

ABSTRACT
Retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) are essential intracellular detectors of viral RNA. They contribute to the type I interferon (IFN) response that is crucial for host defense against viral infections. Given the potent antiviral and proinflammatory activities elicited by the type I IFNs, induction of the type I IFN response is tightly regulated. Members of the tripartite motif (TRIM) family of proteins have recently emerged as key regulators of antiviral immunity. We show that TRIM13, an E3 ubiquitin ligase, is expressed in immune cells and is upregulated in bone marrow-derived macrophages upon stimulation with inducers of type I IFN. TRIM13 interacts with MDA5 and negatively regulates MDA5-mediated type I IFN production in vitro, acting upstream of IFN regulatory factor 3. We generated Trim13−/− mice and show that upon lethal challenge with encephalomyocarditis virus (EMCV), which is sensed by MDA5, Trim13−/− mice produce increased amounts of type I IFNs and survive longer than wild-type mice. Trim13−/− murine embryonic fibroblasts (MEFs) challenged with EMCV or poly(I·C) also show a significant increase in beta IFN (IFN-β) levels, but, in contrast, IFN-β responses to the RIG-I-detected Sendai virus were diminished, suggesting that TRIM13 may play a role in positively regulating RIG-I function. Together, these results demonstrate that TRIM13 regulates the type I IFN response through inhibition of MDA5 activity and that it functions nonredundantly to modulate MDA5 during EMCV infection.
IMPORTANCE The type I interferon (IFN) response is crucial for host defense against viral infections, and proper regulation of this pathway contributes to maintaining immune homeostasis. Retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) are intracellular detectors of viral RNA that induce the type I IFN response. In this study, we show that expression of the gene tripartite motif 13 (Trim13) is upregulated in response to inducers of type I IFN and that TRIM13 interacts with both MDA5 and RIG-I in vitro. Through the use of multiple in vitro and in vivo model systems, we show that TRIM13 is a negative regulator of MDA5-mediated type I IFN production and may also impact RIG-I-mediated type I IFN production by enhancing RIG-I activity. This places TRIM13 at a key junction within the viral response pathway and identifies it as one of the few known modulators of MDA5 activity.
INTRODUCTION
A crucial component of the host defense against viral infections is the activation of the type I interferon (IFN) response, which is initiated by detection of viral RNA or DNA by host pattern recognition receptors (PRRs). The cytosolic proteins retinoic acid-inducible gene I (RIG-I; encoded by Ddx58) and melanoma differentiation-associated gene 5 (MDA5; encoded by Ifih1) are essential intracellular sensors of viral RNA that discriminate between different classes of RNA viruses. For example, RIG-I is responsible for detection of Newcastle disease virus, vesicular stomatitis virus (VSV), influenza A virus (IAV), and Sendai virus (SeV), while MDA5 is important for recognition of picornaviruses, including encephalomyocarditis virus (EMCV) (1). The importance of the type I IFN pathway is underscored by the numerous viral immune evasion strategies that target and interfere with components of the pathway. Tight control of type I IFN production is crucial to the host, as is evident from studies which link dysregulation of the IFN response to autoimmune diseases, such as systemic lupus erythematosus (2). Excessive levels of type I IFN can also lead to defects, such as the loss of CD4 T cells in chronic disease states (3) and defective CD8α-positive dendritic cell (DC) development (4).
Several members of the tripartite motif (TRIM) family of proteins have emerged as central regulators of antiviral immunity. Many TRIM proteins, including TRIM5α and TRIM19 (PML), inhibit the activity of numerous viruses, including HIV and murine leukemia virus (5). In addition to direct interference with viral entry and replication, several TRIM family members are involved in regulating the type I IFN response. Interestingly, two TRIM proteins specifically target the RIG-I pathway: TRIM23 acts as a positive regulator of RIG-I and Toll-like receptor (TLR) 3 activity by polyubiquitinating nuclear factor kappa B (NF-κB) essential modulator (NEMO) (6), and TRIM25 was shown to ubiquitinate RIG-I at K63, a key modification necessary for RIG-I activation (7). In addition, TRIM44 was recently shown to interact with mitochondrial antiviral signaling (MAVS) to positively regulate type I IFN production (8). The expression of many TRIM proteins is upregulated in innate immune cells treated with type I IFNs (9, 10), indicating that additional TRIM proteins are also likely to play important roles in the immune response to viruses. Indeed, recent systematic analyses of human TRIM proteins in vitro have raised the possibility that most are positive modulators of several PRRs (11, 12). However, the in vivo functional relevance for the vast majority of TRIM proteins is yet to be established.
In addition to functioning in antiviral immunity and protein modifications, TRIM proteins are involved in a wide array of cellular processes, such as transcription, microRNA activation, cell cycle regulation, apoptosis, and oncogenesis (13, 14). The TRIM family contains over 60 proteins and is defined by N-terminal domains that are conserved in their spacing, consisting of a RING domain, one or two B-box domains, and a coiled-coil domain. The more varied C termini of TRIM proteins can be used to cluster the family into 11 subgroups on the basis of domain structure and homology (15).
A unique subgroup of TRIM proteins that contain a transmembrane domain in the C-terminal region consists of only two proteins: TRIM13 and TRIM59. TRIM13 is an E3 ubiquitin ligase with autopolyubiquitination properties. It localizes to the nuclear and endoplasmic reticulum (ER) membranes and may play a role in ER-associated degradation, a process that removes unfolded and misfolded proteins from the ER (16). Trim13 was recently shown to negatively regulate the initiation of autophagy, a process by which cells degrade their cellular components, during ER stress (17). Human TRIM13 was found to be a positive regulator of NF-κB activation and the type I IFN response in association with RIG-I in vitro (11, 12). Trim59 and Ift80 encode a joint transcript termed Ift80L, the mutation of which may be associated with some cases of Jeune syndrome, a developmental disease in humans (18). TRIM59 has also been implicated in cancer as a target of c-Myc repression and a biomarker of tumorigenesis (19, 20).
In the immune system, TRIM13, but not TRIM59, is upregulated in human macrophages after stimulation with lipopolysaccharide (LPS) and gamma IFN (IFN-γ), which activates IFN-β production in macrophages (9, 21). As multiple TRIM proteins are known to modulate antiviral responses, we sought to determine the role of TRIM13 in innate immunity and type I IFN production in vivo. Here we show that Trim13 is expressed in multiple innate immune subsets and that it is upregulated in activated bone marrow-derived macrophages (BMDMs). TRIM13 specifically inhibits MDA5-mediated activation of IFN reporters in an interferon regulatory factor 3 (IRF3)-dependent, NF-κB-independent manner. TRIM13 interacts with MDA5 and inhibits its function in vitro and in vivo. Consistent with a role for TRIM13 in the inactivation of MDA5-mediated innate immune responses, mice lacking Trim13 show increased production of type I IFNs and enhanced resistance against a lethal challenge with EMCV. Trim13−/− murine embryonic fibroblasts (MEFs) infected with EMCV or poly(I·C) also show significant increases in IFN-β production, while infection with SeV, which is detected by RIG-I, results in decreased IFN-β production. Infection of Trim13−/− mice with VSV, another virus detected by RIG-I, also shows a decrease in IFN-β production. Collectively, these results demonstrate that TRIM13 not only is a negative regulator of MDA5-mediated IFN-α/β production but also may positively regulate RIG-I function.
MATERIALS AND METHODS
Mice.
Trim13−/− mice were generated as described in Fig. 4. Mice were backcrossed to C57BL/6 mice for 8 to 10 generations. Testing for single nucleotide polymorphisms indicated that the mice were 99.9% backcrossed to C57BL/6 mice (testing was performed by The Jackson Laboratory). Mavs−/− mice were obtained from Z. Chen (22) and backcrossed to C57BL/6 mice for 10 generations. IFN-α and -β receptor 1-deficient (Ifnar1−/−) mice were originally obtained from J. Sprent (23). All mice used in these experiments were housed in a specific-pathogen-free rodent barrier facility. All animal experiments were approved by the University of Massachusetts Medical School Institutional Animal Care and Use Committee (Worcester, MA).
FIG 4.

Generation and characterization of Trim13−/− mice. (a) The Trim13-targeting construct was generated by PCR cloning from genomic DNA extracted from an embryonic stem (ES) cell line (AB2.2). A schematic of the Trim13 endogenous locus and targeting construct (not to scale) is shown. Black boxes, protein-coding exons; white boxes, non-protein-coding exons; black triangles, LoxP sites; neo, neomycin resistance gene; TK, thymidine kinase gene; KO, knockout. (b) Representative Southern blot and PCR to identify Trim13+/+, Trim13+/−, and Trim13−/− mice. Tail DNA was digested with BamHI and subjected to Southern blotting using a probe located in the 5′ homology region (WT allele, ∼16.1 kb; knockout allele, ∼2.6 kb). (c) Representative PCR of genomic tail DNA from Trim13+/+, Trim13+/−, and Trim13−/− mice. The primers used were Trim13-for-5706 (3′-TCC TCT AGT CAA GGT TGA CCT ACA-3′) Trim13-for-7561 (5′-CTT GAT GGG ATT GTT GGA GAA C-3′), and Trim13-rev-104 (5′-CAG CTC ATG TGT CGT AGT TGG T-3′). (d) Semiquantitative RT-PCR to detect the expression of Trim13 in cDNA from the thymuses of embryonic day 18 fetuses of WT and Trim13−/− mice. cDNA was serially diluted 4-fold. The primers used were Trim13-e8-for (5′-GCA TAT ACT TGC CTG GAA CA-3′) and Trim13-594-rev (5′-CGG CGC CAA GTC TCG AAA C-3′). (e, f) Trim13 deficiency does not affect the levels of Trim59 in BMDMs. BMDMs were generated from WT and Trim13−/− mice, cultured in the presence of medium alone or various stimuli for 6 h, and analyzed for Trim13 (e) and Trim59 (f) mRNA expression by qPCR. Data shown are normalized to those for β-actin, are expressed as the mean ± SD from duplicate qPCR analyses, and are representative of those from two independent experiments. Data for expression from WT BMDMs shown in Fig. 1 were regraphed here for comparison with data for samples from Trim13−/− mice. ND, not detected.
Reagents.
The luciferase plasmid p125 (a full-length IFN-β enhancer) and positive regulatory domain III-I (PRDIII-I), PRDII, and PRDIV in the pLuc-MCS vector have been described previously (24). The IRF3-5D plasmid was a gift from John Hiscott (McGill University, Montreal, Canada). TRIM13-V5 and TRIM59-V5 were generated by PCR cloning of the murine coding sequence into the pcDNA3-V5 vector using a pcDNA3.1/V5-his TOPO TA expression kit (Invitrogen). Plasmids containing the sequences for MDA5, MAVS, TANK-binding kinase 1 (TBK1), RIG-I, and AIM2 were described previously (25, 26).
Culture and stimulation of BMDMs.
Bone marrow cells were isolated from C57BL/6 mice and cultured for 8 to 12 days in Dulbecco modified Eagle medium (DMEM; Cellgro) containing 10% heat-inactivated fetal calf serum (FCS; HyClone), ciprofloxacin (University of Massachusetts Medical School Pharmacy), and 20% L929 cell supernatant. The differentiation state of the cells was confirmed by flow cytometric analysis of F4/80 (Caltag) and CD11b (Pharmingen) expression. BMDMs were plated at a concentration of 1 × 106/ml in 4 ml of medium and treated with LPS (100 ng/ml; purified LPS from Sigma), transfected with poly(dA-dT) (Sigma) at 2 μg/well (Lipofectamine; Invitrogen), transfected with poly(I·C) (Amersham) at 2 μg/well, infected with SeV at 300 hemagglutination units (HAU)/ml (Charles River Laboratories), or infected with EMCV-K (a gift from Michael Diamond) at a multiplicity of infection (MOI) of 20:1 for 6 h. RNA was extracted from BMDMs with an RNeasy kit (Qiagen Inc.) according to the manufacturer's instructions. cDNA was synthesized using an iScript cDNA synthesis kit (Bio-Rad).
Flow cytometry and cell sorting and isolation.
Standard culture medium (RPMI 1640 with 10% FCS, 50 μM 2-maercpatoethanol, 2 mM l-glutamine, 20 mM HEPES, and antibiotics) was used for all experiments unless otherwise indicated. Monoclonal fluorochrome-conjugated antibodies to the following were used for flow cytometry and were purchased from eBiosciences or BD Biosciences: CD3 (145-2C11), CD4 (RM4-5), CD8α (53-6.7), T cell receptor γ (TCRγ; UC7-135D), TCRβ (H57-597), NK1.1 (PK136), CD25 (7D4), CD44 (IM7), and CD62L (MEL-14). Cells were sorted from pooled spleens from 5 to 10 C57BL/6 mice (The Jackson Laboratory). For sorting of γδ T, NKT, and NK cells from the spleen, cells were first depleted with CD4, CD8, and B220 magnetic beads (Dynal). For sorting of all other splenic subsets, cells were first depleted with B220 magnetic beads (Dynal). After depletion, cells were stained for surface markers and sorted with a FACSAria (Becton, Dickinson) or MoFlo (Dako Cytomation) cytometer. Intraepithelial lymphocytes (IELs) were prepared using standard protocols. To induce the expansion of CD11c+ DCs, B6 mice were subcutaneously injected with the B16-FLT3L melanoma cell line (from Ulrich von Adrian, Harvard Medical School, Boston, MA, and Glen Dranoff, Dana-Farber Cancer Institute, Boston, MA) at ∼3 × 106 cells/mouse in 300 μl phosphate-buffered saline. Spleens were harvested 14 days after injection, and CD11c+ DCs were isolated using a CD11c selection kit (Stem Cell Technologies) according to the manufacturer's directions.
Quantitative real-time PCR.
RNA was prepared from cell subsets using the TRIzol reagent (Invitrogen), and cDNA was prepared using Sensiscript reverse transcriptase (RT; Qiagen). Quantitative real-time RT-PCR (qPCR) was performed using SYBR green (Applied Biosystems) on a Bio-Rad iCycler and the following primers: Trim13-51-for (5′-AAC GAA CTG GCT CTC TCC AC-3′) and Trim13-143-rev (5′-CTT CTT CAA GCA GCT CCA TTA C-3′), Trim59-179-for (5′-GTC CAG ATC AGG AGA TTG ACA GAC-3′) and Trim59-292-rev (5′-TGT ATG AGA GCA TGG TAG TAC ACG G-3′), and β-actin-for (5′-CTA GGC ACC AGG GTG TGA TGG-3′) and β-actin-rev (5′-TCT CTT TGA TGT CAC GCA CGA-3′).
Luciferase reporter assays.
HEK293T cells were transfected with 40 ng of the luciferase reporter gene indicated below together with 40 ng of a thymidine kinase-driven Renilla luciferase reporter gene (Promega) and expression plasmids in the amounts indicated below using the GeneJuice transfection reagent (Novagen). In all experiments, cell lysates were prepared at 24 h after transfection using 5× lysis buffer (Promega), and reporter gene activity was measured using luciferase substrate (made at the University of Massachusetts Medical School) and coelenterazine (Renilla substrate; Biotium).
Immunoprecipitation and Western blotting.
HEK293T cells were transfected with plasmids containing murine TRIM13-V5, MDA5-Flag, RIG-I–Flag, or AIM2-Flag (1.5 μg each) using the Lipofectamine Plus reagent (Invitrogen) for 24 h. Lysates were made with radioimmunoprecipitation assay (RIPA) lysis buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.5, 1% Nonidet P-40, 0.25% Na-deoxycholate, 0.1% SDS, 1 mM EDTA) with protease inhibitors (1 mM Na3VO4, 2 mM N-ethylmaleimide, 1 mM phenylmethylsulfonyl fluoride, Roche complete protease inhibitor). Lysates were immunoprecipitated overnight with anti-Flag M2 antibody (Sigma), and complexes were pulled down using protein A agarose beads (Roche) and washed with RIPA buffer and RIPA buffer containing 0.5 M NaCl. Samples were resuspended in sample lysis buffer (Bio-Rad). Lysates were resolved by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Membranes were incubated with anti-V5 (Invitrogen) or anti-Flag M2 (Sigma) and the appropriate secondary horseradish peroxidase antibody. Proteins were detected by chemiluminescence (Pierce). The intensities of the bands in the blots were quantified by densitometry using the Image Studio Lite program according to the developer's instructions.
Infections and IFN bioassay.
For survival experiments, Trim13−/− and wild-type (WT) animals were injected with a lethal dose of 1,000 PFU of EMCV-K intravenously and monitored for survival. For IFN production measurements, Trim13−/− and WT mice were injected with a high dose of 1 × 107 PFU of EMCV-K intravenously, and serum was collected 5 h after infection and used in type I IFN functional bioassays as described previously (27). Briefly, mouse sera and control human recombinant IFN-α (PBL Assay Science) were serially diluted (2-fold) in a 96-well flat-bottom plate that had been seeded with 2 × 104 L929 cells (NCTC clone 929). On the following day, cells were infected with 7.5 × 105 PFU VSV. At 2 days postinfection, cell morphology and cytopathic effects were monitored, and the amount of functional IFN was measured as the last dilution of serum or control recombinant IFN-α to provide any protection from VSV-mediated cytopathic effects (termed the endpoint dilution). The log2 values of the reciprocal of the endpoint dilutions were graphed. For IAV infections, Trim13−/−, WT, and Mavs−/− mice were infected via the intratracheal route with 600 HAU (1.5 ×105 PFU) influenza A/PR8 virus (Charles River Laboratories) or saline as a control. After 24 h, mice were sacrificed and lungs were isolated, weighed, and homogenized to measure type I IFN levels by bioassay as described above. For VSV infections, WT, Trim13−/−, and Ifnar1−/− (IFN-α/β receptor−/−) mice were infected by the intranasal route with 2.5 × 107 PFU VSV strain Indiana as previously described (28) and monitored daily for signs of illness.
Culture and stimulation of MEFs.
Embryonic fibroblasts were generated from WT and Trim13−/− mice using standard methods (29) and cultured in DMEM (Cellgro) containing 10% heat-inactivated fetal calf serum (HyClone), 1% penicillin-streptomycin (Corning), and 1% l-glutamine (Corning). MEFs were seeded in a 48-well plate at a concentration of 1 × 105/well in 0.5 ml of medium, transfected with poly(I·C) at 5 μg/well using Lipofectamine (Invitrogen), and infected with Sendai virus at 80 HAU/ml (Charles River Laboratories) or infected with EMCV-2887A; EGFP (a gift from L. Bakkali-Kassimi) at an MOI of 0.001. Supernatants were collected at 6 h or 24 h following challenge and stored at −80°C. At 6 h and 24 h poststimulation, cell morphology and cytopathic effects were monitored. IFN-β was quantified by enzyme-linked immunosorbent assay (ELISA; PBL Assay Science).
Statistical analysis.
Data were analyzed using the Student t test or the Gehan-Breslow-Wilcoxon test (for survival curves). A P value of less than 0.05 was considered significant.
RESULTS
Trim13 expression is increased in BMDMs upon stimulation with inducers of type I IFNs.
Given the similarity in the sequence and structure of TRIM13 and TRIM59, we sought to determine whether these genes are coordinately regulated and possibly redundant in immune cells or whether they have different expression patterns suggestive of distinct functions. We first investigated the expression of Trim13 and Trim59 in sorted murine immune cell subsets by quantitative real-time RT-PCR (qPCR). The expression patterns of Trim13 and Trim59 were distinct in the secondary lymphoid tissues (Fig. 1a). Most notably, Trim13 was expressed in γδ T cells and other peripheral innate immune cell and innate immune cell-like subsets, including NK cells and intraepithelial lymphocytes (IELs), while expression was low in adaptive αβ T cells. In contrast, Trim59 expression was negligible in all peripheral lymphocyte subsets tested except γδ T cells (Fig. 1a).
FIG 1.
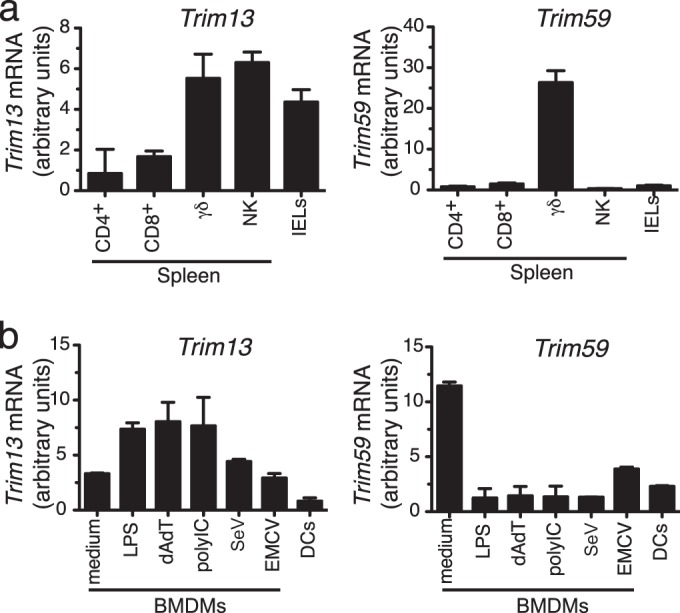
Trim13 and Trim59 have distinct patterns of expression in immune cells. (a) IELs and purified subsets of lymphocytes from the spleen were analyzed for Trim13 and Trim59 mRNA expression by qPCR, and their levels of expression were normalized to the level of β-actin expression. CD4+, CD4+ CD8− CD25− CD44− CD62L+ cells; CD8+, CD4− CD8+ CD44− CD62L+ cells; γδ, TCRδ+ CD3+ cells; NK, NK1.1+ TCRβ− CD3− cells; IELs, total intraepithelial lymphocytes. (b) BMDMs were stimulated with various inducers of type I IFN for 6 h and analyzed for Trim13 and Trim59 mRNA expression by qPCR, and their levels of expression were normalized to the level of β-actin expression. DCs, CD11c+ DCs. Data shown are expressed as the mean ± SD from duplicate qPCR analyses and are representative of those from two independent experiments.
To determine the expression of Trim13 and Trim59 in cells of the myeloid lineage, we generated BMDMs and treated the cells with various inducers of type I IFN. Analysis by qPCR showed that Trim13 and Trim59 have the opposite pattern of expression in macrophages (Fig. 1b). Trim13 was expressed in unstimulated macrophages, and its expression was increased upon stimulation with LPS, poly(dA-dT), and poly(I·C) but was only marginally increased following SeV challenge and was unchanged following EMCV challenge. In contrast, Trim59 expression was the highest in unstimulated BMDMs, and its expression decreased after stimulation with each of these agents. Expression of both genes was low in ex vivo dendritic cells. These results suggest that TRIM13 and TRIM59 may have unique roles during the development and/or function of macrophages and innate lymphocytes.
TRIM13 and TRIM59 inhibit MDA5-mediated type I IFN induction.
Several E3 ubiquitin ligases, including TRIM25, RNF135 (REUL), Triad3A, and RNF125, have been shown to directly regulate type I IFN production in innate immune cells by activating (7, 30) or inhibiting (31, 32) RIG-I function. In contrast, RNF125 is the only E3 ubiquitin ligase shown to also modify MDA5, which it does by ubiquitin conjugation that targets MDA5 for degradation (31). Given that TRIM13 is an E3 ubiquitin ligase that is upregulated in human macrophages (9, 21) and murine BMDMs under type I IFN-stimulating conditions (Fig. 1b), we investigated whether TRIM13 also modulates type I IFN production through the RIG-I or MDA5 pathway. First, we performed luciferase reporter assays in HEK293T cells using a reporter plasmid regulated by the IFN-β enhancer (p125). Expression of TRIM13 alone in HEK293T cells did not activate the IFN-β luciferase reporter (Fig. 2a). Transfection of expression plasmids containing either MDA5, RIG-I, mitochondrial antiviral signaling (MAVS; which is downstream of MDA5 and RIG-I in the type I IFN pathway), or TANK-binding kinase 1 (TBK1; which is downstream of TLR- and RLR-induced type IFN signaling) activated IFN-β luciferase reporters in HEK293T cells, as expected (Fig. 2a and b). TRIM13 and TRIM59 inhibited MDA5-dependent activation of the IFN-β reporter in a dose-dependent manner (Fig. 2a). However, TRIM13 and TRIM59 did not inhibit the ability of TBK1 or MAVS to activate the IFN-β reporter (Fig. 2a and b). Interestingly, TRIM13, but not TRIM59, enhanced RIG-I-mediated induction of the IFN-β reporter in a dose-dependent manner. This finding is consistent with the observed activities of human TRIM13, which was also reported to enhance RIG-I activity in vitro (12). In that study, however, the effect of TRIM13 on MDA5 was not assayed. These data suggest that murine TRIM13 regulates type I IFN production by selectively inhibiting and enhancing the MDA5- and RIG-I-mediated pathways, respectively.
FIG 2.

TRIM13 inhibits MDA5-mediated induction of type I IFN through an IRF3-dependent mechanism. (a) HEK293T cells were transfected with 40 ng of empty vector, MDA5, MAVS, or TBK1 in the presence of increasing amounts of TRIM13 or TRIM59 plasmid (4, 40, or 80 ng), and activation of full-length p125 IFN-β luciferase reporters was monitored. (b) HEK293T cells were transfected with 40 ng empty vector or RIG-I in the presence of increasing amounts of TRIM13 (20, 40, or 60 ng) (left) or TRIM59 (20, 40, or 60 ng) (right) plasmid, and activation of full-length p125 IFN-β luciferase reporters was monitored. (c) HEK293T cells were transfected with 40 ng of empty vector, MDA5, or IRF3-5D in the presence of increasing amounts of TRIM13 plasmid, and activation of the PRDIII-I luciferase reporter was monitored. (d, e) HEK293T cells were transfected with increasing amounts of TRIM13 plasmid in the presence or absence of MDA5, and activation of the PRDII (d) or PRDIV (e) luciferase reporters was monitored. (f) HEK293T cells were transfected with increasing amounts of TRIM13 or TRIM59 plasmid, and induction of NF-κB luciferase reporters was monitored. (g) HEK293T cells were transfected with 40 ng of empty vector, the NF-κB inhibitor IκB-SP, and/or MDA5 plasmids in the presence of increasing amounts of TRIM13 plasmid, and activation of PRDIII-I luciferase reporters was monitored. Numbers above the bars show the percentage of PRDIII-I activation, with the level of activation observed with 20 ng of TRIM13 set at 100%. All data shown are expressed as the mean ± SD for triplicate samples and are normalized to the results for Renilla luciferase. The data are representative of the results from at least three independent experiments. ND, not detected; vector, empty vector.
The IFN-β enhancer contains four positive regulatory domains (PRDs) where transcription factors bind. PRDIII-I contains the binding sites for IRF3 and IRF7, while PRD regions II and IV contain sites for NF-κB and ATF-2/c-Jun, respectively. In order to examine the exact effects of TRIM13 on each regulatory domain of the IFN-β enhancer, we transfected HEK293T cells with luciferase reporter constructs containing each PRD along with TRIM13. TRIM13 inhibited MDA5-mediated activation of the full IFN-β promoter construct (Fig. 2a) and the PRDIII-I construct (Fig. 2c) but did not inhibit MDA5-mediated activation of the PRDII or PRDIV construct (Fig. 2d and e). These results suggest that TRIM13 specifically modulates the activities of IRF3/IRF7. TRIM13 was not able to inhibit PRDIII-I activation in the presence of a constitutively active form of IRF3 (IRF3-5D) (Fig. 2c), implying that TRIM13 interferes with the MDA5 pathway upstream of IRF3.
TRIM13, but not TRIM59, activated PRDII constructs (Fig. 2d and unpublished data) and NF-κB luciferase reporters on its own in this assay (Fig. 2f). Hence, we tested whether the mechanism by which TRIM13 was inhibiting MDA5-mediated IFN-β production required NF-κB activation. Transfection of a mutant form of an inhibitor of NF-κB kinase (IκBα) which acts as a potent inhibitor of NF-κB activation (called the IκBα superrepressor [IκB-SP]) had no effect on the ability of TRIM13 to inhibit MDA5-driven IFN-β responses (Fig. 2g), as the extent of the decrease in PRDIII-I activation caused by TRIM13 was equivalent in the presence and absence of the NF-κB superrepressor. Thus, inhibition of MDA5-mediated type I IFN production by TRIM13 involves inhibition of IRF3 activation and is not dependent on the ability of TRIM13 to drive NF-κB activation.
TRIM13 interacts with MDA5 and RIG-I.
Many of the E3 ubiquitin ligases that regulate the activity of RIG-I have been shown to directly interact with the protein (7, 30, 31). To determine whether TRIM13 binds to or indirectly regulates the intracellular viral sensors to affect type I IFN production, we generated a V5-tagged version of TRIM13 and performed coimmunoprecipitation experiments in HEK293T cells. TRIM13 coimmunoprecipitated with MDA5 and RIG-I but not with AIM2, a cytosolic sensor of double-stranded DNA that is part of the inflammasome response (Fig. 3). This result suggests that TRIM13 negatively regulates MDA5 either through a direct, physical association or as part of a multiprotein complex that involves intermediaries. Given that TRIM13 appears to enhance the RIG-I function (Fig. 2b) (12), the physical interaction per se is unlikely to be sufficient for the specificity of TRIM13 activity, raising the likelihood that additional cofactors are involved.
FIG 3.
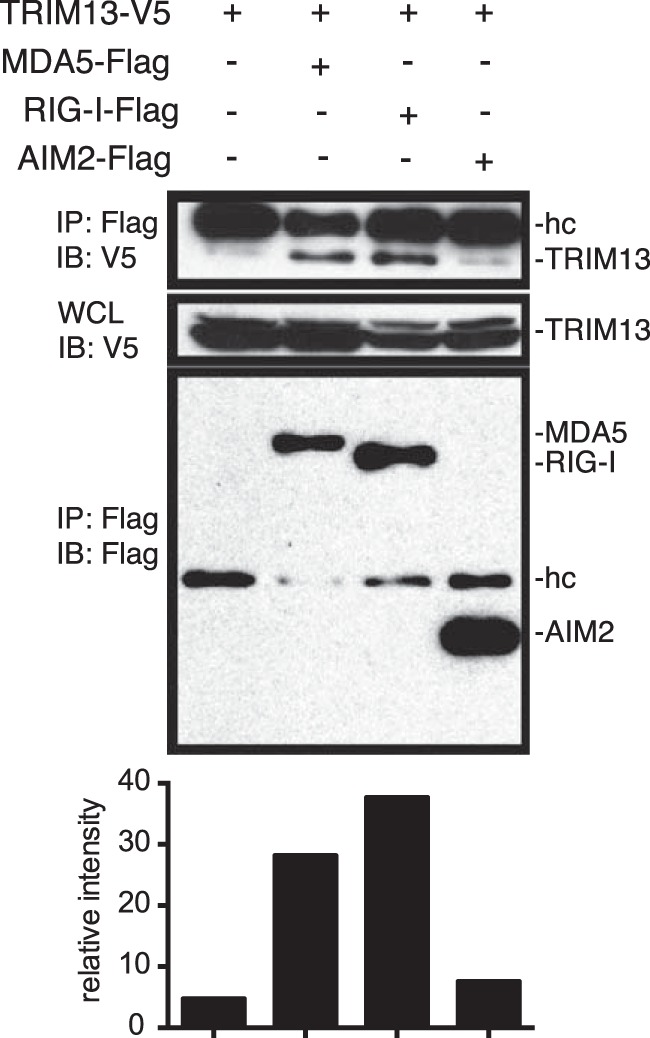
TRIM13 interacts with MDA5 and RIG-I. HEK293T cells were transfected with 1.5 μg of MDA5-Flag, RIG-I–Flag, AIM2-Flag, and/or TRIM13-V5 for 24 h, and lysates were immunoprecipitated (IP) with anti-Flag antibodies and immunoblotted (IB) with anti-Flag and anti-V5 antibodies. Quantification of the immunoprecipitate by densitometry is shown below the blot. Relative band intensities for immunoprecipitated TRIM13 were determined by densitometry and normalized to the TRIM13 levels in whole-cell lysate (WCL) for each immunoprecipitate. The data shown are representative of the results of three independent experiments. hc, Ig heavy chain.
Trim13−/− mice exhibit improved resistance to EMCV infection.
The results from in vitro assays of TRIM13 function suggested that TRIM13 may be a specific negative regulator of MDA5. Trim13−/− (C57BL/6) mice were generated (Fig. 4a to d) to determine whether TRIM13 nonredundantly regulates MDA5 activity in vivo. Trim13−/− mice are viable and fertile, and no gross abnormalities were observed. In humans, Trim13 is located in chromosome 13q14.3, a region that is lost in a number of cancers, and TRIM13 is a candidate tumor suppressor (33). However, no incidence of spontaneous tumorigenesis has been observed in unmanipulated Trim13−/− mice up to 1 year of age.
Trim59 expression was relatively unaltered in Trim13−/− BMDMs under various stimulation conditions (Fig. 4e and f), indicating that the expression of Trim13 and Trim59 is independently regulated. As the expression of TRIM13 inhibited MDA5-dependent type I IFN production in vitro, we investigated whether a lack of Trim13 in vivo would result in enhanced type I IFN production and in improved protection from viral infection. The response to the picornavirus EMCV has been shown to be strictly dependent on MDA5, but not RIG-I (34). Ifih1−/− mice challenged with EMCV died within 3 days of infection, while WT mice survived 2 to 3 days longer (34). Serum IFN-α production from Ifih1−/− mice at 4 h after infection with a high dose of EMCV was completely absent. In contrast, there was no survival difference between Ddx58−/− (RIG-I-deficient) and WT mice upon EMCV infection (34). If Trim13 acts as a negative regulator of MDA5 in vivo, Trim13−/− mice infected with EMCV should exhibit a survival advantage. To test this, we infected WT and Trim13−/− mice with a lethal dose of EMCV intravenously and monitored their survival. Trim13−/− mice survived significantly longer than WT mice (Fig. 5a, left) with a mean survival time of 143.3 h after EMCV infection, whereas the mean survival time was 101.4 h for WT mice (Fig. 5a, right). This improved protection was restricted to the female mice, as there was no significant survival advantage in male mice (data not shown). This difference reflects the reported disparities in the susceptibility of male and female mice to EMCV that occur in a virus dose-dependent manner (35–37). The underlying cause of the sex difference is unknown, but hormones and commensal bacteria are possible contributors (38).
FIG 5.
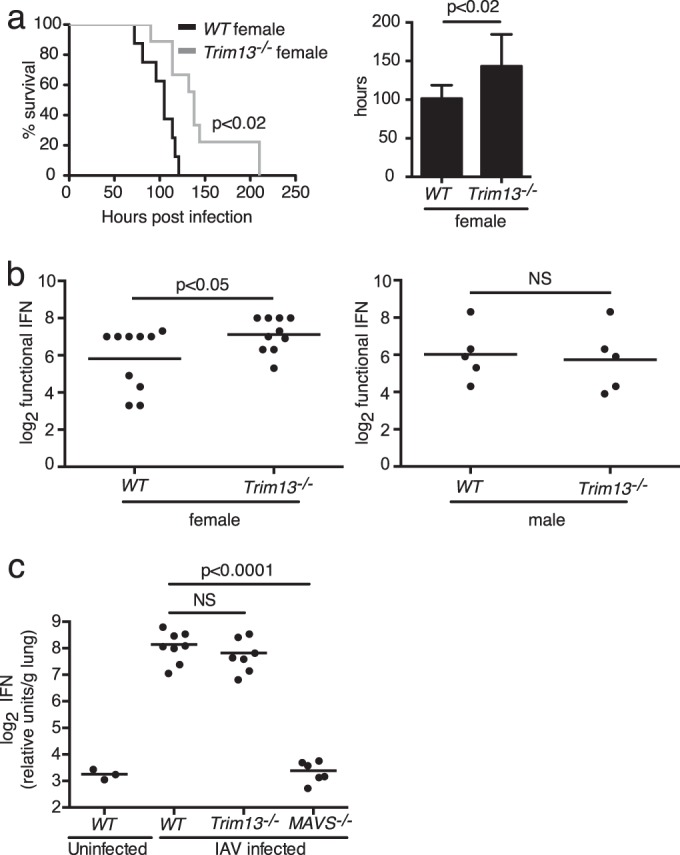
Trim13−/− female mice have extended survival and produce higher levels of type I IFN than WT mice following challenge with EMCV. (a) WT and Trim13−/− mice were infected with 1,000 PFU of EMCV intravenously and monitored for survival (left). One female WT mouse in the infected cohort was removed from the analysis, as it was determined that no virus was injected. The results shown are representative of those from three independent experiments (for WT female mice, n = 9; for Trim13−/− female mice, n = 9; for WT male mice, n = 8; for Trim13−/− male mice, n = 8). P was <0.02 (Gehan-Breslow-Wilcoxon test) for WT female mice versus Trim13−/− female mice. (Right) The mean time to death was calculated for female WT and Trim13−/− mice. P was <0.02 (Student's t test) for WT female mice versus Trim13−/− female mice. (b) WT and Trim13−/− mice were infected with 1 × 107 PFU of EMCV intravenously. Serum was collected at 5 h postinfection, and IFN levels were measured by bioassay for female (left) and male (right) mice. The results shown are the results from two independent experiments combined (for WT female mice, n = 10; for Trim13−/− female mice, n = 10; P < 0.05 [Student's t test] for WT female mice versus Trim13−/− female mice; for WT male mice, n = 5; for Trim13−/− male, n = 5; P was not significant [NS] for WT male mice versus Trim13−/− male mice). (c) WT, Trim13−/−, and Mavs−/−mice were infected via the intratracheal route with 600 HAU (1.5 × 105 PFU) of influenza A/PR8 virus or saline as a control. After 24 h, the mice were sacrificed and lungs were isolated, weighed, and homogenized to measure type I IFN levels by bioassay. A mixture of male and female mice was used (for infected WT mice, n = 8; for Trim13−/− mice, n = 7; for Mavs−/− mice, n = 6). The results shown are representative of those from one of two independent experiments. P was <0.0001 (Student's t test) for WT versus Mavs−/− mice.
We next determined whether the enhanced protective response to EMCV in Trim13−/− mice correlated with increased type I IFN production. Type I IFN is central to the host immune response against EMCV, as evidenced by the significantly increased kinetics of mortality of EMCV-infected Ifnar1−/− mice compared to WT mice. Ifnar1−/− mice die within 2 days of lethal EMCV infection, succumbing to infection even more quickly than Ifih1−/− mice (34). We injected Trim13−/− mice with a high dose of EMCV by the intravenous route and measured functional serum IFN levels at 5 h postinfection using a conventional bioassay. Consistent with our survival data, female Trim13−/− mice showed significantly increased levels of type I IFN compared to WT female mice (Fig. 5b, left), while there was no difference in type I IFN production among male mice (Fig. 5b, right). Together, these results show that female mice lacking Trim13 produce more type I IFN upon EMCV infection, which likely contributes to the improved survival of female Trim13−/− mice. These results support the conclusion that TRIM13 negatively regulates MDA5 in vitro and in vivo. To determine whether the protection of Trim13−/− mice against viral infection was specific to MDA5, we measured type I IFN levels after challenge with IAV, to which IFN responses are predominately mediated by RIG-I (34). Trim13−/− mice infected with IAV showed no significant change in the levels of type I IFN produced, whereas Mavs−/− mice, as expected, were severely impaired in their ability to produce type I IFNs (Fig. 5c). These results further support our in vitro and in vivo data indicating that TRIM13 is a specific negative regulator of MDA5-mediated type I IFN production.
Trim13−/− MEFs produce higher levels of type I IFNs in response to MDA5 agonists.
To validate the observed functional role of TRIM13, we examined the in vitro responses of WT and Trim13−/− MEFs to EMCV (MOI, 0.001), poly(I·C) (5 μg/well), and SeV (80 HAU/ml) at 24 h poststimulation. Administration of the long form of poly(I·C) and EMCV led to the strong induction of IFN-β in Trim13−/− MEFs, and the induction was significantly stronger than that in control WT MEFs (Fig. 6). The cytopathic effect was minimal for EMCV in both WT and Trim13−/− MEFs at this MOI. In contrast, Trim13−/− MEFs exhibited a significantly reduced level of IFN-β in response to challenge with SeV (Fig. 6, right). These results further support the conclusion that TRIM13 specifically inhibits MDA5 and the subsequent MDA5-mediated production of IFN-β.
FIG 6.
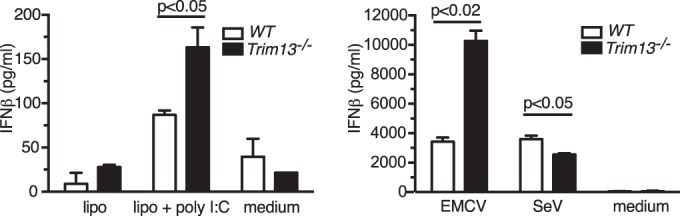
Trim13−/− MEFs produce significantly larger amounts of IFN-β following challenge with poly(I·C) and EMCV but not with SeV. (Left) WT and Trim13−/− MEFs were transfected with poly(I·C) (10 μg/ml) using Lipofectamine (lipo + poly I:C) or treated with Lipofectamine (lipo) or medium alone (medium) for 6 h. Supernatants were collected, and IFN-β levels were quantified by ELISA. (Right) WT and Trim13−/− MEFs were infected with EMCV (MOI, 0.001) or SeV (80 HAU/ml) or treated with medium only for 24 h. Supernatants were collected, and IFN-β levels were quantified by ELISA. Cell cytopathic effects for WT and Trim13−/− MEFs were matched with the doses of virus administered. P values were calculated using Student's t test. Representative data from one of two experiments, each with similar results, are shown.
Survival of Trim13−/− mice following challenge with VSV.
To further study the role of TRIM13 on type I IFN signaling, we infected WT, Trim13−/−, and Ifnar1−/− mice with VSV, which is recognized by RIG-I and is unaffected by the loss of MDA5 (34). VSV infection did not significantly alter the mortality of Trim13−/− mice compared to that of WT mice up to 8 days postinfection (Fig. 7a). In contrast, Ifnar1−/− mice rapidly succumbed to VSV infection, dying within 2 days of infection, which was significantly different from the time to death for WT mice and consistent with previous reports (28). Importantly, VSV-infected Trim13−/− mice had significantly lower levels of circulating type I IFN than WT mice at 20 h after intranasal infection (Fig. 7b). These data suggest that the early type I IFN response is reduced due to the lack of TRIM13 enhancement of RIG-I, an interpretation consistent with the data obtained in Trim13 overexpression studies and with SeV infection of Trim13−/− MEFs.
FIG 7.
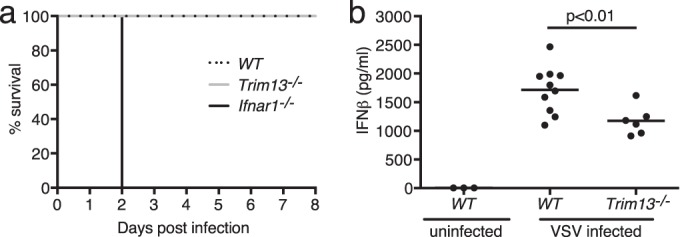
Trim13−/− mice produce smaller amounts of IFN-β than WT mice following infection with VSV. (a) WT, Trim13−/−, and Ifnar1−/− mice were infected with 2.5 × 107 PFU of VSV intranasally and monitored daily for survival. WT and Trim13−/− mice were females, and Ifnar1−/− mice were a mixture of males and females (for WT C57BL/6 mice, n = 10; for Trim13−/− mice, n = 6; for Ifnar1−/− mice, n = 4). P was <0.001 (Gehan-Breslow-Wilcoxon test). (b) WT and Trim13−/− mice were infected intranasally with 2.5 × 107 PFU of VSV, and serum was collected at 20 h postinfection and analyzed for IFN-β by ELISA (for uninfected WT C57BL/6 mice, n = 3; for infected WT C57BL/6 mice, n = 10; for infected Trim13−/− mice, n = 6). P was <0.01 (Student's t test).
DISCUSSION
TRIM13 is an ER- and nuclear membrane-resident E3 ubiquitin ligase that belongs to a family of proteins that are gaining prominence as central regulators of innate and adaptive immune sensors to viruses. RIG-I and MDA5 are the major sensors that detect virus-specific nucleic acid motifs in the cytoplasm of infected cells. While the absolute requirement for posttranslational modifications of RIG-I by accessory factors such as TRIM25 is well established, the identity of the regulatory networks specifically controlling MDA5 activity was unknown. Here we demonstrate that TRIM13 and its closest relative, TRIM59, specifically inhibit MDA5 activity in vitro. TRIM13 and TRIM59 inhibited type I IFN production in the presence of MDA5, whereas TRIM13 appeared to have the opposite effect on RIG-I function (Fig. 2a and b). TRIM13 could physically interact with both MDA5 and RIG-I when overexpressed in HEK293T cells (Fig. 3). Determination of whether TRIM13 is an obligate modifier of RIG-I function will require further studies, as the loss of Trim13 significantly reduced RIG-I-dependent type I IFN production in vitro after SeV stimulation (Fig. 6, right) and in vivo during VSV infection (Fig. 7b). However, the loss of TRIM13 did not significantly impact type I IFN production during influenza virus infection (Fig. 5c).
A systematic analysis of 75 human TRIM proteins in vitro has suggested that at least half are involved in enhancing various innate PRRs at multiple points in the intracellular signaling pathways (12). In this study, human TRIM13, but not TRIM59, was identified to be a positive regulator of innate responses by different type I IFN inducers. However, we observed that Trim13−/− BMDMs treated with a similar set of inducers did not produce antiviral cytokines whose amounts were consistently altered compared to the amounts produced by WT BMDMs (unpublished data). Several explanations for this discrepancy are possible. First, human and mouse TRIM13 may function distinctly. Second, innate immune cells from Trim13−/− mice may have acquired compensatory circuits to retain relatively normal sensing of a pathogen-associated molecular pattern. Third, TRIM13 function may be highly cell type dependent. We currently favor the last explanation, given the precedent of TRIM21, which exhibits a similar context-dependent function: TRIM21 is essential for type I IFN production in embryonic fibroblasts but appears to be redundant in BMDMs (39). In bone marrow-derived dendritic cells, however, it has been shown to negatively regulate the IFN response to double-stranded DNA viruses (40). If the cell type-specific function of TRIM21 and TRIM13 is a general feature of TRIM proteins, caution in the interpretation of results obtained with cell lines is warranted, and where possible, the in vivo impact of each TRIM in a setting of infection should be determined.
Using Trim13−/− mice, we demonstrated that TRIM13 performs a nonredundant function in modulating the MDA5-mediated response to EMCV infection in vivo (Fig. 5a and b) and has a limited effect on the response to infection with IAV (Fig. 5c), a virus not detected by MDA5. Consistent with our in vivo data, we observed that Trim13−/− MEFs exhibited elevated levels of IFN-β in response to the synthetic MDA5 agonist poly(I·C) and EMCV (Fig. 6), further implicating TRIM13 as a negative regulator of MDA5. In vivo, the type I IFN responses of Trim13−/− and WT mice following intravenous challenge with the long form of poly(I·C) were comparable in WT and Trim13−/− mice in serum at 4 h postinjection. However, multiple sensors can detect poly(I·C) in vivo (41), and the IFN-producing cell populations that respond to poly(I·C) may be distinct from those that respond to EMCV in vivo. Hence, the relative contribution of TRIM13 in these particular pathways and populations remains to be determined.
TRIM13 has been characterized as a positive regulator of RIG-I signaling, as it has been demonstrated that overexpression of TRIM13 and RIG-I leads to the increased production of IFN-β- and NF-κB-driven luciferase (12). Using an overexpression system in HEK293T cells, we confirmed the TRIM13 enhancement of RIG-I-dependent type I IFN production (Fig. 2b). We further showed that Trim13−/− MEFs produced significantly less IFN-β in response to SeV stimulation (Fig. 6, right), thereby supporting the suggestion that TRIM13 is a positive regulator of RIG-I in a loss-of-function system. In addition, we infected Trim13−/− mice with VSV and showed that Trim13−/− mice had significantly lower serum IFN-β levels than WT mice (Fig. 7b). Trim13−/− mice did not have significantly altered mortality after VSV infection (Fig. 7a). The lack of a mortality phenotype could be due to either redundant PRRs, or it could occur because the level of reduction in systemic IFN is insufficient to alter the kinetics of viral clearance, resulting in no gross survival difference. We also observed a marginal decrease in type I IFN in the lungs of Trim13−/− mice following infection with IAV (Fig. 5c), another virus recognized by RIG-I. However, this decrease was not statistically significant, which was most likely due to influenza virus's ability to antagonize the RIG-I pathway at multiple steps and specifically block induction of RIG-I-dependent type I IFN (42).
Whether TRIM59 performs a function similar to that of TRIM13 in vivo was not tested, as Trim59−/− mice have not yet been generated. However, given the rapid downmodulation of Trim59 expression in BMDMs after stimulation (Fig. 1b), this possibility appears to be unlikely. Another TRIM protein, TRIM22, also controls EMCV resistance in HeLa cells, but this pathway involves the ubiquitination of a viral protease (43). Hence, different TRIM proteins are predicted to regulate diverse host defensive strategies against a single pathogenic agent. Further, TRIM59 was shown to interact with ECSIT, an adaptor protein in the TLR pathway (44). In agreement with our results (Fig. 2a), TRIM59 was found to inhibit type I IFN production in vitro, possibly by inhibition of phosphorylation or dimerization of IRF3 and IRF7. While TRIM59 was shown to inhibit NF-κB in vitro (44), we and others demonstrated that TRIM13 can enhance NF-κB promoter activity (Fig. 2f) (11, 12). Inhibition of type I IFN production downstream of MDA5 by TRIM13 is, however, independent of the modulation of NF-κB activity. Our results indicate that TRIM13 acts specifically in the MDA5 pathway upstream of IRF3 activity, as TRIM13 could not inhibit MDA5-induced type I IFN in the presence of expression of a constitutively active version of IRF3 (Fig. 2c). In aggregate, it is likely that both TRIM13 and TRIM59 are important for the regulation of the host response to viruses and context-dependent type I IFN activity, although their mechanisms of action are predicted to be distinct.
Over the past several years, it has emerged that the ER is an important site of immune regulation. The ER-resident protein stimulator of interferon genes (STING) has been shown to interact with RIG-I and MAVS to positively regulate type I IFN production and is an important part of the signaling cascade activated in response to cytosolic nucleic acid ligands (45). STING, in turn, is positively regulated by TRIM56 (46). These data link the ER pathway with that of type I IFN induction and, in combination with the ER-resident TRIM13 regulation of MDA5, identify the ER to be a crucial regulatory site and TRIM proteins to be the key modulators of viral RNA sensing and activation of downstream innate effectors. Elucidation of the mechanism of the function of TRIM13 in controlling MDA5 in specific cellular compartments should reveal new facets of the regulatory circuit in innate immune responses.
ACKNOWLEDGMENTS
We thank Raymond Welsh for advice on EMCV infection studies and Leslie Berg and members of the J. Kang and E. Huseby laboratories for helpful discussions and reviews of the manuscript. Expertise and service for cell sorting and generation of gene-deficient mice were provided by the University of Massachusetts Medical School flow cytometry and transgenic animal modeling facilities, respectively. We thank Markus Falk for assistance with initial biochemical studies and Katelyn Sylvia for the maintenance of the Trim13−/− mice.
Core resources supported by a Diabetes Endocrinology Research Center grant were used. This work was supported by grants from the NIH to J.P.W. (AI092105), K.A.F. (AI067497), and J.K. (CA100382).
We have no conflicting financial interests.
Footnotes
Published ahead of print 9 July 2014
REFERENCES
- 1.Wilkins C, Gale M., Jr 2010. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 22:41–47. 10.1016/j.coi.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yap DYH, Lai KN. 2010. Cytokines and their roles in the pathogenesis of systemic lupus erythematosus: from basics to recent advances. J. Biomed. Biotechnol. 2010:365083. 10.1155/2010/365083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herbeuval JP, Shearer GM. 2007. HIV-1 immunopathogenesis: how good interferon turns bad. Clin. Immunol. 123:121–128. 10.1016/j.clim.2006.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Honda K, Mizutani T, Taniguchi T. 2004. Negative regulation of IFN-alpha/beta signaling by IFN regulatory factor 2 for homeostatic development of dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 101:2416–2421. 10.1073/pnas.0307336101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nisole S, Stoye JP, Saib A. 2005. TRIM family proteins: retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 3:799–808. 10.1038/nrmicro1248 [DOI] [PubMed] [Google Scholar]
- 6.Arimoto K, Funami K, Saeki Y, Tanaka K, Okawa K, Takeuchi O, Akira S, Murakami Y, Shimotohno K. 2010. Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc. Natl. Acad. Sci. U. S. A. 107:15856–15861. 10.1073/pnas.1004621107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU. 2007. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446:916–920. 10.1038/nature05732 [DOI] [PubMed] [Google Scholar]
- 8.Yang B, Wang J, Wang Y, Zhou H, Wu X, Tian Z, Sun B. 2013. Novel function of Trim44 promotes an antiviral response by stabilizing VISA. J. Immunol. 190:3613–3619. 10.4049/jimmunol.1202507 [DOI] [PubMed] [Google Scholar]
- 9.Rajsbaum R, Stoye JP, O'Garra A. 2008. Type I interferon-dependent and -independent expression of tripartite motif proteins in immune cells. Eur. J. Immunol. 38:619–630. 10.1002/eji.200737916 [DOI] [PubMed] [Google Scholar]
- 10.Carthagena L, Bergamaschi A, Luna JM, David A, Uchil PD, Margottin-Goguet F, Mothes W, Hazan U, Transy C, Pancino G, Nisole S. 2009. Human TRIM gene expression in response to interferons. PLoS One 4:e4894. 10.1371/journal.pone.0004894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uchil PD, Hinz A, Siegel S, Coenen-Stass A, Pertel T, Luban J, Mothes W. 2013. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 87:257–272. 10.1128/JVI.01804-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Versteeg GA, Rajsbaum R, Sanchez-Aparicio MT, Maestre AM, Valdiviezo J, Shi M, Inn KS, Fernandez-Sesma A, Jung J, Garcia-Sastre A. 2013. The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 38:384–398. 10.1016/j.immuni.2012.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meroni G, Diez-Roux G. 2005. TRIM/RBCC, a novel class of ‘single protein RING finger' E3 ubiquitin ligases. Bioessays 27:1147–1157. 10.1002/bies.20304 [DOI] [PubMed] [Google Scholar]
- 14.Schwamborn JC, Berezikov E, Knoblich JA. 2009. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 136:913–925. 10.1016/j.cell.2008.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ozato K, Shin DM, Chang TH, Morse HC., III 2008. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. 8:849–860. 10.1038/nri2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lerner M, Corcoran M, Cepeda D, Nielsen ML, Zubarev R, Ponten F, Uhlen M, Hober S, Grander D, Sangfelt O. 2007. The RBCC gene RFP2 (Leu5) encodes a novel transmembrane E3 ubiquitin ligase involved in ERAD. Mol. Biol. Cell 18:1670–1682. 10.1091/mbc.E06-03-0248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tomar D, Singh R, Singh AK, Pandya CD, Singh R. 2012. TRIM13 regulates ER stress induced autophagy and clonogenic ability of the cells. Biochim. Biophys. Acta 1823:316–326. 10.1016/j.bbamcr.2011.11.015 [DOI] [PubMed] [Google Scholar]
- 18.Huang W, Kane JK, Li MD. 2008. Identification and characterization of a long isoform of human IFT80, IFT80-L. Biochem. Biophys. Res. Commun. 373:653–658. 10.1016/j.bbrc.2008.06.085 [DOI] [PubMed] [Google Scholar]
- 19.Licchesi JD, Van Neste L, Tiwari VK, Cope L, Lin X, Baylin SB, Herman JG. 2010. Transcriptional regulation of Wnt inhibitory factor-1 by Miz-1/c-Myc. Oncogene 29:5923–5934. 10.1038/onc.2010.322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khatamianfar V, Valiyeva F, Rennie PS, Lu WY, Yang BB, Bauman GS, Moussa M, Xuan JW. 2012. TRIM59, a novel multiple cancer biomarker for immunohistochemical detection of tumorigenesis. BMJ Open 2:e001410. 10.1136/bmjopen-2012-001410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez FO, Gordon S, Locati M, Mantovani A. 2006. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J. Immunol. 177:7303–7311. 10.4049/jimmunol.177.10.7303 [DOI] [PubMed] [Google Scholar]
- 22.Seth RB, Sun L, Ea CK, Chen ZJ. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682. 10.1016/j.cell.2005.08.012 [DOI] [PubMed] [Google Scholar]
- 23.Wang JP, Cerny A, Asher DR, Kurt-Jones EA, Bronson RT, Finberg RW. 2010. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J. Virol. 84:254–260. 10.1128/JVI.00631-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. 2003. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–496. 10.1038/ni921 [DOI] [PubMed] [Google Scholar]
- 25.Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA. 2005. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol. 175:5260–5268. 10.4049/jimmunol.175.8.5260 [DOI] [PubMed] [Google Scholar]
- 26.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. 2009. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518. 10.1038/nature07725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rubinstein S, Familetti PC, Pestka S. 1981. Convenient assay for interferons. J. Virol. 37:755–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou S, Kurt-Jones EA, Fitzgerald KA, Wang JP, Cerny AM, Chan M, Finberg RW. 2007. Role of MyD88 in route-dependent susceptibility to vesicular stomatitis virus infection. J. Immunol. 178:5173–5181. 10.4049/jimmunol.178.8.5173 [DOI] [PubMed] [Google Scholar]
- 29.Kurt-Jones EA, Sandor F, Ortiz Y, Bowen GN, Counter SL, Wang TC, Finberg RW. 2004. Use of murine embryonic fibroblasts to define Toll-like receptor activation and specificity. J. Endotoxin Res. 10:419–424. 10.1179/096805104225006516 [DOI] [PubMed] [Google Scholar]
- 30.Oshiumi H, Matsumoto M, Hatakeyama S, Seya T. 2009. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 284:807–817. 10.1074/jbc.M804259200 [DOI] [PubMed] [Google Scholar]
- 31.Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. 2007. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. U. S. A. 104:7500–7505. 10.1073/pnas.0611551104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakhaei P, Mesplede T, Solis M, Sun Q, Zhao T, Yang L, Chuang TH, Ware CF, Lin R, Hiscott J. 2009. The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog. 5:e1000650. 10.1371/journal.ppat.1000650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Everdink WJ, Baranova A, Lummen C, Tyazhelova T, Looman MW, Ivanov D, Verlind E, Pestova A, Faber H, van der Veen AY, Yankovsky N, Vellenga E, Buys CH. 2003. RFP2, c13ORF1, and FAM10A4 are the most likely tumor suppressor gene candidates for B-cell chronic lymphocytic leukemia. Cancer Genet. Cytogenet. 146:48–57. 10.1016/S0165-4608(03)00126-2 [DOI] [PubMed] [Google Scholar]
- 34.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. 10.1038/nature04734 [DOI] [PubMed] [Google Scholar]
- 35.Friedman SB, Grota LJ, Glasgow LA. 1972. Differential susceptibility of male and female mice to encephalomyocarditis virus: effects of castration, adrenalectomy, and the administration of sex hormones. Infect. Immun. 5:637–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pozzetto B, Gresser I. 1985. Role of sex and early interferon production in the susceptibility of mice to encephalomyocarditis virus. J. Gen. Virol. 66(Pt 4):701–709. 10.1099/0022-1317-66-4-701 [DOI] [PubMed] [Google Scholar]
- 37.Ishikawa R, Bigley NJ. 1990. Sex hormone modulation of interferon (IFN) alpha/beta and gamma production by mouse spleen cell subsets following picornavirus infection. Viral Immunol. 3:225–236. 10.1089/vim.1990.3.225 [DOI] [PubMed] [Google Scholar]
- 38.Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, von Bergen M, McCoy KD, Macpherson AJ, Danska JS. 2013. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 339:1084–1088. 10.1126/science.1233521 [DOI] [PubMed] [Google Scholar]
- 39.Yoshimi R, Chang TH, Wang H, Atsumi T, Morse HC, III, Ozato K. 2009. Gene disruption study reveals a nonredundant role for TRIM21/Ro52 in NF-kappaB-dependent cytokine expression in fibroblasts. J. Immunol. 182:7527–7538. 10.4049/jimmunol.0804121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z, Bao M, Lu N, Weng L, Yuan B, Liu YJ. 2013. The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat. Immunol. 14:172–178. 10.1038/ni.2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Z, Kim T, Bao M, Facchinetti V, Jung SY, Ghaffari AA, Qin J, Cheng G, Liu YJ. 2011. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity 34:866–878. 10.1016/j.immuni.2011.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M, Jr, Garcia-Sastre A. 2007. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J. Virol. 81:514–524. 10.1128/JVI.01265-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eldin P, Papon L, Oteiza A, Brocchi E, Lawson TG, Mechti N. 2009. TRIM22 E3 ubiquitin ligase activity is required to mediate antiviral activity against encephalomyocarditis virus. J. Gen. Virol. 90:536–545. 10.1099/vir.0.006288-0 [DOI] [PubMed] [Google Scholar]
- 44.Kondo T, Watanabe M, Hatakeyama S. 2012. TRIM59 interacts with ECSIT and negatively regulates NF-kappaB and IRF-3/7-mediated signal pathways. Biochem. Biophys. Res. Commun. 422:501–507. 10.1016/j.bbrc.2012.05.028 [DOI] [PubMed] [Google Scholar]
- 45.Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678. 10.1038/nature07317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsuchida T, Zou J, Saitoh T, Kumar H, Abe T, Matsuura Y, Kawai T, Akira S. 2010. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33:765–776. 10.1016/j.immuni.2010.10.013 [DOI] [PubMed] [Google Scholar]