

ABSTRACT
CD8+ T cells are an essential component of successful adaptive immune responses against hepatitis C virus (HCV). A major obstacle to vaccine design against HCV is its inherent viral sequence diversity. Here, we test the hypothesis that different sequence variants of an immunodominant CD8+ T cell epitope, all binding with high affinity to HLA class I, target different T cell receptor repertoires and thereby influence the quality of the CD8+ T cell response. The impacts of sequence differences in the HLA-A*02-restricted HCV NS31406–1415 epitope on in vitro priming of naive CD8+ T cells from seronegative donors and cross-reactivity of primed T cells with other epitope variants were characterized. Although the six epitope variants tested were all high-affinity binders to HLA-A*02:01, substantial differences in priming and cross-reactivity of CD8+ T cells were observed. The variant associated with the most reproducible priming and induction of T cells with broad cross-reactivity was a genotype 1b variant (KLSALGLNAV) that is more common in HCV isolates collected in Asia but is rare in sequences from Europe and North America. The superior immunogenicity and cross-reactivity of this relatively rare epitope variant were confirmed by using HCV-specific memory CD8+ T cells from people who inject drugs, who are frequently exposed to HCV. Collectively, the data suggest that sequence differences at the epitope level between HCV isolates substantially impact CD8+ T cell priming and the degree of cross-reactivity with other epitope variants.
IMPORTANCE The results have important implications for vaccine design against highly variable pathogens and suggest that evidence-based selection of the vaccine antigen sequence may improve immunogenicity and T cell cross-reactivity. Cross-reactive CD8+ T cells are likely beneficial for immune control of transmitted viruses carrying epitope variants and for prevention of immune escape during acute infection. To this end, rare epitope variants and potentially even altered epitope sequences associated with priming of broadly cross-reactive T cell receptors should be considered for vaccine design and need further testing.
INTRODUCTION
Infection with hepatitis C virus (HCV) is one of the leading causes of acute and chronic liver disease. Worldwide, 130 million to 170 million people are chronically infected, representing approximately 2 to 3% of the world's population. Despite the enormous success of new antiviral drugs directly acting against HCV, the high costs of these drugs and barriers to treatment of groups at high risk for HCV infection limit their widespread use in many parts of the world (1, 2). Therefore, development of an effective vaccine to prevent chronic HCV infection still remains a major goal. Both innate and adaptive immunity are essential to control HCV infection; however, only a minority of infected patients achieves spontaneous clearance of the virus, whereas most patients develop chronic hepatitis, associated with the risk of progressive liver disease. In cases where the virus is cleared spontaneously, resolution of reinfection occurs more rapidly (3), indicating that HCV-specific memory immune responses positively impact disease control. Hence, a potent vaccine inducing robust T cell responses could provide significant clinical benefit.
There is strong evidence that CD8+ T cells are an essential component of a successful immune response against HCV during acute infection (3), even though inherent viral sequence diversity is a major obstacle to vaccine design against hepatitis C (4). So far, seven different genotypes and multiple subtypes have been described (5). Moreover, even isolates of the same HCV subtype are highly polymorphic between individuals. In the context of HLA allelic restriction, this high sequence diversity thus represents the main barrier for immune control. Even in conserved regions of the HCV polyprotein, most CD8+ T cell epitopes differ between HCV genotypes (6). Accordingly, the majority of CD8+ T cell responses is directed against one genotype only and shows little cross-reactivity with other genotypes (6). Indeed, the protective effect of beneficial HLA alleles such as HLA-B*27 and HLA-B*57 was limited to certain HCV genotypes and subtypes (7, 8), and there is strong evidence that the sequence of immunodominant CD8 T cell epitopes upon viral transmission impacts the outcome of HCV infection (9).
The CD8+ T cell compartment is characterized by a highly diverse and individualized T cell receptor (TCR) repertoire as a consequence of random gene reassortment. Here, we hypothesized that different sequence variants of an immunodominant CD8+ T cell epitope, all binding with high affinity to HLA class I, target different TCR repertoires and thereby influence the quality of the CD8+ T cell response. By utilizing different peptides corresponding to naturally occurring variants of the HLA-A*02-restricted HCV epitope NS31406–1415 for CD8+ T cell priming in vitro, we addressed whether differences in the TCR repertoire for the respective peptides can be viewed as a reflection of different immunogenic properties associated with different CD8+ T cell cross-reactivities. This epitope was previously reported to be immunodominant in the context of HLA-A*02 (10). The genotype 1a prototype sequence (KLVALGINAV) is the most commonly used sequence in human immunology studies of HCV infection for identification of HCV-specific CD8+ T cells (http://www.iedb.org/) (11), and selection of escape mutations associated with a lack of immunogenicity has been reported for genotype 1a (12). Notably, the epitope region is highly polymorphic in genotypes 1a and 1b. Although CD8+ T cell responses against the most frequent variants of both subtypes have been reported (http://www.iedb.org/) (11), the exact differences in immunogenicity of the epitope variants are unknown. To address this, HCV NS31406–1415 sequence variants were selected from the HCV sequence database (13), including two genotype 1a and three genotype 1b variants. In addition, a fourth genotype 1b variant under reproducible selection pressure by CD8+ T cells from a large single-source outbreak was included (14). Our data demonstrate that there are substantial differences in priming and cross-reactivity of CD8+ T cells between the variants.
MATERIALS AND METHODS
Study subjects.
Buffy coats were obtained from HLA-A*02:01-positive, HCV-seronegative, healthy blood donors at the Institute of Transfusion Medicine, University Hospital Essen, and peripheral blood mononuclear cells (PBMCs) were extracted by Ficoll gradient centrifugation. High-resolution HLA-A typing was performed by using commercially available sequence-specific oligonucleotides (High Definition LABType methodology; One Lambda Inc., Canoga Park, CA, USA). Additional blood samples from people who inject drugs (PWID) were collected from the ward for inpatient detoxification treatment of drug addicts or the clinic for opioid maintenance treatment (OMT) at the Department of Addictive Behavior and Addiction Medicine of the LVR-Hospital Essen, Hospital of the University of Duisburg-Essen. Written informed consent was obtained from all participants, and the study was approved by the ethics committee of the Medical Faculty of the University of Duisburg-Essen. Anti-HCV-seropositive, HLA-A*02-positive PWID (with or without detectable HCV RNA) were selected for analysis. Low-resolution HLA typing of blood samples from PWID was also performed by using LABType methodology (One Lambda Inc., Canoga Park, CA, USA).
Analysis of peptide binding to HLA-A*02:01.
Peptide binding to HLA-A*02:01 was determined at Sanquin Blood Supply (Amsterdam, The Netherlands) as described previously (15). Briefly, peptide exchange was assayed by using the PeliChange p*HLA-A*0201 kit (catalog number M2802; Sanquin) and the PeliScreen HLA class I enzyme-linked immunosorbent assay (ELISA) kit (catalog number M1924; Sanquin). Peptide-exchange reactions were performed by exposure of conditional peptide/HLA (pHLA) complexes to long-wavelength UV by using a 366-nm UV lamp (Camag, Muttenz, Switzerland), in the absence or presence of the indicated peptide (50 μM). Subsequently, peptide-exchange efficiency was analyzed by using the PeliScreen HLA class I ELISA kit. This ELISA determines the presence of intact HLA class I complexes in an exchange reaction mixture. The following control peptides were used: NLVPMVATV, a high-affinity binder to HLA-A*02:01, and CTELKLSDY, an HLA-A*01:01 ligand with no substantial affinity for HLA-A*0201. The absorbance of all peptides was evaluated relative to that of the high-affinity binder, which was set at 100%.
In vitro priming of naive CD8+ T cells.
In vitro CD8+ T cell priming was performed as previously described (16, 17). Briefly, monocytes were isolated by adherence to plastic and differentiated with 1,000 U/ml interleukin-4 (IL-4) and 800 U/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) (Peprotech) in Cellgenix DC medium (Cellgenix) supplemented with 0.4% penicillin-streptomycin (PAA, Austria) and 1% naturally coagulated human serum (PAA, Austria). After 72 h, immature monocyte-derived dendritic cells (moDCs) were matured by using 100 U/ml gamma interferon (IFN-γ) (Peprotech), 100 ng/ml lipopolysaccharide (LPS) (Sigma), and antigenic peptides at a concentration of 2 μg/ml overnight. All peptides were synthesized by EMC Microcollections (Tübingen, Germany), with a purity of ≥95%. Naive CD8+ T cells were isolated by negative selection utilizing a CD8+ T cell isolation kit, followed by depletion of CD57− and CD45RO-positive cells using microbeads (Miltenyi Biotech) according to the manufacturer's instructions. Purified naive CD8+ T cells were supplemented with 5 ng/ml IL-7 (Peprotech) and maintained overnight. The next day, mature moDCs were harvested and exposed to 30 Gy of irradiation. CD8+ T cells cultured overnight were mixed with moDCs at a ratio of 4:1 in T cell medium (Cellgenix DC medium supplemented with 0.4% penicillin-streptomycin and 5% human serum) supplemented with 30 ng/ml IL-21 (Peprotech). Fresh medium containing 5 ng/ml IL-7 and IL-15 (Peprotech) was added after 72 h and subsequently every 2 to 3 days. After 10 and 19 days, cocultures were restimulated by using irradiated, autologous, peptide-pulsed PBMCs. After restimulation, fresh medium supplemented with 100 U/ml IL-2 (Roche) and 5 ng/ml IL-7 and IL-15 was added after 48 h and subsequently every 2 to 3 days. Cells were analyzed after 27 days either by direct staining with HLA-A*02/peptide dextramers (fluorescently labeled phycoerythrin [PE] major histocompatibility complex [MHC] class I dextramers; Immudex, Copenhagen, Denmark) or by functional analysis.
Functional analysis of in vitro-primed CD8+ T cells.
For functional analysis, cells were harvested and stimulated with 1 × 105 autologous, peptide-pulsed monocytes at a 1:2 ratio of monocytes to T cells or with synthetic peptide (10 μg/ml) in the presence of 100 ng/ml brefeldin A. After 5 h of stimulation, cells were stained with viability stain (eFluor780) as well as CD8 (Alexa 700) and CD4 (PE-CF594) antibodies, followed by permeabilization and staining for IFN-γ (fluorescein isothiocyanate [FITC]). For analyses of polyfunctionality, cells were restimulated in the presence of brefeldin A and 1.25 μg/ml anti-CD107a for 5 h. Subsequently, cells were stained for CD4 (allophycocyanin [APC]-eFluor780) and CD8 (eFluor450) and with viability dye (eFluor506). After fixation and permeabilization, the cells were additionally stained with IFN-γ (FITC), IL-2 (peridinin chlorophyll protein [PerCP]-eFlour710), macrophage inflammatory protein 1 beta (MIP1β) (PE-Cy7), and tumor necrosis factor alpha (TNF-α) (APC). Antibodies were purchased from eBioscience (San Diego, CA, USA) or BD Pharmingen (Heidelberg, Germany) and used at optimal staining concentrations. Cells were analyzed by using a FACSCalibur or FACSCanto II flow cytometer (BD Pharmingen) or a Navios flow cytometer (Beckman Coulter) and results were analyzed with FlowJo software (Treestar, Ashland, OR). Polyfunctionality of primed CD8+ T cells was examined in selected experiments. Peptide-specific CD8+ T cells were simultaneously stained with specific antibodies and analyzed by flow cytometry. Data were further analyzed by using Pestle and SPICE software (18).
Expansion of peptide-specific CD8+ T cells in people who inject drugs.
Cryopreserved PBMCs from PWID were used to expand peptide-specific CD8+ T cells. PBMCs were cultured in complete medium (RPMI 1640 containing 10% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, and 100 μM HEPES buffer) supplemented with 1 μg/ml peptide, 25 U/ml IL-2, and 0.1 μg/ml anti-CD28 and anti-CD49d (BD Pharmingen). After 10 days, specific cells were analyzed for IFN-γ secretion by intracellular cytokine staining, as described above.
Analysis of the T cell receptor Vβ repertoire of peptide-specific CD8+ T cells.
Antigen-specific T cells were further analyzed by using an antibody panel directed against 24 different individual variable beta (Vβ) chains of the TCR. The panel contained Vβ1 PE, Vβ2 FITC, Vβ3 FITC, Vβ4 FITC, Vβ5.2 PE, Vβ7.1 PE, Vβ7.2 PE, Vβ11 APC, Vβ12 FITC, Vβ13.6 PE, Vβ14 PE, Vβ16 FITC, Vβ17 PE, Vβ18 PE, Vβ20 FITC, Vβ21.3 FITC, and Vβ22 FITC antibodies (all purchased from Beckman Coulter) as well as Vβ5.1 APC, Vβ5.3 APC, Vβ8 eFluor450, Vβ9 APC, Vβ13.1 FITC, Vβ13.2 PE, and Vβ23 PerCP-eFluor710 antibodies (all obtained from eBioscience), covering approximately 70% of the Vβ repertoire. Cells were stained simultaneously with antibodies against CD4 (APC-eFluor780), CD8 (eFluor450 or APC), and IFN-γ (PE-Cy7), a set of 2 to 3 antibodies directed against TCR Vβ families, as well as a viability stain (eFluor506). CD8+ T cells responding to peptide restimulation were selected based on their IFN-γ secretion and further analyzed for their Vβ usage by using a FACSCanto II instrument.
Sequence analysis of the NS31406–1415 epitope.
All available HCV genotype 1b NS3 sequences were retrieved from the public HCV sequence database (13). Identical sequences and sequences of unknown origin were removed, and additional sequences obtained in Germany and China were added and aligned by using Geneious 7.0.6 (Biomatters, Auckland, New Zealand). A phylogenetic tree was calculated by utilizing the neighbor-joining method and the Jukes-Cantor distance model, as implemented in Geneious 7.0.6 software. The resulting tree was edited with FigTree software (available at http://tree.bio.ed.ac.uk/). Branches were transformed into a cladogram, and sequences from Japan and China are shown in red.
RESULTS
Frequent variants of HCV epitope NS31406–1415 bind with high affinity to HLA-A*02:01.
The most frequent sequence variants of the immunodominant HLA-A*02-restricted CD8+ T cell epitope of HCV (NS31406–1415) were selected from a public HCV sequence database (Table 1). The most frequent sequence of this epitope in genotype 1a is KLVALGINAV (gt1a_1) (43.3% of the genotype 1a sequence entries in the database), which is also the most frequently used variant in studies of HCV immunology (11). The second most frequent genotype 1a variant is KLVALGVNAV (gt1a_2) (31.8% of genotype 1a sequences). Other variants of this epitope are rare in genotype 1a (<4% for individual variants in the HCV sequence database). The most frequent variant of genotype 1b is KLSGLGLNAV (gt1b_1) (42.8% of genotype 1b sequences), followed by KLSSLGLNAV (gt1b_2) (18.4% of genotype 1b sequences) and KLSALGLNAV (gt1b_3) (12.5% of genotype 1b sequences). Other variants of this epitope are rare for genotype 1b (<6% for individual variants in the HCV sequence database). Importantly, CD8+ T cell responses to all five variants have been reported for patients with HCV infection, indicating that all variants are potentially targeted in HLA-A*02-positive humans (http://www.iedb.org/). One additional genotype 1b sequence variant (QLSGLGINAV [gt1b_AD]), which was the source sequence from a large HCV genotype 1b outbreak, was included in this study. A previous study of HLA class I-associated viral evolution in this outbreak suggested that this sequence was under strong immune pressure in HLA-A*02-positive patients (14).
TABLE 1.
Sequence variants of the NS3-A21406-1415 epitope in this study
| Genotype | Variant | Sequence | % frequencya | IC50 for binding to A*02:01b (nM) |
|---|---|---|---|---|
| 1a | gt1a_1 | KLVALGINAV | 43.3 | 36 |
| gt1a_2 | KLVALGVNAV | 31.8 | 58 | |
| 1b | gt1b_1 | KLSGLGLNAV | 42.8 | 26 |
| gt1b_2 | KLSSLGLNAV | 18.4 | 26 | |
| gt1b_3 | KLSALGLNAV | 12.5 | 25 | |
| gt1b_AD | QLSGLGINAVc | NA | 123 |
Data from the HCV sequence database (http://hcv.lanl.gov/). NA, not applicable.
Predicted 50% inhibitory concentration (IC50) for HLA-A*02:01 binding utilizing the artificial neuronal network method.
Inoculum sequence from the East German outbreak (14).
All six variants were predicted to bind with high affinity to HLA-A*02:01 in a prediction algorithm implemented in the Immune Epitope Database (IEDB) (http://tools.immuneepitope.org/mhci/), with the exception of the epitope variant gt1b_AD, which was predicted to bind with intermediate affinity (Table 1). Binding to HLA-A*02:01 was experimentally tested by HLA stabilization assays using UV-induced peptide exchange (Fig. 1). The positive-control peptide used in this assay represents a high-affinity ligand of HLA-A*02:01 (NLVPMVATV). All six variants of the NS31406–1415 epitope showed binding of >100% compared to the positive control, showing that they all bind to HLA-A*02:01 with high affinity.
FIG 1.
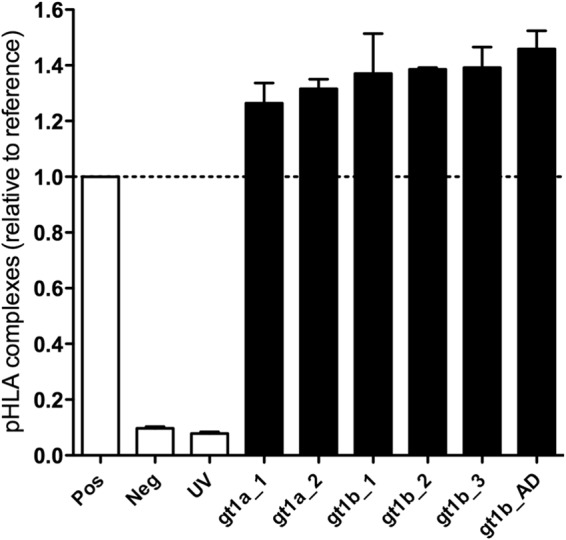
Peptide binding affinity for HLA-A*02:01. Variants of the NS31406–1415 epitope were analyzed for their specific binding to HLA-A*02:01 by a peptide-exchange assay. After induction of degradation of a conditional reference peptide in complex with HLA-A*02:01 by UV light, the peptide-exchange rate with selected ligands was determined by an ELISA that detects intact pHLA complexes. A high-affinity binder to HLA-A*02:01 (NLVPMVATV) and an HLA-A*01:01 ligand with no substantial affinity for HLA-A*02:01 (CTELKLSDY) served as positive (Pos) and negative (Neg) controls, respectively. “UV” indicates the ELISA results for pHLA complexes upon exposure to UV light in the absence of peptide. The absorbance of all peptides was evaluated relative to that of the high-affinity binder, which was set at 100%.
Sequence variants of HCV epitope NS31406–1415 show distinct reproducibility of CD8+ T cell priming in vitro.
Most analyses of HCV-specific CD8+ T cells are focused on memory T cells expanded or isolated from anti-HCV-seropositive donors. In this setting, the exact sequence at the time of transmission is typically unknown; hence, the impact of epitope variants on the recruited CD8+ T cell repertoire is difficult to address. Here, the goal was to prime and expand HCV-specific T cells from an unaltered repertoire of naive CD8+ T cells and to address whether priming and cross-reactivity are influenced by sequence variants. Therefore, peptide-loaded moDCs were used for priming of naive CD8+ T cells from HLA-A*02:01-positive, anti-HCV- and HCV RNA-negative healthy blood donors. After 27 days of culture, including two additional restimulations, the frequency of primed NS31406–1415-specific CD8+ T cells was analyzed by using MHC class I multimers in 6 donors (donors D5 to D11) (Fig. 2A) and by analysis of IFN-γ-secreting CD8+ T cells upon peptide restimulation in all 11 donors (Fig. 2B). The frequencies of successful priming of naive CD8+ T cells differed between the peptide variants tested. Overall, priming was more reproducible with the genotype 1b variants than with the genotype 1a variants. In genotype 1a, the prototype sequence (gt1a_1) showed better priming potential than the gt1a_2 variant. Interestingly, the most frequent genotype 1b variant (gt1b_1) was associated with less reproducible priming than the other three genotype 1b variants. Figure 2C summarizes the frequencies of successful priming (defined as a minimum of 0.5% IFN-γ-secreting CD8+ T cells upon peptide restimulation) for all experiments. Due to limitations in cell yields in some experiments, not all peptides were tested in duplicate throughout all experiments. Priming was most frequently successful when the genotype 1b variants gt1b_3 (55% success rate) and gt1b_AD (40% success rate) were used. Of note, in some cases, there was a drastic discrepancy between the numbers of peptide-specific CD8+ T cells assessed with HLA class I multimers and the percentage of IFN-γ-producing cells (e.g., donor D5 primed with gt1a_1). Therefore, the polyfunctionality of in vitro-primed peptide-specific CD8+ T cell responses directed against the variants gt1a_1 and gt1b_3 was characterized in parallel by analysis of four different cytokines (IFN-γ, TNF-α, IL-2, and MIP1β) and the degranulation marker CD107a in one donor. Figure 2D shows the distribution of specific CD8+ T cells with 1, 2, 3, 4, and 5 effector functions upon stimulation with high peptide concentrations (10 μg/ml). Here, CD8+ T cells primed with the epitope variant gt1b_3 showed superior functionality compared to CD8+ T cells primed with the variant gt1a_1.
FIG 2.

Different epitope variants show distinct priming capacities. Naive CD8+ T cells were primed with different epitope variants, as indicated, for 28 days. All experiments were started in duplicate with cells from 11 different donors. Results from the same donor are presented with the same symbol, as indicated in the key. (A) Cultures from donors D5 to D11 were analyzed by peptide-specific MHC class I multimers. (B) Cultures from donors D1 to D11 were analyzed by intracellular cytokine staining for IFN-γ. (C) Success rate of CD8+ T cell priming, defined as a minimum of 0.5% IFN-γ-secreting CD8+ T cells upon peptide restimulation. (D) Polyfunctionality (secretion of IFN-γ, TNF-α, IL-2, and MIP1β and degranulation) of CD8+ T cells from one donor primed with the gt1a_1 and gt1b_3 variants as determined by Boolean gating (FlowJo software). Pie charts were generated by using Pestle and SPICE software.
CD8+ T cells primed with sequence variants of HCV epitope NS31406–1415 show distinct cross-reactivity.
Next, the cross-reactivity of the in vitro-primed CD8+ T cells against the different sequence variants of the NS31406–1415 epitope was analyzed. Therefore, cultured CD8+ T cells were restimulated with all variants, and the percentage of IFN-γ-producing cells was determined. Figure 3A exemplifies the differences in cross-reactivity obtained for in vitro-primed CD8+ T cells from one donor. CD8+ T cells were primed with peptide gt1b_3, resulting in 1.72% IFN-γ-producing cells upon restimulation with gt1b_3 (Fig. 3A, left). Similar frequencies of IFN-γ-producing cells were observed upon restimulation with the other genotype 1b variants, indicating full cross-reactivity with these genotype 1b variants, even though there was no cross-reactivity with the genotype 1a variants. In contrast, although, in the same donor, high frequencies of CD8+ T cells specific for the variant gt1b_AD were primed (Fig. 3A, right), these cells were not cross-reactive with any other epitope variant. The patterns of cross-reactivity were highly reproducible across different experiments: CD8+ T cells primed with the epitope variant gt1b_AD were never cross-reactive with other variants of the epitope, whereas the highest degree of cross-reactivity was observed for CD8+ T cells primed with the epitope variant gt1b_2 or gt1b_3 (Fig. 3B). Importantly, primed CD8+ T cells seemed to exclusively react with variants of the same subtype. This was most evident for CD8+ T cells primed with gt1b variants, which are almost not reactive with the gt1a variants, indicating that these cells are subtype specific (Fig. 3B).
FIG 3.

Primed peptide-specific CD8+ T cells show distinct cross-reactivity patterns. (A) Examples of cross-reactivity of CD8+ T cells primed with either gt1b_3 (left) or gt1b_AD (right) with all other epitope variants upon restimulation after 28 days. CD8+ T cells primed with gt1b_3 were cross-reactive with other genotype 1b epitope variants, whereas CD8+ T cells primed with gt1b_AD were not cross-reactive. (B) Summary of the cross-reactivity profiles from all priming experiments. The results of 9 to 11 independent experiments utilizing cells of different donors are shown.
Cross-reactive CD8+ T cells primed with gt1b_3 utilize a broader TCR Vβ repertoire.
To address the difference between T cells with high cross-reactivity primed with gt1b_3 and T cells with low cross-reactivity primed with gt1b_AD, TCR Vβ chain profiles were assessed by flow cytometry utilizing specific antibodies. We selected a single donor to prime CD8+ T cells in parallel with either the gt1b_3 variant or the gt1b_AD variant. Consistent with the results described above, in two experiments, CD8+ T cells primed with gt1b_3 were cross-reactive with gt1b_1 (Fig. 4A), whereas, as expected, CD8+ T cells primed with gt1b_AD did not react with gt1b_3 or gt1b_1 (Fig. 4C). Analysis of the TCR Vβ profiles revealed recruitment of multiple TCR Vβ chains for the CD8+ T cell response against the gt1b_3 variant (Fig. 4B). Although there was a large overlap between the two priming experiments with the same peptide variant, some TCR Vβ chains, such as Vβ 20 and Vβ 22, were expanded in one experiment only (Fig. 4B, top and bottom). In contrast, CD8+ T cells primed with the gt1b_AD variant were characterized by a more restricted repertoire (mainly TCR Vβ 3 and Vβ 5.1) (Fig. 4D). Importantly, when CD8+ T cells primed with the gt1b_3 variant were restimulated with the gt1b_1 variant, the same TCR Vβ repertoire was observed, suggesting that cross-reactivity was determined at the level of individual TCR Vβ chains.
FIG 4.

Distinct T cell receptor Vβ repertoire of gt1b_3-primed CD8+ T cells. Naive CD8+ T cells (donor D8) were primed in parallel with gt1b_3 (A and B) or with gt1b_AD (C and D) for 28 days and restimulated with the peptides gt1b_3, gt1b_1, and gt1b_AD in two separate experiments (I and II). (A and C) The frequency of IFN-γ+ CD8+ T cells upon peptide restimulation was determined. (B and D) TCR Vβ+ usage of IFN-γ+ CD8+ T cells upon peptide restimulation was analyzed with a panel of 24 antibodies covering approximately 70% of the TCR Vβ repertoire.
CD8+ T cells specific for gt1b_3 show broader cross-reactivity in PWID exposed to HCV.
We next addressed whether similar cross-reactivity patterns were present in memory CD8+ T cells of HCV-exposed patients. We therefore analyzed the NS31406–1415-specific CD8+ T cell response in 12 HLA-A*02-positive patients from a cohort of PWID, including 6 PWID infected with HCV genotype 1 and 6 anti-HCV-positive PWID without detectable HCV RNA. Previous studies suggested that PWID from this cohort are highly exposed to HCV genotypes 1 and 3, although the exact sequence at the time of transmission and priming of CD8+ T cell responses is unknown. Upon antigen-specific expansion by a standard protocol with the gt1a_1, gt1b_1, or gt1b_3 peptide, CD8+ T cell responses were detectable by intracellular IFN-γ staining in seven PWID (Fig. 5). In four patients, IFN-γ-producing cells were detectable upon expansion with gt1a_1. Notably, these CD8+ T cells showed almost no cross-reactivity with the two other genotype 1b variants tested. Similar to the results of the in vitro priming experiments, IFN-γ-producing CD8+ T cells were most reproducibly expanded in the presence of the variant gt1b_3 (5 of 12 PWID). Furthermore, in all cases, these CD8+ T cells were to some degree cross-reactive with both of the other tested peptides, including the genotype 1a variant. The superiority of the gt1b_3 variant was most obvious in HCV RNA-negative patient IDU172. Here, a strong and fully cross-reactive CD8+ T cell response was detectable after expansion in the presence of gt1b_3, whereas no IFN-γ-producing CD8+ T cells were detected after expansion in the presence of any of the other peptides.
FIG 5.
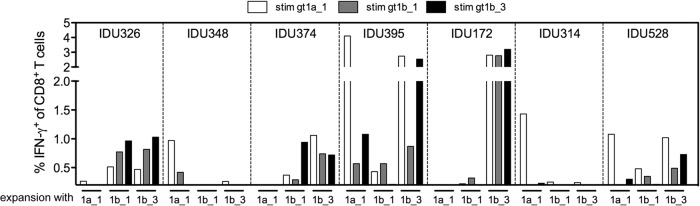
NS31406–1415 variant-specific CD8+ T cell responses in 12 HLA-A*02-positive patients from a cohort of PWID. PBMCs from PWID were expanded in the presence of the epitope variant gt1a_1, gt1b_1, or gt1b_3, as indicated at the bottom of the graph. After 10 days, cultures were restimulated with all three epitope variants and analyzed by intracellular cytokine staining for IFN-γ. The percent frequencies of IFN-γ+ CD8+ T cells upon peptide restimulation are shown.
The gt1b_3 variant is predominantly observed in HCV isolates from Asia.
We next wanted to know if there are population-based differences in the frequencies of sequence variants of the NS31406–1415 epitope. We were particularly interested in the gt1b_3 variant, as this was the most immunogenic epitope variant in our studies. All genotype 1b NS3 sequences of known origin were retrieved from a public database and aligned together with additional sequences obtained from isolates collected in Germany and China. A phylogenetic tree of all genotype 1b sequences revealed distinct clusters (Fig. 6), with most sequences collected in Asia (mainly China and Japan [shown in red in Fig. 6]) branching together. Notably, sequences carrying the variant gt1b_3 (marked with dots in the tree) were highly enriched in HCV genotype 1b isolates of Asian origin. Twenty-six of 129 sequences (20.1%) from Asia carried the gt1b_3 sequence; in contrast, only 12 of 315 sequences (3.8%) collected outside Asia (mainly Europe and North America) carried this variant (P = 2 × 10−7), suggesting that the gt1b_3 variant is more common in Asia.
FIG 6.

The epitope variant gt1b_3 is enriched in HCV genotype 1b isolates from Asia. All available HCV genotype 1b NS3 sequences were retrieved from the public HCV sequence database. Identical sequences and sequences of unknown origin were removed, and additional sequences obtained in Germany and China were added and aligned. A phylogenetic tree was calculated by utilizing the neighbor-joining method and the Jukes-Cantor distance model. Branches were transformed into a cladogram, and sequences from Japan and China are shown in red. Sequences carrying the epitope variant gt1b_3 are indicated by black dots.
DISCUSSION
HCV remains a global health challenge, but effective vaccine design is still hindered by its inherent sequence diversity. The existence of multiple genotypes and subtypes and innumerable variants at the quasispecies level calls for novel strategies to carefully select the optimal antigen for prophylactic immunization strategies. A successful vaccine needs to cope with the enormous antigen diversity, for example, by identifying and targeting the immune response to conserved regions of the virus. However, CD8+ T cell epitopes that are conserved across genotypes are rare in HCV (6). Alternatively, a successful vaccine should activate multiple different CD8+ T cell responses against different sequence variants of important epitopes (19) or should prime CD8+ T cells with highly cross-reactive TCRs that interact with multiple sequence variants. The latter would possibly also be advantageous for prevention of selection of immune escape mutations (20, 21), which strongly contributes to viral persistence during acute infection (22–25).
Here, we show that naturally circulating sequence variants of the immunodominant HLA-A*02-restricted CD8+ T cell epitope NS31406–1415, all binding to HLA-A*02:01 with similar affinities, are associated with reproducible differences regarding the success rate of in vitro priming and the cross-reactivity of primed CD8+ T cells. Different peptide variants in complex with HLA-A*02:01 presumably engage different TCRs. Although not tested here, this could be the consequence of conformational changes, for example, by different peptide folding in complex with HLA class I or direct interactions between replaced amino acids and TCR residues. Importantly, when CD8+ T cells were primed in vitro, there was almost no cross-reactivity between sequence variants of the two distinct HCV subtypes 1a and 1b, suggesting that CD8+ T cell responses directed against the NS31406–1415 epitope are typically specific for one subtype only. The lack of cross-reactivity of HCV-specific CD8+ T cells across genotypes and subtypes is well known in HCV immunology (6, 26) and may be the reason why certain HLA class I alleles are associated with protective immunity against HCV only in the setting of individual genotypes or subtypes (7, 8). Minor sequence differences in immunodominant CD8+ T cell epitopes can have a profound influence on the ability to achieve immune control of HCV (9). For design of a prophylactic vaccine, this suggests that antigen coverage of different genotypes and possibly even different subtypes may be required to achieve broad protection.
Despite the lack of cross-reactivity of the in vitro-primed CD8+ T cells directed against NS31406–1415 between HCV subtypes 1a and 1b, there was substantial cross-reactivity between epitope variants within genotype 1b. Importantly, the degree of cross-reactivity strongly depended on the epitope variant that was used for priming of CD8+ T cells. The epitope variant associated with the highest degree of cross-reactivity was gt1b_3 (KLSALGLNAV). This epitope variant was also associated with the highest level of reproducibility of CD8 T cell priming. The ability to reproducibly prime naive CD8+ T cells and the resulting degree of cross-reactivity were independently attributed to different epitope variants and are not explained by different HLA class I-binding affinities, as all peptides tested here were high-affinity binders to HLA-A*02:01. In contrast to the high degree of cross-reactivity of CD8+ T cells primed with the variant gt1b_3, the variant gt1b_AD was also associated with a high level of reproducibility of successful priming; however, despite up to 20% of the CD8+ T cells being reactive to this variant, these cells were not cross-reactive with any other epitope variant. This suggests that the T cell repertoire recruited for targeting of both variants was distinct. TCR Vβ usage was determined by flow cytometry utilizing a panel of Vβ-specific antibodies. Upon priming with gt1b_AD, analysis of IFN-γ-producing T cells showed a different and narrowly focused Vβ usage compared to that of CD8+ T cells primed with the gt1b_3 variant in the same individual. Here, the immune response was comprised of CD8+ T cells utilizing multiple different Vβs. This indicates that, depending on the epitope variant used for priming, different TCR repertoires were targeted. Notably, cross-reactivity of gt1b_3-specific CD8+ T cells was not a direct consequence of broader Vβ usage, as all individual Vβs were cross-reactive with the gt1b_1 variant. This suggests that cross-reactivity was determined at the level of individual Vβs, either by polyclonal expansion of multiple distinct TCRs utilizing the same Vβ or by oligo- or monoclonal expansion of T cells with a broadly cross-reactive TCR. The plasticity of binding of individual TCRs to peptide-MHC complexes can strongly vary (27). There are reports of individual TCRs binding to a multiplicity of different peptide ligands in complex with HLA-A*02:01 (28–30). Such broadly cross-reactive TCRs would likely be beneficial for protection against highly variable pathogens.
The epitope variant gt1b_3 was also heavily targeted by CD8+ T cells in our cohort of PWID from Germany, although these patients are predominantly exposed to HCV genotypes 1a and 3a, and infections with genotype 1b are less common (data not shown). CD8+ T cells expanded in the presence of gt1b_3 were to some extent cross-reactive with both the most frequent genotype 1a variant, gt1a_1, as well as the most frequent genotype 1b variant, gt1b_1, although cross-reactivity with genotype 1a was uncommon in the in vitro priming experiments. In contrast, CD8+ T cells expanded from HCV-exposed patients in the presence of the genotype 1a variant gt1a_1 showed no or only little cross-reactivity. The protocol used for expansion of CD8+ T cells from HCV-exposed patients targets antigen-specific memory T cells, whereas the more complex in vitro priming experiments targeted the naive CD8+ T cell repertoire. Assuming that the memory T cell responses and cross-reactivity profiles of patients are similarly influenced by the epitope variant at the time of transmission, the high level of reproducibility of specific CD8+ T cell responses directed against gt1b_3 was unexpected, as this epitope variant is rare in German HCV genotype 1b isolates. It is possible that the epitope variant was present as a minority in the quasispecies of infected patients or that priming with a distinct variant not tested here may have driven such CD8+ T cell responses and the observed cross-reactivity profiles. As the exact sequence at the time of transmission is unknown, this is difficult to address in patient samples.
Our results indicate that even if an immune response against the most frequent variant, gt1b_1, is the goal, it is still beneficial to use the gt1b_3 variant for priming. For vaccine design, this implies that the optimal antigen sequence for immunization is not necessarily the most frequent epitope sequence in circulating isolates in a population. The concept of using altered peptide ligands for immunization and priming of cross-reactive CD8+ T cells is well described in cancer immunology (27, 31, 32). Such altered peptide ligands, associated with better priming potential and broader cross-reactivity, are potentially also beneficial for immunization against highly variable pathogens. RNA viruses such as HCV and HIV have the ability to rapidly adapt to CD8+ T cell immune pressure, and there is evidence at the population level that circulating isolates have already adapted to selection pressure by frequent HLA class I alleles (33–36). Accordingly, altering the antigen sequence by reconstruction of ancestral states (37) may enhance immunogenicity, as has been demonstrated for HCV (38). Although not formally tested, it is possible that the most frequent genotype 1b variant (gt1b_1) is already the product of immune selection pressure in the presence of HLA-A*02:01. This would be in line with our observation that the variant associated with the best immunogenicity profile in the context of HLA-A*02:01 was more frequent in Japanese and Chinese isolates. Although HLA-A*02:01 is also frequent in China and Japan (allele frequencies of 0.11 to 0.16 according to the Allele Frequency Net Database [http://www.allelefrequencies.net/]), other subtypes, such as A*02:03, A*02:06, and A*02:07, are collectively as frequent (cumulative allele frequency of 0.12 to 0.17). In Caucasian populations, the frequency of HLA-A*02:01 is two times higher (allele frequency of about 0.27), whereas other subtypes are almost absent. It therefore seems possible that the gt1b_3 epitope variant from Asia was simply not yet fully adapted to HLA*02:01-associated selection pressure and was thus superior in the context of this allele. Unfortunately, when we aimed to perform priming experiments utilizing naive CD8+ T cells from HLA-A*02:non-01 donors, we were unable to identify an individual with such an allele. Therefore, this needs to be addressed in sequence and immunogenicity studies in Asian populations.
Taken together, we show that different naturally occurring variants of the same immunodominant CD8+ T cell epitope in HCV show distinct priming capacities and cross-reactivity profiles. The epitope variant with the best immunogenicity profile of the variants tested was rare in circulating HCV genotype 1b isolates from Europe and North America. For vaccine design against highly variable pathogens such as HCV and HIV that share the ability to adapt to CD8+ T cell immune pressure, the results are highly relevant, as they suggest that cross-reactivity profiles of T cells may be influenced by the antigen sequence used in a vaccine. Our data indicate that besides optimizing antigen processing and HLA class I presentation, both of which are required for an efficient CD8 T cell-based vaccine, a biological assay will provide experimental evidence for the choice of more subtle amino acid exchanges within the sequence. By using one particular HCV epitope as a proof of principle, our study suggests that evidence-based selection of the optimal antigen sequence can enhance both immunogenicity of a vaccine and cross-reactivity of CD8 T cells in humans. To this end, rare epitope variants and potentially even altered epitope sequences associated with priming of broadly cross-reactive TCRs should be considered and need further testing.
ACKNOWLEDGMENT
This work was supported by the Deutsche Forschungsgemeinschaft (DFG TRR60).
Footnotes
Published ahead of print 9 July 2014
REFERENCES
- 1.Callaway E. 2014. Hepatitis C drugs not reaching poor. Nature 508:295–296. 10.1038/508295a [DOI] [PubMed] [Google Scholar]
- 2.Grebely J, Dore GJ. 2014. Can hepatitis C virus infection be eradicated in people who inject drugs? Antiviral Res. 104:62–72. 10.1016/j.antiviral.2014.01.002 [DOI] [PubMed] [Google Scholar]
- 3.Bowen DG, Walker CM. 2005. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature 436:946–952. 10.1038/nature04079 [DOI] [PubMed] [Google Scholar]
- 4.Timm J, Roggendorf M. 2007. Sequence diversity of hepatitis C virus: implications for immune control and therapy. World J. Gastroenterol. 13:4808–4817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. 2014. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment Web resource. Hepatology 59:318–327. 10.1002/hep.26744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giugliano S, Oezkan F, Bedrejowski M, Kudla M, Reiser M, Viazov S, Scherbaum N, Roggendorf M, Timm J. 2009. Degree of cross-genotype reactivity of hepatitis C virus-specific CD8+ T cells directed against NS3. Hepatology 50:707–716. 10.1002/hep.23096 [DOI] [PubMed] [Google Scholar]
- 7.Neumann-Haefelin C, Timm J, Schmidt J, Kersting N, Fitzmaurice K, Oniangue-Ndza C, Kemper MN, Humphreys I, McKiernan S, Kelleher D, Lohmann V, Bowness P, Huzly D, Rosen HR, Kim AY, Lauer GM, Allen TM, Barnes E, Roggendorf M, Blum HE, Thimme R. 2010. Protective effect of human leukocyte antigen B27 in hepatitis C virus infection requires the presence of a genotype-specific immunodominant CD8+ T-cell epitope. Hepatology 51:54–62. 10.1002/hep.23275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim AY, Kuntzen T, Timm J, Nolan BE, Baca MA, Reyor LL, Berical AC, Feller AJ, Johnson KL, Wiesch JS, Robbins GK, Chung RT, Walker BD, Carrington M, Allen TM, Lauer GM. 2011. Spontaneous control of HCV is associated with expression of HLA-B*57 and preservation of targeted epitopes. Gastroenterology 140:686–696.e1. 10.1053/j.gastro.2010.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ziegler S, Ruhl M, Tenckhoff H, Wiese M, Heinemann FM, Horn PA, Spengler U, Neumann-Haefelin C, Nattermann J, Timm J. 2013. Susceptibility to chronic hepatitis C virus infection is influenced by sequence differences in immunodominant CD8+ T cell epitopes. J. Hepatol. 58:24–30. 10.1016/j.jhep.2012.08.016 [DOI] [PubMed] [Google Scholar]
- 10.Schmidt J, Neumann-Haefelin C, Altay T, Gostick E, Price DA, Lohmann V, Blum HE, Thimme R. 2011. Immunodominance of HLA-A2-restricted hepatitis C virus-specific CD8+ T cell responses is linked to naive-precursor frequency. J. Virol. 85:5232–5236. 10.1128/JVI.00093-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward S, Lauer G, Isba R, Walker B, Klenerman P. 2002. Cellular immune responses against hepatitis C virus: the evidence base 2002. Clin. Exp. Immunol. 128:195–203. 10.1046/j.1365-2249.2002.01840.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wolfl M, Rutebemberwa A, Mosbruger T, Mao Q, Li HM, Netski D, Ray SC, Pardoll D, Sidney J, Sette A, Allen T, Kuntzen T, Kavanagh DG, Kuball J, Greenberg PD, Cox AL. 2008. Hepatitis C virus immune escape via exploitation of a hole in the T cell repertoire. J. Immunol. 181:6435–6446. 10.4049/jimmunol.181.9.6435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuiken C, Hraber P, Thurmond J, Yusim K. 2008. The hepatitis C sequence database in Los Alamos. Nucleic Acids Res. 36:D512–D516. 10.1093/nar/gkm962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruhl M, Knuschke T, Schewior K, Glavinic L, Neumann-Haefelin C, Chang DI, Klein M, Heinemann FM, Tenckhoff H, Wiese M, Horn PA, Viazov S, Spengler U, Roggendorf M, Scherbaum N, Nattermann J, Hoffmann D, Timm J. 2011. CD8+ T-cell response promotes evolution of hepatitis C virus nonstructural proteins. Gastroenterology 140:2064–2073. 10.1053/j.gastro.2011.02.060 [DOI] [PubMed] [Google Scholar]
- 15.Toebes M, Coccoris M, Bins A, Rodenko B, Gomez R, Nieuwkoop NJ, van de Kasteele W, Rimmelzwaan GF, Haanen JB, Ovaa H, Schumacher TN. 2006. Design and use of conditional MHC class I ligands. Nat. Med. 12:246–251. 10.1038/nm1360 [DOI] [PubMed] [Google Scholar]
- 16.Wolfl M, Merker K, Morbach H, Van Gool SW, Eyrich M, Greenberg PD, Schlegel PG. 2011. Primed tumor-reactive multifunctional CD62L+ human CD8+ T cells for immunotherapy. Cancer Immunol. Immunother. 60:173–186. 10.1007/s00262-010-0928-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolfl M, Greenberg PD. 2014. Antigen-specific activation and cytokine-facilitated expansion of naive, human CD8+ T cells. Nat. Protoc. 9:950–966. 10.1038/nprot.2014.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roederer M, Nozzi JL, Nason MC. 2011. SPICE: exploration and analysis of post-cytometric complex multivariate datasets. Cytometry A 79:167–174. 10.1002/cyto.a.21015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yusim K, Dilan R, Borducchi E, Stanley K, Giorgi E, Fischer W, Theiler J, Marcotrigiano J, Korber B, Barouch DH. 2013. Hepatitis C genotype 1 mosaic vaccines are immunogenic in mice and induce stronger T-cell responses than natural strains. Clin. Vaccine Immunol. 20:302–305. 10.1128/CVI.00605-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer-Olson D, Shoukry NH, Brady KW, Kim H, Olson DP, Hartman K, Shintani AK, Walker CM, Kalams SA. 2004. Limited T cell receptor diversity of HCV-specific T cell responses is associated with CTL escape. J. Exp. Med. 200:307–319. 10.1084/jem.20040638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ladell K, Hashimoto M, Iglesias MC, Wilmann PG, McLaren JE, Gras S, Chikata T, Kuse N, Fastenackels S, Gostick E, Bridgeman JS, Venturi V, Arkoub ZA, Agut H, van Bockel DJ, Almeida JR, Douek DC, Meyer L, Venet A, Takiguchi M, Rossjohn J, Price DA, Appay V. 2013. A molecular basis for the control of preimmune escape variants by HIV-specific CD8+ T cells. Immunity 38:425–436. 10.1016/j.immuni.2012.11.021 [DOI] [PubMed] [Google Scholar]
- 22.Erickson AL, Kimura Y, Igarashi S, Eichelberger J, Houghton M, Sidney J, McKinney D, Sette A, Hughes AL, Walker CM. 2001. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity 15:883–895. 10.1016/S1074-7613(01)00245-X [DOI] [PubMed] [Google Scholar]
- 23.Timm J, Lauer GM, Kavanagh DG, Sheridan I, Kim AY, Lucas M, Pillay T, Ouchi K, Reyor LL, Schulze zur Wiesch J, Gandhi RT, Chung RT, Bhardwaj N, Klenerman P, Walker BD, Allen TM. 2004. CD8 epitope escape and reversion in acute HCV infection. J. Exp. Med. 200:1593–1604. 10.1084/jem.20041006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cox AL, Mosbruger T, Mao Q, Liu Z, Wang XH, Yang HC, Sidney J, Sette A, Pardoll D, Thomas DL, Ray SC. 2005. Cellular immune selection with hepatitis C virus persistence in humans. J. Exp. Med. 201:1741–1752. 10.1084/jem.20050121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tester I, Smyk-Pearson S, Wang P, Wertheimer A, Yao E, Lewinsohn DM, Tavis JE, Rosen HR. 2005. Immune evasion versus recovery after acute hepatitis C virus infection from a shared source. J. Exp. Med. 201:1725–1731. 10.1084/jem.20042284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fytili P, Dalekos GN, Schlaphoff V, Suneetha PV, Sarrazin C, Zauner W, Zachou K, Berg T, Manns MP, Klade CS, Cornberg M, Wedemeyer H. 2008. Cross-genotype-reactivity of the immunodominant HCV CD8 T-cell epitope NS3-1073. Vaccine 26:3818–3826. 10.1016/j.vaccine.2008.05.045 [DOI] [PubMed] [Google Scholar]
- 27.Sewell AK. 2012. Why must T cells be cross-reactive? Nat. Rev. Immunol. 12:669–677. 10.1038/nri3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borbulevych OY, Santhanagopolan SM, Hossain M, Baker BM. 2011. TCRs used in cancer gene therapy cross-react with MART-1/Melan-A tumor antigens via distinct mechanisms. J. Immunol. 187:2453–2463. 10.4049/jimmunol.1101268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bulek AM, Cole DK, Skowera A, Dolton G, Gras S, Madura F, Fuller A, Miles JJ, Gostick E, Price DA, Drijfhout JW, Knight RR, Huang GC, Lissin N, Molloy PE, Wooldridge L, Jakobsen BK, Rossjohn J, Peakman M, Rizkallah PJ, Sewell AK. 2012. Structural basis for the killing of human beta cells by CD8(+) T cells in type 1 diabetes. Nat. Immunol. 13:283–289. 10.1038/ni.2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wooldridge L, Ekeruche-Makinde J, van den Berg HA, Skowera A, Miles JJ, Tan MP, Dolton G, Clement M, Llewellyn-Lacey S, Price DA, Peakman M, Sewell AK. 2012. A single autoimmune T cell receptor recognizes more than a million different peptides. J. Biol. Chem. 287:1168–1177. 10.1074/jbc.M111.289488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watson AM, Mylin LM, Thompson MM, Schell TD. 2012. Modification of a tumor antigen determinant to improve peptide/MHC stability is associated with increased immunogenicity and cross-priming a larger fraction of CD8+ T cells. J. Immunol. 189:5549–5560. 10.4049/jimmunol.1102221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guevara-Patino JA, Engelhorn ME, Turk MJ, Liu C, Duan F, Rizzuto G, Cohen AD, Merghoub T, Wolchok JD, Houghton AN. 2006. Optimization of a self antigen for presentation of multiple epitopes in cancer immunity. J. Clin. Invest. 116:1382–1390. 10.1172/JCI25591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Timm J, Li B, Daniels MG, Bhattacharya T, Reyor LL, Allgaier R, Kuntzen T, Fischer W, Nolan BE, Duncan J, Schulze zur Wiesch J, Kim AY, Frahm N, Brander C, Chung RT, Lauer GM, Korber BT, Allen TM. 2007. Human leukocyte antigen-associated sequence polymorphisms in hepatitis C virus reveal reproducible immune responses and constraints on viral evolution. Hepatology 46:339–349. 10.1002/hep.21702 [DOI] [PubMed] [Google Scholar]
- 34.Neumann-Haefelin C, Frick DN, Wang JJ, Pybus OG, Salloum S, Narula GS, Eckart A, Biezynski A, Eiermann T, Klenerman P, Viazov S, Roggendorf M, Thimme R, Reiser M, Timm J. 2008. Analysis of the evolutionary forces in an immunodominant CD8 epitope in hepatitis C virus at a population level. J. Virol. 82:3438–3451. 10.1128/JVI.01700-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leslie A, Kavanagh D, Honeyborne I, Pfafferott K, Edwards C, Pillay T, Hilton L, Thobakgale C, Ramduth D, Draenert R, Le Gall S, Luzzi G, Edwards A, Brander C, Sewell AK, Moore S, Mullins J, Moore C, Mallal S, Bhardwaj N, Yusim K, Phillips R, Klenerman P, Korber B, Kiepiela P, Walker B, Goulder P. 2005. Transmission and accumulation of CTL escape variants drive negative associations between HIV polymorphisms and HLA. J. Exp. Med. 201:891–902. 10.1084/jem.20041455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, Gatanaga H, Fujiwara M, Hachiya A, Koizumi H, Kuse N, Oka S, Duda A, Prendergast A, Crawford H, Leslie A, Brumme Z, Brumme C, Allen T, Brander C, Kaslow R, Tang J, Hunter E, Allen S, Mulenga J, Branch S, Roach T, John M, Mallal S, Ogwu A, Shapiro R, Prado JG, Fidler S, Weber J, Pybus OG, Klenerman P, Ndung'u T, Phillips R, Heckerman D, Harrigan PR, Walker BD, Takiguchi M, Goulder P. 2009. Adaptation of HIV-1 to human leukocyte antigen class I. Nature 458:641–645. 10.1038/nature07746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munshaw S, Bailey JR, Liu L, Osburn WO, Burke KP, Cox AL, Ray SC. 2012. Computational reconstruction of Bole1a, a representative synthetic hepatitis C virus subtype 1a genome. J. Virol. 86:5915–5921. 10.1128/JVI.05959-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burke KP, Munshaw S, Osburn WO, Levine J, Liu L, Sidney J, Sette A, Ray SC, Cox AL. 2012. Immunogenicity and cross-reactivity of a representative ancestral sequence in hepatitis C virus infection. J. Immunol. 188:5177–5188. 10.4049/jimmunol.1103008 [DOI] [PMC free article] [PubMed] [Google Scholar]