

Abstract
Hepatitis C virus (HCV), a member of the family Flaviviridae, is a leading cause of chronic liver disease and cancer. Recent advances in HCV therapeutics have resulted in improved cure rates, but an HCV vaccine is not available and is urgently needed to control the global pandemic. Vaccine development has been hampered by the lack of high-resolution structural information for the two HCV envelope glycoproteins, E1 and E2. Recently, Kong and coworkers (Science 342:1090–1094, 2013, doi:10.1126/science.1243876) and Khan and coworkers (Nature 509[7500]:381–384, 2014, doi:10.1038/nature13117) independently determined the structure of the HCV E2 ectodomain core with some unexpected and informative results. The HCV E2 ectodomain core features a globular architecture with antiparallel β-sheets forming a central β sandwich. The residues comprising the epitopes of several neutralizing and nonneutralizing human monoclonal antibodies were also determined, which is an essential step toward obtaining a fine map of the human humoral response to HCV. Also clarified were the regions of E2 that directly bind CD81, an important HCV cellular receptor. While it has been widely assumed that HCV E2 is a class II viral fusion protein (VFP), the newly determined structure suggests that the HCV E2 ectodomain shares structural and functional similarities only with domain III of class II VFPs. The new structural determinations suggest that the HCV glycoproteins use a different mechanism than that used by class II fusion proteins for cell fusion.
INTRODUCTION
Hepatitis C virus (HCV), a member of the Hepacivirus genus of the family Flaviviridae, is a leading cause of chronic hepatitis, cirrhosis, liver failure, and hepatocellular carcinoma (1). The worldwide incidence of people who are chronically infected with HCV numbers over 150 million, presenting a significant public health and economic burden (2). At least three million people in the United States have a chronic HCV infection, placing them at risk for developing serious liver disease or cancer (3). HCV is the underlying cause of about 25% of liver cancers worldwide and is the leading indicator for liver transplants in the United States (4). At least for the moment, HCV has surpassed HIV as a cause of death in the United States (5). While ongoing treatment advances, including telaprevir-, boceprevir-, and sofosbuvir-based therapies, have greatly improved the cure rates of hepatitis C in developed countries (6) and further improvements in hepatitis C treatments are imminent (7), access to HCV therapies depends on the availability of an effective health care infrastructure that is not available to many populations at high risk for HCV infection.
An HCV vaccine is urgently needed to control the worldwide pandemic (8). There are many challenges to HCV vaccine development, foremost of which is the extreme variability of the virus (9). HCV has been recently regrouped into seven major genotypes and 67 subtypes (10). In patients, HCV exists as a complex mixture of genetically distinct, but closely related, variants or quasispecies (11). This variability facilitates chronic HCV infection by enabling evasion of the host immune response (12, 13). Infection with one HCV genotype does not preclude a second infection with another genotype, which is a strong indication that simple vaccine approaches will not succeed. Vaccine development was hampered for many years by the lack of efficient cell culture systems for replicating HCV, a deficiency that has now largely been overcome with the advent of HCV pseudoparticle (HCVpp), HCV cell culture (HCVcc), and other replication systems (14–23). While chimpanzees can support HCV infection (24, 25), development of more accessible animal models for hepatitis C has also been a significant bottleneck for vaccine development. The recent development of humanized mouse models that do not have intact immune systems and support HCV replication is a promising advance (26–28).
Viral surface glycoproteins are the major targets of vaccination strategies for viruses such as HCV that have a lipid-containing viral envelope. The surface glycoproteins mediate host cell receptor binding and membrane fusion, allowing entry of the viral genome into host cells (29, 30). Production of antibodies that block one or more of these processes is essential for a protective humoral immune response. The two HCV envelope glycoproteins, E1 (amino acids [aa] 192 to 383 of the strain H77 polyprotein) and E2 (aa 384 to 746), have C-terminal transmembrane (TM) anchor domains and form a noncovalent heterodimer complex anchored in the virion lipid envelope. It is well established that HCV E2 mediates binding to at least some of the HCV cell receptors (30–32). However, the roles of E1 and E2 in mediating virus membrane-cell membrane fusion remain unclear due in part to the lack of high-resolution X-ray crystallographic structures of E1 and E2. Recently, Kong and coworkers (33) and Khan and coworkers (34) made a major advance in the field by independently solving the structures of constructs consisting of the core residues of the E2 ectodomain. Here, we discuss how the newly determined HCV E2 ectodomain structure allows for reinterpretation of some prior studies and how this key breakthrough will guide future studies.
STRUCTURE OF HCV E2
Expression of the HCV envelope proteins for structural analyses has proven challenging. Both E1 and E2 are heavily glycosylated and have multiple disulfide cross-links. E1 and E2 associate as a heterodimer and require an extended period of time for completion of the folding process in the endoplasmic reticulum, further complicating expression and purification (35). When expressed in eukaryotic cells, a significant proportion of E1 or E2 forms misfolded aggregates with incorrect disulfide cross-links (36, 37). Kong and coworkers (33) used brute force to overcome this challenge. Focusing on E2, they created and expressed 41 different recombinant E2 proteins, deleting various glycosylation sites, loops, or other predicted structures in E2. This effort yielded seven “well-behaved” E2 constructs. Khan and coworkers (34) took a similar approach, but rather than mutagenizing glycosylation sites, they enzymatically removed most of the glycosyl residues from the purified protein. Drawing from the resourceful crystallographer's bag of tricks, both groups “stabilized” their constructs for crystal formation with antigen binding fragments (Fabs) of E2-specific human monoclonal antibodies (huMAbs). Fabs, particularly those that bind to conformational (nonlinear) epitopes, are unlikely to bind misfolded proteins without proper disulfide bonds. Kong and coworkers solved the structure of the E2 ectodomain core (E2c-Kong) in the presence of a Fab from a huMAb designated antigenic region 3C (AR3C). Khan et al. (34) produced crystals of their deglycosylated E2 core (E2c-Khan) in complex with Fab 2A12.
Flexible regions and glycans may deter useful crystal formation. E2c-Kong consists of the “core” of the E2 ectodomain of HCV strain H77 (genotype 1a), spanning aa 412 to 645, with a deletion of the 28 amino acids at the N terminus known as hypervariable region 1 (HVR1). E2c-Kong also carries a substitution with a short linker (glycine-serine-serine-glycine) for HVR2 (aa 460 to 485), a potentially flexible region, and decommissioned glycosylation sites at N448 and N576 and produced crystals diffracting to 2.65 Å (Fig. 1A). Khan and coworkers produced crystals diffracting to a 2.4-Å resolution from a shorter fragment of HCV E2 of a different strain and genotype (HCV strain J6, genotype 2a), which had a deletion of approximately 80 amino acids from the N terminus (aa 456 to 656) (Fig. 1B). Both groups made the same 98-aa truncation of E2c at the C terminus, thus removing the aromatic juxtamembrane domain (called the stem domain in flaviviruses) and the transmembrane anchor domain. Protein crystallization is very difficult in the presence of such hydrophobic, membrane-associated sequences. They have been removed in essentially all viral membrane envelope protein structural determinations, starting with the influenza virus hemagglutinin ectodomain (38), the first viral fusion protein (VFP) structure that was solved.
FIG 1.

Structure of the ectodomain core of HCV envelope protein 2. (A) Ribbon diagram of the construct used to determine the structure of the HCV E2 ectodomain core by Kong et al. (33). β-Sheets are represented by arrows, and helices are represented by cylinders. Glycosylation sites are numbered from the N terminus. Dicysteine bonds are indicated by finely dashed lines. Due to constraints of representing a three-dimensional structure in two dimensions, the length of loops may not correspond to the length in the loops in the HCV E2 ectodomain core structure. Numbering of sheets, helices, and glycosylation sites is from Kong et al. (33). Blue structures represent the central β sandwich, a structural motif present in class II viral fusion proteins. The red loop is the putative CD81 binding domain. Gray structures represent amino acids not present in the construct. Gold structures were present in the construct, but disordered. Glycosylation sites 4 and 9 were mutated. Hypervariable region 2 (HVR2) is deleted from the construct but had an inserted linker (GSSG), which was disordered. Amino acids delineating structural and other features are numbered from the start of the polyprotein. (B) Ribbon diagram of the construct used to determine the structure of the HCV E2 ectodomain core by Khan et al. (34). Symbols and the color scheme are as in panel A, except that glycosylation was mostly removed enzymatically. (C) X-ray crystallographic structure of the HCV E2 ectodomain core by Kong et al. (33). Symbols and the color scheme are as in panel A, except that cysteines are represented as yellow sticks. The dashed circle indicates the fusion loop postulated by Krey et al. (36). (D) X-ray crystallographic structure of the HCV E2 ectodomain core by Khan et al. (34). Symbols and the color scheme are as in panel C.
The core domains of the two structures are highly similar, with a root mean square deviation of 0.8 Å for similar carbon α atoms (34). The E2c architecture features a central sandwich of antiparallel β-sheets, which is analogous to folds found in members of the immunoglobulin protein superfamily (Fig. 1, blue residues) as well as domains of other virus envelope proteins (39–43). The central immunoglobulin (Ig)-like β sandwich scaffold is stabilized by two disulfide cross-links (Fig. 1A and B, dashed lines; Fig. 1 C and D, yellow residues). Front and back layers flank the central β-sheet sandwich in both E2c-Kong and E2c-Khan. High-resolution structures could not be determined for some parts of E2c, namely, aa 412 to 420 (hypervariable region 1 [HVR1]), aa 454 to 491 (HVR2), and aa 586 to 596 (HVR3) in E2c-Kong and aa 456 to 491 (HVR2), aa 523 to 538 (implicated as part of the CD81 binding loop [see below]), and aa 572 to 595 (HVR3) in E2c-Khan (Fig. 1A and B). These disordered, poorly diffracting regions are located in or near HVRs and are presumed to be flexible loops. The cysteine bonds in E2c-Kong and E2c-Khan are the same except for the four cysteines near HVR3. In E2c-Kong, two dicysteine linkages are apparent, but only a single different dicysteine linkage is resolved in the E2c-Khan structure. We speculate that the alternative cysteine bonding could account for why HRV3 is not resolved in E2c-Khan but is resolved in E2c-Kong. Not surprisingly, some of the glycans are also disordered in E2c-Kong and are thus either not visible or not completely resolved in the X-ray structure. However, ordered glycans visible in the E2c-Kong crystal structure and other glycosylation sites in both constructs are clustered on surfaces that are likely to be exposed on the virion (Fig. 1C, purple residues).
Despite the lack of some of the loops and glycans, it is apparent that the structures solved by Kong et al. (33) and Khan et al. (34) represent the essential scaffold of the E2 ectodomain. Kong and coworkers (33) performed cryoelectron microscopy using a construct of E2 with a deletion of only the TM domain (E2ΔTM) in complex with Fab AR3C. A 16-Å resolution density map was obtained. The sequences that are deleted in E2c-Kong but retained in E2ΔTM as well as the disordered sequences fit comfortably into this density map, leading to the conclusion that the overall structure of the E2 ectodomain is globular. Likewise, Khan et al. (34) used solution-based small-angle X-ray scattering (SAXS) to correlate the dimensions of their E2 ectodomain core structure with the dimensions of fully glycosylated E2. The ab initio SAXS envelopes of E2c-Khan and fully glycosylated E2 were similar, with approximately the same radius. The missing glycosylations of the E2c-Khan crystal structure, roughly one-third of the mass, fully accounted for the difference in the SAXS envelopes.
NEUTRALIZATION AND ANTIBODY BINDING
Humoral immunity is likely to be important in vaccine-induced protection from HCV infection (44). Monoclonal antibodies (MAbs) have proven to be invaluable tools in dissecting the role of immune responses to viral pathogens. In particular, human MAbs (huMAbs) can directly indicate which viral epitopes are recognized by the human humoral immune system (45). Kong et al. (33) directly mapped the binding site of huMAb AR3C on E2c in their crystallization studies. One of the mechanisms that HCV employs to evade the immune response is the presence of multiple glycans on its envelope proteins (46–48). E2 is heavily glycosylated, and the newly determined E2 ectodomain structure provides a clear demonstration of how glycans shield large portions of the molecule from antibodies. Although not all of the glycans are resolved in E2c-Kong, modeling of the multiple sugars onto the structure shows that there is only a small area for the binding of a set of neutralizing huMAbs, on a region of E2 that overlaps the CD81 binding site (33). The binding site of AR2A, a nonneutralizing Fab, was mapped to the back “face” of E2c using negative-stain electron microscopy. Khan et al. (34) directly mapped the binding site of another nonneutralizing antibody, 2A12, to the back face of E2c. Determination of the E2c structure will allow for quick mapping of antibodies for which data for binding to peptides or site-directed mutagenesis is available. The E2 ectodomain structure thus enables an essential first step toward obtaining a fine map of the human humoral response to HCV. Further studies are required, since neutralizing antibodies to HCV E1 are also produced, as are epitopes that span E1 and E2 in the virion heterodimer (49). The latter group of antibodies, which recognize quaternary epitopes, are potentially important for broad protection from HCV (50, 51). For example, huMAb AR4A recognizes a discontinuous epitope outside the CD81 binding site on the E1-E2 complex (45). AR4A is exceptional in that it neutralizes HCV from diverse genotypes and protects against heterologous HCV challenge in a small animal model (45, 52).
BINDING OF E2 TO CELLULAR RECEPTORS
E2c-Kong retains more of the N terminus of E2 than E2c-Khan, including the CD81 binding domain. The study by Kong et al. (33) provides the first visualization of how HCV binds one of its major cellular receptors. CD81 is a member of the tetraspanin superfamily and is necessary for infection of primary human hepatocytes or hepatoma cell lines by HCV (53–56). CD81 has short intracellular N and C termini, four transmembrane domains, a small extracellular loop (SEL), and a large extracellular loop (LEL). CD81-specific MAbs or recombinant CD81 protein blocks infection by HCVpp bearing HCV E1 and E2 (57). CD81-negative cells support HCVpp infection when transduced to express CD81 (58). HCV infection is also inhibited when CD81 expression is silenced by the use of small interfering RNAs (59). Several putative CD81 binding regions of HCV E2 have been identified through mutagenesis studies (60–64). The first proposed region spans the second hypervariable domain (HVR2), extending from aa 474 to 492. The second potential CD81 binding region of E2 spans aa 522 to 551, and the third region is from aa 612 to 619. The E2c-Kong crystal structure provides clarity regarding which of these regions directly bind CD81. Most of the region comprising aa 474 to 492 is deleted in E2c-Kong, and the part that is not deleted is adjacent to, but not part of, the CD81 binding region. The observation that aa 474 to 492 can be partly deleted from E2c and still bind CD81 confirms a prior study from S.L.U.'s laboratory indicating that this region was not directly involved in CD81 binding (65). Likewise, aa 612 to 619 are on a different face from the CD81 binding domain. The aa 612 to 619 form a central α-helix, which may be critical for the overall E2 architecture (Fig. 1, α2). Kong et al. (33) visualized the complex of CD81 dimer binding to E2c by negative-stain electron microscopy. With the newly available E2c-Kong structure, contact points between E2c and CD81 can be narrowed to the region of E2c encompassing aa 522 to 551. Consistent with these observations, E2c-Khan has a deletion of the CD81 binding region, and this construct did not bind CD81 (34). CD81 is not thought to be responsible for initial virion host cell binding. Further studies of CD81 binding to E2 are needed, as it remains possible that CD81 binds to other distinct and separate E2 regions, including sites induced by prior binding to other receptors. Besides CD81, scavenger receptor class B member I is also known to interact directly with HCV E2 (66, 67). Tight junction proteins claudin-1 (68, 69) and occludin (70) are required for HCV entry, but it is not established that they bind directly to HCV. Evidence has also been presented for several additional cell surface receptors, coreceptors, or entry factors (65, 68, 69, 71–77). The E2 ectodomain structure can guide studies to identify binding sites for HCV's other receptors and coreceptors or elucidate other ways that cell surface factors can mediate HCV attachment. It is also important to consider the possibility that HCV E2 structural rearrangements may occur during the entry process following interactions with other receptors.
IMPLICATIONS FOR HCV-INDUCED FUSION AND ENTRY: DOES A LOOP IN E1 INITIATE MEMBRANE FUSION?
Knowledge of the structure of the E2 ectodomain may also help to resolve a long-standing controversy regarding which of the two HCV envelope proteins initiates fusion. Following binding to its receptors and coreceptors, HCV, like other enveloped viruses, must fuse its membrane with the host cell membrane to permit internalization of its genome (78). HCV enters cells through an endocytic pathway, and this step is followed by fusion with the vesicular membrane (14, 79). There are three known classes of viral fusion proteins (VFPs) (80–83). Class II VFP includes the envelope glycoproteins (E) of the flaviviruses (39, 84), envelope protein 1 (E1) of the alphaviruses (85), and C-terminal glycoprotein (Gc) of the bunyaviruses (86, 87). The ectodomains of class II VFP are comprised of three domains that are predominantly β-sheets, the structurally central amino terminal domain I, the fusion/dimerization domain II, and the carboxyl-terminal domain III. Domain II contains the fusion loop, which makes the first direct contact with lipids of the cellular membrane (88). Domain III of the members of the flavivirus genus of the Flaviviridae, which contains the Ig-like fold, binds the cellular receptors. In contrast, receptor binding by alphaviruses or bunyaviruses is the function of the VFP companion envelope glycoprotein, E2 or Gn, respectively.
Krey and coworkers (36) modeled HCV E2 as a class II VFP, as previously advanced by Yagnik and coworkers (89). Their structural model was guided by their experimentally determined disulfide cross-link pattern for E2. They concluded that HCV E2 functions as both the receptor binding protein and fusion protein, analogous to E of the flaviviruses. While this model has proven useful for numerous studies (47, 90–92), the newly determined E2 ectodomain structure shows the model to be incorrect. The disulfide bonding pattern determined by Krey and coworkers (36) differs from the disulfide bonds in the recently determined E2c crystal structures. Furthermore, HCV E2 does not assume the overall extended configuration of a class II VFP. Rather, the overall structure of the E2 ectodomain is globular, with similarities only to the Ig-like fold of domain III of class II VFPs. Krey et al. (36) identified E2 aa 502 to 520 as the putative E2 fusion loop. This sequence is part of the Ig-like sandwich, which is constrained in E2c by disulfide bonding and may be further buried by HVR1 residues not present in either E2c-Kong or E2c-Khan (Fig. 1C and D, dashed circles). Based on this information, it can be concluded that the location of aa 502 to 520 in E2c is inconsistent with a role as the fusion loop, the initial contact of the virion with the cellular membrane.
Previously, Flint and coworkers (93) identified a conserved region in HCV E1, at aa 264 to 290, as a potential HCV fusion loop. We (S.D. and R.F.G.) also advanced the possibility that HCV E1 might resemble domain II of class II VFP, on the basis of computational modeling and homology assessments (94). The sequence from aa 264 to 290 has a high propensity to interact with lipid membranes, as determined by the Wimley-White interfacial hydrophobicity scale (WWIHS) (95) codeveloped previously by one of us (W.C.W.). All viral fusion peptides or fusion loops identified to date have positive WWIHS scores. Several investigators have confirmed the importance of E1 aa 264 to 290 in viral infectivity by mutagenesis studies (96–99). Furthermore, peptides corresponding to this putative HCV E1 fusion loop were among the most active peptides derived from a library of overlapping peptides representing E1 and E2 at inducing fusion and disruption of large unilamellar vesicles (also known as liposomes), a property that also supports a role for this sequence as the fusion loop of HCV (78, 100, 101). Synthetic peptides corresponding to the fusion peptides of several enveloped viruses also have been shown to block infectivity (reference 102 and citations therein). Cheng and coworkers (103) reported that peptides (aa 267 to 284 and aa 274 to 291) from the putative E1 fusion peptide were among the most active of those from a large peptide library representing the entire HCV polyprotein in interfering with HCVcc infectivity. Other features of E1 are also consistent with its role as the HCV VFP. E1 contains a number of conserved histidine residues. As is the case for other enveloped viruses (104), protonation of the conserved E1 His residues of HCV has been proposed as a possible mechanism underlying low-pH-dependent refolding of the E1-E2 heterodimer during membrane fusion (105). Proof that E1 represents the HCV fusion protein awaits the determination of its ectodomain structure and the demonstration that its putative fusion loop is the initial point of interaction with cellular membranes. However, it is possible that neither HCV E1 nor E2 contains a canonical fusion loop and that cell fusion is initiated by a process that does not resemble that of other class II VFPs.
OTHER REGIONS OF HCV E2 AND INTERACTIONS WITH E1
Previous studies provide insight into the structures of domains that are deleted in the E2c-Kong and E2c-Khan constructs. Zhang et al. (106) determined the structure of the membrane protein domains of mature dengue virus (DENV) particles to a resolution of 9.5 Å by cryoelectron microscopy. The stem region of flavivirus E (defined as the domain between the ectodomain and the TM anchor) is comprised of two α-helical domains, the first of which is a “leucine zipper” or “heptad repeat,” a leucine or other hydrophobic amino acid in the first and fourth (a and d) positions of a 7-aa periodicity (106–108). Both α-helices in the stem of flavivirus E have positive WWIHS scores and are partly embedded in the lipid bilayer of the virion (106). The stem region of E2 (aa 675 to 703) also appears to be comprised of two α-helices, the most prominent of which is a leucine zipper that is an important determinant of E1-E2 heterodimerization and viral entry (92, 109). The HCV E1 stem also appears to have an α-helical leucine zipper and a second shorter helix, as shown by the nuclear magnetic resonance structural analysis of a synthetic peptide corresponding to this sequence (96). A synthetic peptide with the sequence of the HCV E1 stem strongly partitions into phospholipid membranes, interacts with negatively charged phospholipids, and locates to a shallow position in the membrane (110). Drummer and coworkers (96) demonstrated that mutagenesis of amino acids on the hydrophobic face of the HCV E1 stem had no effect on heterodimerization between E1 and E2 but did result in site-specific defects in viral entry.
The transmembrane anchor region of the class II E protein of dengue virus is much longer than the 19 to 25 residues of a canonical transmembrane helix (111) and likely forms a helical hairpin in the viral envelope, as shown in Fig. 2A. The associated M protein likely also spans the membrane twice. HCV E2, like the dengue virus and tick-borne encephalitis virus (TBEV) E proteins, contains a long (42-aa) segment of mostly hydrophobic aa (aa 701 to 743) that potentially could span the membrane twice. However, the presence of several aromatic (interfacial) amino acids and the charged amino acids in the E2 C terminus in some HCV strains suggest a more shallow membrane interaction for the aa 701 to 714 segment, which is followed by a single transmembrane helix (aa 718 to 742). Similarly, HCV E1 contains a single, C-terminal membrane-spanning helix. Like the first transmembrane domain of TBEV E (TM1), the hydrophobic transmembrane anchors of both HCV E1 (aa 353 to 381) and E2 contain unusual membrane-embedded charged residues (K370 in E1, D728 and R730 in E2) that are critical for assembly of the fusion protein complex (112). These residues (Fig. 2B, blue asterisks) likely mediate lateral interactions between the helices (113). In addition, scanning mutagenesis studies have shown that sequence-specific heteromeric interactions along the lengths of the E1 and E2 transmembrane helices are critical for assembly of the fusion protein complex of HCV and for infectivity (112, 114).
FIG 2.
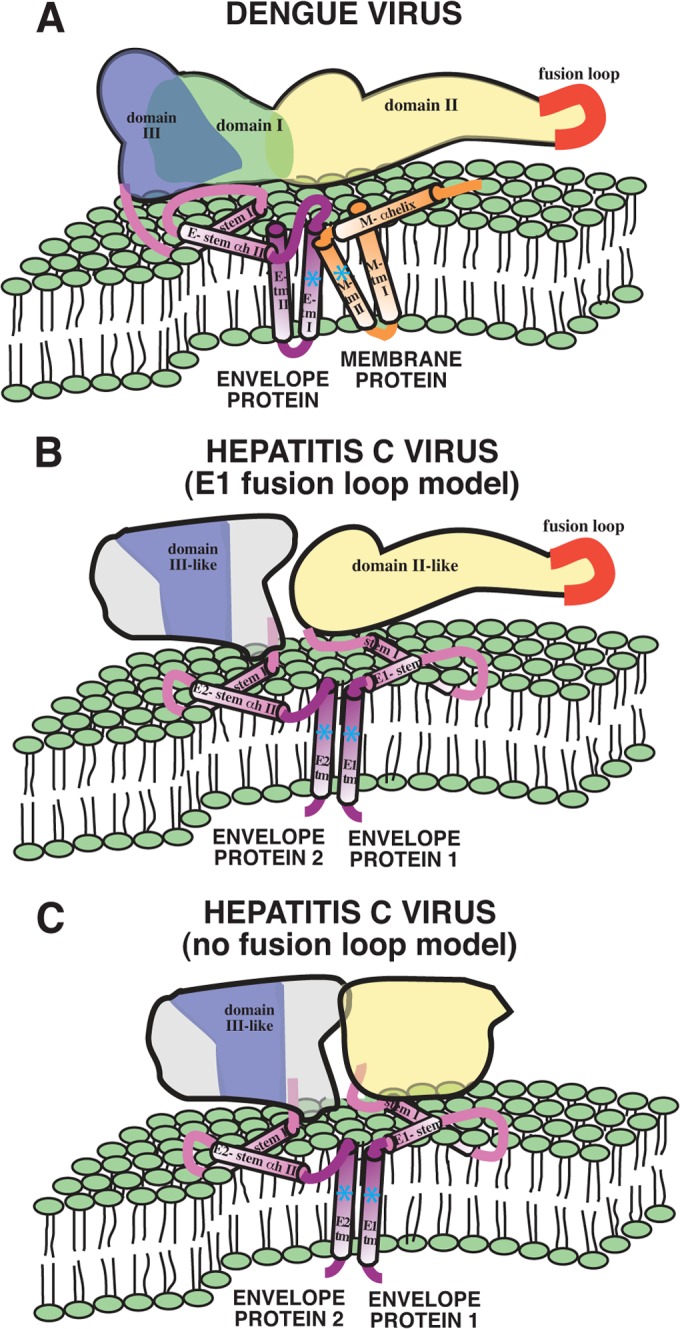
Comparison of the architecture of the dengue virus E/M fusion protein complex with the proposed architecture of the HCV E1/E2 complex. (A) The domain architecture of DENV E is indicated in green (domain I), yellow (domain II), and blue (domain III). The stem and anchor helices of E are violet and blue, respectively. The M protein is depicted in orange. Panel A is reprinted from Nature Structural Biology (106) with permission of the publisher. (B) Model in which the E2 ectodomain structure is combined with a schematic diagram of the ectodomain, stem, and transmembrane domain proteins, based on the presentation by Zhang et al. (106). Also depicted is a speculative model in which HCV E1 contains a fusion loop. Proposed analogous regions of E1 and E2 are colored according to their similarity to corresponding regions of DENV E. This model indicates that it is unlikely that either E1 or E2 folds back on itself to yield the canonical class II postfusion complex. (C) Alternative model for the HCV envelope glycoproteins in which neither E1 nor E2 contains a classical fusion loop.
Given the similarities of the stem and TM domains of HCV E2 and DENV E, it is reasonable to graft the E2 ectodomain structure onto the schematic diagram of the DENV ectodomain, stem, and transmembrane domain proteins presented by Zhang et al. (106) (Fig. 2A). The mechanism of virion-cell membrane fusion mediated by class II VFPs encoded by members of the Flavivirus genus of the Flaviviridae follows a pathway that involves large-scale domain rearrangement of E and a transition from dimers to trimers (42, 115). The new structural determinations suggest that the HCV glycoproteins do not behave like class II fusion proteins and most likely use a different mechanism. By virtue of the lack of a fusion loop in the E2c structure, it is possible that HCV E1 represents the VFP and contains the fusion loop (Fig. 2B). In contrast to low-pH activation of a hinge region yielding a canonical class II postfusion complex, low pH may simply disrupt the E1-E2 interaction in this model, freeing the fusion loop of E1 for insertion into a target membrane. Following insertion of the E1 fusion loop, surfaces of E1 and E2 with high interfacial hydrophobicity may mediate fusion of the virion and host membranes without rearranging into a hairpin configuration. Certainly, this model is highly speculative and requires extensive testing. Alternative fusion models for HCV should also be considered and tested, including those in which neither E1 nor E2 contains a classical fusion loop (Fig. 2C).
The arrangement of the E1-E2 heterodimers on the surface of the HCV virion is unknown, and it is unclear if E1 and E2 form a dimer of heteromers as in other flaviviruses, another pattern, or no specific arrangement at all. A cryoelectron microscopy study of HCV virus-like particles (VLPs) found a herringbone pattern on the VLP surface reminiscent of the arrangement of E dimers on flavivirus virions (116). No obvious patterns were observed in a recent cryoelectron microscopy study of authentic HCV virions (117), suggesting that this arrangement may apply only to VLPs. If the HCV glycoproteins are as dense and patterned as they are on other flaviviruses, several domains, such as the putative fusion loop in E1, would likely not be available for binding membrane components until some glycoprotein conformational change allowed for such access. An additional complication is that HCV virions in patient serum and produced in the HCV cell culture (HCVcc) system exist as complexes with low-density lipoprotein, triglycerides, and β-lipoproteins that appear to be essential for viral entry and fusion (118).
CONCLUSIONS
The determination of the structure of the E2 ectodomain core by Kong and coworkers (33) and Khan and coworkers (34) is a significant advance in HCV research. This structure will guide future work toward an urgently needed HCV vaccine. For example, knowledge of the deletions that result in a stable and crystallizable E2 ectodomain can inform the engineering of constructs that could be used in vaccines to elicit specific, targeted immunological responses (119), such as vaccine constructs that induce optimally binding antibodies. Furthermore, the stable E2 construct could be helpful in solving the structure of the HCV E1-E2 heterodimer, as was done successfully with the E1-E2 heterodimer of alphaviruses (41, 42). Finally, as we have described here, the crystal structure of E2 domain core can inform vaccine design principles even in the absence of a high-resolution E1 structure.
ACKNOWLEDGMENTS
This research was supported by grants DK070551, UC1AI067188, R41AI068230, R21AI097809, R21AI092073, and R56 AI64617 from the National Institutes of Health and RC-0013-07 from the Louisiana Board of Regents.
We thank Jane A. McKeating, Bruno Sainz, Jr., William R. Gallaher, Scott F. Michael, Joshua M. Costin, Yancey M. Hrobowski, Thomas G. Voss, and Russell B. Wilson for informative ongoing discussions on viral VFP. We also thank Richard Kuhn for permission to reproduce previous data from his laboratory as part of Fig. 2 and for helpful and collegial feedback.
All authors assisted with writing this minireview.
Footnotes
Published ahead of print 2 July 2014
REFERENCES
- 1.Shlomai A, de Jong YP, Rice CM. 2014. Virus associated malignancies: the role of viral hepatitis in hepatocellular carcinoma. Semin. Cancer Biol. 26C:78–88. 10.1016/j.semcancer.2014.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dibonaventura MD, Yuan Y, Lescrauwaet B, L'Italien G, Liu GG, Kamae I, Mauskopf JA. 2014. Multicountry burden of chronic hepatitis C viral infection among those aware of their diagnosis: a patient survey. PLoS One 9:e86070. 10.1371/journal.pone.0086070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahajan R, Liu SJ, Klevens RM, Holmberg SD. 2013. Indications for testing among reported cases of HCV infection from enhanced hepatitis surveillance sites in the United States, 2004–2010. Am. J. Public Health 103:1445–1449. 10.2105/AJPH.2013.301211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong RJ, Cheung R, Ahmed A. 2014. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 59:2188–2195. 10.1002/hep.26986 [DOI] [PubMed] [Google Scholar]
- 5.Ly KN, Xing J, Klevens RM, Jiles RB, Ward JW, Holmberg SD. 2012. The increasing burden of mortality from viral hepatitis in the United States between 1999 and 2007. Ann. Intern. Med. 156:271–278. 10.7326/0003-4819-156-4-201202210-00004 [DOI] [PubMed] [Google Scholar]
- 6.Lange CM, Jacobson IM, Rice CM, Zeuzem S. 2014. Emerging therapies for the treatment of hepatitis C. EMBO Mol. Med. 6:4–15. 10.1002/emmm.201303131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hussain S, Barretto N, Uprichard SL. 2012. New hepatitis C virus drug discovery strategies and model systems. Expert Opin. Drug Discov. 7:849–859. 10.1517/17460441.2012.711312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mehta B, Kumar V, Chawla S, Jindal H, Bhatt B. 2014. Hepatitis C: is a vaccine the solution? Hum. Vaccin. Immunother. 10:417–419. 10.4161/hv.26970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindenbach BD, Rice CM. 2003. Evasive maneuvers by hepatitis C virus. Hepatology 38:769–771. 10.1002/hep.510380327 [DOI] [PubMed] [Google Scholar]
- 10.Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, Simmonds P. 2014. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 59:318–327. 10.1002/hep.26744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14:25–27. 10.1038/nm1698 [DOI] [PubMed] [Google Scholar]
- 12.von Hahn T, Yoon JC, Alter H, Rice CM, Rehermann B, Balfe P, McKeating JA. 2007. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology 132:667–678. 10.1053/j.gastro.2006.12.008 [DOI] [PubMed] [Google Scholar]
- 13.Dustin LB, Rice CM. 2007. Flying under the radar: the immunobiology of hepatitis C. Annu. Rev. Immunol. 25:71–99. 10.1146/annurev.immunol.25.022106.141602 [DOI] [PubMed] [Google Scholar]
- 14.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U. S. A. 100:7271–7276. 10.1073/pnas.0832180100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flint M, Logvinoff C, Rice CM, McKeating JA. 2004. Characterization of infectious retroviral pseudotype particles bearing hepatitis C virus glycoproteins. J. Virol. 78:6875–6882. 10.1128/JVI.78.13.6875-6882.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. 10.1126/science.1114016 [DOI] [PubMed] [Google Scholar]
- 17.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 102:9294–9299. 10.1073/pnas.0503596102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindenbach BD, Meuleman P, Ploss A, Vanwolleghem T, Syder AJ, McKeating JA, Lanford RE, Feinstone SM, Major ME, Leroux-Roels G, Rice CM. 2006. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. U. S. A. 103:3805–3809. 10.1073/pnas.0511218103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ploss A, Khetani SR, Jones CT, Syder AJ, Trehan K, Gaysinskaya VA, Mu K, Ritola K, Rice CM, Bhatia SN. 2010. Persistent hepatitis C virus infection in microscale primary human hepatocyte cultures. Proc. Natl. Acad. Sci. U. S. A. 107:3141–3145. 10.1073/pnas.0915130107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sainz B, Jr, TenCate V, Uprichard SL. 2009. Three-dimensional Huh7 cell culture system for the study of Hepatitis C virus infection. Virol. J. 6:103. 10.1186/1743-422X-6-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sabahi A, Marsh KA, Dahari H, Corcoran P, Lamora JM, Yu X, Garry RF, Uprichard SL. 2010. The rate of hepatitis C virus infection initiation in vitro is directly related to particle density. Virology 407:110–119. 10.1016/j.virol.2010.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z, Simeon RL, Chockalingam K, Rice CM. 2010. Creation and characterization of a cell-death reporter cell line for hepatitis C virus infection. Antiviral Res. 86:220–223. 10.1016/j.antiviral.2010.02.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogt A, Scull MA, Friling T, Horwitz JA, Donovan BM, Dorner M, Gerold G, Labitt RN, Rice CM, Ploss A. 2013. Recapitulation of the hepatitis C virus life-cycle in engineered murine cell lines. Virology 444:1–11. 10.1016/j.virol.2013.05.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fernandez J, Taylor D, Morhardt DR, Mihalik K, Puig M, Rice CM, Feinstone SM, Major ME. 2004. Long-term persistence of infection in chimpanzees inoculated with an infectious hepatitis C virus clone is associated with a decrease in the viral amino acid substitution rate and low levels of heterogeneity. J. Virol. 78:9782–9789. 10.1128/JVI.78.18.9782-9789.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morin TJ, Broering TJ, Leav BA, Blair BM, Rowley KJ, Boucher EN, Wang Y, Cheslock PS, Knauber M, Olsen DB, Ludmerer SW, Szabo G, Finberg RW, Purcell RH, Lanford RE, Ambrosino DM, Molrine DC, Babcock GJ. 2012. Human monoclonal antibody HCV1 effectively prevents and treats HCV infection in chimpanzees. PLoS Pathog. 8:e1002895. 10.1371/journal.ppat.1002895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dorner M, Horwitz JA, Robbins JB, Barry WT, Feng Q, Mu K, Jones CT, Schoggins JW, Catanese MT, Burton DR, Law M, Rice CM, Ploss A. 2011. A genetically humanized mouse model for hepatitis C virus infection. Nature 474:208–211. 10.1038/nature10168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dorner M, Horwitz JA, Donovan BM, Labitt RN, Budell WC, Friling T, Vogt A, Catanese MT, Satoh T, Kawai T, Akira S, Law M, Rice CM, Ploss A. 2013. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature 501:237–241. 10.1038/nature12427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uprichard SL. 2014. Establishment of mice with Inheritable susceptibility to productive HCV infection. Hepatology 59:2043–2046. 10.1002/hep.26949 [DOI] [PubMed] [Google Scholar]
- 29.Flint M, McKeating JA. 2000. The role of the hepatitis C virus glycoproteins in infection. Rev. Med. Virol. 10:101–117. [DOI] [PubMed] [Google Scholar]
- 30.Sabahi A. 2009. Hepatitis C Virus entry: the early steps in the viral replication cycle. Virol. J. 6:117. 10.1186/1743-422X-6-117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ploss A, Evans MJ. 2012. Hepatitis C virus host cell entry. Curr. Opin. Virol. 2:14–19. 10.1016/j.coviro.2011.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meredith LW, Wilson GK, Fletcher NF, McKeating JA. 2012. Hepatitis C virus entry: beyond receptors. Rev. Med. Virol. 22:182–193. 10.1002/rmv.723 [DOI] [PubMed] [Google Scholar]
- 33.Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, Hua Y, Dai X, Stanfield RL, Burton DR, Ward AB, Wilson IA, Law M. 2013. Hepatitis C virus E2 envelope glycoprotein core structure. Science 342:1090–1094. 10.1126/science.1243876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khan AG, Whidby J, Miller MT, Scarborough H, Zatorski AV, Cygan A, Price AA, Yost SA, Bohannon CD, Jacob J, Grakoui A, Marcotrigiano J. 2014. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 509(7500):381–384. 10.1038/nature13117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brazzoli M, Helenius A, Foung SK, Houghton M, Abrignani S, Merola M. 2005. Folding and dimerization of hepatitis C virus E1 and E2 glycoproteins in stably transfected CHO cells. Virology 332:438–453. 10.1016/j.virol.2004.11.034 [DOI] [PubMed] [Google Scholar]
- 36.Krey T, d'Alayer J, Kikuti CM, Saulnier A, Damier-Piolle L, Petitpas I, Johansson DX, Tawar RG, Baron B, Robert B, England P, Persson MA, Martin A, Rey FA. 2010. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 6:e1000762. 10.1371/journal.ppat.1000762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whidby J, Mateu G, Scarborough H, Demeler B, Grakoui A, Marcotrigiano J. 2009. Blocking hepatitis C virus infection with recombinant form of envelope protein 2 ectodomain. J. Virol. 83:11078–11089. 10.1128/JVI.00800-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson IA, Skehel JJ, Wiley DC. 1981. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 289:366–373. 10.1038/289366a0 [DOI] [PubMed] [Google Scholar]
- 39.Rey FA, Heinz FX, Mandl C, Kunz C, Harrison SC. 1995. The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 375:291–298. 10.1038/375291a0 [DOI] [PubMed] [Google Scholar]
- 40.DuBois RM, Vaney MC, Tortorici MA, Kurdi RA, Barba-Spaeth G, Krey T, Rey FA. 2013. Functional and evolutionary insight from the crystal structure of rubella virus protein E1. Nature 493:552–556. 10.1038/nature11741 [DOI] [PubMed] [Google Scholar]
- 41.Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, Rey FA. 2010. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468:709–712. 10.1038/nature09555 [DOI] [PubMed] [Google Scholar]
- 42.Li L, Jose J, Xiang Y, Kuhn RJ, Rossmann MG. 2010. Structural changes of envelope proteins during alphavirus fusion. Nature 468:705–708. 10.1038/nature09546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, Wang J, Kanai R, Modis Y. 2013. Crystal structure of glycoprotein E2 from bovine viral diarrhea virus. Proc. Natl. Acad. Sci. U. S. A. 110:6805–6810. 10.1073/pnas.1300524110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Logvinoff C, Major ME, Oldach D, Heyward S, Talal A, Balfe P, Feinstone SM, Alter H, Rice CM, McKeating JA. 2004. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc. Natl. Acad. Sci. U. S. A. 101:10149–10154. 10.1073/pnas.0403519101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, Rice CM, Ploss A, Burton DR, Law M. 2012. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. U. S. A. 109:6205–6210. 10.1073/pnas.1114927109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goffard A, Callens N, Bartosch B, Wychowski C, Cosset FL, Montpellier C, Dubuisson J. 2005. Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J. Virol. 79:8400–8409. 10.1128/JVI.79.13.8400-8409.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Helle F, Vieyres G, Elkrief L, Popescu CI, Wychowski C, Descamps V, Castelain S, Roingeard P, Duverlie G, Dubuisson J. 2010. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J. Virol. 84:11905–11915. 10.1128/JVI.01548-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pantua H, Diao J, Ultsch M, Hazen M, Mathieu M, McCutcheon K, Takeda K, Date S, Cheung TK, Phung Q, Hass P, Arnott D, Hongo JA, Matthews DJ, Brown A, Patel AH, Kelley RF, Eigenbrot C, Kapadia SB. 2013. Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J. Mol. Biol. 425:1899–1914. 10.1016/j.jmb.2013.02.025 [DOI] [PubMed] [Google Scholar]
- 49.Sabo MC, Luca VC, Prentoe J, Hopcraft SE, Blight KJ, Yi M, Lemon SM, Ball JK, Bukh J, Evans MJ, Fremont DH, Diamond MS. 2011. Neutralizing monoclonal antibodies against hepatitis C virus E2 protein bind discontinuous epitopes and inhibit infection at a postattachment step. J. Virol. 85:7005–7019. 10.1128/JVI.00586-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keck ZY, Li SH, Xia J, von Hahn T, Balfe P, McKeating JA, Witteveldt J, Patel AH, Alter H, Rice CM, Foung SK. 2009. Mutations in hepatitis C virus E2 located outside the CD81 binding sites lead to escape from broadly neutralizing antibodies but compromise virus infectivity. J. Virol. 83:6149–6160. 10.1128/JVI.00248-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sautto G, Tarr AW, Mancini N, Clementi M. 2013. Structural and antigenic definition of hepatitis C virus E2 glycoprotein epitopes targeted by monoclonal antibodies. Clin. Dev. Immunol. 2013:450963. 10.1155/2013/450963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pedersen J, Carlsen TH, Prentoe J, Ramirez S, Jensen TB, Forns X, Alter H, Foung SK, Law M, Gottwein J, Weis N, Bukh J. 2013. Neutralization resistance of hepatitis C virus can be overcome by recombinant human monoclonal antibodies. Hepatology 58:1587–1597. 10.1002/hep.26524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941. 10.1126/science.282.5390.938 [DOI] [PubMed] [Google Scholar]
- 54.McKeating JA, Zhang LQ, Logvinoff C, Flint M, Zhang J, Yu J, Butera D, Ho DD, Dustin LB, Rice CM, Balfe P. 2004. Diverse hepatitis C virus glycoproteins mediate viral infection in a CD81-dependent manner. J. Virol. 78:8496–8505. 10.1128/JVI.78.16.8496-8505.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Flint M, von Hahn T, Zhang J, Farquhar M, Jones CT, Balfe P, Rice CM, McKeating JA. 2006. Diverse CD81 proteins support hepatitis C virus infection. J. Virol. 80:11331–11342. 10.1128/JVI.00104-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Farquhar MJ, Harris HJ, McKeating JA. 2011. Hepatitis C virus entry and the tetraspanin CD81. Biochem. Soc. Trans. 39:532–536. 10.1042/BST0390532 [DOI] [PubMed] [Google Scholar]
- 57.Zhao Z, Zhong L, Elrod E, Struble E, Ma L, Yan H, Harman C, Deng L, Virata-Theimer ML, Liu P, Alter H, Grakoui A, Zhang P. 2014. A neutralization epitope in the hepatitis C virus e2 glycoprotein interacts with host entry factor CD81. PLoS One 9:e84346. 10.1371/journal.pone.0084346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bartosch B, Vitelli A, Granier C, Goujon C, Dubuisson J, Pascale S, Scarselli E, Cortese R, Nicosia A, Cosset FL. 2003. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J. Biol. Chem. 278:41624–41630. 10.1074/jbc.M305289200 [DOI] [PubMed] [Google Scholar]
- 59.Molina S, Castet V, Pichard-Garcia L, Wychowski C, Meurs E, Pascussi JM, Sureau C, Fabre JM, Sacunha A, Larrey D, Dubuisson J, Coste J, McKeating J, Maurel P, Fournier-Wirth C. 2008. Serum-derived hepatitis C virus infection of primary human hepatocytes is tetraspanin CD81 dependent. J. Virol. 82:569–574. 10.1128/JVI.01443-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Flint M, Maidens C, Loomis-Price LD, Shotton C, Dubuisson J, Monk P, Higginbottom A, Levy S, McKeating JA. 1999. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol. 73:6235–6244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clayton RF, Owsianka A, Aitken J, Graham S, Bhella D, Patel AH. 2002. Analysis of antigenicity and topology of E2 glycoprotein present on recombinant hepatitis C virus-like particles. J. Virol. 76:7672–7682. 10.1128/JVI.76.15.7672-7682.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Owsianka A, Clayton RF, Loomis-Price LD, McKeating JA, Patel AH. 2001. Functional analysis of hepatitis C virus E2 glycoproteins and virus-like particles reveals structural dissimilarities between different forms of E2. J. Gen. Virol. 82:1877–1883 [DOI] [PubMed] [Google Scholar]
- 63.Roccasecca R, Ansuini H, Vitelli A, Meola A, Scarselli E, Acali S, Pezzanera M, Ercole BB, McKeating J, Yagnik A, Lahm A, Tramontano A, Cortese R, Nicosia A. 2003. Binding of the hepatitis C virus E2 glycoprotein to CD81 is strain specific and is modulated by a complex interplay between hypervariable regions 1 and 2. J. Virol. 77:1856–1867. 10.1128/JVI.77.3.1856-1867.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Owsianka AM, Timms JM, Tarr AW, Brown RJ, Hickling TP, Szwejk A, Bienkowska-Szewczyk K, Thomson BJ, Patel AH, Ball JK. 2006. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J. Virol. 80:8695–8704. 10.1128/JVI.00271-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rothwangl KB, Manicassamy B, Uprichard SL, Rong L. 2008. Dissecting the role of putative CD81 binding regions of E2 in mediating HCV entry: putative CD81 binding region 1 is not involved in CD81 binding. Virol. J. 5:46. 10.1186/1743-422X-5-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21:5017–5025. 10.1093/emboj/cdf529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Catanese MT, Ansuini H, Graziani R, Huby T, Moreau M, Ball JK, Paonessa G, Rice CM, Cortese R, Vitelli A, Nicosia A. 2010. Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. J. Virol. 84:34–43. 10.1128/JVI.02199-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. 10.1038/nature05654 [DOI] [PubMed] [Google Scholar]
- 69.Harris HJ, Davis C, Mullins JG, Hu K, Goodall M, Farquhar MJ, Mee CJ, McCaffrey K, Young S, Drummer H, Balfe P, McKeating JA. 2010. Claudin association with CD81 defines hepatitis C virus entry. J. Biol. Chem. 285:21092–21102. 10.1074/jbc.M110.104836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886. 10.1038/nature07684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grove J, Huby T, Stamataki Z, Vanwolleghem T, Meuleman P, Farquhar M, Schwarz A, Moreau M, Owen JS, Leroux-Roels G, Balfe P, McKeating JA. 2007. Scavenger receptor BI and BII expression levels modulate hepatitis C virus infectivity. J. Virol. 81:3162–3169. 10.1128/JVI.02356-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pohlmann S, Zhang J, Baribaud F, Chen Z, Leslie GJ, Lin G, Granelli-Piperno A, Doms RW, Rice CM, McKeating JA. 2003. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J. Virol. 77:4070–4080. 10.1128/JVI.77.7.4070-4080.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV. 2007. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J. Virol. 81:374–383. 10.1128/JVI.01134-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF. 2011. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 17:589–595. 10.1038/nm.2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kohaar I, Ploss A, Korol E, Mu K, Schoggins JW, O'Brien TR, Rice CM, Prokunina-Olsson L. 2010. Splicing diversity of the human OCLN gene and its biological significance for hepatitis C virus entry. J. Virol. 84:6987–6994. 10.1128/JVI.00196-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sainz B, Jr, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL. 2012. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 18:281–285. 10.1038/nm.2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martin DN, Uprichard SL. 2013. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc. Natl. Acad. Sci. U. S. A. 110:10777–10782. 10.1073/pnas.1301764110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sabahi A. 2009. Early events in hepatitis C virus infection: an interplay of viral entry, decay, and density. Ph.D. dissertation Tulane University, New Orleans, LA [Google Scholar]
- 79.Meertens L, Bertaux C, Dragic T. 2006. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J. Virol. 80:11571–11578. 10.1128/JVI.01717-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gallaher WR, Ball JM, Garry RF, Griffin MC, Montelaro RC. 1989. A general model for the transmembrane proteins of HIV and other retroviruses. AIDS Res. Hum. Retroviruses 5:431–440. 10.1089/aid.1989.5.431 [DOI] [PubMed] [Google Scholar]
- 81.Igonet S, Vaney MC, Vonrhein C, Bricogne G, Stura EA, Hengartner H, Eschli B, Rey FA. 2011. X-ray structure of the arenavirus glycoprotein GP2 in its postfusion hairpin conformation. Proc. Natl. Acad. Sci. U. S. A. 108:19967–19972. 10.1073/pnas.1108910108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Modis Y. 2013. Class II fusion proteins. Adv. Exp. Med. Biol. 790:150–166. 10.1007/978-1-4614-7651-1_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Backovic M, Jardetzky TS. 2011. Class III viral membrane fusion proteins. Adv. Exp. Med. Biol. 714:91–101. 10.1007/978-94-007-0782-5_3 [DOI] [PubMed] [Google Scholar]
- 84.Modis Y, Ogata S, Clements D, Harrison SC. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature 427:313–319. 10.1038/nature02165 [DOI] [PubMed] [Google Scholar]
- 85.Gibbons DL, Vaney MC, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey FA. 2004. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature 427:320–325. 10.1038/nature02239 [DOI] [PubMed] [Google Scholar]
- 86.Garry CE, Garry RF. 2004. Proteomics computational analyses suggest that the carboxyl terminal glycoproteins of Bunyaviruses are class II viral fusion proteins (beta-penetrenes). Theor. Biol. Med. Model. 1:10. 10.1186/1742-4682-1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dessau M, Modis Y. 2013. Crystal structure of glycoprotein C from Rift Valley fever virus. Proc. Natl. Acad. Sci. U. S. A. 110:1696–1701. 10.1073/pnas.1217780110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Allison SL, Schalich J, Stiasny K, Mandl CW, Heinz FX. 2001. Mutational evidence for an internal fusion peptide in flavivirus envelope protein E. J. Virol. 75:4268–4275. 10.1128/JVI.75.9.4268-4275.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yagnik AT, Lahm A, Meola A, Roccasecca RM, Ercole BB, Nicosia A, Tramontano A. 2000. A model for the hepatitis C virus envelope glycoprotein E2. Proteins 40:355–366. [DOI] [PubMed] [Google Scholar]
- 90.Keck ZY, Saha A, Xia J, Wang Y, Lau P, Krey T, Rey FA, Foung SK. 2011. Mapping a region of hepatitis C virus E2 that is responsible for escape from neutralizing antibodies and a core CD81-binding region that does not tolerate neutralization escape mutations. J. Virol. 85:10451–10463. 10.1128/JVI.05259-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rychlowska M, Owsianka AM, Foung SK, Dubuisson J, Bienkowska-Szewczyk K, Patel AH. 2011. Comprehensive linker-scanning mutagenesis of the hepatitis C virus E1 and E2 envelope glycoproteins reveals new structure-function relationships. J. Gen. Virol. 92:2249–2261. 10.1099/vir.0.034314-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Albecka A, Montserret R, Krey T, Tarr AW, Diesis E, Ball JK, Descamps V, Duverlie G, Rey F, Penin F, Dubuisson J. 2011. Identification of new functional regions in hepatitis C virus envelope glycoprotein E2. J. Virol. 85:1777–1792. 10.1128/JVI.02170-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Flint M, Thomas JM, Maidens CM, Shotton C, Levy S, Barclay WS, McKeating JA. 1999. Functional analysis of cell surface-expressed hepatitis C virus E2 glycoprotein. J. Virol. 73:6782–6790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Garry RF, Dash S. 2003. Proteomics computational analyses suggest that hepatitis C virus E1 and pestivirus E2 envelope glycoproteins are truncated class II fusion proteins. Virology 307:255–265. 10.1016/S0042-6822(02)00065-X [DOI] [PubMed] [Google Scholar]
- 95.Wimley WC, White SH. 1996. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 3:842–848. 10.1038/nsb1096-842 [DOI] [PubMed] [Google Scholar]
- 96.Drummer HE, Boo I, Poumbourios P. 2007. Mutagenesis of a conserved fusion peptide-like motif and membrane-proximal heptad-repeat region of hepatitis C virus glycoprotein E1. J. Gen. Virol. 88:1144–1148. 10.1099/vir.0.82567-0 [DOI] [PubMed] [Google Scholar]
- 97.Lavillette D, Pecheur EI, Donot P, Fresquet J, Molle J, Corbau R, Dreux M, Penin F, Cosset FL. 2007. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J. Virol. 81:8752–8765. 10.1128/JVI.02642-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Russell RS, Kawaguchi K, Meunier JC, Takikawa S, Faulk K, Bukh J, Purcell RH, Emerson SU. 2009. Mutational analysis of the hepatitis C virus E1 glycoprotein in retroviral pseudoparticles and cell-culture-derived H77/JFH1 chimeric infectious virus particles. J. Viral Hepat. 16:621–632. 10.1111/j.1365-2893.2009.01111.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li HF, Huang CH, Ai LS, Chuang CK, Chen SS. 2009. Mutagenesis of the fusion peptide-like domain of hepatitis C virus E1 glycoprotein: involvement in cell fusion and virus entry. J. Biomed. Sci. 16:89. 10.1186/1423-0127-16-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Perez-Berna AJ, Moreno MR, Guillen J, Bernabeu A, Villalain J. 2006. The membrane-active regions of the hepatitis C virus E1 and E2 envelope glycoproteins. Biochemistry 45:3755–3768. 10.1021/bi0523963 [DOI] [PubMed] [Google Scholar]
- 101.Perez-Berna AJ, Pabst G, Laggner P, Villalain J. 2009. Biophysical characterization of the fusogenic region of HCV envelope glycoprotein E1. Biochim. Biophys. Acta 1788:2183–2193. 10.1016/j.bbamem.2009.08.002 [DOI] [PubMed] [Google Scholar]
- 102.Sainz B, Mossel EC, Gallaher WR, Wimley WC, Peters CJ, Wilson RB, Garry RF. 2006. Inhibition of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) infectivity by peptides analogous to the viral spike protein. Virus Res. 120:146–155. 10.1016/j.virusres.2006.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cheng G, Montero A, Gastaminza P, Whitten-Bauer C, Wieland SF, Isogawa M, Fredericksen B, Selvarajah S, Gallay PA, Ghadiri MR, Chisari FV. 2008. A virocidal amphipathic {alpha}-helical peptide that inhibits hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 105:3088–3093. 10.1073/pnas.0712380105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Harrison SC. 2008. The pH sensor for flavivirus membrane fusion. J. Cell Biol. 183:177–179. 10.1083/jcb.200809175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boo I, te Wierik K, Douam F, Lavillette D, Poumbourios P, Drummer HE. 2012. Distinct roles in folding, CD81 receptor binding and viral entry for conserved histidine residues of hepatitis C virus glycoprotein E1 and E2. Biochem. J. 443:85–94. 10.1042/BJ20110868 [DOI] [PubMed] [Google Scholar]
- 106.Zhang W, Chipman PR, Corver J, Johnson PR, Zhang Y, Mukhopadhyay S, Baker TS, Strauss JH, Rossmann MG, Kuhn RJ. 2003. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat. Struct. Biol. 10:907–912. 10.1038/nsb990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Allison SL, Stiasny K, Stadler K, Mandl CW, Heinz FX. 1999. Mapping of functional elements in the stem-anchor region of tick-borne encephalitis virus envelope protein E. J. Virol. 73:5605–5612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kaufmann B, Chipman PR, Holdaway HA, Johnson S, Fremont DH, Kuhn RJ, Diamond MS, Rossmann MG. 2009. Capturing a flavivirus pre-fusion intermediate. PLoS Pathog. 5:e1000672. 10.1371/journal.ppat.1000672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Drummer HE, Poumbourios P. 2004. Hepatitis C virus glycoprotein E2 contains a membrane-proximal heptad repeat sequence that is essential for E1E2 glycoprotein heterodimerization and viral entry. J. Biol. Chem. 279:30066–30072. 10.1074/jbc.M405098200 [DOI] [PubMed] [Google Scholar]
- 110.Perez-Berna AJ, Bernabeu A, Moreno MR, Guillen J, Villalain J. 2008. The pre-transmembrane region of the HCV E1 envelope glycoprotein: interaction with model membranes. Biochim. Biophys. Acta 1778:2069–2080. 10.1016/j.bbamem.2008.03.018 [DOI] [PubMed] [Google Scholar]
- 111.White SH, Wimley WC. 1999. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 28:319–365. 10.1146/annurev.biophys.28.1.319 [DOI] [PubMed] [Google Scholar]
- 112.Ciczora Y, Callens N, Montpellier C, Bartosch B, Cosset FL, Op de Beeck A, Dubuisson J. 2005. Contribution of the charged residues of hepatitis C virus glycoprotein E2 transmembrane domain to the functions of the E1E2 heterodimer. J. Gen. Virol. 86:2793–2798. 10.1099/vir.0.81140-0 [DOI] [PubMed] [Google Scholar]
- 113.Li E, Wimley WC, Hristova K. 2012. Transmembrane helix dimerization: beyond the search for sequence motifs. Biochim. Biophys. Acta 1818:183–193. 10.1016/j.bbamem.2011.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Op De Beeck A, Montserret R, Duvet S, Cocquerel L, Cacan R, Barberot B, Le Maire M, Penin F, Dubuisson J. 2000. The transmembrane domains of hepatitis C virus envelope glycoproteins E1 and E2 play a major role in heterodimerization. J. Biol. Chem. 275:31428–31437. 10.1074/jbc.M003003200 [DOI] [PubMed] [Google Scholar]
- 115.Harrison SC. 2005. Mechanism of membrane fusion by viral envelope proteins. Adv. Virus Res. 64:231–261. 10.1016/S0065-3527(05)64007-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yu X, Qiao M, Atanasov I, Hu Z, Kato T, Liang TJ, Zhou ZH. 2007. Cryo-electron microscopy and three-dimensional reconstructions of hepatitis C virus particles. Virology 367:126–134. 10.1016/j.virol.2007.05.038 [DOI] [PubMed] [Google Scholar]
- 117.Catanese MT, Uryu K, Kopp M, Edwards TJ, Andrus L, Rice WJ, Silvestry M, Kuhn RJ, Rice CM. 2013. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. U. S. A. 110:9505–9510. 10.1073/pnas.1307527110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nielsen SU, Bassendine MF, Burt AD, Martin C, Pumeechockchai W, Toms GL. 2006. Association between hepatitis C virus and very-low-density lipoprotein (VLDL)/LDL analyzed in iodixanol density gradients. J. Virol. 80:2418–2428. 10.1128/JVI.80.5.2418-2428.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mirano-Bascos D, Steede NK, Robinson JE, Landry SJ. 2010. Influence of disulfide-stabilized structure on the specificity of helper T-cell and antibody responses to HIV envelope glycoprotein gp120. J. Virol. 84:3303–3311. 10.1128/JVI.02242-09 [DOI] [PMC free article] [PubMed] [Google Scholar]