

ABSTRACT
The genome of nonsegmented negative-strand RNA viruses is tightly embedded within a nucleocapsid made of a nucleoprotein (N) homopolymer. To ensure processive RNA synthesis, the viral polymerase L in complex with its cofactor phosphoprotein (P) binds the nucleocapsid that constitutes the functional template. Measles virus P and N interact through two binding sites. While binding of the P amino terminus with the core of N (NCORE) prevents illegitimate encapsidation of cellular RNA, the interaction between their C-terminal domains, PXD and NTAIL is required for viral RNA synthesis. To investigate the binding dynamics between the two latter domains, the PXD F497 residue that makes multiple hydrophobic intramolecular interactions was mutated. Using a quantitative mammalian protein complementation assay and recombinant viruses, we found that an increase in PXD-to-NTAIL binding strength is associated with a slower transcript accumulation rate and that abolishing the interaction renders the polymerase nonfunctional. The use of a newly developed system allowing conditional expression of wild-type or mutated P genes, revealed that the loss of the PXD-NTAIL interaction results in reduced transcription by preformed transcriptases, suggesting reduced engagement on the genomic template. These intracellular data indicate that the viral polymerase entry into and progression along its genomic template relies on a protein-protein interaction that serves as a tightly controlled dynamic anchor.
IMPORTANCE Mononegavirales have a unique machinery to replicate RNA. Processivity of their polymerase is only achieved when the genome template is entirely embedded into a helical homopolymer of nucleoproteins that constitutes the nucleocapsid. The polymerase binds to the nucleocapsid template through the phosphoprotein. How the polymerase complex enters and travels along the nucleocapsid template to ensure uninterrupted synthesis of up to ∼6,700-nucleotide messenger RNAs from six to ten consecutive genes is unknown. Using a quantitative protein complementation assay and a biGene-biSilencing system allowing conditional expression of two P genes copies, the role of the P-to-N interaction in polymerase function was further characterized. We report here a dynamic protein anchoring mechanism that differs from all other known polymerases that rely only onto a sustained and direct binding to their nucleic acid template.
INTRODUCTION
Non-segmented negative-strand RNA viruses (or Mononegavirales) share a unique transcription and replication machinery. When using naked genomic RNA as a template, the viral polymerase (L protein) displays poor processivity, with neosynthesized RNAs not exceeding a few tens of nucleotides in length even in the presence of the polymerase cofactor, the phosphoprotein P (1). The functional template is the nucleocapsid made of the RNA genome tightly covered by a continuous helical homopolymer of nucleoprotein (N), the structure of which is well conserved within the Mononegavirales order (2–5). Upon binding of the P-L complex to the nucleocapsid template, transcription initiates at the 3′ genomic end, where the polymerase is switched on by recognizing the transcription promoter localized in the leader region. Transcription of the 6 to 10 genes occurs sequentially thanks to the intergenic regions containing stop and start signals (6). Upon their delivery into the cytoplasm, genomic nucleocapsids are immediately transcribed by ready to start transcriptases residing in the incoming virus particles. Linear transcript accumulation during this primary transcription (5 to 6 h) is followed by exponential transcript accumulation consecutive to the recruitment of neosynthesized transcriptases on the same number of genomic nucleocapsid templates for the next 5 to 8 h. When enough encapsidation substrate, called N0P (a soluble complex made of newly synthesized N protein and P), becomes available at ∼12 to 14 h postinfection (hpi), replication (and secondary transcription, i.e., transcription from neosynthesized genomic nucleocapsid) starts (7). The nascent RNA copy is concomitantly encapsidated, a process possibly responsible for the uninterrupted RNA synthesis at the intergenic regions. To ensure an RNA synthesis processive enough to produce mRNAs of up to ∼6.7 kb in length and of the ∼16-kb genome, measles virus (MeV) polymerase has to be anchored in a sustained manner onto the nucleocapsid template. This anchoring is thought to rely on the interaction between the C-terminal domains of P (PXD) and N (NTAIL). This process should be dynamic so as to permit the progression along the nucleocapsid, where sequential and transient opening of every N subunit would allow the polymerase to access to nucleotides (6, 8).
PXD folds into a three antiparallel α-helical bundle delineating a hydrophobic groove into which a molecular recognition element of helical nature (α-MoRE; amino acids [aa] 486 to 504) located within NTAIL dynamically binds (9–11). In the free state, the side chain of PXD residue F497 is involved in a network of intramolecular hydrophobic interactions, with an additional intramolecular contact being established when PXD is bound to the α-MoRE of NTAIL. As such, this residue provides a good target for modulating PXD binding to NTAIL. It was therefore chosen for substitution in view of binding and functional studies. To perform these studies, we implemented a quantitative protein complementation assay working in mammalian cells (12, 13) and designed an assay allowing the analysis of the function of P variants in the context of viral infection. The latter approach is based on the demonstrated ability of a constitutively expressed small interfering RNA (siRNA) to repress the expression of a viral gene (14, 15). We combined this method with duplication of the viral gene of interest and conditional selective silencing of one of the two gene copies and named this process biGene-biSilencing (biG-biS). Thanks to the exclusive expression of the wild-type (wt) copy of the P gene in cells expressing a siRNA targeting the mutated P gene copy, bi-P viruses with a wild-type (wt) phenotype were successfully rescued. Upon infection of another cell line that specifically prevents the expression of the wt P gene copy, the function of the mutated P gene was explored. Through the combination of these methods, we show that the viral mRNA accumulation rate and the kinetics of virus production depend on optimal binding strength between PXD and NTAIL. Furthermore, a PXD point mutant unable to bind NTAIL not only fails to support viral transcription but also blocks primary transcription by incoming transcriptases. These findings thus highlight how a protein-protein interaction serves as a tightly controlled dynamic anchor for the viral polymerase entry and/or progression along its genomic template.
MATERIALS AND METHODS
Plasmid construction.
The pSP-161 lentiviral vectors coding the puromycin resistance gene under the control of the simian virus 40 promoter and the small hairpin RNA (shRNA) targeting either the P mRNA sequence GGACACCTCTCAAGCATCAT or the green fluorescent protein (GFP) mRNA sequence GAACGGCATCAAGGTGAA (14) under the control of the polymerase (Pol) III H1 promoter were built by subcloning synthetic oligonucleotides using Gateway technology (16).
The plasmid p(+) MVNSe (17) with two unique cloning sites (BsiWI and AatII) added into the 5′ untranslated region (5′UTR) of the P gene was used as the MeV genome backbone. This plasmid encodes a laboratory measles virus strain, derived from the Edmonston strain that uses both CD150 and CD46 as a receptor, and hence can be grown in either Vero or VeroSLAM cells, leading to syncytium formation in both cell lines. This strain exhibits a reduced growth rate, possibly because of a mutation in the V protein, and it proved to be very useful since, in preliminary experiments, it was found to be much more sensitive to the silencing effect against P mRNA than the vaccine Moraten strain. MeV genomic plasmids were built either by a two-step subcloning via an intermediate vector as described previously (18) or by direct recombination of one or two PCR fragments using the vaccinia virus-derived recombinase according to the InFusion user manual (Clontech). To build biG-biS recombinant viruses, the P gene was duplicated in P1 and P2 in gene positions 2 and 3, respectively. P1 was rendered resistant to P RNA interference (RNAi) by introducing silent mutations (GGcCACCTtagctcaATCAT [the mutations are indicated by lowercase letters]) and tagged with an N-terminal Flag peptide and three copies of the GFP RNAi target sequence in the 3′UTR of its mRNA. P2 was tagged with an N-terminal hemagglutinin (HA) peptide and three copies of the P RNAi target sequence in the 3′UTR. Mutations into the X domain of P (D497 and A497) were introduced by subcloning PCR-amplified fragments from a prokaryotic PXD vector mutagenized with QuikChange site-directed mutagenesis kit from Stratagene. The previously described pCG-P eukaryotic expression vector (18) was also used as a backbone for the transient expression of P protein and mutants by transfection.
To avoid the inconvenience of unwanted amino acid intrinsically added by the Gateway cloning system, the two original expression vectors used for Gaussia luciferase-based protein complementation assay (12) were modified into pCI-glu1 and pCI-glu2 to allow subcloning into the unique NotI site: the Gateway insert was eliminated without changing the flanking vector sequence in order to keep unchanged the linker bridging Gaussia luciferase (glu) domains and inserts. Pwt, P-D497, and N401-525 and P376-507 fragments were subcloned downstream Gaussia glu1 and/or glu2 domains by InFusion recombination of PCR-amplified fragments. All plasmids and viruses (N, P1, P2, M, and L gene) were verified by sequencing the subcloned PCR fragments or cDNA obtained by reverse transcription-PCR (RT-PCR) performed on virus stocks. All plasmids have been deposited in the Addgene plasmid repository service except the glu1 and glu2 constructs that Addgene cannot accept. Those constructs are available upon request.
Cell lines and viruses.
Cells were cultured in Dulbecco modified Eagle medium (Life Technologies) supplemented with 10% of heat-inactivated (30 min at 56°C) fetal bovine serum, 1% l-glutamine, and 10 μg of gentamicin/ml at 37°C and 5% CO2. A medium of 293-3-46 helper cells (17) was supplemented with 1.2 mg of G418/ml.
Vero (si2) and Vero-SLAM (19) (si1) cells stably expressing shRNA targeting the P and GFP mRNAs, respectively, were derived by transduction using lentivectors. Infectious nonreplicative retroviral particles were produced in 293T cells (20). Briefly, 4 × 106 cells were cotransfected by 8 μg of pSP-161 coding for shRNA anti-P or anti-GFP mRNA, 8 μg of pCMVΔR8.91, and 4 μg of pMD2.VSVG, the two later coding for retroviral packaging and vesicular stomatitis virus envelope G protein, respectively, using the ProFection mammalian transfection system (Promega). Two days later, 3-fold dilutions of the supernatant were used to infect 293T, Vero, and Vero-SLAM cells. Transduced cells were selected by adding puromycin (2.5 μg/ml for 293T and 10 μg/ml for Vero cells) the day after the infection. After cloning by limiting dilutions, one 293T-derived si2, one 293T-derived si1, one Vero-derived si2, and one VeroSLAM-derived si1 clone able to efficiently silence the transient expression of a GFP-P (18) hybrid construct were selected. Attempts to get si2 and si1 on an identical Vero cell background failed since Vero cells were poorly susceptible to transduction by lentivectors. Fortunately, the growth of a recombinant MeV with an RNAi-resistant P gene in si2 and si1 cell lines was comparable.
To rescue recombinant viruses, the helper cell line 293-3-46 stably expressing T7 polymerase, MeV N, and P was transfected by using the ProFection kit with two plasmids coding for the MeV genome and MeV-L protein (pEMCLa) (17). Three days after transfection, the cells were overlaid on either Vero (single P gene virus) or Vero-si2 cells (bi-P virus). Upon appearance, isolated syncytia were picked and individually propagated on relevant Vero (single P virus) or Vero-si2 (bi-P virus) cells. Virus stock was produced after a second passage at a multiplicity of infection (MOI) of 0.03 in the relevant cell line. This stock was checked for lack of mycoplasma contamination, has its N, P1, P2, M, and L genes sequenced, and was titrated on the relevant host cell before use. In some experiments, the previously described MeV-GFP with GFP expressed from an additional transcription unit in first position (21) was also used.
Analysis of virus protein expression and replication.
Parental Vero, si1 and si2 cells were infected at indicated MOI with recombinant viruses with or without addition of 10 μg/ml of fusion inhibitor peptide z-fFG to prevent syncytium formation. Viral protein expression was determined by flow cytometry analysis of cells labeled with the Y503 anti-F monoclonal antibody and/or GFP detection, detection of the expression of viral N (cl25 antibody), P (49.21 antibody), HA-P2 (anti-HA antibody; Sigma), Flag-P1 (anti-Flag antibody; Sigma), and cellular GAPDH (MAb374 antibody; Chemicon) protein by Western blotting revealed by chemiluminescence as detailed previously (22, 23). Unexpectedly, when the P protein was tagged with the HA peptide, its recognition by 49.21 monoclonal antibody anti-P was reduced. Efficient recognition was restored by using a cell lysis buffer supplemented with 6 M urea. Although P denatured after boiling in the Laemmli sample buffer migrates in SDS-PAGE as a doublet, upon denaturation in the presence of urea P migrates as a single band. This explains the differential P migration behavior according to the absence or presence of urea in the lysis buffer. The protein contents of virus particles were also quantified by using a dot blot assay revealed by monoclonal antibodies specific for N, P, M proteins, Flag, and HA peptides as previously described (24, 25). In some experiments, the luminescent signal was quantified from imaging using Quantity-One software (Bio-Rad), and the viral protein levels were given as the percentage of the signal observed for the studied protein expressed from the control bi-Pwt virus infecting the same host cell (Vero, si1, or si2 cells). Virus production was measured after freeze-thaw cycles of infected cells using a 50% tissue culture infective dose (TCID50) titration assay. Pwt, P-D497, and P-A497 variants were also transiently expressed after the transfection of 6 × 105 cells with the corresponding pCG vectors, and their expression was determined by Western blotting. Contamination of virus stock with internal deletion and copyback defective interfering (DI) minigenomes were assessed according to the method of Shingai et al. (26). Quantification of the MeV genome and mRNA contents of infected cells was performed by reverse transcription-quantitative PCR essentially as described previously (27), with the following minor modification. Negative-strand genome was reverse transcribed using sense 5′-tagged N primer (5′-gcagggcaatctcacaatcaggGTGATCAAAGTGAGAATGAGCT-3′), and the cDNA was PCR quantified using sense tag primer (5′-gcagggcaatctcacaatcagg-3′) and antisense N primer (GCTGACCTTCGACTGTCCT) to overcome the nonoptimal efficacy of the L-Tr and L primers used previously. For the genome the results were expressed as copy number/μg RNA, and for transcripts the results were expressed either as the number of polymerized nucleotides/genome copy or as the viral transcript/μg RNA after normalization for the genome copy contents of each sample. For transcomplementation studies, si2 (293T background) cells were infected at an MOI of 1 for 1 h with the addition of 10 μg/ml of fusion inhibitor peptide and then transfected using jetPRIME (Polyplus). Two days later, the percentage of infected cells was determined by flow cytometry analysis of virus-encoded GFP expression or viral F expression labeled with anti-F antibody.
Protein complementation assay.
Gaussia luciferase-based complementation assay and data analysis (normalized luminescent ratio [NLR]) were performed as described previously (12). Original pSPICA-N1-GW and pSPICA-N2-GW plasmids, kindly provided by Y. Jacob, were modified by replacing the Gateway recombination sequences by a unique NotI site in which the constructs were subcloned using the Infusion recombination technology (Clontech). The linker sequences downstream of the glu1 and glu2 domains were unchanged. The NLR was calculated by dividing the luciferase value of the two chimeric partners by the sum of the luciferase value of every chimeric partner mixed with the other “empty” glu domain.
RESULTS
Design of PXD substitutions in view of functional studies.
In the crystal structure of PXD, the aromatic side chain of F497 is part of an intramolecular hydrophobic network involving residues I464, I468, L481, L484, L485, I488, and L501 from the three α-helical segments (Fig. 1, left panels) (9). In the bound form, an additional interaction involving the side chain of L494 occurs (9, 10). We thus reasoned that targeting F497 for substitution with either an aspartic or an alanine residue would perturb the NTAIL-PXD interaction, thus offering the opportunity to assess the impact of an altered NTAIL-PXD interaction strength on polymerase function. Indeed, molecular modeling of PXD variants bearing the F497D and F497A substitutions predicted complete or partial loss of these hydrophobic interactions, respectively (Fig. 1, middle and right panels).
FIG 1.

(A) PXD residues in contact with residue 497 within the PXD structure (PDB 1OKS). In the left panel, F497 makes hydrophobic contacts with PXD residues I464, I468, L481, L484, L485, I488, and L501. The middle and right panels display structural models of the D497 (middle) and A497 (right) PXD variants showing that while residue D497 does not establish any contacts, residue A497 makes a hydrophobic contact with PXD residue I488. (B) Residues contacting PXD residue 497 within the chimeric PXD/NTAIL α-MoRE structure (PDB 1T60, PXD in blue and NTAIL in red). In the left panel, F497 makes an additional hydrophobic contact with PXD residue L494 in addition to those found in the unbound PXD structure (PDB 1OKS). The middle and right panels display structural models of the D497 (middle) and A497 (right) PXD variants in complex with the α-MoRE showing that while PXD residue D497 is not involved in any interaction (middle), residue A497 makes an additional hydrophobic contact with PXD L494 residue in addition to that involving PXD residue I488 occurring in the unbound form. The structural models were obtained by replacing the side chain of the native F497 residue either in the structure of PXD (PDB 1OKS) or in that of the PXD/α-MoRE complex (PDB 1T60) by the side chain (most frequent conformer) of either Asp or Ala. The models were then energy minimized so as to avoid steric clashes by using the GROMOS96 implementation of the Swiss-PDB Viewer with default parameters.
The D497 and A497 PXD variants were found to be not properly expressed in Escherichia coli, thus precluding direct determination of their binding strength toward NTAIL using purified protein domains. In parallel experiments, full-length P proteins harboring these substitutions were readily expressed in mammalian cells, alone or in combination with the N protein (Fig. 2). This led us to switch to another expression system to evaluate the strength of the PXD-NTAIL interaction. We selected an improved version of the Gaussia luciferase protein complementation assay (PCA) (12) because of its broad signal range and because of the possibility of measuring protein-protein interactions at steady state (13).
FIG 2.

Similar expression levels of P-D497 and P-A497 alone (lane ∅) or together with N protein 1 day after transfection of 293T cells. A Western blot revealed by anti-P and anti-N monoclonal antibodies is shown. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) served as a loading control.
In a first step, we addressed the question as to whether this assay intracellularly reproduces known binding properties of P and truncated P proteins toward the N protein. P and N bind to each other via both their N and C termini, hence, N+P exhibited the highest normalized luminescence ratio (NLR) that was taken as the reference (100%) (Fig. 3A). (When only the N-terminal disordered region of P [PNT-sp-glu1] was used, the NLR signal was reduced to 46%, while the use of monomeric P C-terminal region [i.e., linker-XD] led to a much lower binding ability [reduced by 92.2%]. The substitution of the spacer by VCT with the glu domain grafted at its N terminus [glu1-PNT-VCT construct, i.e., V protein] further decreased PNT binding to N by ∼3-fold. Reduced binding is due to the presence of the glu domain at the N terminus since multiple combinations of glu1 or glu2 grafted at the N terminus of PNT invariably reduced by ∼3-fold the luciferase signal upon interaction with N-glu1/2 constructs. This suggests that although the P protein can tolerate extension at its N terminus with the ∼30-kDa GFP [18], such an N-terminal graft could significantly modulate its ability to bind to N.)
FIG 3.

Assessment of protein-protein interaction strength using the Gaussia luciferase-based protein complementation assay (PCA) in human 293T cells. (A) Interaction strength of full-length and truncated MeV P and V proteins (schemed on left) with the MeV N protein. PNT, P N terminus; sp., spacer; PMD, P multimerization domain; XD, X domain; linker, linker region between PMD and XD; VCT, V C terminus. The V protein is made by RNA editing at aa 231; hence, it has a different C-terminal domain (VCT). Shown are the means of two independent experiments performed in triplicates. Similar rankings were obtained in a third experiment. (B to G) Binding properties of PXDwt, PXD-D497, and PXD-A497 to NTAIL. (B) Interaction between monomeric glu1-MeV-NTAIL (aa 401 to 525) with monomeric glu2-MeV-PXD (aa 376 to 507) hybrid proteins with the expression levels of glu2-MeV-XD determined by Western blotting (B, inset). (C) Interaction of glu1-GCN4-MeV-NTAIL with glu2-GCN4-MeV-XD (black bar) or with glu2-PMD-XD from MeV (MeV-P[aa301-507], red bars) or SeV (P[aa316-568], white bar) hybrid proteins with the expression levels of glu2-MeV-PMD-MeV-XD determined by Western blotting (C, inset). (D and E) Interaction of glu1-SeV-PMD-MeV-NTAIL with glu2-GCN4-XD from MeV (P[aa376-507], red bar) or SeV-P (P[aa445-568], white bar) and tetrameric glu2-PMD-XD hybrid proteins from MeV (P[aa301-507], red bars), SeV (P[aa316-568], black bars), or a 1:1 mixture of glu2-MeV-PMD-MeV-XDwt and glu2-MeV-PMD-MeV-XD-D497 (red bar “D497/wt”). (E) Repeat of D with additional testing of the 1:1 mixture of glu2-MeV-PMD-MeV-XDwt (red bar “D497/wt”). (F) Same as E but using glu1-MeV-N as a sparring partner instead of glu1-SeV-PMD-MeV-NTAIL. (G) Interaction of glu1-SeV-PMD-SeV-NTAIL with glu2-GCN4-XD from MeV (P[aa376-507], red bar) or SeV-P (P[aa445-568], white bar) or glu2-PMD-XD hybrid proteins from MeV (P[aa301-507], red bars) or SeV (P[aa316-568], black bar). The NLR values in panels B to G are means of three independent experimental replicates.
When the construct encompassing XD was tetramerized by the addition of the P multimerization domain (PMD; PMD-XD construct) (28, 29), the NLR signal was increased 3.8-fold (as expected because of enhanced avidity), while the PMD domain on its own exhibited only a background signal.
Then, the gene fragments encoding N[aa401-525] (NTAIL) and P[aa376-507] (hereafter called XD for simplicity but corresponding to the C-terminal fragment downstream PMD) were subcloned downstream the glu1 and glu2 halves of the luciferase. Coexpression of glu1-NTAIL and glu2-MeV-XDwt constructs gave a small but reproducible PCA signal, while the glu2-MeV-XD-D497 and glu2-MeV-XD-A497 constructs elicited a very weak and an enhanced signal, respectively (Fig. 3B). However, both glu2-MeV-XD mutants were found to be poorly expressed (Fig. 3B, inset).
Oligomerization signals were added to both PXD and NTAIL domains both to improve PXD expression and to mimic the natural P oligomer to N oligomer interaction. PXD was tetramerized by generating a construct encompassing the natural P multimerization domain (P[aa301-507]) (29), yielding a construct referred to as MeV-PMD-MeV-XD, while NTAIL was either dimerized or tetramerized by grafting a GCN4 peptide (30) or the PMD of the Sendai virus (SeV) P protein (31). All glu2-MeV-PMD-MeV-XD constructs were similarly expressed (Fig. 3C, inset). Together with the comparable expression of wt and A497- and D497-substituted P proteins (Fig. 2), this indicates that both substitutions at 497 position do not significantly interfere with proper folding and stability of the mutants proteins in these cells. The interaction levels of glu2-MeV-PMD-MeV-XDwt, glu2-MeV-PMD-MeV-XD-D497, and glu2-MeV-PMD-MeV-XD-A497 with dimeric (glu1-GCN4-MeV-NTAIL) (Fig. 3C, red bars) or tetrameric NTAIL (glu1-SeV-PMD-MeV-NTAIL) (Fig. 3D and E, red bars) were higher compared to experiments with monomeric interacting pairs (Fig. 3B). Notably, the use of oligomeric partners led to the same profile in that the three PXD forms (i.e., wt, D497, and A497) led to medium, low/undetectable, and high signals, respectively (Fig. 3C, D, and E, red bars). The use of the entire N protein as a binding partner resulted also in a similar profile (Fig. 3F).
There was a remarkable similarity in the NLR fold increase of the glu2 construct encoding the PXD-A497 variant over that encoding PXDwt (average fold increase of 1.5, P < 0.005; Table 1). As controls, all three glu2-MeV-PMD-MeV-XD proteins displayed a low but equivalent background level of luciferase signal when coexpressed with a tetrameric glu1-SeV-PMD-SeV-NTAIL construct (Fig. 3G, red bars) while interaction of the latter with the homologous glu2-GCN4-SeV-XD construct gave a strong signal (Fig. 3G, white bars). Likewise, the heterologous coexpression of glu2-PMD-SeV-XD with oligomeric glu1-GCN4-MeV-NTAIL proteins and that of glu2-GCN4-SeV-XD with oligomeric glu1-SeV-PMD-MeV-NTAIL proteins resulted in background signals (Fig. 3C and D, respectively, white bars). Mixing two partners fused each to a GCN4 dimerization domain or to homologous PMD tetramerization domains resulted in very strong luciferase signals irrespective of whether the NTAIL and PXD partners were homologous or heterologous, indicating that luciferase reassembly was driven (and dominated) by high-affinity coiled-coil interactions (Fig. 3C, D, E, and G, black bars).
TABLE 1.
Transcript accumulation rates, genome contents, and virus production of wt and mutant A497 virus
| Parameter | Pwt |
P-A497 |
Pb | ||
|---|---|---|---|---|---|
| Mean ± SD | na | Mean ± SD | na | ||
| NTAIL ↔ PXD (% of wt, by PCA) | 1 ± 0.22 | 18 | 1.5 ± 0.6 | 11 | <0.005 |
| Genome content (copies/μg of RNA)c | 262 ± 46 | 5 | 272 ± 34 | 5 | >0.35 |
| Transcript accumulation rate (normalized slope)d | 1.29 ± 0.15 | 4 | 0.79 ± 0.088 | 4 | <0.0025 |
| Virus titer, 2 dpi (log2) | 20.6 ± 1.5 | 6 | 10.5 ± 1.8 | 6 | <0.0025 |
| Virus titer, 2 (wt) and 3 (A497) dpi (log2) | 19.8 ± 0.7 | 3 | 17.5 ± 0.3 | 3 | >0.15 |
n, number of independent trials.
As determined by Student t test.
Note that “μg of RNA” stands for μg of total cellular plus viral RNA from infected cells.
That is, the values from N, P, M, and F genes. Slopes were normalized on the N gene value from the corresponding virus to take into account the transcription gradient. The corresponding calculated wt/A497 slope mean ratio is 1.7 ± 0.4.
P-A497 MeV grows slowly, while rescue of P-D497 MeV fails even by transcomplementation.
Recombinant genomes coding for viruses expressing P-D497 or P-A497 were built and transfected into rescue cells, where standard N, P and L proteins are provided in trans (17). Only the P-A497 virus was rescued, but it grew slowly (Fig. 4A). Lower viral protein expression was documented at 24 hpi (Fig. 4B). Virus production was delayed by 1 day, with A497 virus titer at 3 days postinfection (dpi) being similar to that of the wt virus at 2 dpi (Fig. 4C and D; see Table 1 for statistical analysis). Cells infected with either virus displayed equivalent amounts of genome copy/mg of total RNA that remained unchanged during the first 8 hpi (Fig. 4E). Since the infection was performed using identical MOIs, this indicates that the two viruses had equivalent contents of genomic RNA per infectious unit (P > 0.35) (Table 1). The lower replication kinetics of P-A497 MeV correlated with an ∼1.6-fold reduction in the transcript accumulation rate of the polymerase (P < 0.0025) as measured during the linear accumulation of N, P, M, and F transcripts at early times postinfection from this constant number of genome templates according to (7) (Fig. 4F and Table 1). This growth phenotype could be attributed to the P-A497 substitution and not to the acquisition of compensatory mutations, as shown by the sequence identity of N, P, M, and L genes from our viral stock with those from the genomic plasmids used to rescue the viruses.
FIG 4.
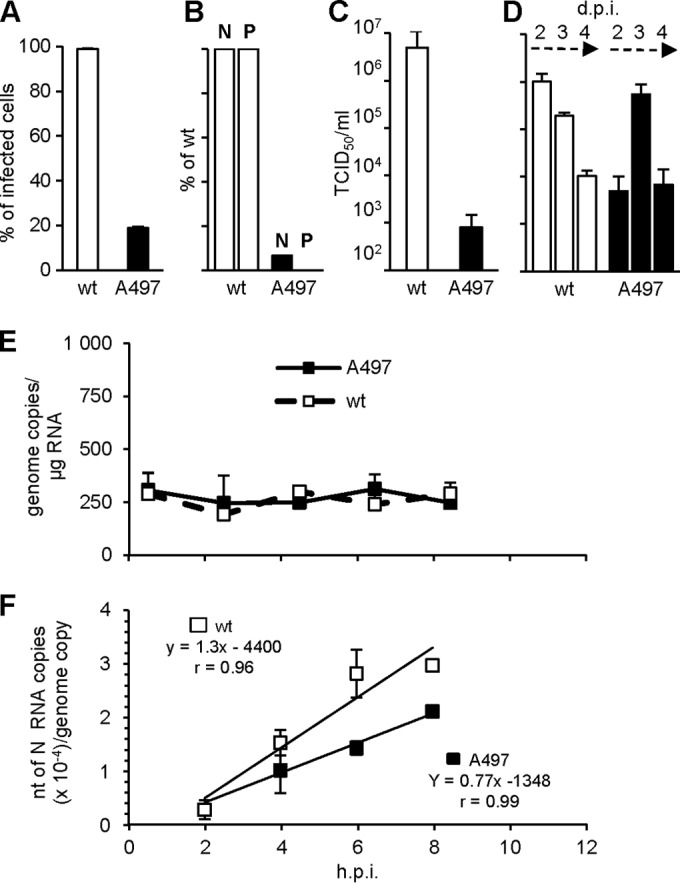
Growth characteristics of wt P and P-A497 virus. (A to D) Percentage of infected cells (A), expression of N and P (with expression of N and P expressed as the percentage of wt N and P as estimated by Western blotting with an anti-N or an anti-P monoclonal antibody, respectively) at 24 hpi with an MOI of 1 (B), and virus production at 2 dpi (C) or 2 to 4 dpi (D). For cytometry analysis of F expression, cell-cell fusion was prevented by adding fusion inhibitor peptide z-fFG after MeV infection. (E and F) Kinetics of RNA accumulation of viral genome (E) and of N mRNA (F) after infection with single P viruses. Error bars indicate the standard deviations based on three experimental replicates. Genomic RNAs in panel E correspond to the virus inputs that remain constant before the replication start (see reference 7 for details). Slopes in panel F correspond to the transcript accumulation rate (according to Plumet et al. [7]). See Table 1 for the genomic inputs of both virus preparations and slope comparisons.
To analyze the P-D497 variant in a viral context, we sought to develop a system based on the transcomplementation of a virus having its P gene expression repressed by RNAi. From an algorithm predictor (from Eurofins-MWG-Operon) and screening, a single P mRNA sequence (+ strand) found to be particularly suitable for RNAi targeting was stably expressed as a shRNA in a 293T cell clone (si2 cells). Unlike the parental 293T cell line, si2 cells resisted infection by a recombinant virus coding for the GFP, as shown by the very small percentage of cells expressing the cell surface F or the GFP proteins (Fig. 5A) and by the undetectable levels of N and P proteins within cells (Fig. 5A, inset) even at 96 hpi. As a control, the virus grew in si1 cells that constitutively express a GFP-specific shRNA targeting the positive-strand (m)RNA, almost as well as in the parental cells (14, 32). However, the onset of GFP expression was delayed reflecting the silencing effect on the viral GFP mRNA (Fig. 5A). Virus production in si2 cells was strongly affected (Fig. 5B). The late virus production in si2 cells at 72 hpi reflected incomplete repression of viral replication through RNA silencing since the viral genome did not exhibit escape mutations within the si2 target region after sequencing the viral genome that was identical to that of the parental virus (GenBank accession number KM054581). Neither MeV N protein expression in cells undergoing active silencing of a cellular protein nor a concurrent irrelevant silencing during MeV infection was found to be modified (Fig. 5C). This suggests that there is little interference between MeV replication and silencing machinery.
FIG 5.
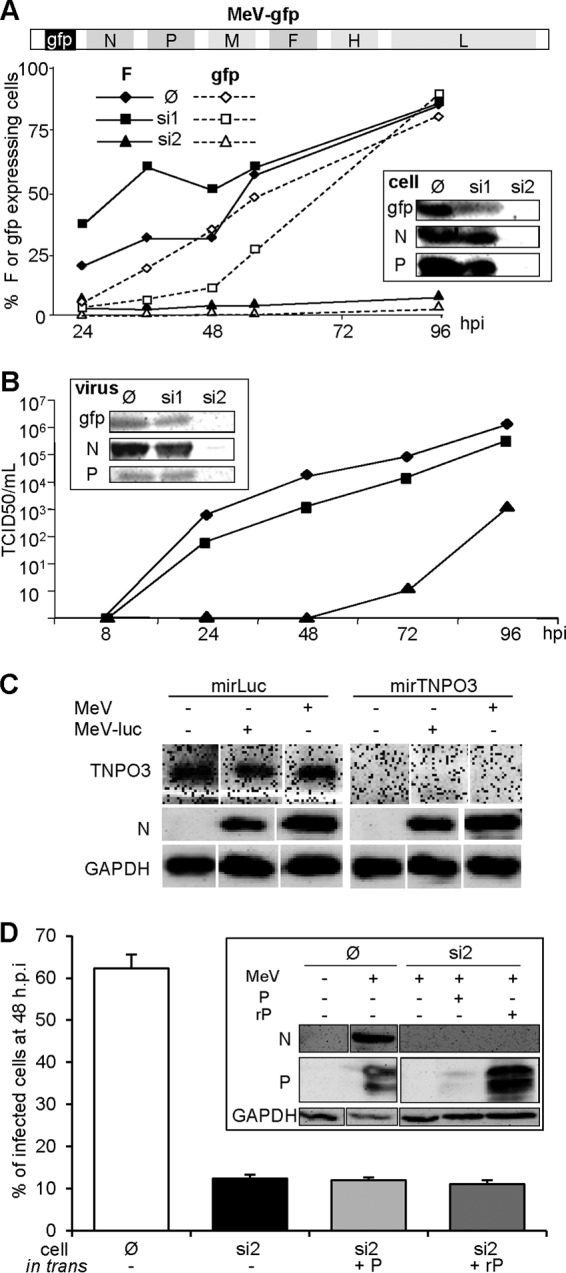
Inhibition of MeV infection by an shRNA targeting the P mRNA (si2) is not alleviated by the expression of an shRNA-resistant P protein in trans. (A) Schematic representation of the MeV genome expressing GFP upstream of the N gene (top scheme). Inhibition of the expression of F (filled symbols, continuous line), gfp (open symbols, dotted line) after infection (MOI = 1) of parental 293T (∅, diamonds), or 293T cells constitutively expressing a shRNA against P (si2, triangles) or GFP (si1, squares) mRNA was assessed. The inset shows the expression of N and P proteins. Protein expression was determined by flow cytometry (curves) and Western blot at 96 hpi (inset). For cytometry analysis of F expression, cell-cell fusion was prevented by adding fusion inhibitor peptide z-fFG after MeV infection. (B) Kinetics of virus production after infection (MOI = 1) of parental (diamonds), si1 (squares), and si2 (triangles) cells and protein contents of purified virions collected at 96 hpi as determined by Western blotting (inset). (C) Ongoing RNA silencing does not affect infection by MeV or MeV expressing luciferase as the reporter gene, nor does MeV infection inhibit an ongoing RNA silencing. Cells constitutively expressing a miRNA-based shRNA targeting the luciferase gene or endogenous TNPO3 gene were infected with MeV or MeV-luc virus (MOI = 1) for 1 day. TNOP3 and virus N protein expression were determined by Western blotting. (D) Inability of P protein transiently expressed from a shRNA resistant transcript (rP) to restore MeV growth in si2 cells (MOI 4). The inset presents the protein expression, as determined by Western blotting, showing resistance and sensitivity to si2 shRNA of the rP and P constructs, respectively. In the absence of urea, P migrates as a doublet due to its phosphorylation heterogeneity.
We then attempted to rescue MeV growth in si2 cells by expressing in trans a P protein harboring silent mutations to resist silencing by si2 cells. This was technically challenging since transfection reduced MeV replication efficiency (Fig. 5D): only 60% of cells transfected with empty vector expressed the F protein at 2 dpi after infection with an MOI of 4. Although well expressed in si2 cells (Fig. 5D, inset), the P construct did not improve virus growth in si2 (Fig. 5D). Similar results were obtained after infection at an MOI of 1 except that the percentage of F-expressing cells in the control was only 45%. Incidentally, this also indicates that the MeV P protein is not naturally endowed with inhibitory properties against the cellular silencing machinery.
Design of a biG-biS cis-complementation system.
We then sought to develop a cis-complementation assay based on a recombinant virus with a duplicated gene of interest. In this system each mRNA (+ strand) copy is artificially made sensitive to unrelated shRNAs (Fig. 6A). We know that silencing of P mRNA tagged in the 3′UTR with one copy of the si1 target is leaky in the context of a recombinant MeV because of overwhelming self-amplifying viral transcription (7). This is illustrated by the modest reduction in GFP expression from MeV-GFP grown into si1 (targeting GFP mRNA) cells (Fig. 5A). Thus, to ensure higher silencing levels, three copies of si1 (Fig. 6A, si1) and si2 (Fig. 6A, si2) target sequences were introduced into the 3′UTR of P1 and P2 copies of the P gene of a recombinant bi-P MeV, respectively. The P1 and P2 proteins were labeled by grafting an N-terminal Flag- and HA-peptide tags, respectively. Indeed, viable MeV tolerates having its P fused downstream to a polypeptide as long as GFP (18), and viable viruses with single P gene tagged with either N-terminal Flag or HA were also easily rescued (data not shown).
FIG 6.
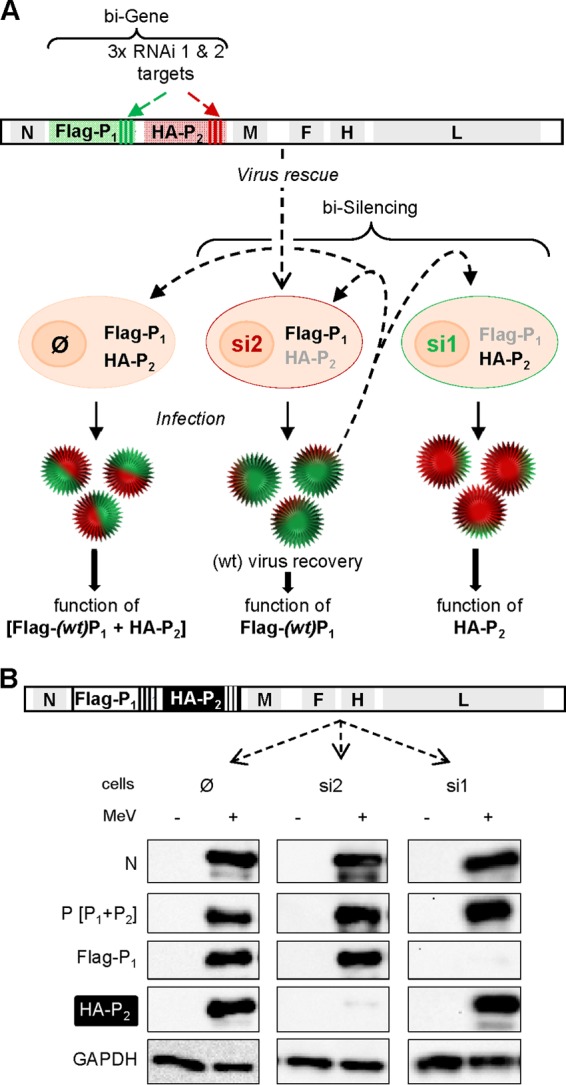
biG-biS assay: principle and proof of concept. (A) Antigenome (+ strand, 5′-3′) organization of recombinant “bi-P” MeV expressing two copies of the P gene: (wt) P1 with mRNA and protein tagged with three copies of the 21-nucleotide target of shRNA 1 (si1) and Flag peptide, respectively; (mutated) P2 with mRNA and protein tagged with three copies of the 21-nucleotide target of shRNA 2 (si2) and HA peptide, respectively. The virus is rescued and amplified in si2 cells (middle) to ensure successful recovery of a wt-like virus thanks to the selective expression of the P1wt gene. This virus can then be used to infect parental (∅, expressing no shRNA, left), si2 (middle), and si1 (right) cells to allow the expression and functional analysis of [Flag-P1 + HA-P2], Flag-P1, and HA-P2, respectively. The gray and black color lettering code for Flag-P1 and HA-P2 illustrates the absence or presence of the protein in the cells, respectively. (B) Efficiency of selective silencing of P1 and P2 from recombinant bi-P MeV in si1 and si2 cells after infection with biP MeV (MOI = 1) revealed by Western blot analysis at 24 hpi.
A virus with two copies (Flag-P1 and HA-P2) of wild-type P gene (bi-Pwt virus) was successfully rescued and amplified in Vero cells, which are very good producers of MeV and unable to produce type I interferon. This virus was then used to infect at an MOI of 1 three Vero cell lines: parental (Fig. 6B, ∅), si1 (Fig. 6B, si1) and si2 cells (Fig. 6B, si2). The three cell lines were equally infected, as shown by a roughly similar expression level of intracellular N protein (Fig. 6B). Although both Flag-P1 and HA-P2 proteins were expressed in parental cells, only HA-P2 and Flag-P1 were detected in si1 (si1, Fig. 6B) and si2 (si2, Fig. 6B), respectively. This illustrates the power of the double silencing system in controlling the selective expression of only one copy of the P gene. When P expression was determined using the 49.21 anti-P antibody that recognizes an epitope located upstream of PXD (33), both Flag-P1 and HA-P2 were detected. In addition, the virus production in si1 and si2 cells reached levels similar to those observed in the parental cells (data not shown).
Functional impact of the A497 and D497 substitutions in PXD.
Both [Flag/P1wt + HA/P2-A497] and [Flag/P1wt + HA/P2-D497] bi-P viruses were successfully rescued in si2 cells, allowing selective expression of the P1wt protein. The sequencing of N, P1, P2, M, and L genes from all viral stocks did not reveal any nucleotide substitution compared to the antigenomic plasmid backbone used for the rescue of each virus, thus excluding the selection of compensatory mutations. Reinfection of si2 cells with these bi-P viruses resulted in normal or slightly increased N expression, reduced expression of P (Fig. 7A, middle panel), slight reduction in the percentage of infected (F-expressing) cells (Fig. 7B, white bars), and virus production levels similar to those observed with bi-Pwt virus (Fig. 7C, white bars). Virions from both recombinant constructs incorporated similar levels of N and P proteins, mostly Flag/P1wt with little HA/P2 proteins, as observed for bi-Pwt virions (Fig. 7D).
FIG 7.

Impact of the D497 and A497 substitutions on P function in the viral context. [Pwt + P-D497] and [Pwt + P-A497] bi-P viruses were rescued and used to infect (MOI = 1) parental (∅, A [left panel] and B [shaded columns]), si2 (A [middle panel] and B [white columns]), and si1 (A [right panel] and B [black columns]) cells. The expression (in percentages of wt/wt virus as estimated by Western blotting with cl55 anti-N and 49.21 anti-P) of N (yellow columns) and P (blue columns) at 24 hpi (A), the percentage of (F-expressing) infected cells at 24 hpi (B), and virus production after infection at an MOI of 0.01 (C) were determined. For cytometry analysis of F expression, cell-cell fusion was prevented by adding fusion inhibitor peptide z-fFG after MeV infection. In panel C, virus production of si1 cells infected by the [Pwt + P-A497] virus was measured after one (p1) and two (p2) successive passages. Error bars indicate the standard deviations based on three experimental replicates. (D and E) Virions produced from si2 cells infected by bi-Pwt, [P1wt + P2-D497], and [P1wt + P2-A497] viruses have similar high Flag-P1 and low HA-P2 protein contents (D), while infection of parental Vero cells by these viruses resulted in the expression of both Flag-P1 and HA-P2 at a similar ratio (E).
When grown in si1 cells, i.e., under conditions of selective expression of the mutated P2, [Flag/P1wt + HA/P2-A497] bi-P MeV grew like the single P-A497 virus: with reduced expression of N and P (Fig. 7A right panel), reduced percentage of F expressing cells (Fig. 7B, black bar), and an ∼1-log reduction in virus production (Fig. 7C [p1], black bar). Because of possible impact of Pwt remnants brought by the incoming virus, a second passage of this virus into si1 cells was performed and resulted in a further ∼1-log decrease in virus production at 2 dpi (Fig. 7C [p2], black bar) that more closely mimicked the phenotype observed with the uni-P-A497 virus. Upon infection of parental Vero (∅) cells that allowed the expression of both P1wt and P2-A497 proteins to a similar extent (Fig. 7E), viral protein expression and virus production were comparable to those observed with the bi-Pwt virus (Fig. 7A, left panel, and Fig. 7B and C, shaded bars). The wt-like and P-A497-like phenotype of the [Flag/P1wt + HA/P2-A497] bi-P MeV when grown into si2 and si1 cells, respectively, underlined the potential usefulness of the biG-biS system.
In agreement with the unsuccessful attempt to rescue the single P-D497 virus, when P2-D497 was selectively expressed from [Flag/P1wt + HA/P2-D497] bi-P virus grown in si1 cells, viral protein expression and virus production were barely detectable: only trace amounts of viral proteins were detected (Fig. 7A, right panel, and Fig. 7B and C, black bars). Notably, the small amount of P1wt that could be produced due to the small leakage of the silencing was not able to overcome this replication block at later times postinfection. Surprisingly, when both P1wt and P2-D497 proteins were coexpressed by infecting parental Vero (∅) cells, significant amounts of N and P protein were detected (Fig. 7A, left panel), with almost every cell expressing the F protein (Fig. 7B, shaded bar). Furthermore, only ∼1.5-log less virus was produced (Fig. 7C, shaded bar) compared to si2 cells allowing restricted expression of P1wt protein or si1 cells infected with bi-Pwt virus.
This intermediate phenotype of the [Flag/P1wt + HA/P2-D497] bi-P virus raised questions about the ability of Pwt and P-D497 to make heterotetramers that could display binding activity toward NTAIL. When assessed through PCA, the two P proteins were found to be able to form hetero-oligomers, as well as homo-oligomers (Fig. 8B, inset). When coexpressed as hetero-oligomers via cotransfection of the two plasmids in a 1:1 ratio, PMD-XDwt/PMD-XD-D497 exhibited a binding ability to oligomeric NTAIL or N proteins similar to that of the homotetrameric PMD-XDwt (Fig. 1E and F). We conclude that the P-D497 variant has a too-low affinity for NTAIL to sustain virus expression on its own but apparently does not exert a dominant-negative effect on Pwt.
FIG 8.
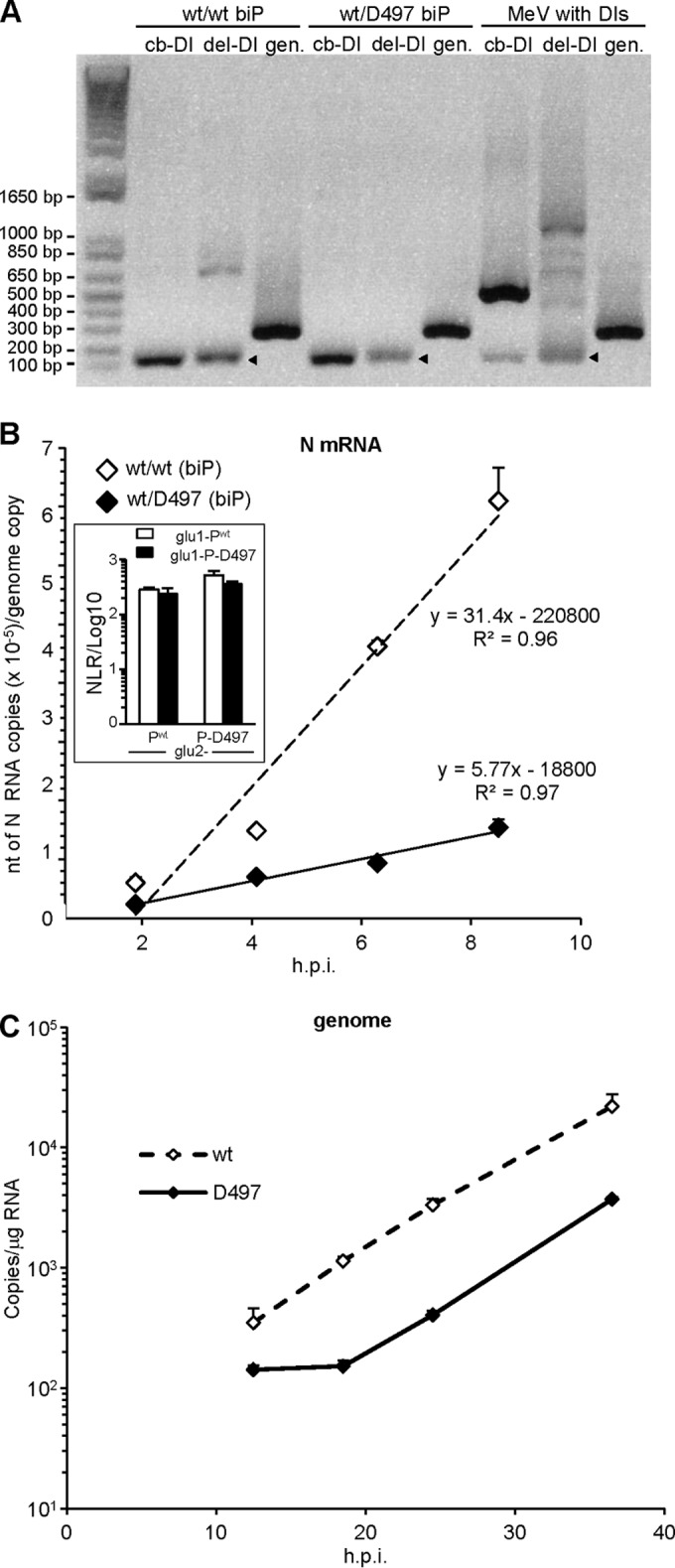
(A) Analysis of biP virus stocks produced in Vero cells for their contamination with deletion (del-DI) and copyback (cb-DI). DI results were detected by RT-PCR. Heavy DI contents (identified by sequencing) of a Moraten MeV stock are shown as DI-positive controls (right lanes). Note the similar intensities of the genomic amplicons (gen) indicative of a similar genomic RNA load for the three viruses. The kinetics of N mRNA (B) and genome (C) accumulation after infection with biP wt/wt and wt/D497 viruses produced in parental Vero cells in which protein is expressed from both P genes (see Fig. 7E) were evaluated. Error bars indicate the standard deviations based on three experimental replicates. Slopes correspond to the transcript accumulation rate (according to Plumet et al. [7]). The genome inputs at earlier times (not shown) for both viruses were similar in range with 83 ± 41 (wt/wt) and 62 ± 18 (wt/D497) copies/μg of RNA (Student t test, P > 0.15). The inset in panel B shows that Pwt and P-D497 make homo- and hetero-oligomers with similar efficiencies, as determined by PCA.
To assess how transcript accumulation rate is influenced by the D497 mutation, the biP viruses were amplified in Vero cells. Virus production peaked at 1.22 × 107 (wt/wt) and 1.02 × 107 (wt/D497) TCID50/ml, with a similar kinetics. HeLa cells were then infected at an MOI of 1, and mRNA accumulation was measured 2, 4, 6, and 8 h after infection. The wt/wt and wt/D497 biP viruses exhibited a remarkably similar genomic content (83 ± 41 and 62 ± 18 copies/μg of total RNA, respectively; P > 0.15). This prompted us to analyze the viral protein contents of virions. Consistent with the similar genomic content, we did not detect any significant difference in the content of N, P, Flag-P1, HA-P2, and M proteins between the two biP viruses (data not shown). Comparison to wt uniP virus revealed a similar protein composition with a slight, but not significant, increase in P content for biP viruses. This similarity, associated with genome content per infectious unit of similar range, indicates that both biP and wt uniP viruses share a similar efficiency in assembling infectious virus. Moreover, both biP viruses were hardly contaminated with defective interfering (DI) nucleocapsids, with only a possible slight contamination of the wt/wt virus with a single internal DI deletion (Fig. 8A). However, the transcript accumulation rate of wt/D497 biP virus measured within the windows of the first transcription step (i.e., 2 to 8 hpi) was reduced to ∼17% of that of the wt/wt biP virus (Fig. 8B). Accordingly, the replication start of wt/D497 biP virus was delayed by ∼6 h as revealed by the kinetics of genome accumulation, which then proceeds with the same rate as that of the wt/wt virus (Fig. 8C).
Negative effect of neo-synthesized D497 transcriptases on preformed Pwt transcriptases.
Upon MeV entry, transcriptases located in the incoming particles transcribe the RNP (see reference 7 and references therein). Thus, we analyzed primary transcription upon infection of si1 cells with wt/D497 biP virus produced in si2 cells. In these conditions, both wt/wt and wt/D497 biP virions are loaded with polymerases that are exclusively and mostly made of Pwt, respectively (Fig. 7D). Indeed, over the first ∼12 h postinfection, the accumulation rates of N mRNA were similar for both biP viruses grown in si1 cells in the absence of viral (and cellular) protein synthesis (Fig. 9, empty symbols and dotted lines, compare panels A and B [note the similar slopes in the equations]). Over this time, the levels of genomic RNA remained unchanged (Fig. 9C and D). When the synthesis of D497 P protein was exclusively allowed from wt/D497 biP virus, the N mRNA transcript accumulation rate was reduced by ∼2-fold as shown by the ratio of the slopes (Fig. 9B, full symbols and lines). In contrast, after the initial linear accumulation rate, N transcript accumulation from wt/wt virus rose exponentially (Fig. 9A, full symbols and lines). The latter phase is due to the recruitment of increasing amounts of neotranscriptases by the constant amount of genome templates at least over the first ∼12 h (Fig. 9C, full symbol and line), i.e., before the onset of wt/wt virus replication observed only at 18.5 hpi. Notably, at this late time point for the three other experimental settings, both N mRNA and cell-associated genome levels dropped to low values illustrating the interruption of the virus replication cycle (Fig. 9C, empty symbols, and Fig. 9D, empty and full symbols that fully overlapped).
FIG 9.

Kinetics of N transcript (A and B) and genome (C and D) accumulation in si1 cells after infection with either wt/wt (A and C) or wt/D497 (B and D) biP viruses in the absence (full symbols, full lines) or presence of 20 μg of cycloheximide/ml (empty symbols and dotted lines). The linear accumulation of N transcripts in the presence of cycloheximide in the 2.5- to 12.5-h time interval (A and B) and in the absence of cycloheximide (B) is shown by the straight lines, equations, and the correlation r factor. Note that at the latest time point (18.5 hpi), the N mRNA (isolated symbols) amount dropped to near the levels detected at 2.5 hpi (A and B), in agreement with the genome contents decrease (C and D) in these three experimental settings, while upon infection with wt/wt virus in the absence of cycloheximide, the N mRNA accumulation sharply increased to reach a level well out of the graphic scale (A) and the genome accumulation started to increase (C). The P1wt gene is indicated in gray to reflect its silencing.
DISCUSSION
The newly developed methodology to study phosphoprotein function described here, together with the study of MeV P497 mutants, yielded insights on how the interaction between P and N could govern MeV transcription. Previously, four different approaches have been used to study the polymerase activity of nonsegmented negative-strand RNA viruses (8, 34). The most popular relies on artificial minireplicons or minigenomes that encode a reporter gene. Spontaneous reconstitution of the first nucleocapsid relies on the complete coverage of a RNA minigenome by a continuous array of N subunits. This event is inefficient, and many subgenomic nucleocapsids are also made, severely limiting mechanistic studies (35, 36). The second assay, which relies on in vitro RNA synthesis with purified viral polymerase components and purified nucleocapsids made in cells as a template, is easily applied only to vesiculoviruses (1, 8, 37–39). The third strategy, consisting in substituting the wild-type (wt) gene with a gene coding for a protein harboring the substitution(s) of interest and in rescuing a mutant virus (see, e.g., reference 24), is limited to modified proteins that are able to sustain a complete virus replication (see, e.g., reference 18). The fourth assay consists in building and rescuing a recombinant virus lacking expression of the P gene in a cell host that provides the lacking viral polymerase component in trans (40, 41). However, transcomplementation results in inefficient viral amplification (40, 41). It remains unclear why transcomplementing polymerase components of paramyxoviruses work inefficiently, and further work will be needed to understand why exogenous viral component cannot be functionally integrated into the putative viral factory (42, 43). We developed here an alternative system to perform structure-function studies of a viral polymerase component, which we name biG-biS. This assay proved to be essential in revealing the inhibitory properties of the nonfunctional D497 P mutant on incoming preloaded transcriptases. Its power has also been documented in a previous study aimed at deciphering the mechanisms of a dominant-negative mutant of the Sendai virus fusion glycoprotein (44).
The F497A substitution enhances the interaction strength with NTAIL by ∼1.5-fold and reduces by 1.6-fold the transcript accumulation rate. This suggests a correlation between the PXD-NTAIL interaction strength and the displacement of the polymerase in the transcription mode along the nucleocapsid template, thereby explaining the slower growth of the A497 virus. We are confident that the interaction strength scale measured by PCA reflects the affinity between these two partners since, in another study, we found a correlation between PCA NLR values and affinity as determined using isothermal titration calorimetry (ITC) and purified NTAIL variants and XD partners (D. Gerlier and S. Longhi, unpublished data). We previously documented that the NTAIL A502D substitution within the α-MoRE, which leads to a 33-fold reduction in the binding affinity toward PXD, causes only a 1.7-fold reduction in the transcript accumulation rate (24). We concluded that there is a poor relationship between the interaction strength of the PXD-NTAIL pair and the transcript accumulation rate (24). While those previous studies suggested a lack of relationship between the interaction strength of the PXD-NTAIL pair and the transcript accumulation rate, we propose here that the tolerance of the polymerase to NTAIL substitutions applies only to a certain range of affinities: in spite of a pronounced drop in the affinity toward PXD brought by the A502D substitution, a PXD-NTAIL interaction with a KD (equilibrium dissociation constant) in the μM range remains strong enough to ensure RNA synthesis by the polymerase. Conversely, an enhanced interaction, such as the one resulting from the PXD F497A substitution, might hinder the polymerase progression along the nucleocapsid template.
Furthermore, according to available structural data and our modeling, the side chain of PXD residue 497 does not contact NTAIL and is rather only involved in intramolecular interactions that likely contribute to stabilize the triple α-helical scaffold of PXD. Thus, the stability of the PXD tertiary structure controls the ability of PXD to accommodate the α-MoRE of NTAIL, which ultimately would result in different levels of anchoring of the polymerase complex onto its template. That substitutions outside the binding interface may affect the binding affinities of protein-ligand interactions through changes in conformational entropy (i.e., fast internal dynamics) is well established (see reference 45 and references therein). Further work is needed to clarify how the dynamic interaction between NTAIL and PXD reflects their different tolerances to amino acid substitution.
When the D497 P protein is produced exclusively or at least in large excess, it interferes with the transcription activity by the preexisting transcriptases interrupting the viral replication cycle. P-D497 cannot bind to N via the PXD-NTAIL interaction, but it can still bind with N via the PNT-NCORE interaction that appears much stronger than the former according to PCA measurements. The current model of Mononegavirales transcription postulates an obligate entry for the P+L polymerase complex at the genomic 3′ end in order to be switched on for RNA synthesis. This is supported by functional evidence (see reference 6 for a review) and the crystal structures of nucleocapsids from rabies (2), vesicular stomatitis (3), and respiratory syncytial (4) viruses, electron microscopy reconstructions of MeV nucleocapsids (46, 47), and docking studies (48). PNT is the best candidate in allowing P+L to be recruited at the 3′ end of the nucleocapsid as inferred from the crystal structures of PNT peptides in complex with the N protein with displacement of the RNA (49) and from functional studies with a P protein having a large deletion within PCT (50). We have previously proposed that the early linear accumulation phase of transcription relies on the very efficient reinitiation of long-living transcriptases over several cycles of transcription along the entire genome (7). That P497D produced in a large excess (i.e., almost exclusively) reduces the primary transcription by preformed Pwt-based polymerases is indicative of a competition between P-D497 and Pwt for binding to the 3′ end of the genomic nucleocapsid and/or of titration of the L component of resident P+L polymerase complexes. It should be stressed here that the transcription was evaluated by measuring N mRNA accumulation and not by directly quantifying the amounts of neosynthesized transcripts. This explained why, during the first 6 to 8 h of the transcription phase, we could only observe a linear accumulation despite the likely recruitment of few neosynthesized P+L, the activity of which remains negligible over that of the resident transcriptases. However, the restricted synthesis of newly transcriptases that are made in small amounts from the inactive P-D497 seem to outcompete the resident molecules, as suggested by the negative effect on transcript accumulation rate. Alternatively, reassortment of Pwt/Pwt and D497/D497 homotetramers into inactive Pwt/D497 heterotetramers could occur. However, this is unlikely owing to the very high cohesion of the PMD domain (28, 29).
Interestingly, the F497D substitution, which by itself no longer supports any transcription and consequently replication to allow virus growth, does not act as a dominant negative over wt P when coexpressed at roughly similar levels. The reduced level of transcript accumulation rate of wt/D497 virus down to only ∼17% that of wt/wt virus is higher than that expected from the 6.25% predicted wt/wt tetramers (random combination probability of 1:16) but lower than that expected from the sum of wt/wt tetramers (6.25%) and 3wt:1D497 heterotetramers (25%). This suggests that most if not all wt/D497 heterotetramers are inactive and that the ratio of wt/wt P homotetramer (6.25%) may be underestimated. This would be the case if there is a higher propensity to homotetramerization of successive nascent P subunits from the same P transcript (i.e., cis-homotetramerization would be favored over trans-tetramerization). Alternatively, we cannot exclude that the transcript accumulation rate is reduced by competition at the 3′ genomic entry site by nonfunctional D497 containing tetramers. Furthermore, the replication of wt/D497 virus proceeds with kinetics similar to that of the wt/wt virus except for an ∼6-h delay, as expected from the lower kinetics of production of functional polymerases.
In conclusion, these results are fully consistent with a model wherein the RNA-dependent RNA polymerase of nonsegmented negative-strand RNA viruses attaches to the 3′ end of genomic nucleocapsid via PNT interaction with NCORE and progresses along its genomic template through a tightly controlled dynamic anchor mediated by the PXD-NTAIL interaction. This is in contrast to all other known polymerases that rely only on a sustained direct binding to their nucleic acid template.
ACKNOWLEDGMENTS
We thank P. Pothier, R. Iggo, L. Roux, and Y. Jacob for providing useful reagents, S. Plumet for pioneering the MeV-P silencing work, C. Lazert and L. Drevet for technical help, and J. Louber for helpful discussions. We are also indebted to the flow cytometry (T. Andrieu and S. Dussurgey) and qPCR (B. Blanquier) facilities of the SFR Biosciences Gerland-Lyon Sud (UMS344/US8).
D.G., S.L., and R.C. designed the research. J.B., D.C., L.-M.B., E.U., and P.D. designed and/or contributed new reagents. J.B., D.C., E.U., L.-M.B., and M.D. performed research, J.B., D.C., E.U., L.-M.B., and D.G. analyzed data. Every author drafted or revised the manuscript, and they all approved the final version.
Footnotes
Published ahead of print 9 July 2014
REFERENCES
- 1.Morin B, Rahmeh AA, Whelan SP. 2012. Mechanism of RNA synthesis initiation by the vesicular stomatitis virus polymerase. EMBO J. 31:1320–1329. 10.1038/emboj.2011.483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albertini AA, Wernimont AK, Muziol T, Ravelli RB, Clapier CR, Schoehn G, Weissenhorn W, Ruigrok RW. 2006. Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 313:360–363. 10.1126/science.1125280 [DOI] [PubMed] [Google Scholar]
- 3.Green TJ, Zhang X, Wertz GW, Luo M. 2006. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 313:357–360. 10.1126/science.1126953 [DOI] [PubMed] [Google Scholar]
- 4.Tawar RG, Duquerroy S, Vonrhein C, Varela PF, Damier-Piolle L, Castagne N, MacLellan K, Bedouelle H, Bricogne G, Bhella D, Eleouet JF, Rey FA. 2009. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 326:1279–1283. 10.1126/science.1177634 [DOI] [PubMed] [Google Scholar]
- 5.Rudolph MG, Kraus I, Dickmanns A, Eickmann M, Garten W, Ficner R. 2003. Crystal structure of the Borna disease virus nucleoprotein. Structure 11:1219–1226. 10.1016/j.str.2003.08.011 [DOI] [PubMed] [Google Scholar]
- 6.Kolakofsky D, Le Mercier P, Iseni F, Garcin D. 2004. Viral RNA polymerase scanning and the gymnastics of Sendai virus RNA synthesis. Virology 318:463–473. 10.1016/j.virol.2003.10.031 [DOI] [PubMed] [Google Scholar]
- 7.Plumet S, Duprex WP, Gerlier D. 2005. Dynamics of viral RNA synthesis during measles virus infection. J. Virol. 79:6900–6908. 10.1128/JVI.79.11.6900-6908.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whelan SP, Barr JN, Wertz GW. 2004. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr. Top. Microbiol. Immunol. 283:61–119 [DOI] [PubMed] [Google Scholar]
- 9.Johansson K, Bourhis JM, Campanacci V, Cambillau C, Canard B, Longhi S. 2003. Crystal structure of the measles virus phosphoprotein domain responsible for the induced folding of the C-terminal domain of the nucleoprotein. J. Biol. Chem. 278:44567–44573. 10.1074/jbc.M308745200 [DOI] [PubMed] [Google Scholar]
- 10.Kingston RL, Hamel DJ, Gay LS, Dahlquist FW, Matthews BW. 2004. Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc. Natl. Acad. Sci. U. S. A. 101:8301–8306. 10.1073/pnas.0402690101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Longhi S. 2012. The measles virus N(TAIL)-XD complex: an illustrative example of fuzziness. Adv. Exp. Med. Biol. 725:126–141. 10.1007/978-1-4614-0659-4_8 [DOI] [PubMed] [Google Scholar]
- 12.Cassonnet P, Rolloy C, Neveu G, Vidalain PO, Chantier T, Pellet J, Jones L, Muller M, Demeret C, Gaud G, Vuillier F, Lotteau V, Tangy F, Favre M, Jacob Y. 2011. Benchmarking a luciferase complementation assay for detecting protein complexes. Nat. Methods 8:990–992. 10.1038/nmeth.1773 [DOI] [PubMed] [Google Scholar]
- 13.Remy I, Michnick SW. 2006. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat. Methods 3:977–979. 10.1038/nmeth979 [DOI] [PubMed] [Google Scholar]
- 14.Mottet-Osman G, Iseni F, Pelet T, Wiznerowicz M, Garcin D, Roux L. 2007. Suppression of the Sendai virus M protein through a novel short interfering RNA approach inhibits viral particle production but does not affect viral RNA synthesis. J. Virol. 81:2861–2868. 10.1128/JVI.02291-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gosselin-Grenet AS, Mottet-Osman G, Roux L. 2010. Sendai virus particle production: basic requirements and role of the SYWST motif present in HN cytoplasmic tail. J. Virol. 405:439–447. 10.1016/j.virol.2010.06.030 [DOI] [PubMed] [Google Scholar]
- 16.Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R. 2003. Induction of an interferon response by RNAi vectors in mammalian cells. Nat. Genet. 34:263–264. 10.1038/ng1173 [DOI] [PubMed] [Google Scholar]
- 17.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dotsch C, Christiansen G, Billeter MA. 1995. Rescue of measles viruses from cloned DNA. EMBO J. 14:5773–5784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devaux P, Cattaneo R. 2004. Measles virus phosphoprotein gene products: conformational flexibility of the P/V protein amino-terminal domain and C protein infectivity factor function. J. Virol. 78:11632–11640. 10.1128/JVI.78.21.11632-11640.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893–897. 10.1038/35022579 [DOI] [PubMed] [Google Scholar]
- 20.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. 1996. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272:263–267. 10.1126/science.272.5259.263 [DOI] [PubMed] [Google Scholar]
- 21.Duprex WP, McQuaid S, Hangartner L, Billeter MA, Rima BK. 1999. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J. Virol. 73:9568–9575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen M, Cortay JC, Logan IR, Sapountzi V, Robson CN, Gerlier D. 2005. Inhibition of ubiquitination and stabilization of human ubiquitin E3 ligase PIRH2 by measles virus phosphoprotein. J. Virol. 79:11824–11836. 10.1128/JVI.79.18.11824-11836.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devaux P, Christiansen D, Plumet S, Gerlier D. 2004. Cell surface activation of the alternative complement pathway by the fusion protein of measles virus. J. Gen. Virol. 85:1665–1673. 10.1099/vir.0.79880-0 [DOI] [PubMed] [Google Scholar]
- 24.Shu Y, Habchi J, Costanzo S, Padilla A, Brunel J, Gerlier D, Oglesbee M, Longhi S. 2012. Plasticity in structural and functional interactions between the phosphoprotein and nucleoprotein of measles virus. J. Biol. Chem. 287:11951–11967. 10.1074/jbc.M111.333088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vincent S, Spehner D, Manie S, Delorme R, Drillien R, Gerlier D. 1999. Inefficient measles virus budding in murine L.CD46 fibroblasts. Virology 265:185–195. 10.1006/viro.1999.0064 [DOI] [PubMed] [Google Scholar]
- 26.Shingai M, Ebihara T, Begum NA, Kato A, Honma T, Matsumoto K, Saito H, Ogura H, Matsumoto M, Seya T. 2007. Differential type I IFN-inducing abilities of wild-type versus vaccine strains of measles virus. J. Immunol. 179:6123–6133. 10.4049/jimmunol.179.9.6123 [DOI] [PubMed] [Google Scholar]
- 27.Plumet S, Gerlier D. 2005. Optimized SYBR green real-time PCR assay to quantify the absolute copy number of measles virus RNAs using gene specific primers. J. Virol. Methods 128:79–87. 10.1016/j.jviromet.2005.03.020 [DOI] [PubMed] [Google Scholar]
- 28.Blocquel D, Habchi J, Durand E, Sevajol M, Ferron F, Erales J, Papageorgiou N, Longhi S. 2014. Coiled-coil deformations in crystal structures: the measles virus phosphoprotein multimerization domain as an illustrative example. Acta Crystallogr. D Biol. Crystallogr. 70:1589–1603. 10.1016/j.virol.2010.06.030 [DOI] [PubMed] [Google Scholar]
- 29.Communie G, Crepin T, Maurin D, Ringkjobing Jensen M, Blackledge M, Ruigrok RW. 2013. Structure of the tetramerization domain of measles virus phosphoprotein. J. Virol. 87:7166–7169. 10.1128/JVI.00487-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Shea EK, Klemm JD, Kim PS, Alber T. 1991. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science 254:539–544. 10.1126/science.1948029 [DOI] [PubMed] [Google Scholar]
- 31.Tarbouriech N, Curran J, Ruigrok RW, Burmeister WP. 2000. Tetrameric coiled coil domain of Sendai virus phosphoprotein. Nat. Struct. Biol. 7:777–781. 10.1038/79013 [DOI] [PubMed] [Google Scholar]
- 32.Bitko V, Barik S. 2001. Phenotypic silencing of cytoplasmic genes using sequence-specific double-stranded short interfering RNA and its application in the reverse genetics of wild-type negative-strand RNA viruses. BMC Microbiol. 1:34. 10.1186/1471-2180-1-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen M, Cortay JC, Gerlier D. 2003. Measles virus protein interactions in yeast: new findings and caveats. Virus Res. 98:123–129. 10.1016/j.virusres.2003.09.003 [DOI] [PubMed] [Google Scholar]
- 34.Curran J, Kolakofsky D. 2008. Nonsegmented negative-strand RNA virus RNA synthesis in vivo. Virology 371:227–230. 10.1016/j.virol.2007.11.022 [DOI] [PubMed] [Google Scholar]
- 35.Rennick LJ, Duprex WP, Rima BK. 2007. Measles virus minigenomes encoding two autofluorescent proteins reveal cell-to-cell variation in reporter expression dependent on viral sequences between the transcription units. J. Gen. Virol. 88:2710–2718. 10.1099/vir.0.83106-0 [DOI] [PubMed] [Google Scholar]
- 36.Conzelmann KK. 2004. Reverse genetics of Mononegavirales. Curr. Top. Microbiol. Immunol. 283:1–41 [DOI] [PubMed] [Google Scholar]
- 37.Rahmeh AA, Li J, Kranzusch PJ, Whelan SP. 2009. Ribose 2′-O methylation of the vesicular stomatitis virus mRNA cap precedes and facilitates subsequent guanine-N-7 methylation by the large polymerase protein. J. Virol. 83:11043–11050. 10.1128/JVI.01426-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang JT, McElvain LE, Whelan SP. 2007. Vesicular stomatitis virus mRNA capping machinery requires specific cis-acting signals in the RNA. J. Virol. 81:11499–11506. 10.1128/JVI.01057-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ogino T, Banerjee AK. 2008. Formation of guanosine(5′)tetraphospho(5′)adenosine cap structure by an unconventional mRNA capping enzyme of vesicular stomatitis virus. J. Virol. 82:7729–7734. 10.1128/JVI.00326-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baron J, Baron MD. 2013. Creation of a completely helper cell-dependent recombinant morbillivirus. J. Gen. Virol. 94(Pt 6):1195–1199. 10.1099/vir.0.050872-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiegand MA, Bossow S, Schlecht S, Neubert WJ. 2007. De novo synthesis of N and P proteins as a key step in Sendai virus gene expression. J. Virol. 81:13835–13844. 10.1128/JVI.00914-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carlos TS, Young DF, Schneider M, Simas JP, Randall RE. 2009. Parainfluenza virus 5 genomes are located in viral cytoplasmic bodies whilst the virus dismantles the interferon-induced antiviral state of cells. J. Gen. Virol. 90:2147–2156. 10.1099/vir.0.012047-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lahaye X, Vidy A, Pomier C, Obiang L, Harper F, Gaudin Y, Blondel D. 2009. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: evidence that NBs are sites of viral transcription and replication. J. Virol. 83:7948–7958. 10.1128/JVI.00554-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Essaidi-Laziosi M, Shevtsova A, Gerlier D, Roux L. 2013. Mutation of the TYTLE motif in the cytoplasmic tail of the Sendai virus fusion protein deeply affects viral assembly and particle production. PLoS One 8:e78074. 10.1371/journal.pone.0078074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tzeng SR, Kalodimos CG. 2012. Protein activity regulation by conformational entropy. Nature 488:236–240. 10.1038/nature11271 [DOI] [PubMed] [Google Scholar]
- 46.Schoehn G, Mavrakis M, Albertini A, Wade R, Hoenger A, Ruigrok RW. 2004. The 12 A structure of trypsin-treated measles virus N-RNA. J. Mol. Biol. 339:301–312. 10.1016/j.jmb.2004.03.073 [DOI] [PubMed] [Google Scholar]
- 47.Bhella D, Ralph A, Yeo RP. 2004. Conformational flexibility in recombinant measles virus nucleocapsids visualized by cryo-negative stain electron microscopy and real-space helical reconstruction. J. Mol. Biol. 340:319–331. 10.1016/j.jmb.2004.05.015 [DOI] [PubMed] [Google Scholar]
- 48.Desfosses A, Goret G, Farias Estrozi L, Ruigrok RW, Gutsche I. 2011. Nucleoprotein-RNA orientation in the measles virus nucleocapsid by three-dimensional electron microscopy. J. Virol. 85:1391–1395. 10.1128/JVI.01459-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leyrat C, Yabukarski F, Tarbouriech N, Ribeiro EA, Jr, Jensen MR, Blackledge M, Ruigrok RW, Jamin M. 2011. Structure of the vesicular stomatitis virus N(0)-P complex. PLoS Pathog. 7:e1002248. 10.1371/journal.ppat.1002248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krumm SA, Takeda M, Plemper RK. 2013. The measles virus nucleocapsid protein tail domain is dispensable for viral polymerase recruitment and activity. J. Biol. Chem. 288:29943–29953. 10.1074/jbc.M113.503862 [DOI] [PMC free article] [PubMed] [Google Scholar]