

ABSTRACT
During the budding process, influenza A viruses (IAVs) incorporate multiple host cell membrane proteins. However, for most of them, their significance in viral morphogenesis and infectivity remains unknown. We demonstrate here that the expression of annexin V (A5) is upregulated at the cell surface upon IAV infection and that a substantial proportion of the protein is present in lipid rafts, the site of virus budding. Western blotting and immunogold analysis of highly purified IAV particles showed the presence of A5 in the virion. Significantly, gamma interferon (IFN-γ)-induced Stat phosphorylation and IFN-γ-induced 10-kDa protein (IP-10) production in macrophage-derived THP-1 cells was inhibited by purified IAV particles. Disruption of the IFN-γ signaling pathway was A5 dependent since downregulation of its expression or its blockage reversed the inhibition and resulted in decreased viral replication in vitro. The functional significance of these results was also observed in vivo. Thus, IAVs can subvert the IFN-γ antiviral immune response by incorporating A5 into their envelope during the budding process.
IMPORTANCE Many enveloped viruses, including influenza A viruses, bud from the plasma membrane of their host cells and incorporate cellular surface proteins into viral particles. However, for the vast majority of these proteins, only the observation of their incorporation has been reported. We demonstrate here that the host protein annexin V is specifically incorporated into influenza virus particles during the budding process. Importantly, we showed that packaged annexin V counteracted the antiviral activity of gamma interferon in vitro and in vivo. Thus, these results showed that annexin V incorporated in the viral envelope of influenza viruses allow viral escape from immune surveillance. Understanding the role of host incorporated protein into virions may reveal how enveloped RNA viruses hijack the host cell machinery for their own purposes.
INTRODUCTION
Influenza is an ineradicable contagious disease that constitutes a major public health problem, occurring as a seasonal epidemic of variable impact or sporadic pandemic outbreaks (1, 2). The etiological agents of the disease, the single-stranded RNA influenza viruses, are classified into three types (A, B, and C), of which influenza A virus (IAV) is clinically the most important. Annually, IAV causes 3 to 5 million clinical infections and 200,000 to 500,000 fatal cases (3). Thus, these viruses are of great concern to human health and impose a considerable socioeconomic burden. Important factors in the pathogenesis of influenza include the efficient replication of the virus in the respiratory tract and the host immune response, traits that are dependent on each other (4–6). While the immune response aims to control the spread of the virus, IAV has developed strategies for subverting host defenses, thereby facilitating their spread (7–10). Further knowledge into how IAV escapes the host immunosurveillance is critical for the design of new treatments that are able to control the disease.
Similarly to other enveloped viruses, IAV exits the host cell by budding from a cellular membrane (11, 12). Thereby, particles released from infected cells can incorporate many host cellular proteins during the assembly and budding steps of morphogenesis. Earlier study identified 36 host-encoded proteins in purified IAV particles in addition to viral virion components (13). Among them, the annexin family of proteins that bind to negatively charged phospholipids is well represented (13, 14). However, the functional significance of host protein incorporation has not been determined yet, except for the role of annexin II, which promotes viral replication when incorporated into a virus particle (14, 15). One protein of interest is annexin V (A5), which has recently been found to play a role in the regulation of the immune response (16, 17). We address here the specific incorporation of A5 into IAV particles and its functional relevance in viral replication.
We found that the host protein A5 was incorporated into IAV particles and inhibited gamma interferon (IFN-γ)-induced signaling and antiviral activity both in vitro and in vivo. Collectively, these results show that incorporation of A5 into IAV virions supports influenza virus escape from immunosurveillance.
MATERIALS AND METHODS
Viruses and reagents.
IAV A/PR/8/34 (H1N1) was a gift from G. F. Rimmelzwaan (Erasmus University, Rotterdam, Netherlands), and A/WSN/33 (H1N1) and A/Udorn/72 (H3N2) IAV were a gift from N. Naffakh (Pasteur Institute, Paris, France). The following reagents were used in the study: small interfering RNA (siRNA) targeting A5 (Santa Cruz Biotechnology), recombinant mouse IFN-γ (Sigma-Aldrich), recombinant human IFN-γ and IFN-α (R&D Systems), trypsin (Becton Dickinson), an enzyme-linked immunosorbent assay (ELISA) kit for IFN-γ-induced 10-kDa protein (IP-10) and interleukin-1β (IL-1β; R&D Systems), cholera toxin B subunit (Sigma-Aldrich, France), monoclonal anti-tubulin (Sigma), polyclonal anti-A5 (Santa Cruz Biotechnology), monoclonal anti-M2 (Santa Cruz Biotechnology), monoclonal anti-hemagglutinin (anti-HA; Santa Cruz Biotechnology), polyclonal anti-ERK (Cell Signaling Technology, Saint Quentin, France), monoclonal anti-Stat1 (Santa Cruz Biotechnology), and polyclonal anti-p-Stat1 (R&D Systems) antibodies. Rabbit polyclonal anti-A/PR/8/34 virus cross-reacting with A/WSN/33 virus proteins (referred to as polyclonal anti-influenza) was a gift from G. F. Rimmelzwaan (Erasmus University). Phorbol myristate acetate (PMA; Sigma) was used for human monocytic cell line (THP-1) differentiation.
Cell culture and raft isolation.
The human monocytic THP-1, human alveolar A549, human epithelial kidney 293T, HeLa, and Madin-Darby canine kidney (MDCK) cell lines used in the present study were obtained from the American Type Culture Collection. MDCK cells were cultured in Eagle minimal essential medium (EMEM; Lonza, France) supplemented with 5% fetal bovine serum (FBS; Lonza), 2 mM l-glutamine, and 100 international units (IU)/ml penicillin-streptomycin (PS). A549 and 293T cells were grown in Dulbecco modified Eagle medium (DMEM; Lonza) supplemented with 10% FBS, 2 mM l-glutamine, and 100 IU/ml PS. THP-1 cells were cultured in RPMI (Lonza) supplemented with 10% FBS, 2 mM l-glutamine, 100 IU/ml PS, 5 ml of pyruvate sodium, 5 ml of amino acids, and β-mercaptoethanol. Raft isolations were performed as previously described (18).
Virus production, titration, and purification.
MDCK cells were seeded at 13 × 106 cells per 175-cm2 tissue culture flask and then incubated at 37°C overnight. The next day, based on previous evaluations, cell confluence was evaluated at 20 × 106 cells per 175 cm2, and the cells were infected with IAV at a multiplicity of infection (MOI) of 10−3 in EMEM containing 1 μg of trypsin/ml. At 2 days postinfection, the supernatant was harvested and then clarified using low-speed centrifugation, and the virus particles were titrated as previously described (19). Briefly, MDCK cells were infected with IAV for 1 h at 37°C. After viral adsorption, the cells were overlaid with medium containing 2% agarose and 1 μg of trypsin/ml, followed by incubation for 3 days at 37°C. Viral plaques were then visualized using bromophenol blue staining. To purify the virus particles, the supernatants were clarified and concentrated 100-fold by ultracentrifugation at 60,000 × g for 105 min at 4°C. Concentrated viruses were then purified by centrifugation for 2 h at 80,000 × g at 4°C in a 20 to 60% sucrose density gradient. The virus particles were then separated into two different tubes for pretreatment with 20 μg of either blocking anti-A5 antibody (referred to as “AV-V”) or isotype control antibody (referred to as “V”)/ml for 1 h at 4°C. Viruses were then washed by ultracentrifugation at 31,000 × g for 2 h and suspended in medium. Infectious virus titers were then evaluated in both virus preparations and used for experiments. AV-V or V particles were then used in the experiments.
Identification and quantification of cell surface proteins by SILAC (stable isotope labeling by amino acids in cell culture)-based mass spectrometric (MS) analysis.
A549 cells were grown in stable isotope-labeled DMEM (SILAC-DMEM, PAA) supplemented with 10% dialyzed FBS (Invitrogen), 2 mM l-glutamine, and antibiotics at 37°C with 5% CO2. Cells were either cultivated in SILAC medium containing light (R0K0: R = 12C6,14N4; K = 12C6,14N2) or heavy (R10K8: R = 13C6, 15N4; K = 13C6, 15N2) arginine and lysine for at least six cell doublings prior to infection. A total of 4 × 107 heavy-labeled cells (R10K8) were infected with IAV A/PR/8/34 (H1N1) at an MOI of 5, while the same number of light-labeled cells (R0K0) served as a mock control. At 16 h postinfection (hpi) cells were washed with phosphate-buffered saline (PBS) and incubated with 1 mg/ml Sulfo-NHS-SS-Biotin (Thermo Fisher Scientific)/PBS for 40 min at 4°C, followed by quenching with 10 mM glycine-PBS buffer. After biotinylation of cell surface proteins, the cell extract of each population (heavy or light) was prepared in 1 ml of lysis buffer (50 mM Tris-HCl [pH 8], 150 mM NaCl, 1% Nonidet P-40, 2 mM Na3VO4, 1 mM Pefabloc) and cleared by centrifugation. The protein concentration of each lysate was determined by a BCA protein assay (Thermo Fisher Scientific), and the lysates were mixed at a 1:1 heavy/light ratio, followed by selection of biotinylated proteins on a streptavidin-agarose resin (Thermo Fisher Scientific) at 4°C for 16 h. The beads were washed once with 50 mM Tris-HCl (pH 7.4)–150 mM NaCl–5 mM EDTA, twice with 50 mM Tris-HCl (pH 7.4)–500 mM NaCl–5 mM EDTA, three times with 20 mM Tris-HCl (pH 7.4)–500 mM NaCl, and once with 10 mM Tris-HCl (pH 7.4). The precipitated proteins were eluted in 4× sodium dodecyl sulfate (SDS) sample buffer–20% β-mercaptoethanol for 30 min at 37°C. Affinity-purified proteins were reduced and alkylated by the addition of 10 mM dithiothreitol (2 min, 95°C) and 50 mM iodacetamid (30 min, 22°C, in the dark), respectively. Proteins were separated by SDS–12.5% PAGE, and the gel lane was cut into six slices, which were then subjected to in-gel tryptic digest using a trypsin profile IGD kit (Sigma). The resulting peptides were separated using a C18 capillary analytical column (10 cm [inner diameter, 75 μm]; Thermo Fisher Scientific) with a linear gradient over 95 min (solvent A = 1% FA, 99% H2O and solvent B = 80% ACN, 1% FA) at a constant flow rate of 300 nl/min using an Easy Nano liquid chromatography II system coupled to an LTQ Orbitrap discovery XL (Thermo Fisher Scientific). Eluting peptides were ionized by electrospray ionization at 1.4 kV and a capillary temperature of 200°C. Mass spectra (m/z range, 300 to 2000) were measured with a resolution of M/ΔM = 30,000 at m/z 400. The top five precursor peptide ions were fragmented by collision-induced dissociation (normalized collision energy, 35%; activation Q, 0.250, activation time, 30 ms) with a dynamic exclusion time of 30 s. The data were acquired using Xcalibur software. Raw data files were evaluated using Proteome Discoverer (PD) software (version 1.4; Thermo Fisher Scientific). Proteins were identified by searching against the UniProt/Swiss-Prot Human and Influenza A/PR/8/34 database (89,454 entries) using SEQUEST algorithm and the following search parameters: carbamidomethylation of cysteine (+57.021) as a fixed modification, oxidation of histidine, methionine, and tryptophan (+15.995); phosphorylation of serine, threonine, and tyrosine (+79.966) and appropriate SILAC labels as variable modifications; tryptic digestion with a maximum of two missed cleavages; a peptide precursor mass tolerance of 10 ppm; and a fragment mass tolerance of 0.8 Da. The decoy database search option was enabled and all peptides were filtered with a maximum false discovery rate (FDR) of 1%. Protein quantification was performed with at least two unique and labeled peptides per protein and a mass precision of 4 ppm. The relative abundance of a protein at cell surface was derived from its heavy/light (H/L) ratio in the differently labeled cell populations. Quantification values outside the range from 0.01 to 100 were recorded as 0.01 (ratio ≤ 0.01) and 100 (ratio ≥ 100). Proteins were grouped by PD annotation software tool and selected according to the gene ontology cellular component (GOCC) categories “membrane,” “cell surface,” or “extracellular” (20). An H/L ratio of >2 for a given protein was considered to signal increased surface abundance.
Depletion of A5 from virions by siRNA-mediated knockdown.
Specific siRNA targeting A5 was used to knock down protein expression in 293T cells. These cells were chosen because of their high transfection efficiency. Nontargeted siRNA was used as a control. According to the manufacturer's instructions, 293T cells (60 to 80% confluence, i.e., 2 × 105 cells per 10 cm2) were transfected with siRNA targeting A5 (1 μg/2 × 105 cells) or control-siRNA, diluted in transfection reagent (confidential lipidic composition from Santa Cruz). AT 24 h posttransfection, DMEM containing 20% fetal calf serum, PS (200 IU/ml), and l-glutamine (4 mM) was added to the cells, followed by incubation at 37°C for additional 48 h. At this step, Western blot analysis was performed to verify the transfection efficiency (data not shown). Alternatively, cells were infected with IAV (MOI = 1), and supernatants containing the virus particles were harvested at 16 hpi. The virus titers were evaluated by plaque assay and used in experiments. Similar ratios of the different viral proteins in both preparations and reduced expression of packaged A5 in the virions released in the supernatant of A5-specific siRNA-treated cells (referred to as A5 siRNA v) compared to control viruses (referred to as Ctl siRNA v) were confirmed by loading 20 μl of the corresponding supernatants on a gel, followed by Western blot analysis. Of note, the downregulation of A5 by siRNAs had no effect on the release of infectious particles (data not shown).
Flow cytometry, immunocytochemistry, and Western blot analysis.
A549 or MDCK cells either were left uninfected or were infected with A/PR8/34, A/Udorn/72, or A/WSN/33 (MOI of 1 or 10) for 24 h, and the expression of A5 was assessed by using flow cytometry analysis or cytochemistry, as previously described (8, 21). For the kinetic experiments, A549 cells were infected with IAV A/WSN/33 (MOI of 10−2) in the presence of trypsin (0.5 μg/ml), and A5 expression was assessed by flow cytometry at 6, 24, and 48 hpi. For experiments assessing virus attachment to the cells, differentiated THP-1 cells were incubated with the indicated IAV (MOI of 1) for 5 min at 37°C or for 30 min at 4°C; the cells were then washed, and virus binding to the cells was analyzed by flow cytometry using anti-HA antibody. For internalization experiments, differentiated-THP1 cells were first incubated with “Ctl siRNA v” or “A5 siRNA v” at 4°C for 30 min. The cells were then shifted to 37°C for 1 h to allow virus internalization. Back at 4°C, the cells were then fixed and permeabilized or not to assess the percentage of internalized versus cell surface bound viruses by flow cytometry using anti-HA antibody. For intracytoplasmic staining, the cells were fixed with 0.5% paraformaldehyde for 10 min and permeabilized 10 min with 0.1% Triton X-100 at 4°C (22). For the Western blot analysis, purified virions or cells were lysed in ice-cold lysis buffer (1% Triton X-100, 100 mM Tris-HCl [pH 7.4], 1.5 M NaCl, and 5 mM EDTA in the presence of a complete proteinase inhibitor mixture), and the proteins were analyzed as previously described (19).
Stat activation experiments.
THP-1 cells were incubated with PMA for 48 h at 37°C (differentiated THP-1 cells). After differentiation into macrophages, the cells were incubated with or without AV-V or V (strain A/WSN/33) or A5 siRNA v or Ctl siRNA v for 5 min, 1 h, or 16 h and either left unstimulated or stimulated with human IFN-α or IFN-γ (1,000 IU) for 5 min at 37°C. Alternatively, HeLa cells were used in the experiments. The cells were then lysed for 45 min on ice, and proteins from the lysate were analyzed by Western blotting. For the dose-response analysis, differentiated THP-1 cells were stimulated for 5 min with the indicated dose of IFN-γ, and the cells were lysed before Western blot analysis.
IP-10 and IL-1β production experiments.
Differentiated THP-1 cells were preincubated with or without AV-V or V (strain A/WSN/33) or A5 siRNA v or Ctl siRNA v at an MOI of 1 for 5 min at 37°C. The cells were then either left unstimulated or stimulated with 1,000 IU of IFN-α or IFN-γ for 3 h or 24 h at 37°C. Subsequently, supernatants were harvested, and IP-10 or IL-1β production was quantified by ELISA.
Immunogold analysis.
Immunogold labeling of A5 was performed on gradient-purified virus particles by the flotation of grids on drops of reactive media. To prevent nonspecific binding, the grids were coated with 1% bovine serum albumin (BSA) in 50 mM Tris-HCl (pH 7.4) for 10 min at room temperature. Thereafter, the grids were incubated for 4 h at 4°C in a wet chamber with a polyclonal antibody raised against A5 (dilution 1/50) in 1% BSA–50 mM Tris–HCl (pH 7.4). The grids were successively washed once in 50 mM Tris–HCl at pH 7.4 and pH 8.2 at room temperature and then incubated in a wet chamber for 45 min at room temperature in 1% BSA–50 mM Tris–HCl (pH 8.2) for 10 min at room temperature. The grids were labeled with a goat anti-rabbit gold-conjugated IgG (10 nM; Tebu Bio) diluted 1:80 in 1% BSA–50 mM Tris–HCl (pH 8.2) and then successively washed once in 50 mM Tris-HCl (pH 8.2) and 50 mM Tris-HCl (pH 7.4) at room temperature and once in filtered distilled water. The grids with the suspension were then labeled with 2% phosphotungstic acid for 2 min and observed on a transmission electron microscope (1400 JEM; JEOL, Tokyo, Japan), equipped with a Gatan camera (Orius 600) and digital micrograph software.
In vitro replication.
To test the susceptibility of differentiated THP-1 cells to IAV infection, cells were infected with A/WSN/33 virus (MOI of 1), and infectious virus titers were determined at the indicated time point postinfection by plaque assay titration. To determine the role of packaged A5 in the antiviral activity mediated by IFN-γ, differentiated THP-1 cells were incubated with either AV-V or V (strain A/WSN/33) or A5 siRNA v or Ctl siRNA v (MOI of 1) for 5 min at 37°C. The cells were then washed, and 1 ml of RPMI medium without serum, containing or not 1,000 IU of rIFN-γ, was added to cells, followed by incubation for 24 h at 37°C. Infectious virus titers were then evaluated by plaque assay in the supernatant of the cells.
Mice.
C57BL/6 Mice were infected intranasally with IAV (500 PFU) in a volume of 25 μl as previously described (23, 24). Once all of the mice were infected, the animals were still anesthetized, and they were then administered intranasally with vehicle or mouse recombinant IFN-γ (8 × 104 IU/25 μl). Mice were sacrificed at 2 days postinfection to sample the lungs. Virus titers in lung homogenates were then determined by plaque assay as described above. Animal experiments were performed according to recommendations of the National Commission of Animal Experiment (CNEA) and the National Committee on the Ethic Reflection of Animal Experiments (CNREEA). Experiments were approved by the Animal Ethics Committee (permit BH2008-13; Lyon University) and carried out under the license accreditation 78-114.
Statistical analysis.
The Mann-Whitney test was used for statistical analysis. The results were considered statistically significant at a P value of <0.05 (*). All bars in the figures represent the mean values ± the standard deviations (SD) from the indicated number of experiments.
RESULTS
An MS-based approach detects increased annexin V levels on the surfaces of IAV-infected cells.
First, changes in cell surface protein composition after IAV virus infection were investigated by using SILAC-based MS analysis. Proteins accessible at the cell surface to amine-reactive thiol-cleavable biotin ester were compared in mock-treated (light amino acids) and influenza A/PR/8/34 virus-infected A549 cells (heavy amino acid) at 16 hpi. Cell lysates were prepared, mixed, and subjected to affinity selection using streptavidin-agarose. Subsequently, proteins were eluted from the matrix and identified by MS analysis. Alterations in cell surface protein expression due to IAV infection correspond to changes in heavy/light (H/L) ratio (Table 1). As expected, the viral surface proteins HA, NA, and M2 were detected exclusively in infected cells. Table 1 also depicts cellular surface and membrane proteins with the most prominent increases in response to virus infection, including A5, as well as four other proteins (annexin 2, ezrin, annexin 1, and alpha-enolase) that were previously also detected as the cell surface increased or in purified influenza virions (13–15, 25).
TABLE 1.
Upregulated cell surface proteins upon IAV infection compared to noninfected cells identified by LC-MS/MSa
| Protein (accession no.) | Description | Score | Coverage (%) | H/L ratio | H/L count | H/L variability (%) |
|---|---|---|---|---|---|---|
| P60903 | Protein S100-A10 (S100-A10) | 31.21 | 36.08 | 5.319 | 6 | 4.9 |
| Q9BQE5 | Apolipoprotein L2 (APOL2) | 8.89 | 13.06 | 4.747 | 3 | 17.5 |
| P07355 | Annexin A2 (ANXA2) | 534.01 | 69.62 | 4.352 | 84 | 12.2 |
| P08758 | Annexin A5 (ANXA5) | 10.30 | 11.25 | 4.072 | 3 | 24.9 |
| Q9HCC0 | Methylcrotonoyl-CoA carboxylase beta chain, mitochondrial (MCCC2) | 17.35 | 18.29 | 2.940 | 8 | 12.6 |
| E7EQR4b | Ezrin (EZR) | 5.77 | 6.14 | 2.872 | 3 | 33.6 |
| O95994 | Anterior gradient protein 2 homolog (AGR2) | 9.26 | 24.57 | 2.743 | 4 | 11.1 |
| O00220 | Tumor necrosis factor receptor superfamily member 10A (TNFRSF10A) | 50.86 | 19.02 | 2.716 | 10 | 7.3 |
| P30510b | HLA class I histocompatibility antigen, Cw-14 alpha chain (HLA-C) | 262.98 | 39.88 | 2.595 | 9 | 5.0 |
| P04083 | Annexin A1 (ANXA1) | 65.75 | 46.82 | 2.444 | 17 | 12.6 |
| O60218 | Aldo-keto reductase family 1 member B10 (AKR1B10) | 91.28 | 60.76 | 2.406 | 21 | 9.8 |
| P06733 | Alpha-enolase (ENO1) | 29.77 | 21.89 | 2.310 | 6 | 10.0 |
| P06821 | Matrix protein 2 {influenza A virus[(A/Puerto Rico/8/34(H1N1)]}) | 176.41 | 39.18 | 100.000 | 7 | 0.0 |
| P03452 | Hemagglutinin {influenza A virus [A/Puerto Rico/8/34(H1N1)]} | 87.22 | 46.83 | 100.000 | 31 | 0.0 |
| P03468 | Neuraminidase {influenza A virus [A/Puerto Rico/8/34(H1N1)]} | 38.28 | 19.38 | 100.000 | 13 | 0.0 |
A549 cells were infected with A/PR/8/34 virus at an MOI of 5 and, at 16 h postinoculation, upregulated cell surface proteins were identified by liquid chromatography-tandem MS (LC-MS/MS). Heavy/light (H/L) ratios of cellular proteins are organized from the potential strongest change in cell surface abundance to minor changes upon IAV infection. The accession numbers, descriptions, and total scores for the cellular and three viral proteins are shown. The total score is the sum of the scores of the individual peptides that identified the protein. “Coverage” indicates the percentage of the protein sequence covered by the identified peptides. The H/L count indicates the number of peptide ratios that were actually used to calculate a particular protein ratio, whereas H/L variability indicates the variability of these peptide ratios from the particular H/L protein ratio.
Uniprot accession number.
Annexin V upregulation at the cell surface upon IAV infection is independent on the strain and the cell type.
In our further analysis we focused on the role of A5. As shown in Fig. 1A, fluorescence-activated cell sorting analysis confirmed increased cell surface expression of A5 after the infection of epithelial A549 cells with A/PR/8/34 (H1N1), A/Udorn/72 (H3N2), or A/WSN/33 (H1N1) viruses. Upregulation of A5 was independent of the cell type, since similar results were also observed after IAV infection of MDCK cells (Fig. 1B). The viral protein M2 was included as a positive control and was only detected after virus infection. Relative to the M2 protein, cells infected with A/WSN/33 virus showed the strongest A5 upregulation in terms of median fluorescence intensity for both cell types, suggesting that A5 upregulation at the cell surface differs between IAV strains. To confirm these results, A549 cells were infected with IAV, and A5 expression was visualized using immunofluorescence confocal microscopy (Fig. 2A). In the IAV-infected cells, A5 expression was mainly observed at the plasma membrane, while in uninfected cells A5 was mainly present in the cytoplasm. As controls, the infected but not uninfected cells displayed detectable HA proteins. Also, nuclei were stained with DAPI, and the merged images are shown (Fig. 2A). We then further investigated whether total A5 expression or simply its localization was affected by the infection. A549 cells were left uninfected or infected with influenza A/WSN/33 virus, and total A5 expression was assessed by flow cytometry analysis on permeabilized cells (Fig. 2B, left panel). The results indicated that total A5 protein levels were similar in infected compared to uninfected cells. In marked contrast, cytometry analysis performed on unpermeabilized cells, which only revealed cell surface protein, confirmed a specific increase of A5 at the cell surface upon IAV infection. These results are highlighted in the right panel of Fig. 2B by the quantification of the mean fluorescence intensity (MFI) of A5 labeling. Altogether, these results indicated that IAV infection induced A5 translocation to the cell surface, without affecting total cellular A5 levels.
FIG 1.

The host cellular protein A5 is upregulated at the cell surface after IAV infection. A549 (A) or MDCK (B) cells were either left uninfected or infected with A/PR/8/34, A/Udorn/72, or A/WSN/33 viruses (MOI of 10). At 24 hpi, flow cytometry analysis was performed with an anti-A5 antibody (closed histograms) or an isotype control (open histograms). The viral protein M2 was used as a positive control for viral infection. The results are representative of two independent experiments.
FIG 2.

The host cellular protein A5 is translocated to the cell surface. (A) A549 cells were either left uninfected or infected with IAV A/WSN/33 virus (MOI of 1). At 24 hpi, cellular A5 or viral HA proteins were visualized by immunofluorescence microscopy, using anti-A5 and anti-HA specific antibodies, respectively. The nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole), and the merged images are shown (original magnification, ×189). The results are representative of two independent experiments. Please note the presence of A5 labeling in the cytoplasm in uninfected cells, which is largely absent in the infected ones (arrows) but rather detected at the plasma membrane (stars). (B) A549 cells were either left uninfected or infected with A/WSN/33 virus (MOI of 10). At 24 hpi, flow cytometry analysis was performed using an anti-A5 antibody (closed histograms) or an isotype control (open histograms). Labeling of A5 was performed either on unpermeabilized cells, showing cell surface A5 proteins, or on permeabilized cells, showing total A5 proteins (left panel). Quantification of the mean fluorescence intensity of A5 expression ± the SD from five independent experiments is shown on the right panel. *, P < 0.05 (NI versus WSN).
Cell surface expression of annexin V is dependent on viral replication.
Although we observed that upon IAV infection all strains increased A5 expression at the cell surface, the levels of A5 translocation differed between IAV strains. Thus, possibly, cell surface A5 translocation was dependent on the rate of IAV replication, which could differ between IAV strains. To test this hypothesis, we investigated whether A5 localization at the cell membrane would increase in a replication-dependent manner. A549 cells were thus infected with IAV A/WSN/33 at a low MOI (10−2) in the presence of trypsin. Cell surface expression of A5 was then assessed by flow cytometry experiments at 6, 24, and 48 hpi (Fig. 3A). The results showed that, in marked contrast to noninfected cells (NI), upon infection (INF) A5 was translocated at the cell surface in a time course-dependent manner, showing that translocation of A5 to the cell surface increases with multiple rounds of replication. In these experiments, M2 expression was assessed as a positive control for IAV infection. Thus, translocation of A5 to the cell surface is dependent on viral replication.
FIG 3.

Kinetic of cell surface expression of A5 after IAV infection and its expression in DRM fractions. (A) Time course experiment of cell surface expression of A5 upon infection of A549 cells with A/WSN/33 virus (MOI of 10−2 in the presence of trypsin). Expression of the viral M2 protein was used as a positive control of viral infection. (B) A549 cells were either left uninfected or infected with A/WSN/33 virus (MOI of 10) for 16 h. Cells were then lysed, and the DRM domains were isolated by sucrose gradient ultracentrifugation. After centrifugation, 1-ml fractions were collected from the top of the tube and characterized by Western blot analysis (fractions 1 to 10). Blots were probed with anti-ERK antibody (ERK), cholera toxin B subunit (GM1), and anti-HA (HA0-HA2), anti-M2 (M2), and anti-A5 (A5) antibodies. Fractions 3 to 5 correspond to the DRMs, whereas the soluble fractions correspond to fractions 7 to 10. The results are representative of two independent experiments.
A substantial proportion of annexin V is present in lipid rafts.
Due to the functional importance of lipid rafts in IAV infection and budding, we then investigated the association between A5 and these domains. Clustered rafts were thus floated by sucrose density gradient centrifugation, which by definition isolates detergent-resistant membrane (DRM or lipid raft) domains, and gradient fractions were analyzed by Western blotting (Fig. 3B). ERK1/2 was present in the detergent-soluble fractions (Fig. 3B, lanes 7 to 10), while the ganglioside GM1, a resident raft component, detected by cholera toxin B subunit, was present in the DRM fractions (Fig. 3B, lanes 3 and 4). Only infected cells displayed detectable viral HA and M2 proteins. HA protein (HA0 or HA2) was found almost exclusively in association with the DRM, while M2 protein was predominantly associated with the soluble membrane fraction. More importantly, A5 was indifferently found in the soluble membrane fraction and with the DRM in uninfected cells or infected cells. Therefore, a substantial proportion of A5 is present in lipid rafts, although influenza virus infection did not alter its localization.
Annexin V is incorporated into virus particles.
IAVs bud from lipid rafts, and a substantial proportion of A5 is located in these domains. Thus, we investigated whether A5 could be packaged into virions when released from the infected cell. To investigate this point, IAV A/PR/8/34, A/Udorn/72, and A/WSN/33 were purified from culture supernatants of infected MDCK cells, and the resulting purified virions were probed by Western blotting with anti-A5, anti-M2, and anti-ERK antibodies (Fig. 4A). MDCK cells were used because of their high susceptibility to infection with various IAV strains, allowing us to obtain a sufficient amount of virus particles in the supernatant for subsequent purification. The results showed the presence of A5 in all purified virions, in addition to the viral protein M2. In contrast, the cytoplasmic protein extracellular signal-regulated kinase (ERK) was not detected in the virions but was present in the lysates of uninfected or A/PR/8/34 virus-infected MDCK cells, excluding a nonspecific incorporation of cellular proteins into virus particles. It is of note that higher quantities of purified A/PR/8/34 and A/Udorn/72 particles were loaded onto the gel to detect A5 within these virions. Most likely, the level of A5 incorporation into virus particles is strain dependent. To confirm that A5 was not a copurified contaminant of cellular origin, electron microscopic immunogold labeling was performed on purified virions with anti-A5 and secondary gold antibodies, followed by negative staining. Immunogold staining confirmed that A5 was indeed associated with the A/PR/8/34, A/Udorn/72, and A/WSN/33 IAV strains (Fig. 4B). Altogether, these data demonstrated that the cellular protein A5 is incorporated into IAV particles.
FIG 4.

Cellular A5 protein is incorporated into IAV particles. (A) A/PR/8/34, A/Udorn/72, and A/WSN/33 viruses, produced on MDCK cells, were purified by sucrose ultracentrifugation and analyzed by Western blotting with anti-A5, anti-M2, and anti-ERK antibodies. Aliquots of total proteins from MDCK cells either left uninfected or infected for 16 h with A/PR/8/34 strain were used as controls. The molecular mass is indicated in kilodaltons. (B) Electron microscopic immunogold labeling was performed on purified virions using A5-specific antibodies or isotype control. Scale bar, 50 nm. The results presented in both panels are representative of three independent experiments.
Packaged A5 inhibits IFN-γ receptor signaling.
A5 associates with the IFN-γ receptor and downregulates its signaling (16). We therefore investigated whether A5 incorporated into IAV particles (A/WSN/33 strain) could modulate the IFN-γ response in differentiated THP-1 macrophages, which express the IFN-γ receptor at the cell surface (data not shown). Although productive infection of IAV by macrophages is a matter of debate (26–28), we found that differentiated THP-1 macrophages were highly susceptible to IAV infection (Fig. 5A). First, stimulation of these cells with recombinant IFN-γ activated the Jak/Stat pathway in a dose-dependent manner, as demonstrated by increased Stat1 phosphorylation by Western blot analysis (Fig. 5B). The maximal effect was observed at around 1,000 IU of IFN-γ, which was the concentration used in subsequent experiments. When differentiated THP-1 cells were preincubated with purified A/WSN/33 virions (V), Stat1 phosphorylation triggered by IFN-γ was strongly inhibited (Fig. 5C). Thus, purified virions inhibited IFN-γ-induced Stat1 phosphorylation. This effect was not observed when A5 on purified virions was masked with a specific neutralizing antibody (AV-V), showing that inhibition of stat phosphorylation was A5 dependent. In the absence of IFN-γ, purified virions had no effect on Stat phosphorylation. Thus, we concluded that A5 incorporated into virus particles inhibits IFN-γ-induced signaling.
FIG 5.

Packaged A5 inhibits IFN-γ receptor signaling. (A) Macrophage-differentiated THP-1 cells were infected with A/WSN/33 virus (MOI of 1), and virus titers were determined in the supernatants of the cells at the indicated time points postinoculation. (B) Macrophage-differentiated THP-1 cells were treated with different doses of human rIFN-γ for 5 min at 37°C. The cells were lysed, and Stat phosphorylation was analyzed by Western blotting with an anti-phospho Stat antibody (p-Stat). Tubulin was used as a control for loading. (C and D) Differentiated THP-1 cells were incubated for 5 min with purified A/WSN/33 particles (MOI of 1), which were either pretreated (AV-V) or not pretreated (V) with an anti-A5 antibody. The cells were then either left unstimulated or stimulated with IFN-γ (1,000 IU). (C) After 5 min, the cells were lysed, and Stat phosphorylation was analyzed by Western blotting. (D) Alternatively, IP-10 release was evaluated in the supernatant at 3 h poststimulation by classical ELISA. *, P < 0.05 (between “−” versus “V” and “V” versus “AV-V”). The results in panels A to D are representative of at least two independent experiments. (E) Differentiated THP-1 cells were incubated for 5 min with purified A/WSN/33 particles (MOI of 1), which were either pretreated (AV-V) or not pretreated (V) with an anti-A5 antibody. The cells were then analyzed for virus binding by flow cytometry with an anti-HA antibody (left panel). The MFI for HA staining was obtained from three replicates (right panel). (F) Infectious titers of V and AV-V preparations.
Signal transduction via the Jak/Stat pathway initiated by IFN-γ receptors leads to the release of C-X-C motif chemokine 10 (CXCL10), also known as IP-10 (29). Therefore, to confirm that A5 blocked IFN-γ receptor signaling, we next investigated whether packaged A5 could also interfere with IFN-γ-induced IP-10 production. As expected, IFN-γ triggered IP-10 production in differentiated THP-1 cells (Fig. 5D). Cells preincubated with purified A/WSN/33 virus particles inhibited this response. However, such an inhibition was not observed in the presence of purified A/WSN/33 viruses, in which packaged A5 was masked with a specific antibody. In the absence of IFN-γ, IP-10 release was barely detectable. Importantly, flow cytometry experiments showed comparable attachment of the cells by the two viruses, V versus AV-V, as revealed by similar HA staining in both groups (Fig. 5E, left panel). Quantification of MFI labeling of HA is shown on the right panel (Fig. 5E). Also, both virus preparations displayed identical infectivity (Fig. 5F). Thus, we concluded that A5 incorporated into virus particles inhibits IFN-γ-induced stat activation and IP-10 release.
These findings were further confirmed by an approach using siRNA, allowing us to obtain viruses with reduced A5 levels (referred to as A5 siRNA v) compared to control viruses (referred to as Ctl siRNA v) (Fig. 6A). Both virus preparations displayed identical infectivity (Fig. 6B) and similar ratios of the different virus proteins, as shown by Western blot analysis with a polyclonal anti-influenza virus antibody (Fig. 6C). When differentiated THP-1 cells were preincubated for 5 min with Ctl siRNA v, Stat phosphorylation triggered by IFN-γ was again inhibited. In marked contrast, no effect was observed in the presence of A5 siRNA v (Fig. 6D). Similar results were obtained in HeLa cells, suggesting that the inhibitory effect of virion-associated A5 was independent of the cell type (Fig. 6E). Packaged A5 also interfered with IFN-γ-induced IP-10 production at 3 h poststimulation, but this effect was lost after 24 h (Fig. 6F). In contrast, no effect of packaged A5 was observed upon IL-1β release (Fig. 6F). Comparable attachment of the cells by the two viruses, A5 siRNA v versus Ctl siRNA v, was confirmed by flow cytometry experiments (Fig. 7A) after binding assays for 5 min at 37°C or 1 h at 4°C. Indeed, quantification of the MFI showed similar A5 labeling (Fig. 7B). Also, after internalization assays for 30 min at 37°C, cell surface-bound viruses decreased, and both viruses showed similar internalization within the cells (Fig. 7C, left panel). Quantification of the MFI of cell surface versus the total (cell surface and internalized) viruses is shown in Fig. 7D. More importantly, inhibition mediated by packaged A5 on IFN-γ receptor signaling was specific, and such an effect was not detected in the presence of IFN-α (Fig. 7E and F). Altogether, these observations strengthen the previous findings showing that A5 incorporated into virus particles specifically blocks intracellular signaling mediated by IFN-γ.
FIG 6.

Packaged A5 inhibits IFN-γ receptor signaling. (A) Western blot analysis of virions produced from 293T cells transfected with nontargeted siRNA (Ctl siRNA v) or specific siRNA targeting A5 (A5 siRNA v), using an anti-A5 antibody. Anti-HA antibody was used as a positive control for virus detection. (B) Infectious titers of Ctl siRNA v and A5 siRNA v preparations. (C) Western blot analysis of control siRNA v and A5 siRNA v, using a polyclonal anti-influenza virus antibody. (D to F) Differentiated THP-1 cells (D) or HeLa cells (E) were incubated for 5 min with Ctl siRNA v or A5 siRNA v (MOI of 1). Cells were then either left unstimulated or stimulated with IFN-γ (, IU). After 5 min, the cells were lysed, and Stat phosphorylation was analyzed by Western blotting. (F) Alternatively, IP-10 release was evaluated in the supernatant at 3 or 24 h poststimulation by classical ELISA. *, P < 0.05 (between “−” versus “Ctl siRNA v” and “Ctl siRNA v” versus “A5 siRNA v”). The results are representative of at least two independent experiments.
FIG 7.

Packaged A5 does not inhibit IFN-α receptor signaling. (A) Differentiated THP-1 cells were incubated with Ctl siRNA v or A5 siRNA v (MOI of 1) for 5 min at 37°C or for 30 min at 4°C. (B) The cells were then analyzed for virus binding by flow cytometry with an anti-HA antibody, and the MFI of HA staining was obtained from three triplicates. (C) Alternatively, cells were incubated with the virus for 30 min at 4°C and with a shift to 37°C to allow virus internalization. Labeling of HA was performed either on unpermeabilized cells, showing cell surface-bound viruses (left panel), or on permeabilized cells, showing total viruses, including the cell surface and internalized ones (right panel). (D) The MFI of HA staining was obtained from three triplicates. (E and F) Differentiated THP-1 cells were incubated for 5 min with Ctl siRNA v or A5 siRNA v (MOI of 1). The cells were then either left unstimulated or stimulated with IFN-α (1,000 IU) or IFN-γ (1,000 IU). (E) After 5 min, the cells were lysed, and Stat1 phosphorylation was analyzed by Western blotting. (F) Alternatively, IP-10 release was evaluated in the supernatant at 3 h poststimulation by classical ELISA. *, P < 0.05 (between “−” versus “Ctl siRNA v” and “Ctl siRNA v” versus “A5 siRNA v”). The results are representative of two independent experiments (B and C).
Virus replication in vitro.
IFN-γ mediates a cellular antiviral state that prevents further viral spread (30). Since packaged A5 inhibits IFN-γ receptor signaling, we then investigated whether it could also block the antiviral activity of IFN-γ and promote viral replication. To address this point, viral growth was evaluated in the supernatant of differentiated THP-1 cells infected with IAV particles (Fig. 8). In the presence of IFN-γ treatment, masking A5 with a specific antibody on A/WSN/33 virus particles inhibited viral replication in differentiated THP-1 cells. Also, A5 siRNA v replicated less efficiently than Ctl siRNA v in the presence of IFN-γ. Altogether, these results showed that A5 incorporated into IAV particles triggers an intracellular process leading to increased virus production in the presence of IFN-γ.
FIG 8.
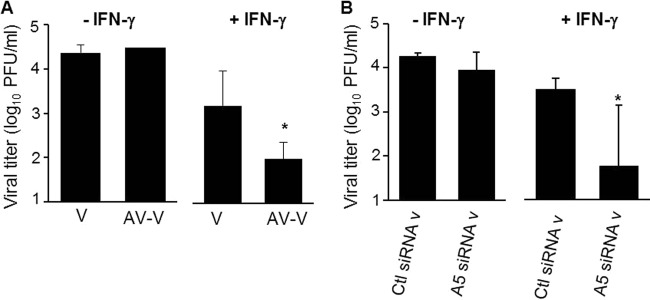
Packaged A5 inhibits the antiviral activity mediated by IFN-γ in vitro. PMA-differentiated THP-1 macrophages were infected with purified A/WSN/33 particles, in which A5 was previously masked (AV-V) or not masked (V) with anti-A5 antibody (A), or the supernatant of A/WSN/33-infected 293T cells, in which expression of A5 was downregulated by siRNA (A5 siRNAv) or not downregulated (Ctl siRNAv) (B). All viruses were used at an MOI of 1. The cells were either left in the presence or in the absence of rIFN-γ. Infectious virus titers were then evaluated in the supernatant of the cells at 24 hpi. The results represent mean virus titers ± the SD from three independent experiments. *, P < 0.05 (between “V” versus “AV-V” and “Ctl siRNA v” versus “A5 siRNA v”). The results are representative of three independent experiments.
Virus replication in vivo.
Next, we investigated whether packaged A5 could also promote viral replication by subverting the IFN-γ response in vivo. First, infectious virus titers were determined in lungs collected from infected mice treated with different concentrations of rIFN-γ. On day 2 postinoculation with IAV, the mean lung virus titers in mice treated with IFN-γ was lower than that of untreated mice, and this effect was dose dependent. A significant inhibition at 8 × 104 IU of rIFN-γ per mouse was observed (Fig. 9A). Thus, in vivo, the administration of rIFN-γ inhibits virus production in mouse lungs. Next, mice were infected with a high dose of purified IAV particles that were preincubated with or without anti-A5 neutralizing antibodies. At 2 days postinfection, the lung virus titers were evaluated. When purified virions (V) were used for infection, IFN-γ treatment inhibited the mean lung virus titers obtained compared to untreated mice. However, this inhibition was much greater when purified virions in which A5 was blocked were used to infect the mice (Fig. 9B). No difference was observed in lung virus titers obtained from mice infected with V or AV-V in the absence of rIFN-γ treatment. Thus, we concluded that A5 incorporated into IAV particles increases lung viral replication in the presence of IFN-γ in vivo.
FIG 9.
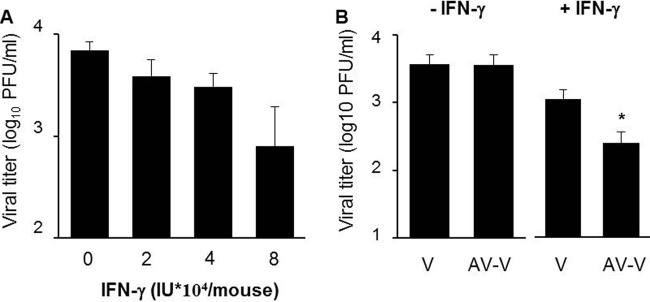
Packaged A5 inhibits the antiviral activity mediated by IFN-γ in vivo. (A) Mice were infected with purified A/PR/8/34 virus (500 PFU) and treated with the indicated quantities of mouse rIFN-γ by intranasal administration. At 2 days postinfection, virus titers were evaluated in the lungs by classical plaque assay. (B) Mice (n = 5 per group) were treated with 8 × 104 IU of rIFN-γ and infected with purified A/PR/8/34 viruses, in which A5 was previously blocked with anti-A5 antibody (AV-V) or not blocked (V). At 2 days postinfection, lung virus titers were evaluated by plaque assay. *, P < 0.05 (between “V” and “AV-V”). The results are representative of two independent experiments.
DISCUSSION
Previous works have shown that virus infection can alter the contingent of proteins exposed at the surface of the host cell (31). It is interesting that 5 of the 12 proteins with the strongest increase in surface abundance in influenza virus-infected cells have been previously detected in purified IAV virions. Therefore, it is tempting to speculate that their augmented display at the cell surface is not merely an incidental event but may be rather stimulated by the infection to support virus propagation. In the present study, we have demonstrated that incorporation of the host cellular protein A5 into IAV particles provided the virus with a means to inhibit IFN-γ signaling and increase its replication in vitro and in vivo. The in vitro data showed increased A5 cell surface expression after IAV infection. Cellular programmed cell death is activated by IAV and, during such event, phosphatidylserine becomes exposed to the cell surface (32). A5 has a strong affinity for phosphatidylserine (33), making it a useful probe for the detection of apoptotic cells (34). Thus, most likely, cellular A5 is translocated from the cytoplasm to the cell surface through phosphatidylserine binding and flip-flop transmembrane translocation of lipids (35). Interestingly, a substantial proportion of A5 was located in cholesterol-rich membrane domains, referred to as lipid rafts. HA was enriched in these domains, whereas M2 and the host ERK molecule were rather excluded, which is in line with other reports (36). It has been demonstrated that these domains are the platforms for IAV assembly and budding (36, 37). Since the viral envelope of IAV is derived from the host cell plasma membrane, it is likely that enveloped viruses incorporate proteins enriched in lipid rafts from the host cell. Accordingly, we were able to detect A5 in highly purified IAV preparations by Western blot analysis, as well as by immunogold labeling, indicating that cellular contaminants are probably not responsible for the detection of A5. Consistently, along with annexin 2, A5 has also been detected by matrix-assisted laser desorption ionization–time of flight analysis of purified IAV particles previously (data not shown; 14). These results are in accordance with results obtained by others and show that A5 is one of the 36 host proteins incorporated into influenza virus particles (13). Interestingly, A5 is also associated with other enveloped viruses, such as human cytomegaloviruses (38), human immunodeficiency viruses (39), herpes simplex viruses (40), vaccinia viruses (41), and porcine reproductive and respiratory syndrome viruses (42). Thus, the acquisition of A5 from the host cell membrane during the budding process is not specific to IAV. However, to our knowledge, the present study is the first to show a functional role for packaged A5 in the context of viral infection. Indeed, our results showed that A5-associated with IAV inhibited IFN-γ receptor signaling and allowed for an increase in viral replication, in vitro, using differentiated THP-1 macrophages and HeLa cells. These results were not observed in epithelial A549 cells, which surprisingly did not express the IFN-γ receptor at the cell surface (data not shown). Interestingly, however, we were able to confirm the role of packaged A5 on virus replication in vivo after 48 h of IFN-γ administration in mice, a period during which its biological activity remains stable (43).
In our study, the role of packaged A5 was detected when the virus was preincubated for 5 min but not 1 or 16 h before IFN-γ treatment (data not shown). Preincubation for 5 min most likely corresponds to virus binding to the cells, whereas after 1 h the virus may be internalized and after 16 h the virus may have undergone replication. Thus, virus binding to the cells, but not endocytosis or replication, was required for inhibition of IFN-γ receptor signaling. These results are consistent with a previous report which showed that A5 associates with the IFN-γ receptor and negatively regulates IFN-γ signaling (16).
IFN-γ plays an important role in recovery from IAV infection by helping to clear the virus (44–46). Thus, the incorporation of A5 into IAV particles provides the virus a way to escape from host immune IFN-γ responses and therefore is an opportunity for the virus to become more infectious. In line with this hypothesis, it has been shown that IAV abrogates the IFN-γ response to evade its antiviral activity (47). Thus, as previously suggested, strategies attempting to restore IFN-γ function may be of interest for therapeutic effects against IAV pathogenesis in humans (46).
We found that downregulation of A5 expression in 293T cells or in A549 epithelial cells had no effect on viral replication (data not shown), showing that A5 has no role in the viral replication cycle, at least in our conditions. These results differ from a previous study, which suggested that A5 could serve as a second receptor for viral entry (48). The precise physiological role of A5 remains to be determined. However, it has been proposed that A5 inhibits blood coagulation by competing for phosphatidylserine binding sites with prothrombin (49–51). Recently, we found that the thrombin protease-activated receptor 1 (PAR1) and hemostasis deregulation play a pivotal role in the inflammation and cytokine storm induced during severe virus infections (5, 23, 24, 52). Thus, the modulating function of A5 during IAV could go beyond IFN-γ. Possibly, by modulating hemostasis, A5 expression may also play a role in the inflammation and cytokine storm that occur during severe cases of influenza.
Altogether, this report suggests that specific incorporation of A5 into virus particles is a strategy adopted by IAV for subverting host defenses, thereby facilitating viral spread. The differential capacity of IAV to upregulate A5 at the surfaces of infected cells and to incorporate A5 during the budding process may be an additional factor for differences in the virulence of IAV.
ACKNOWLEDGMENTS
This study was supported by the Agence Nationale de la Recherche (grant ANR-13-BSV3-0011 HemoFlu to B.R.), the VIROSIGN project funded by the German Ministry of Education and Research (to T.W.), and the German Research Foundation (grant DFG SFB-TR84 to T.W.).
We are grateful to G. F. Rimmelzwaan (Erasmus University, Rotterdam, Netherlands) and N. Naffakh (Pasteur Institute, Paris, France) for the IAV strains and to T. Schwecke (Robert Koch Institute) for initial help with the MS analysis.
Footnotes
Published ahead of print 16 July 2014
REFERENCES
- 1.Fukuyama S, Kawaoka Y. 2011. The pathogenesis of influenza virus infections: the contributions of virus and host factors. Curr. Opin. Immunol. 23:481–486. 10.1016/j.coi.2011.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Webby RJ, Webster RG. 2003. Are we ready for pandemic influenza? Science 302:1519–1522. 10.1126/science.1090350 [DOI] [PubMed] [Google Scholar]
- 3.Stohr K. 2002. Influenza: WHO cares. Lancet Infect. Dis. 2:517. 10.1016/S1473-3099(02)00366-3 [DOI] [PubMed] [Google Scholar]
- 4.Kuiken T, Riteau B, Fouchier RA, Rimmelzwaan GF. 2012. Pathogenesis of influenza virus infections: the good, the bad, and the ugly. Curr. Opin. Virol. 2:276–286. 10.1016/j.coviro.2012.02.013 [DOI] [PubMed] [Google Scholar]
- 5.Berri F, Le VB, Jandrot-Perrus M, Lina B, Riteau B. 2014. Switch from protective to adverse inflammation during influenza: viral determinants and hemostasis are caught as culprits. Cell. Mol. Life Sci. 71:885–898. 10.1007/s00018-013-1479-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foucault ML, Moules V, Rosa-Calatrava M, Riteau B. 2011. Role for proteases and HLA-G in the pathogenicity of influenza A viruses. J. Clin. Virol. 51:155–159. 10.1016/j.jcv.2011.04.013 [DOI] [PubMed] [Google Scholar]
- 7.Kochs G, Garcia-Sastre A, Martinez-Sobrido L. 2007. Multiple anti-interferon actions of the influenza A virus NS1 protein. J. Virol. 81:7011–7021. 10.1128/JVI.02581-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.LeBouder F, Khoufache K, Menier C, Mandouri Y, Keffous M, Lejal N, Krawice-Radanne I, Carosella ED, Rouas-Freiss N, Riteau B. 2009. Immunosuppressive HLA-G molecule is upregulated in alveolar epithelial cells after influenza A virus infection. Hum. Immunol. 70:1016–1019. 10.1016/j.humimm.2009.07.026 [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Sastre A. 2011. Induction and evasion of type I interferon responses by influenza viruses. Virus Res. 162:12–18. 10.1016/j.virusres.2011.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le Gal FA, Riteau B, Sedlik C, Khalil-Daher I, Menier C, Dausset J, Guillet JG, Carosella ED, Rouas-Freiss N. 1999. HLA-G-mediated inhibition of antigen-specific cytotoxic T lymphocytes. Int. Immunol. 11:1351–1356. 10.1093/intimm/11.8.1351 [DOI] [PubMed] [Google Scholar]
- 11.Nayak DP, Balogun RA, Yamada H, Zhou ZH, Barman S. 2009. Influenza virus morphogenesis and budding. Virus Res. 143:147–161. 10.1016/j.virusres.2009.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nayak DP, Hui EK, Barman S. 2004. Assembly and budding of influenza virus. Virus Res. 106:147–165. 10.1016/j.virusres.2004.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw ML, Stone KL, Colangelo CM, Gulcicek EE, Palese P. 2008. Cellular proteins in influenza virus particles. PLoS Pathog. 4:e1000085. 10.1371/journal.ppat.1000085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LeBouder F, Morello E, Rimmelzwaan GF, Bosse F, Pechoux C, Delmas B, Riteau B. 2008. Annexin II incorporated into influenza virus particles supports virus replication by converting plasminogen into plasmin. J. Virol. 82:6820–6828. 10.1128/JVI.00246-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LeBouder F, Lina B, Rimmelzwaan GF, Riteau B. 2010. Plasminogen promotes influenza A virus replication through an annexin 2-dependent pathway in the absence of neuraminidase. J. Gen. Virol. 91:2753–2761. 10.1099/vir.0.023804-0 [DOI] [PubMed] [Google Scholar]
- 16.Leon C, Nandan D, Lopez M, Moeenrezakhanlou A, Reiner NE. 2006. Annexin V associates with the IFN-gamma receptor and regulates IFN-gamma signaling. J. Immunol. 176:5934–5942. 10.4049/jimmunol.176.10.5934 [DOI] [PubMed] [Google Scholar]
- 17.Yan X, Doffek K, Yin C, Krein M, Phillips M, Sugg SL, Johnson B, Shilyansky J. 2012. Annexin V promotes anti-tumor immunity and inhibits neuroblastoma growth in vivo. Cancer Immunol. Immunother. 61:1917–1927. 10.1007/s00262-012-1250-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riteau B, Barber DF, Long EO. 2003. Vav1 phosphorylation is induced by β2 integrin engagement on natural killer cells upstream of actin cytoskeleton and lipid raft reorganization. J. Exp. Med. 198:469–474. 10.1084/jem.20021995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riteau B, de Vaureix C, Lefevre F. 2006. Trypsin increases pseudorabies virus production through activation of the ERK signalling pathway. J. Gen. Virol. 87:1109–1112. 10.1099/vir.0.81609-0 [DOI] [PubMed] [Google Scholar]
- 20.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene ontology: tool for the unification of biology. Nat. Genet. 25:25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riteau B, Moreau P, Menier C, Khalil-Daher I, Khosrotehrani K, Bras-Goncalves R, Paul P, Dausset J, Rouas-Freiss N, Carosella ED. 2001. Characterization of HLA-G1, -G2, -G3, and -G4 isoforms transfected in a human melanoma cell line. Transplant Proc. 33:2360–2364. 10.1016/S0041-1345(01)02021-8 [DOI] [PubMed] [Google Scholar]
- 22.Zilberman S, Schenowitz C, Agaugue S, Benoit F, Riteau B, Rouzier R, Carosella ED, Rouas-Freiss N, Menier C. 2012. HLA-G1 and HLA-G5 active dimers are present in malignant cells and effusions: the influence of the tumor microenvironment. Eur. J. Immunol. 42:1599–1608. 10.1002/eji.201141761 [DOI] [PubMed] [Google Scholar]
- 23.Berri F, Rimmelzwaan GF, Hanss M, Albina E, Foucault-Grunenwald ML, Le VB, Vogelzang-van Trierum SE, Gil P, Camerer E, Martinez D, Lina B, Lijnen R, Carmeliet P, Riteau B. 2013. Plasminogen controls inflammation and pathogenesis of influenza virus infections via fibrinolysis. PLoS Pathog. 9:e1003229. 10.1371/journal.ppat.1003229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khoufache K, Berri F, Nacken W, Vogel AB, Delenne M, Camerer E, Coughlin SR, Carmeliet P, Lina B, Rimmelzwaan GF, Planz O, Ludwig S, Riteau B. 2013. PAR1 contributes to influenza A virus pathogenicity in mice. J. Clin. Invest. 123:206–214. 10.1172/JCI61667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sagara J, Tsukita S, Yonemura S, Tsukita S, Kawai A. 1995. Cellular actin-binding ezrin-radixin-moesin (ERM) family proteins are incorporated into the rabies virion and closely associated with viral envelope proteins in the cell. Virology 206:485–494. 10.1016/S0042-6822(95)80064-6 [DOI] [PubMed] [Google Scholar]
- 26.Mok CK, Lee DC, Cheung CY, Peiris M, Lau AS. 2007. Differential onset of apoptosis in influenza A virus H5N1- and H1N1-infected human blood macrophages. J. Gen. Virol. 88:1275–1280. 10.1099/vir.0.82423-0 [DOI] [PubMed] [Google Scholar]
- 27.Perrone LA, Plowden JK, Garcia-Sastre A, Katz JM, Tumpey TM. 2008. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 4:e1000115. 10.1371/journal.ppat.1000115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu WC, Chan RW, Wang J, Travanty EA, Nicholls JM, Peiris JS, Mason RJ, Chan MC. 2011. Viral replication and innate host responses in primary human alveolar epithelial cells and alveolar macrophages infected with influenza H5N1 and H1N1 viruses. J. Virol. 85:6844–6855. 10.1128/JVI.02200-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu M, Guo S, Hibbert JM, Jain V, Singh N, Wilson NO, Stiles JK. 2011. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 22:121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia-Sastre A, Biron CA. 2006. Type 1 interferons and the virus-host relationship: a lesson in detente. Science 312:879–882. 10.1126/science.1125676 [DOI] [PubMed] [Google Scholar]
- 31.Gudleski-O'Regan N, Greco TM, Cristea IM, Shenk T. 2012. Increased expression of LDL receptor-related protein 1 during human cytomegalovirus infection reduces virion cholesterol and infectivity. Cell Host Microbe 12:86–96. 10.1016/j.chom.2012.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Demchenko AP. 2012. The change of cellular membranes on apoptosis: fluorescence detection. Exp. Oncol. 34:263–268 [PubMed] [Google Scholar]
- 33.van Engeland M, Nieland LJ, Ramaekers FC, Schutte B, Reutelingsperger CP. 1998. annexin V-affinity assay: a review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 31:1–9. [DOI] [PubMed] [Google Scholar]
- 34.Niu G, Chen X. 2010. Apoptosis imaging: beyond annexin V. J. Nucl. Med. 51:1659–1662. 10.2967/jnumed.110.078584 [DOI] [PubMed] [Google Scholar]
- 35.Boon JM, Lambert TN, Sisson AL, Davis AP, Smith BD. 2003. Facilitated phosphatidylserine (PS) flip-flop and thrombin activation using a synthetic PS scramblase. J. Am. Chem. Soc. 125:8195–8201. 10.1021/ja029670q [DOI] [PubMed] [Google Scholar]
- 36.Leser GP, Lamb RA. 2005. Influenza virus assembly and budding in raft-derived microdomains: a quantitative analysis of the surface distribution of HA, NA and M2 proteins. Virology 342:215–227. 10.1016/j.virol.2005.09.049 [DOI] [PubMed] [Google Scholar]
- 37.Rossman JS, Lamb RA. 2011. Influenza virus assembly and budding. Virology 411:229–236. 10.1016/j.virol.2010.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varnum SM, Streblow DN, Monroe ME, Smith P, Auberry KJ, Pasa-Tolic L, Wang D, Camp DG, 2nd, Rodland K, Wiley S, Britt W, Shenk T, Smith RD, Nelson JA. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960–10966. 10.1128/JVI.78.20.10960-10966.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chertova E, Chertov O, Coren LV, Roser JD, Trubey CM, Bess JW, Jr, Sowder RC, II, Barsov E, Hood BL, Fisher RJ, Nagashima K, Conrads TP, Veenstra TD, Lifson JD, Ott DE. 2006. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J. Virol. 80:9039–9052. 10.1128/JVI.01013-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 82:8605–8618. 10.1128/JVI.00904-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jensen ON, Houthaeve T, Shevchenko A, Cudmore S, Ashford T, Mann M, Griffiths G, Krijnse Locker J. 1996. Identification of the major membrane and core proteins of vaccinia virus by two-dimensional electrophoresis. J. Virol. 70:7485–7497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang C, Xue C, Li Y, Kong Q, Ren X, Li X, Shu D, Bi Y, Cao Y. 2010. Profiling of cellular proteins in porcine reproductive and respiratory syndrome virus virions by proteomics analysis. Virol. J. 7:242. 10.1186/1743-422X-7-242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miyakawa N, Nishikawa M, Takahashi Y, Ando M, Misaka M, Watanabe Y, Takakura Y. 2011. Prolonged circulation half-life of interferon gamma activity by gene delivery of interferon gamma-serum albumin fusion protein in mice. J. Pharm. Sci. 100:2350–2357. 10.1002/jps.22473 [DOI] [PubMed] [Google Scholar]
- 44.Karupiah G, Chen JH, Mahalingam S, Nathan CF, MacMicking JD. 1998. Rapid interferon gamma-dependent clearance of influenza A virus and protection from consolidating pneumonitis in nitric oxide synthase 2-deficient mice. J. Exp. Med. 188:1541–1546. 10.1084/jem.188.8.1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wiley JA, Cerwenka A, Harkema JR, Dutton RW, Harmsen AG. 2001. Production of interferon-gamma by influenza hemagglutinin-specific CD8 effector T cells influences the development of pulmonary immunopathology. Am. J. Pathol. 158:119–130. 10.1016/S0002-9440(10)63950-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khoufache K, LeBouder F, Morello E, Laurent F, Riffault S, Andrade-Gordon P, Boullier S, Rousset P, Vergnolle N, Riteau B. 2009. Protective role for protease-activated receptor-2 against influenza virus pathogenesis via an IFN-gamma-dependent pathway. J. Immunol. 182:7795–7802. 10.4049/jimmunol.0803743 [DOI] [PubMed] [Google Scholar]
- 47.Uetani K, Hiroi M, Meguro T, Ogawa H, Kamisako T, Ohmori Y, Erzurum SC. 2008. Influenza A virus abrogates IFN-gamma response in respiratory epithelial cells by disruption of the Jak/Stat pathway. Eur. J. Immunol. 38:1559–1573. 10.1002/eji.200737045 [DOI] [PubMed] [Google Scholar]
- 48.Huang RT, Lichtenberg B, Rick O. 1996. Involvement of annexin V in the entry of influenza viruses and role of phospholipids in infection. FEBS Lett. 392:59–62. 10.1016/0014-5793(96)00783-1 [DOI] [PubMed] [Google Scholar]
- 49.Rand JH, Wu XX, Quinn AS, Taatjes DJ. 2010. The annexin A5-mediated pathogenic mechanism in the antiphospholipid syndrome: role in pregnancy losses and thrombosis. Lupus 19:460–469. 10.1177/0961203310361485 [DOI] [PubMed] [Google Scholar]
- 50.Rand JH. 2000. The pathogenic role of annexin V in the antiphospholipid syndrome. Curr. Rheumatol. Rep. 2:246–251. 10.1007/s11926-000-0086-7 [DOI] [PubMed] [Google Scholar]
- 51.Joseph JE, Harrison P, Mackie IJ, Isenberg DA, Machin SJ. 2001. Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br. J. Haematol. 115:451–459. 10.1046/j.1365-2141.2001.03101.x [DOI] [PubMed] [Google Scholar]
- 52.Aerts LHM, Rhéaume C, Lavigne S, Couture C, Kim W, Susan-Resiga D, Prat A, Seidah NG, Vergnolle N, Riteau B, Boivin G. 2013. Modulation of protease activated receptor 1 influences human metapneumovirus disease severity in a mouse model. PLoS One 28:e72529. 10.1371/journal.pone.0072529 [DOI] [PMC free article] [PubMed] [Google Scholar]