

ABSTRACT
Kaposi's sarcoma-associated herpesvirus (KSHV) is associated with several human malignances. As saliva is likely the major vehicle for KSHV transmission, we studied in vitro KSHV infection of oral epithelial cells. Through infection of two types of oral epithelial cells, normal human oral keratinocytes (NHOKs) and papilloma-immortalized human oral keratinocyte (HOK16B) cells, we found that KSHV can undergo robust lytic replication in oral epithelial cells. By employing de novo lytic infection of HOK16B cells, we studied the functions of two previously uncharacterized genes, ORF18 and ORF30, during the KSHV lytic cycle. For this purpose, an ORF18-deficient virus and an ORF30-deficient virus were generated using a mutagenesis strategy based on bacterial artificial chromosome (BAC) technology. We found that neither ORF18 nor ORF30 is required for immediately early or early gene expression or viral DNA replication, but each is essential for late gene expression during both de novo lytic replication and reactivation. This critical role of ORF18 and ORF30 in late gene expression was also observed during KSHV reactivation. In addition, global analysis of viral transcripts by RNA sequencing indicated that ORF18 and ORF30 control the same set of viral genes. Therefore, we suggest that these two viral ORFs are involved in the same mechanism or pathway that coregulates the viral late genes as a group.
IMPORTANCE While KSHV can infect multiple cell types in vitro, only a few can support a full lytic replication cycle with progeny virions produced. Consequently, KSHV lytic replication is mostly studied through reactivation, which requires chemicals to induce the lytic cycle or overexpression of the viral transcriptional activator, RTA. In this study, we present a robust de novo lytic infection system based on oral epithelial cells. Using this system, we demonstrate the role of two viral ORFs, ORF18 and ORF30, in regulating viral gene expression during KSHV lytic replication. As the major route of KSHV transmission is thought to be via saliva, this new KSHV lytic replication system will have important utility in the field.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8 (HHV-8), belongs to the gamma subfamily of herpesviruses and establishes lifelong persistent infections in humans. KSHV has been shown to be the causative agent of Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease (1–3). Saliva is most likely the major source of transmission for KSHV. KSHV DNA and infectious viral particles are frequently detected in the saliva of KSHV-seropositive individuals, and the level of KSHV DNA in saliva is significantly higher than those in other body fluids (4–9). It is thought that KSHV may infect oral epithelial cells in transit during initial infection. Moreover, studies from Johnson et al. have shown that latent KSHV in the infected oral keratinocytes can be activated and reenters the lytic cycle to produce infectious progeny virions when oral keratinocytes differentiate into mature epithelium (10). The other human gammaherpesvirus, Epstein-Barr virus (EBV), is also transmitted by saliva. Virus shedding into saliva occurs not only during primary infection but also continuously during persistent infection (11–13). While B lymphocytes are the major site for persistent infection of KSHV and EBV, it is generally believed that epithelial cells in the oral cavity are likely the cell type that is lytically infected to produce virions for subsequent infection of naive cells and transmission to a new host. However, evidence supporting such a role of oral epithelial cells in natural history of EBV and KSHV is still limited.
The lytic genes of herpesviruses are expressed in a highly regulated cascade manner and can be classified as immediate early (IE), early (E), and late (L) genes. While the regulation of IE and E gene expression has been studied extensively in herpesviruses, much less is known about the mechanisms controlling L gene expression. Moreover, by definition, late genes are not expressed until viral DNA replication; nevertheless, how these two processes are linked together remains a mystery. Our previous studies in murine gammaherpesvirus 68 (MHV-68) have shown that open reading frame 18 (ORF18), ORF24, ORF30, ORF31, and ORF34 are required for L expression but not for IE or E gene expression or viral DNA replication (14–17; unpublished data). These five viral ORFs play an important role in activating viral late gene promoters, and it has also been shown that ORF30 and -34 are critical for recruiting RNA polymerase II (Pol II). More recently, studies in cytomegalovirus (CMV), a betaherpesvirus, have demonstrated that the UL79, -87, -91, -92, and -95 genes, homologous to MHV-68 ORF18, -24, -30, -31, and -34, respectively, are also essential for viral late gene expression (18–22). The evidence strongly suggests that beta- and gammaherpesviruses share a similar mechanism to regulate late gene expression.
Recently, a new KSHV bacterial artificial chromosome (BAC) plasmid, BAC16, was generated to facilitate the efficient genetic modification of the KSHV genome (23). A high titer of BAC16-derived virus stocks can be obtained with the use of the cell line iSLK-puro, engineered to express a doxycycline (DOX)-inducible immediate early viral protein, RTA (replication and transcription activator), that drives the lytic replication of KSHV (23, 24). By taking advantage of efficient production of infectious KSHV virions using the iSLK-KSHV BAC16 system, we studied KSHV infection in oral epithelial cells with concentrated virus. We found that in two types of oral epithelial cell lines, KSHV undertakes a robust lytic gene expression program, resulting in virion production. This de novo productive infection cell culture system provides a novel model to study the function of KSHV genes during lytic replication and the virus-host interactions that may occur in the oral cavity.
KSHV also encode the homologues of the five MHV-68 regulators of late gene expression, ORF18, -24, -30, -31, and -34, but the functions of these homologs during lytic replication of KSHV remain to be determined. To study the roles of KSHV ORF18 and ORF30, we used KSHV BAC16 to generate an ORF18-deficient virus and an ORF30-deficient virus. In addition to examining the consequences of lacking ORF18 or ORF30 on viral lytic replication, we also globally evaluated the impact on viral gene expression by high-throughput RNA sequencing (RNA-Seq). In conclusion, neither ORF18 nor ORF30 is required for IE or E gene expression or viral DNA replication, but these two ORFs are both essential for efficient late gene expression. Moreover, ORF18 and ORF30 appear to control the same set of viral genes, indicating that these two ORFs are likely involved in the same mechanism or pathway that coregulates the viral late genes as a group.
MATERIALS AND METHODS
Cells.
Human papillomavirus 16 (HPV-16)-immortalized keratinocytes, HOK16B, and normal human oral keratinocytes, NHOK, were described in the previous studies (25). These two oral epithelial cell lines were maintained in keratinocyte growth medium from Lonza or Epilife, respectively. HEK293T, SCC23, Fadu, SLK, iSLK-puro, and iSLK-BAC16 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum. iSLK-puro cells were supplied with 1 μg/ml puromycin and 250 μg/ml G418. iSLK-BAC16 and its derivatives were maintained in the presence of 1 μg/ml puromycin, 250 μg/ml G418, and 1,200 μg/ml hygromycin B (23). To generate a KSHV ORF18 or ORF30 stable expression cell line (293T-kORF18, 293T-kORF30, iSLK-kORF18, or iSLK-kORF30), 293T or iSLK-puro cells were transfected with pMSCV-zeocin-kORF18 or pMSCV-zeocin-kORF30 plasmid and then selected and maintained with 100 μg/ml zeocin. (Puromycin and G418 were also added for iSLK-kORF18 and iSLK-kORF30 cells.)
Construction of KSHV-18S and KSHV-30S BACs.
A stop codon was introduced into ORF18 or ORF30 to generate BAC-18S or BAC-30S as previously described (23, 26). BAC-18S and BAC-30S were further used to generate ORF18 revertant (18R) and ORF30 revertant (30R) BACs. The restriction patterns of BAC plasmids were verified by comparing them with BAC16. Fragments with the mutations in the middle were PCR amplified from the BAC plasmids and sequenced to confirm that all mutations were correct. BAC-18S and BAC-30S were then transfected into the corresponding 293T-kORF18 or 293T-kORF30 cells, followed by selection with 100 μg/ml hygromycin B for 2 weeks. The resulting 293T-kORF18–18S and 293T-kORF30–30S cells were then induced with 1.5 mM sodium butyrate (NaB) plus 20 ng/ml tetradecanoyl phorbol acetate (TPA) for 2 days to induce lytic replication, followed by coculturing with iSLK-puro cells for 3 days. In the end, iSLK-18S and iSLK-30S cells were established by selection with 1,200 μg/ml hygromycin B in addition to puromycin and G418 for 1 month. iSLK-18R and iSLK-30R were generated by the same method, except that 293T cells were used for the transfection. In addition, BAC-18S and BAC-30S were also introduced into the complementary cell lines iSLK-kORF18 and iSLK-kORF30 to generate iSLK-kORF18-18S and iSLK-kORF30-30S cells, respectively.
Production and titration of KSHVs.
Wild-type (WT) KSHV BAC virus (KSHV BAC16) was prepared from iSLK-BAC16 cells as previously described (23, 24). Briefly, iSLK-BAC16 cells were expanded to 30 15-cm-diameter plates and then induced with 1 μg/ml doxycycline plus 1 mM sodium butyrate for 3 days, when 90% of the cells were round and detached from the plates. Supernatant was collected, centrifuged at 1,000 × g for 15 min at 4°C, and then filtered (0.45-μm pore) to clear cells and debris. Viral particles were pelleted by ultracentrifugation in an SW27 rotor (21,000 rpm for 1 h at 4°C), resuspended in 2 ml DMEM with 10% fetal bovine serum (FBS), aliquoted, flash frozen in liquid nitrogen, and stored in a −80°C freezer. 18S and 30S viruses were prepared from iSLK-kORF18–18S or iSLK-kORF30–30S cells, and 18R and 30R viruses were prepared from iSLK-18R or iSLK-30R cells by the same method as WT BAC16 virus. To determine the KSHV titers, virus stocks were diluted in DMEM with 10% FBS and used to infect 293T cells in 96 wells by spinoculation (3,000 × g for 1 h at 30°C). Three days postinfection, green fluorescent protein (GFP)-positive cell clusters containing two or more cells were counted under a fluorescence microscope to determine the titers of KSHVs. Infectious units (IU) are expressed as the number of GFP-positive cell clusters in each well at the time of analysis. The titers of the final KSHV stocks are 1 × 108 to 4 × 108 IU/ml.
De novo KSHV infection.
For de novo infection of KSHV, cells were seeded in 12-well plates. When cells reached 60 to 70% confluence, the cell numbers were determined, and the desired amounts of KSHV were added to the cells in 400 μl culture medium. After incubation for 2 h at 37°C with occasional swirling, the inoculum was removed, and the cells were washed with 1 ml phosphate-buffered saline (PBS) three times, followed by addition of 1 ml cell growth medium before being returned to the incubator.
Real-time PCR.
Total DNA was isolated from cells, and viral DNA copy numbers were determined by quantitative real-time PCR using primers for ORF59. Total RNA was extracted from cells with the Purelink RNA minikit (Ambion), treated with DNase I, and reverse transcribed with the qScript cDNA synthesis kit (Quantas). The sequences of the primers used to quantify DNA and RNA are as follows: GAPDH (glyceraldehyde-3-phosphate dehydrogenase), 5′-TGC ACC ACC AAC TGC TTA GC-3′ and 5′-GGC ATG GAC TGT GGT CAT GAG-3′; RTA, 5′-CAC AAA AAT GGC GC AAG ATG A-3′ and 5′-TGG TAG AGT TGG GCC TTC AGT T-3′; latency-associated nuclear antigen (LANA), 5′-GAA GTG GAT TAC CCT GTT GTT AGC-3′ and 5′-TTG GAT CTC GTC TTC CAT CC-3′; ORF59, 5′-TTG GCA CTC CAA CGA AAT ATT AGA A-3′ and 5′-CGG GAA CCT TTT GCG AAG A-3′; ORF57, 5′-TGG ACA TTA TGA AGG GCA TCC TA-3′ and 5′-CGG GTT CGG ACA ATT GCT-3′; ORF52, 5′-CTT ACG ATG GAA GAC CTA ACC G-3′ and 5′-ATC CCA GTG CTT TCC GAA G-3′; ORF39, 5′-ATC CTT TGC CCA CAT CCA G-3′ and 5′-GAT AGG TAC AGG TTG ACG CAG-3′; ORF27, 5′-ATC AAA GAC GCC TTC CTC AG-3′ and 5′-TGT GAG AAT TTG AGG GCA GG-3′; and ORF26, 5′-AGC CGA AAG GAT TCC ACC AT-3′ and 5′-TCC GTG TTG TCT ACG TCC AG-3′.
Western blotting and antibodies.
Cell lysates were resolved by SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membrane. Viral proteins were detected with the antibodies against K8 (Novus), K8.1A (Advanced Biotechnologies), ORF26 (Novus), FLAG (Sigma), or RTA (polycolonal antibody produced in rabbit).
IFA.
For the immunofluorescence assay, cells were plated on glass slides placed in 24-well plates and infected with KSHV. At 2 days postinfection, cells were fixed with 2% paraformaldehyde, permeabilized with 0.1% Triton X-100, and then blocked with 3% bovine serum albumin (BSA) and 10% FBS. The viral proteins were detected with the antibodies against LANA, K8.1A, or ORF59 (all from Advanced Biotechnologies). Hoechst dye was added for 10 min prior to analysis.
Directional RNA-Seq library preparation and sequencing.
Libraries were prepared according to the method of ScriptSeq mRNA-Seq library preparation kits (Epicentre). Briefly, RNA was isolated from cells with a PureLink RNA minikit (Ambion). Ten micrograms of total RNA was submitted to two rounds of selection with Oligo (dT)25 Dynabeads (Ambion). Poly(A)-enriched RNA was partially fragmented by heating, followed by first-strand DNA synthesis using the SuperScriptIII first-strand synthesis system (Invitrogen) and a random hexamer primer with a tagging sequence (5′-GAC GTG TGC TCT TCC GAT CT-NNNNNN-3′). After reverse transcription, RNA template was removed by RNase H treatment, and the first-strand cDNA was further tagged at the 3′ terminus with an oligonucleotide (5′-ACA CGA CGC TCT TCC GAT CT-NNNNNN/3Phos/-3′) using T4 DNA polymerase. After tagging, the first-strand cDNA was purified with AMPure XP beads (Beckman), and the cDNA tagged at both ends was amplified by 15 cycles of PCR using primers that anneal to the tag sequences (forward, 5′-AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC T-3′; reverse, 5′-CAA GCA GAA GAC GGC ATA CGA GAT-barcode-GTG ACT GG AGT TCA GAC GTG TGC TCT TCC GAT CT-3′). The PCR products were again purified with AMPure XP beads, and their qualities were checked on an Agilent 2000 Bioanalyzer DNA 1000 chip (Agilent Technologies). Multiplex sequencing was performed by 50-bp single-end read with an Illumina HiSeq 2000 machine at UCLA Clinical Microarray Core. Raw reads were aligned to both the KSHV ORF and the viral RNA transcripts using TopHat under the default parameter (27). Results were quantified by reads per kilobase of coding region per million total reads (RPKM).
RESULTS
KSHV infection in different oral epithelial cell cultures.
Previous studies have shown that KSHV readily infects oral epithelial cells in vitro and establishes latent infection in most of the infected cells (9, 10, 28). Here, we reexamined de novo KSHV infection of oral epithelial cells by using a concentrated KSHV viral stock (108 IU/ml) obtained from the iSLK-KSHV BAC16 system (Materials and Methods). In addition, the KSHV BAC16 virus harbors a green fluorescent protein (GFP) expression cassette driven by the cellular EF1α promoter (23), and thus, GFP fluorescence provides an indicator for infection regardless of lytic or latent replication. We tested multiple types of cells derived from oral keratinocytes, including primary NHOK, immortalized HOK16B, tumor-derived SCC23, and Fadu cells, by infecting them with KSHV BAC16 at two different dosages, 3 and 30 IU/cell (referring to multiplicities of infection [MOI] of 3 and 30, respectively). As the epithelial cell line SLK has been used recently to develop the robust KSHV lytic replication system, it was also included in our experiment. An immunofluorescence assay (IFA) with the antibodies against two KSHV lytic antigens, ORF59 (an early gene) and K8.1 (a late gene), was performed to examine lytic replication of KSHV. The graphs in Fig. 1A summarize the results from day 2 after infection. At an MOI of 3, more than 80% of cells were infected, as indicated by GFP fluorescence, in all of the cell lines we tested, except for Fadu. However, very few cells, if any, expressed viral lytic antigens. At an MOI of 30, 100% of NHOK, HOK16B, and SLK cells were infected but with different levels of lytically infected cells. While infection with KSHV in SLK, SCC23, and Fadu cells largely led to latent infection, significant percentages of infected NHOK and HOK16B cells expressed viral lytic proteins. The representative IFA images of infected HOK16B and NHOK cells are shown in Fig. 1B and C, respectively. In addition to the lytic antigens, we also examined expression of the latent antigen, LANA (latency-associated nuclear antigen). For HOK16B cells at an MOI of 3, over 90% of the HOK16B cells were positive for LANA, less than 1% were positive for ORF59, and no cells were positive for K8.1A. At an MOI of 30, all of the cells were positive for LANA with a much higher expression level than at an MOI of 3. Forty-four percent of the cells were ORF59 positive, and 24% were K8.1A positive. For NHOK cells, we observed higher percentages of cells that expressed the two lytic antigens. Furthermore, the lytic gene expression was verified by Western blotting (Fig. 1D). All three viral proteins, LANA, RTA, and K8.1A, were expressed well in HOK16B cells infected at an MOI of 30 compared to SCC23, Fadu, and SLK cells, while at an MOI of 3, only LANA was detected, albeit at a very low level.
FIG 1.

Analysis of KSHV infection in different epithelial cells. (A) Cells of four oral epithelial cell lines (NHOK, HOK16B, SCC23, and Fadu) and an additional epithelial cell line (SLK) were infected with KSHV BAC16 virus at an MOI of 3 or 30, when the confluence of cells reached 60%. Cells were fixed 2 days after infection and stained with antibodies against ORF59 and K8.1A. Images were taken under the fluorescence microscope for quantitation of the percentage of cells expressing GFP, ORF59, and K8.1A. The data shown are means ± standard deviations from three experiments. (B) Immunofluorescence analysis of KSHV latent and lytic proteins in HOK16B cells with antibodies against LANA, ORF59, and K8.1A. (C) Immunofluorescence analysis of KSHV latent and lytic proteins in NHOK cells. (D) KSHV BAC16-infected HOK16B, SCC23, Fadu, and SLK cells were also collected and submitted to Western blotting with antibodies against LANA, RTA, K8.1A, and actin.
KSHV infection in HOK16B cells.
Since primary NOHK cells grow slowly and have a limited life span in vitro, we carried out further characterization of KSHV infection in HOK16B cells. Figure 2A shows at 2 days postinfection, over 90% of the HOK16B cells were infected at an MOI of 3, and 100% of the cells infected at an MOI of 30 showed expression of GFP. Compared to the infection at an MOI of 3, stronger GFP fluorescence and an extensive cytopathic effect (CPE) with cell death in approximately half of the culture were observed at an MOI of 30. We also monitored infection at the high MOI over time (Fig. 2B). The infected cells experienced CPE and stopped proliferating from day 1 to day 4 after infection, and the cells that survived appeared to start growing again at day 5 postinfection.
FIG 2.

Infection of HOK16B cells by KSHV BAC16 recombinant viruses. (A) HOK16B cells were infected with KSHV BAC16 at an MOI of 3 or 30, when the confluence of cells reached 60%. Images were taken 2 days postinfection. (B) HOK16B cells were infected with KSHV BAC16 at an MOI of 30, and photographs of cells were taken on the indicated days postinfection. (C) Infectious progeny virus production from KSHV BAC16-infected HOK16B cells. The supernatants were harvested at the indicated times postinfection, and viral titers were determined on 293T cells as described in Materials and Methods. (D) Quantification of viral DNA replication. HOK16B cells were infected with KSHV BAC16 at an MOI of 30 with 0.1 mM phosphonoacetic acid (PAA) or control vehicle. Total DNA from infected cells was harvested on the indicated days, and viral DNA copy number was determined by quantitative PCR. The data shown are means ± standard deviations from three experiments.
As shown in Fig. 1, infection of HOK16B results in lytic gene expression. We then examined whether lytic KSHV gene expression in oral epithelial cells leads to the production and release of progeny virions into the supernatant. HOK16B cells were infected with KSHV BAC16 at an MOI of 3 or 30 for 2 h, and then the inoculum was removed by being washed with PBS three times. At the indicated time points after infection, the supernatants were collected and replaced with fresh medium. The amount of infectious virions in the supernatant was measured by incubating 293T cells with serial dilutions of the supernatant and then counting GFP-positive clusters. As shown in Fig. 2C, at an MOI of 30, the resulting viral titers in the supernatant reached the peak at day 1 postinfection, decreased gradually thereafter, and lasted for at least 5 days. No progeny KSHV was detected at an MOI of 3, which is consistent with the observation of a very low or undetectable lytic gene expression (Fig. 1). We also showed the increase in viral DNA within first 24 h after infection, and this increase was abolished by treatment with an inhibitor of viral DNA replication, phosphonoacetic acid (PAA) (Fig. 2D). Taken together, these results demonstrate that when infected at an MOI of 30, HOK16B cells can support the completion of KSHV lytic replication with the expression of immediate early, early, and late genes and the release of infectious progeny virions. Such lytic replication resulted in the observed cytopathic effect of infected cells. However, it is also clear that KSHV lytic replication does not occur in all of the infected cells.
Next, we examined the consequences of KSHV infection at an MOI of 3, which led to infection in over 90% of HOK16B cells, and most were latently infected (Fig. 1). Since KSHV BAC16 contains a hygromycin-resistant selection marker, we first selected the infected culture in the presence of 100 μg/ml of hygromycin, starting from the third day of infection for a month. After 1 month of hygromycin selection, all cells were GFP positive and did not show obvious morphological difference from normal, uninfected HOK16B cells. We then removed hygromycin from the growth medium and passaged the infected cells every 2 or 3 days to determine whether KSHV could be stably maintained in HOK16B cells. As shown in Fig. 3, as early as 5 days after hygromycin withdrawal, HOK16B cells started to lose KSHV BAC16, indicated by the decrease in the number of GFP-expressing cells. At 10 days after withdrawal, about 30% of the cells lost GFP fluorescence and LANA staining. The result indicates that similar to what has been previously observed in other cultured cell lines (29), KSHV does not establish stable latency in HOK16B cells.
FIG 3.
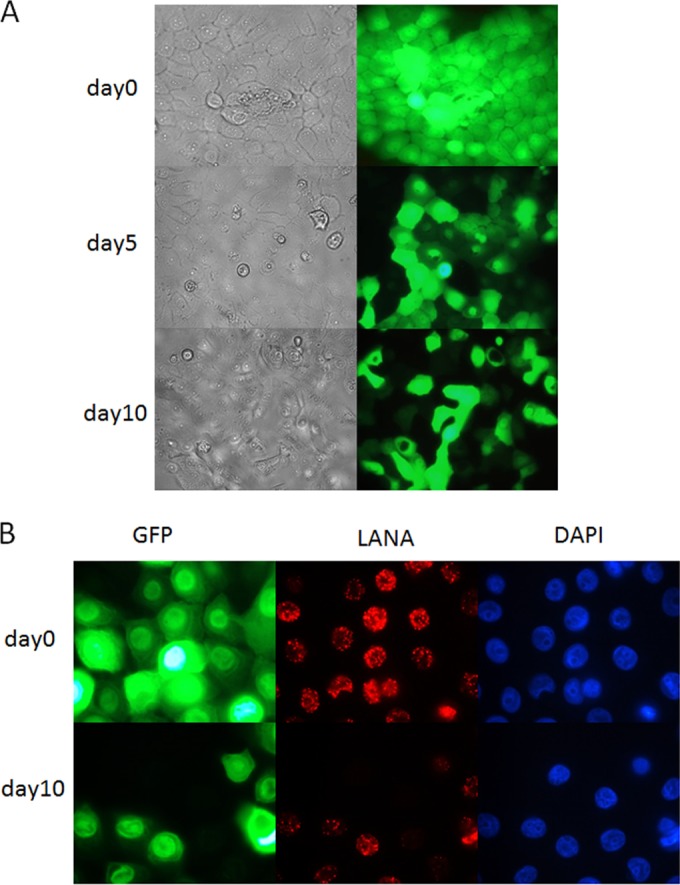
Analysis of KSHV latent infection in HOK16B cells. (A) HOK16B cells were infected with KSHV at an MOI of 3, followed by selection with 100 μg/ml hygromycin for 2 weeks. Then cells were cultured in medium without hygromycin, and images were taken on the indicated days after hygromycin withdrawal. (B) Immunofluorescence analysis of KSHV latent infection in HOK16B cells with antibodies against LANA. DAPI, 4′,6-diamidino-2-phenylindole.
Generation of ORF18- and ORF30-null KSHV mutants.
Like all herpesviruses, KSHV lytic gene expression is tightly regulated in a coordinated cascade fashion (30). Our previous studies in MHV-68 have shown that ORF18 and ORF30 play an important role in regulating late gene expression (15, 16). To study the functions of KSHV ORF18 and ORF30 in the context of viral replication, we constructed the mutant KSHV BAC plasmids to eliminate the expression of either ORF18 or ORF30 by introducing a stop codon near the N terminus of the coding region using the scarless mutagenesis strategy (described in Materials and Methods). A restriction site (XbaI or HindIII) was inserted next to the stop codon to facilitate the screening of the mutants (Fig. 4A). The respective revertants (18R and 30R) were also generated from the 18S and 30S BAC plasmids. All of the BAC plasmids were analyzed by digestions of XbaI or HindIII to ensure their overall integrity without gross changes other than the expected ones (Fig. 4B). Furthermore, direct sequencing of the modified region was also carried out to verify the flanking regions of the introduced Stop codons.
FIG 4.

Construction and analysis of KSHV mutant BACs. (A) Schematic illustration of KSHV BAC16 at the regions containing ORF18 and ORF30. A single stop codon was introduced into the coding region of ORF18 or ORF30. The XbaI and HindIII restriction sites were also underlined. (B) Restriction analysis of KSHV BACs. All BAC plasmids were digested with XbaI or HindIII overnight. Fragments different between WT KSHV BAC16 and mutants are indicated by arrowheads.
ORF18 and ORF30 are required for late gene expression during reactivation.
The BAC plasmids of the WT, null mutants, and revertants were individually introduced into iSLK-puro cells and selected by hygromycin to generate stable cells containing the BAC16-derived KSHV genome. iSLK-puro cells were previously generated to express RTA, the viral immediate early protein that drives the lytic replication of KSHV, upon doxycycline induction (23). Therefore, to produce the virus, the stable iSLK cells were treated with doxycycline and sodium butyrate, a chemical known to induce KSHV reactivation. While the levels of progeny virus production were comparable among iSLK-WT, iSLK-18R, and iSLK-30R cells, there was no detectable infectious virus from iSLK-18S or iSLK-30S cells (Fig. 5A). This result indicates that both ORF18 and ORF30 are required for KSHV virion production. To confirm this interpretation, we also introduced the 18S and 30S BAC plasmids into their respective iSLK-derived complementary cell lines, iSLK-kORF18 and iSLK-kORF30, which were generated to stably express ORF18 and ORF30, respectively. Indeed, virion production of 18S and 30S was rescued from iSLK-kORF18–18S and iSLK-kORF30–30S cells. To confirm that the rescued virions are not revertants, we purified the virion DNA and validated the presence of the desired mutation by both PCR-restriction enzyme digestion and sequencing analyses.
FIG 5.
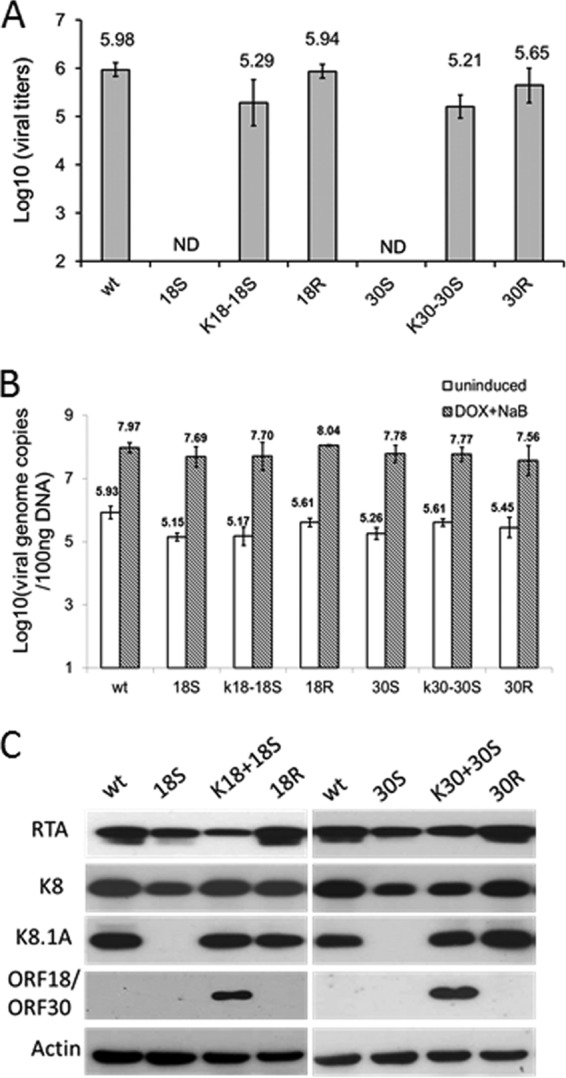
Characterization of KSHV-18S and -30S viruses during reactivation. (A) Virus production during reactivation. iSLK cells harboring WT KSHV BAC16 or mutants (KSHV-18S or -30S) were induced with 1 mM sodium butyrate plus 1 μg/ml doxycycline for 3 days, and titers of infectious virus in supernatant were determined on 293T cells. (B) Total DNA from the infected cells was harvested, and viral DNA copy number was determined by quantitative PCR. (C) Expression of viral proteins in the same set of infected cells was also analyzed by Western blotting with the indicated antibodies.
To determine at which stage of viral lytic replication the 18S and 30S mutants were blocked, we first examined viral DNA replication by real-time PCR to quantify the levels of viral genomes. We found that both 18S and 30S synthesized comparable amounts of viral DNA to the WT and revertant viruses after reactivation. Therefore, neither ORF18 nor ORF30 is required for viral lytic DNA replication (Fig. 5B). We then assessed the expression of viral IE, E, and L genes by Western blotting with antibodies against RTA, K8, and K8.1A. Both 18S and 30S produced levels of RTA and K8 similar to or slightly lower than those of the WT and revertant viruses. In contrast, neither of the mutant viruses expressed detectable K8.1A, which is a viral late gene product and was readily detected from the infection of the WT and the revertant viruses (Fig. 5C). Taken together, the results indicate that in the absence of ORF18 or ORF30, KSHV lytic replication is able to proceed normally with the expression of IE and E genes as well as viral lytic DNA synthesis, but KSHV is severely impaired in the expression of viral late genes, thereby resulting in no detectable virion production.
ORF18 and ORF30 are also required for late gene expression during de novo lytic replication.
As we were able to obtain 18S and 30S viruses from iSLK-kORF18–18S and iSLK-kORF30–30S cells, we sought to examine the function of ORF18 and ORF30 during KSHV de novo lytic replication in HOK16B cells. Cells were infected with the WT or mutant viruses at an MOI of 30, and virus production was analyzed. Both 18S and 30S produced more than 100-fold less infectious virions than the WT virus, and thus, ORF18 and ORF30 were both required for KSHV de novo productive infection in HOK16B cells (Fig. 6A). Since similar levels of expression of LANA were observed among all infections (Fig. 6C), the efficiency of 18S and 30S infection was comparable to that of the WT and revertant viruses. Moreover, lack of either ORF18 or ORF30 did not significantly affect viral lytic DNA synthesis (Fig. 6B) or the expression of RTA (IE) and K8 (E) (Fig. 6C). However, major deficiency in the production of viral late gene products (ORF26 and K8.1A) was seen for infection with 18S or 30S. We further examined the transcripts of viral genes by reverse transcription and real-time PCR. Similar to the protein results, only the transcripts of the viral late genes, including ORF52, ORF39, ORF27, and ORF26, were greatly reduced (Fig. 6D). In summary, ORF18 and ORF30 are required for de novo lytic replication in HOK16B cells and most likely function in regulating late gene transcription after viral DNA synthesis. Importantly, this result also shows that de novo infection of HOK16B can be used as a model to study the roles of KSHV genes during lytic replication, without using artificial chemicals, which potentially induce many signaling changes.
FIG 6.

Characterization of KSHV-18S and -30S viruses during de novo infection. (A) Virus production during de novo infection of HOK16B cells. HOK16B cells were infected with WT KSHV BAC16 virus or mutants (KSHV-18S or -30S) at an MOI of 30. Two days postinfection, titers of the infectious virus in the supernatant were determined on 293T cells. (B) Total DNA from the infected cells was harvested, and the viral DNA copy number was determined by quantitative PCR. (C) Expression of viral proteins in the same set of infected cells was also analyzed by Western blotting with the indicated antibodies. (D) Levels of viral transcripts were also measured by reverse transcription and real-time PCR with the indicated PCR primers, normalized to β-actin mRNA, and then presented as fold change over mRNA level of WT virus at 12 h postinfection. (E) HOK16B cells were infected with the WT KSHV or the two mutants at an MOI of 30 in the presence or absence of PAA. Six or 48 h after infection, titers of the infectious virus in the supernatant were determined on 293T cells. (F) Infected cells at 48 h postinfection were also collected, and levels of viral transcripts were measured by reverse transcription and real-time PCR with the indicated PCR primers, normalized to β-actin mRNA, and then presented as fold change over the mRNA level of WT virus.
While reactivation of 18S and 30S in SLK cells led to no virion production, de novo infection of these two mutant viruses in HOK16B cells still produced some infectious viruses. This result is also different from the observations in human cytomegalovirus (HCMV) and MHV-68 that similar mutants did not yield any virion (14–16, 20, 21). To determine whether the difference is intrinsic to KSHV de novo infection of HOK16B cells, we compared levels of virion production with and without PAA treatment (Fig. 6E and F). PAA is a known inhibitor of herpesviral DNA replication and is often used to define the viral late genes whose expression depends on viral DNA replication and thus is inhibited by PAA. PAA reduced virion production of WT to a level similar to those in the 18S and 30S viruses but did not completely abolish it. In addition, PAA did not further reduce the viral titers from 18S and from 30S infection. We also showed that this low level of virions detected in the presence of PAA or from infection of 18S and 30S is not from the inoculum since there was no infectious virus at 6 h postinfection. The analysis of viral transcripts confirmed that late gene transcription of the WT was indeed greatly diminished by PAA and as low as that during infection with 18S and 30S. Therefore, during both reactivation in iSLK cells and de novo infection of HOK16B cells, transcription of late genes is regulated by viral DNA replication and by ORF18 and ORF30. However, despite the severe reduction in late gene transcription, unlike reactivation in iSLK cells, there appear to be sufficient late gene products synthesized during de novo infection of HOK16B cells to support some virion production.
Genome-wide analysis of viral transcripts during de novo lytic replication and reactivation.
To globally examine how viral gene expression was affected by the lack of ORF18 or ORF30, we performed high-throughput RNA sequencing (RNA-seq). For de novo lytic replication, HOK16B cells were infected with the WT, 18S, or 30S virus (or the corresponding UV-treated viruses as a control) at an MOI of 30. Total RNA was harvested at 12, 24, and 48 h after infection. For reactivation, total RNA was isolated from iSLK-WT, iSLK-18S, or iSLK-30S cells 3 days after being treated with doxycycline plus NaB or the vehicle. The oligo(dT)-selected RNA was subjected to directional cDNA sequencing library construction. To discriminate reads among different samples, a unique barcode was introduced into each cDNA library generated from each RNA sample. Eighteen barcoded libraries were mixed in one lane and submitted to deep sequencing analysis on the Illumina 2000 platform. Raw reads were grouped by barcodes and then aligned to the KSHV ORF library. The transcript levels of individual viral genes were quantified in reads per kilobase of coding region per million total reads (RPKM) in the sample (see Table S1 in the supplemental material).
We first compared the transcriptional profiles of the WT between reactivation at 3 days after induction and de novo lytic replication at 48 h postinfection. As shown in Fig. 7A, reactivation in iSLK-puro cells had more robust lytic gene expression than de novo infection of HOK16B cells, and almost all of the viral genes showed greater expression (∼10-fold higher), except for K4. This gene expression result is consistent with more progeny viruses produced from iSLK-puro cells than infected HOK16B cells (Fig. 5A and 6A). Although the expression levels of individual viral genes are different between de novo infection and reactivation, the relative levels among viral genes are very similar (Fig. 7A and see Fig. 9A), indicating that KSHV employs the same mechanism to regulate its gene expression during de novo lytic replication and reactivation. It is not surprising to find that PAN, as the most abundant viral transcript, overwhelms the transcripts of all the other genes, and the number of reads for PAN is more than half of the total reads for all viral genes.
FIG 7.

Genome-wide analysis of KSHV transcripts by RNA deep sequencing. (A) Comparison of the transcription profiles of KSHV BAC16 during reactivation and de novo infection. Transcriptional levels of viral genes were quantified in reads per kilobase of coding region per million total read numbers (RPKM) in the sample. (B) Fold change of viral gene expression in the KSHV-18S and -30S viruses compared to the WT. The viral genes are ordered based on the decreasing fold reduction. The upper panel shows results from de novo lytic infection in HOK16B cells at 48 h postinfection, and the lower panel shows results from reactivation in iSLK cells.
FIG 9.

Correlation analysis of KSHV gene expression. (A) Comparison of WT, KSHV-18S, or KSHV-30S gene expression during reactivation and de novo infection (48 h). Plotted is the expression pattern correlation for all viral genes in two lytic replication systems. R2 is the coefficient of determination. (B) Comparison of levels of viral gene expression between KSHV-18S and -30S. Plotted is the expression pattern correlation for the fold decrease of all viral genes in KSHV-18S and -30S during reactivation or de novo lytic infection at 48 h.
Next we compared the level of viral gene expression of the 18S or 30S virus with that of the WT virus during both reactivation in iSLK cells and de novo lytic replication in HOK16B cells. The comparisons were displayed as fold changes and arranged according to the magnitude from largest to smallest (Fig. 7B; see Table S2 in the supplemental material). A general reduction in viral gene expression was observed for both the 18S and 30S viruses; however, the degrees varied among genes. The most downregulated viral genes are those encoding envelope glycoproteins, capsid proteins, DNA packaging proteins, and other virion-associated proteins, which are classical viral late genes (Table 1). While this result is consistent with the roles of ORF18 and ORF30 in regulating viral late genes, it is noticeable that the viral early transcripts, though to a lesser extent, were also reduced.
TABLE 1.
Top 20 genes downregulated in ORF18S or ORF30S virus
| Functional category | Gene(s) downregulated during: |
|||
|---|---|---|---|---|
|
De novo infection |
Reactivation |
|||
| KSHV-18S | KSHV-30S | KHSV-18S | KSHV-30S | |
| Envelope glycoproteins | K8.1, ORF53, ORF39, ORF8, ORF27 | K8.1, ORF53, ORF39, ORF8 | K8.1, ORF39, ORF27 | K8.1, ORF39, ORF27 |
| Capsid | ORF26, ORF62, ORF17 | ORF26, ORF17 | ORF26, ORF17, ORF25 | ORF26, ORF17, ORF25 |
| DNA packaging | ORF29, ORF32 | ORF32 | ORF29, ORF32 | ORF29, ORF32 |
| Tegument | ORF52, ORF75, ORF33 | ORF52, ORF75, ORF33, ORF45, ORF38, ORF42 | ORF52, ORF75, ORF33, ORF45, ORF38, ORF42 | ORF52, ORF75, ORF33, ORF45, ORF38 |
| Immune modulation | ORF4, K4.1, K4.2 | K9, ORF4, K4.1, K4.2 | K9, K4.2 | K9, K4.1, K4.2 |
| Others | K15, K14, ORF46, ORF31 | K15, ORF30, ORF31 | K15, ORF46, ORF30, ORF31 | K15, ORF46, ORF30, ORF31 |
Herpesviral genes are expressed in a highly ordered manner. Because it is difficult to synchronize lytic replication during reactivation, we examined the kinetics of viral gene expression during de novo infection of in HOK16B cells (see Table S2 and Fig. S1 in the supplemental material). In the WT-infected cells, some genes had already significant expression at 12 h, while others showed major increases in expression from 12 to 24 h postinfection. After 24 h, the expression of most genes further increased. In contrast, the transcripts of most viral genes in the 18S- or 30S-infected cells did not increase from 12 to 24 h postinfection, and many of the viral transcripts even decreased after 24 h. This probably accounts for the overall reduction in viral gene expression observed at late times for infection with 18S and 30S (Fig. 7B).
To better illustrate the different expression kinetics of viral genes, we created a graph using the percentage of maximal expression by dividing the RPKM value at each time point by the maximum. We classified the viral genes into three groups according to when their expression increased the most during WT infection (Fig. 8A). More than two-thirds of genes have most of their increases in expression before 24 h. Expression of the first group of genes increases rapidly after infection, and most of the increases in expression occur from 0 to 12 h. Group I includes the known viral immediately early and early genes, ORF50, K8, and ORF57, as well as those involved in DNA replication, ORF40, ORF44, ORF6, ORF56, and ORF59. The second group of genes were not expressed much prior to 12 h and had most of their increase in expression from 12 to 24 h. Group II consists mostly of the classical late genes that encode structural components of the virion, such as K8.1, ORF8, ORF25, ORF26, ORF39, ORF52, and ORF65. The third group of genes are those whose expression increased most from 24 to 48 h. Group III include several structural genes and assembly genes, ORF38, ORF29, ORF53, ORF62, ORF67, and ORF68, as well as PAN, K2 (coding for viral interleukin-6 [vIL-6]), K3, and K5. Despite some exceptions, overall, the expression patterns of viral genes are generally consistent with what would be predicted for their functions.
FIG 8.

Viral gene expression kinetics during de novo infection of HOK16B cells. (A) Gene expression levels of WT virus were plotted as percentages of maximal expression, and viral genes were divided into three groups based on when their expression increases the most among three time points—12, 24, or 48 h postinfection. (B) Fold change of viral gene expression in the 18S and 30S viruses compared to the WT in three different time points. The viral genes are ordered the same way as in panel A.
We next compared the gene expression of 18S or 30S virus with that of the WT virus (Fig. 8B). At 12 h postinfection, the differences in viral gene expression were small among three viruses, and for most of the viral genes, the expression was even slightly higher in the 18S- and 30S-infected cells than in WT-infected cells. At 24 h postinfection, the transcripts of several genes were significantly reduced for the 18S and 30S viruses, and importantly, most of the highly affected genes belong to group II. Furthermore, expression of almost every gene in group II was diminished greatly, while only a few genes in group I and group III were moderately affected in expression. In addition, many genes in group I and group III that had higher expression at 12 h postinfection in the 18S- and 30S-infected cells were still expressed more than in WT-infected cells at this time. However, at 48 h postinfection, we observed a global reduction across the three groups of genes. This across-the-board effect is most likely due to the general decrease in viral transcripts that occurred from 24 to 48 h during infection of 18S and 30S, while expression of most of genes still increased for the WT (see Fig. S1 and Table S1 in the supplemental material). Nevertheless, group II genes were still affected more than the genes in the other two groups.
The most downregulated viral genes during lytic replication of 18S and 30S (Fig. 7B) are those encoding envelope glycoproteins, capsid proteins, DNA packaging proteins, and other virion-associated proteins, which are traditionally regarded as the viral late genes (Table 1). Furthermore, most of these genes belong to group II, the expression of which increased largely from 12 to 24 h and which were also most affected by the lack of ORF18 or ORF30. Interestingly, certain viral genes that function to modulate the host immune response (ORF4, K9, K4.1, and K4.2) or angiogenesis (K15) (31–35) were also among the highly downregulated ones.
Both ORF18 and ORF30 play an important role in regulating viral late gene transcription, and the effects from the lack of either of them on viral gene expression appear similar (Fig. 7B). To quantitatively compare the effects from lacking ORF18 and ORF30, we plotted the folds of reduction in the viral transcripts between 18S and 30S infection. We found a correlation in the reduction of viral gene expression between these two mutants during both reactivation and de novo infection (Fig. 9B). Taken together, while neither ORF18 nor ORF30 is required for early gene expression or DNA replication, the RNA-Seq data support the conclusion that ORF18 and ORF30 are important for regulating the expression of a specific subset of viral genes that includes mostly the classical late genes that encode structural components of virions.
DISCUSSION
In this report, we describe a new cell culture model for de novo KSHV lytic replication based on an oral epithelial cell line, HOK16B. Infection of HOK16B cells with KSHV at an MOI of 3 led to latent infection in more than 90% of cells, with very few cells, if any, expressing detectable lytic genes. When HOK16B cells were infected with an MOI of 30, lytic gene expression could be detected in about half of the cells, with more than 20% of the cells expressing the viral late gene, resulting in a relatively large amount of progeny virions released. In addition to HOK16B cells, we also tested the susceptibility of NHOK cells (normal human oral keratinocytes) to KSHV infection. We found that NHOK cells can be infected by KSHV with a high efficiency as well and support lytic replication when infected at a high MOI.
Previous studies have shown that KSHV is able to infect multiple cell types in vitro, including epithelial cells, endothelial cells, monocytes, macrophages, hematopoietic progenitor cells, tonsillar IgMl-expressing B cells, and more, with the result of latent infection (29, 36–40). The lytic gene expression in these cells lines is inefficient after de novo infection, resulting in no or very few progeny virions released. Consequently, KSHV lytic replication is studied through reactivation, which requires one or more chemicals to artificially induce the lytic cycle. Thus, there is still a need for a cell line that can support robust de novo lytic replication of KSHV. The HOK16B cell line was created by human papillomavirus 16 (HPV-16) transfection of normal human oral keratinocytes (NHOK) (25). It is not fully transformed, maintains some features of primary cells, and has been used for studying pathogen-host interaction, innate immune response, differentiation, apoptosis, and tumorigenesis (41–49). As saliva is the major source of transmission for KSHV and epithelial cells may play an important role in producing infectious virions, we sought to identify an oral epithelial cell line that can support robust lytic replication of KSHV. Our studies support the use of HOK16B as a novel cell culture model to study KSHV lytic replication and virus-host interactions in oral epithelial cells, which may generate important knowledge regarding the events during KSHV transmission.
The regulation of IE and E gene expression in herpesviruses has been extensively studied, whereas the mechanisms governing L gene expression, especially how L gene expression is coupled with lytic DNA replication, are still not fully understood. In recent years, our group has identified five ORFs (ORF18, -24, -30, -31, and -34) in MHV-68 as critical regulators of viral late gene expression independent of viral DNA synthesis (14–17, 50). The exact molecular mechanisms of these five viral proteins remain to be determined. The homologue of MHV-68 ORF24 in EBV is BcRF1, and that in HCMV is UL87. Both BcRF1 and UL87 were postulated to be a potential viral TATA-binding protein (TBP) based on bioinformatics analysis (51). Recently, BcRF1 of EBV and MHV-68 ORF24 were both shown to be capable of binding the core promoter sequence of the viral late gene in vitro (52, 53). Previously, we carried out extensive mutagenesis of the viral late promoters of MHV-68 and found that the viral TATA boxes have unconventional requirements for the TATA sequence, supporting a virus-specific recognition of the TATA box (53). Furthermore, we showed that MHV-68 ORF24 is also able to distinguish the TATT sequence of a late gene promoter from the TATA sequence of an early gene promoter for in vitro binding. It is worth noting that although numerous viral proteins are conserved between all three subfamilies of herpesviruses, the homologues of the five late gene regulators identified in MHV-68 are encoded only by the beta and gamma subfamilies, suggesting alphaherpesviruses use a different set of viral genes to regulate late gene expression.
Except for ORF24, the homologues of the other four late gene regulators in human gammaherpesviruses have not been characterized. By employing the novel de novo lytic replication cell culture model described here and KSHV reactivation in iSLK cells, we studied the function of KSHV ORF18 and ORF30. Similar to what was found in MHV-68, both ORFs are not required for the expression of IE and E genes or for lytic viral DNA replication but are essential for viral late gene expression, demonstrating that both ORF18 and ORF30 of KSHV function as viral late gene regulators. Furthermore, this result supports the use of MHV-68 as a model organism for studying gammaherpesviruses. More studies are required to elucidate the underlying detailed mechanisms.
It is noticeable that elimination of the expression of ORF18 or ORF30 from KSHV while completely abolishing virion production during reactivation in iSLK cells still allows some viruses produced from infected HOK16B cells. However, treatment with PAA, a known inhibitor of herpesviral DNA replication, reduced virion production from de novo WT infection of HOK16B cells to a level similar to those in 18S and 30S infections without completely eliminating it. The incomplete block by PAA of virion production during de novo infection of HOK16B cells is an unexpected and yet interesting result, especially because there are less late gene transcripts and viruses in HOK16B cells compared to with reactivation in iSLK cells (Fig. 5A, 6A, and 7A). We further confirmed that PAA indeed greatly diminished late gene transcripts in the infected HOK16B cells, as one would expect. Therefore, the unusual outcome of PAA treatment likely results from the difference in the steps after the viral late genes are transcribed, such as their translation and assembly into virions. Understanding the precise reason will require more work in the future.
The efficient de novo KSHV lytic replication system also allows us to examine the highly ordered pattern of gene expression following infection. We normalized different levels of expression and fold change by displaying the results as percentages of maximal expression (Fig. 8A). We divided the viral genes into three groups based on when their expression increases the most among three time points—12, 24, and 48 h postinfection. The main constituents of group I and group II include classical early and late genes, respectively, validating our grouping analysis. Furthermore, we showed that lack of ORF18 or ORF30 affects the same set of viral transcripts, and many of the genes whose transcripts were reduced the most belong to group II. Previous studies have shown that unconventional TATT boxes are critical for late gene regulation of gammaherpesvirus (53–55). We have further defined the sequence requirement for the viral late gene core promoters using MHV-68 (53). Based on the MHV-68 study, we scanned the KSHV genome for distinct TATT sequences, TATTA/TA or TATATA, within 400 bp upstream of the translation start codon. Many of the genes that have these TATT sequences in their putative promoter regions are classical viral late genes, such as those encoding envelope glycoproteins, capsid proteins, DNA packaging proteins, and tegument proteins (Table 2). Consistently, most of them belong to group II, and their expression is severely affected in expression in 18S- and 30S-infected cells. Taken together, these analyses support a model in which ORF18 and ORF30 are part of the mechanism or pathway that coregulates the viral late genes as a group through ORF24 recognition of the unconventional TATT box. It is also notable that several genes that are not classified as the viral late genes, including ORF4, K9, and K15, also have TATTA/TA or TATATA sequences in their putative promoter regions, and their transcription is severely attenuated in the absence of ORF18 or ORF30. Thus, our result indicates that the regulation of those viral genes' expression may be more complicated than previously thought and should be further characterized in the future.
TABLE 2.
TATT sequences in the putative KSHV promoters
| Sequence type and gene | Sequencea | Location of initial T in TATT sequence |
|---|---|---|
| TATTAAA | ||
| K8.1 | CCGGCAGCAATATTAAAGGGACCGAAG | −48 |
| ORF33 | GGGGTTTTGCTATTAAAGGCCGCTATA | −56 |
| ORF26 | ATGAAGAGACTATTAAAGCTCGGAAAC | −57 |
| ORF4 | GTCGGATACTTATTAAAAGCATTGTCT | −60 |
| ORF38 | GTCAAATCAGTATTAAAGCCCGCTTCA | −348 |
| ORF25 | CACCGTCAATTATTAAACCTTCGCCGC | −307 |
| ORF22 | CCAATCCGGCTATTAAACCCAGGGCCA | −72 |
| ORF64 | TTCCTCCATCTATTAAACGTTCATATT | −156 |
| ORF65 | GAGGCTGCCCTATTAAAGCACCGTGAC | −109 |
| TATTTAA | ||
| ORF52 | TTCAATAAAGTATTTAAAGCTGGGTAT | −79 |
| K15 | TCCTAAAACGTATTTAAAAATCACAAA | −65 |
| ORF17 | GCGTGGAATGTATTTAAATTCTTATTT | −32 |
| ORF62 | TGGGCAACAGTATTTAAAAACTGCTCT | −167 |
| ORF27 | CTCAGTTCTATATTTAATCTTGGCCCC | −104 |
| ORF39 | CCGGGGCGCGTATTTAAAAGTCAACCG | −116 |
| ORF8 | TATGGCTGGATATTTAAAGACCTGTAC | −54 |
| ORF32 | AACAGTATGCTATTTAAATGCACAAAG | −120 |
| ORF19 | CCCCTGCTGATATTTAAGAATCAGAAA | −114 |
| TATATAA/G | ||
| ORF53 | TTGCAGAGTGTATATAAGAGGCGAACG | −61 |
| ORF42 | ACTCTTTGTGTATATAACCGACGTGGG | −52 |
| K9 | CTAGCCGTGATATATAGAGAGCCTGTC | −106 |
The specific TATT sequence type is underlined for clarity.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Basic Research Program of China (973 program, no. 2011CB504800), NSFC 2011CB504803 and 2011CB504300, NIH grants DE15752, CA91791, and AI078133, and the Stop Cancer Foundation.
Footnotes
Published ahead of print 23 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00793-14.
REFERENCES
- 1.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191. 10.1056/NEJM199505043321802 [DOI] [PubMed] [Google Scholar]
- 2.Boshoff C, Weiss R. 2002. AIDS-related malignancies. Nat. Rev. Cancer 2:373–382. 10.1038/nrc797 [DOI] [PubMed] [Google Scholar]
- 3.Corbellino M, Poirel L, Aubin JT, Paulli M, Magrini U, Bestetti G, Galli M, Parravicini C. 1996. The role of human herpesvirus 8 and Epstein-Barr virus in the pathogenesis of giant lymph node hyperplasia (Castleman's disease). Clin. Infect. Dis. 22:1120–1121. 10.1093/clinids/22.6.1120 [DOI] [PubMed] [Google Scholar]
- 4.Boldogh I, Szaniszlo P, Bresnahan WA, Flaitz CM, Nichols MC, Albrecht T. 1996. Kaposi's sarcoma herpesvirus-like DNA sequences in the saliva of individuals infected with human immunodeficiency virus. Clin. Infect. Dis. 23:406–407. 10.1093/clinids/23.2.406 [DOI] [PubMed] [Google Scholar]
- 5.Koelle DM, Huang ML, Chandran B, Vieira J, Piepkorn M, Corey L. 1997. Frequent detection of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) DNA in saliva of human immunodeficiency virus-infected men: clinical and immunologic correlates. J. Infect. Dis. 176:94–102. 10.1086/514045 [DOI] [PubMed] [Google Scholar]
- 6.Pauk J, Huang ML, Brodie SJ, Wald A, Koelle DM, Schacker T, Celum C, Selke S, Corey L. 2000. Mucosal shedding of human herpesvirus 8 in men. N. Engl. J. Med. 343:1369–1377. 10.1056/NEJM200011093431904 [DOI] [PubMed] [Google Scholar]
- 7.Vieira J, Huang ML, Koelle DM, Corey L. 1997. Transmissible Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in saliva of men with a history of Kaposi's sarcoma. J. Virol. 71:7083–7087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Franca TR, de Araujo RA, Ribeiro CM, Leao JC. 2011. Salivary shedding of HHV-8 in people infected or not by human immunodeficiency virus 1. J. Oral Pathol. Med. 40:97–102. 10.1111/j.1600-0714.2010.00959.x [DOI] [PubMed] [Google Scholar]
- 9.Duus KM, Lentchitsky V, Wagenaar T, Grose C, Webster-Cyriaque J. 2004. Wild-type Kaposi's sarcoma-associated herpesvirus isolated from the oropharynx of immune-competent individuals has tropism for cultured oral epithelial cells. J. Virol. 78:4074–4084. 10.1128/JVI.78.8.4074-4084.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson AS, Maronian N, Vieira J. 2005. Activation of Kaposi's sarcoma-associated herpesvirus lytic gene expression during epithelial differentiation. J. Virol. 79:13769–13777. 10.1128/JVI.79.21.13769-13777.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson MP, Kurzrock R. 2004. Epstein-Barr virus and cancer. Clin. Cancer Res. 10:803–821. 10.1158/1078-0432.CCR-0670-3 [DOI] [PubMed] [Google Scholar]
- 12.Sixbey JW, Nedrud JG, Raab-Traub N, Hanes RA, Pagano JS. 1984. Epstein-Barr virus replication in oropharyngeal epithelial cells. N. Engl. J. Med. 310:1225–1230. 10.1056/NEJM198405103101905 [DOI] [PubMed] [Google Scholar]
- 13.Hadinoto V, Shapiro M, Sun CC, Thorley-Lawson DA. 2009. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog. 5:e1000496. 10.1371/journal.ppat.1000496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong E, Wu TT, Reyes N, Deng H, Sun R. 2007. Murine gammaherpesvirus 68 open reading frame 24 is required for late gene expression after DNA replication. J. Virol. 81:6761–6764. 10.1128/JVI.02726-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arumugaswami V, Wu TT, Martinez-Guzman D, Jia Q, Deng H, Reyes N, Sun R. 2006. ORF18 is a transfactor that is essential for late gene transcription of a gammaherpesvirus. J. Virol. 80:9730–9740. 10.1128/JVI.00246-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu TT, Park T, Kim H, Tran T, Tong L, Martinez-Guzman D, Reyes N, Deng H, Sun R. 2009. ORF30 and ORF34 are essential for expression of late genes in murine gammaherpesvirus 68. J. Virol. 83:2265–2273. 10.1128/JVI.01785-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jia Q, Wu TT, Liao HI, Chernishof V, Sun R. 2004. Murine gammaherpesvirus 68 open reading frame 31 is required for viral replication. J. Virol. 78:6610–6620. 10.1128/JVI.78.12.6610-6620.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Omoto S, Mocarski ES. 2013. Cytomegalovirus UL91 is essential for transcription of viral true late (gamma 2) genes J. Virol. 87:8651–8664. 10.1128/JVI.01052-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isomura H, Stinski MF, Murata T, Yamashita Y, Kanda T, Toyokuni S, Tsurumi T. 2011. The human cytomegalovirus gene products essential for late viral gene expression assemble into prereplication complexes before viral DNA replication. J. Virol. 85:6629–6644. 10.1128/JVI.00384-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Omoto S, Mocarski ES. 2014. Transcription of true late (γ2) cytomegalovirus genes requires UL92 function that is conserved among beta- and gammaherpesviruses. J. Virol. 88:120–130. 10.1128/JVI.02983-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chapa TJ, Perng YC, French AR, Yu D. 2014. Murine cytomegalovirus protein pM92 is a conserved regulator of viral late gene expression. J. Virol. 88:131–142. 10.1128/JVI.02684-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chapa TJ, Johnson LS, Affolter C, Valentine MC, Fehr AR, Yokoyama WM, Yu D. 2013. Murine cytomegalovirus protein pM79 is a key regulator for viral late transcription. J. Virol. 87:9135–9147. 10.1128/JVI.00688-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 86:9708–9720. 10.1128/JVI.01019-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J. Virol. Methods 174:12–21. 10.1016/j.jviromet.2011.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park NH, Min BM, Li SL, Huang MZ, Cherick HM, Doniger J. 1991. Immortalization of normal human oral keratinocytes with type 16 human papillomavirus. Carcinogenesis 12:1627–1631. 10.1093/carcin/12.9.1627 [DOI] [PubMed] [Google Scholar]
- 26.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. 634:421–430. 10.1007/978-1-60761-652-8_30 [DOI] [PubMed] [Google Scholar]
- 27.Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cerimele F, Curreli F, Ely S, Friedman-Kien AE, Cesarman E, Flore O. 2001. Kaposi's sarcoma-associated herpesvirus can productively infect primary human keratinocytes and alter their growth properties. J. Virol. 75:2435–2443. 10.1128/JVI.75.5.2435-2443.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lagunoff M, Bechtel J, Venetsanakos E, Roy AM, Abbey N, Herndier B, McMahon M, Ganem D. 2002. De novo infection and serial transmission of Kaposi's sarcoma-associated herpesvirus in cultured endothelial cells. J. Virol. 76:2440–2448. 10.1128/jvi.76.5.2440-2448.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schulz TF, Chang Y. 2007. KSHV gene expression and regulation, chapter 28 In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: http://www.ncbi.nlm.nih.gov/books/NBK47415/ [PubMed] [Google Scholar]
- 31.Bala K, Bosco R, Gramolelli S, Haas DA, Kati S, Pietrek M, Havemeier A, Yakushko Y, Singh VV, Dittrich-Breiholz O, Kracht M, Schulz TF. 2012. Kaposi's sarcoma herpesvirus K15 protein contributes to virus-induced angiogenesis by recruiting PLCgamma1 and activating NFAT1-dependent RCAN1 expression. PLoS Pathog. 8:e1002927. 10.1371/journal.ppat.1002927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spiller OB, Robinson M, O'Donnell E, Milligan S, Morgan BP, Davison AJ, Blackbourn DJ. 2003. Complement regulation by Kaposi's sarcoma-associated herpesvirus ORF4 protein. J. Virol. 77:592–599. 10.1128/JVI.77.1.592-599.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao SJ, Boshoff C, Jayachandra S, Weiss RA, Chang Y, Moore PS. 1997. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene 15:1979–1985. 10.1038/sj.onc.1201571 [DOI] [PubMed] [Google Scholar]
- 34.Coscoy L. 2007. Immune evasion by Kaposi's sarcoma-associated herpesvirus. Nat. Rev. Immunol. 7:391–401. 10.1038/nri2076 [DOI] [PubMed] [Google Scholar]
- 35.Lee HR, Lee S, Chaudhary PM, Gill P, Jung JU. 2010. Immune evasion by Kaposi's sarcoma-associated herpesvirus. Future Microbiol. 5:1349–1365. 10.2217/fmb.10.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao SJ, Deng JH, Zhou FC. 2003. Productive lytic replication of a recombinant Kaposi's sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J. Virol. 77:9738–9749. 10.1128/JVI.77.18.9738-9749.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hassman LM, Ellison TJ, Kedes DH. 2011. KSHV infects a subset of human tonsillar B cells, driving proliferation and plasmablast differentiation. J. Clin. Invest. 121:752–768. 10.1172/JCI44185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bechtel JT, Liang Y, Hvidding J, Ganem D. 2003. Host range of Kaposi's sarcoma-associated herpesvirus in cultured cells. J. Virol. 77:6474–6481. 10.1128/JVI.77.11.6474-6481.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu W, Vieira J, Fiore N, Banerjee P, Sieburg M, Rochford R, Harrington W, Jr, Feuer G. 2006. KSHV/HHV-8 infection of human hematopoietic progenitor (CD34+) cells: persistence of infection during hematopoiesis in vitro and in vivo. Blood 108:141–151. 10.1182/blood-2005-04-1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blackbourn DJ, Lennette E, Klencke B, Moses A, Chandran B, Weinstein M, Glogau RG, Witte MH, Way DL, Kutzkey T, Herndier B, Levy JA. 2000. The restricted cellular host range of human herpesvirus 8. AIDS 14:1123–1133. 10.1097/00002030-200006160-00009 [DOI] [PubMed] [Google Scholar]
- 41.Ji S, Kim Y, Min BM, Han SH, Choi Y. 2007. Innate immune responses of gingival epithelial cells to nonperiodontopathic and periodontopathic bacteria. J. Periodontal Res. 42:503–510. 10.1111/j.1600-0765.2007.00974.x [DOI] [PubMed] [Google Scholar]
- 42.Shin JE, Baek KJ, Choi YS, Choi Y. 2013. A periodontal pathogen Treponema denticola hijacks the Fusobacterium nucleatum-driven host response. Immunol. Cell Biol. 91:503–510. 10.1038/icb.2013.35 [DOI] [PubMed] [Google Scholar]
- 43.Shin JE, Choi Y. 2010. Treponema denticola suppresses expression of human beta-defensin-2 in gingival epithelial cells through inhibition of TNFalpha production and TLR2 activation. Mol. Cells 29:407–412. 10.1007/s10059-010-0048-5 [DOI] [PubMed] [Google Scholar]
- 44.Huang GT, Zhang X, Park NH. 2000. Increased ICAM-1 expression in transformed human oral epithelial cells: molecular mechanism and functional role in peripheral blood mononuclear cell adhesion and lymphokine-activated-killer cell cytotoxicity. Int. J. Oncol. 17:479–486. 10.3892/ijo.17.3.479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park NH, Gujuluva CN, Baek JH, Cherrick HM, Shin KH, Min BM. 1995. Combined oral carcinogenicity of HPV-16 and benzo(a)pyrene: an in vitro multistep carcinogenesis model. Oncogene 10:2145–2153 [PubMed] [Google Scholar]
- 46.Vigneswaran N, Baucum DC, Wu J, Lou Y, Bouquot J, Muller S, Zacharias W. 2007. Repression of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) but not its receptors during oral cancer progression. BMC Cancer 7:108. 10.1186/1471-2407-7-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rey O, Baluda MA, Park NH. 1999. Differential gene expression in neoplastic and human papillomavirus-immortalized oral keratinocytes. Oncogene 18:827–831. 10.1038/sj.onc.1202328 [DOI] [PubMed] [Google Scholar]
- 48.Oh KJ, Kalinina A, Park NH, Bagchi S. 2006. Deregulation of eIF4E: 4E-BP1 in differentiated human papillomavirus-containing cells leads to high levels of expression of the E7 oncoprotein. J. Virol. 80:7079–7088. 10.1128/JVI.02380-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oh JE, Kim RH, Shin KH, Park NH, Kang MK. 2011. DeltaNp63alpha protein triggers epithelial-mesenchymal transition and confers stem cell properties in normal human keratinocytes. J. Biol. Chem. 286:38757–38767. 10.1074/jbc.M111.244939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong Y, Qi J, Gong D, Han C, Deng H. 2011. Replication and transcription activator (RTA) of murine gammaherpesvirus 68 binds to an RTA-responsive element and activates the expression of ORF18. J. Virol. 85:11338–11350. 10.1128/JVI.00561-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wyrwicz LS, Rychlewski L. 2007. Identification of herpes TATT-binding protein. Antiviral Res. 75:167–172. 10.1016/j.antiviral.2007.03.002 [DOI] [PubMed] [Google Scholar]
- 52.Gruffat H, Kadjouf F, Mariame B, Manet E. 2012. The Epstein-Barr virus BcRF1 gene product is a TBP-like protein with an essential role in late gene expression. J. Virol. 86:6023–6032. 10.1128/JVI.00159-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong-Ho E, Wu TT, Davis ZH, Zhang B, Huang J, Gong H, Deng H, Liu F, Glaunsinger B, Sun R. 2014. Unconventional sequence requirement for viral late gene core promoters of murine gammaherpesvirus 68. J. Virol. 88:3411–3422. 10.1128/JVI.01374-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Serio TR, Cahill N, Prout ME, Miller G. 1998. A functionally distinct TATA box required for late progression through the Epstein-Barr virus life cycle. J. Virol. 72:8338–8343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Amon W, Binne UK, Bryant H, Jenkins PJ, Karstegl CE, Farrell PJ. 2004. Lytic cycle gene regulation of Epstein-Barr virus. J. Virol. 78:13460–13469. 10.1128/JVI.78.24.13460-13469.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.