

ABSTRACT
Gammaherpesviruses display tropism for B cells and, like all known herpesviruses, exhibit distinct lytic and latent life cycles. One well-established observation among members of the gammaherpesvirus family is the link between viral reactivation from latently infected B cells and plasma cell differentiation. Importantly, a number of studies have identified a potential role for a CREB/ATF family member, X-box binding protein 1 (XBP-1), in trans-activating the immediate early BZLF-1 or BRLF1/gene 50 promoters of Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV), respectively. XBP-1 is required for the unfolded protein response and has been identified as a critical transcription factor in plasma cells. Here, we demonstrate that XBP-1 is capable of trans-activating the murine gammaherpesvirus 68 (MHV68) RTA promoter in vitro, consistent with previous observations for EBV and KSHV. However, we show that in vivo there does not appear to be a requirement for XBP-1 expression in B cells for virus reactivation. The MHV68 M2 gene product under some experimental conditions plays an important role in virus reactivation from B cells. M2 has been shown to drive B cell differentiation to plasma cells, as well as interleukin-10 (IL-10) production, both of which are dependent on M2 induction of interferon regulatory factor 4 (IRF4) expression. IRF4 is required for plasma cell differentiation, and consistent with a role for plasma cells in MHV68 reactivation from B cells, we show that IRF4 expression in B cells is required for efficient reactivation of MHV68 from splenocytes. Thus, the latter analyses are consistent with previous studies linking plasma cell differentiation to MHV68 reactivation from B cells. The apparent independence of MHV68 reactivation from XBP-1 expression in plasma cells may reflect redundancy among CREB/ATF family members or the involvement of other plasma cell-specific transcription factors. Regardless, these findings underscore the importance of in vivo studies in assessing the relevance of observations made in tissue culture models.
IMPORTANCE All known herpesviruses establish a chronic infection of their respective host, persisting for the life of the individual. A critical feature of these viruses is their ability to reactivate from a quiescent form of infection (latency) and generate progeny virus. In the case of gammaherpesviruses, which are associated with the development of lymphoproliferative disorders, including lymphomas, reactivation from latently infected B lymphocytes occurs upon terminal differentiation of these cells to plasma cells—the cell type that produces antibodies. A number of studies have linked a plasma cell transcription factor, XBP-1, to the induction of gammaherpesvirus reactivation, and we show here that indeed in tissue culture models this cellular transcription factor can trigger expression of the murine gammaherpesvirus gene involved in driving virus reactivation. However, surprisingly, when we examined the role of XBP-1 in the setting of infection of mice—using mice that lack a functional XBP-1 gene in B cells—we failed to observe a role for XBP-1 in virus reactivation. However, we show that another cellular factor essential for plasma cell differentiation, IRF4, is critical for virus reactivation. Thus, these studies point out the importance of studies in animal models to validate findings from studies carried out in cell lines passaged in vitro.
INTRODUCTION
Gammaherpesviruses are lymphotropic viruses that maintain distinct lytic and latent life cycles (1). Primary infections are generally asymptomatic in immunocompetent hosts, although Epstein-Barr virus (EBV) acute infection can result in the self-limiting lymphoproliferative syndrome infectious mononucleosis in young adults. Furthermore, EBV is associated with the development of several cancers in humans—including endemic Burkitt's lymphoma, nasopharyngeal carcinoma, and some Hodgkin's lymphomas (2–8). The other known human gammaherpesvirus, Kaposi's sarcoma-associated herpesvirus (KSHV), has also been linked to various cancers—most notably primary effusion lymphoma (PEL) and multicentric Castleman's disease (9–12). Murine gammaherpesvirus 68 (MHV68) infection of laboratory strains of inbred mice has been extensively studied to characterize basic aspects of gammaherpesvirus pathogenesis in vivo. Similar to the human viruses, MHV68 has been associated with the induction of B cell lymphomas in immunosuppressed mice (13, 14) and has been used to immortalize fetal liver-derived B cells in vitro (15).
Members of the gammaherpesvirus family predominantly infect and maintain latency in B cells (5, 16–18). In fact, persistence of the latency reservoir is postulated to be dependent on viral reactivation from latency. Several reports have indicated a close link between plasma cell (PC) differentiation and viral reactivation from latency (19–21). Previous work from our lab has demonstrated a distinct requirement for Blimp-1-mediated plasma cell differentiation in viral reactivation, long-term maintenance of latency, and persistence of long-term MHV68-specific antibody responses (22). Recently, a strong link between plasma cell transcription factor X-box binding protein 1 (XBP-1) and viral reactivation has been reported for both EBV (23, 24) and KSHV (25–27). These studies demonstrated that overexpression of XBP-1s in latently infected EBV or KSHV cell lines could induce production of the BZLF-1 and ORF50 gene products, respectively, transcription factors which have been shown to induce viral reactivation from latency (28–30). Additionally, these data collectively demonstrate that XBP-1s binds to specific residues in the BZLF-1 and RTA promoters, as well as synergizing with RTA expression.
XBP-1, a basic region leucine zipper (bZIP) transcription factor (31), has been shown to be essential for plasma cell function (32, 33). XBP-1 is a member of the CREB/ATF family of transcription factors that was initially discovered due to its ability to bind cyclic AMP (cAMP) response sequences in the major histocompatibility complex (MHC) class II human gene locus (31). Functionally, it plays an integral role in mediating the unfolded protein response (UPR) in the endoplasmic reticulum. Initially described in yeast, inositol-requiring enzyme 1 (IRE-1) is a transmembrane protein with kinase and endonuclease activity. It is activated in response to the accumulation of unfolded proteins and chaperones during cellular stress. Oligomerization and transautophosphorylation activate the endoribonuclease function of IRE-1. The only identified substrates for IRE-1 processing are the homologs Hac1 (yeast) and XBP-1 (metazoans). IRE-1 activation results in the removal of a 26-nucleotide intron in the XBP-1 transcript, changing it from the unspliced form XBP-1u (inhibitor of the UPR) to the spliced form XBP-1s (activator of the UPR). XBP-1 splicing results in the acquisition of a C-terminal transactivation domain (34–36). As a result, XBP-1s targets expression of UPR genes, such as those for chaperone proteins, that mediate a survival response in the stressed cell. B cell differentiation into a plasma cell results in large quantities of immunoglobulin production, which induces the UPR. Additionally, XBP-1 expression is an integral component of mediating the UPR during plasma cell differentiation and permits immunoglobulin (Ig) secretion (32, 33, 37).
This direct interaction between a plasma cell host transcription factor and the immediate early gene promoter of gene 50 indicates a viral evolutionary adaptation that senses changes in the viability of the host cell and signals an escape response, such as reactivation. However, due to the species-specific nature of the human gammaherpesviruses, there are no direct in vivo studies addressing the role of XBP-1 in plasma cell differentiation-mediated reactivation. As such, using MHV68 as a model system, we have evaluated the in vivo implications of loss of XBP-1 expression specifically in B cells on MHV68 latency and reactivation. A valuable transgenic model which selectively deletes XBP-1 expression from B cells has been previously described (38) and used in this study. Loss of XBP-1 expression in B cells was demonstrated to have no effect on germinal center or plasma cell frequencies but showed a defect in immunoglobulin secretion from plasma cells (38). Here, we report that XBP-1s transactivation of the MHV68 gene 50 proximal promoter can be observed in vitro, consistent with studies on the impact of XBP-1 expression on EBV and KSHV immediate early gene expression (23–27). However, we did not observe a requirement for B cell-specific XBP-1 expression to promote viral reactivation or maintenance of MHV68 latency in vivo. This unexpected observation points to the value of in vivo pathogenesis models, which help reveal the often complex and perhaps redundant network of host transcription factors that can modulate the viral life cycle. Additionally, in contrast to the apparent lack of a role for XBP-1 in MHV68 reactivation, we show that another host transcription factor, IRF4—which is essential for plasma cell differentiation—plays a critical role in MHV68 reactivation from B cell latency. Notably, IRF4 has been implicated in EBV transformation of human B lymphocytes (39). However, the role of IRF4 in reactivation from B cells for the human gammaherpesviruses still remains unknown.
MATERIALS AND METHODS
Promoters and expression vectors.
The proximal ORF50 promoter (position 66142 to 66552 in viral genome) of MHV68 was cloned into a pGL4.10[luc 2] vector from Promega (pGL410-PpORF50). Primers used to amplify and clone the proximal promoter include forward primer 5′TCAGGGATTCAGCCAACAA3′ and reverse primer 5′AAGGTGGTGGTTGCCAGC3′. NheI and EcoRV restriction enzymes were used to clone the promoter upstream of the luciferase expression construct. The XBP-1s expression vector was a kind gift of Xiaozhan Liang and was initially cloned into the murine stem cell virus (MSCV)-internal ribosome entry site (IRES)-green fluorescent protein (GFP) vector (Addgene). For experiments outlined in this work, the XBP-1s insert was cloned into the pCMV.Tag2b (Addgene) vector using the following primers: F-XBP-1s-BglII, 5′ccatcgaagatctATGGTGGTGGTGGCAGCGG3′, and R-XBP-1s-PstI, 5′ccatcgactgcagTTAGAGGCTTGGTGTATACATGGTCA3′, where lowercase letters indicate random nucleotides and restriction enzyme sequences and uppercase letters represent the complement of the gene that is targeted for amplification.
Tissue culture and promoter assays.
The M12 B cell murine lymphoma cell line was used for promoter assays. Cells were plated in 6-well Corning tissue culture dishes at a density of 2.5 × 105 cells/2 ml of medium 12 h prior to transfection. The lipid-based transfection was performed using the Transfectin lipid reagent (Bio-Rad). The protocol was designed based on the manufacturer's instructions. Briefly, a total of 2 μg of DNA (pGL410-PpORF50 plus XBP-1s.pCMV.Tag2b) was added to 7 μl of the lipid reagent and diluted in neat Dulbecco modified Eagle medium (DMEM). The mixture was allowed to incubate at room temperature for 20 min. Five hundred microliters of the master mix was dripped onto the plated B cells and gently swirled. At 12 h posttransfection, cells were treated with 20 ng/ml of 12-O-tetradecanoylphorbol-13-acetate (TPA) and/or a final concentration of 0.5 μM ionomycin. Cells were allowed to incubate for an additional 12 h before harvesting and analysis of luciferase activity (Promega luciferase assay system).
Animals and infections.
XBP-1Flox/Flox CD19CRE/+ C57BL/6 mice were a kind gift of Neal Iwakoshi. The mice were generated as previously described (38) and were bred in-house. In order to confirm the LoxP site at the XBP-1 exon 2 locus, we used the following primers: forward 3′loxS, 5′ACTTGCACCAACACTTGCCATTTC3′, and reverse 3′loxA, 5′CAAGGTGGTTCACTGCCTGTAATG3′. In order to confirm CD19-CRE recombinase insertion, we used the following primers: forward primer CRE-F, 5′GCGGTCTGGCAGTAAAAACTATC3′, and reverse primer CRE-R, 5′GTGAAACAGCATTGCTGTCACTT3′. As littermate (LM) controls in our experiments, we used XBP-1fl/fl CD19+/+ mice that contain the floxed allele without Cre expression.
IRF4fl/fl mice and CD19Cre/Cre mice were purchased from Jackson Labs (catalog numbers 009380 and 006785, respectively). IRF4fl/fl mice were crossed with CD19+/CRE mice to generate Cre-expressing mice that were homozygous for the conditional IRF4 allele. The resulting IRF4fl/fl CD19Cre/+ mice express Cre recombinase under the CD19 promoter, thus lacking IRF4 expression in CD19-expressing B cells. As littermate controls in our experiments, we used IRF4fl/fl CD19+/+ mice that contain the floxed allele without Cre expression. Genotyping was performed as described in the Jackson Labs protocol. Additionally, the IRF4fl/fl mice contain an enhanced GFP (EGFP) cassette flanking the LoxP sites, such that Cre-mediated excision of the floxed alleles results in EGFP expression. Therefore, mice were confirmed by genotyping of ear tissue followed by flow cytometry analysis on peripheral blood to confirm expression of EGFP expression in the knockouts (KOs).
Animals were housed at the Emory University Division of Animal Resources in accordance with guidelines specified by the Institutional Animal Care and Use Committee at Emory University (IACUC; protocol number YER-2002245-031416GN). Mice were infected at 8 to 12 weeks of age. Animals were anesthetized with isoflurane prior to infection. The MHV68-H2bYFP virus (40) was administered intranasally in a 20-μl volume at a dose of 1,000 PFU. For infections of IRF4fl/fl CD19+/Cre and IRF4fl/fl CD19+/+ mice, the M2stop.HY and M2MR.HY viruses described in reference 41 were used as indicated. Animals were sacrificed between days 16 and 18 postinfection for analysis of splenic populations.
Limiting dilution analysis.
Limiting dilution PCR (LD-PCR) was used to quantify latent levels of viral genome as previously described (42). Briefly, frozen splenocytes were thawed (after storage at −80°C in complete DMEM (cDMEM; DMEM to which fetal bovine serum [FBS], l-glutamine, penicillin, and streptomycin have been added) plus 10% dimethyl sulfoxide), washed in an isotonic buffer, and plated in a 96-well PCR plate at a starting density of 104 splenocytes and serially diluted 3-fold on a background of uninfected 3T12 cells. Each dilution underwent 12 PCRs, and each sample was serially diluted 6 times. Samples were digested with proteinase K overnight (8 to 12 h), followed by two rounds of nested PCR. Standards included 0, 0.1, 1, and 10 copies of a control DNA plasmid to determine viral DNA copy number. Samples were then run on a 2% agarose gel and stained with ethidium bromide. Wells positive for viral genome at each dilution were plotted using the Poisson distribution. A limiting dilution reactivation assay was performed to evaluate the frequency of viral genome reactivation from latency (42). Briefly, 5 × 106 splenocytes were explanted on a monolayer of mouse embryonic fibroblasts (MEFs) in a 96-well format. Splenocytes were serially diluted 2-fold in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Cells were plated in 24 wells per dilution and diluted up to 12 dilutions. After 14 to 21 days of incubation, wells were monitored for cytopathic effect (CPE) and frequencies were plotted using the Poisson distribution. As a control, splenocytes were physically disrupted to release preformed virus using 0.5-mm silicon beads in a bead beater (beating for 1 min 4 times and resting on ice after every disruption cycle for 1 min). Homogenates were plated on MEFs as described above.
Antibodies.
Splenocytes were blocked with anti-CD16/32 (BD Bioscience). Surface stains were performed in phosphate-buffered saline (PBS)–2% FBS–1 mM EDTA for 20 min on ice. Markers used were CD138-phycoerythrin (PE) (BD Bioscience), B220-Pacific Blue (BioLegend), CD95-PE-Cy7 (BD Biosciences), GL7-Alexa Fluor 660 (eBioscience), and CD3/4/8-peridinin chlorophyll protein (PerCP) (BD Bioscience). Fixable LIVE/DEAD stains in Zombie Yellow (Pacific Orange) were purchased from BioLegend and used according to the manufacturer's guidelines.
ELISA.
Blood was collected during terminal bleeds. Serum was collected by allowing the blood to clot at 4°C for 1 h. Blood was centrifuged at 14,000 rpm at 4°C for 2 min. Serum was collected and frozen at −80°C for long-term storage. An enzyme-linked immunosorbent assay (ELISA) protocol was used as previously described (43) to measure global IgG and IgM levels. Briefly, 96-well Nunc Immuno MaxiSorp ELISA plates were coated with 0.5 μg/well of goat anti-mouse IgG or IgM antibody (Southern Biotech). Serum was serially diluted (3-fold, beginning at 1:100), and 6 dilutions were plated for each sample. Alkaline phosphatase-conjugated goat anti-mouse IgG or IgM (Southern Biotech) was used as a secondary antibody. Color was developed using p-nitrophenyl phosphate (Sigma) in a diethanolamine substrate buffer. Absorbance at 405 nm was read on a BioTek Synergy HT reader.
RESULTS
Murine XBP-1s can transactivate the MHV68 ORF50 proximal promoter in vitro.
Previous reports indicated the potential for the active spliced form of XBP-1 to transactivate the BZLF-1 promoter in EBV (23, 24) and RTA (encoded by gene 50) in KSHV (25–27). To evaluate whether murine XBP-1s could regulate gene 50/RTA expression in MHV68, we transfected the murine M12 B lymphoma cell line with an expression construct expressing XBP-1s in conjunction with a luciferase reporter construct driven by the proximal gene 50 promoter (PpORF50) (Fig. 1). Cells were incubated for 12 h posttransfection in the presence and absence of treatment with the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA) and/or ionomycin (Fig. 1). Notably, expression of XBP-1s alone was capable of inducing a 10-fold increase in PpORF50 activity relative to basal promoter activity. Treatment of the PpORF50 with only TPA or ionomycin alone modestly increased basal activity by 3- to 5-fold. Interestingly, cells expressing XBP-1s that were treated with either TPA, ionomycin, or both showed synergistic induction of promoter activity up to 50-fold over basal promoter activity (Fig. 1). TPA and ionomycin induce protein kinase C- and Ca2+-mediated signaling, thus promoting B cell proliferation and mimicking the signals induced by B cell receptor (BCR) cross-linking (44). Of note, CD40 and Ig cross-linking has been shown to induce MHV68 reactivation from latency (45). Since XBP-1s reflects a stress signal in a terminally differentiating cell, it is evident that the virus has evolved mechanisms to reactivate from latency when the host cell is at risk of undergoing cell death. Thus, to ensure continuity of the latency reservoir, the immediate early gene product RTA (replication and transcription activator) is synthesized to promote viral reactivation from latency. The requirement for BCR cross-linking, costimulation (CD40), and plasma cell-specific transcription factors (XBP-1s and Blimp-1 [22]) reflects the specificity and timely induction of viral reactivation from latency.
FIG 1.
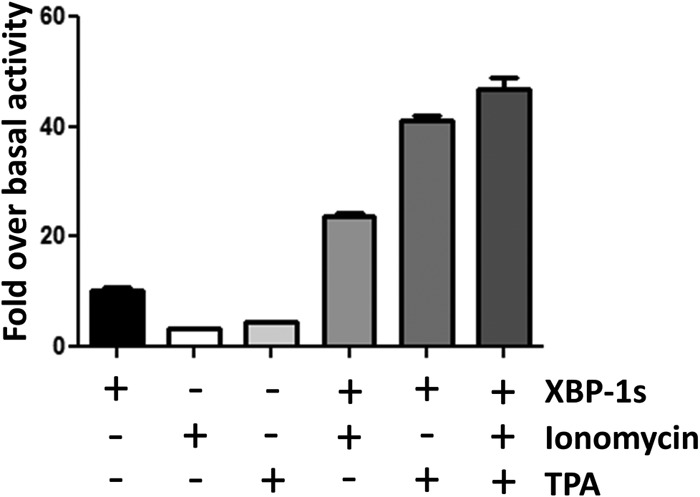
XBP-1s can transactivate the MHV68 gene 50 proximal promoter in M12 cells. The murine B cell lymphoma M12 cell line was transfected with an XBP-1s expression vector (pFlag.XBP1p.CMV2) and a luciferase reporter driven by the proximal gene 50 promoter (PpORF50). Where indicated, cells were treated at 12 h posttransfection with TPA and/or ionomycin and allowed to incubate for another 12 h. Values are reported as fold increase in luciferase activity over basal promoter activity. Error bars indicate standard errors of the means. Each experiment included 3 independent wells per experimental condition and was repeated in triplicate.
MHV68 infection in XBP-1flox/flox CD19CRE/+ cells results in normal germinal center and plasma cell responses but reduced levels of immunoglobulin in the periphery.
We next characterized the course of MHV68 infection in mice that were deficient in XBP-1 expression in B cells. Previous work by Todd et al. (38) demonstrated that mice lacking XBP-1 expression in B cells could generate normal germinal center and plasma cell frequencies in response to challenge with a foreign antigen. Interestingly, they determined that XBP-1 expression occurred after B cells upregulated syndecan-1 (CD138), a receptor commonly used to identify the plasma cell population (46). As such, B220int CD138+ cell frequencies were comparable to those of littermate control mice. The only defect observed in these animals was a significant reduction in global levels of IgG and IgM in the periphery (38). To investigate the role of XBP-1 in MHV68 infection, XBP-1flox/flox CD19CRE/+ mice were infected with 1,000 PFU of the MHV68-H2bYFP virus (40) via the intranasal (i.n.) route. Spleen and blood were collected on day 18 postinfection. Upon analysis of splenic subsets, we observed that germinal center responses, measured by the percentage of CD19+/GL7+ CD95+ cells, were identical in knockout (KO) and littermate (LM) controls (Fig. 2A and B). Additionally, the frequencies of B220int CD138+ plasma cells were also comparable in both KOs and LMs (Fig. 2A and C).We next evaluated the concentration of total IgM and IgG levels from sera collected at day 18 postinfection (Fig. 3). As reported earlier (38), upon MHV68 infection, XBP-1flox/flox CD19CRE/+ animals failed to mount both an IgM and IgG response compared to LM controls (Fig. 3A and B). Taken together, these data suggest that XBP-1 expression in B cells is not required for the generation of germinal center and plasma cell responses during an MHV68 infection but is required for Ig secretion and generation of a virus-specific humoral response.
FIG 2.

MHV68 infection in XBP-1flox/flox CD19CRE/+ mice induces normal germinal center and plasma cell responses. Animals were infected with 1,000 PFU of the recombinant MHV68-H2bYFP virus and sacrificed at day 18 postinfection to analyze splenic lymphoid population. (A) Representative flow cytometry plots illustrating the gating strategy employed to identify the splenic germinal center and plasma cell populations. Cells were gated on CD19+ GL7+ CD95+ to identify germinal center B cells, while cells were gated on CD3− B220int CD138+ to identify plasma cells. (B) Frequencies of germinal center populations in XBP-1 knockouts (KO) and littermate controls (LM). (C) Frequencies of plasma cell subsets in XBP-1 knockouts (KO) and littermate group controls (LM).
FIG 3.
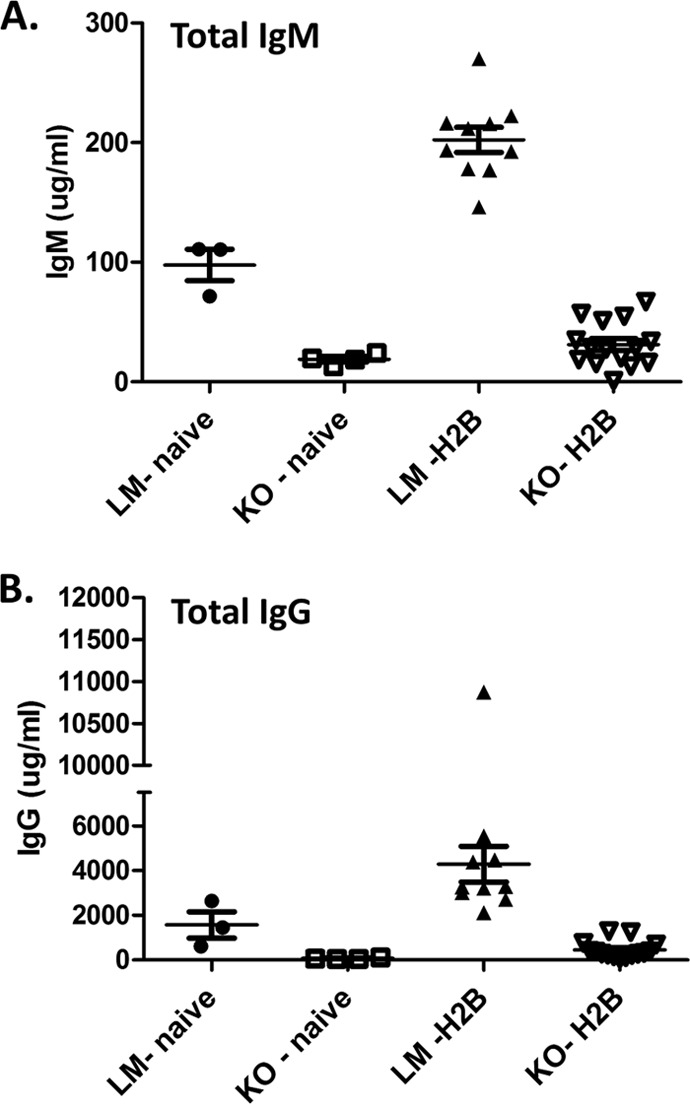
XBP-1flox/flox CD19CRE/+ mice fail to mount an immunoglobulin response following MHV68 infection. Animals were infected with 1,000 PFU of the recombinant MHV68-H2bYFP virus and terminally bled at day 18 postinfection to collect serum. Total IgM (A) and IgG (B) levels in naive and MHV68-infected XBP-1flox/flox CD19CRE/+ and littermate control animals.
Global frequency of MHV68-infected B cells is unaltered in XBP-1flox/flox CD19CRE/+ mice.
Using yellow fluorescent protein (YFP) expression from the MHV68-H2bYFP virus as an indicator of the frequency of infected B cells (40, 47), we evaluated the requirement for XBP-1 expression from B cells during infection and maintenance of latency. Upon evaluation of global levels of YFP expression from the CD3-negative splenic population, we determined that the efficiencies of viral infection were equivalent in KOs and LMs (Fig. 4A and B). Collins et al. (47) demonstrated that greater than 75% of YFP-positive B cells have a germinal center phenotype. Consistently, we detected close to 70% of YFP+ B cells in an XBP-1flox/flox CD19CRE/+ mouse infection with a germinal center phenotype (Fig. 5A and B). Interestingly, the frequency of YFP+ B cells with a plasma cell phenotype was significantly lower in XBP-1flox/flox CD19CRE/+ mice (x̄ = 9%) than in LMs (x̄ = 15%) (Fig. 5A and C). As expected, frequencies of infected germinal center B cells were comparable (LM x̄ = 7.8% and KO x̄ = 7.4%) (Fig. 6A and B). However, when we compared the frequencies of plasma cells that were infected with the virus, we noted a modest decrease in the frequency of infection in KOs compared to LM controls that was not statistically significant (LM x̄ = 6.6% and KO x̄ = 4.5%) (Fig. 6A and C). Since the efficiency of viral infection of the plasma cell repertoire was not significantly compromised in XBP-1flox/flox CD19CRE/+ mouse infections compared to LM controls (Fig. 6C), the defect observed in the frequency of YFP+ cells that have a plasma cell (PC) phenotype during an XBP-1flox/flox CD19CRE/+ mouse infection (Fig. 5C) cannot be explained by inefficient infection of that cellular subset. The basis for this defect is unclear at this point. However, this prompted a key question of whether a reduction in the levels of YFP+ cells with a plasma cell phenotype could affect the frequency of cells reactivating virus.
FIG 4.

Frequencies of virus-infected B cells (YFP+) in XBP-1flox/flox CD19CRE/+ mice are comparable to those in littermate controls. Animals were infected with 1,000 PFU of the recombinant MHV68-H2bYFP virus and sacrificed at day 18 postinfection. Splenocytes were collected to evaluate the frequency of virally infected cells. (A) Representative flow cytometry plots are shown illustrating the gating strategy employed. Cells were gated on the CD3− YFP+ population. (B) Compiled frequencies of virus-infected splenocytes (CD3− YFP+) in littermate controls (LM) and XBP-1 knockout (KO) mice (P = 0.3).
FIG 5.

The frequency of virus-infected cells with a plasma cell phenotype is reduced in MHV68-infected XBP-1flox/flox CD19CRE/+ animals. Mice were infected with 1,000 PFU of the recombinant MHV68-H2bYFP virus and sacrificed at day 18 postinfection. (A) Representative flow cytometry plots illustrating the gating strategy employed to identify the splenic germinal center and plasma cell populations. Cells were first gated on the CD3− YFP+ population and then gated on either the CD19+ GL7+ CD95+ population to identify germinal center B cells or the B220int CD138+ population to identify plasma cells. (B) Compiled frequencies of infected cells with a germinal center phenotype in littermate controls (LM) or XBP-1 knockout (KO) mice. NS, P value = 0.4. (C) Frequencies of the virus-infected cells with a plasma cell phenotype in littermate controls (LM) or XBP-1 knockout (KO) mice. ***, P = 0.003.
FIG 6.

Frequencies of infected germinal center B cells and plasma cells are similar in XBP-1flox/flox CD19CRE/+ and littermate control animals. Mice were infected with 1,000 PFU of MHV68-H2bYFP virus and sacrificed at day 18 postinfection. (A) Either cells were gated on the CD19+ GL7+ CD95+ germinal center population and subsequently gated on YFP+ cells, or they were gated on the CD3− B330int CD138+ plasma cell population with subsequent gating on YFP+ cells. YFP expression from that subset was then determined. Germinal center cells were then evaluated for YFP expression. (B) Frequency of infected germinal center B cells (P = 0.9). (C) Frequency of infected plasma cells (P = 0.3).
XBP-1s expression in B cells is not essential in vivo for viral reactivation or persistence.
Several studies of EBV, KSHV, and MHV68 have suggested a strong link between plasma cell differentiation and virus reactivation (19–21). As discussed above, our in vitro assays strongly suggested a role for XBP-1s in regulating viral reactivation from latency (Fig. 1). Since we observed a significant decrease in the frequency of YFP+ cells with a plasma cell phenotype in KO versus LM controls (Fig. 5C), we wanted to determine if there was a defect in MHV68 reactivation from latently infected splenocytes, where the vast majority of latently infected cells are B cells. As such, we conducted limiting dilution reactivation assays to evaluate the frequency of cells reactivating from latency at day 18 postinfection. Surprisingly, loss of XBP-1 expression in B cells did not alter the efficiency of viral reactivation from latency in XBP-1flox/flox CD19CRE/+ mice (1:10,543) compared to LM controls (1:8,336) (Fig. 7A). As expected, viral genome loads were comparable in KO (1:488) and LM (1:474) animals (Fig. 7B). Notably, however, slightly higher levels of preformed infectious virus were detected in splenocytes recovered from littermate control animals than in those from KO mice (Fig. 7A; see disrupted samples). This difference was statistically significant when lysates from 105 mechanically disrupted splenocytes were plated (P = 0.04). Overall, this raises the possibility that XBP-1 may contribute in vivo to either virus reactivation or replication in B cells—even though this contribution was not apparent upon ex vivo culture of latently infected splenocytes.
FIG 7.
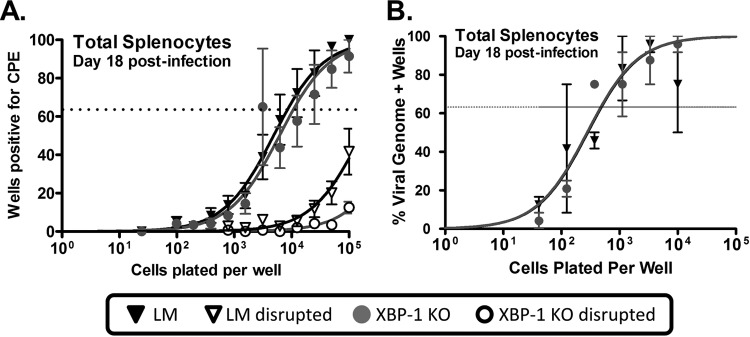
XBP-1 expression is not required for MHV68 reactivation or establishment of latency. (A) Limiting dilution analysis of MHV68 reactivation from total splenocytes isolated at day 18 postinfection. (B) Limiting dilution PCR analyses of MHV68 genome-positive splenocytes isolated at day 18 postinfection.
IRF4 expression in B cells is required for MHV68 reactivation from latency and maintenance of latency.
The interferon response factor 4 (IRF4) is another key transcription factor involved in plasma cell differentiation. B cells deficient in IRF4 expression have diminished germinal center responses and cannot generate a plasma cell response (48, 49). We recently demonstrated that the unique MHV68 gene product, M2, is capable of inducing IRF4 expression via activation of NFAT and promotes interleukin-10 (IL-10) expression from B cells (41). Infection of IRF4fl/fl mice with a recombinant virus that expresses Cre recombinase resulted in a significant defect in the establishment of latency, along with a greater defect in viral reactivation (41). This novel finding suggested a close link between viral reactivation and a requirement for IRF4 expression. However, since IRF4 is expressed in cell types other than B cells and since the virus can infect macrophages and dendritic cells, we could not infer that the requirement for reactivation was contingent on IRF4 expression in B cells. Given that XBP-1 does not appear to be involved in controlling MHV68 reactivation from latently infected B cells, along with our previous demonstration that there is only a moderate requirement for Blimp-1 (22), we hypothesized that IRF4 may be a vital player in reactivation associated with plasma cell differentiation upon MHV68 infection of mice.
To address the requirement for IRF4 in a more direct fashion, we used mice in which IRF4 was specifically deleted in B cells. We have recently shown that M2, a viral gene unique to MHV68, is sufficient to drive plasma cell differentiation in a B cell line (21) and to induce IRF4 expression in B cells (41). Since the phenotype observed with infection of IRF4fl/fl mice with a recombinant MHV68 expressing Cre recombinase closely resembles that of infection of wild-type mice with an M2-null virus, we also hypothesized that M2 induction of IRF4 is a critical step in the viral life cycle. As such, IRF4fl/fl CD19+/Cre (IRF4 KO) or IRF4fl/fl CD19+/+ littermate (LM) control mice were infected with 1,000 PFU of either M2MR.HY or M2stop.HY viruses (described in reference 41). Spleens were harvested on day 16 postinfection and analyzed by flow cytometry. Notably, the percentage of total germinal center B cells was significantly different between the LM and KO mice infected with the M2stop marker rescue virus (MR), suggesting that IRF4 does play a role in the germinal center response to MHV68 infection (Fig. 8C, compare KO-MR and LM-MR infected animals). However, it should be noted that the basal level of germinal center B cells was also substantially lower in IRF4 B cell knockout mice (Fig. 8C, KO-Naive). Indeed, the fold increases in splenic germinal center B cells (naive mice versus MHV68-infected mice) were very similar when comparing littermate control and IRF4 B cell knockout mice (ca. 20-fold for the IRF4 KOs versus ca. 15-fold for the littermate control animals) (Fig. 8C). We next assessed the percentage of total plasma cells, defined by B220int CD138+ cells. As expected, there was a significant difference in the percentage of plasma cells in the IRF4 KO animals compared to littermate controls (Fig. 8D). While in littermate control animals there was a nearly 4-fold increase in the number of splenic plasma cells in marker rescue-infected mice, there was only a 1.3-fold increase in the IRF4 KO animals (Fig. 8D).
FIG 8.

Loss of IRF4 expression in B cells impairs MHV68 reactivation from latency to a similar extent as does disruption of the MHV68 M2 gene. IRF4flox/flox CD19CRE/+ (IRF4 KO) or littermate control (LM) mice were infected with 1,000 PFU i.n. of either MHV68/M2MR.HY or MHV68/M2stop.HY virus. (A and B) Limiting dilution reactivation analyses plating either intact splenocytes (A) or mechanically disrupted splenocytes (B) recovered at day 16 postinfection. (C) Frequencies of germinal center B cells (B220+ GL7+ CD95+) in the spleens of naive and MHV68-infected animals, as indicated. **, P = 0.007. (D) Frequencies of plasma cells (CD3− B220int CD138+) in the spleens of naive and MHV68-infected animals, as indicated. KO, IRF4flox/flox CD19CRE/+; LM, littermate control (IRF4flox/flox CD19+/+); M2stop, MHV68/M2stop.HY; MR, MHV68/M2MR.HY. ***, P = 0.0001.
Given the significant impact of loss of IRF4 expression in B cells on the splenic plasma cell response, we next assessed the impact of loss of IRF4 in B cells on MHV68 reactivation from latency (Fig. 8A). Notably, littermate control animals (LM) infected with M2stop marker rescue virus exhibited the expected frequency of reactivating splenocytes (compared to historical values for wild-type MHV68 reactivation from C57BL/76 splenocytes at day 16 postinfection) (Fig. 8A). In contrast, reactivation of splenocytes from IRF4 KO mice infected with the M2stop marker rescue virus exhibited an approximately 30-fold defect (1 in 3,954 cells reactivating virus in littermate controls versus 1 in 115,877 cells in IRF4 KO mice). Importantly, infection of littermate control animals with the M2 null mutant (M2stop) yielded a very similar reactivation defect (Fig. 8A)—consistent with a major component of M2 function being induction of IRF4 expression in latently infected B cells (41). However, it is also notable that infection of IRF4 knockout mice with the M2 null mutant resulted in a more severe defect in reactivation from latency (1 in 630,957 cells reactivating virus), suggesting that the complete loss of IRF4 is more severe than the loss of IRF4 induction mediated by M2 (i.e., there may be, alternatively, M2-independent mechanisms to induce IRF4 expression in B cells during virus infection). Finally, not surprisingly, the amount of preformed infectious virus present in the day 16 samples mirrored the levels of virus reactivation observed—but did not interfere with accurate assessments of the frequency of splenocytes reactivating virus (Fig. 8B).
DISCUSSION
Here, we have shown that while XBP-1s can activate the MHV68 immediate early gene 50 proximal promoter, which drives expression of the essential transcriptional activator RTA, it is not required for MHV68 reactivation from latently infected B cells as assessed by a limiting dilution ex vivo reactivation analysis of latently infected splenocytes. However, it is possible that XBP-1s plays a role in activation of gene 50 transcription in B cells under other conditions (e.g., in response to specific stimuli) or from other infected cell types. Notwithstanding these possibilities, the data presented here bring into question the significance of studies to date that have demonstrated the ability of XBP-1s to activate EBV and KSHV immediate early gene expression—all of which have exclusively relied on studies in tissue culture models.
Notably, the requirement for IRF4 expression in B cells for efficient MHV68 reactivation from latently infected splenic B cells likely reflects the essential role that IRF4 plays in plasma cell differentiation. This is based on (i) our earlier studies that demonstrated that the vast majority of MHV68 reactivation from splenocytes arises from the infected splenic plasma cell population (21) and (ii) the fact that we have failed to detect IRF4 transactivation of any of the identified gene 50 promoters (data not shown). Taken together with the XBP-1 data, this suggests that there is another plasma cell factor that triggers gene 50 expression upon terminal differentiation of latently infected B cells. One logical possibility is Blimp-1, another cellular factor that is essential for plasma cells. Indeed, the phenotype in Blimp-1 B cell knockout mice is similar to that observed in IRF4 B cell knockout animals (22). It is also possible that the loss of expression of a transcriptional repressor upon terminal differentiation is also involved in the induction of RTA expression. Further studies are required to identify the critical cis elements and trans-acting factors involved in the induction of MHV68 gene 50 transcription upon plasma cell differentiation.
Interestingly, the reactivation defect in the IRF4flox/flox CD19CRE/+ splenocytes is nearly identical to an M2-null virus phenotype. M2 is a unique gene product of MHV68 that is not required for acute viral replication in vivo but has a dose- and route-specific defect in establishment of latency and reactivation (50). Consistent with the requirement of M2 in reactivation, it is also required for the differentiation of infected B cells to become plasma cells (21). Indeed, it was shown that M2 induction of IL-10 from B cells occurs via an NFAT-dependent induction of IRF4 (41). In fact, when overexpressed in B cells, M2 has been shown to induce upregulation of IRF4, Blimp-1, and XBP-1 transcripts (21). EBV has been shown to mimic BCR cross-linking and Ca2+ signaling via the LMP2a protein product and was also shown to encode an IL-10 homolog (v-IL-10) (51, 52). It is evident not only that this family of viruses has evolved to respond to changes in B cell differentiation state but also that they appear capable of modulating B cell differentiation. This likely reflects the requirement for proper expansion of the B cell reservoir in order to efficiently establish viral latency (53).
Finally, SAP−/− (54) and IL21R−/− (C. M. Collins and S. H. Speck, unpublished data) mice infected with MHV68 display severe defects in viral reactivation and establishment of latency. Of note, both of these transgenic models show a defective generation or maintenance of the germinal center response, which also negatively impacts the plasma cell repertoire (55–57). Once more, the loss of this cellular subset further supports viral dependency on this host cell for proper regulation of the viral life cycle. Together with our results presented above, it strongly establishes that the virus takes advantage of a healthy host by utilizing the normal B cell differentiation that occurs in response to viral infection. The ability of MHV68 to induce B cell differentiation, as well as respond to plasma cell-specific transcription factors, reflects a close link between the viral life cycle and the plasma cell niche. Our data suggest a role for multiple redundant plasma cell-specific transcription factors in modulating the viral life cycle. Finally, the data presented here emphasize the importance of in vivo pathogenesis studies in identifying those host cell factors and cellular reservoirs that are required for gammaherpesvirus infection.
Footnotes
Published ahead of print 30 July 2014
REFERENCES
- 1.Barton E, Mandal P, Speck SH. 2011. Pathogenesis and host control of gammaherpesviruses: lessons from the mouse. Annu. Rev. Immunol. 29:351–397. 10.1146/annurev-immunol-072710-081639 [DOI] [PubMed] [Google Scholar]
- 2.Andersson J. 2006. Epstein-Barr virus and Hodgkin's lymphoma. Herpes 13:12–16 [PubMed] [Google Scholar]
- 3.Greenspan JS, Greenspan D, Lennette ET, Abrams DI, Conant MA, Petersen V, Freese UK. 1985. Replication of Epstein-Barr virus within the epithelial cells of oral “hairy” leukoplakia, an AIDS-associated lesion. N. Engl. J. Med. 313:1564–1571. 10.1056/NEJM198512193132502 [DOI] [PubMed] [Google Scholar]
- 4.Henle G, Henle W. 1970. Observations on childhood infections with the Epstein-Barr virus. J. Infect. Dis. 121:303–310. 10.1093/infdis/121.3.303 [DOI] [PubMed] [Google Scholar]
- 5.Henle G, Henle W, Diehl V. 1968. Relation of Burkitt's tumor-associated herpes-type virus to infectious mononucleosis. Proc. Natl. Acad. Sci. U. S. A. 59:94–101. 10.1073/pnas.59.1.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johansson B, Klein G, Henle W, Henle G. 1970. Epstein-Barr virus (EBV)-associated antibody patterns in malignant lymphoma and leukemia. I. Hodgkin's disease. Int. J. Cancer 6:450–462 [DOI] [PubMed] [Google Scholar]
- 7.Nonoyama M, Huang CH, Pagano JS, Klein G, Singh S. 1973. DNA of Epstein-Barr virus detected in tissue of Burkitt's lymphoma and nasopharyngeal carcinoma. Proc. Natl. Acad. Sci. U. S. A. 70:3265–3268. 10.1073/pnas.70.11.3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Raab-Traub N, Flynn K, Pearson G, Huang A, Levine P, Lanier A, Pagano J. 1987. The differentiated form of nasopharyngeal carcinoma contains Epstein-Barr virus DNA. Int. J. Cancer 39:25–29. 10.1002/ijc.2910390106 [DOI] [PubMed] [Google Scholar]
- 9.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. 10.1126/science.7997879 [DOI] [PubMed] [Google Scholar]
- 10.Chen KT. 1984. Multicentric Castleman's disease and Kaposi's sarcoma. Am. J. Surg. Pathol. 8:287–293. 10.1097/00000478-198404000-00006 [DOI] [PubMed] [Google Scholar]
- 11.Collandre H, Ferris S, Grau O, Montagnier L, Blanchard A. 1995. Kaposi's sarcoma and new herpesvirus. Lancet 345:1043. 10.1016/S0140-6736(95)90779-3 [DOI] [PubMed] [Google Scholar]
- 12.Fan W, Bubman D, Chadburn A, Harrington WJ, Jr, Cesarman E, Knowles DM. 2005. Distinct subsets of primary effusion lymphoma can be identified based on their cellular gene expression profile and viral association. J. Virol. 79:1244–1251. 10.1128/JVI.79.2.1244-1251.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sunil-Chandra NP, Arno J, Fazakerley J, Nash AA. 1994. Lymphoproliferative disease in mice infected with murine gammaherpesvirus 68. Am. J. Pathol. 145:818–826 [PMC free article] [PubMed] [Google Scholar]
- 14.Tarakanova VL, Suarez F, Tibbetts SA, Jacoby MA, Weck KE, Hess JL, Speck SH, Virgin HW., IV 2005. Murine gammaherpesvirus 68 infection is associated with lymphoproliferative disease and lymphoma in BALB beta2 microglobulin-deficient mice. J. Virol. 79:14668–14679. 10.1128/JVI.79.23.14668-14679.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang X, Paden CR, Morales FM, Powers RP, Jacob J, Speck SH. 2011. Murine gamma-herpesvirus immortalization of fetal liver-derived B cells requires both the viral cyclin D homolog and latency-associated nuclear antigen. PLoS Pathog. 7:e1002220. 10.1371/journal.ppat.1002220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein G, Svedmyr E, Jondal M, Persson PO. 1976. EBV-determined nuclear antigen (EBNA)-positive cells in the peripheral blood of infectious mononucleosis patients. Int. J. Cancer 17:21–26. 10.1002/ijc.2910170105 [DOI] [PubMed] [Google Scholar]
- 17.Mesri EA, Cesarman E, Arvanitakis L, Rafii S, Moore MA, Posnett DN, Knowles DM, Asch AS. 1996. Human herpesvirus-8/Kaposi's sarcoma-associated herpesvirus is a new transmissible virus that infects B cells. J. Exp. Med. 183:2385–2390. 10.1084/jem.183.5.2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willer DO, Speck SH. 2003. Long-term latent murine gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J. Virol. 77:8310–8321. 10.1128/JVI.77.15.8310-8321.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crawford DH, Ando I. 1986. EB virus induction is associated with B-cell maturation. Immunology 59:405–409 [PMC free article] [PubMed] [Google Scholar]
- 20.Laichalk LL, Thorley-Lawson DA. 2005. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 79:1296–1307. 10.1128/JVI.79.2.1296-1307.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang X, Collins CM, Mendel JB, Iwakoshi NN, Speck SH. 2009. Gammaherpesvirus-driven plasma cell differentiation regulates virus reactivation from latently infected B lymphocytes. PLoS Pathog. 5:e1000677. 10.1371/journal.ppat.1000677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siegel AM, Rangaswamy US, Napier RJ, Speck SH. 2010. Blimp-1-dependent plasma cell differentiation is required for efficient maintenance of murine gammaherpesvirus latency and antiviral antibody responses. J. Virol. 84:674–685. 10.1128/JVI.01306-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhende PM, Dickerson SJ, Sun X, Feng WH, Kenney SC. 2007. X-box-binding protein 1 activates lytic Epstein-Barr virus gene expression in combination with protein kinase D. J. Virol. 81:7363–7370. 10.1128/JVI.00154-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun CC, Thorley-Lawson DA. 2007. Plasma cell-specific transcription factor XBP-1s binds to and transactivates the Epstein-Barr virus BZLF1 promoter. J. Virol. 81:13566–13577. 10.1128/JVI.01055-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dalton-Griffin L, Wilson SJ, Kellam P. 2009. X-box binding protein 1 contributes to induction of the Kaposi's sarcoma-associated herpesvirus lytic cycle under hypoxic conditions. J. Virol. 83:7202–7209. 10.1128/JVI.00076-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson SJ, Tsao EH, Webb BL, Ye H, Dalton-Griffin L, Tsantoulas C, Gale CV, Du MQ, Whitehouse A, Kellam P. 2007. X box binding protein XBP-1s transactivates the Kaposi's sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J. Virol. 81:13578–13586. 10.1128/JVI.01663-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu F, Feng J, Harada JN, Chanda SK, Kenney SC, Sun R. 2007. B cell terminal differentiation factor XBP-1 induces reactivation of Kaposi's sarcoma-associated herpesvirus. FEBS Lett. 581:3485–3488. 10.1016/j.febslet.2007.06.056 [DOI] [PubMed] [Google Scholar]
- 28.Gradoville L, Gerlach J, Grogan E, Shedd D, Nikiforow S, Metroka C, Miller G. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 74:6207–6212. 10.1128/JVI.74.13.6207-6212.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lukac DM, Kirshner JR, Ganem D. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348–9361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Speck SH, Chatila T, Flemington E. 1997. Reactivation of Epstein-Barr virus: regulation and function of the BZLF1 gene. Trends Microbiol. 5:399–405. 10.1016/S0966-842X(97)01129-3 [DOI] [PubMed] [Google Scholar]
- 31.Liou HC, Boothby MR, Finn PW, Davidon R, Nabavi N, Zeleznik-Le NJ, Ting JP, Glimcher LH. 1990. A new member of the leucine zipper class of proteins that binds to the HLA DR alpha promoter. Science 247:1581–1584. 10.1126/science.2321018 [DOI] [PubMed] [Google Scholar]
- 32.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. 2003. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat. Immunol. 4:321–329. 10.1038/ni907 [DOI] [PubMed] [Google Scholar]
- 33.Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH. 2001. Plasma cell differentiation requires the transcription factor XBP-1. Nature 412:300–307. 10.1038/35085509 [DOI] [PubMed] [Google Scholar]
- 34.Shamu CE, Walter P. 1996. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 15:3028–3039 [PMC free article] [PubMed] [Google Scholar]
- 35.Sidrauski C, Walter P. 1997. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90:1031–1039. 10.1016/S0092-8674(00)80369-4 [DOI] [PubMed] [Google Scholar]
- 36.Welihinda AA, Kaufman RJ. 1996. The unfolded protein response pathway in Saccharomyces cerevisiae. Oligomerization and trans-phosphorylation of Ire1p (Ern1p) are required for kinase activation. J. Biol. Chem. 271:18181–18187 [DOI] [PubMed] [Google Scholar]
- 37.Gass JN, Gifford NM, Brewer JW. 2002. Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J. Biol. Chem. 277:49047–49054. 10.1074/jbc.M205011200 [DOI] [PubMed] [Google Scholar]
- 38.Todd DJ, McHeyzer-Williams LJ, Kowal C, Lee AH, Volpe BT, Diamond B, McHeyzer-Williams MG, Glimcher LH. 2009. XBP1 governs late events in plasma cell differentiation and is not required for antigen-specific memory B cell development. J. Exp. Med. 206:2151–2159. 10.1084/jem.20090738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu D, Zhao L, Del Valle L, Miklossy J, Zhang L. 2008. Interferon regulatory factor 4 is involved in Epstein-Barr virus-mediated transformation of human B lymphocytes. J. Virol. 82:6251–6258. 10.1128/JVI.00163-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collins CM, Speck SH. 2012. Tracking murine gammaherpesvirus 68 infection of germinal center B cells in vivo. PLoS One 7:e33230. 10.1371/journal.pone.0033230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rangaswamy US, Speck SH. 2014. Murine gammaherpesvirus M2 protein induction of IRF4 via the NFAT pathway leads to IL-10 expression in B cells. PLoS Pathog. 10:e1003858. 10.1371/journal.ppat.1003858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weck KE, Kim SS, Virgin HW, IV, Speck SH. 1999. B cells regulate murine gammaherpesvirus 68 latency. J. Virol. 73:4651–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sangster MY, Topham DJ, D'Costa S, Cardin RD, Marion TN, Myers LK, Doherty PC. 2000. Analysis of the virus-specific and nonspecific B cell response to a persistent B-lymphotropic gammaherpesvirus. J. Immunol. 164:1820–1828. 10.4049/jimmunol.164.4.1820 [DOI] [PubMed] [Google Scholar]
- 44.Kim KM, Ishigami T, Hata D, Yamaoka K, Mayumi M, Mikawa H. 1992. Regulation of cell division of mature B cells by ionomycin and phorbol ester. J. Immunol. 148:1797–1803 [PubMed] [Google Scholar]
- 45.Moser JM, Upton JW, Gray KS, Speck SH. 2005. Ex vivo stimulation of B cells latently infected with gammaherpesvirus 68 triggers reactivation from latency. J. Virol. 79:5227–5231. 10.1128/JVI.79.8.5227-5231.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rawstron AC. 2006. Immunophenotyping of plasma cells. Curr. Protoc. Cytom. Chapter 6:Unit 6.23. 10.1002/0471142956.cy0623s36 [DOI] [PubMed] [Google Scholar]
- 47.Collins CM, Boss JM, Speck SH. 2009. Identification of infected B-cell populations by using a recombinant murine gammaherpesvirus 68 expressing a fluorescent protein. J. Virol. 83:6484–6493. 10.1128/JVI.00297-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, Ludwig T, Rajewsky K, Dalla-Favera R. 2006. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat. Immunol. 7:773–782. 10.1038/ni1357 [DOI] [PubMed] [Google Scholar]
- 49.Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. 2006. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity 25:225–236. 10.1016/j.immuni.2006.07.009 [DOI] [PubMed] [Google Scholar]
- 50.Herskowitz JH, Jacoby MA, Speck SH. 2005. The murine gammaherpesvirus 68 M2 gene is required for efficient reactivation from latently infected B cells. J. Virol. 79:2261–2273. 10.1128/JVI.79.4.2261-2273.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caldwell RG, Wilson JB, Anderson SJ, Longnecker R. 1998. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity 9:405–411. 10.1016/S1074-7613(00)80623-8 [DOI] [PubMed] [Google Scholar]
- 52.Miyazaki I, Cheung RK, Dosch HM. 1993. Viral interleukin 10 is critical for the induction of B cell growth transformation by Epstein-Barr virus. J. Exp. Med. 178:439–447. 10.1084/jem.178.2.439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moser JM, Upton JW, Allen RD, III, Wilson CB, Speck SH. 2005. Role of B-cell proliferation in the establishment of gammaherpesvirus latency. J. Virol. 79:9480–9491. 10.1128/JVI.79.15.9480-9491.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Collins CM, Speck SH. 2014. Expansion of murine gammaherpesvirus latently infected B cells requires T follicular help. PLoS Pathog. 10:e1004106. 10.1371/journal.ppat.1004106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Al-Alem U, Li C, Forey N, Relouzat F, Fondaneche MC, Tavtigian SV, Wang ZQ, Latour S, Yin L. 2005. Impaired Ig class switch in mice deficient for the X-linked lymphoproliferative disease gene Sap. Blood 106:2069–2075. 10.1182/blood-2004-07-2731 [DOI] [PubMed] [Google Scholar]
- 56.Rasheed MA, Latner DR, Aubert RD, Gourley T, Spolski R, Davis CW, Langley WA, Ha SJ, Ye L, Sarkar S, Kalia V, Konieczny BT, Leonard WJ, Ahmed R. 2013. Interleukin-21 is a critical cytokine for the generation of virus-specific long-lived plasma cells. J. Virol. 87:7737–7746. 10.1128/JVI.00063-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yin L, Al-Alem U, Liang J, Tong WM, Li C, Badiali M, Medard JJ, Sumegi J, Wang ZQ, Romeo G. 2003. Mice deficient in the X-linked lymphoproliferative disease gene sap exhibit increased susceptibility to murine gammaherpesvirus-68 and hypo-gammaglobulinemia. J. Med. Virol. 71:446–455. 10.1002/jmv.10504 [DOI] [PubMed] [Google Scholar]