

ABSTRACT
Plasmacytoid dendritic cells (pDCs) are key components of the innate immune response that are capable of synthesizing and rapidly releasing vast amounts of type I interferons (IFNs), particularly IFN-α. Here we investigated whether pDCs, often regarded as a mere source of IFN, discriminate between various functionally discrete stimuli and to what extent this reflects differences in pDC responses other than IFN-α release. To examine the ability of pDCs to differentially respond to various doses of intact and infectious HIV, hepatitis C virus, and H1N1 influenza virus, whole-genome gene expression analysis, enzyme-linked immunosorbent assays, and flow cytometry were used to investigate pDC responses at the transcriptional, protein, and cellular levels. Our data demonstrate that pDCs respond differentially to various viral stimuli with significant changes in gene expression, including those involved in pDC activation, migration, viral endocytosis, survival, or apoptosis. In some cases, the expression of these genes was induced even at levels comparable to that of IFN-α. Interestingly, we also found that depending on the viral entity and the viral titer used for stimulation, induction of IFN-α gene expression and the actual release of IFN-α are not necessarily temporally coordinated. In addition, our data suggest that high-titer influenza A (H1N1) virus infection can stimulate rapid pDC apoptosis.
IMPORTANCE Plasmacytoid dendritic cells (pDCs) are key players in the viral immune response. With the host response to viral infection being dependent on specific virus characteristics, a thorough examination and comparison of pDC responses to various viruses at various titers is beneficial for the field of virology. Our study illustrates that pDC infection with influenza virus, HIV, or hepatitis C virus results in a unique and differential response to each virus. These results have implications for future virology research, vaccine development, and virology as a whole.
INTRODUCTION
Innate immunity provides a first line of defense for eukaryotic organisms to evade viruses and other pathogens (1). This first line of defense is activated in response to the detection of pathogen-associated molecular patterns (PAMPs) by cellular pattern recognition receptors (PRRs) (2). Once foreign PAMPs are detected, intracellular signaling pathways are activated that result in the initiation of numerous defense mechanisms, including the production of protective cytokines (3). Immune activation through cytokine secretion and subsequent recruitment of immune effector cells of the cellular innate and adaptive immune systems to the site of infection complete the host immune response (4).
Of particular importance are interferons (IFNs), a cytokine family of proteins involved in the activation of immune cells, upregulation of antigen presentation, and signaling of uninfected host cells to resist infection (5). The IFN family of cytokines is composed of type I IFNs (IFN-α, IFN-β, and IFN-ω), a type II IFN (IFN-γ), and a type III IFN (IFN-λ) (6). Following secretion from infected cells, IFNs can act in an autocrine or paracrine manner by binding to specific cell surface receptors to initiate the induction of a growing number of IFN-stimulated genes (>150) via signaling through the Janus protein kinase (JAK)/signal transducer and activator of transcription (STAT) pathway (7).
Dendritic cells are critical components of innate and adaptive immunity that can detect foreign antigens and present them to other effector cells of the immune system (8). Furthermore, a subset of these cells, plasmacytoid dendritic cells (pDCs), can produce copious amounts of IFN-α and other cytokines in response to foreign molecules to further bolster the immune response (9). As a result of this ability, pDCs play a central role in antiviral immunity (10). Though the response of pDCs can vary, depending on the type and magnitude of viral infection, the significance of activated pDCs for early type I IFN production is undisputed (11, 12).
Of particular importance is the role of pDCs during influenza virus infection, a major problem globally with a significant impact on vulnerable populations, including the young, elderly, and immunocompromised. pDCs mitigate influenza virus infection through the early secretion of type I IFN and subsequent antigen presentation (13). Though pDCs have been shown to be dispensable in mice during sublethal influenza virus infection, their role during lethal infection has yet to be investigated (14).
In this study, we investigated the in vitro antiviral responses of human pDCs to various doses of influenza virus by not only considering IFN-α production but also compiling data on gene expression and protein production following infection. Other RNA viruses, namely, human immunodeficiency virus (HIV) and hepatitis C virus (HCV), were also included in this study for comparison and to point out potential analogies and/or differences between pDC antiviral responses. Distinct antiviral responses were observed among the pathogens. Moreover, we observed a dose-dependent response in pDCs specifically to the influenza virus, with our data suggesting that while low-titer infection with H1N1 influenza virus triggers robust production of type I IFN, high-titer H1N1 influenza virus infection can stimulate rapid pDC apoptosis.
MATERIALS AND METHODS
Blood samples, processing, viral stimulation, and ELISA.
Whole blood collected for this study was obtained from healthy donors by the Department of Transfusion Medicine, Clinical Center, National Institutes of Health, over 2 years under an Institutional Review Board-approved research protocol. Buffy coats were subjected to Ficoll-based density gradient centrifugation to obtain peripheral blood mononuclear cells (PBMCs). Samples were enriched for pDCs via magnetic-bead-associated cell sorting with the Miltenyi Biotec (Auburn, CA) human pDC isolation kit for negative selection. pDC purity was ascertained by labeling for BDCA-4, and the samples used were, on average, 92% pure. The total amount of pDCs isolated was used to maximize the total RNA yield for microarray analysis. From 8 × 104 to 2.5 × 105 pDCs were cultured for 18 h in 1 ml of AIM V serum-free medium CTS (Life Technologies, Grand Island, NY) and interleukin-3 (IL-3) by Symansis (Auckland, New Zealand) at 10 ng/ml. After 18 h in culture, pDCs were stimulated with virus for 6 h before RNA isolation or analysis by flow cytometry. Cell culture supernatant was collected after 6 h of viral stimulation and stored at −20°C. An enzyme-linked immunosorbent assay (ELISA) for IFN-α and IFN-β was performed with kits purchased from PBL Interferon Source (Piscataway, NJ). An ELISA for IL-28 was done with kits purchased from R&D Systems (Minneapolis, MN).
Gene expression arrays.
Total RNA was isolated from virus-stimulated samples and untreated controls with Qiagen RNeasy Plus microkits (Qiagen, Valencia, CA) and used for two-color gene expression array experiments. Samples were amplified into antisense RNA (aRNA) by a two-cycle amplification procedure with the MessageAmp II aRNA kit (Ambion, Grand Island, NY) and compared indirectly with a pool of total RNA derived from independent human PBMCs serving as a common reference. Both reference and test aRNA samples were directly labeled with the Kreatech (Durham, NC) ULS aRNA fluorescent labeling kit with Cy3 for reference samples and Cy5 for test samples. Whole-genome human 36K oligonucleotide arrays representing 25,100 unique genes of the Operon Human Genome Array-Ready Oligo Set, version 4.0, were printed in house with oligonucleotides purchased from Operon. The design is based on the Ensembl Human Database build NCBI-35c, with full coverage on the NCBI human RefSeq data set (04/04/2005). Hybridization was carried out in a water bath at +42°C for 18 h, and the arrays were then washed and scanned on a microarray scanner (Agilent, Santa Clara, CA). Human array transcriptional data were uploaded to the mAdb data bank (http://nciarray.nci.nih.gov) and analyzed with BRBArrayTools developed by the Biometric Research Branch, National Cancer Institute (http://linus.nci.nih.gov/BRB-ArrayTools.html) (15) or Partek Genomics Suite software (Partek, St. Louis, MO), as appropriate. Raw data sets were normalized by using the median over the entire array or the robust multiarray average. Absent background values were handled by the k–nearest-neighbor imputation approach at a k value of 15 as described previously (16). Experimental-array batches were merged into a single set of microarrays by distance-weighted discrimination as described elsewhere (17). Analysis of variance (ANOVA) was used to identify genes that were differentially expressed between samples infected with different viruses and their respective controls. Differences were visualized by hierarchical clustering of the genes retrieved from the ANOVA. Functional gene network analysis was performed by Ingenuity pathway analysis (Ingenuity Systems).
Flow cytometry.
Stimulated pDCs were washed twice with 1× phosphate-buffered saline (PBS; Gibco) and then incubated with Miltenyi Biotec (Auburn, CA) FcR Blocking Reagent for 20 min at 4°C. pDCs were then triple stained for annexin V from eBioscience (San Diego, CA), 7-amino-actinomycin D (7AAD) from BD (Franklin Lakes, NJ), and BDCA-4 from R&D Systems (Minneapolis, MN) and incubated for 30 min at 4°C. Cells were then washed once more with Miltenyi Biotec AutoMACS running buffer and then analyzed on a BD FACSCalibur flow cytometer. Data were evaluated by the FlowJo software. Appropriate compensation settings for signal spillover control were determined with BD Calibrite beads. Specific fluorescence signals were calculated by normalization to the appropriate isotype controls.
Viruses.
Direct pelleted HIV-1 IIIB and BaL were purchased from Advanced Biotechnologies Inc. (Columbia, MD). Influenza A/PR/8/34 (H1N1) purified virus was purchased from Charles River Laboratories International Inc. (Wilmington, MA), Advanced Biotechnologies (Columbia, MD), and the American Type Culture Collection (ATCC; Manassas, VA). HCV JFH-1 was provided by T. Jake Liang, Liver Diseases Branch, National Institute of Diabetes and Digestive Diseases, NIH. PB1-F2 knockout (KO) and rescue influenza A/PR/8/34/Mount Sinai (H1N1) viruses were provided by Jonathan W. Yewdell, Laboratory of Viral Diseases, National Institute of Allergy and Infectious Diseases, NIH. Hemagglutination inhibition titers of PB1-F2 KO and rescue viruses were determined with Turkey erythrocytes (Lampire Biological Laboratories) diluted to 1% in 1× PBS for 1 h.
Image stream.
Cell vitality was assayed with the ImageStreamX imaging flow cytometer (Amnis, Seattle, WA) equipped with 405-, 488-, and 642-nm laser excitation (ex) light sources; a 12-channel, dual-camera fluorescence detection system sensitive to fluorophore emission (em) in the violet-to-near-infrared light spectrum; and a 60× 0.9 numerical aperture objective. Cells were stained with the non-membrane-permeating DNA dye Hoechst 33342 (ex, 405 nm; em, 430 to 505 nm) and the non-membrane-permeating DNA dye propidium iodide (PI) (ex, 488 nm; em, 592 to 642 nm) and imaged at a flow rate of ∼500 cells/s, with a total of 10,000 cells acquired for each sample. Cell imagery was processed with IDEAS image analysis software (Amnis). All nucleated cells were labeled with Hoechst 33342. Live cells were identified as PI negative, whereas dead cells were colabeled with both Hoechst 33342 and PI.
Microarray data accession number.
Data from the 95 microarray experiments have been uploaded to the GEO database with accession number GSE59837.
RESULTS
In order to investigate the responses of human pDCs obtained from healthy blood donors to distinct human RNA viruses, pDCs were isolated by negative selection from PBMCs and cultured for 18 h prior to infection as previously described (18). Using this method, we were able to obtain a 92% pure pDC population for our experiments (see Fig. S1 in the supplemental material). With influenza virus-stimulated pDCs shown to produce IFN-α as early as 3 h (19), we chose to investigate a relatively early stage of pDC activation by using the 6-h time point for comparison.
Initially, we sought to characterize the pDC in vitro responses to various doses of H1N1 influenza virus, HIV, and HCV. Four doses each of H1N1 influenza virus and HIV and three doses of HCV were used to investigate pDC responses. The doses used were chosen to cover the range of viral titers typically reported in respective viral infections or doses proven to stimulate pDCs in vitro (13, 19). We treated pDCs from three individual donors with each virus before lysing the cells for RNA isolation and collecting the cell culture supernatant to examine both IFN-α mRNA levels and protein production via microarray analysis and ELISA, respectively (Fig. 1). Distinct responses were observed for each virus at both the mRNA and protein levels.
FIG 1.

Differential regulation of IFN-α in pDCs exposed to H1N1 influenza virus, HIV, and HCV JFH-1. pDCs from three separate donors were exposed to various concentrations of virus. (A) Heat maps generated with microarray data of the IFN-α genes from pDCs stimulated with PBS (control) or influenza virus A/PR/8/34 (H1N1; Charles River Laboratories) at 4, 32, 256, or 2,048 HAU/ml. (B) Heat maps generated with microarray data of the IFN-α genes from pDCs stimulated with PBS (control) or HIV-1 IIIB (Advanced Biotechnologies) at 1.00 ×102, 1.00 ×103, 1.00 × 104, or 1.00 ×105 50% tissue culture infective doses (TCID50)/ml. (C) Heat maps generated with microarray data of the IFN-α genes from pDCs stimulated with PBS (control 1), conditioned medium (control 2), or HCV at 1.00 ×104, 1.00 ×105, or 1.00 × 106 RNA copies/ml. (D) Quantification of IFN-α protein production by ELISA in pDCs stimulated with PBS (control) or influenza virus A/PR/8/34 (H1N1; Charles River Laboratories) at 4, 32, 256, or 2,048 HAU/ml. (E) Quantification of IFN-α protein production by ELISA in pDCs stimulated with PBS (control) or HIV-1 IIIB (Advanced Biotechnologies) at 1.00 ×102, 1.00 ×103, 1.00 × 104, or 1.00 ×105 TCID50/ml. (F) Quantification of IFN-α protein production by ELISA in pDCs stimulated with PBS (control 1), conditioned medium (control 2), or HCV at 1.00 ×104, 1.00 ×105, or 1.00 × 106 RNA copies/ml. *, P < 0.05; **, P < 0.01 (determined by ratio paired t test compared with the corresponding control). NS, not significant. ND, not detectable.
When examining IFN-α mRNA levels following pDC stimulation with various doses of H1N1 influenza virus, maximum IFN-α gene expression was observed at the lowest titer used (4 hemagglutination units [HAU]/ml). Interestingly, a gradual decrease in IFN-α gene expression was detected as the dose was increased. At the highest titer of 2,048 HAU/ml, we observed minimal changes in IFN-α gene expression above the baseline (Fig. 1A) and this corresponded to a broader dampening of the expression of IFN-stimulated genes. In experiments performed with increasing doses of HIV, we observed a corresponding increase in IFN-α gene expression (Fig. 1B). In striking contrast, we observed minimal changes in gene expression regardless of the dose of HCV used (Fig. 1C).
We next sought to compare the changes observed in IFN-α mRNA levels with those observed at the protein level as determined through ELISA. Following stimulation of pDCs with H1N1 influenza virus, we detected the production of large amounts of IFN-α in the supernatant compared to those in the uninfected controls. Regardless of the dose used, large amounts of IFN-α protein were secreted into the cell culture supernatant (Fig. 1D). When we examined IFN-α protein production following pDC stimulation with HIV, we observed a dose-dependent increase in cytokine production in all three donors tested that mirrored the IFN-α mRNA levels (Fig. 1E); however, at the doses used, H1N1 influenza virus appeared to be a more potent stimulus, with approximately 10 times as much IFN-α protein being produced. Similar to what was observed at the mRNA level, we were unable to detect the production of IFN-α protein above the baseline levels following stimulation with HCV (Fig. 1F). The results were similar, as expected, when we corrected for the number of cells present in each experiment (see Fig. S2 in the supplemental material).
To corroborate these findings, we performed additional experiments with pDCs from 10 separate donors and directly compared the type I and III IFN protein levels produced following stimulation with the three distinct viruses at the dose that resulted in maximal IFN-α induction at both the mRNA and protein levels. In addition, a second strain of HIV was included in this analysis to confirm the differences observed between H1N1 influenza virus and HIV and also account for potential differences between R5 and X4 HIV strains in this regard. Figure 2A clearly shows that H1N1 influenza virus is the most potent of these viruses in stimulating IFN-α protein production, with lower levels produced by the two distinct strains of HIV and no IFN-α release observed upon pDC exposure to HCV. Similarly, Fig. 2B shows that H1N1 influenza virus is the most potent of these viruses in stimulating IFN-β protein production, with lower levels produced by the two distinct strains of HIV and no IFN-α release observed upon pDC exposure to HCV. Interestingly, we observed minimal protein production of the type III IFN IL-28 following stimulation by all of the viruses (Fig. 2C).
FIG 2.
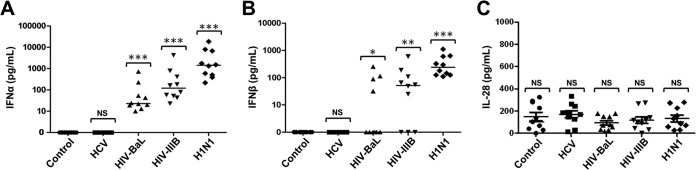
Differential protein production of type I and III IFNs in pDCs treated with H1N1 influenza virus, HIV-1 IIIB, HIV-1 BaL, or HCV. ELISA of supernatants from pDCs from 10 separate donors treated with PBS (control), HCV at 1.00 × 106 RNA copies/ml, HIV-1 BaL 1.00 ×105 TCID50/ml (Advanced Biotechnologies), HIV-1 IIIB at 1.00 ×105 TCID50/ml (Advanced Biotechnologies), or influenza virus A/PR/8/34 (H1N1; Charles River Laboratories) at 4 HAU/ml. (A) IFN-α protein. (B) IFN-β protein. (C) IL-28 protein. *, P < 0.05; **, P < 0.01; ***, P < 0.005 (determined by ratio paired t test compared with the corresponding control). NS, not significant.
Since we observed strong activation of the antiviral response following stimulation with H1N1 influenza virus and HIV, we next further characterized the changes in global gene expression in pDCs following stimulation with these viruses at titers that elicit maximum IFN-α stimulation. Figure S3 in the supplemental material shows that although these viruses stimulate the upregulation and production of type I IFNs (Fig. 1 and 2), they elicit distinct changes in global gene expression. Specifically, when H1N1 influenza virus and HIV-1 IIIB were compared, H1N1 influenza virus induced primarily IFNs and ILs whereas HIV-1 IIIB induced genes related to antigen presentation and regulation of transcription (see Fig. S3A and Table S1A in the supplemental material). When HIV-1 IIIB-induced changes were compared to those found following stimulation with HIV-1 BaL, there were very similar changes in the transcriptome; however, of the few genes that were differentially regulated, it appeared that HIV-1 IIIB elicited a more potent immune response in pDCs (see Fig. S3B and Table S1B). This result mirrors data from Fig. 2A and B, where HIV-1 IIIB triggered greater IFN production than HIV-1 BaL. Figure S3C and Table S1C further demonstrate that HIV-1 BaL was the weaker immune stimulator of the two HIV strains examined. Analysis of the transcriptome following HCV stimulation of pDCs demonstrated minimal differences from the untreated controls (data not shown).
To further characterize the difference in the changes in the transcriptome following stimulation with these viruses, we determined the number of differentially expressed genes. Using ANOVA (change, 1.5-fold; P < 0.05), we found 717, 503, and 177 genes to be differentially expressed between uninfected controls and H1N1 influenza virus-, HIV-1 IIIB-, and HIV-1 BaL-infected pDCs, respectively. Subsequently, we used these gene lists to identify genes whose expression patterns overlap between the samples stimulated with the distinct viruses. Overall, 155 genes were found to be differentially expressed following infection with both H1N1 influenza virus and HIV (Fig. 3A). Ingenuity pathway analysis of these 155 genes revealed an enrichment of the genes in various biological processes. As expected, the antiviral IFN response was the most upregulated pathway observed, with many other affected processes associated mainly with inflammation and ancillary viral responses (Fig. 3B).
FIG 3.

Changes in gene expression and biologic pathways affected following infection with H1N1 influenza virus and HIV. pDCs were stimulated with influenza virus A/PR/8/34 (H1N1; Charles River Laboratories) at 4 HAU/ml, HIV-1 IIIB (Advanced Biotechnologies) at 1.00 ×105 TCID50/ml, or HIV-1 BaL (Advanced Biotechnologies) at 1.00 ×105 TCID50/ml. (A) Venn diagram displaying the numbers of differentially expressed genes in control versus virus-stimulated pDCs. The value next to each virus name is the total number of differentially expressed genes versus the uninfected control. (B) Top canonical biological pathways associated with the overlapping 155 differentially expressed genes in virus-stimulated samples. The stacked bar chart displays for each canonical pathway the number of genes found to be significantly upregulated (red) or downregulated (green) by Bayesian model selection. The molecules or genes in a given pathway that were not found on our list of significantly regulated genes are termed not overlapping with our data set (white). The value at the top of each bar represents the total number of genes or molecules in the canonical pathway. The Benjamini-Hochberg (B-H) method was used to adjust the right-tailed Fisher exact t test P values, which measure how significant each pathway is.
Since we observed that, specifically for H1N1 influenza virus, there was a difference in the antiviral response involving IFN-α gene expression between the low titer of 4 HAU/ml and the high titer of 2,048 HAU/ml, we chose to perform an additional analysis of the data set obtained by microarray analysis of these RNA samples. Figure 4 reports the most significant biological pathways affected and the genes with the greatest changes in gene expression that were specifically altered by low- or high-titer influenza virus infection. The low titer of H1N1 influenza virus clearly elicited a strong antiviral response characterized by the activation of innate immune pathways, specifically, many known IFN-related gene networks (Fig. 4A). Interestingly, the high titer of H1N1 influenza virus stimulated a distinct change in the transcriptome with the absence of induction of the IFN-α family members (Fig. 1D). To explore why IFN-α gene induction was absent when the high titer of H1N1 influenza virus was used, Ingenuity pathway analysis of differentially expressed genes showed gene expression patterns related to apoptosis signaling and mitochondrial dysfunction that can subsequently lead to cell death (Fig. 4B).
FIG 4.

Microarray analysis of pDCs infected with H1N1 influenza virus. Gene lists are based on two-way ANOVAs comparing low-titer H1N1 influenza virus with the control (A) and high-titer H1N1 influenza virus with the control (B). Change, 1.5-fold (P < 0.01).
To further investigate the effect of high-titer influenza virus infection and the possible induction of apoptosis in pDCs, flow cytometry was used in conjunction with cell death markers to investigate this observation. In order to also rule out a contamination effect due to manufacturing, we used H1N1 influenza virus isolates from three different sources (Fig. 5). All three viruses stimulated the production of similar amounts of IFN-α protein by infected pDCs, and the levels of these protein from pDCs isolated from three separate donors were consistently high (Fig. 5A). Interestingly, we observed an enlarged population of pDCs following influenza virus treatment that was not present in untreated samples (Fig. 5B and C). In the influenza virus-treated samples, three distinct populations based on size were present and were designated populations I (cell debris/apoptotic remnants), II, and III (Fig. 5C). Following gating of the samples, we labeled them with the pDC marker BDCA-4 and with the corresponding cell death markers annexin V and 7AAD to distinguish between early and late apoptosis, respectively. Compared with untreated controls, the cells in gate III demonstrated greater staining for early apoptosis markers than those in gate II (Fig. 5D). Similarly, compared to untreated controls, the cells in gate III demonstrated greater staining for late apoptosis than those in gate II (Fig. 5E). Since this experiment was performed with the highest titer of H1N1 influenza virus, we also investigated the effect of the lowest titer of H1N1 influenza virus used on pDC cell size. Figure 6 demonstrates that although there were similar effects on cell size following treatment with both the high and low titers of H1N1 influenza virus, it was only the high titer that resulted in marked induction of apoptosis. This observation was confirmed through the significant increase in cell debris and apoptotic remnants in population I and the decrease in viable cells in population II (Fig. 6B), further confirming the data provided in Fig. 4. To further characterize populations I, II, and III and to validate 7AAD staining, imaging flow cytometry of stimulated pDCs stained with Hoechst 33342 and PI was performed. Figure 6C and D demonstrate that the greater cell death in population III than in population II following treatment with either a high or a low titer is associated with the enlargement of individual dying cells, thus excluding the possibility that the larger size was due to the formation of cell agglomerates. In addition, PI staining confirmed that the cells in gate I were dead cells and debris.
FIG 5.

Increased apoptosis in pDCs infected with a high titer of H1N1 influenza virus. pDCs from three separate donors were stimulated with PBS (control), influenza virus A/PR/8/34 (H1N1-1; Charles River Laboratories) at 2,048 HAU/ml, influenza virus A/PR/8/34 (H1N1-2; Advanced Biotechnologies) at 2,048 HAU/ml, or influenza virus A/PR/8/34 (H1N1-3; ATCC) at 105.2 50% chicken embryo infective doses/ml. (A) Quantitation of IFN-α protein by ELISA of supernatants from pDCs following infection with H1N1-1, H1N1-2, and H1N1-3 influenza viruses. Representative flow cytometry side scatter versus forward scatter plot of untreated pDCs (B) and H1N1 influenza virus-treated pDCs (C). (D) Flow cytometry plots of BDCA- and annexin V-stained pDCs treated with PBS or H1N1-1, H1N1-2, or H1N1-3 influenza virus. (e) Flow cytometry plots of BDCA-4 and 7AAD double-stained pDCs treated with PBS or H1N1-1, H1N1-2, or H1N1-3 influenza virus. ***, P < 0.005 (determined by ratio paired t test compared with the corresponding control). ND, not detectable.
FIG 6.

pDC enlargement in response to treatment with low and high titers of H1N1 influenza virus. pDCs from three separate donors were stimulated with PBS (control) or influenza virus A/PR/8/34 (H1N1-1; Charles River Laboratories) at 4 or 2,048 HAU/ml. (A) Quantitation of IFN-α protein by ELISA of supernatants from pDCs treated with PBS (control) or (H1N1; Charles River Laboratories) at 4 or 2,048 HAU/ml. (B) Flow cytometry side scatter versus forward scatter plot of pDCs treated with PBS (control) or (H1N1; Charles River Laboratories) at 4 or 2,048 HAU/ml. (C) Representative side scatter versus forward scatter plot of pDCs with an ImageStreamX imaging flow cytometer following H1N1 influenza virus stimulation. (D) Representative images of cell viability analysis of pDCs with an ImageStreamX imaging flow cytometer following H1N1 influenza virus stimulation. All nucleated cells were labeled with Hoechst 33342 (green). Dead cells were colabeled with both Hoechst 33342 and PI (red). Live cells were identified as PI negative. *, P < 0.05; **, P < 0.01 (determined by ratio paired t test compared with the corresponding control). SSC, side scatter.
To further investigate the mechanism of influenza virus-induced pDC apoptosis, we also used mutant H1N1 influenza virus strains. Specifically, PB1-F2 KO and rescue H1N1 influenza virus strains were used to determine if the PB1-F2 viral protein, previously found to disrupt mitochondrial function in influenza virus-infected HeLa cells, contributes to the induction of apoptosis in infected pDCs (20). Following stimulation, the levels of IFN-α protein produced were consistently high among all of the H1N1 influenza virus-treated samples, including the mutant and rescue strains determined by ELISA (see Fig. S4A in the supplemental material). In addition, compared with untreated controls, as observed previously, the cells in population III had greater levels of early apoptosis markers in all H1N1 influenza virus-treated samples, including the mutant and rescue strain, than standard-size (population II) pDCs (see Fig. S4B and C). Similarly, the number of cells undergoing late apoptosis because of both the PB1-F2 KO and rescue strains mirrored that of the low- and high-titer wild-type influenza virus-treated samples (see Fig. S4C), indicating that the PB1-F2 protein caused minimally increased apoptosis in pDCs.
DISCUSSION
pDCs are critically important for the initiation and propagation of a successful immune response to viral pathogens (21). One of the mechanisms by which they accomplish this is the robust production of type I IFNs following the detection of viral PAMPs (9). Here, we compared the responses of pDCs to three distinct viral pathogens in order to characterize the manner in which these cells attempt to control these diverse pathogens. Two of these RNA viruses (HIV and HCV) can cause chronic infections, while influenza virus causes an acute infection that is subsequently cleared by the human immune system in most instances (22).
We initiated the study examining the production of the type I IFN, namely, IFN-α. Interestingly, we observed distinct responses in pDCs following stimulation with the viruses tested. As expected, both influenza A virus and HIV stimulated a robust antiviral response in the pDCs that was characterized by massive upregulation of IFN-α mRNA and subsequent protein production (Fig. 1) (19). Interestingly, there were significant differences between these antiviral responses. In the case of HIV, the antiviral response became stronger as the infective dose of HIV used to stimulate the pDCs was increased at both the mRNA and protein levels (Fig. 1B and E). Surprisingly, following exposure of pDCs to influenza virus, we observed the highest production of type I IFN mRNA at the lowest titer used in this study (Fig. 1A). Furthermore, the amount of IFN-α produced when a higher titer of influenza virus was used did not increase significantly, as was observed with HIV (Fig. 1D and E). To confirm the effect of HIV on pDC IFN production, we used two different strains of HIV. In addition, the second strain of HIV was included to account for potential differences between HIV strains R5 and X4. When found in HIV-infected patients, X4-trophic viruses (HIV-1 IIIB) can lead to accelerated disease progression with a rapid decline of CD4+ and CD8+ T cells compared to that in patients infected with R5 strains (HIV-1 BaL) (23, 24). Our data showing increased upregulation of several immune-related genes in HIV-1 IIIB-infected versus HIV-1 BaL-infected pDCs (see Fig. S3b in the supplemental material) may provide insight into these clinical findings; however, further studies are needed to clarify this.
In contrast, of the three viruses, minimal production of IFN was seen in pDCs exposed to high titers of the HCV virus. HCV is known to infect hepatocytes exclusively (25); however; it was predicted that pDCs, with their robust expression of a diverse array or PRRs, would be able to detect distinct components of this virus regardless of whether the cells were directly infected with the subsequent product of replication intermediates. Surprisingly, minimal stimulation was observed following global transcriptome analysis in these cells exposed to HCV (Fig. 1C). On the basis of these data, one can speculate that the HCV virion has potent mechanisms to suppress the activation of pDC innate immune responses that can usually detect most other pathogens. Alternatively, pDCs may simply be incapable of detecting HCV-associated PAMPs unless the contact is mediated through other intermediary mechanisms (26). Stone et al. have recently demonstrated that components of the HCV genome can stimulate IFN production in pDCs (27). This publication is significant because an indirect transfer of viral genetic material may be occurring in vivo between pDCs and HCV-infected hepatocytes that may trigger IFN production from pDCs.
When mRNA levels of the IFN-α family were specifically examined, there was a decrease in mRNA upregulation when the highest titers of influenza virus were used (Fig. 1A). To explore this further, we used Ingenuity pathway analysis of the microarray data, which subsequently demonstrated that pDCs infected with high titers of influenza virus may undergo programmed cell death (Fig. 4B). In order to confirm this, we used flow cytometry and three different isolates of H1N1 influenza virus and saw that, indeed, exposure of pDCs to a high virus titer results in cell death (Fig. 5). To characterize the mechanism by which influenza A virus is able to do this, we used mutant influenza viruses lacking viral components that are known to trigger cell death (20) and we were unable to observe that the PB1-F2 viral protein is important for triggering cell death in pDCs (see Fig. S4 in the supplemental material). However, it was demonstrated that an influenza vaccine can also cause cell death in pDCs, further demonstrating the ability of viral components to trigger this phenomenon (data not shown).
The triggering of influenza virus-activated pDC cell death is biologically relevant since it has been demonstrated that individuals who succumb to influenza virus infection do so following the triggering of a virus-induced cytokine storm (28). This influenza virus-induced apoptotic pDC death may be one mechanism by which the immune system attempts to minimize the overproduction of antiviral and inflammatory cytokines following infection. In addition, a recently published study (29) demonstrated that type I IFN itself can result in decreased numbers of pDCs in mice. Therefore, we propose that pDCs, after the initial detection of a large amount of a viral pathogen and subsequent type I IFN secretion, negatively regulate their own numbers to ensure that only limited amounts of IFN are generated. This may be facilitated both through IFN signaling (30) and also by the more rapid activation of a direct apoptotic program that causes cell enlargement as an intermediary state toward the removal of the pDCs that are infected with a large quantity of virus. We hypothesize that in most individuals, this pathway is sufficient to minimize cytokine responses in instances where a large pathogen inoculum is encountered; however, in some individuals, a cytokine storm is produced from cells of the innate immune system that results in the death of the host (28). In addition, airway epithelium cell death has also been reported to occur during influenza virus infection. This virus-triggered cell death is thought to serve multiple purposes in host protection, including removal of the source of replicating virus. The death of these cells infected with high titers of virus will, in turn, both limit the further spread of the virus in the respiratory tract and decrease the levels of potentially damaging proinflammatory cytokines (31). Further study of this phenomenon in pDCs is warranted, as deficiencies in these and similar pathways to limit an immune response may also result in autoimmunity and perhaps other pathological states in humans.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Jonathan W. Yewdell and T. Jake Liang for their expert consultation toward the completion of this work.
This work was completed through funds from the NIH intramural research program. E.T. acknowledges support from the Miami Center for AIDS Research (CFAR) at the University of Miami Miller School of Medicine funded by an NIH grant (P30AI073961).
We have no conflicts of interest to declare.
Footnotes
Published ahead of print 9 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01501-14.
REFERENCES
- 1.Kumar H, Kawai T, Akira S. 2009. Pathogen recognition in the innate immune response. Biochem. J. 420:1–16. 10.1042/BJ20090272 [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. 2009. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 21:317–337. 10.1093/intimm/dxp017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newton K, Dixit VM. 2012. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 4(3):a006049. 10.1101/cshperspect.a006049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Salazar-Mather TP, Hokeness KL. 2003. Calling in the troops: regulation of inflammatory cell trafficking through innate cytokine/chemokine networks. Viral Immunol. 16:291–306. 10.1089/088282403322396109 [DOI] [PubMed] [Google Scholar]
- 5.Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. 2007. Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 6:975–990. 10.1038/nrd2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gale M, Jr, Sen GC. 2009. Viral evasion of the interferon system. J. Interferon Cytokine Res. 29:475–476. 10.1089/jir.2009.0078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darnell JE., Jr 1998. Studies of IFN-induced transcriptional activation uncover the Jak-Stat pathway. J. Interferon Cytokine Res. 18:549–554. 10.1089/jir.1998.18.549 [DOI] [PubMed] [Google Scholar]
- 8.Zhang Z, Wang FS. 2005. Plasmacytoid dendritic cells act as the most competent cell type in linking antiviral innate and adaptive immune responses. Cell Mol. Immunol. 2:411–417 [PubMed] [Google Scholar]
- 9.Szabo G, Dolganiuc A. 2008. The role of plasmacytoid dendritic cell-derived IFN alpha in antiviral immunity. Crit. Rev. Immunol. 28:61–94. 10.1615/CritRevImmunol.v28.i1.40 [DOI] [PubMed] [Google Scholar]
- 10.Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. 2011. Plasmacytoid dendritic cells: recent progress and open questions. Annu. Rev. Immunol. 29:163–183. 10.1146/annurev-immunol-031210-101345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Swiecki M, Cella M, Alber G, Schreiber RD, Gilfillan S, Colonna M. 2012. Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe 11:631–642. 10.1016/j.chom.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. 2010. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity 33:955–966. 10.1016/j.immuni.2010.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lui G, Manches O, Angel J, Molens JP, Chaperot L, Plumas J. 2009. Plasmacytoid dendritic cells capture and cross-present viral antigens from influenza-virus exposed cells. PLoS One 4(9):e7111. 10.1371/journal.pone.0007111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolf AI, Buehler D, Hensley SE, Cavanagh LL, Wherry EJ, Kastner P, Chan S, Weninger W. 2009. Plasmacytoid dendritic cells are dispensable during primary influenza virus infection. J. Immunol. 182:871–879. 10.4049/jimmunol.182.2.871 [DOI] [PubMed] [Google Scholar]
- 15.Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. 2007. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform. 3:11–17 [PMC free article] [PubMed] [Google Scholar]
- 16.Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R, Botstein D, Altman RB. 2001. Missing value estimation methods for DNA microarrays. Bioinformatics 17:520–525. 10.1093/bioinformatics/17.6.520 [DOI] [PubMed] [Google Scholar]
- 17.Benito M, Parker J, Du Q, Wu J, Xiang D, Perou CM, Marron JS. 2004. Adjustment of systematic microarray data biases. Bioinformatics 20:105–114. 10.1093/bioinformatics/btg385 [DOI] [PubMed] [Google Scholar]
- 18.Chung E, Amrute SB, Abel K, Gupta G, Wang Y, Miller CJ, Fitzgerald-Bocarsly P. 2005. Characterization of virus-responsive plasmacytoid dendritic cells in the rhesus macaque. Clin. Diagn. Lab. Immunol. 12:426–435. 10.1128/CDLI.12.3.426-435.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo CC, Schwartz JA, Johnson DJ, Yu M, Aidarus N, Mujib S, Benko E, Hyrcza M, Kovacs C, Ostrowski MA. 2012. HIV delays IFN-alpha production from human plasmacytoid dendritic cells and is associated with SYK phosphorylation. PLoS One 7(5):e37052. 10.1371/journal.pone.0037052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibbs JS, Malide D, Hornung F, Bennink JR, Yewdell JW. 2003. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J. Virol. 77:7214–7224. 10.1128/JVI.77.13.7214-7224.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Swiecki M, Colonna M. 2010. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol. Rev. 234:142–162. 10.1111/j.0105-2896.2009.00881.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Virgin HW, Wherry EJ, Ahmed R. 2009. Redefining chronic viral infection. Cell 138:30–50. 10.1016/j.cell.2009.06.036 [DOI] [PubMed] [Google Scholar]
- 23.Rosenberg YJ, Anderson AO, Pabst R. 1998. HIV-induced decline in blood CD4/CD8 ratios: viral killing or altered lymphocyte trafficking? Immunol. Today 19:10–17. 10.1016/S0167-5699(97)01183-3 [DOI] [PubMed] [Google Scholar]
- 24.Wilkin TJ, Su Z, Kuritzkes DR, Hughes M, Flexner C, Gross R, Coakley E, Greaves W, Godfrey C, Skolnik PR, Timpone J, Rodriguez B, Gulick RM. 2007. HIV type 1 chemokine coreceptor use among antiretroviral-experienced patients screened for a clinical trial of a CCR5 inhibitor: AIDS Clinical Trial Group A5211. Clin. Infect. Dis. 44:591–595. 10.1086/511035 [DOI] [PubMed] [Google Scholar]
- 25.Thomas E, Gonzalez VD, Li Q, Modi AA, Chen W, Noureddin M, Rotman Y, Liang TJ. 2012. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology 142:978–988. 10.1053/j.gastro.2011.12.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takahashi K, Asabe S, Wieland S, Garaigorta U, Gastaminza P, Isogawa M, Chisari FV. 2010. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. U. S. A. 107:7431–7436. 10.1073/pnas.1002301107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stone AE, Giugliano S, Schnell G, Cheng L, Leahy KF, Golden-Mason L, Gale M, Jr, Rosen HR. 2013. Hepatitis C virus pathogen associated molecular pattern (PAMP) triggers production of lambda-interferons by human plasmacytoid dendritic cells. PLoS Pathog. 9(4):e1003316. 10.1371/journal.ppat.1003316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. 2012. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 76:16–32. 10.1128/MMBR.05015-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swiecki M, Wang Y, Vermi W, Gilfillan S, Schreiber RD, Colonna M. 2011. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J. Exp. Med. 208:2367–2374. 10.1084/jem.20110654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swiecki M, Colonna M. 2011. Type I interferons: diversity of sources, production pathways and effects on immune responses. Curr. Opin. Virol 1:463–475. 10.1016/j.coviro.2011.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanders CJ, Doherty PC, Thomas PG. 2011. Respiratory epithelial cells in innate immunity to influenza virus infection. Cell Tissue Res. 343:13–21. 10.1007/s00441-010-1043-z [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.