

ABSTRACT
Hepatitis C virus (HCV) is a widespread human pathogen causing liver cirrhosis and cancer. Similar to the case for other viruses, HCV depends on host and viral factors to complete its life cycle. We used proteomic and yeast two-hybrid approaches to elucidate host factors involved in HCV nonstructural protein NS5A function and found that MOBKL1B interacts with NS5A. Initial experiments with small interfering RNA (siRNA) knockdown suggesting a role in HCV replication led us to examine the interaction using biochemical and structural approaches. As revealed by a cocrystal structure of a core MOBKL1B-NS5A peptide complex at 1.95 Å, NS5A binds to a hydrophobic patch on the MOBKL1B surface. Biosensor binding assays identified a highly conserved, 18-amino-acid binding site in domain II of NS5A, which encompasses residues implicated in cyclophilin A (CypA)-dependent HCV RNA replication. However, a CypA-independent HCV variant had reduced replication in MOBKL1B knockdown cells, even though its NS5A does not interact with MOBKL1B. These discordant results prompted more extensive studies of MOBKL1B gene knockdowns, which included additional siRNAs and specifically matched seed sequence siRNA controls. We found that reduced virus replication after treating cells with MOBKL1B siRNA was actually due to off-target inhibition, which indicated that the initial finding of virus replication dependence on the MOBKL1B-NS5A interaction was incorrect. Ultimately, using several approaches, we found no relationship of the MOBKL1B-NS5A interaction to virus replication. These findings collectively serve as a reminder to investigators and scientific reviewers of the pervasive impact of siRNA off-target effects on interpretation of biological data.
IMPORTANCE Our study illustrates an underappreciated shortcoming of siRNA gene knockdown technology. We initially identified a cellular protein, MOBKL1B, as a binding partner with the NS5A protein of hepatitis C virus (HCV). MOBKL1B siRNA, but not irrelevant RNA, treatment was associated with both reduced virus replication and the absence of MOBKL1B. Believing that HCV replication depended on the MOBKL1B-NS5A interaction, we carried out structural and biochemical analyses. Unexpectedly, an HCV variant lacking the MOBKL1B-NS5A interaction could not replicate after cells were treated with MOBKL1B siRNA. By repeating the MOBKL1B siRNA knockdowns and including seed sequence-matched siRNA instead of irrelevant siRNA as a control, we found that the MOBKL1B siRNAs utilized had off-target inhibitory effects on virus replication. Collectively, our results suggest that stricter controls must be utilized in all RNA interference (RNAi)-mediated gene knockdown experiments to ensure sound conclusions and a reliable scientific knowledge database.
INTRODUCTION
Hepatitis C virus (HCV) is an enveloped RNA virus in the Flaviviridae family, in which there are seven major genotypes diverging by 30 to 35% at the nucleotide level. Globally, more than 170 million people are infected with HCV, and although 20 to 30% of acute HCV infections are spontaneously cleared, the majority of virus-exposed individuals develop chronic infection, which can lead to liver cirrhosis, end-stage liver disease, and hepatocellular carcinoma (1). Despite new treatment options that are highly effective (2), drug resistance and incomplete genotype coverage are important limitations. Thus, it is important to thoroughly understand the mechanistic activity of inhibitors and to develop additional antivirals directed at different targets.
The HCV genome is an ∼9.6-kb positive-sense single-stranded RNA carrying a single open reading frame, which is translated into a polyprotein precursor. From the polyprotein, three structural proteins (core and envelope proteins E1 and E2) and seven nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) are produced after a series of co- and posttranslational cleavages by both host and viral proteases. The genome is replicated in an altered intracellular membrane-associated replication complex that is composed of viral and cellular factors (reviewed in reference 3).
Along with other HCV proteins, NS5A has been the focus of intensive inhibitor discovery efforts, and highly potent new compounds targeting it have been shown to be clinically efficacious (4). NS5A is a phosphoprotein with multiple roles in the virus life cycle, including virus genome replication and particle assembly (5–7). However, the mechanistic activity of HCV NS5A is not fully understood. In an effort to broaden our understanding of NS5A, our objective was to define its interaction with cellular partners. To this end, we performed a pulldown assay of Huh-7.5 hepatoma cells infected with a genotype 2a virus expressing a tagged NS5A, and we discovered a novel NS5A-interacting protein, Mps one binder kinase activator-like 1B (MOBKL1B, also termed MOB1A). MOBKL1B is a component of the protein kinase cascade of the Salvador/Warts/Hippo (SWH) tumor suppressor pathway, and although it has no known enzymatic activity, it plays an important role in the pathway as an activator of nuclear Dbf2-related (NDR) serine/threonine kinases (8–10).
To test whether the NS5A-MOBKL1B binding was functionally significant for HCV replication, we took a typical approach and performed MOBKL1B small interfering RNA (siRNA) knockdown experiments in permissive hepatoma cells before infection. Results from these initial studies indicated that virus replication required MOBKL1B expression, prompting us to characterize the NS5A-MOBKL1B with biochemical, genetic, and structural studies. We showed that the MOBKL1B binding site on NS5A overlapped with the binding site of cyclophilin A (CypA), a cellular protein that is essential for HCV RNA replication, as its interaction with NS5A enhances HCV RNA binding (11–14). CypA has emerged as the first host factor targeted for anti-HCV therapy, and drugs blocking the CypA-NS5A interaction are currently in various stages of clinical development (15). Mutant HCVs that replicate in the absence of CypA have amino acid substitutions in the CypA binding site of NS5A, and because of the overlap of the MOBKL1B and CypA binding sites, we questioned whether these mutations would also affect NS5A-MOBKL1B interactions. With protein pulldown experiments, we found that a CypA-independent mutant NS5A did not interact with MOBKL1B. However, the CypA-independent virus had reduced replication after cells were treated with MOBKL1B siRNA, which was unanticipated, since a virus lacking the MOBKL1B-NS5A interaction would not be expected to depend on it for replication. This contradiction prompted us to reexamine our MOBKL1B siRNA gene knockdown experiments with wild-type HCV. After including matched seed sequence controls alongside our siRNAs targeting MOBKL1B, we showed off-target inhibitory effects of siRNA treatment on virus replication. Our cumulative results ultimately showed that HCV replication in cultured cells does not depend on an interaction between MOBKL1B and NS5A. We conclude that for accurate interpretation, gene knockdowns should be performed with multiple siRNAs with stringent matched controls, rather than the more commonly used irrelevant (IRR) or nonsilencing siRNA controls.
MATERIALS AND METHODS
Cells and antibodies.
Huh-7.5 cells were maintained in Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) and 0.1 mM nonessential amino acids (NEAA) at 37°C in a 5% CO2 incubator. Rabbit polyclonal antibody against MOBKL1B was purchased from Cell Signaling Technology (Danvers, MA). Mouse anti-CypA antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and mouse anti-NS3 antibody (H23) was from Abcam (Cambridge, United Kingdom). The mouse anti-NS5A antibody 9E10 has been previously described (16).
Plasmid construction. (i) Affinity-tagged Jc1 constructs for the pulldown experiment.
The One-STrEP tag (OST) (IBA GmbH, Gottingen, Germany) was introduced into the Jc1 genome (17) at a position between NS5A residues 383 and 384, which was previously shown to be tolerant to tag insertion (18), to generate Jc1 NS5A-OST. The D316E Y317N (DEYN) CypA-independent mutant virus, termed Jc1 NS5A-OST-DEYN, was constructed using the megaprimer overlapping PCR method. This strategy was also used to introduce the DEYN mutations into Jc1 FLAG(p7-nsGluc2A) (19) to construct Jc1 FLAG(p7-nsGluc2A)DEYN.
(ii) Yeast two-hybrid constructs.
MOBKL1B cDNA (GenBank accession number BC003398) was amplified and cloned into the pGADT7 (activation domain) vector (Clontech, Mountain View, CA) using 5′ NdeI and 3′ BamHI cloning sites. Genes encoding each viral protein were amplified from Jc1 or Con1 (7) genome-containing plasmids and cloned into pGBKT7 (binding domain) vector (Clontech) using 5′ NdeI/3′ BamHI or 5′ EcoRI/3′ BamHI cloning sites. Con1 NS5A domain II serial deletion mutants were subcloned using the genotype 1b HCV replicon (7), and JFH1 NS5A alanine substitution mutants were constructed using the megaprimer method.
(iii) MOBKL1B bacterial expression constructs.
MOBKL1B cDNA was amplified with primers containing 5′ BamHI and 3′ XhoI restriction sites and cloned into pET-SUMO vector (Invitrogen, Carlsbad, CA).
(iv) Glutathione S-transferase (GST)-fused NS5A peptide constructs.
5′-phosphorylated sense and antisense oligonucleotides encoding NS5A residues 310 to 335, 310 to 327, 310 to 324, 310 to 320, 314 to 329, 305 to 316, and 308 to 327 were synthesized with 5′ EcoRI/3′ NotI linkers (IDT, Coralville, IA). Oligonucleotides were annealed and cloned into the pGEX-6P-1 vector (GE Healthcare, Uppsala, Sweden) using 5′ EcoRI/3′ NotI cloning sites.
Protein pulldown and MS identification.
To identify cellular proteins interacting with viral proteins, in vitro-transcribed Jc1 NS5A-OST RNA was electroporated into Huh-7.5 cells (10 plates of 150-mm dishes) as described previously (16). Cells were harvested at 72 h postelectroporation by incubation in 1 ml of lysis buffer (LB) (20 mM Tris [pH 8.0], 150 mM NaCl, 2 mM MgCl2, and 1% Triton X-100) supplemented with EDTA-free protease inhibitor cocktail (Roche, Basel, Switzerland), DNase I (50 U/ml), and RNase A (10 μg/ml). Lysates were clarified by centrifugation (16,000 × g, 20 min, 4°C) and incubated with 50 μl of Strep-Tactin Sepharose (IBA GmbH, Gottingen, Germany) for 2 h at 4°C. The matrix was washed 5 times in LB. Bound proteins were released by boiling in SDS-PAGE loading buffer, separated by 4 to 20% SDS-PAGE, and visualized by Coomassie blue staining. Proteins in SDS-PAGE were digested with trypsin, extracted, and identified by liquid chromatography with online tandem mass spectrometry (LC-MS/MS). Peptide mass fingerprinting analysis was performed using Mascot Search (Matrix Science, Boston, MA) against the Swiss-Prot database (downloaded in August 2008).
RNA interference (RNAi).
siRNAs corresponding to MOBKL1B coding nucleotides (nt) 241 to 259 (1, 5′-GAAGCAAGCUGUCCAGUCA-3′), nt 91 to 111 (2, 5′-CAUGCAGAAGCAACUCUAGGA-3′), or nt 292 to 310 (3, 5′-GCAGAUGGUACUAAUAUUA-3′), their seed sequence-matched siRNA controls (1 C911, 5′-GAAGCAAGGACUCCAGUCA-3′; 2 C1012, 5′-CAUGCAGAACGUACUCUAGGA-3′; and 3 C911, 5′-GCAGAUGGAUGUAAUAUUA-3′), and pools of MOBKL1B, CypA-targeting siRNAs (ON-TARGETplus SMARTpools) and irrelevant siRNA designed for minimal targeting of human genes (ON-TARGETplus nontargeting pool) were purchased from Dharmacon (Lafayette, CO). For the depletion of target proteins, ∼3.0 × 104 Huh-7.5 cells per well were seeded in 24-well plates 1 day before transfection. Cells were transfected twice at 24-h intervals with 50 nM siRNA using 1.5 μl of Lipofectamine RNAiMAX following the manufacturer's instructions (Invitrogen).
HCV replication and production assays.
HCV Jc1 virus stock was generated by electroporation of Huh-7.5 cells with in vitro-transcribed RNA as previously described (16), and its infectious titer was determined by limiting-dilution assay followed by NS5A staining (16). To initiate infection with RNA, wild-type or DEYN mutant Jc1FLAG(p7-nsGluc2A) plasmid was linearized by XbaI digestion, and RNA was generated by in vitro transcription using the T7 RiboMAX transcription kit according to the manufacturer's instructions (Promega, Madison, WI). Viral RNAs (500 ng per well of a 24-well plate) were transfected into Huh-7.5 cells in DMEM supplemented with 1% FBS and 0.1 mM NEAA using 1 μl of Lipofectamine 2000 (Invitrogen). Plates were centrifuged at 1,000 × g for 30 min at 37°C. Media were replaced 6 h later with DMEM containing 10% FBS and 0.1 mM NEAA. Viral RNA replication was assayed by measuring luciferase released into the medium over time (at 6, 24, 48, and 72 h posttransfection) using the Renilla luciferase assay system (Promega).
To analyze infectious virus production from HCV Jc1FLAG(p7-nsGluc2A), naive Huh-7.5 cells were incubated with cell culture supernatant containing infectious virus for 4 h and exchanged with fresh medium. Detection of luciferase in the supernatant was carried out 2 days after infection with the Renilla luciferase assay system (Promega).
J6/JFH-1-based NS5A recombinant virus (20) was produced by transfecting in vitro-transcribed RNA into Huh-7.5 cells, followed by determination of the infectious titer by limiting-dilution assay staining for NS3.
Western blotting.
Cells were lysed in phosphate-buffered saline (PBS) containing 1% Triton X-100, 0.05% SDS, 0.5% deoxycholate, and EDTA-free protease inhibitor cocktail (Roche) for 10 min on ice. Lysates were clarified by centrifugation (16,000 × g, 10 min, 4°C), and proteins were separated on 4 to 12% SDS-polyacrylamide gels. Proteins were transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA). Blots were incubated with primary antibodies against MOBKL1B (1:1,000; Cell Signaling Technology), CypA (1:1,000; Santa Cruz Biotechnology), or β-actin (1:10,000; Sigma), followed by horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse secondary antibody (1:10,000; Jackson ImmunoResearch, West Grove, PA). Proteins were detected using Super Signal West Pico substrate (Thermo Scientific, Waltham, MA).
Cytotoxicity assay.
Toxicity of the MOBKL1B knockdown was evaluated using the CellTiter-Glo luminescent cell viability assay according to the manufacturer's instructions (Promega). Huh-7.5 cells were transfected twice at 24-h intervals with three individual MOBKL1B-specific siRNAs (1, 2, and 3), their mismatch siRNAs (1 C911, 2 C1012, and 3 C911), or irrelevant (IRR) siRNA. Cells were lysed in CellTiter-Glo reagent at 48 h after the second siRNA transfection and assessed for the luminescent signal.
Yeast two-hybrid binding assays.
Yeast two-hybrid binding assays were performed as described previously (21) using the Matchmaker GAL4 yeast two-hybrid 3 system (Clontech).
Purification of recombinant proteins.
Full-length MOBKL1B and MOBKL1B33-216 were expressed as His6-Smt3 N-terminal fusion proteins in Rosetta (DE3) cells (Merck KGaA, Darmstadt, Germany). The cells were grown to reach an A600 of ∼0.8 and induced for the protein expression with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside). After incubation for 5 h at 25°C, cells were harvested by centrifugation and lysed for 30 min at 4°C using a lysis buffer consisting of 20 mM Tris (pH 8.0), 50 mM imidazole, 500 mM NaCl, and EDTA-free protease inhibitor cocktail (Roche) containing 10 mg/ml lysozyme, followed by sonication. Lysates were clarified by centrifugation (35,000 × g, 30 min, 4°C) and passed through an Ni-Sepharose 6 Fast Flow (GE Healthcare, Uppsala, Sweden) column. Unbound and nonspecifically bound materials were washed off using wash buffer (20 mM Tris [pH 8.0], 50 mM imidazole, and 25 mM NaCl), and bound proteins were eluted with the same buffer containing 250 mM imidazole. The His6-Smt3 tag was removed as described previously (22) from eluted proteins by incubation with Ulp1 protease (100 μg/10 mg protein, 4 h, 4°C). The proteins were further purified by ion-exchange chromatography (HiTrap Q HP; GE Healthcare), a second pass over Ni-Sepharose 6 Fast Flow to remove free His6-Smt3 tag, and finally size exclusion chromatography (Superdex 75; GE Healthcare) in buffer containing 20 mM Tris (pH 7.5), 50 mM NaCl, 2 mM dithiothreitol (DTT), and 10% glycerol. MOBKL1B proteins were concentrated to 25 mg/ml using an Amicon Ultra-15 centrifugal filter unit (Millipore).
Biosensor analyses.
Biosensor binding experiments were performed using BIACORE2000 (GE Healthcare) and ForteBio Octet Red 384 (ForteBio, Menlo Park, CA) instruments. For the experiments using the BIACORE2000 instrument, ∼5,000 response units (RU) of anti-GST antibody was immobilized on flow cells of the research-grade CM5 sensor chip using amine-coupling chemistry (23). GST-NS5A peptides or GST alone was captured from soluble Escherichia coli lysates on the antibody surface at densities of 1.5 to 2.5 kRU. Pure full-length MOBKL1B protein at the designated concentrations was injected in triplicate in running buffer (20 mM sodium phosphate [pH 7.2], 150 mM NaCl, 0.1 mg/ml bovine serum albumin [BSA], and 0.01% Tween 20) at a flow rate of 50 μl/min at 25°C. Data were collected at a rate of 2 Hz during 50-s association and dissociation phases. All interactions reached equilibrium rapidly and dissociated completely within seconds. Equilibration constants were obtained by fitting the responses at 25 s to a simple one-to-one binding interaction isotherm (24). The experiments using ForteBio Octet Red 384 were performed as described above except that the running temperature was 30°C.
Crystallization.
Terminus-blocked (N, acetyl; C, amide group) NS5A308-327 peptide was synthesized with the sequence 308ALPAWARPDYNPPLVESWRR327. Core MOBKL1B (residues 33 to 216) and NS5A308-327 peptide were mixed to a protein/peptide molar ratio of ∼1:1.2 at a final core MOBKL1B concentration of ∼15 mg/ml. Crystals of core MOBKL1B-NS5A308-327 complex were grown by hanging-drop vapor diffusion at 4°C. The reservoir solution was composed of 0.1 M HEPES (pH 7.5), 0.1 M potassium chloride, and 15% (wt/vol) polyethylene glycol (PEG) 5000. Crystals were cryoprotected by transfer to the reservoir solution with 20% (vol/vol) glycerol and frozen in liquid nitrogen.
Data collection and refinement.
Data were collected at beamline X29 at the Brookhaven National Laboratory's National Synchrotron Light Source. The data set was processed with DENZO, SCALEPACK, and CCP (25, 26). Molecular replacement experiments were carried out with MOLREP and PHASER (CCP4 program suite). The structure of core MOBKL1B alone (PDB code 1PI1) (27) was used as a searching model in the initial molecular replacement experiment. Rigid-body refinement and simulated annealing (28) were carried out sequentially. Model building was done with the O program (29). Noncrystallographic symmetry (NCS) restraints were applied to the two complexes in the asymmetric unit. No NCS was applied in the final round of refinement. For statistics, see Table 2.
TABLE 2.
Crystallographic data and refinement statistics
| Parameter | Value for core MOBKL1B–NS5A(x-y) complex (PDB ID 4J1V)a |
|---|---|
| Data collection statistics | |
| Source | NSLS X29 |
| Wavelength (Å) | 1.075 |
| Resolution (Å) | 50-1.95 (1.97-1.95) |
| Space group | P21 |
| Unit cell | |
| a, b, c (Å) | 50.1, 54.5, 86.6 |
| α, β, γ (°) | 90, 89.9, 90 |
| No. of observations | 120,225 |
| No. of unique reflections | 34,680 |
| Redundancy | 3.5 (3.0) |
| Completeness (%) | 99.2 (97.4) |
| Mean I/σI | 24.5 |
| Rmerge on Ib | 0.04 (0.08) |
| Cutoff criteria I/σI | 0 |
| Refinement statistics | |
| Resolution limits (Å) | 50-1.95 (1.97-1.95) |
| No. of reflections | 34083 |
| Completeness (%) | 99.5 |
| Cutoff criteria I/σI | 0 |
| No. of atoms | |
| Protein | 3146 |
| Solvent | 387 |
| Rcrystc | 0.196 (0.219) |
| Rfree (5% of data) | 0.225 (0.252) |
| Bonds (Å)/angles (°)d | 0.0056/1.02 |
| Mean B value (Å2) | 30.0 |
| Ramachandran plot | |
| Most favored (%) | 93.3 |
| Additionally allowed (%) | 6.7 |
| Generously allowed (%) | 0 |
| Disallowed (%) | 0 |
Numbers in parentheses indicate the range of the highest-resolution bin and statistics for data in this resolution bin.
Rmerge = ∑hkl∑i|I(hkl)i − 〈I(hkl)〉 /∑hkl∑i〈I(hkl)i〉.
Rcryst = ∑hkl |Fo(hkl)-Fc(hkl)|/∑hkl|Fo(hkl)|, where Fo and Fc are observed and calculated structure factors, respectively.
Values indicate root mean square deviations in bond lengths and bond angles.
RESULTS
Identification of HCV NS5A-interacting cellular proteins.
To identify HCV NS5A-interacting cellular proteins, we carried out a pulldown assay in Huh-7.5 cells using Jc1 HCV, a fully infectious genotype 2a virus (17) that expressed One-STrEP tag (OST)-tagged NS5A (Jc1 NS5A-OST), followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Two independent analyses were performed using the same experimental conditions, and nine cellular proteins with MASCOT scores higher than 100 were identified (Table 1). The possible roles in virus replication of the identified host proteins were further evaluated by siRNA knockdown. Except for the previously known HCV replication factors CypA (11–14) and phosphatidylinositol-4-kinase IIIα (PIK4CA) (30) and newly identified MOBKL1B, siRNA knockdown of the other 6 NS5A-interacting host proteins did not affect HCV replication (not shown). We therefore focused our attention on MOBKL1B, a component of the SWH tumor suppressor pathway not previously implicated in HCV replication. To confirm the interaction between MOBKL1B and NS5A, we performed yeast two-hybrid interaction assays with MOBKL1B and each of the individual HCV proteins and found that MOBKL1B bound to HCV NS5A, confirming the pulldown results (Fig. 1). Moreover, NS5A was the only HCV protein with which an interaction with MOBKL1B was detected. Thus, we further characterized the MOBKL1B-NS5A interaction and its possible role in HCV replication.
TABLE 1.
Cellular proteins copurified with HCV NS5A identified by LC-MS/MS analyses
| Protein name | Entrez gene ID | Mass (Da) | MASCOT scorea | No. of observed peptidesa | Cellular localization |
|---|---|---|---|---|---|
| BIN1 | 274 | 64,887 | 7,490, 8,165 | 371, 361 | Cytoplasm/membrane/nucleus |
| NAP1L1 | 4,673 | 45,631 | 2,470, 2,233 | 122, 115 | Cytoplasm/nucleus |
| NAP1L4 | 4,676 | 42,968 | 1,908, 1,253 | 167, 167 | Cytoplasm/nucleus |
| PPIA | 5,478 | 18,229 | 1,203, 894 | 75, 75 | Cytoplasm/membrane |
| PIK4CA | 5,297 | 233,595 | 412, 684 | 34, 48 | Membrane |
| AMPH | 273 | 76,381 | 405, 305 | 37, 24 | Cytoplasm/membrane |
| MOBKL1B | 55,233 | 25,246 | 285, 413 | 19, 15 | Cytoplasm |
| USP19 | 10,869 | 153,242 | 261, 391 | 20, 38 | Membrane |
| PDIA6 | 10,130 | 48,490 | 243, 111 | 11, 7 | Membrane |
Values represent the results of two independent analyses.
FIG 1.
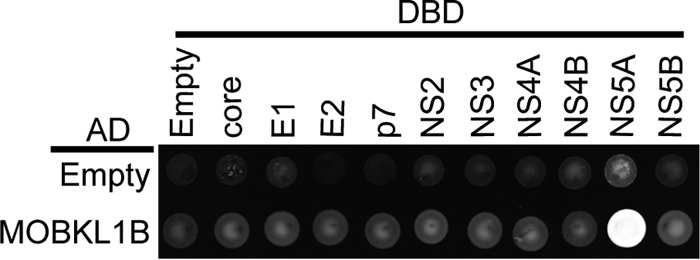
MOBKL1B interacts with NS5A. Yeast two-hybrid analysis of the interaction between MOBKL1B and 10 HCV proteins was performed. MOBKL1B-AD (GAL4 activation domain) fusion or a control AD construct (empty) was coexpressed with core, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, or NS5B-DBD (GAL4 DNA binding domain) fusions or control DBD construct (empty) and tested for positive yeast two-hybrid interactions under selective nutritional conditions (lacking leucine, tryptophan, histidine, and adenosine). Each viral protein was derived from the Jc1 sequence.
RNA interference implicates MOBKL1B in HCV RNA replication.
Given the robust interaction between NS5A and MOBKL1B, we used a depletion strategy to investigate the importance of MOBKL1B for HCV growth in cell culture. We depleted the endogenous protein in Huh-7.5 cells using a pool of MOBKL1B-targeting siRNAs (Dharmacon SMARTpool) before infection with genotype 2 HCV Jc1. Measured at 72 h postinfection, siRNA-treated cells had no detectable MOBKL1B, which was associated with a 10- to 20-fold reduction in viral progeny released into the medium compared to the case for control cells treated with irrelevant siRNA (Fig. 2A). Since a defect following infection may represent a block to viral entry, RNA replication, or infectious particle production, we next investigated the stage of the viral life cycle impacted by MOBKL1B depletion. To test the importance of MOBKL1B for RNA replication, we used an Huh-7.5-derived cell line lacking the HCV entry factor, CD81 (31); this allowed detection of single-cycle replication without contributions from viral spread. These cells were treated with MOBKL1B-specific individual siRNA 1 or 2 and then transfected with in vitro-transcribed Jc1 RNA encoding a luciferase reporter [Jc1FLAG(p7-nsGluc2A)] (19). We observed that viral RNA replication, as assessed by luciferase expression, was decreased ∼95% and ∼65% in cells treated with MOBKL1B siRNAs 1 and 2, respectively, as measured at 48 h posttransfection (Fig. 2B). These results indicate that treatment with siRNA targeting MOBKL1B affected the RNA replication step of the HCV life cycle. Since siRNA 1 more strongly reduced RNA replication and MOBKL1B expression than siRNA 2, we selected it for subsequent experiments to achieve efficient endogenous MOBKL1B knockdown.
FIG 2.

Treatment with MOBKL1B siRNA inhibits HCV RNA replication. (A) HCV growth in culture is inhibited by MOBKL1B depletion. Huh-7.5 cells were transfected twice with MOBKL1B-specific or irrelevant (IRR) siRNA at 24-h intervals and were infected with HCV Jc1 at 48 h after the second transfection. Depletion of endogenous MOBKL1B protein was analyzed by Western blotting (top panel); β actin levels are shown as a control. The results shown are representative of three independent experiments. (B) MOBKL1B depletion blocks viral RNA replication. Depletion of MOBKL1B in CD81 knockdown Huh-7.5 cells was performed, as described above. Two individual MOBKL1B-specific siRNAs (1 and 2) were used. Jc1FLAG(p7-nsGluc2A) RNA was transfected at 48 h after the second siRNA transfection. At each indicated time point, released luciferase was measured to analyze viral RNA replication (mean of n = 3; error bars, SD). RLU, relative light units. (C) MOBKL1B knockdown does not affect the entry of pseudoparticles. After siRNA 1 treatment, Huh-7.5 cells were infected with luciferase reporter pseudoparticles at 48 h after the second siRNA transfection. Luciferase was measured at 48 h after infection. Values are normalized to RLU measured in irrelevant-siRNA-treated cells (mean of n = 3; error bars, SD). VSV-G, vesicular stomatitis virus glycoprotein; H77-HCV, HCV strain H77 glycoproteins. (D) MOBKL1B is also required for the replication of J6/JFH1-based HCV NS5A recombinants. MOBKL1B depletion and viral genome transfection were performed as described above. Recombinant HCV variants were as follows: 1a, J6/JFH1(H77-NS5A); 1b, J6/JFH1(J4-NS5A)R867H, C1185S; 2a, J6/JFH1(J6-NS5A)F772S; 3a, J6/JFH1(S52-NS5A)D1975G; 4a, J6/JFH1(ED43-NS5A)F772S, Y1644H, E2267G; 5a, J6/JFH1(SA13-NS5A)R1978G, S2416G; 6a, J6/JFH1(HK6a-NS5A)I2268N; 7a, J6/JFH1(QC69-NS5A). The results shown are representative of two independent experiments. The dashed line indicates the quantitation limit of the assay.
We believed that the HCV RNA replication phenotype observed under conditions of MOBKL1B silencing was unlikely to be due to off-target effects since (i) three different siRNAs targeting MOBKL1B gave the same HCV replication phenotype (SMARTpool and two individual siRNAs) (Fig. 2A and B), (ii) different levels of endogenous, presumably fully functional, MOBKL1B achieved by differing efficacy of two different siRNAs correlated with the efficacy of HCV RNA replication (Fig. 2B), and (iii) we had strong evidence that MOBKL1B was capable of physically interacting with the HCV NS5A protein. We therefore proceeded to characterize the role of MOBKL1B in HCV replication in greater detail.
To investigate whether in addition to having effects on RNA replication, MOBKL1B might affect HCV entry, we utilized HIV-based luciferase-expressing pseudoparticles (HCVpp) expressing genotype 1a (H77) envelope proteins or G glycoprotein from vesicular stomatitis virus (VSV-G) as described previously (32). Knockdown of MOBKL1B did not affect the entry of HCV, suggesting that this host factor is not required for HCV entry (Fig. 2C). Next, we tested the importance of MOBKL1B for its effect on the replication of other HCV genotypes. Since authentic viral replication systems for most HCV genotypes are not yet available, we used infectious chimeric recombinant HCV variants bearing NS5A from genotypes 1 to 7 in a genotype 2 J6/JFH1 genome background (20). Although virus titers of some recombinant HCV variants were low, across all genotypes we observed a >5-fold reduction in infectious virus release into the medium after MOBKL1B knockdown in Huh-7.5 cells compared to that with an irrelevant RNA knockdown control (Fig. 2D). Taken together, the data suggest that the host factor MOBKL1B positively influences the RNA replication of all HCV genotypes.
Mapping the minimal MOBKL1B binding motif in NS5A.
Given the likely role of MOBKL1B in HCV RNA replication and its interaction with NS5A, we set out to characterize the physical interaction in greater detail. Yeast two-hybrid assays had shown that NS5A was the only HCV protein that specifically bound MOBKL1B (Fig. 1). To identify the MOBKL1B binding motif on NS5A, we carried out further yeast two-hybrid analyses using serially deleted mutants and alanine substitution mutants (Fig. 3A and B). Deletion of residues 314 to 322 or 323 to 337 reduced the MOBKL1B-NS5A interaction, and alanine substitution further pinpointed residues in the region of amino acids 315 to 324 in NS5A domain II as required for the interaction (Fig. 3A and B). To further map the minimal interaction sequence and to quantify the MOBKL1B-NS5A binding affinity, we performed biosensor assays using a series of serially deleted NS5A peptides (Fig. 3C). These experiments indicated that the minimal NS5A motif for MOBKL1B interaction (amino acids 310 to 327) bound with an apparent dissociation constant (Kd) of 18 ± 1 μM (mean ± standard deviation [SD]) (Fig. 3C, black curve). Interestingly, this binding motif is highly conserved in all seven HCV genotypes (Fig. 3D), consistent with the effect of MOBKL1B silencing on chimeric viruses containing NS5A from each of the seven genotypes (Fig. 2D).
FIG 3.

Mapping the MOBKL1B binding site in NS5A. (A) Yeast two-hybrid mapping of the MOBKL1B binding motif in NS5A. An MOBKL1B-AD fusion or a control AD construct (empty) was coexpressed with serially deleted NS5A-DBD fusion or control DBD construct (empty) and tested for positive yeast two-hybrid interactions under selective nutritional conditions (lacking leucine, tryptophan, histidine, and adenosine). NS5A deletion mutants were derived from genotype 1b, Con1. Numbers indicate the Con1 NS5A amino acid positions. (B) A schematic of NS5A is shown in the top panel. JFH1 NS5A amino acid positions are indicated. Yeast two-hybrid interactions are shown in the bottom panel. Numbers indicate the JFH1 NS5A amino acid positions. Note that JFH1 NS5A residues 310 to 335 are equivalent to Con1 NS5A residues 314 to 339. (C) Biosensor binding isotherms for full-length MOBKL1B binding to GST-JFH1 NS5A peptides. Triplicate data points are shown for each condition. Apparent binding affinities (Kds) derived after fitting to a simple one-to-one binding model were as follows: NS5A310-335, 24 ± 1 μM; NS5A310-327, 18 ± 1 μM; NS5A310-324, 182 ± 39 μM; NS5A310-320, 830 ± 100 μM; NS5A314-329, no binding detected (means ± SD). Note that adaptive mutations in 1b, 2a, 3a, 4a, 5a, and 6a recombinant HCV variants (Fig. 2D) are not located in the NS5A MOBKL1B binding motif. (D) Sequence alignment of the MOBKL1B minimal binding motif of NS5A for the seven HCV genotypes. Asterisks indicate residues absolutely conserved in all genotypes. NS5A amino acid positions (JFH1 numbering) are indicated.
Structure of the core MOBKL1B-NS5A308-327 complex.
To elucidate the molecular details of the MOBKL1B-NS5A interaction, we determined the cocrystal structure of protease-resistant core MOBKL1B (residues 33 to 216) (27) complexed with an NS5A peptide encompassing the MOBKL1B binding site (residues 308 to 327) at 1.95-Å resolution to an Rfree value of 0.225 (Rcryst = 0.196) (Fig. 4 and Table 2). The overall structure of core MOBKL1B in the complex is superimposable with that of the ligand-free protein. The structure showed two NS5A peptides binding to one MOBKL1B molecule (Fig. 4A). The peptide was resolved from L309 to E323 in one interaction (designated interaction 1) and from A308 to D316 in the other interaction (designated interaction 2). Interaction 1 is likely the relevant MOBKL1B-NS5A association detected in vitro based on the biosensor binding assay (Fig. 4B). The NS5A peptide (residues 305 to 316) encompassing interaction 2 showed no reliable binding to full-length MOBKL1B (Fig. 4B, green curve). In contrast, the structural properties of interaction 1 were consistent with the biochemical binding results (Fig. 3 and Fig. 4B), as described below.
FIG 4.

Structure of core MOBKL1B-NS5A308-327 complex. (A) Core MOBKL1B (residues 33 to 216) with the NS5A308-327 peptide depicted in ribbon representation as two views related by a 180° rotation. Nine α-helices and two β-strands are numbered as previously reported (27). There are two MOBKL1B-NS5A peptide interactions, interaction 1 and interaction 2, in which NS5A308-327 peptide was determined from L309 to E323 in interaction 1 and from A308 to D316 in interaction 2. H, α-helix; S, β-strand; N, N terminus; C, C terminus. (B) Biosensor binding isotherms for full-length MOBKL1B binding to GST-NS5A peptides. Triplicate data points are shown for each condition. No binding was detected for NS5A308-327 W312A and Y317A or for NS5A305-316 Wt in interaction 2. (C) Close-up views of interaction 1, showing the NS5A peptide (stick) binding to the shallow hydrophobic patch of MOBKL1B in space-filling models or ribbon representation (left and right panels, respectively). Amino acid residues participating in the MOBKL1B-NS5A interaction are highlighted by showing their side chains and labeled with the corresponding amino acid sequence number. Left panel, NS5A residues labeled in black. Right panel, MOBKL1B residues in blue. Black arrows in the left panel point out trans conformational amide bonds at P319 and P320 NS5A residues that are putative CypA substrates.
The MOBKL1B-NS5A interaction consists of both hydrogen bonds and van der Waals surface contacts. NS5A residues W312 and Y317 are adjacent to each other in space, with the residues between them bulging out and covered by weak electron density (Fig. 4C). The side chain of W312 is embedded in a shallow hydrophobic depression formed by the well-conserved MOBKL1B residues Y163, H164, F167, N180, L204, and L207. The W312 side chain is recognized by a hydrogen bond with the MOBKL1B Y163 main-chain carbonyl oxygen (Fig. 4C). The hydroxyl oxygen of NS5A Y317 has bifurcated hydrogen bond interactions with the side chains of MOBKL1B E176 and N180. These intermolecular hydrogen bonds are likely secured by the MOBKL1B N180 side chain orientation through a hydrogen bond with MOBKL1B Y163 that exists in the absence of NS5A peptide (27) (Fig. 4C). These structural observations explain the severely impaired MOBKL1B-NS5A interaction caused by individual alanine substitutions of NS5A peptide residues W312 and Y317, for which no binding was detected (Fig. 4B). Similarly, full-length MOBKL1B did not bind to the NS5A305-316 peptide, a region of NS5A found in the NS5A-MOBKL1B interaction 2. In contrast, we calculated that MOBKL1B bound to NS5A308-327 peptide, a region found in NS5A-MOBKL1B interaction 1, with a Kd of 6.74 ± 1 μM (Fig. 4B).
In addition, residues P319 to V322 of NS5A contribute to the interaction by multiple van der Waals surface contacts and one hydrogen bond between the main-chain nitrogen atom of NS5A V322 and the side chain of MOBKL1B T181 (Fig. 4C). This explains the ∼18-fold-reduced binding affinity of the NS5A310-320 peptide to the full-length MOBKL1B, compared to that of the NS5A 310-327 peptide (Fig. 3C). It is worth noting that the NS5A peptide contains internal stacking interactions through the W312 main chain and a β-carbon atom, the Y317 side chain, the P319 residue, and the N318 carbonyl group (Fig. 4C). Such stacking likely facilitates establishment of the interactions with MOBKL1B.
CypA-independent HCV unexpectedly reveals off-target replication inhibition mediated by MOBKL1B siRNA treatment.
Similar to the case for MOBKL1B, the host peptidyl-prolyl isomerase CypA has been reported to interact with NS5A and is critically important for HCV RNA replication (11–14). Although its detailed function remains to be defined, NS5A mutations (D316E and Y317N, here designated DEYN) that permit CypA-independent replication have been reported (13). Interestingly, these mutations are located within the NS5A motif required for MOBKL1B binding. Because of this, and since the NS5A Y317 residue is critical for MOBKL1B-NS5A interaction in vitro (Fig. 4B, blue curve), we wondered whether DEYN NS5A from the CypA-independent virus would interact with MOBKL1B. We therefore examined the interaction of NS5A and MOBKL1B in the context of HCV replication after transfection of Huh-7.5 cells with wild-type HCV (Jc1 NS5A-OST) or the CypA-independent mutant (Jc1 NS5A-OST DEYN) genomic RNA. After lysis, a coprecipitation experiment was performed using antibodies to the OST tag in NS5A. Consistent with the previous in vitro experiment, MOBKL1B was coprecipitated with wild-type NS5A. Precipitation of the DEYN mutant NS5A, however, failed to coprecipitate MOBKL1B (Fig. 5A), as expected given the loss of residue Y317, which is critical for the NS5A-MOBKL1B interaction.
FIG 5.
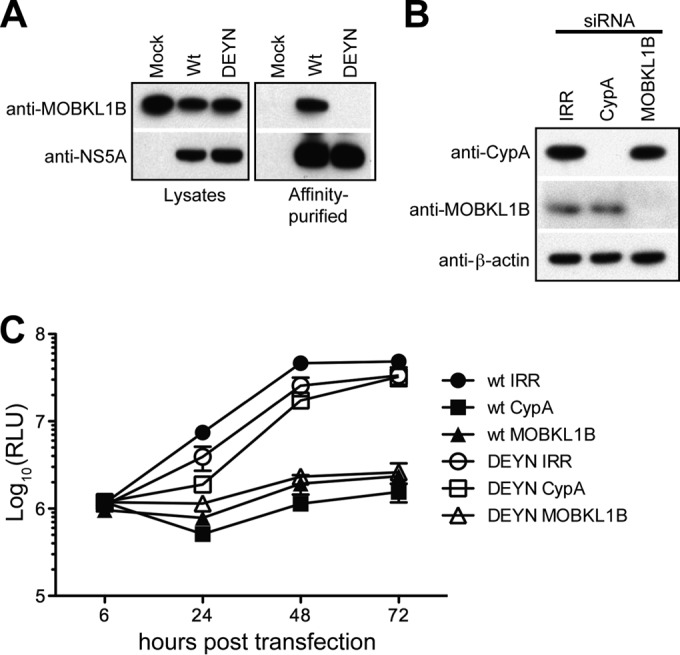
MOBKL1B depletion reduces CypA-independent HCV replication despite lack of MOBKL1B-NS5A interaction. (A) NS5A bearing the DEYN mutation does not bind to MOBKL1B. One-STrEP-tag (OST)-fused NS5A was purified using Strep-Tactin Sepharose from Jc1 NS5A-OST wild-type- or DEYN mutant-transfected Huh-7.5 cell lysates. Left panels, Western blot signals of MOBKL1B and NS5A in the input lysates using anti-MOBKL1B and anti-NS5A antibodies. Right panels, Western blot signals following affinity purification of NS5A. (B) Depletion of endogenous CypA or MOBKL1B protein in CD81 knockdown Huh-7.5 cells was analyzed by Western blotting; β-actin is included as a control. (C) Viral genome replication following CypA or MOBKL1B knockdown. At each indicated time point, released luciferase was measured to analyze viral RNA replication (mean of n = 3; error bars, SD). RLU, relative light units.
Because there was no interaction between the DEYN mutant NS5A and MOBKL1B, we expected that replication of such a mutant virus would be independent of MOBKL1B and would be unaffected by MOBKL1B depletion. To test this, we constructed a CypA-independent mutant luciferase reporter virus [Jc1FLAG(p7-nsGluc2A)DEYN] and tested the effect of MOBKL1B and CypA depletion (Fig. 5B) on virus replication (Fig. 5C). Wild-type Jc1FLAG(p7-nsGluc2A) replication was inhibited by the depletion of either CypA (∼40-fold reduction at 48 h posttransfection) or MOBKL1B (∼20-fold reduction). As expected, Jc1FLAG(p7-nsGluc2A)DEYN replication was not affected by CypA depletion. In contrast, and quite unexpectedly, its replication was impaired ∼15-fold after MOBKL1B depletion (Fig. 5C). It was unanticipated that replication of this CypA-independent virus would be reduced in MOBKL1B siRNA-treated cells, given that its NS5A did not interact with MOBKL1B in protein pulldown assays. These results were discordant from our initial finding that MOBKL1B-NS5A interactions were associated with efficient virus replication, and they led us to question whether MOBKL1B was implicated in virus replication indirectly and not via direct interaction with NS5A. As well, these findings caused us to reexamine our siRNA gene knockdown strategy and to explore whether off-target inhibitory effects might be blocking virus replication.
Seed sequence-matched siRNA controls do not deplete MOBKL1B.
To reinvestigate the importance of MOBKL1B for HCV growth in cell culture, we redesigned our siRNA approach using two previously used MOBKL1B-specific siRNAs (siRNAs 1 and 2), and a new siRNA (siRNA 3) to deplete endogenous MOBKL1B (Fig. 6A). In addition, we included both irrelevant (IRR) and recently reported C911/C1012 negative-control siRNAs (33) for each MOBKL1B-specific siRNA to more carefully investigate whether effects of silencing could be due to off-target effects of the siRNAs. C911/C1012 control siRNAs for 19-mer and 21-mer siRNAs, respectively, are 3-base internal mismatch controls where bases 9 to 11 or 10 to 12 are switched to the complementary nucleotides of the target mRNA-specific siRNA. Compared to the targeting siRNAs, such control siRNAs do not lead to degradation of the siRNA target mRNA and usually exert a much less pronounced knockdown effect. However, they retain the same seed sequences (bases 2 to 8 of the siRNA antisense and sense strands) and therefore have the potential to retain any seed-dependent off-target effects (33, 34).
FIG 6.
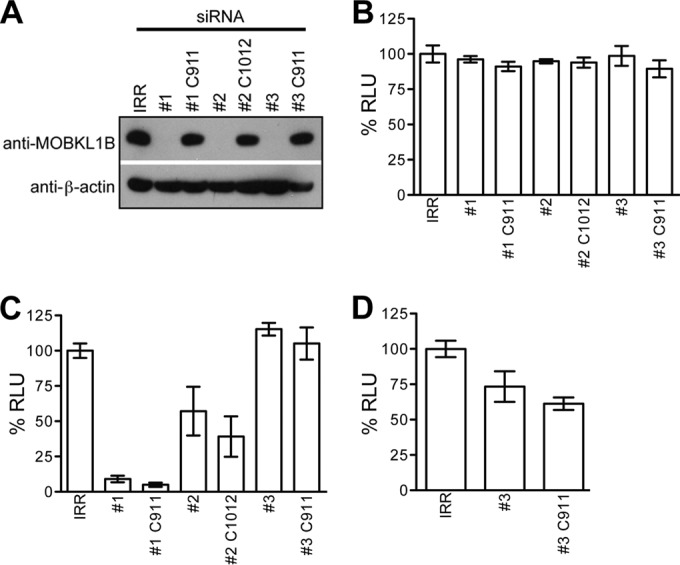
Seed sequence-matched siRNA shows that MOBKL1B is not required for HCV RNA replication in Huh-7.5 cells. (A) Three MOBKL1B-specific siRNAs (1, 2, and 3), their seed sequence-matched control siRNAs (1 C911, 2 C1012, and 3 C911), or irrelevant (IRR) siRNA was transfected into Huh-7.5 cells as described for Fig. 2. Depletion of endogenous MOBKL1B protein was analyzed by Western blotting. β-Actin levels are shown as a control. (B) MOBKL1B siRNA treatment is not cytotoxic. Huh-7.5 cells were transfected twice with three individual MOBKL1B-specific siRNAs (1, 2, and 3), their seed sequence-matched siRNAs (1 C911, 2 C1012, and 3 C911), or irrelevant (IRR) siRNA at 24-h intervals. Toxicity of the MOBKL1B knockdown was evaluated with a luminescent cell viability assay (mean of n = 3; error bars, SD). RLU, relative light units. (C) MOBKL1B depletion does not inhibit HCV genome replication. Depletion of MOBKL1B in CD81 knockdown Huh-7.5 cells was performed, as described above. Jc1FLAG(p7-nsGluc2A) was transfected at 48 h after the second siRNA transfection. Medium was collected 48 h later for luciferase quantitation. (D) MOBKL1B depletion and infectious virus production. Naive Huh-7.5 cells were infected from the media from cells treated with IRR, siRNA 3, and siRNA 3 C911 (shown in panel C), and luciferase was measured to analyze infectious virus production. Values are normalized to RLU measured in irrelevant siRNA-treated cells (mean of n = 3; error bars, SD).
Endogenous MOBKL1B was efficiently depleted in cells treated with three different MOBKL1B-specific siRNAs but not with the IRR control or their corresponding C911/C1012 control siRNAs, as expected (Fig. 6A). As a control for cytotoxicity, we showed that transfection with all siRNAs did not affect cell viability (Fig. 6B).
Seed sequence-matched siRNA controls show that MOBKL1B is not required for HCV replication in Huh-7.5 cells.
To retest the effect of MOBKL1B on viral RNA replication and infectious particle production, we first transfected Jc1FLAG(p7-nsGluc2A) RNA into CD81 knockdown Huh-7.5 cells that had been treated with the siRNAs and quantified luciferase expression in the supernatant (Fig. 6C). We observed that although viral RNA replication was decreased by ∼90% in cells treated with siRNA 1, this was likely an off-target effect because its control (1 C911) had a comparable effect (Fig. 6C), despite normal MOBKL1B protein expression (Fig. 6A). The results were similar, but to a lesser degree, for siRNA 2, where transfection of its control (2 C1012) also reduced virus genome replication relative to that with the irrelevant RNA control. In the absence of the seed sequence-matched controls for both siRNAs 1 and 2, the resultant reduction in virus genome replication (Fig. 2B) was easily misinterpreted as being related to MOBKL1B depletion. In contrast, transfection of siRNA 3, which efficiently reduced MOBKL1B levels, and its control (3 C911), which did not, both failed to affect HCV RNA replication, suggesting that MOBKL1B depletion via this siRNA veritably had no effect on virus genome replication (Fig. 6C). This result observed for siRNA 3 contrasts directly with results initially obtained with siRNAs 1 and 2 (Fig. 2B), where levels of virus replication correlated with MOBKL1B levels. Because the latter experiment includes a seed sequence control matching the targeting siRNA, interpretation is simplified, and we conclude that HCV RNA replication is independent from MOBKL1B expression.
To be complete, we tested whether MOBKL1B knockdown affected infectious particle production. The supernatant from the siRNA 3-transfected cells shown in Fig. 6C was added to Huh-7.5 cells, and infection was quantified by luciferase detection. As shown in Fig. 6D, infectious particle production was decreased to about 70% in cells treated with siRNA 3 relative to that with irrelevant RNA. However, infectious particle production was even lower from cells treated with the siRNA 3 C911 control, where MOBKL1B levels were unaffected and thus should not be expected to affect infectivity. Thus, although siRNA 3 legitimately had no effect on HCV RNA replication (Fig. 6C), its effect on the generation of infectious particles was most likely due to off-target effects, since similar results were obtained with its C911 matched control (Fig. 6D). Similarly, MOBKL1B depletion in immune-suppressed human fetal liver cells (35) using siRNA 3 did not affect HCV RNA replication or infectious particle production (data not shown). Taken together, these more stringent siRNA knockdown experiments showed that there is no functional requirement for the MOBKL1B-NS5A interaction in HCV genome replication in cell culture. Furthermore, they suggest that all siRNA experiments should be carried out with matched seed sequence controls to differentiate false- and true-positive results (Fig. 2B versus Fig. 6C, Fig. 6D).
DISCUSSION
In this report, we identified MOBKL1B, a component of the SWH tumor suppressor pathway, as an HCV NS5A binding partner. On the basis of initial MOBKL1B siRNA knockdown experiments, we believed that the MOBKL1B-NS5A interaction was essential for HCV genome replication, and we committed significant time and resources to characterize the interaction. However, we ultimately found that this interpretation was incorrect and that our original MOBKL1B siRNAs had off-target inhibitory effects on HCV RNA replication. Although we tested three MOBKL1B siRNAs, all had off-target effects on RNA replication or virion production. The use of many more siRNAs of different sequence targeting the same gene might have been one way to diminish false-positive results (33). Compensatory expression of the target protein after siRNA treatment and the restoration of the phenotype also constitute a valuable method to verify that a phenotype is due to depletion of the factor under study. However, this method can potentially fail, due to multiple reasons, such as suboptimal protein restoration, erroneous posttranslational modifications or localization, incorrect timing of competing silencing and restoration, and undefined effects on other interacting proteins.
In fact, while we were able to restore MOBKL1B protein levels to near-endogenous levels in the presence of siRNA 1 using an siRNA-resistant MOBKL1B construct, expecting to restore HCV RNA replication, we saw no corresponding rescue of HCV replication (data not shown). As an additional control, we studied the depletion and restoration of large tumor suppressor kinase 1 (LATS1), a component of the SWH tumor suppressor pathway whose expression depends on MOBKL1B (36). Indeed, MOBKL1B depletion resulted in codepletion of LATS1. However, the restoration of MOBKL1B did not rescue LATS1 expression to pre-MOBKL1B knockdown levels (data not shown). Thus, at the time we reasoned that failure to restore HCV replication in the setting of the rescue experiment might be because the MOBKL1B was unable to function properly due to alterations in other pathway components, which had not yet been properly restored during the time frame of the experiment. In retrospect, we, and likely other investigators, were too quick to dismiss a failure to restore function upon successful factor reconstitution.
The subcellular localization of MOBKL1B in the presence and absence of HCV would have been an additional useful way to analyze the interaction between NS5A and MOBKL1B. However, despite using various anti-MOBKL1B antibodies, we had difficulty obtaining reliable MOBKL1B-specific signals in immunofluorescence experiments (data not shown), precluding the use of this approach.
Of all the methods to discern whether off-target effects are contributing to a phenotype under study, we would like to note the efficacy of the C911 and C1012 control siRNAs in distinguishing between true- and false-positive results after siRNA treatment. Although siRNA is a potent molecular tool to study the function of target genes and also a potential therapeutic tool for various diseases, off-target effects of siRNA can lead to deceptive discoveries and hamper the therapeutic usage of RNAi (37, 38). On the basis of our observations, we suggest that C911 and C1012 matched control siRNAs should be adopted for RNA knockdown experiments as a more stringent control than IRR or scrambled RNAi. Collectively, these results have demonstrated that multiple approaches should be used to define functional protein interactions. These could include but not be limited to gene knockdowns consisting of diverse siRNAs with seed sequence-matched controls, protein colocalization, and compensatory expression of the siRNA target protein. Performed early on, they would reduce or eliminate the need for further studies if the effect of gene knockdown is not verified.
Although MOBKL1B is not apparently required for HCV replication in cell culture, the interaction with NS5A does occur, and we were able to characterize the peptide complex structurally in what is, to our knowledge, the first reported cocrystal structure involving NS5A and a cellular binding partner. NS5A is a membrane-anchored phosphoprotein composed of three domains (I, II, and III) separated by low-complexity sequences (39). Among the three domains, NS5A domain I is highly conserved across all HCV genotypes, with an N-terminal amphipathic α-helix that is responsible for membrane association (40). NS5A domain II is intrinsically unstructured, lacking discrete secondary structures (41), and may be inherently flexible, allowing NS5A to interact with multiple biological partners (42–44). Although the NS5A peptide in the complex structure is short, it highlights the flexible nature of NS5A domain II (41). We found that the NS5A peptide binds to MOBKL1B without forming α-helical or β-strand elements, allowing discontinuous amino acid residues to contact the MOBKL1B surface. These results bear on how the structural flexibility of NS5A domain II contributes to the broad virus-host interactions needed for virus replication. This observation is highlighted by our finding that MOBKL1B and a previously described HCV replication factor, CypA, share overlapping NS5A binding motifs.
In the core MOBKL1B-NS5A peptide complex structure, the peptide contains three putative CypA substrate proline residues (P315, P319, and P320) (13), each of which adopted only trans conformational amide bonds (Fig. 4C, black arrows [P319 and P320] and data not shown [P315]). Furthermore, P319 and P320 stabilize the MOBKL1B-NS5A interaction via van der Waals contacts, and P319 participates in internal stacking. It is therefore conceivable that cis conformations of peptidyl-prolyl bonds at these positions will inhibit MOBKL1B-NS5A binding. The minimal MOBKL1B binding domain of NS5A adopts a loop shape in our cocrystal structure, suggesting that MOBKL1B-NS5A interactions may restrain the inherent flexibility of NS5A domain II to produce a transiently stable conformation. Despite characterization of a structural interaction between NS5A and MOBKL1B, its relevance to HCV genome and virus replication has not been demonstrated. However, the identification of MOBKL1B as an NS5A binding protein may shed light on the pathogenesis of hepatitis C. To our knowledge, this is the first report showing the direct connection between a cancer-associated virus and the SWH tumor suppressor pathway. Previous studies have shown that the phosphorylated active form of MOBKL1B is frequently lost in human hepatocarcinoma and that interruption of the SWH tumor suppressor pathway in mice can promote liver tumors (45–47). It is worth noting that a MOBKL1B residue, T181 (48), that is important for the normal function of this pathway makes a hydrogen bond with the NS5A peptide. Taken together with previous reports, our findings suggest that the connection between MOBKL1B-NS5A binding, the SWH tumor suppressor pathway, and HCV-induced hepatocellular carcinoma should be further examined.
ACKNOWLEDGMENTS
We thank the staff of the Rockefeller Proteomics Resource Center for peptide synthesis and mass spectrometry analyses, Jens Bukh (University of Copenhagen, Denmark) for kindly providing recombinant HCV variants, David G. Myszka (Biosensor Tools LLC, Salt Lake City, UT) for assistance with the biosensor analyses, and Mayla Hsu for editing. We also thank the staff of the Rockefeller Structural Biology Resource Center and the X29 beamline (National Synchrotron Light Source).
The research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number R01CA057973 (C.M.R.). Additional financial support was provided by the Greenberg Medical Research Institute and the Starr Foundation (to C.M.R). H.C. was supported in part by a Merck postdoctoral fellowship at the Rockefeller University.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 16 July 2014
REFERENCES
- 1.Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. 2000. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann. Intern. Med. 132:296–305. 10.7326/0003-4819-132-4-200002150-00008 [DOI] [PubMed] [Google Scholar]
- 2.Schinazi R, Halfon P, Marcellin P, Asselah T. 2014. HCV direct-acting antiviral agents: the best interferon-free combination. Liver Int. 34:69–78. 10.1111/liv.12423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tellinghuisen TL, Rice CM. 2002. Interaction between hepatitis C virus proteins and host cell factors. Curr. Opin. Microbiol. 5:419–427. 10.1016/S1369-5274(02)00341-7 [DOI] [PubMed] [Google Scholar]
- 4.Pawlotsky JM. 2013. NS5A inhibitors in the treatment of hepatitis C. J. Hepatol. 59:375–382. 10.1016/j.jhep.2013.03.030 [DOI] [PubMed] [Google Scholar]
- 5.Blight KJ, Kolykhalov AA, Rice CM. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972–1974. 10.1126/science.290.5498.1972 [DOI] [PubMed] [Google Scholar]
- 6.Appel N, Zayas M, Miller S, Krijnse-Locker J, Schaller T, Friebe P, Kallis S, Engel U, Bartenschlager R. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C virus infectious particle assembly. PLoS Pathog. 4:e1000035. 10.1371/journal.ppat.1000035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tellinghuisen TL, Foss KL, Treadaway JC, Rice CM. 2008. Identification of residues required for RNA replication in domain II and III of the hepatitis C virus NS5A protein. J. Virol. 82:1073–1083. 10.1128/JVI.00328-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao B, Tumaneng K, Guan KL. 2011. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 13:877–883. 10.1038/ncb2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng Q, Hong W. 2008. The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell 13:188–192. 10.1016/j.ccr.2008.02.011 [DOI] [PubMed] [Google Scholar]
- 10.Harvey K, Tapon N. 2007. The Salvador-Warts-Hippo pathway—an emerging tumour-suppressor network. Nat. Rev. Cancer 7:182–191. 10.1038/nrc2070 [DOI] [PubMed] [Google Scholar]
- 11.Kaul A, Stauffer S, Berger C, Pertel T, Schmitt J, Kallis S, Zayas M, Lohmann V, Luban J, Bartenschlager R. 2009. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 5:e1000546. 10.1371/journal.ppat.1000546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanoulle X, Badillo A, Wieruszeski JM, Verdegem D, Landrieu I, Bartenschlager R, Penin F, Lippens G. 2009. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J. Biol. Chem. 284:13589–13601. 10.1074/jbc.M809244200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang F, Robotham JM, Grise H, Frausto S, Madan V, Zayas M, Bartenschlager R, Robinson M, Greenstein AE, Nag A, Logan TM, Bienkiewicz E, Tang H. 2010. A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS Pathog. 6:e1001118. 10.1371/journal.ppat.1001118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foster TL, Gallay P, Stonehouse NJ, Harris M. 2011. Cyclophilin A interacts with domain II of hepatitis C virus NS5A and stimulates RNA binding in an isomerase-dependent manner. J. Virol. 85:7460–7464. 10.1128/JVI.00393-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baugh JM, Garcia-Rivera JA, Gallay PA. 2013. Host-targeting agents in the treatment of hepatitis C: a beginning and an end? Antiviral Res. 100:555–561. 10.1016/j.antiviral.2013.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. 10.1126/science.1114016 [DOI] [PubMed] [Google Scholar]
- 17.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. U. S. A. 103:7408–7413. 10.1073/pnas.0504877103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaller T, Appel N, Koutsoudakis G, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2007. Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J. Virol. 81:4591–4603. 10.1128/JVI.02144-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marukian S, Jones CT, Andrus L, Evans MJ, Ritola KD, Charles ED, Rice CM, Dustin LB. 2008. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 48:1843–1850. 10.1002/hep.22550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scheel TKH, Gottwein JM, Mikkelsen LS, Jensen TB, Bukh J. 2011. Recombinant HCV variants with NS5A from genotypes 1–7 have different sensitivities to an NS5A inhibitor but not interferon-α. Gastroenterology 140:1032–1043. 10.1053/j.gastro.2010.11.036 [DOI] [PubMed] [Google Scholar]
- 21.Langelier C, von Schwedler UK, Fisher RD, De Domenico I, White PL, Hill CP, Kaplan J, Ward D, Sundquist WI. 2006. Human ESCRT-II complex and its role in human immunodeficiency virus type 1 release. J. Virol. 80:9465–9480. 10.1128/JVI.01049-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mossessova E, Lima CD. 2000. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Mol. Cell 5:865–876. 10.1016/S1097-2765(00)80326-3 [DOI] [PubMed] [Google Scholar]
- 23.Johnsson B, Lofas S, Lindquist G. 1991. Immobilization of proteins to a carboxymethyldextran-modified gold surface for biospecific interaction analysis in surface plasmon resonance sensors. Anal. Biochem. 198:268–277. 10.1016/0003-2697(91)90424-R [DOI] [PubMed] [Google Scholar]
- 24.Myszka DG. 1999. Improving biosensor analysis. J. Mol. Recognit. 12:279–284 10556875 Year does not match (PubMed did not provide). [DOI] [PubMed] [Google Scholar]
- 25.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 276:307–326. 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 26.CCP4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50:760–763. 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
- 27.Stavridi ES, Harris KG, Huyen Y, Bothos J, Verwoerd PM, Stayrook SE, Pavletich NP, Jeffrey PD, Luca FC. 2003. Crystal structure of a human Mob1 protein: toward understanding Mob-regulated cell cycle pathways. Structure 11:1163–1170. 10.1016/S0969-2126(03)00182-5 [DOI] [PubMed] [Google Scholar]
- 28.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D 54:905–921. 10.1107/S0907444998003254 [DOI] [PubMed] [Google Scholar]
- 29.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47:110–119. 10.1107/S0108767390010224 [DOI] [PubMed] [Google Scholar]
- 30.Tai AW, Benita Y, Peng LF, Kim SS, Sakamoto N, Xavier RJ, Chung RT. 2009. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 5:298–307. 10.1016/j.chom.2009.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Witteveldt J, Evans MJ, Bitzegelo J, Koutsoudakis G, Owsianka AM, Angus AG, Keck ZY, Foung SK, Pietschmann T, Rice CM, Patel AH. 2009. CD81 is dispensable for hepatitis C virus cell-to cell transmission in hepatoma cells. J. Gen. Virol. 90:48–58. 10.1099/vir.0.006700-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wölk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. 10.1038/nature05654 [DOI] [PubMed] [Google Scholar]
- 33.Buehler E, Chen Y, Martin S. 2012. C911: A bench-level control for sequence specific siRNA off-target effects. PLoS One 7:e51942. 10.1371/journal.pone.0051942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Birmingham A, Anderson EM, Reynolds A, Ilsley-Tyree D, Leake D, Fedorov Y, Baskerville S, Maksimova E, Robinson K, Karpilow J, Marshall WS, Khvorova A. 2006. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat. Methods 3:199–204. 10.1038/nmeth854 [DOI] [PubMed] [Google Scholar]
- 35.Andrus L, Marukina S, Jones CT, Catanese MT, Sheahan TP, Schoggins JW, Barry WT, Dustin LB, Trehan K, Ploss A, Bhatia SN, Rice CM. 2011. Expression of paramyxovirus V proteins promotes replication and spread of hepatitis C virus in cultures of primary human fetal liver cells. Hepatology 54:1901–1912. 10.1002/hep.24557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishio M, Hamada K, Kawahara K, Sasaki M, Noguchi F, Chiba S, Mizuno K, Suzuki SO, Dong Y, Tokuda M, Morikawa T, Hikasa H, Eggenschwiler J, Yabuta N, Nojima H, Nakagawa K, Hata Y, Nishina H, Mimori K, Mori M, Sasaki T, Mak TW, Nakano T, Itami S, Suzuki A. 2012. Cancer susceptibility and embryonic lethality in Mob1a/1b double-mutant mice. J. Clin. Invest. 122:4505–4518. 10.1172/JCI63735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohr S, Bakal C, Perrimon N. 2010. Genomic screening with RNAi: results and challenges. Annu. Rev. Biochem. 79:37–64. 10.1146/annurev-biochem-060408-092949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arbuthnot P, Carmona S, Ely A. 2005. Exploiting the RNA interference pathway to counter hepatitis B virus replication. Liver Int. 25:9–15. 10.1111/j.1478-3231.2004.0966.x [DOI] [PubMed] [Google Scholar]
- 39.Tellinghuisen TL, Marcotrigiano J, Gorbalenya AE, Rice CM. 2004. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J. Biol. Chem. 279:48576–48587. 10.1074/jbc.M407787200 [DOI] [PubMed] [Google Scholar]
- 40.Brass V, Bieck E, Montserret R, Wölk B, Hellings JA, Blum HE, Penin F, Moradpour D. 2002. An amino-terminal amphipathic alpha-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J. Biol. Chem. 277:8130–8139. 10.1074/jbc.M111289200 [DOI] [PubMed] [Google Scholar]
- 41.Liang Y, Ye H, Kang CB, Yoon HS. 2007. Domain 2 of nonstructural protein 5A (NS5A) of hepatitis C virus is natively unfolded. Biochemistry 46:11550–11558. 10.1021/bi700776e [DOI] [PubMed] [Google Scholar]
- 42.Tan SL, Nakao H, Vijaysri S, Neddermann P, Jacobs BL, Mayer BJ, Katze MG. 1999. NS5A, a nonstructural protein of hepatitis C virus, binds growth factor receptor-bound protein 2 adaptor protein in a Src homology 3 domain/ligand-dependent manner and perturbs mitogenic signaling. Proc. Natl. Acad. Sci. U. S. A. 96:5533–5538. 10.1073/pnas.96.10.5533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pflugheber J, Fredericksen B, Sumpter R, Jr, Wang C, Ware F, Sodora DL, Gale M., Jr 2002. Regulation of PKR and IRF-1 during hepatitis C virus RNA replication. Proc. Natl. Acad. Sci. U. S. A. 99:4650–4655. 10.1073/pnas.062055699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shirota Y, Luo H, Qin W, Kaneko S, Yamashita T, Kobayashi K, Murakami S. 2002. Hepatitis C virus (HCV) NS5A binds RNA-dependent RNA polymerase (RdRP) NS5B and modulates RNA-dependent RNA polymerase activity. J. Biol. Chem. 277:11149–11155. 10.1074/jbc.M111392200 [DOI] [PubMed] [Google Scholar]
- 45.Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, Halder G, Finegold MJ, Lee JS, Johnson RL. 2010. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. U. S. A. 107:1437–1442. 10.1073/pnas.0911427107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, Chen Y, Park O, Chang J, Simpson RM, Wang CY, Gao B, Jiang J, Yang Y. 2010. Mammalian MstI and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc. Natl. Acad. Sci. U. S. A. 107:1431–1436. 10.1073/pnas.0911409107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasier W, Lee JT, Avruch J, Bardeesy N. 2009. MstI and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16:425–438. 10.1016/j.ccr.2009.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hirabayashi S, Nakagawa K, Sumita K, Hidaka S, Kawai T, Ikeda M, Kawata A, Ohno K, Hata Y. 2008. Threonine 74 of MOB1 is a putative key phosphorylation site by MST2 to form the scaffold to activate nuclear Dbf2-related kinase 1. Oncogene 21:4281–4292. 10.1038/onc.2008.66 [DOI] [PubMed] [Google Scholar]