

ABSTRACT
The paramyxoviruses human respiratory syncytial virus (hRSV), human metapneumovirus (hMPV), and human parainfluenza virus type 3 (hPIV3) are responsible for the majority of pediatric respiratory diseases and inflict significant economic loss, health care costs, and emotional burdens. Despite major efforts, there are no vaccines available for these viruses. The conserved region VI (CR VI) of the large (L) polymerase proteins of paramyxoviruses catalyzes methyltransferase (MTase) activities that typically methylate viral mRNAs at positions guanine N-7 (G-N-7) and ribose 2′-O. In this study, we generated a panel of recombinant hMPVs carrying mutations in the S-adenosylmethionine (SAM) binding site in CR VI of L protein. These recombinant viruses were specifically defective in ribose 2′-O methylation but not G-N-7 methylation and were genetically stable and highly attenuated in cell culture and viral replication in the upper and lower respiratory tracts of cotton rats. Importantly, vaccination of cotton rats with these recombinant hMPVs (rhMPVs) with defective MTases triggered a high level of neutralizing antibody, and the rats were completely protected from challenge with wild-type rhMPV. Collectively, our results indicate that (i) amino acid residues in the SAM binding site in the hMPV L protein are essential for 2′-O methylation and (ii) inhibition of mRNA cap MTase can serve as a novel target to rationally design live attenuated vaccines for hMPV and perhaps other paramyxoviruses, such as hRSV and hPIV3.
IMPORTANCE Human paramyxoviruses, including hRSV, hMPV, and hPIV3, cause the majority of acute upper and lower respiratory tract infections in humans, particularly in infants, children, the elderly, and immunocompromised individuals. Currently, there is no licensed vaccine available. A formalin-inactivated vaccine is not suitable for these viruses because it causes enhanced lung damage upon reinfection with the same virus. A live attenuated vaccine is the most promising vaccine strategy for human paramyxoviruses. However, it remains a challenge to identify an attenuated virus strain that has an optimal balance between attenuation and immunogenicity. Using reverse genetics, we generated a panel of recombinant hMPVs that were specifically defective in ribose 2′-O methyltransferase (MTase) but not G-N-7 MTase. These MTase-defective hMPVs were genetically stable and sufficiently attenuated but retained high immunogenicity. This work highlights a critical role of 2′-O MTase in paramyxovirus replication and pathogenesis and a new avenue for the development of safe and efficacious live attenuated vaccines for hMPV and other human paramyxoviruses.
INTRODUCTION
Human metapneumovirus (hMPV) is a relatively new human pathogen that was first described in 2001 in the Netherlands. It was isolated from infants and children experiencing respiratory tract infections with symptoms similar to those of infections caused by human respiratory syncytial virus (hRSV) (1). Since its discovery in 2001, hMPV has been recognized worldwide to be one of the leading causes of lower respiratory infections, second only to hRSV (2, 3). hMPV infections are observed in all age groups, with a high prevalence and severity among infants, children, the elderly, and immunocompromised patients (2, 3). Clinical symptoms associated with hMPV infection are similar to those caused by hRSV infection and range from asymptomatic infection to severe bronchiolitis and pneumonia. hMPV is a nonsegmented negative-sense (NNS) RNA virus belonging to the family Paramyxoviridae, the subfamily Pneumovirinae, and the genus Metapneumovirus (1). There are at least two lineages of hMPV circulating in human populations. These are designated A and B, which can be further divided into A1, A2, B1, and B2 on the basis of their surface glycoproteins and antigenicity (4, 5). The only other member in the genus Metapneumovirus is avian metapneumovirus (aMPV), also known as avian pneumovirus or turkey rhinotracheitis virus, which causes respiratory diseases in turkeys (6, 7).
Despite major efforts, no vaccines or antiviral drugs with activity against hMPV are available. Generally, the two most common strategies used in vaccine development are inactivated and live attenuated vaccines. For safety reasons, an inactivated vaccine is often preferred. However, inactivated vaccines developed for human paramyxoviruses have resulted in serious complications when tested in human clinical trials. In the early 1960s, not only did the vaccination of infants with a formalin-inactivated (FI) RSV vaccine fail to protect the recipients against RSV disease during the following RSV season, but also many of the vaccine recipients developed enhanced disease upon infection with RSV, resulting in increased rates of severe pneumonia and two deaths (8). Enhanced lung damage was also observed when an FI vaccine for human parainfluenza virus type 3 (PIV3), another important human paramyxovirus, was used (9). In 2007, Yim et al. reported that an FI hMPV vaccine induced virus-specific immune responses but resulted in enhanced lung damage upon reinfection in cotton rats (10), similar to what had been previously described in the hRSV clinical trial. These results demonstrate that an FI vaccine is not advised for hMPV, RSV, or PIV3, all of which can cause serious respiratory tract infections in the same populations, namely, infants, children, the elderly, and the immunocompromised.
Live attenuated vaccines are the most promising vaccine candidates against human paramyxoviruses, since enhanced lung damage has not been observed in animal models or human clinical trials (11). Importantly, live attenuated vaccines are capable of inducing robust and prolonged immune responses since they mimic a natural virus infection. Soon after the discovery of hMPV, cold-passaged (cp) and temperature-sensitive (ts) hMPV strains were isolated by randomly passing the virus in cell culture at reduced temperatures (12). These cp and ts hMPVs showed attenuation in replication in the upper and lower respiratory tracts and induced protection against challenge with hMPV strains (12). Recently, a reverse genetics system that has made manipulation of the hMPV genome possible has been developed (13, 14). A variety of recombinant hMPVs have been generated by deleting nonessential genes, such as the G, SH, M2-1, and M2-2 genes (15–19). In addition, chimeric hMPV and aMPV isolates were also successfully recovered from molecular clones (20). However, it has been a challenge to identify an hMPV vaccine strain that has a satisfactory balance between attenuation and immunogenicity. Recombinant hMPV (rhMPV) with a G-gene deletion (rhMPV-ΔG) demonstrates reduced immunogenicity because G protein is one of the major surface glycoproteins and plays an important role in modulating the innate immune response (16, 17, 21). rhMPV with an SH gene deletion (rhMPV-ΔSH) was not sufficiently attenuated in an animal model (17). rhMPV with a an M2-1 gene deletion (rhMPV-ΔM2-1) was highly attenuated in vivo, but it was not able to trigger a neutralizing antibody response or protective immunity in animal models (19). The only attenuated hMPV currently in phase I human clinical trials is a chimeric rhMPV in which the P gene is derived from aMPV (20). The genetic stability, safety, and immunogenicity of these vaccine candidates in humans are currently not known.
Like all other NNS RNA viruses, hMPV produces capped, methylated, and polyadenylated mRNAs (reviewed in reference 22). Recent evidence suggests that the mRNA capping and methylation in NNS RNA viruses have evolved a mechanism that is distinct to their hosts. Using vesicular stomatitis virus (VSV), a prototypical NNS RNA virus, as a model, it was found that capping of VSV mRNA is achieved by a novel polyribonucelotidyltransferase (PRNTase), located in CR V of the large (L) polymerase protein (Fig. 1A), which transfers a monophosphate RNA onto a GDP acceptor through a covalent L-protein RNA intermediate (23–25). In contrast, cellular mRNA capping is catalyzed by an RNA guanylyltransferase (GTase) that reacts with GTP to form a covalent enzyme-GMP intermediate and transfers GMP to diphosphate RNA via a 5′-5′ triphosphate linkage (26). In addition, mRNA cap methylation in NNS RNA viruses is also unique, in that both guanine N-7 (G-N-7) and ribose 2′-O (2′-O) methyltransferase (MTase) activities are catalyzed by a single peptide, CR VI, within the L protein (Fig. 1A) and both methylases share the same binding site for the methyl donor, S-adenosylmethionine (SAM) (27–30). In contrast, the cellular mRNA G-N-7 and 2′-O MTase activities are catalyzed by two separate enzymes, each containing its own SAM binding site (31). Although a detailed characterization of paramyxovirus capping and methylase activities and the mechanism involved in these reactions has not been described, sequence alignment showed that the capping motif [GxxT(n)HR] and MTase signature motif are conserved in CR V and CR VI of the L proteins of all paramyxoviruses, respectively, suggesting that the general mechanism of capping and methylation found in VSV may be conserved in all paramyxoviruses (24, 25).
FIG 1.

Sequence alignment of conserved domain VI of L proteins and modeling with two known 2′-O MTase structures, VP39 and RRMJ. (A) CRs in the L proteins of paramyxoviruses. Amino acid sequence alignment identified six CRs, numbered I to VI, in L proteins. The function of CRs III, V, and VI has been mapped to the VSV L protein. (B) Sequence alignment of CR VI in L proteins. STR, structure of RRMJ and VP39. Predicted or known alpha-helical regions are shown as cylinders, and the β-sheet regions are shown as arrows. The conserved motifs (motifs X and I to VIII) corresponding to the SAM-dependent MTase superfamily are indicated. The predicted MTase active site (K-D-K-E tetrad) is shown by yellow boxes. The predicted SAM binding site (GXGXG … D) is shown by gray boxes. The conserved aromatic amino acid resides are shown by red boxes. The sequences of representative members of the Paramyxoviridae (HMPV, human metapneumovirus; AMPVC, avian metapneumovirus subtype C; HRSV, human respiratory syncytial virus; BRSV, bovine respiratory syncytial virus; PVM, pneumonia virus of mice; PIV3, parainfluenza virus type 3; NDV, Newcastle disease virus), Filoviridae (EBOM, Ebola virus), and Rhabdoviridae (VSIV, vesicular stomatitis virus Indiana serotype) are shown.
It appears that the entire mRNA capping and methylation machinery in NNS RNA viruses is distinct from that of their hosts. This difference, coupled with the fact that the replication of NNS RNA viruses occurs in the cytoplasm, makes mRNA cap formation an excellent target for antiviral drug discovery and vaccine development. We hypothesize that inhibition of viral mRNA cap MTase can serve as a new approach to rationally attenuate hMPV for the development of live attenuated vaccines. The rationale for this hypothesis is that G-N-7 methylation is essential for efficient protein translation, whereas 2′-O methylation is required to escape the interferon (IFN)-mediated innate immune response (32, 33). Thus, inhibition of the viral MTase will likely downregulate viral gene expression and, in turn, viral replication and spread, and ultimately, it will result in viral attenuation. To test this hypothesis, we generated a panel of recombinant hMPVs carrying mutations in the SAM binding site in the L protein. We found that these recombinant viruses were specifically defective in 2′-O methylation but not G-N-7 methylation. These MTase-defective hMPVs were highly attenuated in vitro and in vivo and provided complete protection against challenge with wild-type rhMPV. Thus, our study highlights a new strategy to attenuate hMPV and perhaps other paramyxoviruses for vaccine purposes.
MATERIALS AND METHODS
Ethics statement.
The animal study was conducted in strict accordance with USDA regulations and the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Research Council (34) and was approved by The Ohio State University Institutional Animal Care and Use Committee (IACUC; animal protocol no. 2009A0221). The animals were housed within the University Laboratory Animal Resources (ULAR) facilities of The Ohio State University according to the guidelines of the Institutional Animal Care and Use Committee (IACUC). The animal care facilities at The Ohio State University are AAALAC accredited. Every effort was made to minimize potential distress, pain, or discomfort to the animals throughout all experiments.
Cell lines.
LLC-MK2 (ATCC CCL-7) cells were maintained in Opti-MEM medium (Life Technologies, Bethesda, MD) supplemented with 2% fetal bovine serum (FBS). Vero E6 cells (ATCC CRL-1586) and BHK-SR19-T7 cells (kindly provided by Apath, LLC, Brooklyn, NY) were grown in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 10% FBS. The medium for the BHK-SR19-T7 cells was supplemented with 10 μg/ml puromycin (Life Technologies) during every other passage to select for T7 polymerase-expressing cells.
Plasmids and site-directed mutagenesis.
Plasmids encoding the hMPV minigenome, the full-length genomic cDNA of hMPV strain NL/1/00, and support plasmids expressing hMPV N protein (pCITE-N), P protein (pCITE-P), L protein (pCITE-L), and M2-1 protein (pCITE-M2-1) were kindly provided by Ron A. M. Fouchier at the Department of Virology, Erasmus Medical Center, Rotterdam, The Netherlands (14). The F cleavage site in the genome of hMPV NL/1/00 was modified to a trypsin-independent F cleavage site, as described previously (35). The L-protein CR VI mutations were introduced into the full-length genome of wild-type hMPV NL/1/100 carrying the F-cleavage-site mutations. A QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) was utilized following the manufacturer's recommendations. Mutations were confirmed by DNA sequencing.
Minigenome assay.
The minigenome assay was performed in BHK-SR19-T7 cells, which stably express the T7 RNA polymerase. Briefly, confluent BHK-SR19-T7 cells were transfected with 1.0 μg of the minigenome plasmid together with 0.8 μg of pCITE-N, 0.4 μg of pCITE-P, 0.4 μg of pCITE-M2-1, and 0.4 μg of pCITE-L or pCITE-L mutants using the Lipofectamine 2000 reagent (Life Technologies). Transfections were performed overnight following the manufacturer's recommendations. At 24 h posttransfection, the medium was replaced with DMEM containing 5% FBS. At day 3 posttransfection, the expression of green fluorescent protein (GFP) in the transfected cells was visualized by use of a fluorescence microscope.
Recovery of rhMPVs from the full-length cDNA clones.
rhMPVs were rescued using a reverse genetics system as described previously (13, 14). Briefly, BHK-SR19-T7 cells (kindly provided by Apath LLC), which stably express T7 RNA polymerase, were transfected with 5.0 μg of plasmid phMPV carrying the full-length hMPV genome, 2.0 μg of pCITE-N, 2.0 μg of pCITE-P, 1.0 μg of pCITE-L, and 1.0 μg of pCITE-M2-1 using Lipofectamine 2000 (Life Technologies). At day 6 posttransfection, the cells were harvested using scrapers and were cocultured with LLC-MK2 cells at 50 to 60% confluence. When an extensive cytopathic effect (CPE) was observed, the cells were subjected to three freeze-thaw cycles, followed by centrifugation at 3,000 × g for 10 min. The supernatant was subsequently used to infect new LLC-MK2 cells. The successful recovery of the rhMPVs was confirmed by immunostaining, agarose overlay plaque assay, and reverse transcription (RT)-PCR.
Purification of hMPV.
The rhMPV stocks used in animal studies were grown in LLC-MK2 cells and further purified by ultracentrifugation. Briefly, confluent LLC-MK2 cells in 10 T150 flasks were infected with each rhMPV at a multiplicity of infection (MOI) of 0.01 in a volume of 2 ml of DMEM. After 1 h of adsorption with constant shaking, 20 ml of Opti-MEM medium (supplemented with 2% FBS) was added to the cultures, and infected cells were incubated at 37°C for 6 days. When an extensive CPE was observed, cells were harvested by scraping. The cell suspension was clarified by low-speed centrifugation at 3,000 × g for 20 min at 4°C in a Beckman Coulter Allegra 6R centrifuge. The cell pellet was resuspended in 2 ml of Opti-MEM medium and was subjected to three freeze-thaw cycles. The mixture was clarified by low-speed centrifugation, and the supernatants were combined. The virus was pelleted by ultracentrifugation at 28,000 × g in a Beckman Ty 50.2 rotor for 2 h. The final virus pellet was resuspended in 0.3 ml of Opti-MEM medium, aliquoted, and stored at −80°C. One vial of virus was thawed, and the titer was determined by an immunostaining assay.
RT-PCR and sequencing.
All plasmids, viral stocks, and virus isolates from the nasal turbinates and lungs of cotton rats were sequenced. Viral RNA was extracted from 200 μl of each recombinant virus using an RNeasy minikit (Qiagen, Valencia, CA) following the manufacturer's recommendations. A 1.4-kb DNA fragment spanning CR VI of the hMPV L-protein gene was amplified by RT-PCR using primers designed to anneal to nucleotide positions 11759 and 13199 (the numbering is based on the genome sequence of hMPV strain NL/1/00), primers hMPV-L-11759-Foward (5′-TATATAGGGTTTAAGAATTGG-3′) and hMPV-L-13199-Reverse (5′-ATCATTTTTTACTTACAAGC-3′), respectively.
The PCR products were purified and sequenced at The Ohio State University Plant Microbe Genetics Facility to confirm the presence of the designed mutations using a sequencing primer, hMPV-L-12113-Forward (5′-GCTAAAGGAAAGCTAAC-3′). To eliminate possible contamination with the original plasmid DNA, the initially recovered virus stocks were digested with DNase I (Invitrogen), followed by RNA extraction and RT-PCR. In addition, PCR alone (without the RT step) was performed to confirm the complete digestion of plasmid DNA.
Immunostaining plaque assay.
Vero E6 cells were seeded in 24-well plates and infected with serial dilutions of rhMPV. At day 6 postinfection, the supernatant was removed and cells were fixed in a prechilled acetone-methanol solution (ratio, 3:2) at room temperature for 15 min. Cells were permeabilized in phosphate-buffered saline (PBS) containing 0.4% Triton X-100 at room temperature for 10 min and blocked at 37°C for 1 h using 1% bovine serum albumin (BSA) in PBS. The cells were then labeled with an anti-hMPV N-protein primary monoclonal antibody (Millipore, Billerica, MA) at a dilution of 1:1,000, followed by incubation with horseradish peroxidase (HRP)-labeled rabbit antimouse secondary antibody (Thermo Scientific, Waltham, MA) at a dilution of 1:5,000. After incubation with 3-amino-9-ethylcarbazole (AEC) chromogen substrate (Sigma, St. Louis, MO), positive cells were visualized under a microscope. The viral titer was calculated as the number of PFU per ml.
Viral replication kinetics in LLC-MK2 cells.
Confluent LLC-MK2 cells in 35-mm dishes were infected with wild-type rhMPV or mutant rhMPV at an MOI of 0.01. After 1 h of adsorption, the inoculum was removed and the cells were washed three times with PBS. Fresh DMEM (supplemented with 2% FBS) was added, and the infected cells were incubated at 37°C. At different time points postinfection, the supernatant and cells were harvested by three freeze-thaw cycles, followed by centrifugation at 1,500 × g at room temperature for 15 min. The virus titer was determined by an immunostaining assay in Vero E6 cells.
Analysis of hMPV N-protein expression by Western blotting.
Confluent LLC-MK2 cells were infected with each rhMPV mutant at an MOI of 1.0. At days 3, 5, and 7 postinfection, the cell culture supernatant was removed and the cells were lysed in 200 μl of radioimmunoprecipitation assay lysis buffer (25 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS). Twenty microliters of the cell lysate was denatured at 99°C for 5 min and analyzed on a 12% polyacrylamide bis-Tris gel. The separated protein was transferred to a Hybond-P polyvinylidene difluoride membrane (Amersham Biosciences, Pittsburgh, PA) using a Trans-Blot SD semidry transfer cell (Bio-Rad, Hercules, CA). Membranes were blocked with 5% skim milk in PBS and subsequently probed with anti-hMPV N-protein monoclonal antibody (Millipore) diluted 1:200 in PBS-milk, followed by incubation with horseradish peroxidase-conjugated anti-mouse IgG monoclonal antibody (Thermo Scientific) diluted to 1:2,000 in PBS-milk. Membranes were developed with a chemiluminescence substrate (Thermo Scientific) and exposed to Biomax MR film (Kodak) for visualization of the hMPV N protein.
Quantification of viral genomic RNA replication and mRNA synthesis by real-time RT-PCR.
Confluent LLC-MK2 cells were infected with each rhMPV mutant at an MOI of 1.0. At days 1, 3, 5, and 7 postinfection, total RNA was isolated from virus-infected cells using the TRIzol reagent (Life Technologies). Viral genomic RNA copies were quantified by real-time RT-PCR using two primers specifically targeting the hMPV leader sequence and N-protein gene. Poly(A)-containing viral mRNA was isolated from total RNA using a Dynabeads mRNA isolation kit (Life Technologies) according to the manufacturer's recommendations. Using the viral mRNA as the template, the N-protein mRNA copies were quantified by real-time RT-PCR using two primers targeting the viral N-protein gene.
Genetic stability of rhMPV mutants in cell culture.
Confluent LLC-MK2 cells in 150-mm dishes were infected with each rhMPV mutant at an MOI of 0.1. At day 3 postinfection, the cell culture supernatant was harvested and used for the next passage in LLC-MK2 cells. Using this method, each rhMPV mutant was repeatedly passaged 10 times in LLC-MK2 cells. At each passage, CR VI of the L-protein gene was amplified by RT-PCR and sequenced. At passage 10, the entire genome of each recombinant virus was amplified by RT-PCR and sequenced.
In vitro trans-methylation assay for hMPV.
Confluent Vero E6 cells in 150-mm dishes were mock infected or infected with wild-type or mutant rhMPV at an MOI of 0.1. At day 3 postinfection, total RNA was isolated from virus-infected cells using the TRIzol reagent (Life Technologies) and dissolved in 10 mM Tris-HCl buffer (pH 7.5). Subsequently, poly(A)-containing RNA was isolated from total RNA using a Dynabeads mRNA isolation kit (Life Technologies) according to the manufacturer's recommendations. hMPV-specific mRNA (N mRNA) and cellular mRNA (GAPDH mRNA) were then quantified by real-time RT-PCR. To determine whether rhMPV mutants were defective in G-N-7 methylation, 500 ng of mRNA was incubated with 10 units of vaccinia virus G-N-7 MTase supplied by an m7G capping system (Cellscript, Madison, WI) in the presence of 15 μCi [3H]SAM (85 Ci/mmol; PerkinElmer) for 4 h. After the methylation reaction, RNA was purified using an RNeasy minikit (Qiagen), and the methylation of the mRNA cap structure was measured by determination of the level of 3H incorporation using a 1414 series scintillation counter (PerkinElmer). The 3H incorporation (CCPM) from RNA from mock-infected cells was subtracted from the CCPM of wild-type and mutant hMPV samples. 3H incorporation was normalized to the level of viral mRNA quantified by real-time RT-PCR. Similarly, a trans-ribose 2′-O methylation assay was performed by incubating 500 ng of mRNA with 10 units of vaccinia virus 2′-O MTase supplied by a vaccinia virus 2′-O-methyltransferase kit (Cellscript, Madison, WI) in the presence of 15 μCi [3H]SAM (85 Ci/mmol; PerkinElmer). After the methylation reaction, RNA was purified, and the level of 2′-O methylation was measured by determination of the level of 3H incorporation using a scintillation counter. The CCPM was normalized by virus-specific mRNA. The ratio of 3H-SAM incorporation between each mutant and wild-type virus was calculated.
In vitro trans-methylation assay for VSV.
VSV mRNAs synthesized from in vitro transcription using highly purified VSV or VSV mRNAs purified from VSV-infected cells were used for the trans-methylation assay.
(i) Reconstitution of VSV mRNA synthesis in vitro.
Transcription of VSV mRNA in vitro was performed using 10 μg of purified virus (recombinant VSV [rVSV] or rVSV with the K1651A, G1670A, G1674A, and D1735A mutations) as described previously (27, 28, 36). Reactions were performed in the presence of 1 mM ATP; 0.5 mM CTP, GTP, and UTP; 1 mM SAM; and 25% (vol/vol) rabbit reticulocyte lysate (Promega). Where indicated, reaction mixtures were supplemented with 1 mM S-adenosylhomocysteine (SAH). After incubation at 30°C for 5 h, RNA was purified with an RNeasy minikit (Qiagen). Poly(A)-containing VSV mRNA was isolated from total RNA using a Dynabeads mRNA isolation kit (Invitrogen).
(ii) Isolation of VSV RNA from virus-infected cells.
Confluent BHK-21 cells in 150-mm dishes were mock infected or infected with wild-type or mutant rVSVs at an MOI of 0.1. At 10 h postinfection, total RNA was isolated from virus-infected cells using the TRIzol reagent (Invitrogen). Poly(A)-containing VSV mRNA was isolated from total RNA using a Dynabeads mRNA isolation kit. Equal amounts of mRNAs synthesized in vitro or purified from VSV-infected cells were used for the trans-methylation assay, as described above for hMPV.
Replication and pathogenesis of rhMPV in cotton rats.
For the replication and pathogenesis study, 25 4-week-old female specific-pathogen-free (SPF) cotton rats (Harlan Laboratories, Indianapolis, IN) were randomly divided into five groups (5 cotton rats per group). The cotton rats were housed within the ULAR facilities of The Ohio State University according to IACUC policies and guidelines (animal protocol no. 2009A0221). Each inoculated group was separately housed in rodent cages under biosafety level 2 conditions. Prior to virus inoculation, the cotton rats were anesthetized with isoflurane. The cotton rats in group 1 were inoculated with 2.0 × 105 PFU of wild-type rhMPV and served as positive controls. The cotton rats in groups 2 to 5 were inoculated with 2.0 × 105 PFU of three MTase-defective rhMPV mutants (the rhMPV G1696A, D1755A, and D1700A mutants). The cotton rats in group 6 were mock infected with 0.1 ml of Opti-MEM medium and served as uninfected controls. Each cotton rat was inoculated intranasally with a volume of 100 μl. After inoculation, the animals were evaluated on a daily basis for mortality and the presence of any respiratory symptoms. At day 4 postinfection, the cotton rats were sacrificed, and lungs and nasal turbinates were collected for both virus isolation and histological analysis.
Immunogenicity of rhMPVs in cotton rats.
For the immunogenicity study, 30 cotton rats (Harlan Laboratories, Indianapolis, IN) were randomly divided into six groups (5 cotton rats per group). The cotton rats in group 1 were mock infected with Opti-MEM medium and used as an uninfected control, and those in groups 2 to 6 were intranasally inoculated with 2.0 × 105 PFU of wild-type rhMPV or MTase-defective hMPVs in 0.1 ml Opti-MEM medium. The cotton rats in group 7 were inoculated with DMEM and served as the unimmunized challenged control. After immunization, the cotton rats were evaluated daily for mortality and the presence of any symptoms of hMPV infection. Blood samples were collected from each rat weekly by facial vein retro-orbital bleeding, and serum was isolated for neutralizing antibody detection. At week 4 postimmunization, the cotton rats in groups 2 to 7 were challenged intranasally with wild-type rhMPV at a dose of 1.0 × 106 PFU per cotton rat. After challenge, the animals were evaluated twice every day for mortality and the presence of any symptoms of hMPV infection. At day 4 postchallenge, all cotton rats from each group were euthanized by CO2 asphyxiation. The lungs and nasal turbinates from each cotton rat were collected for virus isolation and histological evaluation. The immunogenicity of the MTase-defective hMPVs was evaluated using the following methods: (i) humoral immunity was determined by analysis of serum neutralization of virus using an endpoint dilution plaque reduction assay. (ii) The viral titers in the nasal turbinates and lungs were determined by an immunostaining plaque assay, and viral genomic RNA was quantified by real-time RT-PCR. (iii) Pulmonary histopathology and viral antigen distribution were determined using the procedure described below. The protection was evaluated with respect to viral replication, antigen distribution, and pulmonary histopathology.
Pulmonary histology.
After sacrifice, the right lung of each animal was removed, inflated, and fixed with 4% neutral buffered formaldehyde. Fixed tissues were embedded in paraffin and sectioned at 5 μm. Slides were then stained with hematoxylin-eosin (H&E) for the examination of histological changes by light microscopy. The pulmonary histopathological changes were reviewed by 2 to 3 independent pathologists. Histopathological changes were scored to include the extent of inflammation (focal or diffuse), the pattern of inflammation (peribronchiolar, perivascular, interstitial, alveolar), and the nature of the cells making up the infiltrate (neutrophils, eosinophils, lymphocytes, macrophages).
Immunohistochemical (IHC) staining.
The right lung of each animal was fixed in 10% neutral buffered formaldehyde and embedded in paraffin. Five-micrometer sections were cut and placed onto positively charged slides. After deparaffinization, sections were incubated with target retrieval solution (Dako, Carpinteria, CA) for antigen retrieval. After antibody block, a primary mouse anti-hMPV monoclonal antibody (Virostat, Portland, ME) was added and incubated for 30 min at room temperature, followed by incubation with a biotinylated horse antimouse secondary antibody (Vector Laboratories, Burlingame, CA). Slides were further incubated with ABC Elite complex to probe biotin (Vector Laboratories), and the slides were then developed using a 3,3′-diaminobenzidine (DAB) chromogen kit (Dako) and hematoxylin as a counterstain. Lung sections from hMPV-infected and uninfected samples were used as positive and negative controls, respectively.
Determination of hMPV-neutralizing antibody.
hMPV-specific neutralizing antibody titers were determined using a plaque reduction neutralization assay. Briefly, cotton rat sera were collected by retro-orbital bleeding weekly until challenge. The serum samples were heat inactivated at 56°C for 30 min. Twofold dilutions of the serum samples were mixed with an equal volume of DMEM containing approximately 100 PFU/well hMPV NL/1/00 in a 96-well plate, and the plate was incubated at room temperature for 1 h with constant rotation. The mixtures were then transferred to confluent Vero E6 cells in a 96-well plate in triplicate. After 1 h of incubation at 37°C, the virus-serum mixtures were removed and the cells were overlaid with 0.75% methylcellulose in DMEM and incubated for another 4 days before virus plaque titration. The plaques were counted, and 50% plaque reduction titers were calculated as the hMPV-specific neutralizing antibody titers.
Determination of viral titer in lung and nasal turbinate.
The nasal turbinate and the left lung from each cotton rat were removed, weighed, and homogenized in 1 ml of PBS solution using a Precellys 24 tissue homogenizer (Bertin, MD) following the manufacturer's recommendations. The presence of infectious virus was determined by an immunostaining plaque assay in Vero cells, as described above.
Sensitivity of recombinant hMPVs to IFN-α and IFN-β treatment.
Confluent Vero E6 cells in 24-well plates were treated with DMEM or DMEM containing the indicated amounts (2 or 20 U) of alpha interferon (IFN-α) and IFN-β (PBL, Piscataway, NJ) at 37°C for 4 h. The cells were then washed three times with DMEM and infected with wild-type rhMPV or the rhMPV mutants at an MOI of 0.5. After 1 h adsorption, cells were washed three times with DMEM before adding fresh medium. At 48 h postinfection, the supernatant from each well was harvested and the viral titers were determined by immunostaining using Vero E6 cells.
Statistical analysis.
Quantitative analysis was performed by either densitometric scanning of autoradiographs or by using a phosphorimager (Typhoon; GE Healthcare, Piscataway, NJ) and ImageQuant TL software (GE Healthcare, Piscataway, NJ). Statistical analysis was performed by one-way multiple comparisons using SPSS (version 8.0) statistical analysis software (SPSS Inc., Chicago, IL). A P value of <0.05 was considered statistically significant.
RESULTS
Sequence analysis of the putative MTase domain in hMPV L protein.
To begin to utilize MTase as the target to attenuate hMPV, we first performed a detailed bioinformatics analysis of CR VI within the hMPV L protein. Our aim was to predict critical amino acids that may be essential for mRNA cap methylation by comparing the amino acid sequence of the hMPV L protein with those of other MTases and the structural homology of the hMPV L protein with the available crystal structures of MTases, such as those of vaccinia virus VP39 (37) and Escherichia coli RRMJ (38). The S-adenosylmethionine (SAM)-dependent MTase superfamily contains six motifs involved either in SAM binding (motifs I, III, and IV) or in the catalytic reaction (motifs IV, VI, VIII, and X) (Fig. 1B) (39, 40). Sequence alignment and structural modeling identified these motifs in CR VI of paramyxovirus L proteins (Fig. 1B). Typically, a ribose 2′-O MTase contains a K-D-K-E tetrad whose residues function as the catalytic residues for methylation (Fig. 1B). As predicted, this K-D-K-E tetrad is highly conserved in CR VI of the L proteins of all known NNS RNA viruses, with the exception of Borna disease virus, which replicates in the nucleus. Amino acid sequence alignments suggest that residues K1673, D1779, K1817, and E1848 of the hMPV L protein correspond to the catalytic K-D-K-E tetrad. In methylation reactions, a G-rich motif and an acidic residue (D) are involved in binding the methyl donor SAM. Indeed, this GXGXG … D motif is conserved in all paramyxoviruses and other NNS RNA viruses. Sequence alignments suggested that the SAM binding site residues of hMPV L protein include G1696, G1698, G1700, and D1755 (Fig. 1B).
Examination of the function of mutants with mutations in the MTase region of L protein using a minigenome assay.
We performed mutagenesis in the MTase region of the hMPV L protein, individually changing each of the amino acids in the predicted KDKE motif and in the SAM binding site to alanine by site-directed mutagenesis. A minigenome assay was performed to examine whether these L-protein mutants were functional. To do this, BHK-SR19-T7 cells were transfected with the hMPV minigenome (pSP72-hMPV-GFP) together with support plasmids pCITE-N, pCITE-P, pCITE-M2-1, pCITE-L, or pCITE-L carrying a mutated L protein. At 48 h postinfection, the cells expressing GFP were observed by fluorescence microscopy. As shown in Fig. 2, strong GFP expression was observed when wild-type pCITE-L was used for transfection. No GFP signal was detected when pCITE-L was omitted from the transfection. Cotransfection of pCITE-L carrying any of the mutations in the predicted MTase catalytic and SAM binding sites decreased GFP expression. This result suggests that the amino acids predicted to be important in MTase catalytic and SAM binding sites were critical because the mutations conferred a defect in replication, gene expression, or both.
FIG 2.

Mutations to CR VI of the hMPV L gene diminished GFP expression in a minigenome assay. Confluent BHK-SR19-T7 cells were transfected with the minigenome plasmid together with pCITE-N, pCITE-P, pCITE-M2.1, pCITE-L, or pCITE-L mutant using Lipofectamine 2000. GFP expression was visualized by a fluorescence microscope at 48 h postinfection.
Recovery of rhMPV carrying mutations in the SAM binding sites.
Previously, we demonstrated that rVSV carrying mutations in the KDKE tetrad had 2- to 3-log-unit reductions in viral titer, whereas rVSV carrying mutations in the SAM binding site had 1- to 2-log-unit reductions (27, 28). Thus, we decided to focus on the SAM binding site with the goal of generating rhMPVs that are attenuated but grow reasonably well in cell culture. Since L-protein mutants were functional in the minigenome replication assay, it was assumed to be feasible to recover recombinant hMPVs carrying mutations in SAM binding sites. To do this, the amino acids in the predicted GXGXG … D/E motif were individually mutated to alanine in the full-length genomic cDNA of hMPV lineage A strain NL/1/00 (phMPV). Using a reverse genetics system, we recovered three recombinant viruses (the rhMPV G1696A, G1700A, and D1755A mutants) carrying mutations in the SAM binding site. As shown in Fig. 3, all rhMPV mutants were positive for viral N-protein expression in the immunostaining assay using a monoclonal antibody against N protein. However, the immunospots formed by these rhMPV mutants in LLC-MK2 cells were much smaller than those formed by wild-type rhMPV. To further confirm the phenotype of these recombinant hMPVs, a direct agarose overlay plaque assay was performed in LLC-MK2 cells. As shown in Fig. 3, the plaque sizes of the rhMPV G1696A, G1700A, and D1755A mutants were 0.6 ± 0.1 mm, 0.5 ± 0.1 mm, and 0.7 ± 0.2 mm, respectively, which were significantly smaller than the plaque size of wild-type rhMPV (1.2 ± 0.1 mm) (P < 0.05). This suggests that rhMPVs carrying mutations in the SAM binding site had defects in cell-cell spread and/or viral replication. To confirm that the mutant rhMPVs contained the desired mutations, virus stocks were treated with DNase I to remove possible contamination with plasmid DNA from transfection, followed by RNA extraction and RT-PCR. A 1.4-kb DNA fragment spanning CR VI of the L gene was amplified by RT-PCR from each rhMPV mutant (data not shown). However, no DNA band was observed when PCR was performed without the RT step (data not shown), suggesting that the plasmid DNA was completely removed by DNase treatment. Sequencing results showed that all rhMPV mutants contained the desired mutation in CR VI of the L-protein gene. Finally, the entire genomes of these recombinants were sequenced to confirm that no additional mutation(s) had been introduced.
FIG 3.

Recovery of recombinant hMPVs carrying mutations in the SAM binding site. (Top) Immunostaining spots formed by recombinant hMPVs. LLC-MK2 cells were infected with recombinant hMPV mutants and incubated at 37°C for 1 h. At day 4 postinfection, the supernatant was removed and the cells were fixed. The cells were then labeled with an anti-hMPV N-protein primary monoclonal antibody, followed by incubation with HRP-labeled rabbit antimouse secondary antibody. After incubation with AEC chromogen substrate, positive cells with immunostaining spots were visualized under a microscope. (Bottom) Plaque morphology of recombinant hMPVs. An agarose overlay plaque assay was performed with an LLC-MK2 cell monolayer. Viral plaques were developed at day 6 postinfection.
Recombinant hMPVs carrying mutations in the SAM binding site have a delay in viral replication.
The replication kinetics of these rhMPV mutants in LLC-MK2 cells was determined. Briefly, LLC-MK2 cells were infected with each recombinant virus at an MOI of 0.01. At the indicated time points, the supernatant of infected LLC-MK2 cell cultures was collected and the virus yield was determined on LLC-MK2 cells, using the immunostaining assay. Figure 4A shows the growth curves of these rhMPV mutants. The rhMPV G1696A, G1700A, and D1755A mutants were significantly delayed in viral replication compared to wild-type rhMPV (P < 0.05). The peak viral titer of the hMPV G1755A mutant was 4.6 log10 PFU/ml, which was 1.4 log units lower than that of wild-type rhMPV. The peak titers of the rhMPV G1696A and D1700A mutants were 5.5 and 5.6 log10 PFU/ml at day 6 postinfection, respectively; these values were only 0.5 log unit less than the value for wild-type rhMPV (Fig. 4A). Since the rhMPV D1755A mutant had a 1.5-log-unit reduction in titer, we also tested the growth of this rhMPV mutant at higher MOIs. As shown in Fig. 4B, the viral titer significantly increased when LLC-MK2 cells were infected with the rhMPV D1755A mutant at a higher MOI (P < 0.05). At an MOI of 1.0, the titer of the rhMPV D1755A mutant was approximately 0.4 log unit less than that of wild-type rhMPV. The progression of the CPE was recorded in LLC-MK2 cells. Wild-type rhMPV caused an extensive CPE at day 3 postinoculation. The CPEs caused by the rhMPV mutants were significantly delayed. The onset of the CPE caused by the rhMPV G1696A mutant was delayed by 3 days, whereas that of the CPE caused by the rhMPV G1700A and D1755A mutants was delayed by 1 day (Fig. 5). Collectively, these results demonstrate that rhMPVs carrying mutations in the SAM binding site had a delay in viral replication and were attenuated in cell culture.
FIG 4.

Single-step growth curve of recombinant hMPVs carrying mutations in the SAM binding site. LLC-MK2 cells in 35-mm dishes were infected with each recombinant hMPV at an MOI of 0.01. After adsorption for 1 h, the inocula were removed and the infected cells were washed 3 times with Opti-MEM medium. Then, fresh Opti-MEM medium containing 2% FBS was added and the cells were incubated at 37°C for various times. Aliquots of the cell culture fluid were removed at the indicated intervals. The viral titer was determined by an immunostaining assay in Vero E6 cells. DPI, day postinfection. (A) Growth curves for wild-type rhMPV and the rhMPV G1696A, G1700A, and D1755A mutants; (B) viral titers for wild-type rhMPV and the rhMPV D1755A mutant at different MOIs.
FIG 5.

CPEs caused by hMPVs carrying mutations in the SAM binding site. LLC-MK2 cells were infected with each recombinant hMPV at an MOI of 1.0. The CPE was monitored on a daily basis. Pictures were taken at days 3, 4, 5, 6, and 7 postinfection.
Recombinant hMPVs carrying mutations in the SAM binding site have defects in viral protein synthesis.
We determined whether these recombinant viruses had defects in viral protein synthesis by using Western blotting for the N protein. Figure 6A shows the kinetics of viral N-protein synthesis. The rhMPV G1700A and D1755A mutants had a significant delay in N-protein synthesis (P < 0.05). Quantitative analysis of protein bands showed that the rhMPV G1700A and D1755A mutants had approximately 70 to 80% and 50 to 70% reductions in N-protein expression, respectively, compared with that of wild-type rhMPV at days 1 and 3 postinfection. In comparison, the rhMPV G1696A mutant showed moderate (10 to 30%) reductions in N-protein expression compared to wild-type rhMPV at days 1 and 3 postinfection (Fig. 6A). The level of N protein in wild-type rhMPV-infected cells decreased at days 5 and 7 postinfection because many cells started to lyse. In contrast, N-protein expression in rhMPV G1700A and D1755A mutant-infected cells continued to increase at days 5 and 7 postinfection (Fig. 6A).
FIG 6.

Recombinant hMPVs carrying mutations in the SAM binding site were defective in gene expression and RNA synthesis. (A) N-protein synthesis. LLC-MK2 cells were infected with wild-type rhMPV or rhMPV mutants at an MOI of 1.0 for various periods of time, as indicated. Total cell lysates were harvested and subjected to Western blotting using a monoclonal antibody against hMPV N protein. (B) N mRNA synthesis. Viral mRNA was further purified using a Dynabeads mRNA isolation kit and subsequently quantified by real-time PCR using primers annealing to the N gene. (C) Viral genomic RNA replication. LLC-MK2 cells were infected with wild-type rhMPV or rhMPV mutants at an MOI of 0.1 for various periods of time. Total RNA was isolated using the TRizol reagent, and genomic RNA was quantified by real-time RT-PCR using specific primers annealing to the hMPV leader sequence and N gene. The results are the averages of three independent experiments and are expressed as means ± SEs of the numbers of copies of the transcribed N mRNA or viral genomic RNA. (D) Ratio between mRNA and genomic RNA. The ratio between N mRNA and genomic RNA was calculated for each recombinant hMPV. Data are the averages of three independent experiments.
Recombinant hMPVs carrying mutations in the SAM binding site have defects in viral genomic RNA replication and mRNA synthesis.
We also determined viral genomic RNA replication and mRNA synthesis for each recombinant virus. Since we used a primer annealing to the hMPV leader sequence, viral genomic RNA could be directly quantified by real-time RT-PCR using total RNA isolated from virus-infected cells. At day 5 postinfection, the amount of genomic RNA in rhMPV G1696A, G1700A, and D1755A mutant-infected cells was 1.6-, 0.8-, and 1.7-fold less than that in wild-type rhMPV, respectively (Fig. 6B). We also separated viral mRNA from total RNA by poly(A) binding, since viral genomic RNA is not polyadenylated. At day 5 postinfection, the rhMPV G1696A, G1700A, and D1755A mutants had 1.1-, 0.5-, and 3.3-fold less N-protein mRNA than wild-type rhMPV, respectively (Fig. 6C). Thus, all three rhMPV mutants had significant defects in viral mRNA synthesis and genomic RNA replication (P < 0.05). In addition, the ratio between mRNA and genomic RNA for each hMPV mutant was calculated. As shown in Fig. 6D, the ratio of viral mRNA/genomic RNA for each mutant was not significantly altered from that for wild-type rhMPV (P > 0.05).
Genetic stability of recombinant hMPV in vitro.
To determine whether the rhMPV mutants were genetically stable in cell culture, each rhMPV mutant was passaged 10 times in LLC-MK2 cells. The region containing the respective mutation in the virus from each passage was sequenced. It was found that all rhMPV mutants retained their mutation in CR VI of the L gene. At passage 10, the entire genome of each recombinant virus was sequenced. No mutations other than the desired mutation in the MTase region in the L gene were found in the genome. This result suggests that these rhMPV mutants are genetically stable.
Recombinant hMPVs carrying mutations in the SAM binding site are defective in 2′-O methylation but not G-N-7 methylation.
Analysis of mRNA cap methylation of paramyxoviruses has been hampered because most of them, including hMPV, cannot synthesize viral mRNA in vitro. We have developed a trans-methylation assay that allows us to analyze the cap methylation of viral mRNA in virus-infected cells. Briefly, Vero E6 cells were mock infected or infected with wild-type or mutant rhMPVs at an MOI of 0.1, and mRNAs for each recombinant virus were harvested and purified. To determine whether the rhMPV mutants were defective in G-N-7 methylation, equal amounts of mRNA from each mutant were incubated with 10 units of vaccinia virus G-N-7 MTase in the presence of 15 μCi [3H]SAM. After the methylation reaction, RNA was purified, and the methylation of the mRNA cap structure was quantified by determining the level of 3H incorporation with a scintillation counter. As shown in Fig. 7A, left, rhMPV mRNAs were not methylated by vaccinia virus G-N-7 MTase, which is consistent with this site already being methylated by the rhMPV enzymes. Similarly, the level of [3H]SAM incorporation of mRNAs produced by the rhMPV G1696A, G1700A, and D1755A mutants was indistinguishable from that of mRNA produced by wild-type rhMPV (P > 0.05), suggesting that the mRNA of these mutants was not methylated by the vaccinia virus G-N-7 MTase. Therefore, this result indicates that the rhMPV G1696A, G1700A, and D1755A mutants are not defective in G-N-7 methylation.
FIG 7.
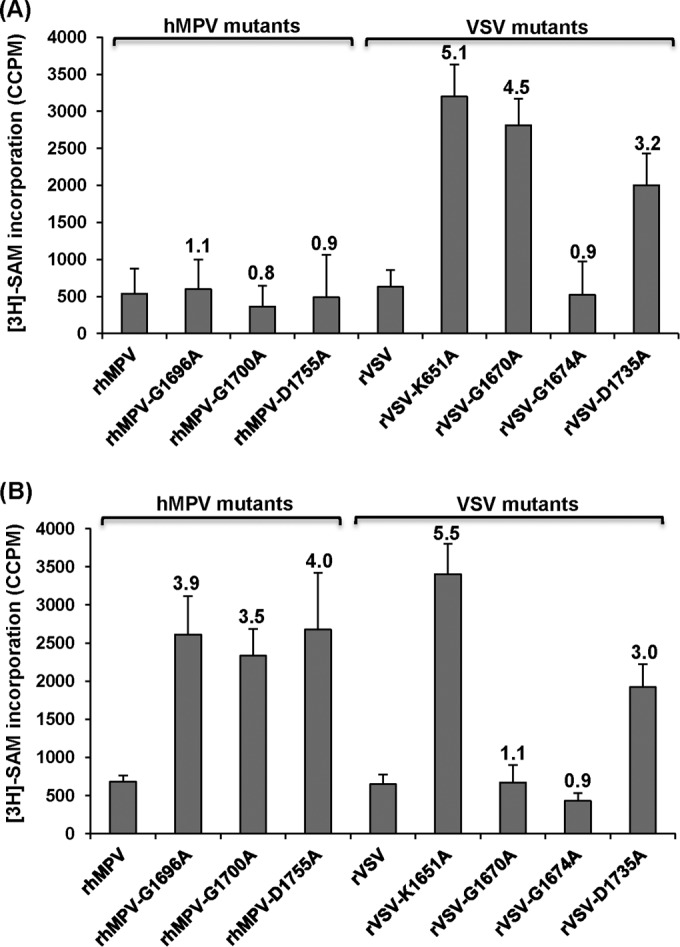
Analysis of hMPV and VSV mRNA cap methylation by in vitro trans-methylation assay. (A) trans-G-N-7 methylation assay for hMPV and VSV. Five hundred nanograms of mRNA was isolated from virus-infected Vero E6 cells and was trans-methylated by vaccinia virus G-N-7 MTase in the presence of 15 μCi [3H]SAM as described in Materials and Methods. The number of corrected counts per minute (CCPM) of [3H]SAM incorporation from RNA from mock-infected cells was subtracted from the CCPM of each hMPV mutant. Subsequently, the CCPM was normalized by the level of viral mRNA quantified by real-time RT-PCR. The number on the top of each column indicates the ratio of [3H]SAM incorporation of viral mRNAs between mutant and wild-type virus. The averages of four independent experiments are shown. (B) trans-2′-O methylation assay for hMPV and VSV. Five hundred nanograms of mRNA was isolated from virus-infected cells and was subsequently premethylated by vaccinia virus G-N-7 MTase in the presence of 1 mM cold SAM. The RNAs were purified and further trans-methylated by vaccinia virus 2′-O MTase in the presence of 15 μCi [3H]SAM. The number of CCPM of [3H]SAM incorporation for each virus is shown. The number on the top of each column indicates the ratio of [3H]SAM incorporation of viral mRNAs between mutant and wild-type virus. The averages of four independent experiments are shown.
A similar strategy was used to determine 2′-O methylation in mRNA. Since G-N-7 methylation is required for 2′-O methylation in vaccinia virus (37, 41), we first incubated the RNA with cold SAM and the vaccinia virus G-N-7 MTase to methylate the G-N-7 position. In this way, if there was a lack of G-N-7 methylation in the original RNA, it would not inhibit the methylation of the 2′-O position. The mRNAs were purified and further methylated by 10 units of vaccinia virus 2′-O MTase in the presence of 15 μCi [3H]SAM. After the methylation reaction, the RNA was purified and the level of 2′-O methylation was measured by determination of the level of 3H incorporation using a scintillation counter. As shown in Fig. 7B, left, the rhMPV mRNAs were not methylated by vaccinia virus 2′-O MTase, consistent with the fact that rhMPV produces 2′-O-methylated mRNA. The level of 3H incorporation of the rhMPV G1696A, G1700A, and D1755A mutants was 3.5- to 4-fold higher than that of wild-type rhMPV (P < 0.05), suggesting that the rhMPV G1696A, G1700A, and D1755A mutants had defects in 2′-O methylation. Therefore, these results indicate that rhMPVs carrying mutations in the SAM binding site are specifically defective in 2′-O methylation but not G-N-7 methylation.
To verify the results for hMPV described above, we performed a trans-methylation assay using VSV as a control. VSV mRNA cap MTase is the best-characterized NNS RNA virus MTase because purified VSV can reconstitute viral mRNA synthesis in vitro. Furthermore, purified VSV L protein can methylate VSV-specific mRNA in vitro. We chose the following four VSV L-protein mutants for this assay: the rVSV K1651A mutant, carrying a point mutation in the MTase catalytic site which abolishes both G-N-7 and 2′-O methylations, and the rVSV G1670A, rVSV G1674A, and rVSV D1735A mutants, each of which carries a mutation in the SAM binding site equivalent to the rhMPV G1696A, G1700A, and D1755A mutations, respectively. Previously, we had found that the rVSV G1670A mutant is specifically defective in G-N-7 methylation but not 2′-O methylation, the rVSV G1674A mutant is fully methylated at G-N-7 and 2′-O at high (1 mM) SAM concentrations, and the rVSV D1735A mutant inhibits both G-N-7 and 2′-O methylations by approximately 70% (27–29).
Using the exact same trans-methylation assay developed for hMPV, we determined the methylation status of VSV mRNAs isolated from virus-infected cells. As shown in Fig. 7A, right, rVSV and rVSV G1674A mutant mRNAs were not methylated by vaccinia virus G-N-7 MTase, which is consistent with the fact that they produce fully G-N-7-methylated mRNA in the presence of 1 mM SAM. In contrast, the mRNAs of the rVSV K1651A, G1670A, and D1735A mutants were efficiently methylated by vaccinia virus G-N-7 MTase. The level of 3H incorporation of the rVSV K1651A, G1670A, and D1735A mutants was approximately 5.1-, 4.5-, and 3.2-fold higher than that of wild-type rVSV, respectively. This is consistent with the fact that the rVSV K1651A and G1670A mutants lacked G-N-7 methylation and the rVSV D1735A mutant had a 70% deficiency in G-N-7 methylation (27, 28). Using the pre-G-N-7-methylated mRNAs as the substrates, we performed a trans-2′-O methylation assay. As shown in Fig. 7B, right, mRNAs synthesized from the rVSV K1651A and D1735A mutants but not those synthesized from wild-type rVSV or the rVSV G1674A and rVSV G1670A mutants could be efficiently methylated by vaccinia virus 2′-O MTase. The level of 3H incorporation of the rVSV K1651A and D1735A mutants was approximately 5.5- and 3.0-fold higher than that of wild-type rVSV, respectively. This is consistent with our previous results showing that the rVSV K1651A mutant completely lacked both G-N-7 and 2′-O methylations, the rVSV G1670A mutant lacked G-N-7 methylation but not 2′-O methylation, and the rVSV D1735A mutant was partially defective in both G-N-7 and 2′-O methylations (27, 28).
Replication and pathogenesis of MTase-defective rhMPVs in cotton rats. (i) Viral replication in cotton rats.
To determine whether the MTase-defective rhMPVs are attenuated in vivo, cotton rats were inoculated with recombinant wild-type and mutant viruses and viral replication and pathogenesis were examined. None of the cotton rats inoculated with recombinant viruses exhibited detectable clinical symptoms of respiratory tract infection. No distinguishable body weight change was observed in any treatment group (data not shown). At day 4 postinfection, cotton rats were sacrificed, and viral replication in nasal turbinates and lungs and pulmonary histology were determined.
As shown in Table 1, wild-type rhMPV replicated efficiently in the nasal turbinates and lungs of all five cotton rats. Average viral titers of 5.24 ± 0.35 log10 PFU/g and 3.02 ± 0.46 log10 PFU/g were found in the nasal turbinate and lung, respectively. All MTase-defective rhMPVs had reduced viral replication in the nasal turbinate and lung. For the rhMPV G1700A mutant, 4 out of 5 cotton rats had detectable virus in the nasal turbinate, with an average titer of log 2.69 ± 0.34 log10 PFU/g, and only 1 out of 5 rats had detectable virus in lung tissue, with a titer of log 2.02 log10 PFU/g. For the rhMPV G1696A and G1755A mutants, there was no detectable infectious virus in either nasal turbinate or lung tissue. This result demonstrated that the rhMPV G1696A and G1755A mutants were completely defective in viral replication in both the upper and lower respiratory tracts in cotton rats, whereas the rhMPV G1700A mutant had low levels of viral replication only in the upper respiratory tract. These results demonstrate that MTase-defective hMPVs are attenuated in viral replication in vivo.
TABLE 1.
Replication of MTase-defective rhMPVs in cotton ratsh
| rhMPV mutant a | Viral replication in nasal turbinateb |
Viral replication in lungc |
Lung histology scored | Lung IHC scoree | ||||
|---|---|---|---|---|---|---|---|---|
| % of infected animals | Viral titer (log10 PFU/g) | Viral RNA load (log10 GRCf/g) | % of infected animals | Viral titer (log10 PFU/g) | Viral RNA load (log10 GRC/g) | |||
| Wild type | 100 | 5.24 ± 0.35A | 7.54 ± 0.32A | 100 | 3.02 ± 0.46A | 8.54 ± 0.34A | 1.5A | 3.0A |
| G1696A | 0 | NDg | 6.34 ± 0.72B | 0 | ND | 6.72 ± 0.24B | 0.3B | 0.2B |
| G1700A | 80 | 2.69 ± 0.34B | 6.95 ± 0.18B | 60 | 2.27 ± 0.28B | 7.46 ± 1.10B | 0.5B | 0.5B |
| D1755A | 0 | NDA | 5.27 ± 0.77B | 0 | ND | 6.07 ± 1.55B | 0.4B | 0.2B |
| DMEM | 0 | ND | ND | 0 | ND | ND | 0 | 0 |
Cotton rats were inoculated intranasally with DMEM or 2 × 105 PFU of wild-type rhMPV or an rhMPV mutant. At day 4 postimmunization, animals were euthanized for pathology study.
The viral titer was determined by an immunostaining assay. For the rhMPV G1700A mutant, 4 out of 5 cotton rats had detectable virus with an average titer of 2.69 log units. Viral RNA was determined by real-time RT-PCR.
For the rhMPV G1700A mutant, 3 out of 5 cotton rats had detectable virus with a titer of 2.27 log units.
The severity of lung histology was scored for each lung tissue specimen. The average score for each group is shown. 0, no change; 1, mild change; 2, moderate change; 3, severe change.
The amount of hMPV antigen expressed in lung tissue was scored. The average score for each group is shown. 0, no antigen; 1, small amount of antigen; 2, moderate amount of antigen; 3, large amount of antigen.
GRC, genomic RNA copies.
ND, not detected.
Five cotton rats were tested in each group. Values within a column followed by different capital letters (A and B) are significantly different (P < 0.05).
Since no infectious virus was detected in some of the MTase-defective rhMPV-inoculated animals, real-time RT-PCR was performed to determine whether viral genomic RNA was present in nasal turbinate and lung tissues. All virus-infected cotton rats had detectable viral genomic RNA in their tissues, even though they were negative for infectious virus (Table 1). In cotton rats inoculated with wild-type rhMPV, 7.54 and 8.54 log10 genomic RNA copies/g tissue were detected in nasal turbinate and lung tissues, respectively. The viral RNA level was significantly reduced in the upper and lower respiratory tracts of all animals infected with the MTase-defective rhMPVs. In animals infected with the rhMPV G1696A, G1700A, and D1755A mutants, 6.34, 6.95, and 5.27 log10 genomic RNA copies/g tissue were detected in the nasal turbinates, respectively. In addition, 6.72, 7.46, and 6.07 log10 genomic RNA copies per gram of lung tissue were detected in animals infected with the rhMPV G1696A, G1700A, and D1755A mutants, respectively. These levels of genomic RNA production are approximately 10- to 100-fold lower than the level for wild-type rhMPV. Therefore, all MTase-defective rhMPVs had significant defects in viral genome replication in cotton rats.
(ii) Pulmonary histopathology.
Half of the lung tissue was inflated with formaldehyde and subjected to H&E staining and histopathological examination. Each lung tissue specimen was scored on a scale of from 0 (no change) to 3 (severe change) (Table 1). Wild-type rhMPV caused moderate pulmonary histopathological changes, including interstitial pneumonia, peribronchial lymphoplasmocytic infiltrates, mononuclear cell infiltrates, and edematous thickening of the bronchial submucosa (Fig. 8). The rhMPV G1700A mutant similarly induced interstitial pneumonia but caused fewer epithelial changes and less bronchial exudate and mononuclear cell infiltration (Fig. 8). The rhMPV G1696A and D1755A mutants caused mononuclear inflammation similar to that caused by wild-type rhMPV but caused significantly fewer pulmonary histopathological changes. No pathological changes were found in the lungs of cotton rats inoculated with DMEM. These results similarly indicate that MTase-defective hMPVs are attenuated in cotton rats.
FIG 8.

Lung histological changes in cotton rats infected with MTase-defective rhMPVs. The right lung from each cotton rat was preserved in 4% (vol/vol) phosphate-buffered paraformaldehyde. Fixed tissues were embedded in paraffin, sectioned at 5 μm, and stained with H&E for the examination of histological changes by light microscopy. Arrows, lymphocyte infiltrates.
(iii) Viral antigen distribution in lung tissues.
To determine the viral antigen distribution in lung tissues, IHC was performed using an antibody against the hMPV matrix protein. As shown in Fig. 9 and Table 1, large numbers of viral antigen-positive cells were detected at the luminal surfaces of the bronchial epithelial cells in the lungs of wild-type rhMPV-infected cotton rats. The pattern of viral antigen was discontinuous, and antigen appeared in clusters of adjacent cells. In some cases, luminal cellular debris that may have included both epithelial cell debris and macrophages also stained positive for hMPV antigen. Significantly fewer viral antigen-positive cells were detected in the bronchial epithelial cells of the rhMPV G1700A mutant-infected group. No or little viral antigen was detected in lung tissues from the groups infected with the rhMPV G1696A and D1755A mutants. Consistently, these data demonstrate that MTase-defective hMPVs had significant defects in viral replication and antigen expression in the lower respiratory tract.
FIG 9.

IHC staining of lung tissue from cotton rats infected with MTase-defective rhMPVs. Lung tissues were fixed in 4% (vol/vol) phosphate-buffered paraformaldehyde. Deparaffinized sections were stained with monoclonal antibody against hMPV matrix protein (Virostat, Portland, ME) to determine the distribution of viral antigen.
(iv) Genetic stability of MTase-defective rhMPVs in vivo.
To determine whether MTase-defective rhMPVs were genetically stable in cotton rats, we amplified CR VI of the L gene from total RNA extracted from each lung. Sequencing of these PCR products showed that all MTase-defective rhMPVs retained the desired mutation in CR VI of the L gene. No additional mutation was found in this region. This result suggests that MTase-defective rhMPVs are genetically stable in cotton rats.
Immunogenicity of MTase-defective hMPVs in cotton rats.
Since MTase-defective rhMPVs were attenuated in vitro and in vivo, we subsequently determined their immunogenicity in cotton rats. Briefly, cotton rats were inoculated intranasally with the rhMPV mutants. Serum samples were collected weekly for detection of a humoral immune response. At week 4 postinoculation, animals were challenged with 106 PFU of rhMPV. At day 4 postchallenge, all the animals were sacrificed and nasal turbinate and lung tissue samples were collected for virus detection and pathological examination.
Serum antibody was determined by a plaque reduction neutralization assay. As shown in Fig. 10, all of the MTase-defective hMPVs elicited high levels of neutralizing antibody in cotton rats. Antibody was detectable at week 1 postimmunization, and the levels gradually increased during weeks 2 to 4. Overall, the antibodies generated by MTase-defective hMPVs were comparable to those generated after wild-type rhMPV immunization (P > 0.05). In contrast, no hMPV-specific antibody was detected in the unvaccinated control. This result demonstrates that MTase-defective rhMPVs are not only sufficiently attenuated but also capable of triggering high levels of antibody.
FIG 10.
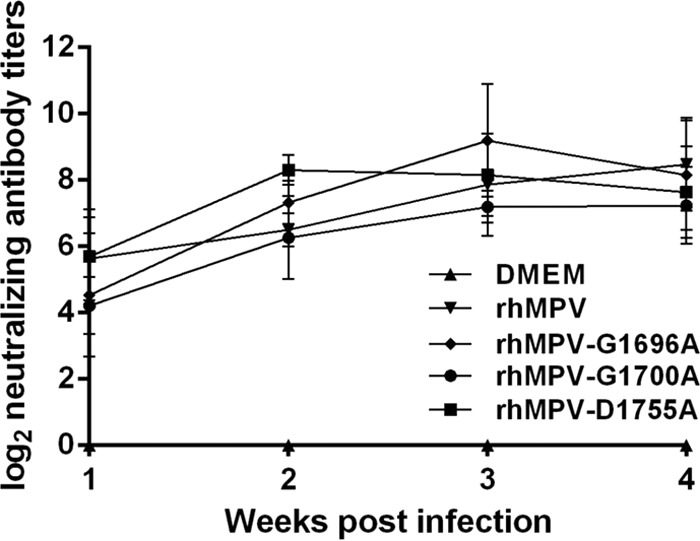
Recombinant hMPVs triggered a high level of neutralizing antibody titer in cotton rats. Cotton rats were immunized with each recombinant hMPV intranasally at a dose of 2.0 × 105 PFU per rat. Blood samples were collected from each rat weekly by retro-orbital bleeding. The hMPV-neutralizing antibody titer was determined using a plaque reduction neutralization assay, as described in Materials and Methods.
Viral replication in the nasal turbinates and lungs of cotton rats after challenge was examined by immunostaining. As shown in Table 2, cotton rats vaccinated with wild-type rhMPV or MTase-defective rhMPVs did not have any detectable infectious virus in either the nasal turbinate or lung tissue after challenge with rhMPV. In contrast, unvaccinated challenged controls had average titers of 5.59 ± 0.49 and 4.76 ± 0.29 log10 PFU/g in the nasal turbinate and lung, respectively. These results demonstrate that infection with the rhMPV G1696A, G1700A, and D1755A mutants provided complete protection from challenge with wild-type rhMPV, preventing viral replication in both the upper and lower respiratory tracts.
TABLE 2.
Immunogenicity of MTase-defective rhMPVs in cotton ratsh
| rhMPV mutant a | Nasal turbinateb |
Lungc |
Lung histology scored | Lung IHC scoree | Protection ratef (%) | |||
|---|---|---|---|---|---|---|---|---|
| % of infected animals | Viral titer (log10 PFU/g) | % of infected animals | Viral titer (log10 PFU/g) | Viral RNA load (log10 GRC/g) | ||||
| DMEM | 100 | 5.59 ± 0.49 | 100 | 4.76 ± 0.29 | 5.65 ± 0.22A | 1.6A | 3.0A | 0 |
| Wild type | 0 | NDg | 0 | ND | 2.54 ± 0.35B | 0.6B | 0.5B | 100 |
| G1696A | 0 | ND | 0 | ND | 2.68 ± 0.22B | 0.8B | 0.5B | 100 |
| G1700A | 0 | ND | 0 | ND | 2.50 ± 0.28B | 0.5B | 0.5B | 100 |
| D1755A | 0 | ND | 0 | ND | 2.46 ± 0.23B | 0.3B | 0.5B | 100 |
Animals were immunized intranasally with DMEM or 2 × 105 PFU of wild-type rhMPV or an rhMPV mutant. At day 28 postimmunization, animals were challenged with 106 PFU of wild-type rhMPV.
The viral titer was determined by an immunostaining assay.
The viral RNA titer was determined by real-time RT-PCR. GRC, genomic RNA copies.
The severity of lung histology was scored for each lung tissue specimen. The average score for each group was shown. 0, no change; 1, mild change; 2, moderate change; 3, severe change.
The amount of hMPV antigen expressed in lung tissue was scored. The average score for each group is shown. 0, no antigen; 1, small amount of antigen; 2, moderate amount of antigen; 3, large amount of antigen.
The protection rate was calculated on the basis of the level of viral replication in lungs and nasal turbinates.
ND, not detected.
Five cotton rats were tested in each group. Values within a column followed by different capital letters (A and B) are significantly different (P < 0.05).
After challenge, lung histology was evaluated for each cotton rat. As expected, unvaccinated challenged controls had moderate pulmonary histological changes, characterized by interstitial pneumonia, mononuclear cell infiltration, and edematous thickening of the bronchial submucosa, and bronchial epithelium changes (Fig. 11 and Table 2). In contrast, significantly fewer histological changes were found in the lungs of cotton rats vaccinated with wild-type rhMPV and the rhMPV G1696A, G1700A, and D1755A mutants (Table 2; Fig. 11). No enhanced lung damage was observed in the vaccinated and challenged groups. No histological changes were found in the unvaccinated and unchallenged controls. These results demonstrate that MTase-defective rhMPV provides protection against lung damage from virus challenge.
FIG 11.

Lung histological changes in cotton rats vaccinated with MTase-defective rhMPVs followed by challenge with rhMPV. The right lung from each cotton rat was preserved in 4% (vol/vol) phosphate-buffered paraformaldehyde. Fixed tissues were embedded in paraffin, sectioned at 5 μm, and stained with H&E for the examination of histological changes by light microscopy. Moderate to severe histological changes were observed in the unimmunized challenged controls. Mild histological changes were observed in vaccinated challenged controls.
For lung tissues from unvaccinated challenged controls, large numbers of viral antigens were found at the luminal surface of the bronchial epithelial cells (Fig. 12). Interestingly, lung tissues from the vaccinated and challenged groups exhibited different antigen distribution patterns (Fig. 12). Antigen was found within bronchial tissue but not on the luminal surface of bronchial epithelial cells, which may be related to viral clearance. No antigen was found in the unvaccinated unchallenged group.
FIG 12.

IHC staining of lungs from cotton rats vaccinated with MTase-defective rhMPVs followed by rhMPV challenge. Lung tissues were fixed in 4% (vol/vol) phosphate-buffered paraformaldehyde. Deparaffinized sections were stained with monoclonal antibody against hMPV matrix protein (Virostat, Portland, ME) to determine the distribution of viral antigen. Arrows, antigen-positive cells.
2′-O MTase-defective hMPVs are highly sensitive to IFN-α and IFN-β treatment.
Recent work on West Nile virus (33), dengue virus (42), mouse hepatitis virus (MHV) (32), and severe acute respiratory syndrome coronavirus (43) found that the mechanism of attenuation of the 2′-O MTase-defective viruses is attributable to enhanced sensitivity to the antiviral action of IFN. However, whether this mechanism is conserved in nonsegmented negative-sense RNA viruses (such as hMPV) is not known. We have now determined the sensitivity of 2′-O MTase-defective hMPVs to IFN treatment in Vero E6 cells, which are defective in type I interferon production but express IFN receptors. Thus, Vero E6 cells can respond to exogenous type I IFN. Briefly, Vero E6 cells were pretreated with the different amounts (2 and 20 units) of IFN-α or IFN-β for 4 h, followed by washing the treated cells with DMEM three times, and the cells were then infected with each hMPV mutant at an MOI of 0.5. At 48 h postinfection, supernatant was harvested and the viral titer was determined by immunostaining assay. Treatment of Vero E6 cells with 2 or 20 units of IFN-α (Fig. 13A) or IFN-β (Fig. 13B), followed by infection with each recombinant hMPV mutant, resulted in a reduction in the viral titer of 2 to 4 log10 PFU. However, there was no significant reduction for wild-type hMPV when Vero E6 cells were treated with IFN-α or IFN-β at the same doses (P > 0.05). Therefore, all 2′-O MTase-defective hMPVs were highly sensitive to both IFN-α and IFN-β inhibition, whereas wild-type hMPV was not sensitive to IFN inhibition. This result suggests that the mechanism of attenuation of 2′-O defective viruses may also be conserved in hMPV.
FIG 13.
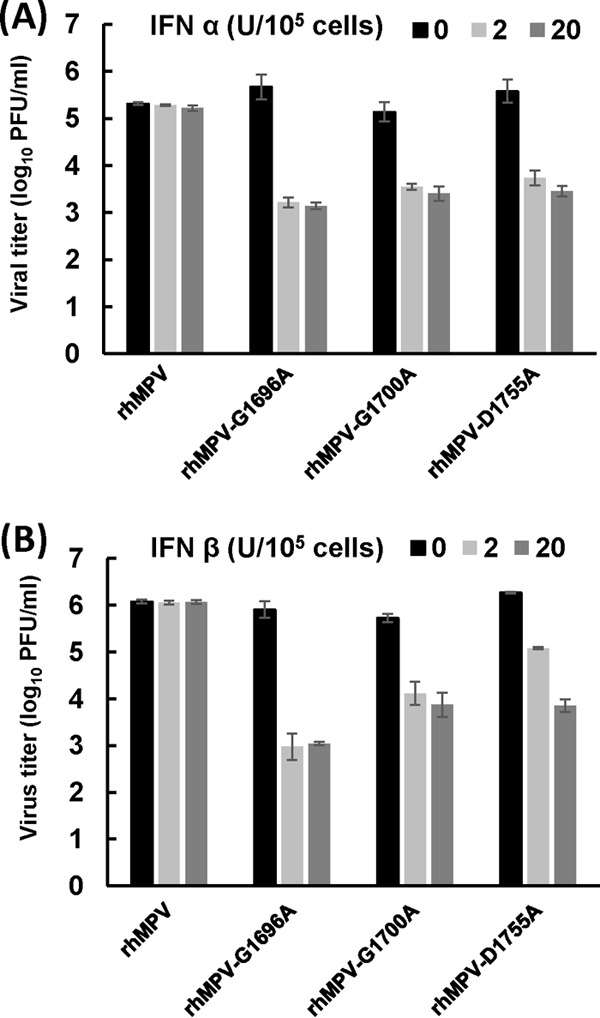
Sensitivity of recombinant hMPVs to IFN-α (A) and IFN-β (B) treatment. Confluent Vero E6 cells in 24-well plates were pretreated with DMEM containing 2 or 20 U of IFN-α or IFN-β at 37°C for 4 h. Cells were then washed three times with DMEM and infected with each rhMPV mutant at an MOI of 0.5. After 1 h of adsorption, cells were washed three times with DMEM and fresh medium was added. At 48 h postinfection, supernatant from each well was harvested and the viral titers were determined by immunostaining using Vero E6 cells. Data are the averages of three experiments.
DISCUSSION
Paramyxoviruses, such as RSV, hMPV, and PIV3, are responsible for the majority of pediatric respiratory tract disease and inflict significant economic loss, health care costs, and emotional burdens. Despite tremendous efforts, no vaccine is currently available for these viruses. In this study, we found that MTase-defective rhMPVs were sufficiently attenuated but retained high immunogenicity in cotton rats. These results demonstrate that MTase-defective rhMPVs are excellent live vaccine candidates.
MTase as a new target for attenuation of hMPV.
A live attenuated vaccine is the most promising vaccine for human paramyxoviruses. Live attenuated vaccines offer many advantages: (i) enhanced lung damage has not been observed either after vaccination with live attenuated viruses or after natural reinfection (44, 45), (ii) intranasal administration of live vaccines induces balanced immune responses that closely resemble those induced after natural virus infection, and (iii) intranasal vaccination induces better local immunity than the intramuscular injection of subunit vaccines. However, it has been technically challenging to identify vaccine strains that have an optimal balance between attenuation and immunogenicity. Previously, cold-passaged (cp), temperature-sensitive (ts) live attenuated hMPV, hMPVs from which nonessential genes were deleted, and chimeric hMPVs have been generated (15–20). Many of these approaches either affect immunogenicity or genetic stability or lead to insufficient attenuation or poor virus growth in vitro. To date, only one live vaccine candidate, which is a chimeric rhMPV carrying the aMPV P gene, has been approved for use in a phase I clinical trial (20). The safety and efficacy of this live attenuated hMPV vaccine in humans are not known. Therefore, exploration of new approaches to attenuate hMPV is urgently needed.
We found that inhibition of viral mRNA cap MTase can serve as a new approach to attenuate hMPV and perhaps other paramyxoviruses for vaccine purposes. Using a reverse genetics system, we generated three rhMPVs carrying mutations in the SAM binding site and showed that these recombinant viruses were specifically defective in 2′-O methylation but not G-N-7 methylation. These MTase-defective rhMPVs produced smaller plaques and had delayed growth kinetics, lower peak titers, and less protein synthesis in cell culture than wild-type rhMPV. In cotton rats, the rhMPV G1696A and D1755A mutants were highly attenuated, based on the fact that no infectious virus was found in the upper or lower respiratory tract. The rhMPV G1700A mutant was moderately attenuated, based on the evidence that 4 out of 5 animals had low levels of virus replication in the nasal turbinates and 1 out of 5 animals had low levels of virus replication in the lungs. In addition, all MTase-defective rhMPVs induced significantly fewer lung histopathological changes, and no or little viral antigen expression in the epithelial cells on the surface of bronchi was detected. An exciting finding was that despite the attenuation, all MTase-defective rhMPVs elicited high titers of neutralizing antibody comparable to those induced by wild-type rhMPV. Furthermore, all immunized animals were completely protected from viral replication in the upper and lower respiratory tracts after challenge with rhMPV. The amount of viral genomic RNA in the lower respiratory tract of the immunized animals showed at least a 1,000-fold reduction. In addition, these recombinant viruses were genetically stable in vitro and in vivo. By optimization of growth conditions, we found that the yield of these recombinant viruses in cell culture was reduced by only approximately 0.5 log unit. Thus, it should be economically feasible to produce vaccine candidates. One of the major concerns in paramyxovirus vaccine development is the potential for enhanced disease upon reinfection, a lesson learned from a clinical trial of a formalin-inactivated (FI) RSV vaccine in the 1960s (8). To date, enhanced disease has not been caused by live attenuated vaccines (cold-adapted chimeric viruses with gene deletions) and subunit vaccine candidates for hMPV (15). In our study, we also evaluated the histopathological changes and viral antigen distribution in the lungs of the immunized animals upon challenge (reinfection) with wild-type rhMPV. No enhanced lung damage was found after vaccination with MTase-defective hMPV followed by challenge. In fact, vaccination protected the lungs from the histological changes caused by rhMPV challenge.
To date, a large number of studies have shown that cotton rats are the best small-animal model of infection with human paramyxoviruses, including RSV, hMPV, and PIV3 (46–48). For example, the permissiveness of cotton rats to infection with RSV was over 100-fold higher than that of mice (48). Recently, we and others showed that cotton rats are more permissive to hMPV infection than hamsters and mice, with the titers in cotton rat lungs being up to 5 log10 PFU higher than those in hamster and mouse lungs (49, 50). Cotton rats have been used as an animal model not only to study the pathogenesis and efficacy of vaccines against RSV, hMPV, and PIV3 but also to evaluate the efficacy and safety of prophylactic antibody RespiGam and Synagis treatment for severe RSV disease (46, 47). Previously, several RSV live vaccine candidates were initially shown to be promising in rodent models (51, 52). However, they lacked sufficient immunogenicity when tested in human clinical trials. Certainly, this is a complicated issue, since many host and viral factors can affect the efficacy of an RSV vaccine in human clinical trials. For instance, unlike hMPV, RSV encodes two addition nonstructural proteins (NS1 and NS2) which can inhibit the innate immunity and subsequently impair the adaptive immunity (53). Although our study showed that MTase-defective hMPVs are highly efficacious in a cotton rat model, it will be interesting to determine whether these vaccine candidates can provide long-term immunity in nonhuman primates and human clinical trials.
MTase as a target for attenuation of other RNA viruses.
Recently, we presented the concept of using MTase-defective viruses as vaccine candidates for VSV (54). Specifically, we showed that rVSVs (K1651A, D1762A, and E1833Q MTase catalytic site mutants) lacking both G-N-7 and 2′-O methylations were highly attenuated, whereas viruses defective in G-N-7 methylation but not 2′-O methylation (rVSV G1670A and G1672A SAM binding site mutants) retained low levels of virulence in a mouse model (54). However, in the case of hMPV, viruses lacking 2′-O methylation alone were sufficiently attenuated. Similarly, flaviviruses (such as Japanese encephalitis virus and dengue virus) lacking 2′-O methylation were also attenuated in animal models (42, 55). There are several advantages to using MTase as the target for virus attenuation. First, viruses lacking MTase would likely not affect immunogenicity, since the MTase is located in the L protein, which is not a target for neutralizing antibodies. Second, the mRNA of paramyxoviruses typically possesses two MTases; thus, recombinant viruses specifically defective in G-N-7 and/or ribose 2′-O can be generated. Third, different amino acid substitutions in the MTase domain will have variable impacts on mRNA cap methylation, which will enable the generation of recombinant viruses with different degrees of attenuation. This will allow us to select vaccine strains that have a proper balance between attenuation and immunogenicity. Finally, the MTase domain is highly conserved in L proteins of all NNS RNA viruses, with the exception of those in the family Bornaviridae (39, 40). Thus, this novel attenuation strategy can be employed for other NNS RNA viruses. By combining multiple substitutions in the MTase region of the L protein, it should be possible to generate an attenuated virus that is genetically stable, because reversion to wild type at any single amino acid would not provide a return to wild type.
Mechanism of mRNA cap methylation in NNS RNA viruses.
To date, the mechanism of mRNA cap methylation in paramyxoviruses is poorly understood because most of them, including hMPV, lack robust mRNA synthesis in vitro. In addition, it has been a challenge to express and purify a functional L protein that can modify virus-specific mRNA in vitro. Our current understanding of mRNA cap methylation in NNS RNA viruses comes from studies of VSV, because the genetic and biochemical tools have been well established for VSV. First, mutations to a single MTase active site in CR VI of the VSV L protein abolished both G-N-7 and 2′-O methylations (28, 29), demonstrating that both G-N-7 and 2′-O MTases are catalyzed by CR VI of L protein. Second, mutations to the SAM binding site in VSV L protein diminished both G-N-7 and 2′-O methylations for some mutations or specifically diminished G-N-7 methylation but not 2′-O methylation for other mutations (27). Third, VSV mRNA cap methylation is achieved in a sequential order in which the 2′-O methylation precedes and facilitates the G-N-7 methylation (29). However, all known cellular and other viral mRNA caps are first methylated at the G-N-7 position, followed by the 2′-O position (26, 31, 41). We compared the roles of amino acid residues in VSV and hMPV SAM binding sites in methylation (Table 3). In VSV, the G1670A mutation specifically abolished G-N-7 methylation, the mutant with the G1674A mutation was fully methylated but sensitive to the SAM concentration, and the D1735A mutation inhibited both G-N-7 and 2′-O methylation by 70%. Using an in vitro methylation assay, we found that mutants with the equivalent mutations in the SAM binding site of hMPV (the rhMPV G1696A, G1700A, and D1755A mutants) were specifically defective in 2′-O methylation but not G-N-7 methylation. This suggests that the role of the SAM binding site in methylation and the order of mRNA cap methylation in hMPV are different from those in VSV. Specifically, 2′-O methylation is not required for G-N-7 methylation in hMPV, whereas 2′-O methylation occurs prior to G-N7 methylation in VSV. From this perspective, methylation of the hMPV mRNA cap structure may be similar to that described for flaviviruses (such as West Nile virus and dengue virus), in that G-N-7 methylation precedes 2′-O methylation by a single peptide at the N terminus of the NS protein (56–58). Interestingly, several other studies support the conclusion that the order of mRNA cap methylation in other paramyxoviruses differs from that in VSV. In the early 1990s, it was reported that the mRNA of RSV lacks 2′-O methylation (59). However, more recent studies have found that RSV mRNA is G-N-7 methylated at a low SAM concentration but could further be chased to have a doubly methylated cap at higher SAM concentrations (60), suggesting that the mRNA cap methylation of RSV first occurs at position G-N-7 and then occurs at position 2′-O. In addition, a fragment of Sendai virus L protein that includes CR VI was able to methylate short Sendai virus-specific RNA sequences in vitro at the G-N-7 position (61), suggesting that G-N-7 methylation of the Sendai virus mRNA cap can occur without premethylation of the 2′-O position.
TABLE 3.
Summary of phenotypes with mutations in the SAM binding site in hMPV and VSV L proteins
| Virus family | Mutant | Methylation | Attenuation in cell culturea | Attenuation in animal modelb | Immunogenicityc |
|---|---|---|---|---|---|
| Paramyxoviridae | rhMPV wild type | WTd | WT | Virulent | WT |
| rhMPV G1696A | Defective in 2′-O | Attenuated | Highly attenuated | WT | |
| rhMPV G1700A | Defective in 2′-O | Attenuated | Attenuated | WT | |
| rhMPV D1755A | Defective in 2′-O | Attenuated | Highly attenuated | WT | |
| Rhabdoviridae | rVSV wild type | WT | WT | Virulent | WT |
| rVSV G1670A | Lacked G-N-7 but not 2′-O | Attenuated | Low virulence | WT | |
| rVSV G1674A | Sensitive to SAM concn | Not attenuated | Virulent | NDe | |
| rVSV D1735A | Diminished G-N-7 and 2′-O | Attenuated | ND | ND |
Attenuation was judged by plaque size, infectivity, and growth curve. Data for VSV are from reference 27.
For hMPV, attenuation was judged by viral replication and pathology in nasal turbinate and lung tissue in cotton rats; for VSV, attenuation was judged by clinical symptoms, body weight loss, viral replication, and pathology in lung and brain tissues in mice. Data for VSV are from reference 54.
Immunogenicity was judged by the antibody level and protection rate after challenge with virulent virus.
WT, wild-type level.
ND, not done.
Biological role of 2′-O methylation.
In this study, we successfully generated a panel of rhMPV mutants specifically defective in 2′-O methylation which will provide an essential tool to study the biological function of 2′-O methylation in NNS RNA viruses. It is firmly established that G-N-7 methylation is essential for mRNA stability as well as efficient translation (62, 63). However, determination of the role(s) of ribose 2′-O methylation has proved more elusive, even though it was discovered more than 4 decades ago. Using 2′-O MTase-defective vaccinia virus, coronaviruses, and flaviviruses, it was recently found that 2′-O methylation of viral RNA provides a molecular signature for the discrimination of self and non-self mRNA (32, 33, 64). In addition, it was found that the mechanism of attenuation of 2′-O MTase-defective viruses is enhanced sensitivity to the antiviral action of IFN (32, 33). In this study, we found that all 2′-O MTase-defective hMPVs were highly sensitive to IFN-α and IFN-β treatment, whereas wild-type hMPV was not sensitive to interferon. This suggests that the general mechanism of attenuation of virus that lacks 2′-O methylation may be conserved in hMPV and perhaps other NNS RNA viruses, although the detailed signaling pathway is still unknown. However, it appears that NNS RNA viruses possess many unique characteristics in mRNA cap methylation. For example, the order of mRNA cap methylation in VSV is reversed compared to that for all known cellular and viral mRNA cap methylations (27, 29). In addition, we recently found that rVSV carrying four mutations in the SAM binding site (the rVSV G4A mutant) produces unmethylated mRNA yet retains considerable virulence, whereas other rVSV mutants (MTase catalytic site mutants) that produce unmethylated mRNA were highly attenuated (54). Furthermore, the mRNA of Newcastle disease virus, an avian paramyxovirus, naturally lacks 2′-O methylation (65). These observations suggest that the functions of 2′-O methylation in NNS RNA viruses may be more complicated than originally thought.
In conclusion, this study highlights a new and novel strategy for the development of live attenuated hMPV vaccines. Our results show that single amino acid substitutions in the SAM binding site of the hMPV L protein can significantly reduce viral replication and pathogenesis in cotton rats but allow retention of wild-type levels of immunogenicity. Since amino acid residues essential for MTase activity are highly conserved, this novel vaccine strategy can be employed with other human paramyxoviruses, such as RSV and PIV3.
ACKNOWLEDGMENTS
This study was supported by grants from the NIH (R01AI090060) and a pilot grant from the OSU Center for Clinical and Translational Science (CCTS) to J.L. Yu Zhang is a fellow of the Center for RNA Biology at The Ohio State University.
We thank Ron A. M. Fouchier for the infectious cDNA clone of hMPV. We thank Sean Whelan for proving VSV mutants for this study. We thank Steven Krakowka for his help on histology. We are grateful to members of the J. Li laboratory for critical readings of the manuscript.
Footnotes
Published ahead of print 23 July 2014
REFERENCES
- 1.van den Hoogen BG, de Jong JC, Groen J, Kuiken T, de Groot R, Fouchier RA, Osterhaus AD. 2001. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 7:719–724. 10.1038/89098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schildgen V, van den Hoogen B, Fouchier R, Tripp RA, Alvarez R, Manoha C, Williams J, Schildgen O. 2011. Human metapneumovirus: lessons learned over the first decade. Clin. Microbiol. Rev. 24:734–754. 10.1128/CMR.00015-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feuillet F, Lina B, Rosa-Calatrava M, Boivin G. 2012. Ten years of human metapneumovirus research. J. Clin. Virol. 53:97–105. 10.1016/j.jcv.2011.10.002 [DOI] [PubMed] [Google Scholar]
- 4.van den Hoogen BG, Herfst S, Sprong L, Cane PA, Forleo-Neto E, de Swart RL, Osterhaus AD, Fouchier RA. 2004. Antigenic and genetic variability of human metapneumoviruses. Emerg. Infect. Dis. 10:658–666. 10.3201/eid1004.030393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biacchesi S, Skiadopoulos MH, Boivin G, Hanson CT, Murphy BR, Collins PL, Buchholz UJ. 2003. Genetic diversity between human metapneumovirus subgroups. Virology 315:1–9. 10.1016/S0042-6822(03)00528-2 [DOI] [PubMed] [Google Scholar]
- 6.Govindarajan D, Buchholz UJ, Samal SK. 2006. Recovery of avian metapneumovirus subgroup C from cDNA: cross-recognition of avian and human metapneumovirus support proteins. J. Virol. 80:5790–5797. 10.1128/JVI.00138-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wei Y, Feng K, Yao X, Cai H, Li J, Mirza AM, Iorio RM, Li J. 2012. Localization of a region in the fusion protein of avian metapneumovirus that modulates cell-cell fusion. J. Virol. 86:11800–11814. 10.1128/JVI.00232-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim HW, Canchola JG, Brandt CD, Pyles G, Chanock RM, Jensen K, Parrott RH. 1969. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am. J. Epidemiol. 89:422–434 [DOI] [PubMed] [Google Scholar]
- 9.Ottolini MG, Porter DD, Hemming VG, Prince GA. 2000. Enhanced pulmonary pathology in cotton rats upon challenge after immunization with inactivated parainfluenza virus 3 vaccines. Viral Immunol. 13:231–236. 10.1089/vim.2000.13.231 [DOI] [PubMed] [Google Scholar]
- 10.Yim KC, Cragin RP, Boukhvalova MS, Blanco JC, Hamlin ME, Boivin G, Porter DD, Prince GA. 2007. Human metapneumovirus: enhanced pulmonary disease in cotton rats immunized with formalin-inactivated virus vaccine and challenged. Vaccine 25:5034–5040. 10.1016/j.vaccine.2007.04.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins PL, Murphy BR. 2002. Respiratory syncytial virus: reverse genetics and vaccine strategies. Virology 296:204–211. 10.1006/viro.2002.1437 [DOI] [PubMed] [Google Scholar]
- 12.Herfst S, de Graaf M, Schrauwen EJ, Sprong L, Hussain K, van den Hoogen BG, Osterhaus AD, Fouchier RA. 2008. Generation of temperature-sensitive human metapneumovirus strains that provide protective immunity in hamsters. J. Gen. Virol. 89:1553–1562. 10.1099/vir.0.2008/002022-0 [DOI] [PubMed] [Google Scholar]
- 13.Biacchesi S, Skiadopoulos MH, Tran KC, Murphy BR, Collins PL, Buchholz UJ. 2004. Recovery of human metapneumovirus from cDNA: optimization of growth in vitro and expression of additional genes. Virology 321:247–259. 10.1016/j.virol.2003.12.020 [DOI] [PubMed] [Google Scholar]
- 14.Herfst S, de Graaf M, Schickli JH, Tang RS, Kaur J, Yang CF, Spaete RR, Haller AA, van den Hoogen BG, Osterhaus AD, Fouchier RA. 2004. Recovery of human metapneumovirus genetic lineages A and B from cloned cDNA. J. Virol. 78:8264–8270. 10.1128/JVI.78.15.8264-8270.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buchholz UJ, Nagashima K, Murphy BR, Collins PL. 2006. Live vaccines for human metapneumovirus designed by reverse genetics. Expert Rev. Vaccines 5:695–706. 10.1586/14760584.5.5.695 [DOI] [PubMed] [Google Scholar]
- 16.Biacchesi S, Pham QN, Skiadopoulos MH, Murphy BR, Collins PL, Buchholz UJ. 2005. Infection of nonhuman primates with recombinant human metapneumovirus lacking the SH, G, or M2-2 protein categorizes each as a nonessential accessory protein and identifies vaccine candidates. J. Virol. 79:12608–12613. 10.1128/JVI.79.19.12608-12613.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biacchesi S, Skiadopoulos MH, Yang L, Lamirande EW, Tran KC, Murphy BR, Collins PL, Buchholz UJ. 2004. Recombinant human metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: deletion of G yields a promising vaccine candidate. J. Virol. 78:12877–12887. 10.1128/JVI.78.23.12877-12887.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schickli JH, Kaur J, Macphail M, Guzzetta JM, Spaete RR, Tang RS. 2008. Deletion of human metapneumovirus M2-2 increases mutation frequency and attenuates growth in hamsters. Virol. J. 5:69. 10.1186/1743-422X-5-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buchholz UJ, Biacchesi S, Pham QN, Tran KC, Yang L, Luongo CL, Skiadopoulos MH, Murphy BR, Collins PL. 2005. Deletion of M2 gene open reading frames 1 and 2 of human metapneumovirus: effects on RNA synthesis, attenuation, and immunogenicity. J. Virol. 79:6588–6597. 10.1128/JVI.79.11.6588-6597.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pham QN, Biacchesi S, Skiadopoulos MH, Murphy BR, Collins PL, Buchholz UJ. 2005. Chimeric recombinant human metapneumoviruses with the nucleoprotein or phosphoprotein open reading frame replaced by that of avian metapneumovirus exhibit improved growth in vitro and attenuation in vivo. J. Virol. 79:15114–15122. 10.1128/JVI.79.24.15114-15122.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naylor CJ, Ling R, Edworthy N, Savage CE, Easton AJ. 2007. Avian metapneumovirus SH gene end and G protein mutations influence the level of protection of live-vaccine candidates. J. Gen. Virol. 88:1767–1775. 10.1099/vir.0.82755-0 [DOI] [PubMed] [Google Scholar]
- 22.Whelan SP, Barr JN, Wertz GW. 2004. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 283:61–119. 10.1007/978-3-662-06099-5_3 [DOI] [PubMed] [Google Scholar]
- 23.Ogino T, Banerjee AK. 2007. Unconventional mechanism of mRNA capping by the RNA-dependent RNA polymerase of vesicular stomatitis virus. Mol. Cell 25:85–97. 10.1016/j.molcel.2006.11.013 [DOI] [PubMed] [Google Scholar]
- 24.Li J, Rahmeh A, Morelli M, Whelan SP. 2008. A conserved motif in region V of the large polymerase proteins of nonsegmented negative-sense RNA viruses that is essential for mRNA capping. J. Virol. 82:775–784. 10.1128/JVI.02107-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogino T, Yadav SP, Banerjee AK. 2010. Histidine-mediated RNA transfer to GDP for unique mRNA capping by vesicular stomatitis virus RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 107:3463–3468. 10.1073/pnas.0913083107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shuman S. 2001. Structure, mechanism, and evolution of the mRNA capping apparatus. Prog. Nucleic Acids Res. Mol. Biol. 66:1–40. 10.1016/S0079-6603(00)66025-7 [DOI] [PubMed] [Google Scholar]
- 27.Li J, Wang JT, Whelan SP. 2006. A unique strategy for mRNA cap methylation used by vesicular stomatitis virus. Proc. Natl. Acad. Sci. U. S. A. 103:8493–8498. 10.1073/pnas.0509821103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Fontaine-Rodriguez EC, Whelan SP. 2005. Amino acid residues within conserved domain VI of the vesicular stomatitis virus large polymerase protein essential for mRNA cap methyltransferase activity. J. Virol. 79:13373–13384. 10.1128/JVI.79.21.13373-13384.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rahmeh AA, Li J, Kranzusch PJ, Whelan SP. 2009. Ribose 2′-O methylation of the vesicular stomatitis virus mRNA cap precedes and facilitates subsequent guanine-N-7 methylation by the large polymerase protein. J. Virol. 83:11043–11050. 10.1128/JVI.01426-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grdzelishvili VZ, Smallwood S, Tower D, Hall RL, Hunt DM, Moyer SA. 2005. A single amino acid change in the L-polymerase protein of vesicular stomatitis virus completely abolishes viral mRNA cap methylation. J. Virol. 79:7327–7337. 10.1128/JVI.79.12.7327-7337.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bisaillon M, Lemay G. 1997. Viral and cellular enzymes involved in synthesis of mRNA cap structure. Virology 236:1–7. 10.1006/viro.1997.8698 [DOI] [PubMed] [Google Scholar]
- 32.Zust R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, Siddell SG, Ludewig B, Thiel V. 2011. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 12:137–143. 10.1038/ni.1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong H, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M, Jr, Shi PY, Diamond MS. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456. 10.1038/nature09489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.National Institutes of Health. 2002. Public Health Service policy on humane care and use of laboratory animals. Office of Laboratory Animal Welfare, National Institutes of Health, Bethesda, MD [Google Scholar]
- 35.Zhang Y, Wei Y, Li J, Li J. 2012. Development and optimization of a direct plaque assay for human and avian metapneumoviruses. J. Virol. Methods 185:61–68. 10.1016/j.jviromet.2012.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J, Chorba JS, Whelan SP. 2007. Vesicular stomatitis viruses resistant to the methylase inhibitor sinefungin upregulate RNA synthesis and reveal mutations that affect mRNA cap methylation. J. Virol. 81:4104–4115. 10.1128/JVI.02681-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hodel AE, Gershon PD, Shi X, Quiocho FA. 1996. The 1.85 Å structure of vaccinia protein VP39: a bifunctional enzyme that participates in the modification of both mRNA ends. Cell 85:247–256. 10.1016/S0092-8674(00)81101-0 [DOI] [PubMed] [Google Scholar]
- 38.Hager J, Staker BL, Bugl H, Jakob U. 2002. Active site in RrmJ, a heat shock-induced methyltransferase. J. Biol. Chem. 277:41978–41986. 10.1074/jbc.M205423200 [DOI] [PubMed] [Google Scholar]
- 39.Bujnicki JM, Rychlewski L. 2002. In silico identification, structure prediction and phylogenetic analysis of the 2′-O-ribose (cap 1) methyltransferase domain in the large structural protein of ssRNA negative-strand viruses. Protein Eng. 15:101–108. 10.1093/protein/15.2.101 [DOI] [PubMed] [Google Scholar]
- 40.Ferron F, Longhi S, Henrissat B, Canard B. 2002. Viral RNA-polymerases—a predicted 2′-O-ribose methyltransferase domain shared by all Mononegavirales. Trends Biochem. Sci. 27:222–224. 10.1016/S0968-0004(02)02091-1 [DOI] [PubMed] [Google Scholar]
- 41.Lockless SW, Cheng HT, Hodel AE, Quiocho FA, Gershon PD. 1998. Recognition of capped RNA substrates by VP39, the vaccinia virus-encoded mRNA cap-specific 2′-O-methyltransferase. Biochemistry 37:8564–8574. 10.1021/bi980178m [DOI] [PubMed] [Google Scholar]
- 42.Zust R, Dong H, Li XF, Chang DC, Zhang B, Balakrishnan T, Toh YX, Jiang T, Li SH, Deng YQ, Ellis BR, Ellis EM, Poidinger M, Zolezzi F, Qin CF, Shi PY, Fink K. 2013. Rational design of a live attenuated dengue vaccine: 2′-O-methyltransferase mutants are highly attenuated and immunogenic in mice and macaques. PLoS Pathog. 9:e1003521. 10.1371/journal.ppat.1003521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Menachery VD, Yount BL, Jr, Josset L, Gralinski LE, Scobey T, Agnihothram S, Katze MG, Baric RS. 2014. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-O-methyltransferase activity. J. Virol. 88:4251–4264. 10.1128/JVI.03571-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wright PF, Karron RA, Belshe RB, Thompson J, Crowe JE, Jr, Boyce TG, Halburnt LL, Reed GW, Whitehead SS, Anderson EL, Wittek AE, Casey R, Eichelberger M, Thumar B, Randolph VB, Udem SA, Chanock RM, Murphy BR. 2000. Evaluation of a live, cold-passaged, temperature-sensitive, respiratory syncytial virus vaccine candidate in infancy. J. Infect. Dis. 182:1331–1342. 10.1086/315859 [DOI] [PubMed] [Google Scholar]
- 45.Wright PF, Shinozaki T, Fleet W, Sell SH, Thompson J, Karzon DT. 1976. Evaluation of a live, attenuated respiratory syncytial virus vaccine in infants. J. Pediatr. 88:931–936. 10.1016/S0022-3476(76)81044-X [DOI] [PubMed] [Google Scholar]
- 46.Boukhvalova MS, Blanco JC. 2013. The cotton rat Sigmodon hispidus model of respiratory syncytial virus infection. Curr. Top. Microbiol. Immunol. 372:347–358. 10.1007/978-3-642-38919-1_17 [DOI] [PubMed] [Google Scholar]
- 47.Boukhvalova MS, Prince GA, Blanco JC. 2009. The cotton rat model of respiratory viral infections. Biologicals 37:152–159. 10.1016/j.biologicals.2009.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Green MG, Huey D, Niewiesk S. 2013. The cotton rat (Sigmodon hispidus) as an animal model for respiratory tract infections with human pathogens. Lab. Anim. (NY) 42:170–176. 10.1038/laban.188 [DOI] [PubMed] [Google Scholar]
- 49.Williams JV, Tollefson SJ, Johnson JE, Crowe JE., Jr 2005. The cotton rat (Sigmodon hispidus) is a permissive small animal model of human metapneumovirus infection, pathogenesis, and protective immunity. J. Virol. 79:10944–10951. 10.1128/JVI.79.17.10944-10951.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, Niewiesk S, Li J. 2014. Animal model for human metapneumovirus: cotton rats are more permissive than hamsters and mice. Pathogens 3:633–655. 10.3390/pathogens3030633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karron RA, Wright PF, Crowe JE, Jr, Clements-Mann ML, Thompson J, Makhene M, Casey R, Murphy BR. 1997. Evaluation of two live, cold-passaged, temperature-sensitive respiratory syncytial virus vaccines in chimpanzees and in human adults, infants, and children. J. Infect. Dis. 176:1428–1436. 10.1086/514138 [DOI] [PubMed] [Google Scholar]
- 52.Collins PL, Graham BS. 2008. Viral and host factors in human respiratory syncytial virus pathogenesis. J. Virol. 82:2040–2055. 10.1128/JVI.01625-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barik S. 2013. Respiratory syncytial virus mechanisms to interfere with type 1 interferons. Curr. Top. Microbiol. Immunol. 372:173–191. 10.1007/978-3-642-38919-1_9 [DOI] [PubMed] [Google Scholar]
- 54.Ma Y, Wei Y, Zhang X, Zhang Y, Cai H, Zhu Y, Shilo K, Oglesbee M, Krakowka S, Whelan SP, Li J. 2014. Messenger RNA cap methylation influences pathogenesis of vesicular stomatitis virus in vivo. J. Virol. 88:2913–2926. 10.1128/JVI.03420-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li SH, Dong H, Li XF, Xie X, Zhao H, Deng YQ, Wang XY, Ye Q, Zhu SY, Wang HJ, Zhang B, Leng QB, Zuest R, Qin ED, Qin CF, Shi PY. 2013. Rational design of a flavivirus vaccine by abolishing viral RNA 2′-O methylation. J. Virol. 87:5812–5819. 10.1128/JVI.02806-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ray D, Shah A, Tilgner M, Guo Y, Zhao Y, Dong H, Deas TS, Zhou Y, Li H, Shi PY. 2006. West Nile virus 5′-cap structure is formed by sequential guanine N-7 and ribose 2′-O methylations by nonstructural protein 5. J. Virol. 80:8362–8370. 10.1128/JVI.00814-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dong H, Ren S, Zhang B, Zhou Y, Puig-Basagoiti F, Li H, Shi PY. 2008. West Nile virus methyltransferase catalyzes two methylations of the viral RNA cap through a substrate-repositioning mechanism. J. Virol. 82:4295–4307. 10.1128/JVI.02202-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou Y, Ray D, Zhao Y, Dong H, Ren S, Li Z, Guo Y, Bernard KA, Shi PY, Li H. 2007. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 81:3891–3903. 10.1128/JVI.02704-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barik S. 1993. The structure of the 5′ terminal cap of the respiratory syncytial virus mRNA. J. Gen. Virol. 74(Pt 3):485–490. 10.1099/0022-1317-74-3-485 [DOI] [PubMed] [Google Scholar]
- 60.Liuzzi M, Mason SW, Cartier M, Lawetz C, McCollum RS, Dansereau N, Bolger G, Lapeyre N, Gaudette Y, Lagace L, Massariol MJ, Do F, Whitehead P, Lamarre L, Scouten E, Bordeleau J, Landry S, Rancourt J, Fazal G, Simoneau B. 2005. Inhibitors of respiratory syncytial virus replication target cotranscriptional mRNA guanylylation by viral RNA-dependent RNA polymerase. J. Virol. 79:13105–13115. 10.1128/JVI.79.20.13105-13115.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ogino T, Kobayashi M, Iwama M, Mizumoto K. 2005. Sendai virus RNA-dependent RNA polymerase L protein catalyzes cap methylation of virus-specific mRNA. J. Biol. Chem. 280:4429–4435. 10.1074/jbc.M411167200 [DOI] [PubMed] [Google Scholar]
- 62.Muthukrishnan S, Morgan M, Banerjee AK, Shatkin AJ. 1976. Influence of 5′-terminal m7G and 2′-O-methylated residues on messenger ribonucleic acid binding to ribosomes. Biochemistry 15:5761–5768. 10.1021/bi00671a012 [DOI] [PubMed] [Google Scholar]
- 63.Furuichi Y, LaFiandra A, Shatkin AJ. 1977. 5′-Terminal structure and mRNA stability. Nature 266:235–239. 10.1038/266235a0 [DOI] [PubMed] [Google Scholar]
- 64.Szretter KJ, Daniels BP, Cho H, Gainey MD, Yokoyama WM, Gale M, Jr, Virgin HW, Klein RS, Sen GC, Diamond MS. 2012. 2′-O methylation of the viral mRNA cap by West Nile virus evades Ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 8:e1002698. 10.1371/journal.ppat.1002698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Colonno RJ, Stone HO. 1976. Newcastle disease virus mRNA lacks 2′-O-methylated nucleotides. Nature 261:611–614. 10.1038/261611a0 [DOI] [PubMed] [Google Scholar]