser residues induced a conformational change in NS5B to release alternative phosphorylation sites that are otherwise hidden within NS5B.
FIG 4.

The Ser29 and Ser42 residues of HCV NS5B are its two major phosphorylation sites. (A) The HCV NS5B protein (570 amino acids [aa]) lacking 21 C-terminal hydrophobic amino acids is illustrated schematically. The region containing the two phosphorylation sites (Ser29 and Ser42) is expanded below the schematic to show the substituted amino acids (boldface) used in the in vitro kinase assays. (B) In vitro kinase assays were performed with wild-type (WT) NS5B and the indicated NS5B mutants as for Fig. 2B. The bottom gel shows equivalent amounts of NS5B used for the assays. Phosphorylated, radiolabeled NS5B signals were quantified using a phosphorimager and normalized to NS5B protein levels. The results shown below the autoradiogram represent the means ± standard deviations (SD) of three experiments, from which one representative autoradiogram is shown. (C) Huh7 cells transfected with plasmids expressing the indicated NS5B proteins were metabolically labeled with [32P]orthophosphate. The labeled NS5B proteins were immunoprecipitated (IP), resolved by SDS-PAGE, and analyzed by autoradiography (top) or Western blot analysis (bottom). Phosphorylated-NS5B levels were quantified as for panel B. (D) Experiments similar to those shown in panel C were performed with or without silencing of PRK2 expression. Knockdown of PRK2 in cells transfected with siPRK2 was verified by Western blotting.
We also analyzed the levels of phosphorylated NS5B in metabolically labeled Huh7 cells transiently expressing wild-type NS5B or its phosphorylation-defective derivatives. As shown in Fig. 4C, the Ser29 and Ser42 residues served as two main phosphorylation sites; Ser29, again, was preferentially phosphorylated. We observed no detectable levels of radiolabeled NS5B in the cells expressing the NS5B_S29AS42A mutant. When PRK2 expression was silenced with the PRK2-specific siRNA (siPRK2) for 48 h prior to metabolic labeling, we observed a substantial reduction in radiolabeled NS5B levels (45% ± 5.5% of the control) (Fig. 4D), demonstrating that PRK2 was responsible for the phosphorylation of NS5B. The results from our MS analysis and NS5B phosphorylation assays in vitro and in cell culture strongly indicate that Ser29 and Ser42 are the two major residues phosphorylated by PRK2.
HCV replication is impaired in HCV subgenomic replicons encoding phosphorylation-defective NS5B proteins.
We assessed the impacts of Ser29 and Ser42 mutations on HCV RNA replication in order to better understand the role(s) of NS5B phosphorylation in HCV replication. We generated mutant HCV subgenomic replicons encoding NS5B proteins with Ala, Glu, or Asp substitutions at the Ser phosphorylation sites (Fig. 5A). In vitro transcripts of the resulting HCV subgenomic replicons were electroporated into Huh7 cells, and cells resistant to G418 due to HCV RNA replication were selected after 3 weeks. As shown in Fig. 5B and C, NS5B_S29A-harboring replicons did not produce G418-resistant cell colonies. Similarly, the Ser42-to-Ala mutation (NS5B_S42A) significantly impaired colony formation activity (26% of the wild-type control). In addition, stable colonies (pool) selected with the S42A mutant contained intracellular HCV genome copy numbers that were reduced to 13% of the wild-type replicon RNA level (Fig. 5D). We found that the Ala mutation remained unchanged at the Ser42 residue in at least 10 independent sequenced clones (data not shown). These results suggest Ser29 might be more critical than Ser42 in NS5B phosphorylation-mediated regulation of HCV RNA replication. This interpretation was further confirmed by showing that the additional Ala substitution at Ser29 on the replicon harboring the NS5B_S42A mutation abolished its ability to replicate.
FIG 5.

Colony-forming ability of HCV subgenomic replicons expressing phosphorylation-defective NS5B proteins. (A) Schematic of the HCV subgenomic replicon. The substituted amino acid sequences are shown below the corresponding expanded NS5B region; ΔC and Neo represent the N-terminal part of the Core protein and neomycin phosphotransferase, respectively. Nonstructural (NS) proteins essential for HCV replication are shown. (B and C) Huh7 cells were electroporated with the indicated in vitro transcripts of subgenomic HCV replicons and seeded in 10-cm dishes. Colonies were selected by culturing with 0.5 mg/ml G418 for 3 weeks, fixed, and stained with crystal violet solution. The results represent the means (±SD) of three independent experiments (B), from which one representative set of stained cells is shown (C). ND, not detected. (D) Steady-state intracellular HCV subgenomic RNA levels of R-1 and the Huh7-derived stable cell line established with the NS5B_S42A subgenomic replicon were measured by real-time qRT-PCR. The results represent the means (±SD) of three independent experiments. Comparisons among the groups were made using the Student t test. *, P < 0.01.
Furthermore, replacement of Ser29 or Ser42 with Asp or Glu to mimic phosphoamino acids did not allow the replicons to form G418-resistant colonies, suggesting these phosphoamino acid surrogates did not faithfully mimic pSer. This result is likely due to the carboxylate group on the side chains of these amino acids, which is functionally significantly different from the pSer side chains in size, number of oxygen atoms, and number of negative charges at neutral pH (1). Collectively, the reverse-genetics experiments demonstrated that the two identified phosphate acceptor residues, Ser29 and Ser42, are critical for HCV RNA replication.
Phosphorylation-defective NS5B leads to inhibition of HCV replication in HCV genome-transfected cells.
The cell colony formation assays carried out with HCV subgenomic replicons might not allow us to estimate their RNA replication potential, given that adaptive mutations might occur during the selection procedure. In addition, the resulting cell colonies generated by the S42A mutant might represent a subpopulation of Huh7 cells with this particular replicon. To exclude such possibilities, the effects of NS5B phosphorylation site mutations on HCV replication were further evaluated by a transient-replication assay using in vitro RNA transcripts derived from the full-length HCV genotype 2a infectious clone pJFH1, which produces infectious viruses in Huh7 cells (30), and from its derivatives with mutations at the NS5B phosphorylation sites. The phosphorylation sites Ser29 and/or Ser42 were mutated to Ala, Asp, or Glu, in the JFH1 plasmid. Huh7 cells were transfected with each of the viral RNAs to measure HCV genome levels at 48 h posttransfection by Northern blotting. As shown in Fig. 6A and B, replacement of Ser29 or Ser42 with Ala resulted in a noticeable reduction in HCV RNA levels (38% ± 15.5% or 71% ± 5.7% of the wild-type JFH1 control, respectively), similar to levels observed in the colony formation assay. When both Ser residues were mutated to Ala in JHF1-NS5B_S29AS42A, no detectable HCV genomic RNA was observed, as with JFH1-NS5B_GND, encoding an inactive NS5B. We also found that replacement of Ser29 or Ser42 with Asp or Glu resulted in loss of replication activity, which was consistent with the results observed from corresponding subgenomic replicons.
FIG 6.

Effects of NS5B phosphorylation site mutations on HCV replication. (A) Wild-type HCV (JFH1) and mutant JFH1 HCV in vitro transcripts were transfected into Huh7 cells. After 48 h, cells were harvested to determine the HCV genomic RNA level by Northern blotting using the in vitro-transcribed HCV RNA genome as a standard. The 28S rRNA detected by ethidium bromide staining is shown as a loading control. (B) Relative HCV genome levels quantified using a phosphorimager (means ± SD of three experiments). *, P < 0.01; **, P < 0.05 (Student's t test). (C) Immunostaining of Core protein in Huh7 cells infected with wild-type HCV or the indicated HCV mutants. DAPI, nuclear staining.
From HCV RNA-transfected cells at 3 days posttransfection, infectious HCV titers were measured, and titers of 4.86 × 103 fluorescence focus-forming units/ml of viral infectivity (243 cells staining HCV Core positive among 1,000 cells examined) were observed in the JFH1 control, whereas no replication was observed in cells infected with the JFH1 mutants expressing phosphorylation-defective NS5B proteins (NS5B_S29A, NS5B_S42A, and NS5B_S29AS42A) or JFH1 expressing inactive NS5B (NS5B_GND) (Fig. 6C).
The phosphate acceptor sites Ser29 and Ser42 are important for both de novo and primer-dependent RNA synthesis.
To evaluate if the RdRp activities of the phosphorylation-defective NS5B mutants are correlated with their abilities to replicate the viral genome in cell culture, we measured the RdRp activities of NS5B_S29A, NS5B_S42A, and NS5B_S29AS42A expressed in E. coli (Fig. 7A) using a poly(A)/oligo(U)20 substrate directing primer-dependent RNA synthesis and an HCV 3′ UTR or 3′(−)341-nt template directing de novo RNA synthesis. We found that the mutation of Ser29 to Ala reduced RdRp activity to 13.9% ± 4.3% that of wild-type NS5B in primer-dependent RNA synthesis (Fig. 7B). Similarly, reduced activity was observed with the 3′ UTR and 3′(−)341-nt templates (7.7% ± 1.5% and 9.3% ± 8.2% that of wild-type NS5B, respectively). Although the NS5B_S42A mutant exhibited greater activity than the S29A mutant, it showed only approximately 25 to 53% of wild-type NS5B activity for the RNA templates tested. However, replacement of both Ser29 and Ser42 with Ala resulted in complete loss of RNA polymerase activity. The inactive NS5B mutant carrying the GAA mutation at the GDD motif, which was used as a control for the assay, did not display any RNA polymerase activity. These RdRp assay results underscore the importance of the two phosphate acceptor Ser residues in the N-terminal region of NS5B for in vitro RNA synthesis and suggest that any conformational change introduced by local amino acid structure alterations at those two Ser residues can affect NS5B function.
FIG 7.

Effect of NS5B phosphorylation site mutations on RdRp activity. (A) Recombinant wild-type and NS5B proteins carrying the indicated mutations were resolved by SDS-PAGE and visualized by Coomassie blue staining. The arrowhead indicates purified NS5B protein. (B) RdRp assays were performed with the indicated NS5B mutants using a poly(A)/oligo(U)20, HCV 3′-UTR, or 3′(−)341-nt template. Reaction products were resolved by electrophoresis on an 8 M urea-5% polyacrylamide gel and stained with ethidium bromide (EtBr) to locate template positions (indicated by arrowheads) and to verify that equal amounts of RNA templates were used in the assays prior to autoradiography. Band intensities were quantified using a phosphorimager. Relative enzyme activities compared to that of the wild-type NS5B are shown. The results represent the means ± SD of three experiments, from which one representative autoradiogram is shown.
Structural analysis of phosphorylated NS5B based on predicted 3D models.
To understand the molecular mechanisms of NS5B phosphorylation-mediated regulation of HCV RNA synthesis, we built in silico molecular models for the phosphorylated form of NS5B based on a crystal structure of NS5B previously reported (PDB 1GX5) (35). As depicted in the molecular models in Fig. 8A, the Ser29 and Ser42 residues are located in the Δ1 finger loop I region (Ile11-Ser46) of NS5B, which is composed of finger, palm, and thumb domains. The Δ1 loop is involved in keeping the enzyme in a closed conformation via strong hydrophobic interaction of Leu30 with the thumb subdomain (Fig. 8B and C) (46). The Δ1 loop, which forms two α-helical turns and is packed closely against the helices of the thumb subdomains, was proposed to undergo conformational change (i.e., from open to closed conformation upon template RNA binding for de novo RNA synthesis initiation) (47, 48). The Ser29 located at the tip of the Δ1 loop stretches away from the finger subdomain (Fig. 8C), and the loop may be involved in the binding of ribo-GTP (rGTP) in a pocket located between the fingers and thumb (35). As presented in Fig. 9A and F, phosphorylation of the Ser29 and Ser42 residues results in a negative electrostatic surface potential. Phosphorylated Ser29 (pSer29) forms hydrogen bonds with Arg503 and His428 (Fig. 8C and 9A). These interactions are absent in NS5B with nonphosphorylated Ser29 and in NS5B with an Ala, Asp, or Glu substitution at Ser29 (Fig. 9B to E).
FIG 8.

Structural analysis of Ser29 and Ser42 phosphorylation sites on a 3D structural model of phosphorylated NS5B. (A) An in silico molecular model of phosphorylated NS5B was generated as described in Materials and Methods. In the ribbon representation of the predicted 3D structure, the two phosphorylation sites, Ser29 and Ser42, are marked by black arrows on the Δ1 loop region, colored raspberry. The NS5B polymerase structure is colored as follows: finger domain, red; palm domain, yellow; thumb domain, blue. (B) Molecular GRASP (55) surface representation of the electrostatic potential of the HCV NS5B finger-thumb connecting region showing the phosphate acceptor residues Ser29 (pSer29) and Ser42 (pSer42). The black arrows indicate the pSer29 and pSer42 residues (red groove) of HCV NS5B polymerase on the GRASP surface. The surface electrostatic potentials are colored red for negative (kBT, −10), blue for positive (kBT, +10), and white for neutral, where kB and T are the Boltzmann constant and temperature (Kelvin), respectively. (C) Combined ribbon and stick models showing the pSer29 and pSer42 residues on the Δ1 loop. In the magnified images, pSer29 (left) and pSer42 (right) residues are depicted as red sticks. Neighboring residues are marked and depicted as green sticks. The dashed magenta lines indicate hydrogen bonds and salt bridges.
FIG 9.

Structural analysis of NS5B proteins with amino acid substitutions at Ser29 and Ser42 phosphorylation sites. Combined ribbon and stick models (left) and GRASP surface representation (right) displaying pSer29 and pSer42 residues (A and F), their nonphosphorylated forms (B and G), or other indicated amino acid substituents (Ala substitution [C and H], Asp substitution [D and I], and Glu substitution [E and J]). The residues interacting with residues 29 and 42 in the thumb subdomain and within the Δ1 loop emerging from the finger domain, respectively, are shown. In panels A and B, pSer29 and pSer42 are depicted as red sticks. Neighboring residues are marked and depicted as green sticks. Hydrogen bonds and salt bridges are indicated by dashed lines (magenta). The GRASP surface representation of pSer29 and pSer42 displays a groove with a negative surface charge. The surface is colored as in Fig. 8B.
We also found that Ser42 phosphorylation can lead to additional interactions with nearby residues within the Δ1 loop. Phosphorylated Ser42 (pSer42) forms a salt bridge with Arg43 (Fig. 8C and 9F) and interacts with Thr41 via a hydrogen bond at the base of the Δ1 loop. In contrast, nonphosphorylated Ser42 and Ala, Asp, or Glu substitutions did not allow these interactions (Fig. 9G to J). These connections of Ser42 at the base of the Δ1 finger loop are surrounded by a hydrophobic pocket formed with the side chains of residues Ile11, Thr12, Pro13, Glu17, Thr44, Ala140, and Asn142.
DISCUSSION
We identified two PRK2 phosphorylation sites, Ser29 and Ser42, on HCV NS5B and demonstrated the critical role of NS5B phosphorylation in HCV RNA replication by a reverse-genetics approach using phosphorylation site mutants of two HCV genotypes. The two identified phosphorylation sites are highly conserved in all HCV genomes. Among 460 HCV sequences in the HCV database (http://hcv.lanl.gov/content/sequence/HCV/ToolsOutline.html), the Ser29 residue is conserved in 98.5% of NS5B sequences from various HCV genotypes (Thr in 6 sequences, GenBank accession numbers AF145454, AF064490, Y13184, DQ314806, D63822, and EF108306, and Ala in 1 sequence, D84263), and the Ser42 residue is 97% conserved (Thr in 13 sequences, EU155370, EU155371, NC_009823, Y11604, DQ418782, DQ418783, DQ418784, DQ418787, DQ418788, DQ418789, DQ314805, DQ835761, DQ835769, and D84264, and Ala in 1 sequence, AF238481) (see Table S1 in the supplemental material), suggesting the importance of these residues in the phosphorylation-mediated regulation of HCV NS5B functions.
HCV RdRp, like other RNA virus RdRps, forms a structure that resembles a closed right hand consisting of fingers, palm, and thumb domains. The palm domain contains the RdRp active site, and extensive finger-thumb interactions shield the palm domain to form a closed conformation, even in the absence of an RNA replication template (47, 49). This closed conformation may cause a template channel to form that is too narrow to accommodate partially duplexed RNA molecules formed during HCV RNA synthesis, suggesting an open-to-closed conformational transition after RNA synthesis initiation. The Δ1 loop, comprised of Ile11-Ser46 and protruding from the finger subdomain of NS5B, plays an important role in determining the active state of HCV RNA polymerase (19, 46). The loop, and particularly its tip, was shown to regulate RdRp conformation; specifically, deletion of five amino acid residues (residues 26 to 30) in the tip region induced an open conformation. Furthermore, previous studies have reported that Ser29 is crucial for thumb-finger interactions to maintain the structural integrity of NS5B (19, 48). Phosphorylation of the Ser29 and Ser42 residues within the loop is likely to result in local perturbation of its electrical charge, causing the region to adopt different secondary structures. If so, regulation of HCV NS5B phosphorylation would have important implications for dynamic conformational changes in viral RNA polymerase, which may be essential to initiation and elongation of RNA synthesis (19).
Crystallographic studies identified the NS5B finger domain residues (Ser29 and Arg32) and the thumb domain residues (Pro495, Pro496, Val499, and Arg503) that form an rGTP-binding pocket at the interface between the finger and thumb subdomains (50). The Ser29 residue at the C terminus of α-helix A, located at the tip of the Δ1 finger loop, interacts with the ribose of rGTP via water-mediated hydrogen bonding (35, 50). This allosteric site is located on the enzyme's surface, 30 Å away from the catalytic site, and may modulate the NS5B conformation after rGTP binding. The Ser29 and Arg32 residues in α-helix A of the fingertip form a network of interactions between the fingers and thumb through rGTP (46). Mutational analyses demonstrated that the rGTP-binding sites mentioned above are important for HCV RNA replication in cell culture (50). We showed that the S29A mutation significantly impaired the replication of HCV subgenomic and genomic RNA. Furthermore, we demonstrated that mutant HCV subgenomic replicons with a mutation of Ser29 to a phosphoamino acid surrogate, such as Asp and Glu, failed to enable the mutant replicons to form cell colonies. Likewise, a change of Ser29 to Ala, Asp, or Glu dramatically reduced HCV genome replication levels in the transient-replication assay. These results highlight the essential role of Ser29 in the HCV RNA replication process. Because Ser29 can interact with guanine through a water-mediated hydrogen bond, phosphorylation of this residue affects rGTP binding to the shallow rGTP pocket at the surface of NS5B (35). Our results and previous findings together suggest that pSer29 may aid in stabilizing the rGTP-binding pocket and enhance de novo RNA synthesis by inducing a closed conformation through stabilization of the Δ1 loop and thumb subdomain interactions. Our molecular-modeling study predicted that only pSer29, and not nonphosphorylated Ser29, Ala29, Asp29, or Glu29, forms a hydrogen bond with Arg503, implying that Ser29 phosphorylation may stabilize the closed conformation to facilitate de novo RNA synthesis initiation (19, 48). The importance of Ser29 and its phosphorylation status in RNA synthesis initiation is supported by the finding that the NS5B inhibitors benzimidazole and indole, which target the interface of the Δ1 loop and thumb domain, block RNA synthesis initiation (19, 51, 52).
To date, few studies have addressed the functional role of Ser42 in HCV RNA replication. We showed the S42A mutation in HCV subgenomic replicon RNA still yielded G418-resistant colonies, although both colony formation efficiency and intracellular subgenomic RNA levels were significantly decreased. Ala substitutions on both Ser29 and Ser42 resulted in complete loss of replication activity of the HCV replicons, suggesting that prevention of Ser29 phosphorylation causes a more marked effect on HCV replication. Transient HCV replication assays also revealed that a mutation of Ser42 to Ala, Asp, or Glu interferes with HCV RNA replication. The last two mutations, generated to mimic the constitutively phosphorylated state, were even more detrimental to HCV replication, suggesting that adapting a fixed conformation with those phosphoamino acids surrogates may inhibit, rather than stimulate, HCV replication. As observed with the subgenomic replicon mutants, the HCV genome with the S42A mutation replicated better than the mutant with the S29A mutation; however, both mutants showed no detectable levels of infectivity when HCV recovered from HCV RNA-transfected cells was used in infection assays, indicating that both Ser residues contribute to the productive replication of HCV.
Our results from the colony formation and transient-replication assays clearly demonstrated that mutations of Ser29 or Ser42 to Ala, Asp, or Glu all led to decreased HCV RNA replication efficiency. We propose that a specific secondary structure within the Δ1 finger loop may undergo dynamic structural changes, depending on the phosphorylation status at the Ser29 and Ser42 residues, which shifts NS5B to a more replication-competent RNA polymerase in vivo. The importance of maintaining the proper structure in the Δ1 finger loop carrying the two identified phosphorylation sites is further highlighted by the RdRp assay results, which showed that both primer-dependent and de novo RNA synthesis are substantially impaired in the NS5B mutants with S29A and S42A mutations. In addition, the NS5B mutant carrying both S29A and S42A substitutions was completely inactive. These results also suggest that decreased RdRp activity is one explanation for the lower replication efficiency of the subgenomic HCV replicons and full-genome mutants with an S29A or S42A mutation in cell culture.
A final and critical question addressed in our study relates to how phosphorylated NS5B regulates HCV replication. The specific mechanism remains unknown; however, one possibility is that enzymatic functions of NS5B itself, including RNA synthesis initiation and elongation, may be regulated by phosphorylation, because NS5B can certainly affect its ability to bind the RNA templates, as discussed above. We also cannot exclude the possibility that phosphorylation of NS5B may affect its ability to form an active RNA replicase complex with other viral nonstructural proteins and/or cellular proteins due to conformational changes at the N-terminal Δ1 loop exposed on the enzyme's surface (35, 47). For example, cellular proteins, such as the tumor suppressor retinoblastoma protein (53) and cyclophilin, which interacts with HCV RdRp to affect its ability to bind RNA (54), may require a conformational change of NS5B for its interaction with these cellular proteins. Presently, we do not know which viral and/or cellular proteins lose or acquire the ability to bind phosphorylated NS5B. Finally, NS5B dimerization, which may be RdRp's de novo initiation-competent conformation (19, 48), might also be affected by phosphorylation, because the Δ1 loop and its interaction with the thumb could influence contact between RdRp subunits.
In summary, many possible models could explain the effects of NS5B phosphorylation, as follows: (i) phosphorylation could influence the uptake of RNA or facilitate the release of nascent RNA synthesized from the replicative complex, (ii) the RNA synthesis initiation and elongation modes of NS5B are likely controlled by dynamic regulation of NS5B phosphorylation states, (iii) NS5B dimerization might be facilitated by phosphorylation, or (iv) phosphorylation of Ser29 and Ser42 of NS5B may affect its interaction with other viral and/or cellular proteins to alter its subcellular localization or functions. Our results nevertheless underscore that Ser29 and Ser42 are two major phosphorylation sites that play critical roles in HCV replication. Future studies should focus on the quantification of phosphorylated NS5B to determine how NS5B phosphorylation is regulated during the HCV life cycle. In addition, it remains unclear how phosphorylation and dephosphorylation of NS5B are regulated during HCV replication and the progression of liver diseases, as the NS5B phosphorylation state can be dynamically reprogrammed by PRK2 activation signals.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the National Research Foundation of Korea (NRF-2009-0092959, NRF-2010-0025982, NRF-2013-063182, and NRF-2014R1A2A2A1005522) and in part by the Technology Innovation Program (MKE 10035159) funded by the Ministry of Knowledge Economy (MKE), South Korea.
Footnotes
Published ahead of print 16 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01826-14.
REFERENCES
- 1.Tarrant MK, Cole PA. 2009. The chemical biology of protein phosphorylation. Annu. Rev. Biochem. 78:797–825. 10.1146/annurev.biochem.78.070907.103047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imai T, Sagou K, Arii J, Kawaguchi Y. 2010. Effects of phosphorylation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase in vivo and in vitro. J. Virol. 84:153–162. 10.1128/JVI.01447-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hwang LN, Englund N, Das T, Banerjee AK, Pattnaik AK. 1999. Optimal replication activity of vesicular stomatitis virus RNA polymerase requires phosphorylation of a residue(s) at carboxy-terminal domain II of its accessory subunit, phosphoprotein P. J. Virol. 73:5613–5620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen C, Wang JC, Zlotnick A. 2011. A kinase chaperones hepatitis B virus capsid assembly and captures capsid dynamics in vitro. PLoS Pathog. 7:e1002388. 10.1371/journal.ppat.1002388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Law LM, Everitt JC, Beatch MD, Holmes CF, Hobman TC. 2003. Phosphorylation of rubella virus capsid regulates its RNA binding activity and virus replication. J. Virol. 77:1764–1771. 10.1128/JVI.77.3.1764-1771.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jakubiec A, Tournier V, Drugeon G, Pflieger S, Camborde L, Vinh J, Hericourt F, Redeker V, Jupin I. 2006. Phosphorylation of viral RNA-dependent RNA polymerase and its role in replication of a plus-strand RNA virus. J. Biol. Chem. 281:21236–21249. 10.1074/jbc.M600052200 [DOI] [PubMed] [Google Scholar]
- 7.Kim SJ, Kim JH, Kim YG, Lim HS, Oh JW. 2004. Protein kinase C-related kinase 2 regulates hepatitis C virus RNA polymerase function by phosphorylation. J. Biol. Chem. 279:50031–50041. 10.1074/jbc.M408617200 [DOI] [PubMed] [Google Scholar]
- 8.Kim SJ, Kim JH, Sun JM, Kim MG, Oh JW. 2009. Suppression of hepatitis C virus replication by protein kinase C-related kinase 2 inhibitors that block phosphorylation of viral RNA polymerase. J. Viral Hepat. 16:697–704. 10.1111/j.1365-2893.2009.01108.x [DOI] [PubMed] [Google Scholar]
- 9.Kapoor M, Zhang L, Ramachandra M, Kusukawa J, Ebner KE, Padmanabhan R. 1995. Association between NS3 and NS5 proteins of dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J. Biol. Chem. 270:19100–19106. 10.1074/jbc.270.32.19100 [DOI] [PubMed] [Google Scholar]
- 10.Hericourt F, Blanc S, Redeker V, Jupin I. 2000. Evidence for phosphorylation and ubiquitinylation of the turnip yellow mosaic virus RNA-dependent RNA polymerase domain expressed in a baculovirus-insect cell system. Biochem. J. 349:417–425. 10.1042/0264-6021:3490417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim SH, Palukaitis P, Park YI. 2002. Phosphorylation of cucumber mosaic virus RNA polymerase 2a protein inhibits formation of replicase complex. EMBO J. 21:2292–2300. 10.1093/emboj/21.9.2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eden JS, Sharpe LJ, White PA, Brown AJ. 2011. Norovirus RNA-dependent RNA polymerase is phosphorylated by an important survival kinase, Akt. J. Virol. 85:10894–10898. 10.1128/JVI.05562-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Das SC, Pattnaik AK. 2004. Phosphorylation of vesicular stomatitis virus phosphoprotein P is indispensable for virus growth. J. Virol. 78:6420–6430. 10.1128/JVI.78.12.6420-6430.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lavanchy D. 2009. The global burden of hepatitis C. Liver Int. 29(Suppl 1):S74–S81. 10.1111/j.1478-3231.2008.01934.x [DOI] [PubMed] [Google Scholar]
- 15.Bartenschlager R, Lohmann V. 2000. Replication of hepatitis C virus. J. Gen. Virol. 81:1631–1648 http://vir.sgmjournals.org/content/81/7/1631.full.pdf+html [DOI] [PubMed] [Google Scholar]
- 16.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. 2007. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9:1089–1097. 10.1038/ncb1631 [DOI] [PubMed] [Google Scholar]
- 17.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, Walther P, Antony C, Krijnse-Locker J, Bartenschlager R. 2012. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 8:e1003056. 10.1371/journal.ppat.1003056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ago H, Adachi T, Yoshida A, Yamamoto M, Habuka N, Yatsunami K, Miyano M. 1999. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure 7:1417–1426. 10.1016/S0969-2126(00)80031-3 [DOI] [PubMed] [Google Scholar]
- 19.Chinnaswamy S, Murali A, Li P, Fujisaki K, Kao CC. 2010. Regulation of de novo-initiated RNA synthesis in hepatitis C virus RNA-dependent RNA polymerase by intermolecular interactions. J. Virol. 84:5923–5935. 10.1128/JVI.02446-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang SM, Choi JK, Kim SJ, Kim JH, Ahn DG, Oh JW. 2009. Regulation of hepatitis C virus replication by the core protein through its interaction with viral RNA polymerase. Biochem. Biophys. Res. Commun. 386:55–59. 10.1016/j.bbrc.2009.05.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamamoto I, Nishimura Y, Okamoto T, Aizaki H, Liu M, Mori Y, Abe T, Suzuki T, Lai MM, Miyamura T, Moriishi K, Matsuura Y. 2005. Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J. Virol. 79:13473–13482. 10.1128/JVI.79.21.13473-13482.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bain J, McLauchlan H, Elliott M, Cohen P. 2003. The specificities of protein kinase inhibitors: an update. Biochem. J. 371:199–204. 10.1042/BJ20021535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goh PY, Tan YJ, Lim SP, Tan YH, Lim SG, Fuller-Pace F, Hong W. 2004. Cellular RNA helicase p68 relocalization and interaction with the hepatitis C virus (HCV) NS5B protein and the potential role of p68 in HCV RNA replication. J. Virol. 78:5288–5298. 10.1128/JVI.78.10.5288-5298.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okamoto T, Nishimura Y, Ichimura T, Suzuki K, Miyamura T, Suzuki T, Moriishi K, Matsuura Y. 2006. Hepatitis C virus RNA replication is regulated by FKBP8 and Hsp90. EMBO J. 25:5015–5025. 10.1038/sj.emboj.7601367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palmer RH, Parker PJ. 1995. Expression, purification and characterization of the ubiquitous protein kinase C-related kinase 1. Biochem. J. 309:315–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim MG, Moon JS, Kim EJ, Lee SH, Oh JW. 2012. Destabilization of PDK1 by Hsp90 inactivation suppresses hepatitis C virus replication through inhibition of PRK2-mediated viral RNA polymerase phosphorylation. Biochem. Biophys. Res. Commun. 421:112–118. 10.1016/j.bbrc.2012.03.126 [DOI] [PubMed] [Google Scholar]
- 27.Yanagi M, St Claire M, Shapiro M, Emerson SU, Purcell RH, Bukh J. 1998. Transcripts of a chimeric cDNA clone of hepatitis C virus genotype 1b are infectious in vivo. Virology 244:161–172. 10.1006/viro.1998.9092 [DOI] [PubMed] [Google Scholar]
- 28.Zhu Q, Guo JT, Seeger C. 2003. Replication of hepatitis C virus subgenomes in nonhepatic epithelial and mouse hepatoma cells. J. Virol. 77:9204–9210. 10.1128/JVI.77.17.9204-9210.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oh JW, Ito T, Lai MM. 1999. A recombinant hepatitis C virus RNA-dependent RNA polymerase capable of copying the full-length viral RNA. J. Virol. 73:7694–7702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796. 10.1038/nm1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oh JW, Sheu GT, Lai MM. 2000. Template requirement and initiation site selection by hepatitis C virus polymerase on a minimal viral RNA template. J. Biol. Chem. 275:17710–17717. 10.1074/jbc.M908781199 [DOI] [PubMed] [Google Scholar]
- 32.Boyle WJ, van der Geer P, Hunter T. 1991. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods Enzymol. 201:110–149. 10.1016/0076-6879(91)01013-R [DOI] [PubMed] [Google Scholar]
- 33.Kim JY, Lee JH, Park GW, Cho K, Kwon KH, Park YM, Cho SY, Paik YK, Yoo JS. 2005. Utility of electrophoretically derived protein mass estimates as additional constraints in proteome analysis of human serum based on MS-MS analysis. Proteomics 5:3376–3385. 10.1002/pmic.200401220 [DOI] [PubMed] [Google Scholar]
- 34.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 309:1577–1581. 10.1126/science.1113329 [DOI] [PubMed] [Google Scholar]
- 35.Bressanelli S, Tomei L, Rey FA, De Francesco R. 2002. Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J. Virol. 76:3482–3492. 10.1128/JVI.76.7.3482-3492.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 37.Sanner MF, Olson AJ, Spehner JC. 1996. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers 38:305–320 [DOI] [PubMed] [Google Scholar]
- 38.Krivov GG, Shapovalov MV, Dunbrack RL., Jr 2009. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 77:778–795. 10.1002/prot.22488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis IW, Murray LW, Richardson JS, Richardson DC. 2004. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32:W615–W619. 10.1093/nar/gkh398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. 2006. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65:712–725. 10.1002/prot.21123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hinsen K. 2000. The molecular modeling toolkit: a new approach to molecular simulations. J. Comput. Chem. 21:79–85. [DOI] [Google Scholar]
- 42.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A. 98:10037–10041. 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li L, Li C, Sarkar S, Zhang J, Witham S, Zhang Z, Wang L, Smith N, Petukh M, Alexov E. 2012. DelPhi: a comprehensive suite for DelPhi software and associated resources. BMC Biophys. 5:9. 10.1186/2046-1682-5-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. 2006. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34:W116–W118. 10.1093/nar/gkl282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palmer RH, Ridden J, Parker PJ. 1995. Cloning and expression patterns of two members of a novel protein-kinase-C-related kinase family. Eur. J. Biochem. 227:344–351. 10.1111/j.1432-1033.1995.tb20395.x [DOI] [PubMed] [Google Scholar]
- 46.Labonte P, Axelrod V, Agarwal A, Aulabaugh A, Amin A, Mak P. 2002. Modulation of hepatitis C virus RNA-dependent RNA polymerase activity by structure-based site-directed mutagenesis. J. Biol. Chem. 277:38838–38846. 10.1074/jbc.M204657200 [DOI] [PubMed] [Google Scholar]
- 47.Bressanelli S, Tomei L, Roussel A, Incitti I, Vitale RL, Mathieu M, De Francesco R, Rey FA. 1999. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 96:13034–13039. 10.1073/pnas.96.23.13034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chinnaswamy S, Yarbrough I, Palaninathan S, Kumar CT, Vijayaraghavan V, Demeler B, Lemon SM, Sacchettini JC, Kao CC. 2008. A locking mechanism regulates RNA synthesis and host protein interaction by the hepatitis C virus polymerase. J. Biol. Chem. 283:20535–20546. 10.1074/jbc.M801490200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. 1999. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 6:937–943. 10.1038/13305 [DOI] [PubMed] [Google Scholar]
- 50.Cai Z, Yi M, Zhang C, Luo G. 2005. Mutagenesis analysis of the rGTP-specific binding site of hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 79:11607–11617. 10.1128/JVI.79.18.11607-11617.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kukolj G, McGibbon GA, McKercher G, Marquis M, Lefebvre S, Thauvette L, Gauthier J, Goulet S, Poupart MA, Beaulieu PL. 2005. Binding site characterization and resistance to a class of non-nucleoside inhibitors of the hepatitis C virus NS5B polymerase. J. Biol. Chem. 280:39260–39267. 10.1074/jbc.M506407200 [DOI] [PubMed] [Google Scholar]
- 52.Di Marco S, Volpari C, Tomei L, Altamura S, Harper S, Narjes F, Koch U, Rowley M, De Francesco R, Migliaccio G, Carfi A. 2005. Interdomain communication in hepatitis C virus polymerase abolished by small molecule inhibitors bound to a novel allosteric site. J. Biol. Chem. 280:29765–29770. 10.1074/jbc.M505423200 [DOI] [PubMed] [Google Scholar]
- 53.Munakata T, Nakamura M, Liang Y, Li K, Lemon SM. 2005. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 102:18159–18164. 10.1073/pnas.0505605102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Z, Robida JM, Chinnaswamy S, Yi G, Robotham JM, Nelson HB, Irsigler A, Kao CC, Tang H. 2009. Mutations in the hepatitis C virus polymerase that increase RNA binding can confer resistance to cyclosporine A. Hepatology 50:25–33. 10.1002/hep.22987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nicholls A, Sharp KA, Honig B. 1991. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11:281–296. 10.1002/prot.340110407 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.