

ABSTRACT
Dendritic cells (DCs) are fundamental for the initiation of immune responses and are important players in AIDS immunopathogenesis. The modulation of DC functional activities represents a strategic mechanism for HIV-1 to evade immune surveillance. Impairment of DC function may result from bystander effects of HIV-1 envelope proteins independently of direct HIV-1 infection. In this study, we report that exposure of immature monocyte-derived DCs (MDDCs) to HIV-1 R5 gp120 resulted in the CCR5-dependent production of interleukin-6 (IL-6) via mitogen-activated protein kinase (MAPK)/NF-κB pathways. IL-6 in turn activated STAT3 by an autocrine loop. Concomitantly, gp120 promoted an early activation of STAT3 that further contributed to IL-6 induction. This activation paralleled a concomitant upregulation of the STAT3 inhibitor PIAS3. Notably, STAT3/IL-6 pathway activation was not affected by the CCR5-specific ligand CCL4. These results identify STAT3 as a key signaling intermediate activated by gp120 in MDDCs and highlight the existence of a virus-induced dysregulation of the IL-6/STAT3 axis. HIV-1 gp120 signaling through STAT3 may provide an explanation for the impairment of DC function observed upon HIV exposure.
IMPORTANCE This study provides new evidence for the molecular mechanisms and signaling pathways triggered by HIV-1 gp120 in human DCs in the absence of productive infection, emphasizing a role of aberrant signaling in early virus-host interaction, contributing to viral pathogenesis. We identified STAT3 as a key component in the gp120-mediated signaling cascade involving MAPK and NF-κB components and ultimately leading to IL-6 secretion. STAT3 now is recognized as a key regulator of DC functions. Thus, the identification of this transcription factor as a signaling molecule mediating some of gp120's biological effects unveils a new mechanism by which HIV-1 may deregulate DC functions and contribute to AIDS pathogenesis.
INTRODUCTION
Dendritic cells (DCs) play a pivotal role in linking innate and adaptive immunity by their ability to induce appropriate immune responses upon recognition of invading pathogens (1). Because of their central role in the induction of immune responses, modulation of DC function represents a strategic mechanism for a pathogen to evade immune surveillance (2). In human immunodeficiency virus type 1 (HIV-1) infection, DCs are among the first cells to encounter HIV-1 at mucosal sites, where they are coopted by HIV-1 to facilitate transmission (3–6). Once infection is established, HIV-1 directly and indirectly modulates DC function to hinder the formation of effective adaptive immunity and promote immune activation (7, 8). Among the events following HIV-1 exposure that may be unrelated to infection, the greatest effects have been attributed to the envelope glycoprotein gp120. Besides facilitating viral entry, gp120 binding to chemokine receptors in several cell types, including monocytes/macrophages and DCs, also initiates signaling events that may have important implications for pathogenesis by affecting postentry stages of infection or by modulating cellular functions apart from infection (8). Cellular signal transduction pathways have been shown to be perturbed by HIV infection; conversely, their activation can regulate the replicative capacity of HIV-1 or dramatically affect cell functions (7). Although chemokine receptors and their ligands play central roles in both HIV infection and immune regulation, how signaling pathways mediated by CCR5 and CXCR4 contribute to the immunopathogenesis of HIV infection has been poorly investigated in DCs. Exposure of these cells to gp120 has been shown to induce a Pyk2-dependent signaling pathway that mediates DC migration, facilitating HIV-1 dissemination, as well as to activate mitogen-activated protein kinases (MAPKs), which act as a central pathway in the signaling network of the host cell (9, 10).
STAT3 is now recognized as a critical regulator of DC physiology, exerting different and apparently opposite effects on DC development and activation. Furthermore, STAT3 represents an important intermediate in the signal transduction pathways triggered through cytokine receptors, and it is involved in the transcriptional activation of some cytokine genes as well as microRNAs (11, 12). Early studies demonstrated that STAT3 deletion in hematopoietic progenitors led to profound deficiency in the DC compartment (13), impaired the expansion of DC progenitors (14), and affected both DC differentiation from hematopoietic precursors (15) and maturation (16). Subsequently, it was found that the constitutive activation of STAT3 in tumor-infiltrating DCs is responsible for the impairment of DC differentiation and functional maturation (16–18). Accordingly, inhibition of JAK2/STAT3 signaling dramatically improved differentiation and activation of murine and human DCs (19, 20), and conditional knockout (KO) mice with STAT3 deletion in CD11c+ DCs exhibited an altered immune homeostasis and chronic inflammation (21). Conversely, very little is known about STAT3 involvement in DC response to HIV-1.
In this work, we identify STAT3 as a key signaling intermediate activated by gp120 in MDDCs as part of a regulatory circuit involving MAPK–NF-κB pathways and IL-6 secretion. Reconstruction of the signaling pathway triggered by gp120 in MDDCs by pharmacologic inhibition of selected pathway components and blocking of IL-6 biological activity unravels a biphasic activation of STAT3.
These results highlight the existence of a novel regulatory network involving STAT3 and IL-6, suggesting that in MDDCs, HIV-1 gp120 signaling through STAT3 contributes to their functional impairment and favors HIV infection.
MATERIALS AND METHODS
Ethics statements.
Healthy donor buffy coats were obtained from Centro Trasfusionale University of Rome Sapienza. Buffy coats were not obtained specifically for this study. Informed consent has not been asked because data were analyzed anonymously. Data from healthy donors have been treated by Centro Trasfusionale according to the Italian law on personal data management, “Codice in materia di protezione dei dati personali” (Testo Unico D.L., 30 June 2003, no. 196).
Cell separation and culture.
Monocytes were isolated from peripheral blood mononuclear cells (PBMCs) obtained from healthy donor buffy coats by immunomagnetic selection using CD14 microbeads (MACS monocyte isolation kit from Miltenyi Biotec, Auburn, CA) according to the manufacturer's instructions. This procedure yields a ≥98% pure population of monocytes, as assessed by fluorescence-activated cell sorting (FACS) analysis of lineage-specific surface markers (CD1a, CD14, CD3, CD19, and CD56). To obtain immature MDDCs, monocytes were cultured at 1 × 106 cells/ml in RPMI 1640 medium (Life Technologies, Gaithersburg, MD) containing 10% fetal bovine serum (FBS) in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) (50 ng/ml) and IL-4 (500 U/ml). Cytokines were added to the cultures every 3 days. Both cytokines were kindly provided by Schering-Plough (Dardilly, France). On day 6, MDDCs were stimulated with lipopolysaccharide (LPS; 10 ng/ml), gp120 (5 μg/ml, unless otherwise indicated), AT-2-inactivated HIV-1 BaL (46.783 ng/ml p24) (22), or CCL4 (100 nM). Monocyte-derived macrophages (MDMs) were obtained after 6 days of in vitro culture in endotoxin-free Iscove's medium containing 10% FBS as previously described (23).
Virus inactivation procedure.
The HIV-1 BaL virus preparation were purchased from ABI (Columbia, MD) and inactivated as previously described (22).
Reagents.
All culture reagents were purchased from BioWhittaker as endotoxin-free lots. Ultrapure LPS from Escherichia coli (serotype EH100, rough mutant a [Ra] Toll-like receptor grade) was purchased from Alexis Biochemicals (Nottingham, United Kingdom). Recombinant gp120 from HIV-1 strain CN54, soluble CD4 (sCD4), and the CCR5 inhibitor Tak779 were obtained from the national AIDS Research and Reference Reagent Program (Bethesda, MD). Recombinant HIV-1 gp120 (ADA strain) was kindly provided by G. Gao. Recombinant CCL4 and neutralizing antibody to human CD4 were purchased from R&D Systems. Anti-IL-6 neutralizing antibody or IgG isotype control were purchased from Sigma and used at a concentration of 5 μg/ml (1 h pretreatment). To test the effect of specific inhibitors, cells were treated prior to and during gp120 exposure with SB 202190, BAY 117082 (Calbiochem, San Diego, CA), N-p-tosyl-l-phenylalanine chloromethyl ketone (TPCK), 4-(4-fluorophenyl)-2-(4-nitrophenyl)-5-(4-pyridyl)-1H-imidazole (PD 169316), AG490, JSI 124, Stattic (Sigma, St. Louis, MO), and S3I 201 (Selleckchem). None of the inhibitors utilized in the experiments exhibited any toxicity at the concentrations used, as assessed by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (data not shown).
Measurement of IL-6.
The IL-6 level (detection limit, 7.8 pg/ml) in culture supernatants was measured by enzyme-linked immunosorbent assay (ELISA) kits purchased from BioLegend (San Diego, CA) according to the manufacturer's instructions.
Immunoblotting.
DCs were subjected to the different treatments for the indicated time periods. Cell were lysed in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris-Cl [pH 7.5], 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS) containing a cocktail of protease and phosphatase inhibitors, and protein extracts were resolved by 8 to 12% SDS-PAGE, transferred to a nitrocellulose membrane, and subjected to immunoblot analysis with antibodies specific for the total or phosphorylated forms of STAT-1, STAT-3, p65 NF-κB, IkBα, p38 MAPK, and PIAS3 (all from Cell Signaling Technology, Beverly, MA), as well as SOCS3 and STAT-2 (from Santa Cruz Biotechnology, Santa Cruz, CA). Levels of phosphorylated and nonphosphorylated proteins were quantified using ImageJ software (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD).
Real-time quantitative PCR (qPCR) analysis.
For gene expression assays, total RNA was isolated with the Qiagen RNeasy minikit (Hilden, Germany), treated with DNase I (Qiagen), and retrotranscribed into cDNA by using pd(N)6 (GE Healthcare, Pittsburg, PA). Real-time PCR was performed on an ABI-Prism 7700 PCR cycler (Applied Biosystems, Foster City, CA) using the TaqMan universal master mix (Applied Biosystems) according to the manufacturer's instructions. Validated PCR primers and TaqMan MGB probe (6-carboxyfluorescein [FAM]-labeled) for IL-6 and for human β actin as an endogenous control were used (Applied Biosystems). Relative quantification was performed by using the comparative cycle threshold (CT) method (24).
Statistical analysis.
Statistical comparison between various groups was performed by Student's t test. Comparisons were made between means from several experiments. Differences were considered significant when P values were <0.05. Statistical significance is indicated by one asterisk for P < 0.05 and two asterisks for P < 0.005.
RESULTS
Engagement of CCR5 by HIV-1 gp120, but not CCL4, upmodulates IL-6 expression.
IL-6 has been involved in the induction of semimature DCs endowed with tolerogenic properties (25), and its induction by gp120 has been shown in some cell models (26–30). Recently, HIV-1-induced IL-6 has been reported to contribute to myeloid-derived suppressor cell (MDSC) expansion (31). As shown in Fig. 1A, concentration-dependent secretion of IL-6 was observed upon exposure of immature MDDCs (iMDDCs) to gp120 at 18 h following treatment. Although IL-6 secretion was already observed at the gp120 concentration of 2 μg/ml, a stronger effect was observed at the higher concentration (5 μg/ml), while no effects were found at lower concentrations (0.5 μg/ml). Conversely, markedly higher secretion of IL-6 was observed when LPS, a classical maturation stimulus, was used. Therefore, all subsequent experiments aimed at defining the kinetics of IL-6 production were performed at the gp120 concentration of 5 μg/ml. IL-6 secretion (Fig. 1B) generally was not found at early time points (1 h) but started to be detected at 4 h, with the maximal amount measured after 18 h of treatment. Similar results were achieved upon exposure of MDDCs to AT-2-inactivated HIV-1 BaL virus, although to a lower extent (Fig. 1B), as well as upon exposure to the R5 gp120 ADA strain (Fig. 1B). Conversely, no IL-6 secretion was observed upon MDDC treatment with heat-inactivated gp120, excluding endotoxin contamination of gp120 preparation (data not shown). Experiments then were conducted to investigate whether the gp120-induced IL-6 upmodulation was associated with an enhanced accumulation of the corresponding mRNA. As shown in Fig. 1C, IL-6 mRNA accumulation already was induced after 1 h of gp120 treatment, peaked at 4 h, and was not further increased at 6 h of treatment. To probe CCR5 involvement in the gp120-mediated IL-6 induction, we used the CCR5 antagonist Tak779, which blocks both HIV-1 coreceptor function and CCL4 signaling. As shown in Fig. 1D, Tak779 almost completely inhibited gp120-induced IL-6 production. In contrast, the gp120-mediated effect was independent of its interaction with CD4, as preincubation with soluble CD4 or neutralizing anti-CD4 antibody did not abrogate IL-6 induction. Moreover, triggering of CCR5 by its most specific natural ligand, CCL4, did not stimulate any IL-6 secretion, supporting the existence of gp120-specific signaling (Fig. 1D).
FIG 1.

HIV gp120, but not CCL4, upmodulates IL-6 expression via CCR5. MDDCs at day 6 of culture were treated with different concentrations of gp120 and LPS (10 ng/ml) (A) or for different time periods with 5 μg/ml gp120 (CN54 and ADA strain) and BaL-AT2 (46.783 ng/ml p24) (B and C). To test the specificity of gp120-induced effects, MDDCs were preincubated for 1 h with or without sCD4 (2 μg/ml), anti-CD4 (2 μg/ml), or Tak779 (5 μM) and then exposed to gp120 (5 μg/ml) for 18 h or treated with CCL4 (100 nM) for the indicated times (D). For panels A, B, and D, supernatants were collected and IL-6 content was measured by ELISA. Data are expressed as means ± standard errors (SE) from six (A) or four (B and D) independent experiments. P values were calculated by Student's t test, and statistical significance is indicated. *, P < 0.05; **, P < 0.005 (gp120 versus the untreated control and Tak779 plus gp120 versus gp120 alone). (C) Total RNA was extracted and reverse transcribed, and real-time PCR was performed using primers specific for the human IL-6 gene and normalized with β actin. Data are expressed as means ± SE from five independent experiments performed. Results, analyzed by the relative quantification method (2−ΔΔCT method), are shown as fold increase (FC) of IL-6 gene expression versus that of the untreated control. P values were calculated by Student's t test, and statistical significance is indicated (P < 0.05 [*] and P < 0.005 [**] for gp120 versus the untreated control).
HIV-1 gp120-induced IL-6 secretion is dependent on p38 MAPK and NF-κB activation.
Previous studies showed that gp120 activates MAPK pathways in DCs, and that this initial activation is responsible for the gp120-induced migration (9, 10). To examine the involvement of this pathway in gp120-induced IL-6 secretion, iMDDCs were incubated with gp120 and processed thereafter for the analysis of phospho-p38 MAPK. In keeping with the results achieved with other gp120 strains (10), p38 MAPK was found to be activated by the gp120 CN54 strain (Fig. 2A). Conversely, ERK1/2 and JNK were not activated by gp120 in iMDDCs (data not shown). IL-6 expression strongly depends on NF-κB activation in different cell types, and binding of NF-κB proteins at the IL-6 promoter has been reported to enhance the transcription of this gene (32–34). Furthermore, the signaling cascade involving MAPK leads to downstream activation of NF-κB. As shown in Fig. 2A, the phosphorylation of IkBα was upmodulated already after 5 min of gp120 treatment and remained high at later time points. Consistent with this result, p65 NF-κB also was phosphorylated after 30 min of gp120 treatment (Fig. 2B). To further investigate the role of the MAPK/NF-κB signaling cascade in gp120-triggered IL-6 secretion, iMDDCs were treated with the p38 MAPK inhibitors SB 202190 and PD 169316 or p65 NF-κB inhibitors BAY 117082 and TPCK prior to gp120 stimulation and assessed 18 h later for their capacity to produce IL-6. At the concentration used, these inhibitors did not affect cell viability (data not shown). As shown in Fig. 2C, blocking gp120-induced p38 MAPK or p65 NF-κB activation strongly reduced IL-6 secretion in the culture medium. Conversely, IL-6 basal levels were not affected by the inhibitors in the absence of gp120 stimulation.
FIG 2.

p38 MAPK, IkBα, and p65 NF-κB are involved in HIV gp120-induced secretion of IL-6. MDDCs were stimulated with gp120 (5 μg/ml) for the indicated time periods. Cell lysates were resolved by 10% to 12% SDS-PAGE, transferred to a nitrocellulose membrane, and subjected to immunoblot analysis with antibodies specific for the total or phosphorylated (P) forms of p38 MAPK (phospho-p38 MAPK [T183/Y185]) and IkBα (phospho-IkBα [Ser32]) (A) or p65 NF-κB (phospho-p65 NF-κB [Ser536]) (B) antibody. Blots shown are from one donor and are representative of three to five experiments carried out with different donors that yielded similar results. Values below the lanes show the intensities of the respective bands. The increase in phosphorylation was estimated by calculating the ratios of the phosphorylated form to total form of proteins. Actin expression is shown as a gel loading control. (C) MDDCs were preincubated for 1 h with or without the specific p38 MAPK inhibitors SB 202190 (1 μg/ml) and PD 169316 (10 μM), or NF-κB inhibitors BAY 117082 (10 μM) and TPCK (2 μM), before gp120 exposure or were left untreated. After 18 h of culture, supernatants were harvested and frozen for IL-6 determination. Data are expressed as means ± SE from four independent experiments performed. P values were calculated by Student's t test and statistical significance is indicated. *, P < 0.05 (gp120 plus inhibitor versus gp120 alone).
HIV-1 gp120 promotes biphasic activation of STAT3.
Because IL-6 is a strong activator of STAT3 and, vice versa, STAT3 mediates transcriptional activation of the IL-6 gene (11), we initially evaluated whether IL-6 secretion paralleled a concomitant activation of STAT3. As shown in Fig. 3A, gp120 induced STAT3 activation in a concentration- and time-dependent manner. At the maximal concentration, both CN54 (Fig. 3A) and ADA gp120 strains (Fig. 3B) consistently induced STAT3 activation after 6 h of treatment. Interestingly, parallel experiments were carried out to assess whether gp120 also induced IL-6 secretion and STAT3 activation in MDMs. As shown in Fig. 4, MDMs exhibited a stronger response to gp120 as assessed by the levels of IL-6 secretion (Fig. 4A) and STAT3 phosphorylation (Fig. 4B) upon stimulation with gp120 at a lower concentration (2 μg/ml) and with more rapid kinetics (3 h).
FIG 3.

HIV gp120, but not CCL4, induces STAT3 activation via IL-6 production. MDDCs were stimulated with gp120 (concentration ranging from 0.5 to 5 μg/ml) or CCL4 (100 nM) for the indicated time periods, or with LPS (10 ng/ml), for 18 h in the presence or absence of anti-IL-6 antibody (5 μg/ml, 1 h of pretreatment). Cell lysates were resolved by 8% SDS-PAGE, transferred to a nitrocellulose membrane, and subjected to immunoblot analysis with antibodies specific for the total or phosphorylated forms of STAT3. Actin expression is shown as a gel loading control. Data from one representative experiment out of four (A and B) or two (C and D) analyzed are shown. Graphs show the ratio of phosphorylated to nonphosphorylated protein as determined by densitometry, where each sample was normalized to total STAT3. Fold phosphorylation was calculated relative to that of the untreated control, and the average fold from four (A) and two (C) independent experiments was represented graphically, along with the standard deviations (SD). Statistical significance is indicated versus the untreated control.
FIG 4.
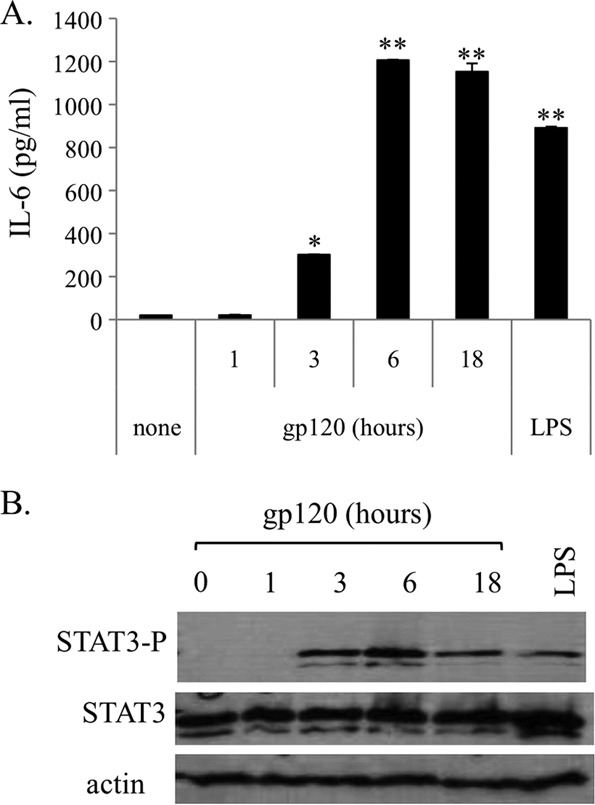
HIV gp120 promotes IL-6 secretion and induces STAT3 phosphorylation in MDMs. MDMs at day 6 of culture were treated for different time periods with gp120 (2 μg/ml) or LPS (10 ng/ml) or were left untreated. (A) At the indicated time points, supernatants were collected and IL-6 contents measured by ELISA. Data are expressed as means ± SE from four independent experiments performed. P values were calculated by Student's t test, and statistical significance is indicated (P < 0.05 and P < 0.005 for gp120 versus the untreated control). (B) Cell lysates were resolved by 8% SDS-PAGE, transferred to a nitrocellulose membrane, and subjected to immunoblot analysis with antibodies specific for the total or phosphorylated forms of STAT3. Actin expression is shown as a gel loading control. Data from one representative experiment out of three performed are shown.
We then determined whether IL-6 secretion was indeed responsible for STAT3 activation. As shown in Fig. 3C, neutralization of IL-6 biological activity by specific antibodies drastically reduced the gp120-induced STAT3 activation at 6 and 18 h. The contribution of other cytokines, including IL-10 and IL-23, known activators of STAT3, was excluded, as they were not secreted by gp120-stimulated MDDCs (data not shown). Conversely, STAT3 activation observed at 18 h upon LPS treatment was not abolished in the presence of antibodies to IL-6, suggesting that different factors induced by LPS or gp120 with specific kinetics can mediate STAT3 activation. In keeping with the effect on IL-6 secretion, CCL4 failed to activate STAT3 phosphorylation at all times tested, further supporting the specificity of gp120 signaling in DCs (Fig. 3D).
It has been described previously that STAT3 activation by epidermal growth factor (EGF) can occur in a biphasic way involving an early direct activation by EGF followed by a later second wave of activation mediated by EGF-induced IL-6 secretion (35). To evaluate whether a similar scenario could account for the gp120-induced STAT3 activation in MDDCs, we assessed STAT3 at very early time points upon gp120 exposure, when IL-6 mRNA and protein were not yet induced. As shown in Fig. 5A, early activation of STAT3 was found after 15 min of treatment. However, some variability in the time point of STAT3 activation was observed among donors, ranging from 5 to 30 min. To further demonstrate that early activation of STAT3 was directly mediated by gp120 and did not involve IL-6, experiments were carried out by using a specific neutralizing antibody to IL-6. As shown in Fig. 5B, blocking the biological activity of IL-6 does not affect gp120-induced early activation of STAT3. These results suggested that STAT3 activation occurring early upon gp120 exposure was responsible for the later upmodulation of the IL-6 transcript leading to protein secretion. To address this issue, Stattic, the first developed nonpeptidic small molecule able to bind STAT3 directly and to selectively inhibit its activation, dimerization, and nuclear translocation, was used (36). As shown in Fig. 5C, Stattic blocked IL-6 mRNA accumulation already at 1 h, and this inhibition was maintained at later time points. Likewise, Stattic markedly blocked the IL-6 secretion induced by gp120 (Fig. 5D), providing additional evidence for the existence of a STAT3/IL-6 regulatory loop.
FIG 5.

HIV gp120-induced early activation of STAT3 is responsible for IL-6 production. (A and B) MDDCs were stimulated with gp120 (5 μg/ml) for the indicated time periods in the presence or absence of anti-IL-6 antibody (5 μg/ml, 1 h of pretreatment). Cell lysates were resolved by 8% SDS-PAGE, transferred to a nitrocellulose membrane, and subjected to immunoblot analysis with antibodies specific for the total or phosphorylated forms of STAT3. Actin expression is shown as a gel loading control. Graphs in panels A and B show the ratio of phosphorylated to nonphosphorylated protein as determined by densitometry, where each sample was normalized to total STAT3. Fold phosphorylation was calculated relative to that of the untreated control, and the average fold change from four (A) and two (B) independent experiments was represented graphically, along with the SD. Statistical significance is indicated versus the untreated control. (C) MDDCs were pretreated for 1 h with the STAT3 inhibitor Stattic (10 μM) and stimulated with gp120 (5 μg/ml) for the indicated times. Total RNA was extracted and reverse transcribed, and real-time PCR was performed using primers specific for the human IL-6 gene and normalized with β actin. Data are expressed as means ± SE from three independent experiments performed. Results, analyzed by the relative quantification method (2−ΔΔCT method), are shown as fold increase of IL-6 gene expression versus the untreated control. P values were calculated by Student's t test, and statistical significance is indicated (*, P < 0.05 for gp120 plus inhibitor versus gp120 alone). (D) MDDCs were pretreated for 1 h with the STAT3 inhibitor Stattic (10 μM) and stimulated with gp120 (5 μg/ml). After 18 h, supernatants were collected and IL-6 production was assessed by ELISA. Data are represented as means ± SE from five independent experiments. Statistical significance is indicated. **, P < 0.005 for gp120 plus inhibitor versus gp120 alone.
However, as we previously reported that Stattic can inhibit, in MDDCs, the LPS- or IFN-β-induced activation of STAT1 and STAT2 in addition to STAT3 (37), the capacity of gp120 to activate these members of the STAT family was evaluated. As shown in Fig. 6A, exposure of MDDCs to gp120 indeed induced STAT1 and STAT2 phosphorylation. However, this activation was observed only at late time points, 6 and 18 h after stimulation. To exclude that gp120-mediated late STAT1 and STAT2 activation played a role in IL-6 upmodulation, iMDDCs were exposed to Stattic before or after gp120 stimulation and tested for IL-6 secretion. As shown in Fig. 6B, preincubation of cells with Stattic completely inhibited IL-6 production at all tested time points. Conversely, when inhibitor addition followed gp120 stimulation, allowing early signaling via STAT3, only a partial inhibition of IL-6 secretion was observed at late time points (18 h).
FIG 6.

HIV gp120-induced late STAT1 and STAT2 activation is not responsible for IL-6 production. (A) MDDCs were stimulated with gp120 (5 μg/ml) or LPS (10 ng/ml) for the indicated time periods. Cell lysates were resolved by 8% SDS-PAGE, transferred to a nitrocellulose membrane, and subjected to immunoblot analysis with antibodies specific for the total or phosphorylated forms of STAT1 and STAT2. Data from one representative experiment out of three analyzed are shown. (B) MDDCs were treated with the STAT3 inhibitor Stattic (10 μM) before (1 h) or after (2 h) gp120 (5 μg/ml) exposure. At the indicated time points, supernatants were collected and IL-6 production was assessed by ELISA. Data are represented as means ± SE from four independent experiments. Statistical significance is indicated. P < 0.05 (*) and P < 0.005 (**) for gp120 plus inhibitor versus gp120 alone. (C) MDDCs were pretreated for 1 h with STAT3 inhibitors Stattic (10 μM), JSI 124 (0.5 μM), AG490 (100 μM), and S3I 201 (250 μM) and stimulated with gp120 (5 μg/ml). After 18 h, supernatants were collected and IL-6 production was assessed by ELISA. Data are represented as means ± SE from four independent experiments. Statistical significance is indicated. *, P < 0.05 for gp120 plus inhibitor versus gp120 alone.
We recently reported that efficient STAT3 silencing in primary DCs is limited, and that transfection per se induces cellular activation (38). Therefore, validation of STAT3 involvement in gp120-induced IL-6 production by STAT3 knockdown was not successful (data not shown). However, to further exclude any involvement of STAT1 and STAT2 activation in the gp120-mediated IL-6 expression, the effects of a panel of STAT3 inhibitors, reported to inhibit STAT3 activation and dimerization at different levels, were tested.
As shown in Fig. 6C, JSI 124, AG490, and S3I 201 blocked the IL-6 secretion induced by gp120 to an extent similar to that of Stattic. Overall, these results strongly suggest that early STAT3 activation by gp120 is responsible for the first wave of IL-6 induction.
HIV gp120 activates PIAS3 but not SOCS3.
The extent and duration of STAT3 signaling is finely regulated (39). Suppressor of cytokine signaling 3 (SOCS3) and protein inhibitor of activated STAT3 (PIAS3) have been characterized as main negative regulators of the STAT3 signaling pathway. To determine whether SOCS3 and PIAS3 expression was modulated as a consequence of gp120 exposure, their expression was analyzed by Western blotting. As shown in Fig. 7, while SOCS3 basal expression was not significantly modified after MDDC exposure to gp120 (Fig. 7A), the levels of PIAS3 increased at all time points tested upon gp120 treatment (Fig. 7B), suggesting that the negative regulation of the IL-6/STAT3 pathway is dysfunctional.
FIG 7.
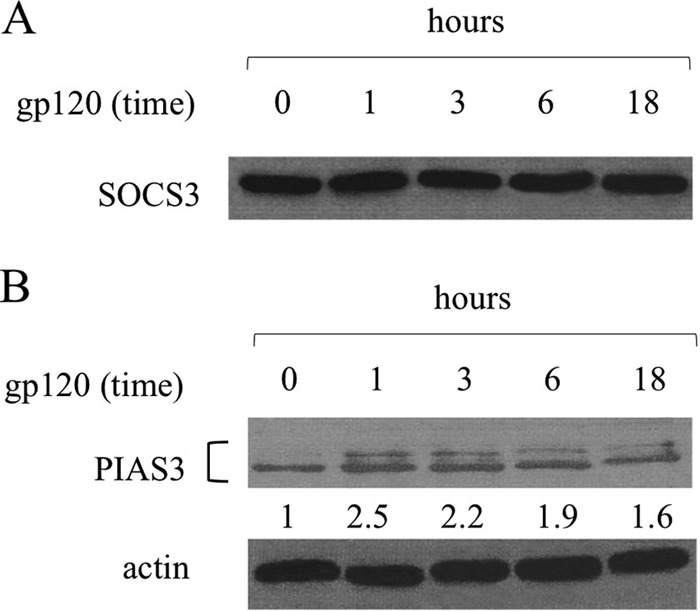
HIV gp120 upregulates PIAS3, but not SOCS3, expression. MDDCs were stimulated with gp120 (5 μg/ml) for the indicated time periods. Cell lysates were resolved by 10% SDS-PAGE, transferred to a nitrocellulose membrane, and subjected to immunoblot analysis with antibodies specific for SOCS3 (A) and PIAS3 (B). Values below the lanes show band intensities of the respective bands normalized to actin expression. Data from one representative experiment out of three (A) or five (B) analyzed are shown.
DISCUSSION
DCs are key regulators of the host response to HIV-1 infection. Thus, modulation of their functional activity represents a strategic mechanism for HIV-1 to evade host immune surveillance. DCs play multiple roles at different stages of HIV infection and are critically involved in HIV pathogenesis. Although these cells are poorly infected by HIV-1, the dynamics and functionality of different DC subsets are strongly affected in the course of infection (40). HIV-1 Env has been shown to modulate functions of several immune cells by engaging chemokine receptor entry coreceptors. In addition to mediating viral entry, coreceptor engagement also may initiate signaling events that alter cell functions or even affect postentry stages of infection. Studies to date indicate that HIV-1 gp120 participates in several signaling pathways through these receptors, including those involving G protein stimulation, ionic signaling, and protein phosphorylation (41). DCs are important targets for HIV-1 in vivo and express both CCR5 and CXCR4, the principal coreceptors for HIV-1 entry. Engagement of HIV coreceptors by gp120 has been reported to exhibit various effects on DCs, including the activation of Pyk2, p38 MAPK, and ERK1/2 signaling pathways (9, 10, 42). The activation of some of these pathways has been linked to the gp120-induced migration of DCs (10), immunosuppression (42), and autophagy exhaustion (43). In addition, we have previously demonstrated that HIV-1 gp120 interaction with MDDCs induces abnormal maturation and functional alterations of these cells, resulting in the reduced secretion of IL-12 and allostimulatory capacity (22). In keeping with our observations, other groups reported altered expression of surface markers and secretion of cytokines upon exposure of DCs to gp120 or HIV infection (29, 44, 45).
In the present study, we further investigated the molecular mechanisms of gp120-triggered signaling pathways in MDDCs. We demonstrate, for the first time, that CCR5 engagement by R5 gp120 promotes STAT3 activation in a concentration- and time-dependent manner, ultimately leading to IL-6 secretion. Likewise, STAT3 activation and IL-6 secretion also are observed in MDM upon gp120 exposure, although with different kinetics and outcomes. Previous studies reported alterations in the expression and phosphorylation of STAT proteins, including STAT3, in other cell models, such as PBMCs and monocyte/macrophage lineage cells, during HIV infection or upon exposure to recombinant HIV proteins (46–48). However, these studies did not relate STAT3 activation with the production of cytokines potentially controlling HIV infection or located this activation in a more complex regulatory circuit. The functional consequences of HIV-mediated activation of STAT3 have been analyzed in subsequent in vitro and in vivo studies of HIV/simian immunodeficiency virus (SIV) infection. In particular, Che and colleagues reported that activation of the p38 MAPK/STAT3 pathway in T lymphocytes primed by HIV-1-exposed DCs negatively affects T cell response, as assessed by upregulation of inhibitory molecules and reduced cell proliferation (49). However, the activation of this signaling pathway does not depend on IL-6. Furthermore, Mohan and colleagues reported that in an in vivo model of SIV infection of Rhesus macaques, the gastrointestinal pathology associated with SIV infection is characterized by the presence of activated STAT3 in lymphocytes and macrophages of the gut mucosa, as well as increased IL-6 production and SOCS3 overexpression (50). Lastly, STAT3 is critically involved in the regulation of DC function, and its activation in tumor-infiltrating DCs is considered a hallmark for their impaired activity favoring immune escape of tumor cells. Thus, hyperactivation of STAT3 may allow HIV-1 to shut down DC-mediated immune responses. In keeping with this hypothesis, it has been reported recently that exposure of PBMCs to gp120 leads to MDSC expansion via gp120-induced IL-6 and STAT3 activation (51). Furthermore, some studies, including ours, have shown that blocking STAT3 activity enhances DC allostimulatory capacity (20, 38) and renders these cells more resistant to immunosuppressive environments, such as that generated at tumor sites (20).
Overall, these observations suggest that HIV-induced STAT3 activation plays a role in the persistent local inflammation and immune activation favoring viral replication and disease progression. Thus, STAT3 activation in DCs may represent a double-edge sword, favoring on one side chronic immune activation and on the other the knockdown of DC function, leading to the profound impairment of the immune system driving AIDS pathogenesis. However, the existence of combined effects of soluble viral products concomitantly present in vivo can further modify the final outcome of the relationship between DC and HIV-1. In this regard, it is of interest that gp120 downregulates Nef-dependent DC-SIGN-mediated IL-6 secretion via IL-10 in DCs (29), adding further complexity to the signaling networks activated by HIV upon interaction with these cells.
Interestingly, we provide evidence for a biphasic activation of STAT3 directly triggered by gp120 within minutes and indirectly via IL-6 at later time points. In keeping with our results, biphasic STAT3 activation has been reported previously in other cell models (35) as a mechanism of temporal regulation of the activity of this transcription factor (39). The abrogation of gp120-mediated STAT3 late activation by IL-6 neutralizing antibody provides compelling evidence that extracellular gp120 signaling results in the activation of STAT3 via IL-6. IL-6 production in turn is dependent upon activation of a signaling cascade involving p38 MAPK and two members of the NF-κB family, IkBα and p65, as demonstrated by the use of specific inhibitors. Importantly, the IL-6 transcript accumulation and protein secretion in response to gp120 also is reversed in the presence of various STAT3 inhibitors, providing support for a role of early STAT3 activation in the production of IL-6 in our cell model. We also show that STAT3 activation parallels a concomitant upmodulation of PIAS3, a factor playing a major role in the inhibition of STAT3 and NF-κB activation (39, 52, 53). Persistent STAT3 activation despite PIAS3 accumulation further suggests that the JAK/STAT pathway is dysregulated upon MDDC exposure to gp120. In keeping with this hypothesis, constitutive STAT3 activation was not counteracted by the high levels of SOCS3 expression in gut mucosa tissue of SIV-infected macaques (50).
In this study, we also demonstrate that gp120-induced STAT3-mediated IL-6 secretion is not elicited by CCL4. Previous studies aimed at characterizing signal transduction pathways triggered by gp120 upon coreceptor engagement revealed that while some of them are activated by both gp120 and CCL4, others are uniquely triggered by gp120 (54–60). In this respect, it was reported that gp120, as well as CCL4, opened calcium-activated potassium-, chloride-, and calcium-permeant nonselective cation channels in macrophages. However, nonselective cation channel activation was unique to gp120 (56). Furthermore, we have demonstrated that gp120 engagement of CCR5, but not CCL4, affects phosphatidylcholine-specific phospholipase C (PC-PLC) cellular distribution and enzymatic activity, as well as CCL2 secretion in primary macrophages (57). Likewise, in the same cell model, nuclear phosphoinositide-specific PLC-β1 activation through CCR5 is triggered by both gp120 and CCL4 but via a different mechanism (58). Altogether, these results add further evidence that the interaction of HIV-1 gp120 with CCR5 evokes complex and distinct signaling responses, and gp120-evoked signals may differ from those activated by the coreceptors' chemokine ligands. This suggests that gp120-specific signals triggered upon engagement of CCR5 in DCs play a role in viral pathogenesis.
The new data provided by the present study are summarized in the schematic model depicted in Fig. 8. We propose a model whereby as a result of binding to CCR5, gp120 directly activates STAT3 and triggers the p38 MAPK/NF-κB signaling cascade. This represents a central intermediate, driving the activation of the downstream molecules involved in this pathway, such as NF-κB. However, the existence of cross talk between STAT3 and the MAPK/NF-κB pathway, suggested in other cell models, remains to be determined (49, 61–63). Thus, the activation of these two signaling pathways would be responsible for IL-6 production. IL-6, via a self-reinforcing pathway, is a key molecule in the late activation of STAT3 by gp120. This gp120-driven signaling pathway is uniquely activated by gp120 but not by the natural CCR5 chemokine ligand CCL4. Concomitant to the activation of the STAT3/IL-6 axis, negative regulators of STAT3 activation, such as PIAS3, also are upmodulated.
FIG 8.

Schematic model of gp120-induced signaling pathways in MDDCs. The cascade of signals triggered upon CCR5 engagement by R5 HIV-1 gp120 in MDDCs is schematically shown.
Together, our results highlight a novel pathway that may contribute to the DC dysfunction observed during the course of HIV disease. A better understanding of the mechanisms underlying the capacity of HIV-1 to induce aberrant signaling in these cells may be instrumental in the development of new therapeutic approaches.
ACKNOWLEDGMENTS
This work was supported by a grant from the Italian Ministry of Health, Bando Nazionale AIDS 2009-2010, 3H/31, to S.G. We also thank Bambino Gesù Children's Hospital for financial support to A.M. and L.D.S. (RC-201302G003019).
We are indebted to the AIDS Reagents and Reference Program, Division of AIDS, NIAID, for providing HIV-1 CN54 gp120, Tak779, sCD4. We thank Fabiola Diamanti and Daniela Diamanti for excellent technical assistance.
Footnotes
Published ahead of print 9 July 2014
REFERENCES
- 1.Pulendran B, Palucka K, Banchereau J. 2001. Sensing pathogens and tuning immune responses. Science 293:253–256. 10.1126/science.1062060 [DOI] [PubMed] [Google Scholar]
- 2.Rinaldo CR, Jr, Piazza P. 2004. Virus infection of dendritic cells: portal for host invasion and host defense. Trends Microbiol. 12:337–345. 10.1016/j.tim.2004.05.003 [DOI] [PubMed] [Google Scholar]
- 3.Spira AI, Marx PA, Patterson BK, Mahoney J, Koup RA, Wolinsky SM, Ho DD. 1996. Cellular targets of infection and route of viral dissemination after an intravaginal inoculation of simian immunodeficiency virus into rhesus macaques. J. Exp. Med. 183:215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller CJ, Hu J. 1999. T cell-tropic simian immunodeficiency virus (SIV) and simian-human immunodeficiency viruses are readily transmitted by vaginal inoculation of rhesus macaques, and Langerhans' cells of the female genital tract are infected with SIV. J. Infect. Dis. 179(Suppl 3):S413–S417. 10.1086/314795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stahl-Hennig C, Steinman RM, Tenner-Racz K, Pope M, Stolte N, Matz-Rensing K, Grobschupff G, Raschdorff B, Hunsmann G, Racz P. 1999. Rapid infection of oral mucosal-associated lymphoid tissue with simian immunodeficiency virus. Science 285:1261–1265. 10.1126/science.285.5431.1261 [DOI] [PubMed] [Google Scholar]
- 6.Hu J, Gardner MB, Miller CJ. 2000. Simian immunodeficiency virus rapidly penetrates the cervicovaginal mucosa after intravaginal inoculation and infects intraepithelial dendritic cells. J. Virol. 74:6087–6095. 10.1128/JVI.74.13.6087-6095.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller E, Bhardwaj N. 2013. Dendritic cell dysregulation during HIV-1 infection. Immunol. Rev. 254:170–189. 10.1111/imr.12082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chougnet C, Gessani S. 2006. Role of gp120 in dendritic cell dysfunction in HIV infection. J. Leukoc. Biol. 80:994–1000. 10.1189/jlb.0306135 [DOI] [PubMed] [Google Scholar]
- 9.Wilflingseder D, Mullauer B, Schramek H, Banki Z, Pruenster M, Dierich MP, Stoiber H. 2004. HIV-1-induced migration of monocyte-derived dendritic cells is associated with differential activation of MAPK pathways. J. Immunol. 173:7497–7505. 10.4049/jimmunol.173.12.7497 [DOI] [PubMed] [Google Scholar]
- 10.Anand AR, Prasad A, Bradley RR, Deol YS, Nagaraja T, Ren X, Terwilliger EF, Ganju RK. 2009. HIV-1 gp120-induced migration of dendritic cells is regulated by a novel kinase cascade involving Pyk2, p38 MAP kinase, and LSP1. Blood 114:3588–3600. 10.1182/blood-2009-02-206342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu H, Pardoll D, Jove R. 2009. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9:798–809. 10.1038/nrc2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haghikia A, Hoch M, Stapel B, Hilfiker-Kleiner D. 2012. STAT3 regulation of and by microRNAs in development and disease. Jak-Stat 1:143–150. 10.4161/jkst.19573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laouar Y, Welte T, Fu XY, Flavell RA. 2003. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity 19:903–912. 10.1016/S1074-7613(03)00332-7 [DOI] [PubMed] [Google Scholar]
- 14.Esashi E, Wang YH, Perng O, Qin XF, Liu YJ, Watowich SS. 2008. The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity 28:509–520. 10.1016/j.immuni.2008.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Onai N, Obata-Onai A, Tussiwand R, Lanzavecchia A, Manz MG. 2006. Activation of the Flt3 signal transduction cascade rescues and enhances type I interferon-producing and dendritic cell development. J. Exp. Med. 203:227–238. 10.1084/jem.20051645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mule J, Kerr WG, Jove R, Pardoll D, Yu H. 2005. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 11:1314–1321. 10.1038/nm1325 [DOI] [PubMed] [Google Scholar]
- 17.Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, Jove R, Gabrilovich D. 2004. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J. Immunol. 172:464–474. 10.4049/jimmunol.172.1.464 [DOI] [PubMed] [Google Scholar]
- 18.Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, Dalton W, Jove R, Pardoll D, Yu H. 2004. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 10:48–54. 10.1038/nm976 [DOI] [PubMed] [Google Scholar]
- 19.Nefedova Y, Cheng P, Gilkes D, Blaskovich M, Beg AA, Sebti SM, Gabrilovich DI. 2005. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J. Immunol. 175:4338–4346. 10.4049/jimmunol.175.7.4338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iwata-Kajihara T, Sumimoto H, Kawamura N, Ueda R, Takahashi T, Mizuguchi H, Miyagishi M, Takeda K, Kawakami Y. 2011. Enhanced cancer immunotherapy using STAT3-depleted dendritic cells with high Th1-inducing ability and resistance to cancer cell-derived inhibitory factors. J. Immunol. 187:27–36. 10.4049/jimmunol.1002067 [DOI] [PubMed] [Google Scholar]
- 21.Melillo JA, Song L, Bhagat G, Blazquez AB, Plumlee CR, Lee C, Berin C, Reizis B, Schindler C. 2010. Dendritic cell (DC)-specific targeting reveals Stat3 as a negative regulator of DC function. J. Immunol. 184:2638–2645. 10.4049/jimmunol.0902960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fantuzzi L, Purificato C, Donato K, Belardelli F, Gessani S. 2004. Human immunodeficiency virus type 1 gp120 induces abnormal maturation and functional alterations of dendritic cells: a novel mechanism for AIDS pathogenesis. J. Virol. 78:9763–9772. 10.1128/JVI.78.18.9763-9772.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fantuzzi L, Borghi P, Ciolli V, Pavlakis G, Belardelli F, Gessani S. 1999. Loss of CCR2 expression and functional response to monocyte chemotactic protein (MCP-1) during the differentiation of human monocytes: role of secreted MCP-1 in the regulation of the chemotactic response. Blood 94:875–883 [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta CT) method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 25.Frick JS, Grunebach F, Autenrieth IB. 2010. Immunomodulation by semi-mature dendritic cells: a novel role of Toll-like receptors and interleukin-6. Int. J. Med. Microbiol. 300:19–24. 10.1016/j.ijmm.2009.08.010 [DOI] [PubMed] [Google Scholar]
- 26.Shah A, Verma AS, Patel KH, Noel R, Rivera-Amill V, Silverstein PS, Chaudhary S, Bhat HK, Stamatatos L, Singh DP, Buch S, Kumar A. 2011. HIV-1 gp120 induces expression of IL-6 through a nuclear factor-kappa B-dependent mechanism: suppression by gp120 specific small interfering RNA. PLoS One 6:e21261. 10.1371/journal.pone.0021261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang B, Akhter S, Chaudhuri A, Kanmogne GD. 2009. HIV-1 gp120 induces cytokine expression, leukocyte adhesion, and transmigration across the blood-brain barrier: modulatory effects of STAT1 signaling. Microvasc. Res. 77:212–219. 10.1016/j.mvr.2008.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeshita S, Breen EC, Ivashchenko M, Nishanian PG, Kishimoto T, Vredevoe DL, Martinez-Maza O. 1995. Induction of IL-6 and IL-10 production by recombinant HIV-1 envelope glycoprotein 41 (gp41) in the THP-1 human monocytic cell line. Cell Immunol. 165:234–242. 10.1006/cimm.1995.1210 [DOI] [PubMed] [Google Scholar]
- 29.Sarkar R, Mitra D, Chakrabarti S. 2013. HIV-1 gp120 protein downregulates Nef induced IL-6 release in immature dendritic cells through interplay of DC-SIGN. PLoS One 8:e59073. 10.1371/journal.pone.0059073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruno R, Galastri S, Sacchi P, Cima S, Caligiuri A, DeFranco R, Milani S, Gessani S, Fantuzzi L, Liotta F, Frosali F, Antonucci G, Pinzani M, Marra F. 2010. gp120 modulates the biology of human hepatic stellate cells: a link between HIV infection and liver fibrogenesis. Gut 59:513–520. 10.1136/gut.2008.163287 [DOI] [PubMed] [Google Scholar]
- 31.Garg A, Spector SA. 2014. HIV type 1 gp120-induced expansion of myeloid derived suppressor cells is dependent on interleukin 6 and suppresses immunity. J. Infect. Dis. 209:441–451. 10.1093/infdis/jit469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Craig R, Larkin A, Mingo AM, Thuerauf DJ, Andrews C, McDonough PM, Glembotski CC. 2000. p38 MAPK and NF-kappa B collaborate to induce interleukin-6 gene expression and release. Evidence for a cytoprotective autocrine signaling pathway in a cardiac myocyte model system. J. Biol. Chem. 275:23814–23824. 10.1074/jbc.M909695199 [DOI] [PubMed] [Google Scholar]
- 33.Kobayashi S, Nagino M, Komatsu S, Naruse K, Nimura Y, Nakanishi M, Sokabe M. 2003. Stretch-induced IL-6 secretion from endothelial cells requires NF-kappaB activation. Biochem. Biophys. Res. Commun. 308:306–312. 10.1016/S0006-291X(03)01362-7 [DOI] [PubMed] [Google Scholar]
- 34.Sehgal PB. 1992. Regulation of IL6 gene expression. Res. Immunol. 143:724–734. 10.1016/0923-2494(92)80011-9 [DOI] [PubMed] [Google Scholar]
- 35.Lee KS, Park JH, Lee S, Lim HJ, Choi HE, Park HY. 2007. HB-EGF induces delayed STAT3 activation via NF-kappaB mediated IL-6 secretion in vascular smooth muscle cell. Biochim. Biophys. Acta 1773:1637–1644. 10.1016/j.bbamcr.2007.07.001 [DOI] [PubMed] [Google Scholar]
- 36.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. 2006. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 13:1235–1242. 10.1016/j.chembiol.2006.09.018 [DOI] [PubMed] [Google Scholar]
- 37.Sanseverino I, Purificato C, Gauzzi MC, Gessani S. 2012. Revisiting the specificity of small molecule inhibitors: the example of stattic in dendritic cells. Chem. Biol. 19:1213–1216. 10.1016/j.chembiol.2012.08.021 [DOI] [PubMed] [Google Scholar]
- 38.Sanseverino I, Purificato C, Varano B, Conti L, Gessani S, Gauzzi MC. 2014. STAT3-silenced human dendritic cells have an enhanced ability to prime IFNgamma production by both alphabeta and gammadelta T lymphocytes. Immunobiology 219:503–511. 10.1016/j.imbio.2014.02.012 [DOI] [PubMed] [Google Scholar]
- 39.Groner B. 2012. Determinants of the extent and duration of STAT3 signaling. Jak-Stat 1:211–215. 10.4161/jkst.21469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manches O, Frleta D, Bhardwaj N. 2014. Dendritic cells in progression and pathology of HIV infection. Trends Immunol. 35:114–122. 10.1016/j.it.2013.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conti L, Fantuzzi L, Del Corno M, Belardelli F, Gessani S. 2004. Immunomodulatory effects of the HIV-1 gp120 protein on antigen presenting cells: implications for AIDS pathogenesis. Immunobiology 209:99–115. 10.1016/j.imbio.2004.02.008 [DOI] [PubMed] [Google Scholar]
- 42.Shan M, Klasse PJ, Banerjee K, Dey AK, Iyer SP, Dionisio R, Charles D, Campbell-Gardener L, Olson WC, Sanders RW, Moore JP. 2007. HIV-1 gp120 mannoses induce immunosuppressive responses from dendritic cells. PLoS Pathog. 3:e169. 10.1371/journal.ppat.0030169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blanchet FP, Moris A, Nikolic DS, Lehmann M, Cardinaud S, Stalder R, Garcia E, Dinkins C, Leuba F, Wu L, Schwartz O, Deretic V, Piguet V. 2010. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 32:654–669. 10.1016/j.immuni.2010.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams MA, Trout R, Spector SA. 2002. HIV-1 gp120 modulates the immunological function and expression of accessory and co-stimulatory molecules of monocyte-derived dendritic cells. J. Hematother. Stem Cell Res. 11:829–847. 10.1089/152581602760404630 [DOI] [PubMed] [Google Scholar]
- 45.Smed-Sorensen A, Lore K, Walther-Jallow L, Andersson J, Spetz AL. 2004. HIV-1-infected dendritic cells up-regulate cell surface markers but fail to produce IL-12 p70 in response to CD40 ligand stimulation. Blood 104:2810–2817. 10.1182/blood-2003-07-2314 [DOI] [PubMed] [Google Scholar]
- 46.Pericle F, Pinto LA, Hicks S, Kirken RA, Sconocchia G, Rusnak J, Dolan MJ, Shearer GM, Segal DM. 1998. HIV-1 infection induces a selective reduction in STAT5 protein expression. J. Immunol. 160:28–31 [PubMed] [Google Scholar]
- 47.Kohler JJ, Tuttle DL, Coberley CR, Sleasman JW, Goodenow MM. 2003. Human immunodeficiency virus type 1 (HIV-1) induces activation of multiple STATs in CD4+ cells of lymphocyte or monocyte/macrophage lineages. J. Leukoc. Biol. 73:407–416. 10.1189/jlb.0702358 [DOI] [PubMed] [Google Scholar]
- 48.Briggs SD, Scholtz B, Jacque JM, Swingler S, Stevenson M, Smithgall TE. 2001. HIV-1 Nef promotes survival of myeloid cells by a Stat3-dependent pathway. J. Biol. Chem. 276:25605–25611. 10.1074/jbc.M103244200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Che KF, Shankar EM, Muthu S, Zandi S, Sigvardsson M, Hinkula J, Messmer D, Larsson M. 2012. p38 mitogen-activated protein kinase/signal transducer and activator of transcription-3 pathway signaling regulates expression of inhibitory molecules in T cells activated by HIV-1-exposed dendritic cells. Mol. Med. 18:1169–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mohan M, Aye PP, Borda JT, Alvarez X, Lackner AA. 2007. Gastrointestinal disease in simian immunodeficiency virus-infected rhesus macaques is characterized by proinflammatory dysregulation of the interleukin-6-Janus kinase/signal transducer and activator of transcription3 pathway. Am. J. Pathol. 171:1952–1965. 10.2353/ajpath.2007.070017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garg A, Spector SA. 2013. HIV type 1 gp120-induced expansion of myeloid derived suppressor cells is dependent on interleukin 6 and suppresses immunity. J. Infect. Dis. 209:441–451. 10.1093/infdis/jit469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jang HD, Yoon K, Shin YJ, Kim J, Lee SY. 2004. PIAS3 suppresses NF-kappaB-mediated transcription by interacting with the p65/RelA subunit. J. Biol. Chem. 279:24873–24880. 10.1074/jbc.M313018200 [DOI] [PubMed] [Google Scholar]
- 53.Liu Y, Bridges R, Wortham A, Kulesz-Martin M. 2012. NF-kappaB repression by PIAS3 mediated RelA SUMOylation. PLoS One 7:e37636. 10.1371/journal.pone.0037636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fantuzzi L, Canini I, Belardelli F, Gessani S. 2001. HIV-1 gp120 stimulates the production of beta-chemokines in human peripheral blood monocytes through a CD4-independent mechanism. J. Immunol. 166:5381–5387. 10.4049/jimmunol.166.9.5381 [DOI] [PubMed] [Google Scholar]
- 55.Del Corno M, Liu QH, Schols D, de Clercq E, Gessani S, Freedman BD, Collman RG. 2001. HIV-1 gp120 and chemokine activation of Pyk2 and mitogen-activated protein kinases in primary macrophages mediated by calcium-dependent, pertussis toxin-insensitive chemokine receptor signaling. Blood 98:2909–2916. 10.1182/blood.V98.10.2909 [DOI] [PubMed] [Google Scholar]
- 56.Liu QH, Williams DA, McManus C, Baribaud F, Doms RW, Schols D, De Clercq E, Kotlikoff MI, Collman RG, Freedman BD. 2000. HIV-1 gp120 and chemokines activate ion channels in primary macrophages through CCR5 and CXCR4 stimulation. Proc. Natl. Acad. Sci. U. S. A. 97:4832–4837. 10.1073/pnas.090521697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fantuzzi L, Spadaro F, Purificato C, Cecchetti S, Podo F, Belardelli F, Gessani S, Ramoni C. 2008. Phosphatidylcholine-specific phospholipase C activation is required for CCR5-dependent, NF-kB-driven CCL2 secretion elicited in response to HIV-1 gp120 in human primary macrophages. Blood 111:3355–3363. 10.1182/blood-2007-08-104901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spadaro F, Cecchetti S, Purificato C, Sabbatucci M, Podo F, Ramoni C, Gessani S, Fantuzzi L. 2013. Nuclear phosphoinositide-specific phospholipase C beta1 controls cytoplasmic CCL2 mRNA levels in HIV-1 gp120-stimulated primary human macrophages. PLoS One 8:e59705. 10.1371/journal.pone.0059705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tomkowicz B, Lee C, Ravyn V, Cheung R, Ptasznik A, Collman RG. 2006. The Src kinase Lyn is required for CCR5 signaling in response to MIP-1beta and R5 HIV-1 gp120 in human macrophages. Blood 108:1145–1150. 10.1182/blood-2005-12-012815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cheung R, Ravyn V, Wang L, Ptasznik A, Collman RG. 2008. Signaling mechanism of HIV-1 gp120 and virion-induced IL-1beta release in primary human macrophages. J. Immunol. 180:6675–6684. 10.4049/jimmunol.180.10.6675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bode JG, Schweigart J, Kehrmann J, Ehlting C, Schaper F, Heinrich PC, Haussinger D. 2003. TNF-alpha induces tyrosine phosphorylation and recruitment of the Src homology protein-tyrosine phosphatase 2 to the gp130 signal-transducing subunit of the IL-6 receptor complex. J. Immunol. 171:257–266. 10.4049/jimmunol.171.1.257 [DOI] [PubMed] [Google Scholar]
- 62.Ehlting C, Ronkina N, Bohmer O, Albrecht U, Bode KA, Lang KS, Kotlyarov A, Radzioch D, Gaestel M, Haussinger D, Bode JG. 2011. Distinct functions of the mitogen-activated protein kinase-activated protein (MAPKAP) kinases MK2 and MK3: MK2 mediates lipopolysaccharide-induced signal transducers and activators of transcription 3 (STAT3) activation by preventing negative regulatory effects of MK3. J. Biol. Chem. 286:24113–24124. 10.1074/jbc.M111.235275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schaljo B, Kratochvill F, Gratz N, Sadzak I, Sauer I, Hammer M, Vogl C, Strobl B, Muller M, Blackshear PJ, Poli V, Lang R, Murray PJ, Kovarik P. 2009. Tristetraprolin is required for full anti-inflammatory response of murine macrophages to IL-10. J. Immunol. 183:1197–1206. 10.4049/jimmunol.0803883 [DOI] [PMC free article] [PubMed] [Google Scholar]