

ABSTRACT
B and CD4+ T lymphocytes are natural targets of murine leukemia virus (MLV). Migrating lymphocytes adopt a polarized morphology with a trailing edge designated the uropod. Here, we demonstrate that MLV Gag localizes to the uropod in polarized B cells and CD4+ T cells. The uropod localization of MLV Gag was dependent on plasma membrane (PM) association and multimerization of Gag but independent of the viral glycoprotein Env. Basic residues in MA that are required for MLV Gag recruitment to virological synapses between HEK293 and XC cells were dispensable for uropod localization in migrating B cells. Ultrastructural studies indicated that both wild-type and basic-residue mutant Gag localized to the outer surface of the PM at the uropod. Late-domain mutant virus particles were seen at the uropod in form of budding-arrested intermediates. Finally, uropods mediated contact between MLV-infected B cells and uninfected T cells to form virological synapses. Our results suggest that MLV, not unlike HIV, accumulates at the uropod of primary lymphocytes to facilitate viral spreading through the formation of uropod-mediated cell-cell contacts.
IMPORTANCE Viruses have evolved mechanisms to coordinate their assembly and budding with cell polarity to facilitate their spreading. In this study, we demonstrated that the viral determinants for MLV Gag to localize to the uropod in polarized B cells are distinct from the requirements to localize to virological synapses in transformed cell lines. Basic residues in MA that are required for the Gag localization to virological synapses between HEK293 and XC cells are dispensable for Gag localization to the uropod in primary B cells. Rather, plasma membrane association and capsid-driven multimerization of Gag are sufficient to drive MLV Gag to the uropod. MLV-laden uropods also mediate contacts between MLV-infected B cells and uninfected T cells to form virological synapses. Our results indicate that MLV accumulates at the uropod of primary lymphocytes to facilitate viral spreading through the formation of uropod-mediated cell-cell contacts.
INTRODUCTION
Retroviral assembly is driven by the viral precursor polyprotein Gag that consists of matrix (MA), capsid (CA), and nucleocapsid (NC) (1–4). Each domain serves distinct functions during the viral assembly process. The MA domain mediates binding of Gag to the plasma membrane (PM). The CA domain mediates Gag-Gag interaction required to form immature and mature viral particles. CA consists of two domains, an N-terminal domain (CA-NTD) and a C-terminal domain (CA-CTD) (5, 6). The CA-NTD facilitates the oligomerization into the hexameric and pentameric rings within the capsid structure. CA-CTD dimers and trimers form the contact of neighboring hexamers and pentamers and are critical for Gag oligomerization and particle formation (7, 8). NC mediates packaging of genomic RNA into the viral core and initiates the oligomerization of Gag. Gag of various retroviruses encodes additional proteins that play important roles in assembly and release. For instance, p12 from murine leukemia virus (MLV) and p6 from the human immunodeficiency virus (HIV) contain late-domain motifs that are required for the viral particle to pinch off from the PM (9–13). The PPPY motif in MLV p12 recruits NEDD4-like E3 ligases to promote virus release via a pathway that is dependent on the vacuolar protein sorting 4 (VPS4) (9–11).
It is generally accepted that the assembly of MLV and HIV predominantly occurs at the PM or its invaginations (14–20). Both HIV Gag and MLV Gag have also been observed to associate with late endosomes and multivesicular bodies (MVBs) in HeLa, HEK293, and T cells and macrophages (21–25). The endosome/MVB pathway has further been suggested to play a role in viral trafficking to assembly sites (26–28). Gag has also been observed to colocalize with exosomes/microvesicles (EMVs) that are secreted as exosomes (29, 30). In migrating lymphocytes, the association of Gag proteins with EMV facilitates their polarization, which may further promote the polarization of the cells (30). The localization of MLV Gag in B and CD4+ T cells, physiologically relevant cell types for MLV infection, has not been characterized extensively.
MA is the primary viral determinant responsible for targeting Gag to the PM. It mediates Gag-PM association via a covalently linked myristoyl group at its N terminus and basic charges (31–33). Basic charges are thought to interact with acidic phospholipids that are enriched at the inner leaflet of the PM (14, 34–38). Neutralization of basic residues in the polybasic cluster leads to a relocalization of Gag to intracellular compartments and severely reduces viral release in HeLa cells and HEK293 cells (35, 36, 39–43). However, one such HIV basic-domain mutant, K29/31E, although still targeted to intracellular membranes, assembles and releases relatively efficiently in macrophages and T cells (42, 44).
Viruses exploit and manipulate existing cellular structures and cell-cell adhesion for the purpose of efficient spreading (45–48). Retroviral cell-to-cell transmission has been shown to occur through broad virological synapses or thin filopodial bridges and nanotubes (47, 49, 50). In most cases, the interaction between the viral glycoprotein Env and its receptor leads to the establishment of cell-cell contacts. Consequently, Env and viral receptor accumulate at the cell-cell interface (49, 51–54). In transformed cell lines, the establishment of adhesion between MLV-infected and uninfected cells is followed by the polarized assembly of particles at the virological synapse (51). A single tyrosine residue in the cytoplasmic tail of Env is critically involved in polarizing assembly (51, 55). Basic residues in matrix domain (MA) and Gag multimerization are then needed for MLV Gag recruitment to the virological synapse (56).
Immune cells are the natural host cells for most retroviruses, including MLV, which infects T and B cells (52, 57). Migrating leukocytes, including T and B lymphocytes, display a polarized morphology. The front extension of a migrating cell is referred to as the leading edge, while the rear protrusion is referred to as the uropod (58–61). Uropods are enriched in the adhesion molecules intercellular adhesion molecule 1 (ICAM-1), ICAM-2, and ICAM-3, P-selectin glycoprotein ligand-1 (PSGL-1), CD43 and CD44, cholesterol, and the ganglioside GM1 (62–65). The microtubule-organizing center (MTOC) and various organelles, including the endoplasmic reticulum (ER), Golgi apparatus, and mitochondria, locate toward the uropod (59, 66, 67). Uropods are also characterized by actin-rich PM projections such as microspikes and microvilli (59, 66, 68).
The uropod has also been implicated in viral spreading. In polarized HIV-infected T cells, viral Gag and genome localize to the uropod (54, 69, 70). MLV has also been observed in association with the uropod of polarized lymphocytes (71). In addition, Gag-laden uropods preferentially mediate contacts between infected and uninfected T cells to form virological synapses (54, 69). Interference with T cell polarity using myosin inhibitors blocks subsequent HIV cell-to-cell transmission (69). Mechanistically, HIV uropod localization requires Gag multimerization primarily mediated by the NC domain (69). Gag first associates with PM microdomains, which then laterally migrate to the uropod. Gag localization to these microdomains depends on basic residues within the polybasic region in the Gag MA domain (72). The prevailing model for a role of the uropod in HIV transmission is that it serves as a platform for HIV assembly and mediates the formation of virological synapses (69, 72). Whether MLV assembles and buds at the uropod in polarized murine lymphocytes has not been studied in detail.
Here we demonstrate that MLV Gag localizes to the uropod of polarized B cells and CD4+ T cells. Unlike the polarization of MLV Gag to virological synapses in transformed cell lines, the recruitment of Gag to the uropod was Env independent. Also, basic residues that are required for localization of Gag to virological synapses in transformed cell lines were dispensable for Gag uropod localization. Rather, PM association and Gag multimerization drove uropod localization of MLV Gag. Ultrastructural studies demonstrated that MLV particles accumulate at the outer surface of the uropod in polarized B cells. Finally, we provide evidence that uropods mediate contact between MLV-infected B cells and target T cells, leading to the formation of virological synapses. Our results support a model in which MLV, like HIV, accumulates at the uropod to facilitate viral spreading through uropod-mediated cell-cell contacts.
MATERIALS AND METHODS
Plasmids and reagents.
Proviral pLRB303 vectors expressing wild-type or mutant Friend MLV (F-MLV) Gag-green fluorescent protein (Gag-GFP), Gag-mCherry, or Gag-cyan fluorescent protein (Gag-CFP) (ΔPol) were previously described (55, 56). An F-MLV Gag-GFP/ΔEnv construct was generated by introducing two stop codons in Env. F-MLV Gag-GFP/Env-mCherry was generated by inserting the mCherry sequence into the position of env corresponding to amino acid (aa) 273. In the CA-CTD mutant, the C-terminal domain of capsid (from GQYQ to RHRE) has been deleted. Yellow fluorescent protein (YFP) fusion proteins of Lamp1, Rab7, Syt7, and VPS4 (73) were introduced into the retrovirus-based vector pLZRS to transduce primary cells. Moloney MLV Gag-YFP was previously described (21).
Virus preparation.
Viruses were generated in HEK293 cells by transfection of plasmid pLRB303 or pLZRS, plasmid encoding F-MLV GagPol (to compensate for the absence of Pol), and F-MLV Env (to increase virus infectivity). Fugene6 (Promega) was used for transfection of HEK293 cells. Cell supernatant was filtered after 24 and 48 h using a 0.45-μm-pore-size nylon membrane filter and stored at −80°C.
Primary cell infection.
Isolation and in vitro transduction of primary mouse CD4+ T cells and B cells were described previously (52). To remove residual virus, cells were treated with trypsin for 5 min at 37°C 8 to 12 h postspinoculation. Eight hours after trypsin treatment, cells were plated on coverslips coated with 0.5 μg/ml of recombinant mouse ICAM-1 (R&D systems) and incubated for 1 to 2 h before fixation with 4% paraformaldehyde (PFA) or coculturing with target cells for live-cell imaging.
Live-cell imaging.
A polyclonal S49.1 T cell line stably expressing mCAT-1-mCherry was generated and used as target cells. Primary murine B cells expressing Gag-GFP were cocultured with target cells, and the formation of F-MLV synapses was monitored using time-lapse spinning disc confocal video microscopy as described previously (51, 55, 56). The formation of virological synapses was observed 1 h after initiation of cocultures. The images were analyzed using Volocity software (PerkinElmer).
Immunostaining.
For immunostaining, primary antibodies against the following were used: Lamp1 (ID4B) (21), CD43 (S7; BD Pharmingen), CD44 (IM7; BD Pharmingen), or goat anti-mCherry (Biorbyt). Cells were incubated with the primary antibody for 30 min, washed three times with phosphate-buffered saline (PBS)–1% bovine serum albumin (BSA), and then incubated with Alexa Fluor 569- or Alexa Fluor 647-conjugated secondary antibodies. After fixation with 4% PFA, cells were analyzed using a spinning disc confocal microscope. The Gag-uropod polarization index was calculated as the ratio of Gag-GFP mean fluorescence intensity (MFI) at the uropod relative to Gag-GFP MFI outside the uropod. Both MFIs were normalized to background GFP MFI outside cells.
Electron microscopy.
Correlative fluorescence and scanning electron microscopy (SEM) was performed as previously described (49). A diamond pencil was used to mark regions of interest for easy reidentification under the SEM. Transmission electron microscopy (TEM) was performed as previously described (49, 74).
RESULTS
MLV Gag localizes to the uropod in polarized primary CD4+ T and B lymphocytes.
T and B lymphocytes are the natural targets of MLV (52, 57). To determine the distribution of MLV Gag in polarized T and B cells, we infected primary mouse CD4+ T and B cells with full-length F-MLV encoding GFP-labeled Gag (Gag-GFP). Following the infection, cells were treated with trypsin to remove residual input virus. Cells were plated on ICAM-1-coated coverslips to induce cell migration and polarization. Cells were fixed at 1 h postplating, and Gag-GFP distribution was investigated by spinning disc confocal microscopy. We found that MLV Gag-GFP accumulated at the uropod in both infected CD4+ T cells and B cells (Fig. 1A and B). In primary CD4+ T cells, Gag showed a strong colocalization with the uropod markers CD43 and CD44 (Fig. 1A). In primary B cells, Gag similarly colocalized with the uropod marker CD44 (Fig. 1B). Most splenic B cells do not express CD43 (75). To quantify the extent of polarization, we calculated a polarization index defined as the ratio of Gag-GFP mean fluorescence intensity (MFI) found at the uropod relative to Gag-GFP's MFI outside the uropod. We defined polarization as an index value greater than 3 for an analysis of at least 10 cells (Fig. 1A and B). The theoretical value for the lack of polarity at the uropod (random distribution) would be 1.
FIG 1.

MLV Gag localizes to the uropod in polarized primary T and B lymphocytes. (A and B) Primary CD4+ T cells (A) and primary B cells (B) were infected with F-MLV Gag-GFP (green). Sixteen to 20 h after infection, cells were plated on ICAM-1-coated coverslips, immunostained for the uropod marker CD43 or CD44 (red), and examined by spinning disc confocal microscopy. Depicted images represent the merged extended-focus view of an entire Z-stack. The mean polarization indices of 10 images that quantify the accumulation of Gag-GFP at the uropod are shown at the upper right corners of the Gag-GFP images. (C) Primary B cells were infected with viruses labeled with Gag-YFP (green) and containing full-length Gag-mCherry genomes (red). Eight to 12 h postinfection, cells were treated with trypsin as indicated and monitored by spinning disc confocal microscopy. Scale bars, 10 μm.
To confirm that the observed Gag signal represented de novo-synthesized Gag and not the residual input virus, we generated viruses in HEK293 cells by cotransfecting plasmids expressing Gag-YFP independently of other viral factors, with full-length F-MLV expression plasmids encoding MLV Gag-mCherry. Following infection of B cells, a YFP signal would represent residual input virus, whereas an mCherry signal independent of YFP would indicate de novo-synthesized Gag. Without trypsin treatment, only a few YFP-labeled input viruses were observed, some of which localized to the uropod. However, following trypsin treatment, YFP signal was not detected (Fig. 1C). In both cases, Gag-mCherry was the predominant signal and localized to the uropod in polarized cells (Fig. 1C). Collectively, these results indicate that de novo-synthesized MLV Gag localizes to the uropod of polarized lymphocytes.
Env is not required for MLV Gag localization to the uropod.
Since B cells express MLV proteins to higher levels than primary CD4+ T cells and are able to form virological synapses in vivo (52), we concentrated our subsequent studies on primary B cells. To determine if MLV Env also localizes to the uropod, we infected primary B cells with F-MLV encoding both Gag-GFP and Env-mCherry. In infected cells, Env-mCherry was observed at the uropod, where it colocalized with Gag-GFP (Fig. 2A). Intracellular Env signal that did not colocalize with Gag could be detected near the base of the uropod, likely representing the trans-Golgi network as well as some ER-associated foci (Fig. 2A). In transformed cell lines, MLV Env is essential for the establishment of virological synapses and the subsequent recruitment of Gag to virological synapses (51). To test if Env is required for MLV Gag localization to the uropod, we infected primary B cells with F-MLV Gag-GFP/ΔEnv virus lacking Env. In the absence of Env, Gag still localized to the uropod, indicating that the localization of Gag to the uropod is an intrinsic feature of Gag (Fig. 2B). To test if MLV Env localizes to uropod in the absence of Gag, we infected primary B cells with F-MLV Env-mCherry/ΔGag virus lacking Gag. In the absence of Gag, Env still localized to the uropod as well as some intracellular compartments near the base of the uropod (Fig. 2C).
FIG 2.

The uropod localization of Gag is independent of Env. (A) Primary B cells were infected with full-length F-MLV encoding Gag-GFP (green) and Env-mCherry (red) and monitored by spinning disc confocal microscopy. The mean polarization index of Env-mCherry or Gag-GFP accumulation at uropods is shown at the upper right corner. (B) Primary B cells infected with F-MLV Gag-GFP (green) lacking envelope (ΔEnv). The mean polarization index of Gag-GFP accumulation at uropods is shown at the upper right corner. (C) Primary B cells infected with F-MLV Env-mCherry (red) (ΔGag) lacking Gag. The mean polarization index of Env-mCherry accumulation at uropods is shown at the upper right corner. Scale bars, 10 μm.
MLV Gag PM binding and multimerization are required for localization to the uropod.
To identify viral determinants within Gag that are responsible for uropod localization, we examined a panel of Gag mutants depicted in Fig. 3 (55, 56). A G2A mutant that abrogates myristoylation of Gag was evenly distributed in the cytoplasm, indicating that myristoylation-mediated membrane binding was required for MLV Gag uropod localization (Fig. 3). However, the MA domain was expendable for uropod targeting provided that MA was replaced with a heterologous membrane targeting domain (MTD), in this instance from Src kinase. MLV Gag p12 and NC domains were also not required for uropod targeting (Fig. 3), in contrast to CA and CA-CTD mutants that no longer targeted the uropod despite clearly binding to the PM and additional intracellular membranes. The polarization indices of CA and CA-CTD deletion mutants are slightly higher than the theoretical value of 1, presumably because the uropod is a membrane-rich structure and many organelles localize to this area.
FIG 3.

PM binding and Gag multimerization target MLV Gag to the uropod. The upper scheme displays tested wild-type (wt) MLV Gag and mutants of MLV Gag. All mutations were made in a full-length virus construct with GFP fused to the C terminus of Gag. “LZ” represents the trimeric leucine zipper domain. Localization of indicated Gag-GFP mutants (green) within polarized primary B cells was determined as described for Fig. 1B. Scale bars, 10 μm.
Considering the requirement for CA-CTD, we hypothesized that Gag multimerization was critical for Gag uropod targeting. While deletion of either MA or NC alone was insufficient to abolish Gag uropod targeting, deletion of both (SrcΔMAΔNC) blocked Gag uropod targeting in the context of an intact CA-CTD domain. The ability of SrcΔMAΔNC to localize to the uropod was restored by introducing a trimeric leucine zipper domain (LZ) at the position of NC that stimulates Gag-Gag interactions independently of Gag-RNA binding (56, 76). The observation that SrcΔMAΔNC (= Src-p12-CA) does not localize to the uropod reveals that the CA-CTD is necessary but not sufficient for Gag uropod targeting. Taken together, these data support a model in which multiple Gag multimerization signals are involved in regulating Gag targeting to the uropod.
Basic residues in MA are not required for MLV Gag uropod localization.
We have previously shown that basic residues, irrespective of whether they are contributed by the endogenous polybasic region of MA or come from heterologous MTDs such as Src MTD, are strictly required for the polarized recruitment of Gag to virological synapses in transformed tissue culture cell lines (56). This conclusion was based on several previous observations. First, replacing Gag MA with the Lck MTD (LckΔMA) lacking basic residues supported Gag membrane targeting but not recruitment to virological synapses. In contrast, Gag ΔMA mutants were targeted efficiently to virological synapses by the Src MTD (SrcΔMA) that contains a myristoylation site and 4 basic residues. Second, a basic patch mutant (bm) of Gag in which the net charge of the polybasic region in the MA was neutralized by replacing two basic residues with acidic residues (K31KRR34 to EKER) assembled intracellularly and did not polarize to the virological synapses in transformed cell lines. Third, N-terminally adding the MTDs of Src but not Lck in front of the MA bm mutant (Srcbm and Lckbm) restored the polarization of MLV to virological synapses (56).
To determine if basic residues play a similar role in MLV Gag localization to the uropod, we examined the localization of SrcΔMA, LckΔMA, Srcbm, Lckbm, and bm Gag-GFP constructs in primary B cells. SrcΔMA, LckΔMA, Srcbm, and Lckbm Gag-GFP all displayed strong uropod localization, suggesting that MLV Gag uropod localization in primary B cells is independent of the basic residues in MA (Fig. 4; Table 1). To our surprise, the bm mutant, which is mislocalized to intracellular compartments in HEK293 cells, showed a clear uropod localization in primary B cells (Fig. 4; Table 1). However, the bm mutant appears to be somewhat impaired in membrane binding compared to the wild type. This is visible in its increased cytoplasmic fluorescence and also reflected in the relatively low polarization index, 3.45. Still, Gag uropod targeting in primary lymphocytes is fundamentally different from Gag virological synapse targeting in transformed tissue culture cell lines. Gag uropod targeting requires Gag N-terminal myristoylation and Gag multimerization, in contrast to targeting to virological synapses, which requires these signals in addition to basic residues in Gag's N terminus and upstream Env signaling.
FIG 4.

Basic residues in MA are not required for MLV Gag uropod localization. Upper scheme displays tested wild-type (wt) MLV Gag and mutants of MLV Gag. Localization of indicated Gag-GFP mutants (green) within polarized primary B cells was determined as described for Fig. 1B. Scale bars, 10 μm.
TABLE 1.
Summary of polarization phenotypes of MLV mutants in virological synapses (VS) in fibroblasts and at uropod
| MLV | Polarization |
Polarization index (uropod localization) (n = 10) |
||
|---|---|---|---|---|
| VS in fibroblastsa | Uropod | Mean | SEM | |
| Wild type (T cells) | + | + | 6.75 | 2.44 |
| Wild type (B cells) | + | + | 9.63 | 1.46 |
| ΔEnv | − | + | 10.90 | 1.80 |
| G2A | − | − | 0.91 | 0.06 |
| SrcΔMA | + | + | 5.38 | 1.03 |
| ΔP12 | + | + | 44.32 | 5.50 |
| ΔCA | + | − | 1.43 | 0.05 |
| ΔCA-CTD | + | − | 1.37 | 0.12 |
| ΔNC | + | + | 3.53 | 0.56 |
| SrcΔMAΔNC | − | − | 1.41 | 0.06 |
| SrcΔMAΔNCLZ | + | + | 7.92 | 1.75 |
| SrcNC | + | − | 1.39 | 0.03 |
| LckΔMA | − | + | 8.03 | 2.28 |
| Srcbm | + | + | 6.56 | 1.29 |
| Lckbm | − | + | 7.42 | 0.94 |
| bm | − | + | 3.45 | 0.46 |
| ΔPPPY | + | + | 14.18 | 2.50 |
Determined by Li et al. (56). VS, virological synapses.
Wild-type Gag and bm Gag do not localize to late endosomes in primary B cells.
It has been previously reported that neutralization of the basic residues in the polybasic cluster of MLV interferes with PM association and leads to reduced viral release in NIH 3T3 and HEK293 cells (36). In these cell lines, the bm mutant localizes to intracellular structures, presumably late endosome/MVBs. Because the bm mutant was targeted to the uropod in primary B cells, we asked if wild-type and bm Gag in primary B cells might localize to late endosomes in close proximity to the uropod. We first analyzed the distribution of wild-type and bm Gag-GFP F-MLVs in HEK293 cells. Wild-type MLV Gag was targeted to both the PM and late endosomes/MVBs labeled by Lamp1 and Rab7 (Fig. 5A), consistent with our previous findings (21). bm Gag almost exclusively colocalized with the late endosome markers Lamp1 and Rab7 (Fig. 5A), confirming previous reports (36). MLV-infected B cells were immunostained with an antibody against Lamp1 or cotransduced to express the late endosomal marker YFP-Rab7. Although Lamp1- and Rab7-positive vesicles localized to the base of the uropod, little colocalization was observed with wild-type or bm Gag and either of these markers (Fig. 5B). Thus, unlike with the aforementioned cell lines, neither wild-type nor bm Gag localizes to late endosomes/MVBs in primary B cells.
FIG 5.

Wild-type Gag and bm Gag do not localize to late endosome/MVB in primary B cells. (A) HEK293 cells were transfected with plasmids expressing Lamp1-YFP or YFP-Rab7 (yellow) as well as a plasmid encoding F-MLV Gag-CFP (cyan) and examined 24 h posttransfection by spinning disc confocal microcopy. A single plane of a Z-stack is displayed. (B) Primary B cells were transduced with a viral construct expressing F-MLV Gag-CFP (cyan) and immunostained for Lamp1 (red) or cotransduced with viral constructs expressing YFP-Rab7 (yellow). Images depict an entire Z-stack merged into a single extended-focus view. Scale bars, 10 μm.
Although neither wild-type nor bm Gag localized to late endosomes/MVBs in primary B cells, it was still possible that they would localize to other intracellular compartments just beneath the PM. To address this issue, we developed an assay based on MLV Env accessibility. The rationale is that if the MLV virions localize intracellularly, Env of the virions should not be accessible to antibodies against the Env ectodomain without permeabilization of the cellular membranes. We tested this assay in HEK293 cells, in which wild-type and bm virions accumulate intracellularly (Fig. 6A). Cells were transfected with plasmids encoding F-MLV Gag-GFP/Env-mCherry. After 24 h, cells were fixed and incubated with Alexa Fluor 647-conjugated anti-mCherry antibody and examined by epifluorescence microscopy. The GFP signal and mCherry signal represented the total Gag and total Env, respectively. Indeed, wild-type or bm Gag-GFP/Env-mCherry viruses accumulating in late endosomes/MVB in HEK293 cells were both mCherry and Gag positive but Alexa Fluor 647 negative in the absence of membrane permeabilization (Fig. 6A). In contrast, Env-mCherry expressed at the PM and Env-mCherry colocalizing with wild-type Gag-GFP puncta at the PM were accessible to the Alexa Fluor 647-conjugated anti-mCherry antibody without permeabilization (Fig. 6A). When we applied this assay to primary B cells, all Env-mCherry signals colocalizing with wild-type or bm Gag-GFP puncta were accessible to the Alexa Fluor 647-labeled anti-mCherry Env antibody without permeabilization, indicating surface accessibility for both wild-type and bm virions (Fig. 6B).
FIG 6.
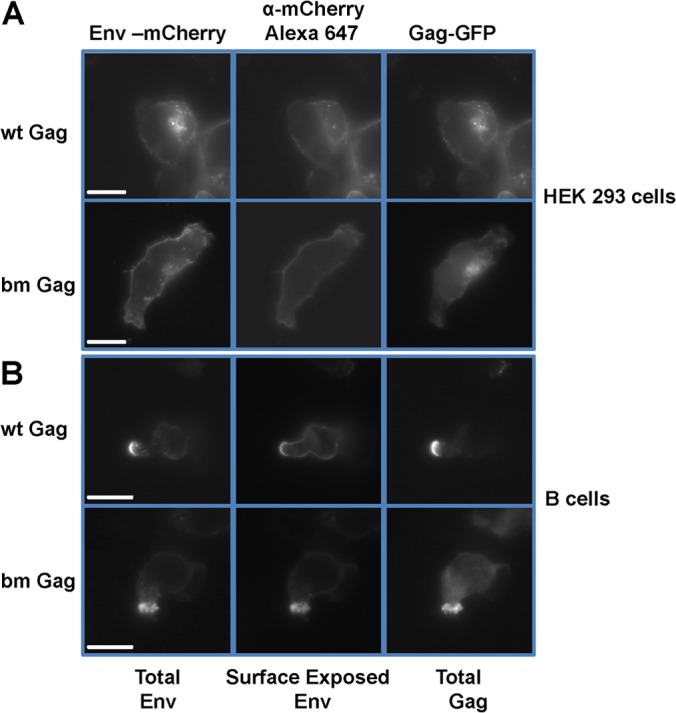
Virions produced by wild-type and bm Gag accumulate at the outer surface of the PM in primary B cells. (A) HEK293 cells were transfected to express wild-type and bm mutant F-MLV Gag-GFP/Env-mCherry. One day later, cells were fixed, incubated with Alexa Fluor 647-conjugated anti-mCherry antibody, and examined by epifluorescence microscopy. The GFP and mCherry signals represent the total Gag and total Env, respectively. The Alexa Fluor 647 signal represents the surface-exposed Env. (B) An experiment as in panel A was performed with primary B cells. Single Z-planes are displayed. Scale bar, 10 μm.
PM of the uropod is a site of active virion production for both wild-type and bm Gag.
The resolution of light microscopy could not determine if the PM of the uropod was the site of active virion assembly, as opposed to Gag accumulating in an unassembled form at the cytoplasmic face of the plasma membrane. To determine if virion formation occurred at the PM of the uropod, we performed transmission electron microscopy (TEM) of primary B cells expressing wild-type or bm Gag. To preserve a normal morphology of viruses, primary B cells were transduced to express wild-type GagPol in addition to F-MLV Gag-GFP virus. To assess the role of the mutant bm Gag in uropod localization, this mutant was also introduced into the GagPol context of F-MLV. Virus-generating B cells were fixed and examined by confocal microscopy before being processed for TEM. The addition of GagPol or bm GagPol did not alter the uropod localization of wild-type or bm Gag-GFP, respectively (data not shown). Uropods could be easily identified under TEM based on the polarized morphology of B cells. In polarized B cells, cellular organelles such as the ER, Golgi apparatus, lysosomes, and mitochondria were polarized toward the uropod (Fig. 7A and B). Importantly, electron-dense virions containing wild-type and bm Gag localized outside the PM and associated thin membrane protrusions or microvilli at the uropod (Fig. 7A and B). No intracellular virions were observed in B cells examined, supporting the absence of virus assembly on intracellular compartments like MVBs in primary B cells (Fig. 7). For wild-type viruses, on average, 3.5 viral particles were at the uropod and 0.03 viral particle was outside the uropod (Fig. 7C). The bm mutant showed a similar distribution (Fig. 7C). It is worth noting that TEM thin sections are typically 50 to 70 nm thick, while the uropod is typically 2 to 5 μm thick. Consequently, 3.5 uropod-localized viral particles in TEM micrographs equals hundreds of viral particles on the surface of one uropod in three-dimensional space.
FIG 7.

Wild-type and bm viruses localize to the surface of uropods in primary B cells. (A and B) Primary B cells were infected with wild-type (A) or bm (B) full-length F-MLV Gag-GFP viruses in the additional presence of wild-type and bm mutant viruses carrying GagPol. One day after infection, B cells were fixed and processed for TEM. The images to the right are magnified views of regions marked by dashed squares. The inset at the right bottom corners are magnified virus particles from indicated areas. Arrows indicate the positions of virions. Scale bars, 10 μm. (C) Quantification of uropod-localized and non-uropod-localized viral particles for wt Gag, bm Gag, and ΔPPPY Gag in TEM micrographs. Sixty, 27, and 28 polarized B cells with viral particles were analyzed for wt Gag, bm Gag, and ΔPPPY Gag, respectively. Error bars represent the standard errors of the means.
To investigate the localization of MLV at the uropod in greater detail, we performed correlative fluorescence and scanning electron microscopy (SEM). The samples were prepared as described above, and MLV Gag-GFP-expressing B cells were first identified by fluorescence microscopy and then visualized by SEM. Correlated micrographs revealed that uropods of uninfected B cells have abundant thin membrane protrusions or microvilli (Fig. 8A). However, infected primary B cells with a strong Gag-GFP signal at the uropod exhibited spherical particles uniform in size (∼150 nm) and morphology in SEM micrographs for both wild-type and bm Gag (Fig. 8B and C). The abundance and the close proximity of the viruses at the uropod do not permit the identification of single Gag-GFP-labeled virus particles. However, the distribution pattern of spherical ∼150-nm structures observed in SEM broadly matched the distribution pattern of Gag-GFP fluorescence, suggesting that these spherical particles are MLV virions (Fig. 8B and C). Thus, the SEM data also support the notion that both wild-type and bm virions undergo active assembly and budding at the uropod.
FIG 8.

SEM electrographs of polarized primary B cells reveal surface accumulation of wild-type and mutant bm Gag particles. Samples were prepared as described for TEM in the legend to Fig. 7. After fixation, virally infected Gag-GFP-labeled (green) polarized B cells were identified using fluorescence microscopy and marked using a diamond pen, and the identical cells were visualized by SEM. Images of uninfected B cells (A) and B cells infected with wild-type (B) and mutant bm Gag (C) F-MLV are displayed. The images to the right are magnifications of indicated regions. The bottom left image of each panel is the corresponding fluorescence image of MLV Gag-GFP virions.
To directly visualize the localization of budding intermediates, we expressed the MLV late-domain mutant (ΔPPPY) in B cells. This mutant still localized to the uropod (Fig. 9A), similar to the earlier phenotype observed for the p12 deletion that contains the PPPY motif (Fig. 3). Both Gag mutants appeared to accumulate at the uropod, resulting in very high polarization indices, consistent with an inability of late-domain mutant virus to pinch off from the membrane. Indeed, TEM revealed that 15.7% of viral particles were still connected with the PM, probably representing budding intermediates of MLV virions (Fig. 9B and C). In contrast, very few such structures were observed in samples containing virus expressing wild-type or bm Gag (Fig. 9C). Taken together, these data suggested that PM of the uropod is a site of active virus production.
FIG 9.
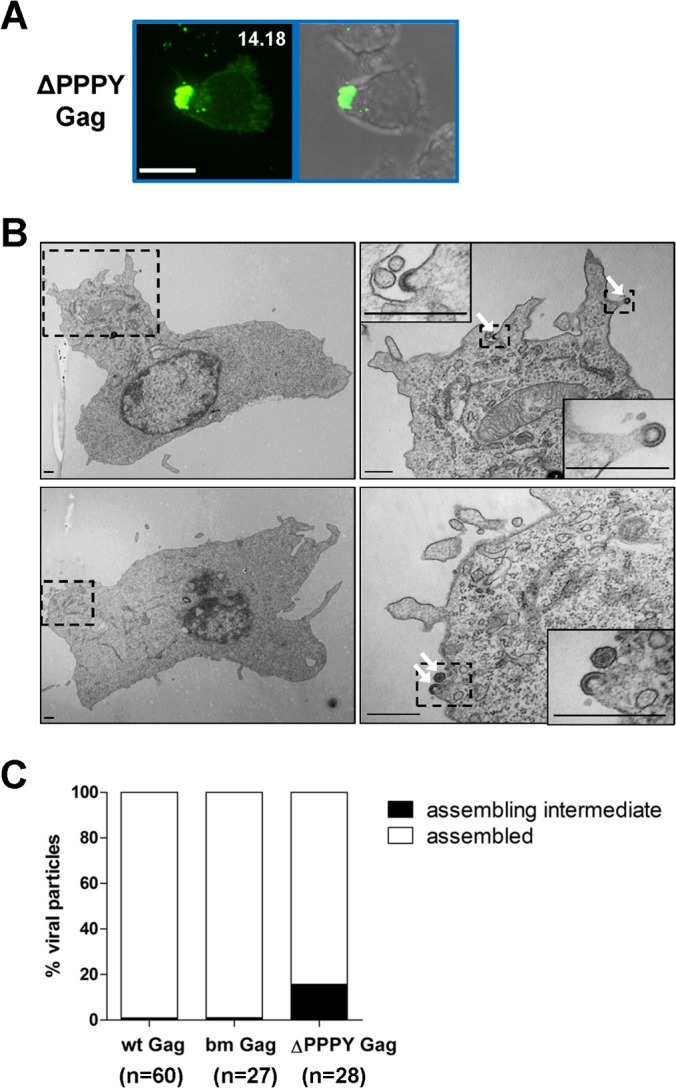
Late-domain mutant virus accumulates at the uropod but fails to pinch off from the PM. (A) Primary B cells infected with F-MLV Gag-GFP ΔPPPY as described for Fig. 1B. Scale bar, 10 μm. (B) Samples were prepared as described for TEM in the legend to Fig. 7 to characterize primary B cells infected with late-domain mutant MLV (ΔΔPPPY Gag). Scale bar, 500 nm. (C) Quantification of assembly intermediates and assembled particles for wt Gag, bm Gag, and ΔPPPY Gag in TEM micrographs. Sixty, 27, and 28 polarized B cells were analyzed for wt Gag, bm Gag, and ΔPPPY Gag, respectively.
Uropods mediate contact between MLV-infected B cells and target cells.
Uropod-associated HIV has been proposed to serve as a preformed platform for the formation of virological synapses to facilitate virus transmission to uninfected cells (69, 72). We performed live-cell imaging to test if uropod-associated MLV also participates in virological synapse formation and MLV cell-to-cell transmission. Primary B cells expressing Gag-GFP labeled virions were cocultured with S49.1 mouse T cells stably expressing mCAT-1-mCherry. The formation of MLV virological synapses was monitored by time-lapse microscopy. Virological synapses between infected B cells and target T cells exhibited a strong accumulation of Gag-GFP and mCAT-1-mCherry at the cell-cell interface (Fig. 10). We often observed two distinct phenotypes. First, uropod-mediated contact between primary B cells and target T cells was long-lived, maintained for >30 min (Fig. 10). Donor and target cells often remained dynamic, with their leading edges migrating into different directions, while the uropod-mediated contact remained unchanged. Second, due to the migratory dynamics of cells, uropod-mediated contacts could transiently stretch into long filopodium-like contacts (Fig. 11).
FIG 10.
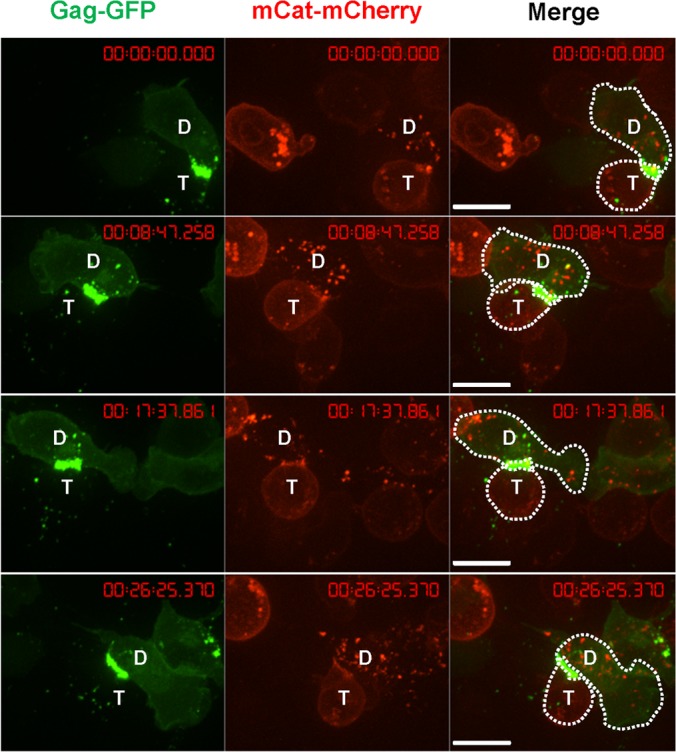
An example of uropod-mediated stable virological synapses between primary B cells and T cells. Primary B cells infected with wild-type F-MLV Gag-GFP (green) as for Fig. 1B were cocultured with S49.1 T cells stably expressing receptor mCAT-1-mCherry (red) and examined by live-cell microscopy. Three image series with 8-min intervals are shown. “D” marks the donor cell and “T” marks the target cell. Scale bar, 10 μm.
FIG 11.

Examples of transitions between filopodia and uropod-mediated virological synapses between primary B cells and T cells. Samples were prepared as described in the legend to Fig. 10. “D” marks the donor cell and “T” marks the target cell. Scale bars, 10 μm.
Uropod-mediated contacts were also observed between infected and uninfected primary B cells in TEM and SEM micrographs (Fig. 12). In thin-section images, viral particles were observed to localize within or around the narrow synaptic cleft (Fig. 12A). Thin membrane extensions were abundant around or within the synaptic cleft. Taken together, these results suggest that uropods can participate in the formation of virological synapses between MLV-infected B cells and target T cells.
FIG 12.

Electron micrographs of uropod-mediated virological synapses between primary B cells. (A and B) Samples were prepared as for Fig. 7 and 8 and examined by TEM (A) and correlative SEM (B). The images to the right in panel A are magnifications of indicated regions. Arrows in panel A point to positions of virions. Insets in panel B are the bright-field and fluorescence images taken by confocal microscopy. “D” marks the donor cell and “T” marks the target cell. The arrow in panel B indicates the position of the uropod-mediated contact.
DISCUSSION
Viruses have evolved mechanisms to coordinate their assembly and budding with cell polarity to facilitate their spreading (47). Cell polarization can be cell adhesion dependent or independent (47). In this study, we demonstrated that the viral determinants for MLV Gag to localize to virological synapses in transformed tissue culture cell lines (cell adhesion, Env receptor dependent) and the uropod in polarized B cells (cell adhesion independent) are different. Basic residues in MA that are required for the Gag localization to virological synapses in transformed cell lines are dispensable for Gag localization to the uropod in primary B cells. Basic residues in MA of HIV Gag have been reported to play a critical role in the association of HIV Gag with uropod-directed microdomains (UDMs) before they comigrate to the uropod (72). A similar role may be played by basic residues in MLV MA during the targeting to an putative acidic interface organized by MLV Env at the virological synapses in transformed cell lines (56). However, MLV and HIV MA proteins are both dispensable for Gag uropod localization in polarized lymphocytes (69). Consequently, basic residues of MA seem to mediate Gag association with specific PM microdomains but are not necessarily required for Gag uropod localization.
The basic patch (bm) mutant of MLV Gag can no longer efficiently bind to the PM and accumulates in late endosomal/MVB compartments in HEK293 cells (Fig. 5) (36). In contrast, bm Gag is targeted to the uropod and viral particles accumulate at the outer surface. These observations recall previous observations with HIV, for which specific basic mutants were similarly relocalized to late endosomal membranes in HEK293T and HeLa cells but were released in macrophages and primary T cells (42, 44). Apparently, primary lymphocytes contain alternative trafficking and release pathways that are distinct from those in transformed cell lines.
Gag multimerization is required for both MLV Gag localization to virological synapses in transformed cell lines and Gag uropod localization in polarized B cells. However, MLV NC plays a more important role in Gag localization to virological synapses in transformed cell lines, whereas MLV CA plays a more important role in Gag localization to the uropod. The reason for this difference is unclear.
Previous studies had proposed that any protein can be targeted to the posterior pole, the precursor of uropod, on the basis of PM binding and higher-order oligomerization (29, 30). The biogenesis of the poster pole primes cells for later signal induced biogenesis of uropod. Our data provide some support for this model, as we observed that Gag derivatives that do not form fluorescent puncta and displayed a diffused distribution on the PM did not localize to the uropod. In contrast, Gag derivatives that multimerize into fluorescent puncta accumulate at the uropod. Late-domain mutant virus also accumulated at uropods and budding arrested particles unable to pinch off from the PM were clearly visible at the uropod. While we cannot exclude surface movement of budding intermediates initiated elsewhere at the PM, these data are consistent with a model in which the uropod represents an active area of virus assembly and release.
MLV Gag localization to the uropod of primary B cells resembles HIV Gag localization to the uropod of primary T cells in many ways. They both are independent of Env expression and represent an intrinsic feature of the retroviral Gag protein (69). They both require Gag multimerization involving several domains of Gag. HIV- and MLV-laden uropods both appear to mediate the formation of virological synapses between infected donor cells and uninfected target cells (69). However, NC is required for HIV Gag uropod localization, while it is dispensable for MLV Gag uropod localization (69). This is unexpected since in HEK293T cells, HIV Gag lacking NC can assemble into somewhat aberrant particles (77), while MLV Gag lacking NC does not assemble at all (78). Indeed, when we transfected HEK293 cells with our full-length MLV construct expressing ΔNC Gag-GFP, no viral particles were observed (55). However, when we infected primary B cells with MLV ΔNC Gag-GFP, GFP puncta could be readily detected in polarized cells, and they accumulated at the uropod. These observations indicate that the dependence of MLV assembly on NC is cell type specific.
The contribution of MLV-laden uropods to the formation of virological synapses has not been previously addressed. Long-term live-cell imaging studies described in this report have shed some light on this process and further add to the original model proposed by the Ono group (69) (Fig. 13). If uropods can make the initial contact with a target cell, it will likely engage target cells to form virological synapses, as previously proposed (69). In contrast, if the leading edge of a migrating lymphocyte makes the initial contact with a target cell, the leading edge will continue to migrate and bypass the target cells. However, uropods, with their many adhesion proteins and Env-laden virions, adhere to the receptor-expressing target cells, while the leading edge continues to drive cellular polarization of the migrating cells. We observed that uropod-mediated contact in this tug of war between migration and cell-cell adhesion is highly dynamic and can stay in a dynamic equilibrium between long filopodium-like contacts and broad uropod contacts.
FIG 13.
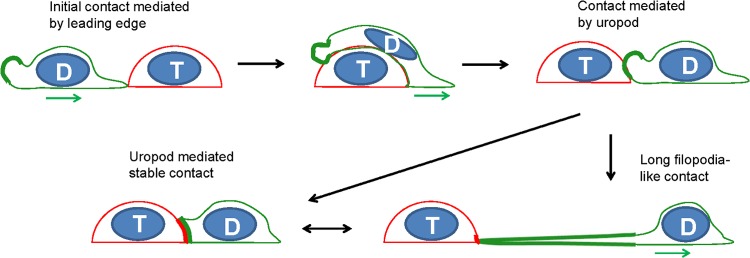
Model for uropod-mediated formation of virological synapses in migrating lymphocytes. The leading edge of the migrating donor cell (green) makes an initial contact with a target cell (red), continues to migrate, and passes the target cell until a uropod-mediated contact is established. Continued migratory forces can lead to long filopodial contacts. “D” depicts the donor cell and “T” the target cell. The green and red thick lines represent the enrichment of Gag-labeled viral particles (green) and viral receptor (red), respectively. The green arrow denotes the direction of migration for donor cells.
In sum, our data provide confirmatory evidence that MLV in addition to HIV can utilize uropods for viral spreading via virological synapses. We report differences with respect to the role of Gag multimerization in the targeting to uropods between both viruses and document the distinct role for basic residues in cell adhesion-induced polarity in virological synapses in transformed cell lines and migration-induced polarity in primary B cells. We also contribute to the characterization of the ultrastructural morphology of polarized lymphocytes infected with MLV and uropod-mediated virological synapses. Live-cell imaging supports the model that uropods participate in the formation of virological synapses between primary cells.
ACKNOWLEDGMENTS
We thank Christin Herrmann for molecular cloning, Zhenting Jiang and Barry Piekos for assistance with scanning electron microscopy, and Xinran Liu for assistance with transmission electron microscopy.
This research was supported by grant R01 CA098727 from the National Institutes of Health to W.M. and Leopoldina Fellowship LPDS2009-21 to X.S.
We declare that no competing interests exist.
Footnotes
Published ahead of print 25 June 2014
REFERENCES
- 1.Morita E, Sundquist WI. 2004. Retrovirus budding. Annu. Rev. Cell Dev. Biol. 20:395–425. 10.1146/annurev.cellbio.20.010403.102350 [DOI] [PubMed] [Google Scholar]
- 2.Balasubramaniam M, Freed EO. 2011. New insights into HIV assembly and trafficking. Physiology (Bethesda) 26:236–251. 10.1152/physiol.00051.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gottlinger HG. 2001. The HIV-1 assembly machine. AIDS 15:S13–S20. 10.1097/00002030-200100005-00003 [DOI] [PubMed] [Google Scholar]
- 4.Briggs JAG, Krausslich HG. 2011. The molecular architecture of HIV. J. Mol. Biol. 410:491–500. 10.1016/j.jmb.2011.04.021 [DOI] [PubMed] [Google Scholar]
- 5.Ehrlich LS, Agresta BE, Carter CA. 1992. Assembly of recombinant human immunodeficiency virus type 1 capsid protein in vitro. J. Virol. 66:4874–4883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gamble TR, Yoo S, Vajdos FF, von Schwedler UK, Worthylake DK, Wang H, McCutcheon JP, Sundquist WI, Hill CP. 1997. Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278:849–853. 10.1126/science.278.5339.849 [DOI] [PubMed] [Google Scholar]
- 7.Coffin JM, Hughes SH, Varmus H. 1997. Retroviruses. Cold Spring Harbor Laboratory Press, Plainview, NY: [PubMed] [Google Scholar]
- 8.Mortuza GB, Haire LF, Stevens A, Smerdon SJ, Stoye JP, Taylor IA. 2004. High-resolution structure of a retroviral capsid hexameric amino-terminal domain. Nature 431:481–485. 10.1038/nature02915 [DOI] [PubMed] [Google Scholar]
- 9.Martin-Serrano J, Eastman SW, Chung W, Bieniasz PD. 2005. HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J. Cell Biol. 168:89–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin-Serrano J, Zang T, Bieniasz PD. 2001. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 7:1313–1319. 10.1038/nm1201-1313 [DOI] [PubMed] [Google Scholar]
- 11.Göttlinger HG, Dorfman T, Sodroski JG, Haseltine WA. 1991. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. U. S. A. 88:3195–3199. 10.1073/pnas.88.8.3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin-Serrano J, Zang T, Bieniasz PD. 2003. Role of ESCRT-I in retroviral budding. J. Virol. 77:4794–4804. 10.1128/JVI.77.8.4794-4804.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin-Serrano J, Neil SJD. 2011. Host factors involved in retroviral budding and release. Nat. Rev. Microbiol. 9:519–531. 10.1038/nrmicro2596 [DOI] [PubMed] [Google Scholar]
- 14.Ono A. 2009. HIV-1 assembly at the plasma membrane: Gag trafficking and localization. Future Virol. 4:241–257. 10.2217/fvl.09.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, Bieniasz PD. 2006. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 4:e435. 10.1371/journal.pbio.0040435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finzi A, Orthwein A, Mercier J, Cohen EA. 2007. Productive human immunodeficiency virus type 1 assembly takes place at the plasma membrane. J. Virol. 81:7476–7490. 10.1128/JVI.00308-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudner L, Nydegger S, Coren LV, Nagashima K, Thali M, Ott DE. 2005. Dynamic fluorescent imaging of human immunodeficiency virus type 1 gag in live cells by biarsenical labeling. J. Virol. 79:4055–4065. 10.1128/JVI.79.7.4055-4065.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Welsch S, Keppler OT, Habermann A, Allespach I, Krijnse-Locker J, Krausslich HG. 2007. HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 3(3):e36. 10.1371/journal.ppat.0030036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deneka M, Pelchen-Matthews A, Byland R, Ruiz-Mateos E, Marsh M. 2007. In macrophages, HIV-1 assembles into an intracellular plasma membrane domain containing the tetraspanins CD81, CD9, and CD53. J. Cell Biol. 177:329–341. 10.1083/jcb.200609050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suomalainen M, Hultenby K, Garoff H. 1996. Targeting of Moloney murine leukemia virus gag precursor to the site of virus budding. J. Cell Biol. 135:1841–1852. 10.1083/jcb.135.6.1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Ingmundson A, Horner SM, Cicchetti G, Allen PG, Pypaert M, Cunningham JM, Mothes W. 2003. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 4:785–801. 10.1034/j.1600-0854.2003.00135.x [DOI] [PubMed] [Google Scholar]
- 22.Grigorov B, Arcanger F, Roingeard P, Darlix JL, Muriaux D. 2006. Assembly of infectious HIV-1 in human epithelial and T-lymphoblastic cell lines. J. Mol. Biol. 359:848–862. 10.1016/j.jmb.2006.04.017 [DOI] [PubMed] [Google Scholar]
- 23.Nydegger S, Foti M, Derdowski A, Spearman P, Thali M. 2003. HIV-1 egress is gated through late endosomal membranes. Traffic 4:902–910. 10.1046/j.1600-0854.2003.00145.x [DOI] [PubMed] [Google Scholar]
- 24.Pelchen-Matthews A, Kramer B, Marsh M. 2003. Infectious HIV-1 assembles in late endosomes in primary macrophages. J. Cell Biol. 162:443–455. 10.1083/jcb.200304008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jolly C, Welsch S, Michor S, Sattentau QJ. 2011. The regulated secretory pathway in CD4(+) T cells contributes to human immunodeficiency virus type-1 cell-to-cell spread at the virological synapse. PLoS Pathog. 7:e1002226. 10.1371/journal.ppat.1002226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Batonick M, Favre M, Boge M, Spearman P, Honing S, Thali M. 2005. Interaction of HIV-1 Gag with the clathrin-associated adaptor AP-2. Virology 342:190–200. 10.1016/j.virol.2005.08.001 [DOI] [PubMed] [Google Scholar]
- 27.Camus G, Segura-Morales C, Molle D, Lopez-Verges S, Begon-Pescia C, Cazevieille C, Schu P, Bertrand E, Berlioz-Torrent C, Basyuk E. 2007. The clathrin adaptor complex AP-1 binds HIV-1 and MLV Gag and facilitates their budding. Mol. Biol. Cell 18:3193–3203. 10.1091/mbc.E06-12-1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong XH, Li H, Derdowski A, Ding LM, Burnett A, Chen XM, Peters TR, Dermody TS, Woodruff E, Wang JJ, Spearman P. 2005. AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell 120:663–674. 10.1016/j.cell.2004.12.023 [DOI] [PubMed] [Google Scholar]
- 29.Fang Y, Wu N, Gan X, Yan WH, Morrell JC, Gould SJ. 2007. Higher-order oligomerization targets plasma membrane proteins and HIV gag to exosomes. PLoS Biol. 5(6):e158. 10.1371/journal.pbio.0050158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen BY, Fang Y, Wu N, Gould SJ. 2011. Biogenesis of the posterior pole is mediated by the exosome/microvesicle protein-sorting pathway. J. Biol. Chem. 286:44162–44176. 10.1074/jbc.M111.274803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou W, Resh MD. 1996. Differential membrane binding of the human immunodeficiency virus type 1 matrix protein. J. Virol. 70:8540–8548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murray PS, Li Z, Wang J, Tang CL, Honig B, Murray D. 2005. Retroviral matrix domains share electrostatic homology: models for membrane binding function throughout the viral life cycle. Structure 13:1521–1531. 10.1016/j.str.2005.07.010 [DOI] [PubMed] [Google Scholar]
- 33.Spearman P, Wang JJ, Vander Heyden N, Ratner L. 1994. Identification of human immunodeficiency virus type 1 Gag protein domains essential to membrane binding and particle assembly. J. Virol. 68:3232–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamard-Peron E, Muriaux D. 2011. Retroviral matrix and lipids, the intimate interaction. Retrovirology 8:15. 10.1186/1742-4690-8-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. 2004. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. U. S. A. 101:14889–14894. 10.1073/pnas.0405596101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamard-Peron E, Juillard F, Saad JS, Roy C, Roingeard P, Summers MF, Darlix JL, Picart C, Muriaux D. 2010. Targeting of murine leukemia virus gag to the plasma membrane is mediated by PI(4,5)P2/PS and a polybasic region in the matrix. J. Virol. 84:503–515. 10.1128/JVI.01134-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saad JS, Ablan SD, Ghanam RH, Kim A, Andrews K, Nagashima K, Soheilian F, Freed EO, Summers MF. 2008. Structure of the myristylated human immunodeficiency virus type 2 matrix protein and the role of phosphatidylinositol-(4,5)-bisphosphate in membrane targeting. J. Mol. Biol. 382:434–447. 10.1016/j.jmb.2008.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. 2006. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. U. S. A. 103:11364–11369. 10.1073/pnas.0602818103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soneoka Y, Kingsman SM, Kingsman AJ. 1997. Mutagenesis analysis of the murine leukemia virus matrix protein: identification of regions important for membrane localization and intracellular transport. J. Virol. 71:5549–5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Granowitz C, Goff SP. 1994. Substitution mutations affecting a small region of the Moloney murine leukemia-virus MA gag protein block assembly and release of virion particles. Virology 205:336–344. 10.1006/viro.1994.1650 [DOI] [PubMed] [Google Scholar]
- 41.Freed EO, Orenstein JM, Bucklerwhite AJ, Martin MA. 1994. Single amino acid changes in the human-immunodeficiency virus type 1 matrix protein block virus particle production. J. Virol. 68:5311–5320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ono A, Freed EO. 2004. Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J. Virol. 78:1552–1563. 10.1128/JVI.78.3.1552-1563.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ono A, Freed EO. 1999. Binding of human immunodeficiency virus type 1 Gag to membrane: role of the matrix amino terminus. J. Virol. 73:4136–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joshi A, Ablan SD, Soheilian F, Nagashima K, Freed EO. 2009. Evidence that productive human immunodeficiency virus type 1 assembly can occur in an intracellular compartment. J. Virol. 83:5375–5387. 10.1128/JVI.00109-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson DC, Huber MT. 2002. Directed egress of animal viruses promotes cell-to-cell spread. J. Virol. 76:1–8. 10.1128/JVI.76.1.1-8.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hope TJ. 2007. Bridging efficient viral infection. Nat. Cell Biol. 9:243–244. 10.1038/ncb0307-243 [DOI] [PubMed] [Google Scholar]
- 47.Sattentau Q. 2008. Avoiding the void: cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 6:815–826. 10.1038/nrmicro1972 [DOI] [PubMed] [Google Scholar]
- 48.Mothes W, Sherer NM, Jin J, Zhong P. 2010. Virus cell-to-cell transmission. J. Virol. 84:8360–8368. 10.1128/JVI.00443-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sherer NM, Lehmann MJ, Jimenez-Soto LF, Horensavitz C, Pypaert M, Mothes W. 2007. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 9:310–315. 10.1038/ncb1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sowinski S, Jolly C, Berninghausen O, Purbhoo MA, Chauveau A, Kohler K, Oddos S, Eissmann P, Brodsky FM, Hopkins C, Onfelt B, Sattentau Q, Davis DM. 2008. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 10:211–219. 10.1038/ncb1682 [DOI] [PubMed] [Google Scholar]
- 51.Jin J, Sherer NM, Heidecker G, Derse D, Mothes W. 2009. Assembly of the murine leukemia virus is directed towards sites of cell-cell contact. PLoS Biol. 7:e1000163. 10.1371/journal.pbio.1000163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sewald X, Gonzalez DG, Haberman AM, Mothes W. 2012. In vivo imaging of virological synapses. Nat. Commun. 3:1320. 10.1038/ncomms2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. 2004. HIV-1 cell to cell transfer across an env-induced, actin-dependent synapse. J. Exp. Med. 199:283–293. 10.1084/jem.20030648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen P, Hubner W, Spinelli MA, Chen BK. 2007. Predominant mode of human immunodeficiency virus transfer between T cells is mediated by sustained Env-dependent neutralization-resistant virological synapses. J. Virol. 81:12582–12595. 10.1128/JVI.00381-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jin J, Li F, Mothes W. 2011. Viral determinants of polarized assembly for the murine leukemia virus. J. Virol. 85:7672–7682. 10.1128/JVI.00409-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li F, Jin J, Herrmann C, Mothes W. 2013. Basic residues in the matrix domain and multimerization target murine leukemia virus Gag to the virological synapse. J. Virol. 87:7113–7126. 10.1128/JVI.03263-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Browne EP. 2011. Toll-like receptor 7 controls the anti-retroviral germinal center response. PLoS Pathog. 7:e1002293. 10.1371/journal.ppat.1002293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fais S, Malorni W. 2003. Leukocyte uropod formation and membrane/cytoskeleton linkage in immune interactions. J. Leukoc. Biol. 73:556–563. 10.1189/jlb.1102568 [DOI] [PubMed] [Google Scholar]
- 59.Sánchez-Madrid F, Serrador JM. 2009. Bringing up the rear: defining the roles of the uropod. Nat. Rev. Mol. Cell Biol. 10:353–359. 10.1038/nrm2680 [DOI] [PubMed] [Google Scholar]
- 60.Gómez-Moutón C, Manes S. 2007. Establishment and maintenance of cell polarity during leukocyte chemotaxis. Cell Adh. Migr. 1:69–76. 10.4161/cam.1.2.4547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krummel MF, Macara I. 2006. Maintenance and modulation of T cell polarity. Nat. Immunol. 7:1143–1149. 10.1038/ni1404 [DOI] [PubMed] [Google Scholar]
- 62.Alonso-Lebrero JL, Serrador JM, Dominguez-Jimenez C, Barreiro O, Luque A, del Pozo MA, Snapp K, Kansas G, Schwartz-Albiez R, Furthmayr H, Lozano F, Sanchez-Madrid F. 2000. Polarization and interaction of adhesion molecules P-selectin glycoprotein ligand 1 and intercellular adhesion molecule 3 with moesin and ezrin in myeloid cells. Blood 95:2413–2419 http://bloodjournal.hematologylibrary.org/content/95/7/2413.long?sso-checked=1 [PubMed] [Google Scholar]
- 63.Itoh S, Susuki C, Takeshita K, Nagata K, Tsuji T. 2007. Redistribution of P-selectin glycoprotein ligand-1 (PSGL-1) in chemokine-treated neutrophils: a role of lipid microdomains. J. Leukoc. Biol. 81:1414–1421. 10.1189/jlb.0606398 [DOI] [PubMed] [Google Scholar]
- 64.Mañes S, Martinez AC. 2004. Cholesterol domains regulate the actin cytoskeleton at the leading edge of moving cells. Trends Cell Biol. 14:275–278. 10.1016/j.tcb.2004.04.008 [DOI] [PubMed] [Google Scholar]
- 65.Gómez-Moutón C, Abad JL, Mira E, Lacalle RA, Gallardo E, Jimenez-Baranda S, Illa I, Bernad A, Manes S, Martinez-A C. 2001. Segregation of leading-edge and uropod components into specific lipid rafts during T cell polarization. Proc. Natl. Acad. Sci. U. S. A. 98:9642–9647. 10.1073/pnas.171160298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McFarland W. 1969. Microspikes on the lymphocyte uropod. Science 163:818–820. 10.1126/science.163.3869.818 [DOI] [PubMed] [Google Scholar]
- 67.Ratner S, Sherrod WS, Lichlyter D. 1997. Microtubule retraction into the uropod and its role in T cell polarization and motility. J. Immunol. 159:1063–1067 [PubMed] [Google Scholar]
- 68.Bhalla DK, Braun J, Karnovsky MJ. 1979. Lymphocyte surface and cytoplasmic changes associated with translational motility and spontaneous capping of Ig. J. Cell Sci. 39:137–147 [DOI] [PubMed] [Google Scholar]
- 69.Llewellyn GN, Hogue IB, Grover JR, Ono A. 2010. Nucleocapsid promotes localization of HIV-1 Gag to uropods that participate in virological synapses between T cells. PLoS Pathog. 6(10):e1001167. 10.1371/journal.ppat.1001167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hatch SC, Sardo L, Chen J, Burdick R, Gorelick R, Fivash MJ, Jr, Pathak VK, Hu WS. 2013. Gag-dependent enrichment of HIV-1 RNA near the uropod membrane of polarized T cells. J. Virol. 87:11912–11915. 10.1128/JVI.01680-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schumacher HR, Stass SA, Creegan WJ, Weislow OS. 1979. Uropod-bearing lymphocytes (hand mirror cells) in a virus-induced murine lymphoma. JNCI 63:1051–1055 [PubMed] [Google Scholar]
- 72.Llewellyn GN, Grover JR, Olety B, Ono A. 2013. HIV-1 Gag associates with specific uropod-directed microdomains in a manner dependent on its MA highly basic region. J. Virol. 87:6441–6454. 10.1128/JVI.00040-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Czibener C, Sherer NM, Becker SM, Pypaert M, Hui EF, Chapman ER, Mothes W, Andrews NW. 2006. Ca2+ and synaptotagmin VII-dependent delivery of lysosomal membrane to nascent phagosomes. J. Cell Biol. 174:997–1007. 10.1083/jcb.200605004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lehmann MJ, Sherer NM, Marks CB, Pypaert M, Mothes W. 2005. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 170:317–325. 10.1083/jcb.200503059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perez-Andres M, Grosserichter-Wagener C, Teodosio C, van Dongen JJM, Orfao A, van Zelm MC. 2011. The nature of circulating CD27(+)CD43(+) B cells. J. Exp. Med. 208:2565–2566. 10.1084/jem.20112203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Accola MA, Strack B, Gottlinger HG. 2000. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1 capsid-p2 and a late assembly domain. J. Virol. 74:5395–5402. 10.1128/JVI.74.12.5395-5402.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ott DE, Coren LV, Chertova EN, Gagliardi TD, Nagashima K, Sowder RC, Poon DTK, Gorelick RJ. 2003. Elimination of protease activity restores efficient virion production to a human immunodeficiency virus type 1 nucleocapsid deletion mutant. J. Virol. 77:5547–5556. 10.1128/JVI.77.10.5547-5556.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muriaux D, Costes S, Nagashima K, Mirro J, Cho E, Lockett S, Rein A. 2004. Role of murine leukemia virus nucleocapsid protein in virus assembly. J. Virol. 78:12378–12385. 10.1128/JVI.78.22.12378-12385.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]