

ABSTRACT
Bluetongue virus (BTV) is a double-stranded RNA (dsRNA) virus that causes an economically important disease in ruminants. BTV infection is a strong inducer of type I interferon (IFN-I) in multiple cell types. It has been shown recently that BTV and, more specifically, the nonstructural protein NS3 of BTV are able to modulate the IFN-I synthesis pathway. However, nothing is known about the ability of BTV to counteract IFN-I signaling. Here, we investigated the effect of BTV on the IFN-I response pathway and, more particularly, the Janus tyrosine kinase (JAK)/signal transducer and activator of transcription protein (STAT) signaling pathway. We found that BTV infection triggered the expression of IFN-stimulated genes (ISGs) in A549 cells. However, when BTV-infected cells were stimulated with external IFN-I, we showed that activation of the IFN-stimulated response element (ISRE) promoter and expression of ISGs were inhibited. We found that this inhibition involved two different mechanisms that were dependent on the time of infection. After overnight infection, BTV blocked specifically the phosphorylation and nuclear translocation of STAT1. This inhibition correlated with the redistribution of STAT1 in regions adjacent to the nucleus. At a later time point of infection, BTV was found to interfere with the activation of other key components of the JAK/STAT pathway and to induce the downregulation of JAK1 and TYK2 protein expression. Overall, our study indicates for the first time that BTV is able to interfere with the JAK/STAT pathway to modulate the IFN-I response.
IMPORTANCE Bluetongue virus (BTV) causes a severe disease in ruminants and has an important impact on the livestock economy in areas of endemicity such as Africa. The emergence of strains, such as serotype 8 in Europe in 2006, can lead to important economic losses due to commercial restrictions and prophylactic measures. It has been known for many years that BTV is a strong inducer of type I interferon (IFN-I) in vitro and in vivo in multiple cell types. However, the ability of BTV to interact with the IFN-I system remains unclear. Here, we report that BTV is able to modulate the IFN-I response by interfering with the Janus tyrosine kinase (JAK)/signal transducer and activator of transcription protein (STAT) signaling pathway. These findings contribute to knowledge of how BTV infection interferes with the host's innate immune response and becomes pathogenic. This will also be important for the design of efficacious vaccine candidates.
INTRODUCTION
Bluetongue (BT) is a disease affecting ruminants that is caused by bluetongue virus (BTV), a pathogen belonging to the Orbivirus genus of the Reoviridae family (1–3). The BTV genome is composed of 10 segments of double-stranded RNA (dsRNA) that encode seven structural (VP1 to VP7) and five nonstructural (NS1 to NS4 and NS3A) proteins (4–6). Worldwide, 26 serotypes (BTV-1 to BTV-26) have been identified (7). BTV is transmitted by blood-feeding midges of the genus Culicoides and infects wild and domestic ruminants (8, 9). Sheep are more sensitive than cattle to the disease, with the exception of serotype 8 of BTV (BTV-8) that is able to cause disease and mortality in cattle (3, 10). BTV is endemic in many parts of the world but emerged in Europe recently (1, 11). Since 1998, several BTV serotypes (serotypes 1, 2, 4, 9, and 16) have been detected in the Mediterranean basin or in Northern Europe (serotypes 6, 8, and 11) (1, 12). Surprisingly, BTV-8 emerged in Belgium and the Netherlands in 2006 and spread rapidly to Central and Western European countries, causing significant economical losses due to vaccination campaigns and exportation bans (3, 13).
The innate immune response is the first line of defense against viral infections. This antiviral response is activated upon recognition of pathogen-associated molecular patterns (PAMPs) by host pattern recognition receptors (PRRs) and results in the production of type I interferon (IFN-I) and other proinflammatory cytokines that help to control the infection (14–18). For RNA viruses, the two major PAMPs are dsRNAs and single-stranded RNAs (ssRNAs) present in viral genomes or generated during viral replication. PRRs include Toll-like receptors (TLRs) and other sensors such as members of the retinoic acid-inducible gene I (RIG-I)-like receptor (RLR) family (18, 19). Activation of these receptors triggers different signaling cascades that lead to the production of IFN-I, which is composed mainly of IFN-α and -β. Secretion of these cytokines triggers the Janus tyrosine kinase (JAK)/signal transducer and activator of transcription protein (STAT) signaling pathway in infected and neighboring uninfected cells. This activation starts with the binding of IFN-I to the cell surface IFN-α/β receptor (IFNAR) and leads to the phosphorylation of tyrosine kinase 2 (TYK2) and JAK1 (20–22). These activated kinases then phosphorylate tyrosine residues located in the cytoplasmic domain of the IFNAR subunits (22–26). This leads to the recruitment of STAT1 and STAT2 and their phosphorylation by the JAK kinases (22, 27). Activated STAT1 and STAT2 form heterodimers that are released from the IFNAR complex and associate in the cytoplasm with IFN response factor 9 (IRF9) to form the transcription factor IFN-stimulated gene (ISG) factor 3 (ISGF3). This complex then migrates to the nucleus, where it binds to specific promoter elements called IFN-stimulated response elements (ISREs). This leads to the expression of more than 380 ISG-encoded proteins (28) that contribute to the establishment of a rapid and robust antiviral state within the cell via the modulation of cellular processes involved in the innate and adaptive immune system, cellular proliferation, and survival.
It has been known for many years that BTV is a strong inducer of IFN-I in many in vivo and in vitro models from various different tissues and host species (29–35). Recently, our group has shown that the RLR pathway controls both the sensing and the antiviral response to BTV in nonhematopoietic target cells (36). In contrast, it has been shown that BTV activates the IFN-I signaling pathway in plasmacytoid dendritic cells (pDCs) via the MyD88 adaptor, independently of TLR7 and TLR8 (TLR7/8), and via a mechanism implicating the dsRNA-activated protein kinase (PKR) and Jun N-terminal protein kinase (JNK) (37). BTV is then able to induce the production of IFN-I through different pathways, depending on the type of cells involved. It is well known that most viruses are able to counteract the IFN system in order to establish an infection. Our group has shown recently that BTV is able to modulate the IFN-I production pathway (38). This inhibition depends on the viral protein NS3 that interferes with the IFN-I synthesis pathway downstream of RIG-I and upstream of TBK1/IKKε activation (38). However, nothing is known about the ability of BTV to modulate the antiviral response triggered by the secretion of IFN-I. In this study, we aimed to study the effect of BTV infection on the IFN-I response pathway to determine whether BTV has also evolved strategies to counteract this response. We show that BTV is able to modulate the IFN-I response in nonhematopoietic cells and that BTV interferes with STAT1 phosphorylation and nuclear translocation upon IFN-I treatment. Our results suggest that this modulation involves different mechanisms that are dependent on the time of infection. After overnight infection, BTV induced the redistribution of STAT1 in perinuclear areas, and at later time points after infection, the virus affected the expression of TYK2 and JAK1.
MATERIALS AND METHODS
Cells.
Human alveolar epithelial A549 cells, HeLa, 293T, and Vero cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal calf serum (FCS), 1% nonessential amino acids (NEAA) (A549 and Vero cells only), 1% pyruvate, and 100 IU/ml penicillin and 100 μg/ml streptomycin (P-S). BHK-21 cells were grown in minimal essential medium (MEM) supplemented with 10% FCS, 1% pyruvate, 1% NEAA, and P-S. Cells were maintained at 37°C in 95% air–5% CO2.
Reagents and antibodies.
Recombinant human IFN-α 2B and IFN-β 1a were purchased from PBL Interferon Source. Mouse monoclonal antibody against VP5 of African horse sickness virus (that cross-reacts with VP5 of several BTV serotypes including BTV-1 and BTV-8) and VP7 of BTV-2 were from Ingenasa (Madrid, Spain) and IdVet (Montpellier, France), respectively. Rabbit polyclonal antibodies against NS3, VP7, and NS1 were kindly provided by Frederick Arnaud (39). A mouse anti-actin monoclonal antibody (clone AC-40) was from Sigma-Aldrich. Polyclonal antibodies against STAT1 (catalog number 06-501), phospho-STAT1 (Tyr701) (catalog number 07-307), and phospho-STAT2 (Tyr689) (catalog number 07-224) were from Millipore. Rabbit antibodies against JAK1 (6G4; catalog number 3344) and TYK2 (catalog number 9312) were from Cell Signaling Technology. A rabbit polyclonal antibody against STAT2 (N-17; catalog number sc-839) was from Santa Cruz Biotechnology. A rabbit polyclonal antibody against phospho-TYK2 (pTyr1054/1055; catalog number PA5-17898) was from Pierce Biotechnology. A mouse monoclonal antibody against RIG-I (clone Alme-1) and a rabbit anti-MDA5 (clone AT113) polyclonal antibody were from Alexis Biochemicals.
Viral infections.
Wild-type field strains of BTV serotypes 4 and 8 (BTV-4 and BTV-8) are from the National Reference Laboratory collection (ANSES, Maisons-Alfort, France) and were isolated in France, in Corsica (2003) and in the Ardennes (2006), respectively. The live vaccine strain of BTV-4 (BTV-4 VAC) used in the field in Corsica (in 2004 and 2005) was derived from a wild virus isolated in South Africa and passaged 50 to 60 times on BHK-21 cells. BTV stocks were prepared as described previously (36). Fifty percent tissue culture infectious dose (TCID50) values were estimated by endpoint titration on BHK-21 cells (method of Spearman-Karber) using a previously described protocol (30). Infections were performed on subconfluent cells as described previously (36). More than 60 and 90% of A549 cells were infected when 0.05 and 0.1 TCID50/cell of BTV-8 were used to infect the cells, respectively (data not shown). Culture supernatant from noninfected BHK-21 cells was used as an inoculum for mock-infected cells. Inactivated virus was prepared by exposing live virus to 254-nm UV; inactivation was completed in 20 min at a UV dose of 2.3 J/cm2.
Immunoblot analysis.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris-HCl, pH 8.8, 50 mM NaCl, 0.5% Nonidet P-40, and 0.1% sodium dodecyl sulfate supplemented with cocktails of protease and phosphatase inhibitors according to the manufacturer's instructions [Roche Molecular Biochemicals]). Insoluble material was centrifuged at 16,000 × g for 20 min at 4°C and discarded. Total protein concentration of the soluble fraction was determined by a Micro bicinchoninic acid (BCA) assay (Interchim). An equal amount of protein extract was reduced by heating in the presence of β-mercaptoethanol and resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer to nitrocellulose membrane (Hybond-ECL; Amersham). Membranes were blocked with phosphate-buffered saline (PBS) containing 5% dry milk and 0.05% Tween 20. The membrane was then incubated with the required dilution of specific antibodies. Bound primary antibodies were detected using horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies (Dako) and an enhanced luminol-based chemiluminescent detection system.
Reporter gene assay.
293T cells were seeded in 24-well plates and mock infected or infected with live BTV-8 or UV-inactivated BTV-8. Six hours later, cells were transfected with 1 μl of JetPRIME transfection reagent (Polyplus Transfection SA) and 100 ng of firefly luciferase ISRE reporter plasmid (pISRE-Luc) or 100 ng of Renilla luciferase cytomegalovirus (CMV) reporter plasmid (pCMV-Luc) and 50 ng of control vector (pRSV–β-Gal; RSV is Rous sarcoma virus and β-Gal is β-galactosidase). Eighteen hours later, the supernatant was removed and replaced with fresh complete medium containing 1,000 IU/ml of IFN-β. Seven hours later, cells were lysed in luciferase lysis buffer (25 mM Tris, pH 7.8, 0.8 mM MgCl2, 0.1% Triton X-100, 15% glycerol) or Renilla luciferase assay lysis buffer (Promega). Firefly and Renilla luciferase activities were determined using a Bright-Glo luciferase assay system (Promega) and a Renilla luciferase assay system (Promega), respectively. β-Galactosidase activity was determined by mixing 50 μl of cell lysate with 50 μl of β-galactosidase assay buffer (200 mM Na2HPO4, pH 7.3, 1.33 mg/ml ortho-nitrophenyl-β-d-galactopyranoside [ONPG], 2 mM MgCl2, and 0.1 M β-mercaptoethanol) and measuring the optical density of the mixture at 450 nm.
Immunostaining and fluorescence microscopy.
Cells were seeded onto 12-mm-diameter coverslips and fixed with 4% paraformaldehyde in PBS. Cells were permeabilized with 0.2% Triton X-100 in PBS and incubated in blocking buffer (0.5% bovine serum albumin [BSA] in PBS). The appropriate dilution of primary antibodies was then added for 1 h at room temperature. Cells were then washed several times in PBS, and Alexa-Fluor 488 anti-mouse and Alexa-Fluor 555 anti-rabbit secondary antibodies (Molecular Probes) were used to detect bound primary antibodies. Samples were mounted in Mowiol containing 4,6-diamidine-2-phenylindole dihydrochloride (DAPI) (Sigma-Aldrich). Microscopy was carried out with an Axio observer Z1 fluorescence microscope (Zeiss), and images were acquired using the AxioVision, release 4.8, or Zen 2012 software.
Statistical analyses.
Data obtained for infected treated samples were compared with those of mock-infected treated samples using an unpaired t test. Differences were considered to be significant if the P value was <0.01.
RESULTS
IFN-I prevents BTV infection.
In order to better understand the importance of IFN-I in the control of BTV infection, we first determined whether BTV infection is sensible to the antiviral effect of IFN-I. Human alveolar epithelial A549 cells were used as this cell line is a good model to study the IFN-I system upon BTV infection (36). A549 cells were treated with IFN-β for 5 h prior to infection with a strain of BTV-8 isolated in Ardennes, France, in 2006. As shown in Fig. 1A, pretreatment with IFN-β led to a dramatic decrease in the expression of the structural viral proteins VP5 and VP7 and the nonstructural proteins NS1 and NS3. The same results were obtained with another cell line (HeLa) and with another serotype of BTV (BTV-4 isolated in Corsica, France, in 2003) (data not shown). This result suggests that IFN-I can efficiently prevent BTV infection.
FIG 1.

BTV is sensitive to IFN-I and inhibits the IFN-I response. (A) Effect of IFN-I on the expression of BTV proteins. A549 cells were treated for 5 h with IFN-β (250 and 1,000 IU/ml) and infected with 0.05 TCID50 of BTV-8/cell for 24 h. Cells were then lysed in RIPA buffer and analyzed by Western blotting. BTV VP7, VP5, NS1, and NS3 expression was detected to assess viral infection, and β-actin was detected as an internal control. (B and C) A549 (B) and Vero (C) cells were infected with 0.02 to 0.1 TCID50 of BTV-8/cell for 18 h and left untreated (NT, not treated) or treated for 6 h with IFN-β (250 IU/ml). Cell lysates were extracted and used for detection of MDA5 and RIG-I. BTV NS3 expression was detected as a control for viral infection, and β-actin served as an internal control. Note that double bands were detected with the antibody raised against BTV NS3, as shown previously (39). These bands correspond to NS3 (higher band) and NS3A (lower band), a truncated form of NS3 that is synthesized from a second in-frame translation initiation codon within the BTV segment 10. (D and E) 293T cells were mock infected, infected with 0.001 or 0.01 TCID50 of live BTV-8/cell, or infected with UV-inactivated BTV-8 (BT8-UV) using the same volume of inoculum as cells infected with 0.01 TCID50 of live BTV-8/cell. Six hours later, cells were transfected with an ISRE reporter plasmid (pISRE-Luc) (D) or a CMV reporter plasmid (pCMV-Luc) (E) and a control vector (pRSV–β-Gal) to normalize for transfection efficiency. Eighteen h later, cells were left untreated (NT) or treated with IFN-β (1,000 IU/ml) for 7 h and lysed to determine β-galactosidase and luciferase activity. Mean ratios between luciferase and β-galactosidase activities of triplicate samples (± standard deviations) are presented. Results are representative of one experiment and were reproduced in two (CMV) or three (ISRE) independent experiments. *, P < 0.01; **, P < 0.001, compared to mock-infected cells for treated samples (unpaired t tests).
BTV is able to modulate the IFN-I response.
As IFN-I pretreatment has an antiviral effect on BTV infection, we then wanted to determine whether BTV is able to counteract the IFN-I response in order to promote an infection. A549 cells were infected with BTV-8 for 18 h, and the cells were treated with IFN-β for 6 h. The expression of two ISGs, MDA5 and RIG-I (40–42), was then analyzed by immunoblotting. As shown in Fig. 1B, MDA-5 and RIG-I protein levels were higher in BTV-8-infected cells than in mock-infected cells prior to IFN-β treatment. This result is in accordance with our previous study showing that BTV induces IFN-I synthesis in A549 cells and the expression of ISGs (36). However, after stimulation with IFN-β, the levels of MDA5 and RIG-I in BTV-infected cells were lower or equivalent to the ones detected in mock-infected cells. To distinguish the effect of IFN-I synthesized during infection from a putative ability of BTV to modulate the IFN-I response, we performed the same experiment in Vero cells in which the IFN-β gene is defective. As expected in this cell line, the expression levels of MDA-5 and RIG-I in BTV-infected cells were similar to those in mock-infected cells prior to IFN-β treatment (Fig. 1C). After stimulation with IFN-β, an increase in MDA5 and RIG-I protein levels was observed in mock-infected cells. However, this increase was visibly impaired in BTV-infected cells, suggesting that BTV infection interferes with the IFN-I response. To confirm this result, we performed a luciferase reporter assay. 293T cells were used in this assay to achieve high transfection efficiency. Mock-infected cells or cells infected with UV-inactivated BTV-8 or live BTV-8 were transfected with an ISRE reporter plasmid (pISRE-Luc) and a control vector (pRSV–β-Gal) to normalize for transfection efficiency and treated 18 h later with IFN-β for 7 h. As shown in Fig. 1D, stimulation with IFN-β activated the ISRE promoter in mock-infected cells, whereas this activation was significantly reduced in cells infected with BTV-8. Infection with UV-inactivated BTV-8 did not affect ISRE promoter activation significantly, suggesting that replication is required for this inhibition. To ensure that the inhibition of ISRE promoter activation observed in BTV-infected cells was specific, we performed the same assay using a CMV reporter plasmid (pCMV-Luc). As shown in Fig. 1E, no inhibition of CMV promoter activation was recorded in BTV-infected cells in comparison to mock-infected cells in untreated and treated cells. Thus, this result strongly suggests that BTV inhibits specifically ISRE promoter activity and that BTV is able to modulate the IFN-I response.
BTV inhibits the JAK/STAT pathway.
To understand better the mechanisms involved in the modulation of the IFN-I response mediated by BTV, we then assessed the ability of BTV to interfere with the JAK/STAT pathway. First, we wanted to determine whether STAT1 nuclear translocation was affected in BTV-infected cells after IFN-I treatment. As shown in Fig. 2, treatment of mock-infected cells with IFN-β led to the translocation of STAT1 into the nucleus, as expected. However, STAT1 distribution remained mostly unchanged when BTV-infected cells were stimulated under the same condition. This result indicates that BTV interferes with STAT1 nuclear translocation. To understand better at which step of the JAK/STAT pathway BTV is acting, we then assessed the phosphorylation status of STAT1 in mock- versus BTV-infected cells after IFN-β treatment. The level of phosphorylated STAT1 generated upon IFN-β treatment was greatly impaired in A549 cells infected with BTV-8 in comparison to mock-infected cells (Fig. 3A). This inhibition was not due to a decrease in the level of STAT1 as bands of similar intensities were obtained when an antibody against total STAT1 was used. The same result was observed in HeLa cells (Fig. 3B). STAT1 phosphorylation was also impaired in BTV-infected Vero cells (Fig. 3C), showing that this inhibition is not the consequence of BTV-induced IFN-I secretion that could have rendered the cells resistant to further stimulation with IFN-I. Moreover, BTV was also able to interfere with STAT1 phosphorylation in cells infected with a wild-type strain of BTV-4 (Fig. 3D), suggesting that the modulation of the IFN-I response by BTV is not serotype dependent.
FIG 2.

BTV infection interferes with STAT1 translocation. A549 cells were mock infected or infected with 0.05 TCID50 of BTV-8/cell for 18 h and left untreated (NT) or treated with IFN-β (1,000 IU/ml) for 30 min. Cells were then washed, fixed, and stained with primary antibodies specific for VP7 and STAT1, followed by fluorescent dye-conjugated secondary antibodies. Intracellular localization of DAPI-stained nuclei (blue), VP7 (green), and STAT1 (red) was visualized by immunofluorescence microscopy (magnification, ×20). Scale bar, 50 μm.
FIG 3.

BTV interferes with STAT1 phosphorylation. (A to C) A549 (A), HeLa (B), or Vero (C) cells were mock infected or infected with 0.01 to 0.1 TCID50 of BTV-8/cell for 18 to 20 h and then left untreated (NT) or treated with IFN-α or IFN-β (1,000 IU/ml) for 30 min. (D) HeLa cells were infected with 0.05 TCID50 of BTV-4/cell for 16 h and then left untreated (not treated, NT) or treated with IFN-β (1,000 IU/ml) for 30 min. For experiments shown in all panels, cells lysates were extracted and used for detection of total STAT1 and phospho-STAT1 (p-STAT1). Note the presence of double bands corresponding to the alpha form (around 91 kDa) and the beta form (around 84 kDa) of STAT1. BTV VP5 or NS3 expression was detected as a control for viral infection, and β-actin served as an internal control.
BTV replication is required to inhibit the JAK/STAT pathway.
As we showed that a wild-type but not a UV-inactivated strain of BTV-8 is able to inhibit ISRE promoter activation (Fig. 1D), we wanted to confirm that BTV replication is required to modulate the JAK/STAT pathway. A549 cells were infected with a wild-type or UV-inactivated BTV-8 strain, and STAT1 phosphorylation was analyzed by immunoblotting after treatment with IFN-β. As shown in Fig. 4A, UV inactivation led to complete inhibition of the expression of the viral protein NS3. The levels of phosphorylated STAT1 were comparable in mock-infected cells and cells infected with UV-inactivated BTV-8 after stimulation, whereas the phosphorylated STAT1 level was lower in cells infected with live BTV-8. Moreover, inhibition of STAT1 phosphorylation by BTV occurred after 24 h infection but not after 3 or 6 h (Fig. 4B). Interestingly, it was shown in a previous study that BTV RNA can be detected from 9 to 15 h postinfection in infected A549 cells (36). Overall, these results strongly suggest that BTV replication and/or viral protein synthesis is required for the inhibition of the JAK/STAT pathway by BTV.
FIG 4.

BTV replication is required for the inhibition of STAT1 phosphorylation. (A) A549 cells were mock infected, infected with 0.1 TCID50/cell of BTV-8 cell, or infected with UV-inactivated BTV-8 (BT8-UV) using the same volume of inoculum as cells infected with 0.1 TCID50 of live BTV-8/cell. Seventeen hour later, cells were left untreated (NT) or treated with IFN-β (1,000 IU/ml) for 30 min. (B) A549 cells were mock infected or infected with 0.1 TCID50/cell of BTV-8 for 3 to 24 h and left untreated (NT) or treated with IFN-β (1,000 IU/ml) for 30 min. For experiments shown in both panels, cells lysates were extracted and used for detection of phospho-STAT1. BTV NS3 expression was detected as a control for viral infection, and β-actin served as an internal control. Note the presence of multiple bands detected with the NS3 antibody corresponding to NS3, NS3a, and their glycosylated forms.
BTV interferes specifically with STAT1 phosphorylation.
In order to determine at which step of the JAK/STAT pathway BTV is acting, we then looked at the expression level and activation of key components of this pathway that are activated upstream of STAT1 phosphorylation. A549 and Vero cells were infected with BTV-8 for 17 h, treated with IFN-β for 15 min, and lysed for analysis by immunoblotting. As shown in Fig. 5, upon IFN-β treatment, similar levels of phosphorylated TYK2 and STAT2 were detected in mock- and BTV-infected A549 and Vero cells. Unfortunately, we could not obtain clear immunoblots for phosphorylated JAK1 although several antibodies were used (data not shown). In contrast, STAT1 phosphorylation was reduced in BTV-infected A549 and Vero cells after stimulation. Thus, these results suggest that BTV interferes specifically with the activation of STAT1 but not with other components of the JAK/STAT pathway after overnight infection.
FIG 5.
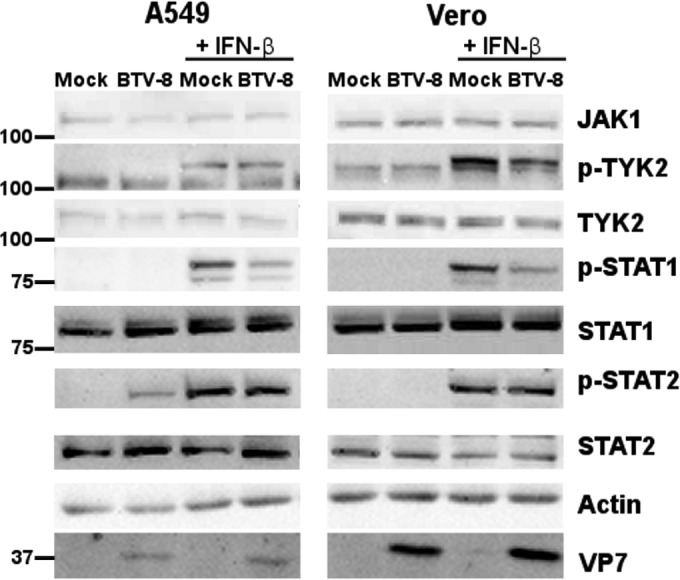
BTV interferes specifically with the phosphorylation of STAT1. A549 cells and Vero cells were mock infected or infected with 0.1 TCID50/cell of BTV-8 for 17 h and treated with IFN-β (1,000 IU/ml) for 15 min. Cells lysates were extracted and used for detection of total JAK1, phosphorylated TYK2 (p-TYK2), total TYK2, p-STAT1, total STAT1, phosphorylated STAT2 (p-STAT2), and total STAT2. BTV VP7 was detected as a control for viral infection, and β-actin served as an internal control.
BTV infection alters the distribution of STAT1.
The next step of our study was to determine the mechanism responsible for the inhibition of STAT1 phosphorylation by BTV. First, we investigated whether BTV is able to interfere with the distribution of STAT1. Cells were infected for 18 h with BTV, and STAT1 localization was assessed after immunolabeling under a fluorescence microscope at high magnification. In untreated uninfected and mock-infected cells (Fig. 6A and data not shown), STAT1 was distributed throughout the cell. In contrast, we observed that a pool of STAT1 accumulated in a region adjacent to the nucleus in untreated BTV-infected cells. This observation was made not only in cells infected with BTV-8 but also in cells infected with a wild-type and a vaccine strain of BTV-4, suggesting that this phenomenon is conserved among BTV strains. Upon treatment with IFN-β (Fig. 6B), STAT1 disappeared from the cytoplasm and concentrated in the nucleus in mock-infected cells. In contrast, in cells infected with BTV-8 or a wild-type or vaccine strain of BTV-4, STAT1 distribution remained unchanged upon IFN-I treatment, and an accumulation of STAT1 was detected in regions adjacent to the nucleus, as observed in untreated cells. These results suggest that BTV induces the redistribution of a pool of STAT1 in perinuclear areas, thus interfering with STAT1 activation and nuclear translocation upon IFN-I treatment.
FIG 6.

BTV interferes with STAT1 distribution. A549 cells were mock infected or infected with 0.05 TCID50/cell of BTV-8, BTV-4, or a vaccine strain of BTV-4 (BTV-4 VAC) for 18 h and left untreated (A) or treated with IFN-β (1,000 IU/ml) for 30 min (B). Cells were then washed, fixed, and stained with primary antibodies specific for VP7 and STAT1, followed by fluorescent dye-conjugated secondary antibodies. Intracellular localization of DAPI-stained nuclei (blue), STAT1 (red), and VP7 (green) was visualized by immunofluorescence microscopy (magnification, ×63). Examples of STAT1 accumulation in perinuclear regions are indicated by white arrows. Scale bar, 20 μm.
BTV alters the expression of specific components of the JAK/STAT pathway late during infection.
To determine whether the inhibition of STAT1 activation by BTV is maintained during infection, we infected A549 and Vero cells with BTV-8 for 42 h, treated the cells with IFN-β, and looked at the activation of different components of the JAK/STAT pathway (Fig. 7A). Phosphorylation of TYK-2, STAT1, and STAT2 was clearly detected in mock-infected cells upon IFN-β stimulation, as expected. However, in contrast to the results obtained after overnight infection (Fig. 5), the levels of phosphorylated STAT1 and also TYK2 and STAT2 were reduced or undetectable in infected cells in comparison to mock-infected cells (Fig. 7A). A diminution of total JAK1 and TYK2 protein levels was also detected in A549 and Vero cells after 42 h of infection. However, total STAT1 and STAT2 protein expression increased in A549 cells or remained constant in Vero cells in comparison to levels in mock-infected cells. As Vero cells do not produce IFN-I, these results suggest that the downregulation of TYK2 and JAK1 protein levels is not a consequence of BTV-induced IFN-I secretion in contrast to the upregulation of STAT1 and STAT2 observed in A549 cells. Interestingly, we noticed that the inhibition of STAT1 phosphorylation was more complete after 42 h of infection (Fig. 7A) than after overnight infection (Fig. 5). This result suggests that the inhibition of STAT1 activation by BTV is more efficient at 42 h postinfection than at an earlier time point (overnight infection). To confirm that BTV infection affects JAK1 and TYK2 protein levels, their expression was analyzed in A549 cells infected with BTV-8 for different times (Fig. 7B). A clear decrease in TYK2 and JAK1 protein expression was detected in BTV-infected cells after 24 h of infection and was maintained until 48 h of infection. In contrast, RIG-I, STAT1, and STAT2 protein levels increased in BTV-infected cells in comparison to levels in mock-infected cells. This increase was most likely a consequence of the secretion of IFN-I in A549 cells (36) as this cytokine upregulates the expression of RIG-I, STAT1, and STAT2 (40, 43). Overall, these results show that BTV infection impairs the activation of STAT1 and other components of the JAK/STAT pathway at a later time during infection (42 h postinfection) and affects the level of expression of JAK1 and TYK2.
FIG 7.
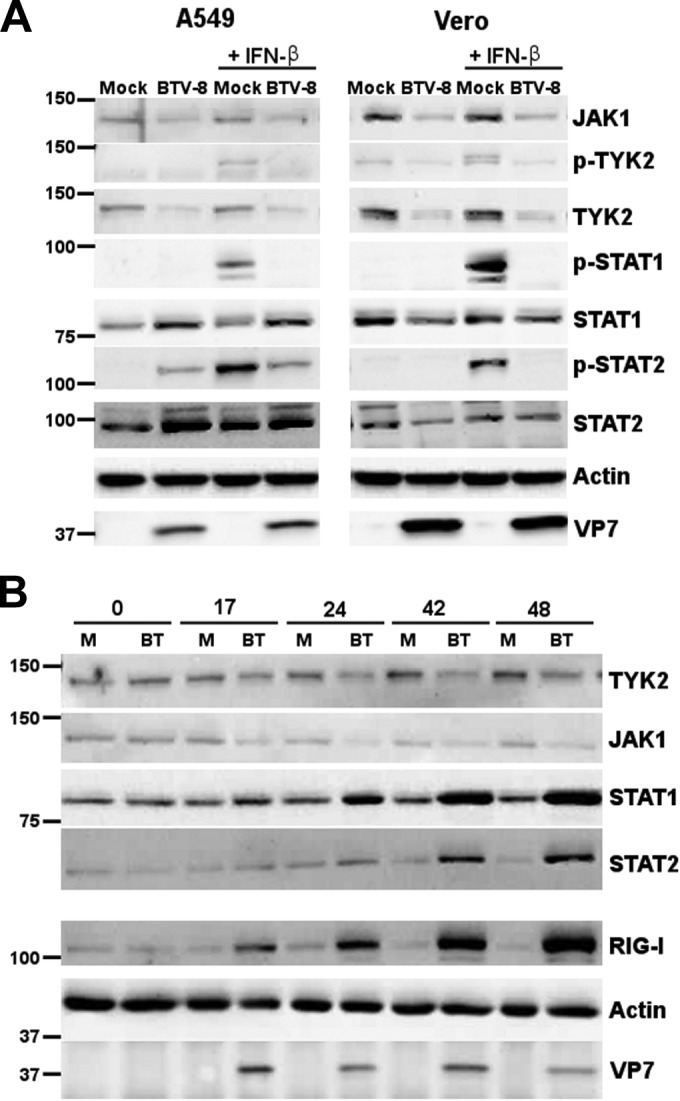
BTV downregulates TYK2 and JAK1 protein levels. (A) A549 cells and Vero cells were mock infected or infected for 42 h with 0.1 or 0.05 TCID50/cell of BTV-8, respectively, and treated with IFN-β (1,000 IU/ml) for 15 min. Cells lysates were extracted and used for detection of total JAK1, p-TYK2, total TYK2, p-STAT1, total STAT1, p-STAT2, and total STAT2. BTV VP7 was detected as a control for viral infection, and β-actin served as an internal control. (B) A549 cells were mock infected (M) or infected with 0.05 TCID50/cell of BTV-8 (BT) for 17 to 48 h. Cells lysates were extracted and used for detection of total TYK2, JAK1, STAT1, STAT2, and RIG-I. BTV VP7 was detected as a control for viral infection, and β-actin was used as an internal control.
DISCUSSION
Many viruses have evolved multiple strategies to counteract host antiviral responses and successfully evade and replicate in host cells. In a previous study, we have shown that BTV modulates IFN-I synthesis in epithelial cells and that this inhibition is mediated by the viral protein NS3 that interferes with the IFN-I synthesis pathway downstream of RIG-I and upstream of TBK1/IKKε activation (38). In the present study, we report novel findings showing that BTV has evolved additional strategies to interfere with the IFN-I system by modulating IFN-I signaling and the JAK/STAT pathway in epithelial cells. We found that BTV interferes specifically with STAT1 phosphorylation after overnight infection and that this inhibition correlates with an alteration in the distribution of STAT1 in infected host cells. By interfering with the normal distribution of STAT1, BTV most likely disturbs its recruitment to phosphorylated STAT2 on the IFNAR complex and its subsequent phosphorylation upon activation of the JAK/STAT pathway. This is in accordance with our findings that, after overnight infection, phosphorylation of STAT1 but not TYK2 and STAT2 is affected by BTV infection. Many viruses are able to interfere with the JAK/STAT pathway using different strategies (44), and previous reports have shown that some viruses counteract this pathway via the alteration of STAT1 distribution. For example, a study has reported that in cells infected with human parainfluenza virus type 1 (HPIV1), STAT1 accumulates around the nucleus in granules and does not translocate to the nucleus after IFN-I treatment. It was suggested that this inhibition is mediated by the C proteins of HPIV1 as these viral proteins interact and colocalize with STAT1 in perinuclear granules (45). Other viral proteins such as the V protein of Hendra virus and Nipah virus are also able to bind and alter STAT1 protein distribution to inhibit the IFN response (46, 47). Work is now being carried out to determine the mechanisms and the viral proteins involved in the relocalization of STAT1 in BTV-infected cells. Preliminary experiments could not identify any protein encoded by BTV able to block IFN-mediated STAT1 nuclear translocation when these viral proteins (VP1 to VP7 and NS1 to NS4) were expressed individually in noninfected cells (data not shown). It is possible that a redistribution of STAT1 occurs only in the context of BTV infection or requires the contribution of two or more viral proteins.
Interestingly, we found that at a later time point after infection (42 h postinfection), BTV is able to interfere with the phosphorylation of other components of the JAK/STAT pathway such as STAT2 and TYK2. We showed that this inhibition coincides with a decrease in total JAK1 and TYK2 protein levels in A549 or Vero cells. This alteration was not induced by the production of IFN-I in infected cells and was specific to these components of the JAK/STAT pathway. BTV replication is actively ongoing at this time point in A549 cells, and these cells are able to survive several days of BTV infection (36). It is then unlikely that the results observed were caused by extreme cell damage or death. By reducing the amount of JAK1 and TYK2 present in infected cells, BTV most likely interferes with the activation of these molecules and the downstream phosphorylation of STAT1 and STAT2. We are currently investigating whether this decrease of JAK1 and TYK2 protein levels observed in BTV-infected cells occurs at the level of transcription or translation or whether BTV has an impact on the stability of these proteins. It has been reported previously that BTV induces a protein shutdown early after infection, but the underlying mechanism has not yet been characterized (48). This phenomenon could potentially explain why we observed an alteration in the levels of JAK1 and TYK2 in our study. However, the fact that this decreased expression is specific to JAK1 and TYK2 and not other components of the JAK/STAT pathway rather suggests that BTV modulates specifically the expression of these proteins to hijack host cellular responses and promote infection. Other viruses have already been shown to counteract the JAK/STAT pathway by modulating the expression of JAK1 and/or TYK2 (44). For example, adenovirus type 5 is able to downregulate the expression of JAK1 to inhibit the IFN-I response pathway (49), and human metapneumovirus inhibits this antiviral response by downregulating JAK1 and TYK2 cellular levels (50).
It is also interesting that BTV-8 induced the expression of ISGs at 24 h postinfection in A549 cells (Fig. 1B), most likely via the secretion of IFN-I. However, there was no evidence of STAT1 phosphorylation or nuclear translocation at this time point or earlier in our experiments. We have shown previously that the level of IFN-β mRNA remains very low in A549 cells between 15 and 24 h postinfection and then increases greatly between 24 and 36 h to peak at 48 h postinfection (36). It is then possible that up to 24 h postinfection, a low level of BTV-induced IFN-I activated only weakly and transiently the JAK/STAT pathway, making this phenomenon difficult to detect. Nevertheless, this activation would be sufficient to trigger the synthesis of some ISGs as BTV is not able to block STAT1 phosphorylation early after infection (before 16 h postinfection). Later during infection, when blockage of the JAK/STAT pathway by BTV is undergoing but not complete, more ISGs will be expressed at low levels and will accumulate in infected cells. This could explain why expression of the ISGs STAT1, STAT2, and RIG-I increased over time in BTV-infected A549 cells (Fig. 7B). The fact that high expression of STAT1, STAT2, and IRF9 can lead to the prolonged expression of a subset of ISGs in the absence of ongoing IFN-I signaling and phosphorylation of JAKs and STATs (51) could also explain how some ISGs are expressed even if activation of the JAK/STAT pathway is blocked. A balance most likely exists between the ability of the host to induce and respond to IFN-I and the ability of viruses to block this antiviral pathway. It is then possible that infected cells would produce more ISGs and trigger a stronger antiviral response if BTV were not able to interfere with the JAK/STAT pathway. The efficiency of this inhibition might also depend on certain conditions, such as the cell type infected and the level of IFN-I present in the extracellular compartment.
Overall, the results reported in this study suggest that BTV has evolved two distinct mechanisms occurring at different times after infection, i.e., redistribution of STAT1 and loss of JAK1 and TYK2 protein expression, to inhibit the JAK/STAT pathway. This would allow the virus to interfere more efficiently and durably with the IFN-I response, as suggested by our finding that the inhibition of STAT1 phosphorylation is more pronounced in BTV-infected cells at later time points after infection, when viral replication is fully active. Together with its ability to modulate IFN synthesis (38), BTV would then ensure a powerful inhibition of the cellular response. Several studies have suggested that IFN-I is an important factor to limit BTV replication and dissemination and control infection (52–54). By inhibiting both IFN-I synthesis and signaling pathways, BTV would then promote the establishment of an infection.
Other viruses use multiple strategies at different times during infection to counteract host antiviral responses. For example, human CMV (hCMV) has developed several mechanisms to inhibit the IFN-II response and ensure that this pathway is sufficiently blocked (55). After 6 h of infection, hCMV downregulates major histocompatibility complex (MHC) class II expression by inhibiting class II transactivator (CIITA) mRNA expression downstream of the JAK/STAT pathway (56). After 16 h of infection, the same virus is able to activate Src homology 2 domain-containing phosphatase 2 (SHP2) to inhibit IFN-II-induced STAT1 phosphorylation (55). Additionally, at 72 h postinfection, hCMV inhibits the IFN-II-stimulated response by interfering with the activation of the JAK/STAT pathway via the degradation of JAK1 (57). Moreover, by inhibiting STAT1 phosphorylation and by degrading JAK1, hCMV acts upstream in the JAK/STAT pathway and most likely affects broader host responses (55). Similarly, BTV acts upstream in the JAK/STAT pathway by inhibiting STAT1 phosphorylation. It is then possible that the virus affects other cellular responses, such as the IFN-II-mediated pathway, to modulate further the intensity of the immune response.
In conclusion, our data provide evidence that BTV inhibits the IFN-I response by targeting different steps of the JAK/STAT pathway according to the time of infection. A better understanding of these evasion strategies evolved by BTV to counteract the host cellular response will give essential insights into the pathogenesis associated with BTV infection and will help to provide better control measures against this virus.
ACKNOWLEDGMENTS
This work was funded by ANSES, INRA, ENVA, and the Agence Nationale pour la Recherche through the EMIDA ORBINET consortium (K1303206). E.C. received a Ph.D. grant from ANSES and INRA.
We thank Frederick Arnaud and Pierre-Olivier Vidalain for providing reagents and for useful discussions.
Footnotes
Published ahead of print 9 July 2014
REFERENCES
- 1.Maclachlan NJ. 2011. Bluetongue: history, global epidemiology, and pathogenesis. Prev. Vet. Med. 102:107–111. 10.1016/j.prevetmed.2011.04.005 [DOI] [PubMed] [Google Scholar]
- 2.Mertens PPC, Diprose J, Maan S, Singh KP, Attoui H, Samuel AR. 2004. Bluetongue virus replication, molecular and structural biology. Vet. Ital. 40:426–437 [PubMed] [Google Scholar]
- 3.Schwartz-Cornil I, Mertens PPC, Contreras V, Hemati B, Pascale F, Bréard E, Mellor PS, MacLachlan NJ, Zientara S. 2008. Bluetongue virus: virology, pathogenesis and immunity. Vet. Res. 39:46. 10.1051/vetres:2008023 [DOI] [PubMed] [Google Scholar]
- 4.Belhouchet M, Mohd Jaafar F, Firth AE, Grimes JM, Mertens PPC, Attoui H. 2011. Detection of a fourth orbivirus non-structural protein. PLoS One 6:e25697. 10.1371/journal.pone.0025697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ratinier M, Caporale M, Golder M, Franzoni G, Allan K, Nunes SF, Armezzani A, Bayoumy A, Rixon F, Shaw A, Palmarini M. 2011. Identification and characterization of a novel non-structural protein of bluetongue virus. PLoS Pathog. 7:e1002477. 10.1371/journal.ppat.1002477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roy P. 2005. Bluetongue virus proteins and particles and their role in virus entry, assembly, and release. Adv. Virus Res. 64:69–123. 10.1016/S0065-3527(05)64004-3 [DOI] [PubMed] [Google Scholar]
- 7.Maan S, Maan NS, Nomikou K, Veronesi E, Bachanek-Bankowska K, Belaganahalli MN, Attoui H, Mertens PPC. 2011. Complete genome characterisation of a novel 26th bluetongue virus serotype from Kuwait. PLoS One 6:e26147. 10.1371/journal.pone.0026147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mellor PS, Boorman J, Baylis M. 2000. Culicoides biting midges: their role as arbovirus vectors. Annu. Rev. Entomol. 45:307–340. 10.1146/annurev.ento.45.1.307 [DOI] [PubMed] [Google Scholar]
- 9.Tabachnick WJ. 2004. Culicoides and the global epidemiology of bluetongue virus infection. Vet. Ital. 40:144–150 [PubMed] [Google Scholar]
- 10.Elbers ARW, Backx A, Ekker HM, Van der Spek AN, Van Rijn PA. 2008. Performance of clinical signs to detect bluetongue virus serotype 8 outbreaks in cattle and sheep during the 2006-epidemic in The Netherlands. Vet. Microbiol. 129:156–162. 10.1016/j.vetmic.2007.10.034 [DOI] [PubMed] [Google Scholar]
- 11.Walton TE. 2004. The history of bluetongue and a current global overview. Vet. Ital. 40:31–38 [PubMed] [Google Scholar]
- 12.Purse BV, Mellor PS, Rogers DJ, Samuel AR, Mertens PPC, Baylis M. 2005. Climate change and the recent emergence of bluetongue in Europe. Nat. Rev. Microbiol. 3:171–181. 10.1038/nrmicro1090 [DOI] [PubMed] [Google Scholar]
- 13.Saegerman C, Mellor P, Uyttenhoef A, Hanon J-B, Kirschvink N, Haubruge E, Delcroix P, Houtain J-Y, Pourquier P, Vandenbussche F, Verheyden B, De Clercq K, Czaplicki G. 2010. The most likely time and place of introduction of BTV8 into Belgian ruminants. PLoS One 5:e9405. 10.1371/journal.pone.0009405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoneyama M, Fujita T. 2009. RNA recognition and signal transduction by RIG-I-like receptors. Immunol. Rev. 227:54–65. 10.1111/j.1600-065X.2008.00727.x [DOI] [PubMed] [Google Scholar]
- 15.Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47. 10.1099/vir.0.83391-0 [DOI] [PubMed] [Google Scholar]
- 16.Meylan E, Tschopp J, Karin M. 2006. Intracellular pattern recognition receptors in the host response. Nature 442:39–44. 10.1038/nature04946 [DOI] [PubMed] [Google Scholar]
- 17.Kawai T, Akira S. 2008. Toll-like receptor and RIG-I-like receptor signaling. Ann. N. Y. Acad. Sci. 1143:1–20. 10.1196/annals.1443.020 [DOI] [PubMed] [Google Scholar]
- 18.Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783–801. 10.1016/j.cell.2006.02.015 [DOI] [PubMed] [Google Scholar]
- 19.Beutler B, Eidenschenk C, Crozat K, Imler J-L, Takeuchi O, Hoffmann JA, Akira S. 2007. Genetic analysis of resistance to viral infection. Nat. Rev. Immunol. 7:753–766. 10.1038/nri2174 [DOI] [PubMed] [Google Scholar]
- 20.Silvennoinen O, Ihle JN, Schlessinger J, Levy DE. 1993. Interferon-induced nuclear signalling by Jak protein tyrosine kinases. Nature 366:583–585. 10.1038/366583a0 [DOI] [PubMed] [Google Scholar]
- 21.Gauzzi MC, Velazquez L, McKendry R, Mogensen KE, Fellous M, Pellegrini S. 1996. Interferon-alpha-dependent activation of Tyk2 requires phosphorylation of positive regulatory tyrosines by another kinase. J. Biol. Chem. 271:20494–20500. 10.1074/jbc.271.34.20494 [DOI] [PubMed] [Google Scholar]
- 22.Krishnan K, Yan H, Lim JT, Krolewski JJ. 1996. Dimerization of a chimeric CD4-interferon-alpha receptor reconstitutes the signaling events preceding STAT phosphorylation. Oncogene 13:125–133 [PubMed] [Google Scholar]
- 23.Platanias LC, Colamonici OR. 1992. Interferon alpha induces rapid tyrosine phosphorylation of the alpha subunit of its receptor. J. Biol. Chem. 267:24053–24057 [PubMed] [Google Scholar]
- 24.Abramovich C, Shulman LM, Ratovitski E, Harroch S, Tovey M, Eid P, Revel M. 1994. Differential tyrosine phosphorylation of the IFNAR chain of the type I interferon receptor and of an associated surface protein in response to IFN-alpha and IFN-beta. EMBO J. 13:5871–5877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Platanias LC, Uddin S, Colamonici OR. 1994. Tyrosine phosphorylation of the alpha and beta subunits of the type I interferon receptor. Interferon-beta selectively induces tyrosine phosphorylation of an alpha subunit-associated protein. J. Biol. Chem. 269:17761–17764 [PubMed] [Google Scholar]
- 26.Zhao W, Lee C, Piganis R, Plumlee C, De Weerd N, Hertzog PJ, Schindler C. 2008. A conserved IFN-alpha receptor tyrosine motif directs the biological response to type I IFNs. J. Immunol. 180:5483–5489. 10.4049/jimmunol.180.8.5483 [DOI] [PubMed] [Google Scholar]
- 27.Yan H, Krishnan K, Greenlund AC, Gupta S, Lim JT, Schreiber RD, Schindler CW, Krolewski JJ. 1996. Phosphorylated interferon-alpha receptor 1 subunit (IFNaR1) acts as a docking site for the latent form of the 113 kDa STAT2 protein. EMBO J. 15:1064–1074 [PMC free article] [PubMed] [Google Scholar]
- 28.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. 10.1038/nature09907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foster NM, Luedke AJ, Parsonson IM, Walton TE. 1991. Temporal relationships of viremia, interferon activity, and antibody responses of sheep infected with several bluetongue virus strains. Am. J. Vet. Res. 52:192–196 [PubMed] [Google Scholar]
- 30.Hemati B, Contreras V, Urien C, Bonneau M, Takamatsu H-H, Mertens PPC, Bréard E, Sailleau C, Zientara S, Schwartz-Cornil I. 2009. Bluetongue virus targets conventional dendritic cells in skin lymph. J. Virol. 83:8789–8799. 10.1128/JVI.00626-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huismans H. 1969. Bluetongue virus-induced interferon synthesis. Onderstepoort J. Vet. Res. 36:181–185 [PubMed] [Google Scholar]
- 32.Jameson P, Schoenherr CK, Grossberg SE. 1978. Bluetongue virus, an exceptionally potent interferon inducer in mice. Infect. Immun. 20:321–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacLachlan NJ, Thompson J. 1985. Bluetongue virus-induced interferon in cattle. Am. J. Vet. Res. 46:1238–1241 [PubMed] [Google Scholar]
- 34.Russell H, O'Toole DT, Bardsley K, Davis WC, Ellis JA. 1996. Comparative effects of bluetongue virus infection of ovine and bovine endothelial cells. Vet. Pathol. 33:319–331. 10.1177/030098589603300309 [DOI] [PubMed] [Google Scholar]
- 35.Vitour D, Doceul V, Ruscanu S, Chauveau E, Schwartz-Cornil I, Zientara S. 2014. Induction and control of the type I interferon pathway by Bluetongue virus. Virus Res. 182:59–70. 10.1016/j.virusres.2013.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chauveau E, Doceul V, Lara E, Adam M, Breard E, Sailleau C, Viarouge C, Desprat A, Meyer G, Schwartz-Cornil I, Ruscanu S, Charley B, Zientara S, Vitour D. 2012. Sensing and control of bluetongue virus infection in epithelial cells via RIG-I and MDA5 helicases. J. Virol. 86:11789–11799. 10.1128/JVI.00430-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruscanu S, Pascale F, Bourge M, Hemati B, Elhmouzi-Younes J, Urien C, Bonneau M, Takamatsu H, Hope J, Mertens P, Meyer G, Stewart M, Roy P, Meurs EF, Dabo S, Zientara S, Breard E, Sailleau C, Chauveau E, Vitour D, Charley B, Schwartz-Cornil I. 2012. The double-stranded RNA bluetongue virus induces type I interferon in plasmacytoid dendritic cells via a MYD88-dependent TLR7/8-independent signaling pathway. J. Virol. 86:5817–5828. 10.1128/JVI.06716-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chauveau E, Doceul V, Lara E, Breard E, Sailleau C, Vidalain P-O, Meurs EF, Dabo S, Schwartz-Cornil I, Zientara S, Vitour D. 2013. NS3 of bluetongue virus interferes with the induction of type I interferon. J. Virol. 87:8241–8246. 10.1128/JVI.00678-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shaw AE, Veronesi E, Maurin G, Ftaich N, Guiguen F, Rixon F, Ratinier M, Mertens P, Carpenter S, Palmarini M, Terzian C, Arnaud F. 2012. Drosophila melanogaster as a model organism for bluetongue virus replication and tropism. J. Virol. 86:9015–9024. 10.1128/JVI.00131-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu M, Tong JH, Mao M, Kan LX, Liu MM, Sun YW, Fu G, Jing YK, Yu L, Lepaslier D, Lanotte M, Wang ZY, Chen Z, Waxman S, Wang YX, Tan JZ, Chen SJ. 1997. Cloning of a gene (RIG-G) associated with retinoic acid-induced differentiation of acute promyelocytic leukemia cells and representing a new member of a family of interferon-stimulated genes. Proc. Natl. Acad. Sci. U. S. A. 94:7406–7411. 10.1073/pnas.94.14.7406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang D-C, Gopalkrishnan RV, Lin L, Randolph A, Valerie K, Pestka S, Fisher PB. 2004. Expression analysis and genomic characterization of human melanoma differentiation associated gene-5, mda-5: a novel type I interferon-responsive apoptosis-inducing gene. Oncogene 23:1789–1800. 10.1038/sj.onc.1207300 [DOI] [PubMed] [Google Scholar]
- 42.Kang D, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. 2002. MDA-5: an interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. U. S. A. 99:637–642. 10.1073/pnas.022637199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lehtonen A, Matikainen S, Julkunen I. 1997. Interferons up-regulate STAT1, STAT2, and IRF family transcription factor gene expression in human peripheral blood mononuclear cells and macrophages. J. Immunol. 159:794–803 [PubMed] [Google Scholar]
- 44.Versteeg GA, García-Sastre A. 2010. Viral tricks to grid-lock the type I interferon system. Curr. Opin. Microbiol. 13:508–516. 10.1016/j.mib.2010.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schomacker H, Hebner RM, Boonyaratanakornkit J, Surman S, Amaro-Carambot E, Collins PL, Schmidt AC. 2012. The C proteins of human parainfluenza virus type 1 block IFN signaling by binding and retaining Stat1 in perinuclear aggregates at the late endosome. PLoS One 7:e28382. 10.1371/journal.pone.0028382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodriguez JJ, Parisien J-P, Horvath CM. 2002. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J. Virol. 76:11476–11483. 10.1128/JVI.76.22.11476-11483.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodriguez JJ, Wang L-F, Horvath CM. 2003. Hendra virus V protein inhibits interferon signaling by preventing STAT1 and STAT2 nuclear accumulation. J. Virol. 77:11842–11845. 10.1128/JVI.77.21.11842-11845.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mertens PPC, Brown F, Sangar DV. 1984. Assignment of the genome segments of bluetongue virus type 1 to the proteins which they encode. Virology 135:207–217. 10.1016/0042-6822(84)90131-4 [DOI] [PubMed] [Google Scholar]
- 49.Shi L, Ramaswamy M, Manzel LJ, Look DC. 2007. Inhibition of Jak1-dependent signal transduction in airway epithelial cells infected with adenovirus. Am. J. Respir. Cell Mol. Biol. 37:720–728. 10.1165/rcmb.2007-0158OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren J, Kolli D, Liu T, Xu R, Garofalo RP, Casola A, Bao X. 2011. Human metapneumovirus inhibits IFN-β signaling by downregulating Jak1 and Tyk2 cellular levels. PLoS One 6:e24496. 10.1371/journal.pone.0024496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, Rice CM, Jackson MW, Junk DJ, Stark GR. 2013. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 32:2751–2763. 10.1038/emboj.2013.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodríguez-Calvo T, Rojas J-M, Martín V, Sevilla N. 2014. Type I interferon limits the capacity of bluetongue virus to infect hematopoietic precursors and dendritic cells in vitro and in vivo. J. Virol. 88:859–867. 10.1128/JVI.02697-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dhanasekaran S, Vignesh AR, Raj GD, Reddy YKM, Raja A, Tirumurugaan KG. 2013. Comparative analysis of innate immune response following in vitro stimulation of sheep and goat peripheral blood mononuclear cells with bluetongue virus—serotype 23. Vet. Res. Commun. 37:319–327. 10.1007/s11259-013-9579-5 [DOI] [PubMed] [Google Scholar]
- 54.Maclachlan NJ, Henderson C, Schwartz-Cornil I, Zientara S. 2014. The immune response of ruminant livestock to bluetongue virus: from type I interferon to antibody. Virus Res. 182:71–77. 10.1016/j.virusres.2013.09.040 [DOI] [PubMed] [Google Scholar]
- 55.Baron M, Davignon J-L. 2008. Inhibition of IFN-gamma-induced STAT1 tyrosine phosphorylation by human CMV is mediated by SHP2. J. Immunol. 181:5530–5536. 10.4049/jimmunol.181.8.5530 [DOI] [PubMed] [Google Scholar]
- 56.Le Roy E, Mühlethaler-Mottet A, Davrinche C, Mach B, Davignon JL. 1999. Escape of human cytomegalovirus from HLA-DR-restricted CD4+ T-cell response is mediated by repression of gamma interferon-induced class II transactivator expression. J. Virol. 73:6582–6589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller DM, Rahill BM, Boss JM, Lairmore MD, Durbin JE, Waldman JW, Sedmak DD. 1998. Human cytomegalovirus inhibits major histocompatibility complex class II expression by disruption of the Jak/Stat pathway. J. Exp. Med. 187:675–683. 10.1084/jem.187.5.675 [DOI] [PMC free article] [PubMed] [Google Scholar]