

ABSTRACT
Following reactivation from latency, there are two distinct steps in the spread of herpes simplex virus (HSV) from infected neurons to epithelial cells: (i) anterograde axonal transport of virus particles from neuron bodies to axon tips and (ii) exocytosis and spread of extracellular virions across cell junctions into adjacent epithelial cells. The HSV heterodimeric glycoprotein gE/gI is important for anterograde axonal transport, and gE/gI cytoplasmic domains play important roles in sorting of virus particles into axons. However, the roles of the large (∼400-residue) gE/gI extracellular (ET) domains in both axonal transport and neuron-to-epithelial cell spread have not been characterized. Two gE mutants, gE-277 and gE-348, contain small insertions in the gE ET domain, fold normally, form gE/gI heterodimers, and are incorporated into virions. Both gE-277 and gE-348 did not function in anterograde axonal transport; there were markedly reduced numbers of viral capsids and glycoproteins compared with wild-type HSV. The defects in axonal transport were manifest in neuronal cell bodies, involving missorting of HSV capsids before entry into proximal axons. Although there were diminished numbers of mutant gE-348 capsids and glycoproteins in distal axons, there was efficient spread to adjacent epithelial cells, similar to wild-type HSV. In contrast, virus particles produced by HSV gE-277 spread poorly to epithelial cells, despite numbers of virus particles similar to those for HSV gE-348. These results genetically separate the two steps in HSV spread from neurons to epithelial cells and demonstrate that the gE/gI ET domains function in both processes.
IMPORTANCE An essential phase of the life cycle of herpes simplex virus (HSV) and other alphaherpesviruses is the capacity to reactivate from latency and then spread from infected neurons to epithelial tissues. This spread involves at least two steps: (i) anterograde transport to axon tips followed by (ii) exocytosis and extracellular spread from axons to epithelial cells. HSV gE/gI is a glycoprotein that facilitates this virus spread, although by poorly understood mechanisms. Here, we show that the extracellular (ET) domains of gE/gI promote the sorting of viral structural proteins into proximal axons to begin axonal transport. However, the gE/gI ET domains also participate in the extracellular spread from axon tips across cell junctions to epithelial cells. Understanding the molecular mechanisms involved in gE/gI-mediated sorting of virus particles into axons and extracellular spread to adjacent cells is fundamentally important for identifying novel targets to reduce alphaherpesvirus disease.
INTRODUCTION
Alphaherpesviruses, such as herpes simplex virus (HSV) and varicella-zoster virus (VZV), have evolved specialized mechanisms enabling virus spread in epithelial and neuronal tissues. Primary infection involves entry into skin or mucosal epithelial cells, followed by rapid virus spread between these cells. During this phase of virus replication and spread, viruses enter sensory neurons by fusion of the virion envelope with neuronal membranes so that capsids are delivered into the cytoplasm. Capsids undergo retrograde axonal transport on microtubules toward neuronal cell bodies or nuclei in ganglia, where latency is established. Later, following stimulation of neurons, latent virus reactivates and there is production of virus particles that undergo fast axonal transport on microtubules in the anterograde direction from cell bodies to axon tips. This anterograde axonal transport requires kinesin motors that transport viral capsids or fully enveloped virions toward axon tips (reviewed in references 1, 2, 3 to 4). We and others concluded that the majority of anterograde transport of HSV particles involves capsids moving separately from vesicles containing viral glycoproteins (5, 6), while others have observed that enveloped HSV particles are the primary form in anterograde transport (7, 8).
Capsids arriving at axon termini become enveloped by membranes containing viral glycoproteins, and the enveloped virions escape into the extracellular space by exocytic mechanisms. Most enveloped particles outside cells remain attached to neuron surfaces. These particles are likely to be in direct contact with epithelial cells that form cell-cell junctions with neurons. The subsequent entry of viruses into epithelial cells involves fusion of the virion envelope with epithelial cell membranes. Once inside highly permissive epithelial cells, viruses can replicate to high titers and rapidly spread, causing local ulceration and amplifying virus that can spread to other hosts.
HSV expresses two membrane proteins, gE/gI and US9, which are key to understanding the combined processes of anterograde axonal transport and extracellular cell-to-cell spread (reviewed in reference 2). gE/gI is a heterodimer formed from two polypeptides, gE and gI. The heterodimer forms cotranslationally or very shortly after synthesis, both polypeptides are required for endoplasmic reticulum (ER) export, all (or the vast majority) of the gE in HSV-infected cells and virions is bound by gI, and both gE/gI are required for cell-to-cell spread (9–13). Thus, we consider HSV gE/gI one protein. gE and gI each contain ∼400-amino-acid (aa) extracellular (ET) domains and ∼100-aa cytoplasmic (CT) domains. HSV and the related pig pseudorabies virus (PRV) gE/gI CT domains encompass multiple membrane-trafficking motifs, including acidic clusters and dileucine and tyrosine motifs that cause the protein to extensively localize to the trans-Golgi network (TGN) (14–19). In polarized epithelial cells, HSV gE/gI-null mutants show markedly reduced cell-to-cell spread, and within the corneal epithelium, HSV spread was ∼5% compared with wild-type (wt) HSV (11, 15, 20). Defects in cell-to-cell spread of gE/gI mutants are the outcome of reduced accumulation of HSV at cell-cell junctions (13). This sorting of virions to cell-cell junctions requires the gE CT domain, suggesting that gE/gI must localize to the TGN (through TGN sorting motifs) in order to direct the egress of HSV particles to epithelial cell junctions. Similar types of gE/gI-mediated sorting may also occur in neurons. HSV and PRV US9 proteins are different in structure from gE/gI molecules. US9 proteins are type II membrane proteins, tail anchored, with no significant extracellular domains but, like gEgI, contain CT domains that contain TGN localization motifs (21–23). HSV US9 plays no role in spread between epithelial cells but specifically functions in neurons, and apparently only in anterograde axonal transport (22).
HSV and PRV gE-, gI-, and US9-null mutants all display significant defects in the transport of capsids, glycoproteins, and enveloped virions in neuronal axons (24–29). These studies fit well with highly reduced spread of HSV and PRV gE/gI and US9 mutants within the nervous system and from sensory ganglia to the corneal epithelium (17, 22, 30–33). HSV gE/gI and US9 possess overlapping or additive effects in anterograde axonal transport; mutants lacking either gE, gI, or US9 displayed significantly reduced transport of capsids and glycoproteins in more distal sections of axons (50 to 90%), but deletion of both gE and US9 produced nearly zero levels of capsids and glycoproteins, even in proximal axons (27, 28). PRV appears different in this regard; gE/gI contributes to efficient anterograde transport, but US9 is essential for anterograde transport (29, 34). As with sorting of virions in epithelial cells, HSV and PRV gE, gI, and US9 CT domain TGN sorting motifs contribute to anterograde axonal transport (16, 17, 21, 27, 32).
Observations that HSV gE/gI and US9 can promote the anterograde axonal transport of unenveloped capsids, which are not obviously membrane associated, provided some clues as to how gE/gI and US9 (both membrane proteins) might function (6, 27, 28). We proposed the “loading hypothesis,” which suggests that HSV gE/gI and US9 promote TGN accumulation of capsids and membrane vesicles containing glycoproteins and enveloped virions in neuron bodies (6, 27). TGN-like membranes are likely sites for loading of virus structural components onto kinesin motors for subsequent transport into axons. gE/gI and other HSV glycoproteins can interact with tegument proteins that in turn interact with capsids (reviewed in reference 2). Thus, by promoting the sorting of viral structural components, including tegument-coated capsids, to TGN-like membranes, gE/gI and US9 might enhance loading onto kinesin motors. Consistent with this hypothesis, an HSV gE− US9− double mutant did not transport glycoproteins or capsids into proximal axons and instead accumulated HSV structural proteins in cell bodies (28).
The second step in alphaherpesvirus spread from neurons to epithelial cells involves exocytosis of enveloped virus particles at axon tips, followed by entry into adjacent epithelial cells. The model described above suggests that enveloped virions are shed onto the surfaces of axons, specifically at neuron-epithelial cell junctions, so that virus particles are directly in contact with epithelial cells. Supporting this model, the PRV membrane glycoprotein gB is not required for anterograde axonal transport but is essential for virus spread from neurons to adjacent cells (35). Alphaherpesvirus gB molecules are the membrane fusion proteins required for virus entry (reviewed in reference 36). These results demonstrate the requirement for viral fusion machinery to promote entry of extracellular virions into epithelial cells.
While the CT domains of gE/gI clearly play important roles in anterograde axonal transport and cell-to-cell spread, the gE/gI heterodimer has ET domains that constitute ∼80% of the protein. There is no information on how the gE/gI ET domains function in either axonal transport or extracellular virus spread, largely related to difficulties in truncating the N-terminal domains while retaining any semblance of ER folding and export and heterodimer formation. During axonal transport, the gE/gI ET domains are present within the lumens of vesicles, and thus, it is not clear how these domains would affect interactions with kinesin motors, adaptors, or other cytoplasmic proteins. However, once virions are in the extracellular space, gE/gI ET domains are exposed on virion surfaces and might play a role in neuron-to-epithelial cell spread. Here, we characterized the capacities of two HSV gE ET domain mutants, gE-277 and gE-348, in neurons to mediate axonal transport and extracellular spread to epithelial cells. Mutants 277 and 348 were both defective in anterograde transport; the numbers of HSV capsids and gB-containing vesicles in distal axons were reduced to numbers similar to those of a gE-null mutant. Surprisingly, despite reduced numbers of virus particles in distal axons, HSV gE-348 could spread normally (as well as wild-type HSV) from neurons to epithelial cells. In contrast, HSV gE-277 was unable to mediate this spread.
MATERIALS AND METHODS
Cells and viruses.
Vero and HaCaT cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum. Vero cells were used to propagate and determine the titer of HSV. HaCaT cells are a spontaneously transformed keratinocyte line (15, 37). HSV F-gEβ is a gE-null mutant that was described previously (11). F-BAC gE-256-5 (here denoted gE-256), F-BAC gE-277, and F-BAC gE-348-4 (here denoted gE-348) contain 4- or 5-amino-acid insertions in the extracellular domain of gE (20). These HSVs have bacterial artificial chromosome (BAC) sequences inserted into the HSV thymidine kinase (TK) gene (38). For replication of these mutant viruses in neurons, the TK gene was repaired by cotransfecting Vero cells with viral DNAs extracted from cytoplasmic nucleocapsids (39) and a plasmid, pTK173, containing the HSV TK gene (40) using the CaPO4 transfection method (41). Viruses derived from these transfections were plated on 143TK− cells (a gift from S. Bacchetti, McMaster University, Ontario, Canada), which were treated with 0.1 mM hypoxanthine, 1 μM aminopterin, and 40 μM thymidine (HAT medium) to select for TK+ viruses (42). TK+ viruses were plaque purified in 3 rounds using HAT selection, and then the presence of the mutations was verified by PCR and by immunoprecipitation with a TK-specific antibody (data not shown). The TK+ viruses produced were denoted HSV gE-256, HSV gE-277, and HSV gE-348. These mutant viruses were repaired for gE expression by preparing viral DNA and cotransfecting Vero cells with this DNA and a plasmid (pUC-US7/8) containing the wild-type gE gene (19). The viruses produced by this transfection were screened using HaCaT cells, selecting large plaques that were then plaque purified 3 times. The repaired viruses, HSV gE-277R and HSV gE-348R, were analyzed by PCR to confirm the wild-type gE sequences.
Neuronal cell cultures.
Superior cervical ganglia (SCG) were dissected from day 18 embryos removed from pregnant Sprague-Dawley rats and dissociated by incubation in 0.25% trypsin in Hibernate A medium lacking calcium (Brainbits, LLC) at 37°C for 10 min. The ganglia were then incubated in 1% soy bean trypsin inhibitor in Hibernate A-Ca for 5 min at 37°C and then transferred into Neurobasal medium supplemented with 2% B27 (a serum-free supplement; Invitrogen) containing 50 ng/ml murine NGF 2.5S subunit (Invitrogen). The ganglia were then mechanically dissociated by repeated passage through a fire-polished Pasteur pipette. In order to measure HSV axonal transport, the neurons were characterized following the extension of axons in SND450 microfluidic devices (Xona Microfluidics) that were mounted on glass coverslips or glass slides (28, 43, 44). The glass surfaces were first prepared by overnight incubation in poly-d-lysine (1 μg/ml) in 0.1 M Na-borate, pH 8.5. The glass was washed twice in water, dried, and then incubated for 2 h in poly-d-lysine (30 μg/ml)–laminin (2 μg/ml) in phosphate-buffered saline (PBS). The glass was then washed twice in water and air dried. SCG neurons were plated in the somal compartments (∼40,000/device) of microfluidic chambers; then, after 2 days, cytosine arabinoside (AraC) (2 μM) was added to the medium for 2 days to kill nonneuronal cells. The neurons produced axons extending from the somal chambers into channels connecting to the axonal compartments. The neurons were infected after 6 to 7 days by adding HSV (8 PFU/cell) to the somal compartments. For experiments measuring HSV spread to nonneuronal cells, ∼40,000 HaCaT cells were plated in the axonal compartments of microfluidic devices 24 h before infection, and 0.1% human gamma globulin (a source of HSV-neutralizing antibodies; Baxter Healthcare) was added to the axonal compartments at the time of virus addition to the somal compartments.
Antibodies.
Rabbit polyclonal anti-VP26 sera were kindly provided by Prashant Desai (Johns Hopkins University, Baltimore, MD). Mouse monoclonal antibody (MAb) specific for ICP4 (58S) was a gift of Roger Everett (University of Glasgow). Mouse anti-gB MAb (SS10) was kindly provided by Gary Cohen (University of Pennsylvania, Philadelphia, PA). A guinea pig anti-tau antibody was purchased from Synaptic Systems. DyLight fluorescent secondary antibodies were from Jackson ImmunoResearch.
Immunofluorescence microscopy of HSV-infected neurons in the axonal chamber.
Microfluidic chambers containing SCG neurons and HaCaT cells were disassembled and fixed in 4% paraformaldehyde in PBS for 10 min at 20°C and permeabilized in 0.1% Triton X-100 for 10 min. In some cases, cells were fixed in acetone for 5 min at −20°C and then washed extensively with PBS. The cells were incubated with primary antibodies in PBS containing 0.1% Tween 20 and 2% normal donkey serum for 1 to 2 h, and then the cells were washed in PBS containing 0.1% Tween 20 and incubated with secondary fluorescent antibodies in PBS containing 0.1% Tween 20 and 2% normal donkey serum for 1 to 2 h. In some cases, nuclei were stained with 600 nM DAPI (4′,6-diamidino-2-phenylindole) for 5 min. Microscopy was performed in the Oregon Health and Sciences University Advanced Light Microscopy Core using a Deltavision CoreDV Widefield Deconvolution microscope. Images were captured with a 60× (numerical aperture, 1.42) Plan Apo N objective in three channels: 488, 549, and 649 nm. For quantification of VP26 and gB puncta, 10 10,551-μm2 images per slide were captured with a minimum of six 0.3-μm z-sections. After deconvolution, the images were processed with ImageJ software by setting VP26 and gB puncta to a minimum intensity threshold of 1,500 (at this threshold, uninfected cells yield approximately 0 VP26 and gB fluorescence) and a minimum area of at least 7 square pixels (6.4 × 10−3 μm2). The puncta quantified by using ImageJ software were also compared to manual counts of gB and capsid puncta in representative images using softWoRxs Explorer software (Applied Precision). Counts of capsid and glycoprotein puncta produced using ImageJ software were also corrected for the relative numbers of axons present in different fields by using tau-specific antibodies to stain axons. HSV-infected HaCaT cells expressing the immediate-early ICP4 protein were immunostained with an ICP4 MAb and manually counted in 10 axonal compartments involving 3 separate experiments.
Analyses of HSV replication in SCG neurons.
SCG neurons were plated in 24-well tissue culture dishes (40,000 cells/well) coated with poly-d-lysine and laminin and then treated with AraC for 2 days to kill nonneuronal cells. Six days after plating, the cells were infected with HSV using 5 PFU/cell. After 2 h, the cells were washed once in Neurobasal medium, and some cells and medium were immediately harvested (at the 2-h time point), while other dishes were incubated for 22 or 30 h. Cells were scraped from the dishes into cell culture medium; this mixture was frozen and sonicated, and HSV titers were determined using Vero cells.
RESULTS
HSV mutants with insertion mutations in the gE ET domain.
Previously, we constructed a panel of HSV mutants that express gE molecules with 4- or 5-aa insertions within the gE ET domain (20). Of special interest were mutants F-BAC gE-277 and F-BAC gE-348-4 (here denoted gE-348), which replicated as well as wild-type HSV in HaCaT epithelial cells and produced normal amounts of gE, which formed heterodimers with gI, and these gE/gI complexes reached cell surfaces and were incorporated into extracellular virus particles. Mutant F-BAC gE-256 produced a misfolded form of gE that dimerized with gI but remained stuck in the ER, and this mutant was used in some experiments as an additional negative control. F-BAC gE-277 and F-BAC gE-348 were unable to mediate spread between epithelial cells and corneal epithelial cells (20). Here, these mutants were used to test whether HSV gE/gI ET domains are important for axonal transport and spread from neurons to epithelial cells.
Before the HSV gE ET domain mutants described above could be used to infect neurons, it was necessary to repair the TK gene in these viruses. F-BAC gE-256, F-BAC gE-277, and F-BAC gE-348 were constructed using an HSV BAC copy of HSV DNA in which the BAC was inserted into the TK gene (38). TK is required for HSV replication in neurons. Viral DNA was cotransfected, along with a plasmid containing the HSV TK gene (40), into Vero cells, and TK+ viruses were selected using HAT medium in 143TK− cells, producing TK-repaired mutants denoted HSV gE-256, HSV gE-277, and HSV gE-348. Replication of HSV gE-277 and HSV gE-348 SCG neurons derived from 18-day-old embryos (6, 26) was similar to that of wt HSV (Fig. 1). Note that these neuronal cultures contain no nonneuronal cells following our modified protocol of cytosine arabinoside treatment (6, 28). Previous studies have shown that HSV gE-null mutants have no defects in replication in neurons and exocytosis into neuron culture supernatants (11, 20). We later produced gE-repaired versions of HSV gE-277 and gE-348 (gE-277R and gE-348R), but because there were no defects in replication of HSV gE-277 and gE-348, replication of the repaired viruses was not shown in Fig. 1.
FIG 1.
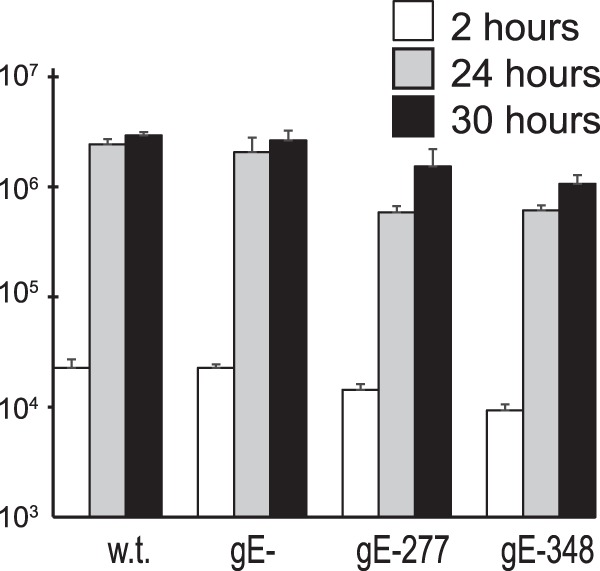
Replication of HSV gE-277 and HSV gE-348 mutants in rat neurons. Rat SCG neurons growing in 24-well dishes (∼40,000 neurons per well) were infected with wt HSV, HSV gE-null, HSV gE-277, or HSV gE-348 using 5 PFU/cell for 2 h; then, the cells were washed once with medium. At that time (2 h) or after 22 or 30 h, the cells were scraped into the culture medium, frozen, and sonicated, and then viral titers were determined using plaque assays with Vero cells. The experiment was performed in triplicate, and the mean titers are shown with standard deviations.
Anterograde axonal transport of HSV capsids and gB into distal axons.
To characterize whether the HSV gE ET domain mutants were defective in anterograde axonal transport, we used rat SCG neurons that were plated in microfluidic chambers (6, 28, 43). These embryonic rat neurons were introduced into the somal compartments of microfluidic devices, so neuron bodies reside in these chambers and extend axons through 450-μm microchannels connecting to axonal compartments, where distal axons and axon tips are found. After 6 to 7 days in culture, these neurons were infected with wt HSV, a gE-null mutant (F-gEβ) (11), HSV gE-277, HSV gE-256, and HSV gE-348, HSV gE-277R, and HSV gE-348R by adding virus (8 PFU/neuron) to the somal chambers. After 18 h of infection, distal axons in axonal chambers were simultaneously stained with rabbit VP26-specific (capsid protein) polyclonal antibodies, a gB mouse MAb, and tau-specific (cellular axon protein) antibodies. There were numerous capsid puncta (green) and gB puncta (red) in distal axons (stained with tau antibodies in blue) derived from wt HSV-infected neurons and fewer capsids and gB puncta in axons derived from HSV gE-null-infected neurons (Fig. 2, top row). Both unenveloped (separate) and enveloped (married) capsids were observed, as in our previous studies (6, 28). Certain axons were out of the plane of focus or not intensely stained with tau, so it appeared some puncta might be outside axons, but a closer examination showed that this was not the case, as we previously described (6, 28). Axons derived from neurons infected with HSV mutants gE-277 and gE-348 also displayed few gB puncta and capsids, whereas axons from neurons infected with the gE-repaired viruses contained numerous capsids and gB puncta (Fig. 2, middle and bottom rows). We quantified these puncta by capturing 10 images from each of 3 axonal chambers and processing the images with ImageJ software to produce the total numbers of puncta per field. These analyses showed substantially reduced numbers of capsids and gB puncta in distal axons from neurons infected with HSV gE-null, HSV gE-256, HSV gE-277, and HSV gE-348 (Fig. 3). Four other experiments produced similar data (not shown). HSV gE-repaired recombinants, HSV gE-277R and HSV gE-348R, were like wt HSV. We concluded that the mutant gE molecules expressed by HSV gE-277 and HSV gE-348 cannot promote efficient axonal transport.
FIG 2.
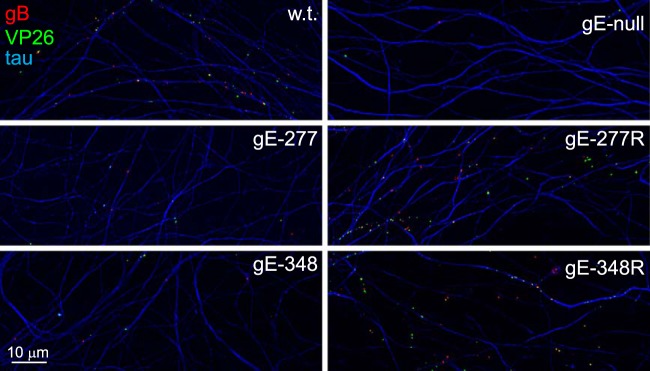
Capsids and gB puncta present in distal axons following infection with HSV gE-277 and HSV gE348. Rat SCG neurons were infected with wt HSV, HSV gE-null, HSV gE-277, HSV gE-277R, HSV gE-348, or HSV gE-348R using 8 PFU/cell by adding virus to the somal compartments of microfluidic devices and then incubated for 18 h. The devices were disassembled, and axons in the axonal chambers were fixed with paraformaldehyde and simultaneously immunostained with antibodies specific for VP26 (green; capsids), gB (red), and the microtubule-associated protein tau (blue) and then with secondary fluorescent antibodies.
FIG 3.
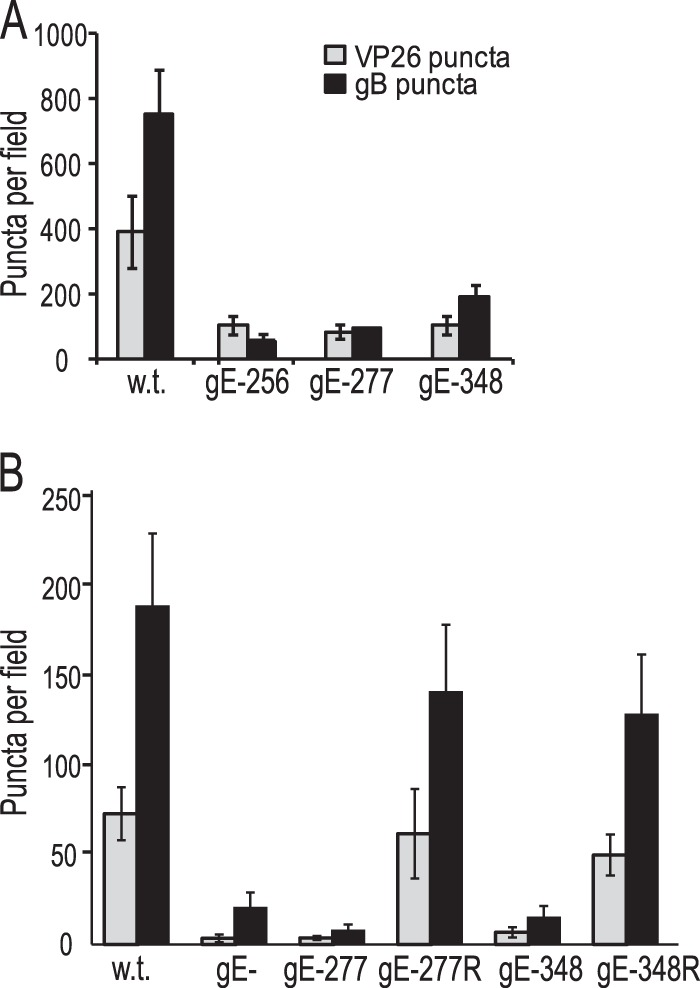
Numbers of capsids and gB puncta in distal axons of SCG neurons. Rat SCG neurons growing in microfluidic devices were infected with wt HSV, HSV gE-256, HSV gE-277, HSV gE-348, HSV gE-277R, or HSV gE-348R using 8 PFU/cell by adding virus to the somal chambers. After 18 h, the devices were disassembled, and the axons in the axonal chambers were fixed with paraformaldehyde and then permeabilized with 0.1% Triton X-100 (A) or fixed and permeabilized with acetone (B) and simultaneously immunostained with antibodies specific for VP26, gB, and the microtubule-associated protein tau and then with secondary fluorescent antibodies. ImageJ software was used to count capsids and gB puncta in 10 distinct 10,551-μm2 fields of the axonal compartments from three separate wells. The error bars represent standard deviations.
HSV gE-277 and HSV gE-348 mutants produce mislocalized capsids in neuron bodies.
While HSV gE-277 and HSV gE-348 exhibited few capsids and gB in distal axons, it was not clear whether this was related to reduced transport within axons or reduced entry of HSV structural components into axons, i.e., defects manifested in neuron bodies. To characterize this, we imaged proximal axons present within the somal chambers of microfluidic chambers. Wt HSV-infected neurons exhibited numerous gB puncta (red) and capsids (VP26; green) within proximal axons (Fig. 4A). These puncta were much more difficult to ascribe to individual axons because of the much higher density of axons in somal chambers than in axonal chambers. However, it was clear that neurons infected with HSV gE-null, HSV gE-277, and HSV gE-348 displayed markedly fewer capsids and gB puncta than wt HSV (Fig. 4A to D). When the microscope was focused on neuron bodies, fluorescence was intense over the cell bodies, related to the concentration of capsids and glycoproteins there. In the case of these cell bodies, the tau (blue) fluorescence was subtracted from the images to allow better focus on the cytoplasmic distribution of capsids and gB. In the bodies of neurons infected with HSV gE-null, HSV gE-277, and HSV gE-348, there were marked concentrations of capsids that were largely localized at one pole of the neuronal cytoplasm, producing green fluorescence (Fig. 4F, G, and H). In these gE-null-, gE-277-, and gE-348-infected neurons, gB was distributed more uniformly throughout the cytoplasm. In contrast, in wt HSV-infected neurons there was more extensive colocalization of capsids with gB and the capsids were much more uniformly distributed over the entire cytoplasm, producing yellow and orange fluorescence (Fig. 4E). There was also some clustering of capsids in clumps (green fluorescence) in the wt-infected neurons, but more of the capsids were observed randomly dispersed throughout the cytoplasm than in HSV gE-277- and HSV ge-348-infected neurons (Fig. 4E to H). These studies demonstrated that capsids were extensively mislocalized in cell bodies of neurons infected with HSV gE-null, HSV gE-277, and HSV gE-348.
FIG 4.
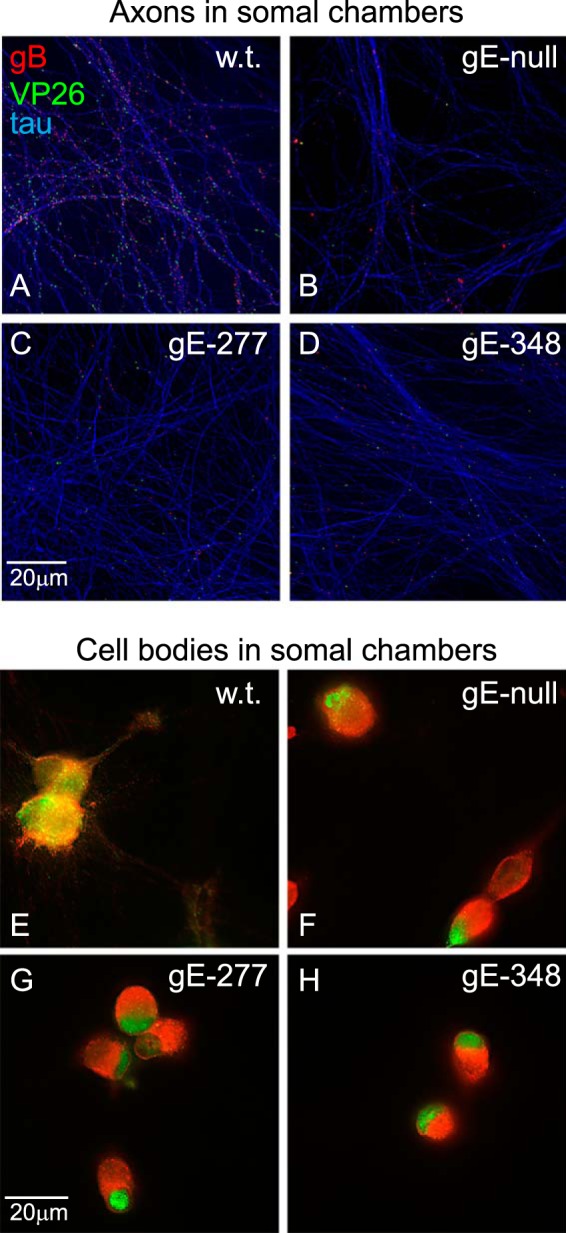
Capsids and gB puncta in proximal axons and cell bodies. SCG neurons were infected as described in the legend to Fig. 2 for 18 h. The microfluidic devices were disassembled, and the neuron bodies and axons in the somal chambers were fixed with paraformaldehyde, permeabilized, and simultaneously immunostained with antibodies specific for VP26 (capsids; green), gB (red), and the microtubule-associated protein tau (purple-blue) and then with secondary fluorescent antibodies. (A to D) Images of neuronal axons, with efforts to exclude cell bodies. (E to H) Images of neuronal cell bodies and their associated proximal axons, with the tau-specific fluorescence subtracted.
Neuron-to-epithelial cell spread of HSV gE-277 and HSV gE-348.
To characterize the role of the gE ET domain in extracellular spread from neuronal axons to adjacent epithelial cells, we plated HaCaT epithelial cells in the axonal chambers of microfluidic devices that already contained distal axons. HaCaT cells are a keratinocyte line (37), and HSV gE mutants display major (8-fold) defects in spread between HaCaT cells (15). Given that the aim of these studies was to characterize only the spread from infected neuronal axons to epithelial cells and not the spread between epithelial cells, we initially performed a time course experiment to determine when the first HaCaT cells became infected by neuron-derived HSV. Human gamma globulin, which contains HSV-neutralizing antibodies, was added to the axonal chambers to ensure that virus spread was limited to spread across cell-cell junctions in a form that is resistant to neutralizing antibodies, as shown in reference 13. Neurons were infected in somal compartments with wt HSV or HSV gE-null, and HaCaT cells were stained with anti-HSV ICP4 antibodies. ICP4 is an HSV immediate-early protein found in the nucleus and not in axons. At 18 h, we detected primarily (>75%) single HaCaT cells infected with wt HSV (stained with ICP4 antibodies) and in contact with tau antibody-stained axons (Fig. 5). Pairs of infected HaCaT cells were seen in the vast majority of cases involving more than a single infected HaCaT cell, and some of these cells were clearly the result of spread from axons. After 20 h, there were clumps of infected HaCaT cells, frequently involving 8 to 12 cells, consistent with significant spread beyond a single infected cell. After 22 h, wt HSV had spread to a large group of HaCaT cells in axonal chambers, and by 24 h, the majority of cells were infected (Fig. 5). After 16 h, there were very few (<5%) ICP4-expressing HaCaT cells with wt HSV (not shown). Compared with cultures infected with wt HSV, there was much less spread of the HSV gE-null mutant to HaCaT cells at 18 h and little spread between HaCaT cells at 20 and 22 h (Fig. 5). Thus, by characterizing the spread of HSV mutants from neurons to epithelial cells at 18 h, we could focus on the process of HSV spread from axons to adjacent epithelial cells rather than spread between epithelial cells.
FIG 5.
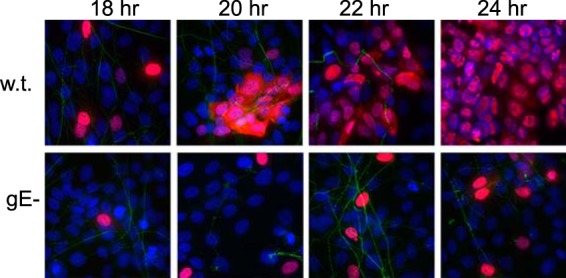
Spread of wt and gE-null HSV from distal axons to adjacent epithelial cells. SCG neurons growing in microfluidic chambers were allowed to produce axons, which extended into the axonal compartment for 6 days, and then HaCaT cells were plated in the axonal compartments for 24 h. The neurons were then infected with wt HSV or HSV gE-null (using 8 PFU/cell) by adding virus to the somal compartments. After 2 to 4 h, 0.1% human gamma globulin (a source of HSV-neutralizing antibodies) was added to the axonal chambers. At the indicated time points, the devices were disassembled and cells in the axonal chambers were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and immunostained with antibodies specific for tau (axons; green) and the HSV immediate-early protein ICP4 (HaCaT cells; red), and then with secondary fluorescent antibodies and the nuclear dye DAPI (purple-blue).
Neurons were infected in somal chambers with HSV mutants gE-256, gE-277, and gE-348 and repaired viruses; then, ICP4+ HaCaT cells in axonal chambers were counted at 18 h. HSV mutants gE-256 and gE-277 displayed fewer infected HaCaT cells than wt HSV-infected cultures or with the repaired viruses HSV gE-277R and HSV gE-348R (Fig. 6A and B). Even if one assumes that pairs of infected HaCaT cells represent spread between HaCaT cells and not spread from neurons, the differences between the wt and HSV gE-277 were still substantial (10-fold). In contrast, HSV gE-348 spread well to infect as many HaCaT cells as wt HSV and the repaired viruses (Fig. 6A and B). These results were surprising, yet they were highly reproducible; in 7 experiments, HSV gE-348 spread as well as wt HSV, while HSV gE-277 did not. Therefore, even under conditions where HSV gE-348 transported only 5 to 20% of the normal number of capsids and gB puncta into distal axons, HSV gE-348 was able to spread as well as wt HSV from neurons to epithelial cells. Although similar numbers of capsids and gB puncta were observed in axons derived from HSV gE-277-infected neurons, the gE-277/gI mutant protein was unable to mediate axon-to-epithelial cell spread.
FIG 6.
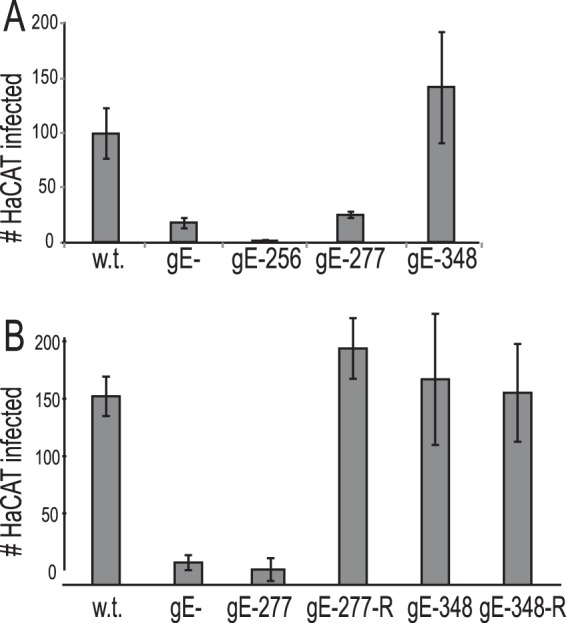
Spread of F, gE−, gE-256, gE-277, and gE-348 mutants from distal axons to adjacent nonneuronal cells. SCG neurons and HaCaT cells were plated in microfluidic chambers and infected with HSV as described in the legend to Fig. 5. Cells in the axonal chambers were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and immunostained with ICP4-specific antibodies and the nuclear dye DAPI. (A) ICP4+ HaCaT cells in 10 axonal compartments involving 3 separate wells were manually counted, and the total numbers of ICP4+ HaCaT cells/axonal compartment are shown with standard deviations. (B) Another experiment was performed as for panel A but including repaired HSV gE-277R and HSV gE348R.
DISCUSSION
The roles of HSV, PRV, and VZV gE/gI molecules in anterograde axonal transport are well established. Loss of either the gE or gI polypeptide disrupts the gE/gI heterodimer and reduces axonal transport of all viral structural proteins. Our observations with an HSV gE− US9− double mutant produced the conclusion that most of the defects in anterograde transport involve defects in sorting of viral structural components within cell bodies before the entry of viral proteins into axons (28). These sorting decisions require TGN-like trafficking motifs in the gE and gI CT domains that also contribute to virus envelopment and sorting in epithelial cells (14, 16, 27, 45, 46). How the relatively large gE and gI ET domains contribute to axonal transport has not been addressed before. Our results here show that HSV gE-277 and gE-348 mutants cannot function in anterograde transport better than a gE-null mutant. These small insertion mutations in the gE ET domain allow gE to fold relatively normally, to form heterodimers with gI, and to become incorporated into the virion (20).
Models explaining how the gE/gI ET domains function in axonal transport are more complex than representations of how the gE/gI CT domains participate (6, 27, 28). Given that the gE/gI and US9 CT domains are present in the cytoplasm, these sequences can potentially interact with kinesins, kinesin adaptors, or cytoplasmic sorting proteins in neuron bodies or in axons. The loading hypothesis suggests that the gE/gI and US9 CT domains participate in sorting of gE/gI, US9, other viral membrane proteins, and tegument proteins into discrete cytoplasmic membrane compartments, e.g., TGN-like membranes, where loading onto kinesin motors occurs. However, the gE/gI ET domains are present inside cytoplasmic vesicles, cannot directly interact with cytoplasmic proteins, and must instead interact with luminal or ET domains of viral or cellular proteins. One possibility is that the gE/gI ET domains interact with the ET domains of other HSV glycoproteins (gB, gD, gH/gL, and gM/gN), leading to their sorting or assembly into TGN-like membranes and promoting loading of essential virion components onto kinesin motors and entry into axons (Fig. 7A). By this model, HSV gE-null, gE-277, and gE-348 mutants fail in sorting decisions, so that membrane proteins are loaded less efficiently onto kinesins (Fig. 7B).
FIG 7.
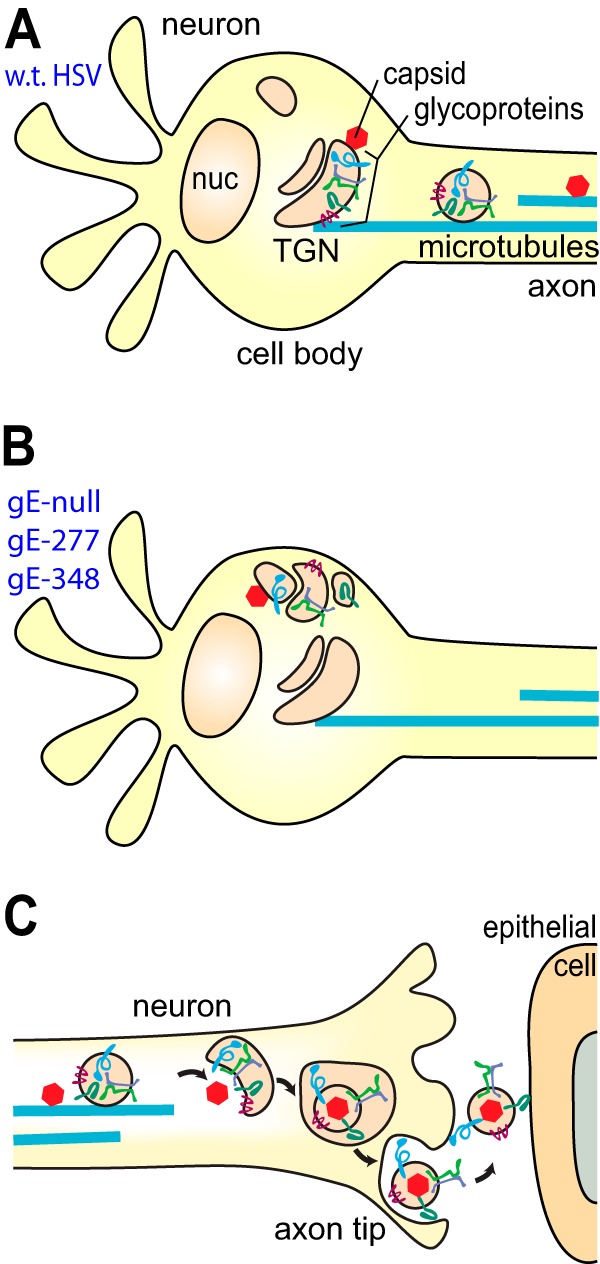
Cartoons depicting models for how gE-277 and gE-348 might fail to promote axonal transport and neuron-to-epithelial cell spread. (A) In wt HSV-infected neurons, wt HSV gE/gI ET domains promote the assembly of other glycoproteins and tegument-coated capsids into TGN-like membranes, where there is loading of viral structural proteins onto kinesin motors and microtubules. (B) In neurons infected with HSV gE-null, HSV gE-277, or HSV gE-348, viral glycoproteins and tegument-coated capsids are missorted, so assembly occurs at intracellular sites distant from where loading onto kinesins occurs. (C) Unenveloped capsids and vesicles containing glycoproteins arrive at axon tips, and secondary envelopment produces enveloped virions. Virions inside membrane vesicles must be sorted to the surfaces of axon tips, where exocytosis occurs. Extracellular virions that remain bound on axon surfaces (at axon-epithelial cell junctions) interact with epithelial cells, promoting rapid entry into epithelial cells.
The gE-277 and gE-348 mutations also substantially reduced axonal transport of capsids. Capsids accumulated in a band at one pole of the neuronal cytoplasm, whereas capsids produced by wt HSV were more uniformly spread throughout the cytoplasm. The effects of the gE ET domain mutations were probably indirect, because the gE/gI ET domain cannot interact directly with cytoplasmic capsids or tegument proteins. gE/gI, gD, and other glycoproteins interact with certain outer tegument proteins, some that are bound to the surfaces of cytoplasmic membranes and others bound to capsids, and this involves the CT domains of HSV glycoproteins and affects trafficking of tegument proteins (46–50). Given that outer tegument proteins coat the surfaces of capsids, we anticipate that gE/gI and other HSV glycoproteins might determine the cytoplasmic localization of unenveloped capsids. This fits with evidence that HSV gE/gI and gD possess redundant functions in secondary envelopment, a process involving interactions between glycoproteins and tegument proteins (46, 51). Thus, in this modified loading hypothesis, the effects of gE ET domain mutations are to cause mislocalization of gE/gI and other HSV membrane and tegument proteins, leading to missorting of tegument-coated unenveloped capsids, so that capsids are inefficiently loaded onto kinesin motors (Fig. 7A and B).
There was a recent report that PRV gE/gI promotes anterograde transport by facilitating the binding of US9 onto kinesin-3 motors (29). The authors concluded that PRV US9 is essential for anterograde transport (34), while gE/gI is important but not essential for axonal transport, acting upstream of US9 to promote loading of US9 onto kinesins (29). In contrast, HSV gE− and US9− mutants each displayed reduced axonal transport of 60 to 90%, but a gE− US9− double mutant was completely blocked for transport of capsids and glycoproteins into proximal axons (27, 28). We concluded that HSV gE/gI and US9 act by redundant or overlapping mechanisms rather than in a linear pathway where one protein acts upstream of the other.
The markedly reduced numbers of capsids and gB puncta in the distal axons of HSV gE-277- and gE-348-infected neurons predicted that there would be diminished spread to adjacent epithelial cells. Surprisingly, there was efficient spread of HSV gE-348, and as many HaCaT epithelial cells were infected as with wt HSV. We estimated that HSV gE-348 produced ∼9,000 capsids in an entire axonal chamber, and ∼100 to 150 HaCat cells were infected at 18 h. Therefore, even with the reduced numbers of HSV gE-348 capsids and glycoproteins in distal axons, there were numerous infectious HSV particles in these axons, sufficient to spread to 100 to 150 HaCaT cells. There was no evidence that HSV gE-348 could spread better than wild-type HSV; it was more likely that reduced numbers of particles did not compromise spread. In contrast, HSV gE-277 was unable to mediate neuron-to-epithelial cell spread, despite levels of capsids and glycoproteins in distal axons similar to those for HSV gE-348. We concluded that the gE-277 mutation abolishes the capacity of gE-277/gI to mediate axon-to-epithelial cell spread, whereas gE-348/gI is normal in this process. Axon-to-epithelial cell spread might be considered to include two steps (Fig. 7C). First, enveloped virions are produced by secondary envelopment at axon tips. The virions are encased in membrane vesicles that must be sorted to axon-epithelial cell junctions and exocytosed. In the second step, extracellular particles adhering to neuron surfaces can simultaneously interact with epithelial cells at cell-cell junctions and then enter the epithelial cells. HSV virions are present at epithelial cell-cell junctions in direct contact with both cells in a compartment that resists neutralizing antibodies (13). HSV gE-277 fails in one or both of these steps, while HSV gE-348 retains the capacity to mediate the spread.
In summary, we genetically separated two stages in the gE/gI-mediated spread of HSV from neurons to adjacent epithelial cells: (i) anterograde axonal transport that was defective for both gE-277 and gE-348 and(ii) spread from axons to adjacent epithelial cells that was not mediated by gE-277.
ACKNOWLEDGMENTS
We are especially indebted to Aurelie Snyder at the Advance Light Microscopy Core at the Jungers Center, OHSU, for her extensive efforts and skill in performing deconvolution microscopy and image analyses.
This work was supported by a grant from the National Institutes of Health (RO1 EY018755 to D.C.J.).
Footnotes
Published ahead of print 16 July 2014
REFERENCES
- 1.Diefenbach RJ, Miranda-Saksena M, Douglas MW, Cunningham AL. 2008. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 18:35–51. 10.1002/rmv.560 [DOI] [PubMed] [Google Scholar]
- 2.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394. 10.1038/nrmicro2559 [DOI] [PubMed] [Google Scholar]
- 3.Kratchmarov R, Taylor MP, Enquist LW. 2012. Making the case: married versus separate models of alphaherpes virus anterograde transport in axons. Rev. Med. Virol. 22:378–391. 10.1002/rmv.1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cunningham A, Miranda-Saksena M, Diefenbach R, Johnson D. 2013. Letter in response to: Making the case: married versus separate models of alphaherpes virus anterograde transport in axons. Rev. Med. Virol. 23:414–418. 10.1002/rmv.1760 [DOI] [PubMed] [Google Scholar]
- 5.Saksena MM, Wakisaka H, Tijono B, Boadle RA, Rixon F, Takahashi H, Cunningham AL. 2006. Herpes simplex virus type 1 accumulation, envelopment, and exit in growth cones and varicosities in mid-distal regions of axons. J. Virol. 80:3592–3606. 10.1128/JVI.80.7.3592-3606.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wisner TW, Sugimoto K, Howard PW, Kawaguchi Y, Johnson DC. 2011. Anterograde transport of herpes simplex virus capsids in neurons by both separate and married mechanisms. J. Virol. 85:5919–5928. 10.1128/JVI.00116-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antinone SE, Zaichick SV, Smith GA. 2010. Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. J. Virol. 84:13019–13030. 10.1128/JVI.01296-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Negatsch A, Granzow H, Maresch C, Klupp BG, Fuchs W, Teifke JP, Mettenleiter TC. 2010. Ultrastructural analysis of virion formation and intraaxonal transport of herpes simplex virus type 1 in primary rat neurons. J. Virol. 84:13031–13035. 10.1128/JVI.01784-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson DC, Frame MC, Ligas MW, Cross AM, Stow ND. 1988. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J. Virol. 62:1347–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanke T, Graham FL, Lulitanond V, Johnson DC. 1990. Herpes simplex virus IgG Fc receptors induced using recombinant adenovirus vectors expressing glycoproteins E and I. Virology 177:437–444. 10.1016/0042-6822(90)90507-N [DOI] [PubMed] [Google Scholar]
- 11.Dingwell KS, Brunetti CR, Hendricks RL, Tang Q, Tang M, Rainbow AJ, Johnson DC. 1994. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 68:834–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dingwell KS, Johnson DC. 1998. The herpes simplex virus gE-gI complex facilitates cell-to-cell spread and binds to components of cell junctions. J. Virol. 72:8933–8942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson DC, Webb M, Wisner TW, Brunetti C. 2001. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 75:821–833. 10.1128/JVI.75.2.821-833.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alconada A, Bauer U, Sodeik B, Hoflack B. 1999. Intracellular traffic of herpes simplex virus glycoprotein gE: characterization of the sorting signals required for its trans-Golgi network localization. J. Virol. 73:377–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wisner T, Brunetti C, Dingwell K, Johnson DC. 2000. The extracellular domain of herpes simplex virus gE is sufficient for accumulation at cell junctions but not for cell-to-cell spread. J. Virol. 74:2278–2287. 10.1128/JVI.74.5.2278-2287.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tirabassi RS, Enquist LW. 1999. Mutation of the YXXL endocytosis motif in the cytoplasmic tail of pseudorabies virus gE. J. Virol. 73:2717–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tirabassi RS, Enquist LW. 2000. Role of the pseudorabies virus gI cytoplasmic domain in neuroinvasion, virulence, and posttranslational N-linked glycosylation. J. Virol. 74:3505–3516. 10.1128/JVI.74.8.3505-3516.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McMillan TN, Johnson DC. 2001. Cytoplasmic domain of herpes simplex virus gE causes accumulation in the trans-Golgi network, a site of virus envelopment and sorting of virions to cell junctions. J. Virol. 75:1928–1940. 10.1128/JVI.75.4.1928-1940.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wisner TW, Johnson DC. 2004. Redistribution of cellular and herpes simplex virus proteins from the trans-golgi network to cell junctions without enveloped capsids. J. Virol. 78:11519–11535. 10.1128/JVI.78.21.11519-11535.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polcicova K, Goldsmith K, Rainish BL, Wisner TW, Johnson DC. 2005. The extracellular domain of herpes simplex virus gE is indispensable for efficient cell-to-cell spread: evidence for gE/gI receptors. J. Virol. 79:11990–12001. 10.1128/JVI.79.18.11990-12001.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brideau AD, Eldridge MG, Enquist LW. 2000. Directional transneuronal infection by pseudorabies virus is dependent on an acidic internalization motif in the Us9 cytoplasmic tail. J. Virol. 74:4549–4561. 10.1128/JVI.74.10.4549-4561.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polcicova K, Biswas PS, Banerjee K, Wisner TW, Rouse BT, Johnson DC. 2005. Herpes keratitis in the absence of anterograde transport of virus from sensory ganglia to the cornea. Proc. Natl. Acad. Sci. U. S. A. 102:11462–11467. 10.1073/pnas.0503230102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lyman MG, Kemp CD, Taylor MP, Enquist LW. 2009. Comparison of the pseudorabies virus Us9 protein with homologs from other veterinary and human alphaherpesviruses. J. Virol. 83:6978–6986. 10.1128/JVI.00598-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomishima MJ, Enquist LW. 2001. A conserved alpha-herpesvirus protein necessary for axonal localization of viral membrane proteins. J. Cell Biol. 154:741–752. 10.1083/jcb.200011146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ch'ng TH, Enquist LW. 2005. Efficient axonal localization of alphaherpesvirus structural proteins in cultured sympathetic neurons requires viral glycoprotein E. J. Virol. 79:8835–8846. 10.1128/JVI.79.14.8835-8846.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ch'ng TH, Enquist LW. 2005. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J. Virol. 79:10875–10889. 10.1128/JVI.79.17.10875-10889.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snyder A, Polcicova K, Johnson DC. 2008. Herpes simplex virus gE/gI and US9 proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J. Virol. 82:10613–10624. 10.1128/JVI.01241-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howard PW, Howard TL, Johnson DC. 2013. Herpes simplex virus membrane proteins gE/gI and US9 act cooperatively to promote transport of capsids and glycoproteins from neuron cell bodies into initial axon segments. J. Virol. 87:403–414. 10.1128/JVI.02465-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kratchmarov R, Kramer T, Greco TM, Taylor MP, Ch'ng TH, Cristea IM, Enquist LW. 2013. Glycoproteins gE and gI are required for efficient KIF1A-dependent anterograde axonal transport of alphaherpesvirus particles in neurons. J. Virol. 87:9431–9440. 10.1128/JVI.01317-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whealy ME, Card JP, Robbins AK, Dubin JR, Rziha HJ, Enquist LW. 1993. Specific pseudorabies virus infection of the rat visual system requires both gI and gp63 glycoproteins. J. Virol. 67:3786–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dingwell KS, Doering LC, Johnson DC. 1995. Glycoproteins E and I facilitate neuron-to-neuron spread of herpes simplex virus. J. Virol. 69:7087–7098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tirabassi RS, Townley RA, Eldridge MG, Enquist LW. 1997. Characterization of pseudorabies virus mutants expressing carboxy-terminal truncations of gE: evidence for envelope incorporation, virulence, and neurotropism domains. J. Virol. 71:6455–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brideau AD, Card JP, Enquist LW. 2000. Role of pseudorabies virus Us9, a type II membrane protein, in infection of tissue culture cells and the rat nervous system. J. Virol. 74:834–845. 10.1128/JVI.74.2.834-845.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor MP, Kramer T, Lyman MG, Kratchmarov R, Enquist LW. 2012. Visualization of an alphaherpesvirus membrane protein that is essential for anterograde axonal spread of infection in neurons. mBio 3:e00063–12. 10.1128/mBio.00063-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Curanovic D, Enquist LW. 2009. Virion-incorporated glycoprotein B mediates transneuronal spread of pseudorabies virus. J. Virol. 83:7796–7804. 10.1128/JVI.00745-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heldwein EE, Krummenacher C. 2008. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 65:1653–1668. 10.1007/s00018-008-7570-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106:761–771. 10.1083/jcb.106.3.761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horsburgh BC, Hubinette MM, Qiang D, MacDonald ML, Tufaro F. 1999. Allele replacement: an application that permits rapid manipulation of herpes simplex virus type 1 genomes. Gene Ther. 6:922–930. 10.1038/sj.gt.3300887 [DOI] [PubMed] [Google Scholar]
- 39.Ligas MW, Johnson DC. 1988. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by beta-galactosidase sequences binds to but is unable to penetrate into cells. J. Virol. 62:1486–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Longnecker R, Roizman B. 1987. Clustering of genes dispensable for growth in culture in the S component of the HSV-1 genome. Science 236:573–576. 10.1126/science.3033823 [DOI] [PubMed] [Google Scholar]
- 41.Graham FL, van der Eb AJ. 1973. Transformation of rat cells by DNA of human adenovirus 5. Virology 54:536–539. 10.1016/0042-6822(73)90163-3 [DOI] [PubMed] [Google Scholar]
- 42.Bacchetti S, Graham FL. 1977. Transfer of the gene for thymidine kinase to thymidine kinase-deficient human cells by purified herpes simplex viral DNA. Proc. Natl. Acad. Sci. U. S. A. 74:1590–1594. 10.1073/pnas.74.4.1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu WW, Goodhouse J, Jeon NL, Enquist LW. 2008. A microfluidic chamber for analysis of neuron-to-cell spread and axonal transport of an alpha-herpesvirus. PLoS One 3:e2382. 10.1371/journal.pone.0002382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park JW, Vahidi B, Taylor AM, Rhee SW, Jeon NL. 2006. Microfluidic culture platform for neuroscience research. Nat. Protoc. 1:2128–2136. 10.1038/nprot.2006.316 [DOI] [PubMed] [Google Scholar]
- 45.Farnsworth A, Johnson DC. 2006. Herpes simplex virus gE/gI must accumulate in the trans-Golgi network at early times and then redistribute to cell junctions to promote cell-cell spread. J. Virol. 80:3167–3179. 10.1128/JVI.80.7.3167-3179.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farnsworth A, Wisner TW, Johnson DC. 2007. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J. Virol. 81:319–331. 10.1128/JVI.01842-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O'Regan KJ, Murphy MA, Bucks MA, Wills JW, Courtney RJ. 2007. Incorporation of the herpes simplex virus type 1 tegument protein VP22 into the virus particle is independent of interaction with VP16. Virology 369:263–280. 10.1016/j.virol.2007.07.020 [DOI] [PubMed] [Google Scholar]
- 48.Stylianou J, Maringer K, Cook R, Bernard E, Elliott G. 2009. Virion incorporation of the herpes simplex virus type 1 tegument protein VP22 occurs via glycoprotein E-specific recruitment to the late secretory pathway. J. Virol. 83:5204–5218. 10.1128/JVI.00069-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yeh PC, Han J, Chadha P, Meckes DGJ, Ward MD, Semmes OJ, Wills JW. 2011. Direct and specific binding of the UL16 tegument protein of herpes simplex virus to the cytoplasmic tail of glycoprotein E. J. Virol. 85:9425–9436. 10.1128/JVI.05178-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han J, Chadha P, Starkey JL, Wills JW. 2012. Function of glycoprotein E of herpes simplex virus requires coordinated assembly of three tegument proteins on its cytoplasmic tail. Proc. Natl. Acad. Sci. U. S. A. 109:19798–19803. 10.1073/pnas.1212900109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Farnsworth A, Goldsmith K, Johnson DC. 2003. Herpes simplex virus glycoproteins gD and gE/gI serve essential but redundant functions during acquisition of the virion envelope in the cytoplasm. J. Virol. 77:8481–8494. 10.1128/JVI.77.15.8481-8494.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]