

ABSTRACT
The herpes simplex virus 1 (HSV-1) UL12 protein (pUL12) is a nuclease that is critical for viral replication in vitro and neurovirulence in vivo. In this study, mass spectrometric analysis of pUL12 and phosphate-affinity SDS-polyacrylamide gel electrophoresis analysis identified tyrosine at pUL12 residue 371 (Tyr-371) as a pUL12 phosphorylation site: Tyr-371 is conserved in pUL12 homologs in herpesviruses in all Herpesviridae subfamilies. Replacement of Tyr-371 with phenylalanine (Y371F) in pUL12 (i) abolished its exonuclease activity in HSV-1-infected Vero, HEL, and A549 cells, (ii) reduced viral replication, cell-cell spread, and pUL12 expression in infected cells in a cell type-dependent manner, (iii) led to aberrant subcellular localization of pUL12 in infected cells in a cell type-dependent manner, and (iv) reduced HSV-1 neurovirulence in mice. The effects of the pUL12 Y371F mutation in cell cultures and mice were similar to those of a nuclease-dead double mutation in pUL12, although the Y371F mutation reduced viral replication severalfold more than the nuclease-dead double mutation in a cell type- and multiplicity-of-infection-dependent manner. Replacement of Tyr-371 with glutamic acid, which mimics constitutive phosphorylation, restored the wild-type phenotype in cell cultures and mice. These results suggested that phosphorylation of pUL12 Tyr-371 was essential for pUL12 to express its nuclease activity in HSV-1-infected cells and that this phosphorylation promoted viral replication and cell-cell spread in cell cultures and neurovirulence in mice mainly by upregulating pUL12 nuclease activity and, in part, by regulating the subcellular localization and expression of pUL12 in HSV-1-infected cells.
IMPORTANCE Herpesviruses encode a considerable number of enzymes for their replication. Like cellular enzymes, the viral enzymes need to be properly regulated in infected cells. Although the functional aspects of herpesvirus enzymes have gradually been clarified, information on how most of these enzymes are regulated in infected cells is lacking. In the present study, we report that the enzymatic activity of the herpes simplex virus 1 alkaline nuclease pUL12 was regulated by phosphorylation of pUL12 Tyr-371 in infected cells and that this phosphorylation promoted viral replication and cell-cell spread in cell cultures and neurovirulence in mice, mainly by upregulating pUL12 nuclease activity. Interestingly, pUL12 and tyrosine at pUL12 residue 371 appeared to be conserved in all herpesviruses in the family Herpesviridae, raising the possibility that the herpesvirus pUL12 homologs may also be regulated by phosphorylation of the conserved tyrosine residue.
INTRODUCTION
The herpes simplex virus 1 (HSV-1) UL12 gene encodes a protein (pUL12) that is phosphorylated in infected cells (1–3). The UL12 phosphoprotein has both endo- and exonuclease activities, with an alkaline pH optimum, and is conserved in herpesviruses in all Herpesviridae subfamilies (3–5). pUL12 has been reported to play a critical role in HSV-1 replication in vitro and in HSV-1 virulence in vivo, based on studies showing that recombinant UL12-null mutant viruses have 100- to 10,000-fold less viral growth in cell cultures and about a 1,000-fold reduction in neurovirulence in mice following intracerebral inoculation (6–8). Since HSV-1 pUL12 has long been recognized primarily as a nuclease, most research on pUL12 to date has concentrated on possible pUL12 functions based on its nuclease activities (5, 9–13). However, we recently reported that a recombinant HSV-1 strain carrying a nuclease-dead double mutation in pUL12 that abolished its endo- and exonuclease activities (5) had only a small (i.e., severalfold) though consistent reduction in its progeny virus yield in Vero simian kidney epithelial cells, while a recombinant UL12-null mutant virus had about a 10,000-fold reduction in its progeny virus yield (8). In contrast, the recombinant virus with the nuclease-dead double mutation in pUL12 showed a 100-fold reduction in neurovirulence in mice following intracerebral inoculation, while the recombinant UL12-null mutant virus showed about a 1,000-fold reduction in virulence (8). These observations suggested that the nuclease activities of pUL12 played a minor but consistent role in viral replication in cell cultures and that a pUL12 function(s) unrelated to its nuclease activities played a major role in this process. pUL12 nuclease activities have also been suggested to be critical for viral neurovirulence in vivo. Thus, pUL12 appears to have two distinct kinds of activities, nuclease activities and unrelated activities, although the latter are unknown at present.
While the functional roles of pUL12 have gradually been clarified as described above, information on the mechanism(s) by which pUL12 is regulated in HSV-1-infected cells is lacking. The intracellular activity of cellular and viral enzymes is usually tightly regulated: they can be turned on or off by phosphorylation, the binding of a regulatory subunit(s), the presence of small molecules, and/or their subcellular localization. For example, it has been reported that the catalytic activity of the HSV-1 protein kinase Us3 was tightly regulated by autophosphorylation of its serine 147 residue and that the regulation of Us3 activity by this autophosphorylation played a critical role in HSV-1 replication in vivo and in HSV-1 pathogenesis (14). Therefore, data on both the mechanism(s) by which an enzyme's activity is regulated and the downstream effects of the enzyme's regulation are necessary for understanding of the overall features of the enzyme. In the studies presented here, we investigated whether the enzymatic activity of pUL12 was regulated by phosphorylation in HSV-1-infected cells. Using liquid chromatography-tandem mass spectrometry (LC–MS-MS) analysis, we identified three phosphorylation sites in pUL12. Of these, we focused on tyrosine at pUL12 residue 371 (Tyr-371), since it is conserved in UL12 homologs in the herpesviruses of all Herpesviridae subfamilies (5, 13). Our studies of the effects of pUL12 Tyr-371 phosphorylation showed that it was essential for the expression of pUL12 exonuclease activity in HSV-1-infected cells and that it was required for efficient viral replication, cell-cell spread, and proper steady-state expression and subcellular localization of pUL12 in a cell type-dependent manner. We also showed that this phosphorylation was required for efficient viral neurovirulence in mice following intracerebral inoculation. These results suggested that the nuclease activity of pUL12 was regulated by its phosphorylation at Tyr-371 and that this regulation played an important role in viral replication and pathogenesis.
MATERIALS AND METHODS
Cells and viruses.
Vero, 293T, HEL, and A549 cells have been described previously (8, 15–17). 6-5 cells (6) are permissive for UL12-null mutant viruses and were kindly provided by S. Weller. The following virus strains have been described previously: the wild-type strain, HSV-1(F); recombinant virus YK655 (ΔUL12), a UL12-null mutant virus in which the UL12 gene was disrupted by replacing UL12 codons 70 to 375 with a kanamycin resistance gene; recombinant virus YK656 (ΔUL12-repair), in which the UL12-null mutation in YK655 was repaired; recombinant virus YK665 (UL12G336A/S338A), encoding a nuclease-inactive UL12 mutant in which the amino acids glycine and serine at pUL12 residues 336 and 338 were replaced with alanine (G336A S338A); and recombinant virus YK666 (UL12GA/SA-repair), in which the UL12 G336A S338A double mutation in YK665 was repaired (8, 16) (Fig. 1). All viruses used in this study were propagated and titrated using 6-5 cells.
FIG 1.
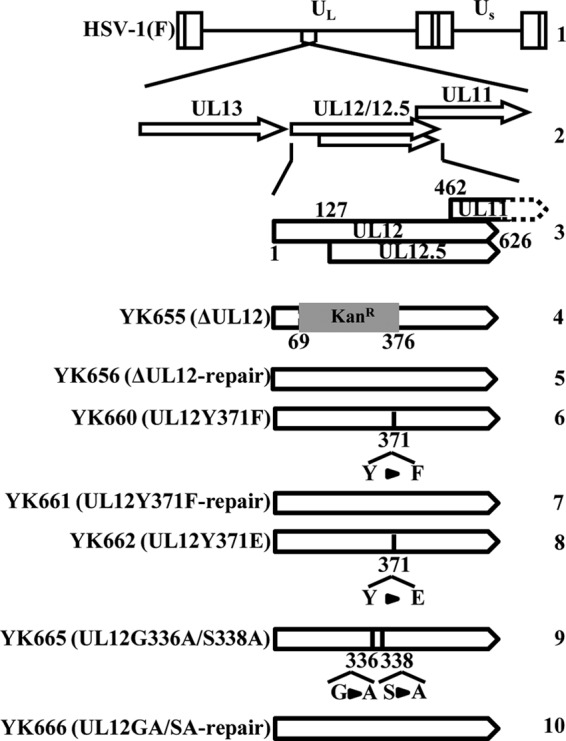
Schematic of the genome structures of the wild-type virus HSV-1(F) and the relevant domains of the recombinant viruses used in this study. Line 1, wild-type HSV-1(F) genome; line 2, domains containing ORFs UL11 to UL13; line 3, domains containing ORFs UL11, UL12, and UL12.5; lines 4 to 10, domains in recombinant virus genomes with mutations in UL12.
Plasmids.
To construct pcDNA-MEF-UL12, an expression plasmid for pUL12 fused to an MEF (Myc epitope–tobacco etch virus [TEV] protease cleavage site–Flag epitope) tag (18), the entire UL12 open reading frame (ORF) was amplified by PCR from pBC1012 (19) and was cloned into pcDNA-MEF (20) in frame with MEF (Fig. 2A). pcDNA-MEF-UL12Y371F and pcDNA-MEF-UL12G336A/S338A, expression plasmids for MEF-tagged pUL12 with a phenylalanine replacement of Tyr-371 and alanine replacements of glycine 336 and serine 338 in the double mutant, respectively, were generated by amplifying the UL12 ORF by PCR from the YK660 (UL12Y371F) and YK665 (UL12G336A/S338A) genomes, respectively, which were purified as described previously (16), and cloning the DNA fragments into pcDNA-MEF in frame with MEF.
FIG 2.
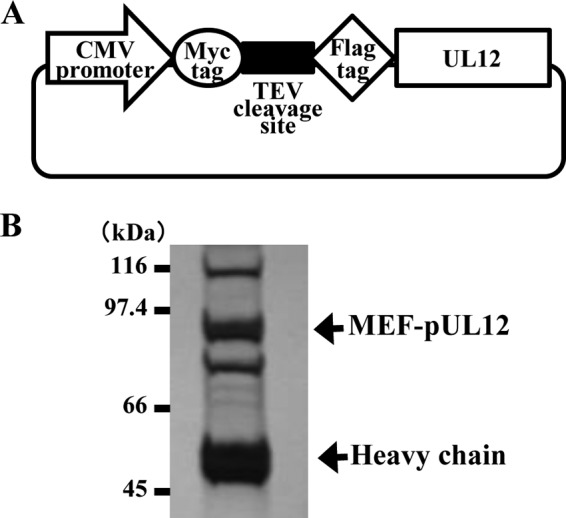
(A) Schematic of expression plasmid pcDNA-MEF-UL12, carrying UL12 tagged with MEF. (B) 293T cells were transfected with pcDNA-MEF-UL12, harvested, and immunoprecipitated with anti-Myc and anti-Flag antibodies. Immunoprecipitates were separated in a denaturing gel and were silver stained. The upper and lower arrows indicate MEF-pUL12 and an immunoglobulin heavy chain, respectively. Molecular mass markers are indicated on the left.
Identification of phosphorylation sites in pUL12.
293T cells were transfected with pcDNA-MEF-UL12 (Fig. 2A) using polyethylenimine as described previously (21), harvested at 48 h posttransfection, and lysed in 0.5% NP-40 buffer (0.5% NP-40, 120 mM NaCl, 50 mM Tris-HCl [pH 8.0], and 50 mM NaF) containing protease and phosphatase inhibitor cocktails (Nacalai Tesque). After centrifugation, the supernatants were immunoprecipitated with an anti-Myc monoclonal antibody, and the immunoprecipitates were reacted with AcTEV protease (Invitrogen). After another centrifugation, the supernatants were immunoprecipitated with an anti-Flag monoclonal antibody, and these immunoprecipitates were electrophoretically separated in a denaturing polyacrylamide gel and were visualized by silver staining. A protein band corresponding to pUL12 (Fig. 2B) was excised from the denaturing gel, digested in the gel with trypsin, and analyzed by nano-LC–MS-MS as described previously (22). For this analysis, we used the QSTAR Elite LC–MS-MS system (AB Sciex) coupled with the DiNa system (KYA Technologies). Proteins were identified by analyzing the MS and MS-MS data against the 68,711 protein sequences in the RefSeq human protein database (National Center for Biotechnology Information [NCBI]) and the 74 virus protein sequences based on the complete genome sequence of human herpesvirus 1 strain F (GenBank accession number GU734771) using the Mascot algorithm (version 2.4.1; Matrix Science) with the following parameters: variable modifications, oxidation (Met), protein N-terminal acetylation, pyroglutamination (Gln), phosphorylation (Ser, Thr, and Tyr); maximum missed cleavages, 2; peptide mass tolerance, 100 ppm; and MS-MS tolerance, 0.5 Da. Protein identification was based on the criterion of having at least one MS-MS data signal with a Mascot score that exceeded the threshold (P, <0.05). The locations of phosphorylated sites in the peptides were determined using the MD scores (23).
Antibodies, immunoblotting, and immunofluorescence.
Rabbit polyclonal antibodies to VP22 and pUL12 have been described previously (8, 24). The antibody to pUL12 reacts with pUL12 but not with pUL12.5, an amino-terminally truncated UL12 protein (4). Commercial mouse monoclonal antibodies against ICP8 (HB-8180 [ATCC] and 10A3 [Millipore]), the Flag epitope (M2; Sigma), α-tubulin (DM1A; Sigma), and Myc (PL14; MBL) were used in this study. The two mouse monoclonal antibodies against ICP8 (HB-8180 and 10A3) were used for immunofluorescence and immunoblotting, respectively. Immunoblotting and immunofluorescence were performed as described previously (19).
Phos-tag SDS-PAGE analysis.
Phos-tag (phosphate affinity) SDS-polyacrylamide gel electrophoresis (PAGE) analysis was performed according to the standard protocol of Wako Chemicals as described previously (25). Briefly, 293T cells were transfected with pcDNA-MEF-UL12, pcDNA-MEF-UL12G336A/S338A, or pcDNA-MEF-UL12Y371F using polyethylenimine as described above, harvested at 48 h posttransfection, solubilized in a buffer for λ protein phosphatase (λ-PPase) (New England BioLabs) containing 0.3% NP-40, 1 mM MnCl2, and a protease inhibitor cocktail (Nacalai Tesque), and analyzed by electrophoresis in denaturing gels containing 290 μM MnCl2 and 145 μM Phos-tag acrylamide (Wako) (26). After electrophoresis, the gels were soaked in standard transfer buffer containing 1 mM EDTA for 10 min to remove Mn2+. The separated proteins in the denaturing gels were then transferred to polyvinylidene difluoride membranes and were analyzed by immunoblotting.
Phosphatase treatment.
The lysates from 293T cells transfected with pcDNA-MEF-UL12, prepared as described above, were treated with λ-PPase (New England BioLabs) as described previously (27).
Mutagenesis of viral genomes in Escherichia coli and generation of recombinant HSV-1.
Recombinant virus YK660 (UL12Y371F), carrying a Y371F mutation in pUL12 (Fig. 1), was constructed by the two-step Red-mediated mutagenesis procedure using Escherichia coli GS1783 containing pYEbac102, a full-length infectious HSV-1(F) clone (16), as described previously (28) except for the use of primers 5′-GACCCCCCTAGCCTTTTACGAGGTCAAATGCCGGGCCAAGTTCGCTTTCGACCCCATGGACCCAGGATGACGACGATAAGTAGGG-3′ and 5′-CGGAGGCCGTGGGGTCGCTGGGGTCCATGGGGTCGAAAGCGAACTTGGCCCGGCATTTGACCTCAACCAATTAACCAATTCTGATTAG-3′. Recombinant virus YK661 (UL12Y371F-repair), in which the pUL12 Y371F mutation in YK660 was repaired (Fig. 1), was generated as described previously (28) except for the use of primers 5′-GACCCCCCTAGCCTTTTACGAGGTCAAATGCCGGGCCAAGTACGCTTTCGACCCCATGGACCCAGGATGACGACGATAAGTAGGG-3′ and 5′-CGGAGGCCGTGGGGTCGCTGGGGTCCATGGGGTCGAAAGCGTACTTGGCCCGGCATTTGACCTCAACCAATTAACCAATTCTGATTAG-3′ and E. coli GS1783 carrying the YK660 genome. Recombinant virus YK662 (UL12Y371E), encoding pUL12 with a glutamic acid substituted for Tyr-371 (Fig. 1), was generated as described previously (28) except for the use of primers 5′-GACCCCCCTAGCCTTTTACGAGGTCAAATGCCGGGCCAAGGAGGCTTTCGACCCCATGGACCCAGGATGACGACGATAAGTAGGG-3′ and 5′-CGGAGGCCGTGGGGTCGCTGGGGTCCATGGGGTCGAAAGCGTCCTTGGCCCGGCATTTGACCTCAACCAATTAACCAATTCTGATTAG-3′ and E. coli GS1783 carrying the YK660 genome. YK660 (UL12Y371F), YK661 (UL12Y371F-repair), and YK662 (UL12Y371E) were propagated and titrated in 6-5 cells.
Alkaline nuclease assays.
Extracts from mock-infected and HSV-1-infected cells were prepared as described previously (7, 29). Exonuclease assays were performed as described previously (7, 29).
Determination of plaque size.
Vero, HEL, and A549 cells were infected with each of the recombinant viruses at multiplicities of infection (MOI) of 0.0001, 0.001, and 0.00001, respectively. After adsorption for 1 h, the inoculum was removed, and the cell monolayers were overlaid with medium 199 containing 1% fetal calf serum and 160 μg pooled human immunoglobulin (Sigma)/ml. Two days postinfection, 25 plaques produced by each of the recombinant viruses were analyzed using a microscope equipped with a digital DP80 camera (Olympus) and cellSens software (Olympus).
Animal experiments.
Three-week-old female ICR mice were purchased from Charles River and were infected intracerebrally with serial 10-fold dilutions of each mutant virus in groups of 3 mice per dilution, as described previously (16). Mortality was monitored for 14 days postinfection, and 50% lethal dose (LD50) values were calculated by the Behrens-Karber method. All animal experiments were carried out in accordance with the Guidelines for Proper Conduct of Animal Experiments, Science Council of Japan. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Medical Science, The University of Tokyo (IACUC protocol approval number 19-26).
RESULTS
Identification of Tyr-371 as a phosphorylation site in pUL12.
To identify phosphorylation sites in pUL12, we used tandem affinity purification of transiently expressed pUL12 in 293T cells, coupled with mass spectrometry-based technology (20, 22). As shown in Fig. 2B, MEF-tagged pUL12 (MEF-pUL12) was copurified with some cellular proteins, suggesting that pUL12 was tightly associated with the cellular proteins. Then the purified MEF-pUL12 was subjected to mass spectrometric analyses, and three phosphorylation sites in pUL12, at Tyr-371, Thr-474, and Ser-604, were identified (Table 1). Phosphorylation of pUL12 Ser-604 in HSV-1-infected cells has been reported previously (30). Of these three phosphorylation sites, we focused on Tyr-371 because it, but not Thr-474 or Ser-604, is conserved in pUL12 homologs in herpesviruses in all Herpesviridae subfamilies (Fig. 3).
TABLE 1.
pUL12 phosphopeptides and phosphorylation sites identified by LC–MS-MSa
| Phosphorylation site | Peptide sequence | Mascot score |
|---|---|---|
| Y371 | pYAFDPMDPSDPTASAYEDLMAHR | 65 |
| T474 | HpTISPVSWSSGDLVR | 49 |
| S604 | pSPGPGPAAAETTSSSPTTGR | 121 |
In the peptide sequences, p indicates phosphorylated amino acid. The Mascot score is −10 × log(P), where P is the probability that the observed match was a random event. A score of >37 indicated identity or extensive homology (P, <0.05).
FIG 3.

Sequence alignment of motif III of pUL12 homologs conserved in members of the three herpesvirus subfamilies. HSV-1 pUL12 Tyr-371 and the corresponding residues of other herpesviruses are boxed and indicated by an arrow. pUL12 and pUL12 homolog sequences are labeled with their NCBI gene identification numbers and virus names. HSV-2, herpes simplex virus 2; VZV, varicella-zoster virus; BHV-1, bovine herpesvirus 1; EHV-1, equine herpesvirus 1; PRV, pseudorabies virus; HCMV, human cytomegalovirus; HHV-6, human herpesvirus 6; HHV-7, human herpesvirus 7; EBV, Epstein-Barr virus; KSHV, Kaposi's sarcoma-associated herpesvirus; HVS, herpesvirus saimiri; EHV-2, equine herpesvirus 2; GHV-2, gallid herpesvirus 2. HSV-1, HSV-2, VZV, BHV-1, EHV-1, and PRV are in the subfamily Alphaherpesvirinae; HCMV, HHV-6, and HHV-7 are in the Betaherpesvirinae; and EBV, KSHV, HVS, EHV-2, and GHV-2 are in the Gammaherpesvirinae.
To confirm the phosphorylation of pUL12 at Tyr-371, we carried out phosphate affinity (Phos-tag) SDS-PAGE analysis of 293T cells transfected with pcDNA-MEF-UL12, pcDNA-MEF-UL12G336A/S338A, or pcDNA-MEF-UL12Y371F. Phos-tag forms a specific complex with phosphorylated amino acids and therefore enables the visualization of some, but not necessarily all, phosphorylated proteins as distinct band shifts relative to the unphosphorylated protein (26). As shown in Fig. 4A, MEF-pUL12 from 293T cells transfected with pcDNA-MEF-UL12 was detected as three bands (Fig. 4, bands a, b, and c) with distinct mobility differences in Phos-tag-positive [Phos-tag(+)] SDS-PAGE: bands a and b were dominant, and band c was very faint. After phosphatase treatment of the lysate from 293T cells transfected with pcDNA-MEF-UL12, bands a and b were barely detectable, while the amount of protein in band c increased dramatically (Fig. 4A). This result indicated that bands a and b represented phosphorylated forms of pUL12 with different numbers of phosphorylated residues. In contrast, band c appeared to represent the unphosphorylated form of pUL12, although we cannot eliminate the possibility that the pUL12 detected as band c was phosphorylated. As shown in Fig. 4B, in lysates of 293T cells transfected with pcDNA-MEF-UL12Y371F and analyzed by Phos-tag(+) SDS-PAGE, MEF-pUL12Y371F was predominantly in band b, and bands a and c were barely detectable. In contrast, in lysates of 293T cells transfected with pcDNA-MEF-UL12G336A/S338A and analyzed by Phos-tag(+) SDS-PAGE, the pattern of MEF-pUL12G336A/S338A bands was similar to that of MEF-pUL12 bands (Fig. 4B). Taken together, these results indicated that pUL12 Tyr-371 was phosphorylated in cell cultures.
FIG 4.
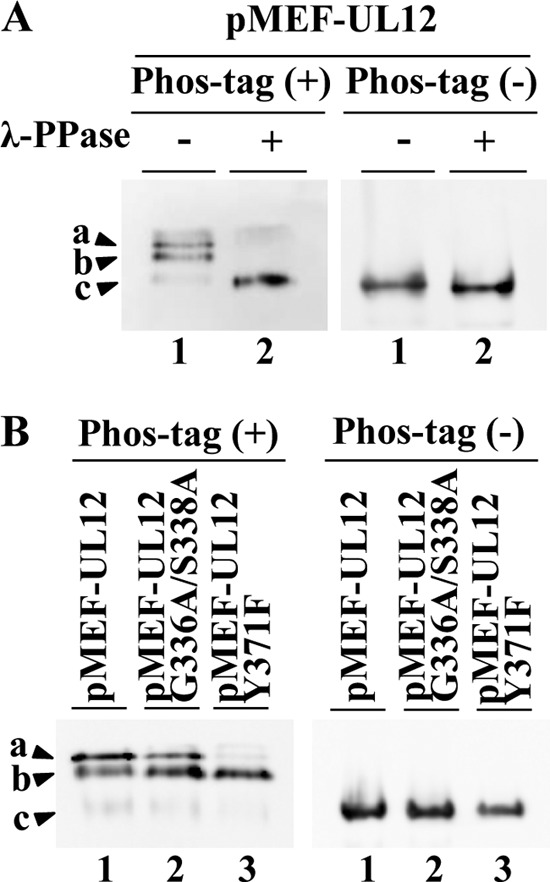
Phosphorylation of pUL12 Y371 in cell cultures. (A) 293T cells were transfected with plasmid pcDNA-MEF-UL12 (diagramed in Fig. 2A), harvested at 48 h posttransfection, and lysed. The lysates were either mock treated (lanes 1) or treated with λ-PPase (lanes 2), analyzed on a Phos-tag(+) (left) or a Phos-tag(−) (right) SDS-PAGE gel, and immunoblotted with an anti-Myc antibody. (B) 293T cells were transfected with plasmid pcDNA-MEF-UL12 (lanes 1), pcDNA-MEF-UL12G336A/S338A (lanes 2), or pcDNA-MEF-UL12Y371F (lanes 3), harvested at 48 h posttransfection, and lysed. The lysates were analyzed on a Phos-tag(+) (left) or a Phos-tag(−) (right) SDS-PAGE gel. The gels were immunoblotted with an anti-Myc antibody. The pUL12 bands detected in the Phos-tag(+) SDS-PAGE gels are labeled a, b, and c.
Effect of pUL12 Tyr-371 phosphorylation on UL12 expression.
To investigate the effect of Tyr-371 phosphorylation on UL12 expression in HSV-1-infected cells, we generated recombinant virus YK660 (UL12Y371F), encoding a mutant pUL12 in which Tyr-371 was replaced with phenylalanine, its repaired virus YK661 (UL12Y371F-repair), and YK662 (UL12Y371E), encoding a mutant pUL12 in which Tyr-371 was replaced with glutamic acid (Fig. 1). Replacement of the phosphorylation site with an acidic amino acid, such as glutamic acid or aspartic acid, is a phosphomimetic mutation (a mutation that mimics the negative charges produced by phosphorylation) (31–33). We first inquired whether phosphorylation of pUL12 Tyr-371 had an effect on UL12 expression in HSV-1-infected cells. For these experiments, Vero cells, HEL human lung fibroblasts, and A549 human lung epithelial cells were either mock infected or infected with either wild-type HSV-1(F), the UL12-null mutant virus YK655 (ΔUL12), its repaired virus YK656 (ΔUL12-repair), YK660 (UL12Y371F), its repaired virus YK661 (UL12Y371F-repair), YK662 (UL12Y371E), the UL12 nuclease-dead double mutant virus YK665 (UL12G336A/S338A), or its repaired virus YK666 (UL12GA/SA-repair) at an MOI of 3. At 12 h postinfection, cell lysates were analyzed by immunoblotting. As shown in Fig. 5, HEL cells infected with YK660 (UL12Y371F) produced much less pUL12 than HEL cells infected with wild-type HSV-1(F) or YK661 (UL12Y371F-repair), but HEL cells infected with YK662 (UL12Y371E) produced pUL12 at a level similar to that of HEL cells infected with wild-type HSV-1(F). These results indicated that an amino acid at pUL12 residue 371 that was negatively charged, due either to Tyr-371 phosphorylation or to the phosphomimetic Y371E substitution, was required for efficient expression of pUL12 in infected HEL cells.
FIG 5.
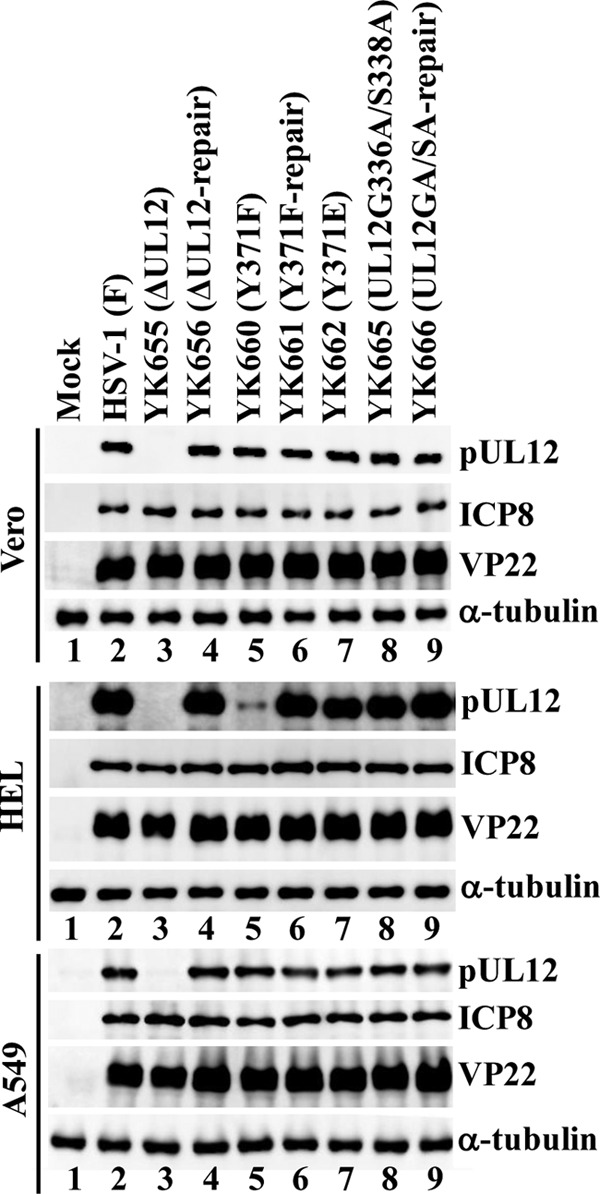
Effects of mutations in UL12 on steady-state expression levels of pUL12 and ICP8 in infected cells. Vero, HEL, and A549 cells were either mock infected (lanes 1) or infected with either wild-type HSV-1(F) (lanes 2), YK655 (ΔUL12) (lanes 3), YK656 (ΔUL12-repair) (lanes 4), YK660 (UL12Y371F) (lanes 5), YK661 (UL12Y371F-repair) (lanes 6), YK662 (UL12Y371E) (lanes 7), YK665 (UL12G336A/S338A) (lanes 8), or YK666 (UL12GA/SA-repair) (lanes 9) at an MOI of 3, harvested at 12 h postinfection, lysed, and analyzed by immunoblotting with antibodies to pUL12, ICP8, VP22, and α-tubulin.
In contrast, the levels of pUL12 expression in Vero and A549 cells infected with YK660 (UL12Y371F) were almost identical to those in these cells infected with wild-type HSV-1(F) or YK661 (UL12Y371F-repair) (Fig. 5). In agreement with our previous report (8), the UL12 nuclease-dead double mutation (UL12G336A/S338A) had no effect on UL12 expression in any of the cell types tested (Fig. 5). These results indicated that phosphorylation of UL12 at Tyr-371 was required for proper steady-state expression levels of pUL12 in HSV-1-infected cells in a cell type-dependent manner.
Effect of Tyr-371 phosphorylation on pUL12 enzymatic activity.
To examine whether the enzymatic activity of pUL12 was regulated by Tyr-371 phosphorylation in HSV-1-infected cells, we assayed exonuclease activity in Vero, HEL, and A549 cells that were either mock infected or infected at an MOI of 3 with either wild-type HSV-1(F), YK655 (ΔUL12), YK656 (ΔUL12-repair), YK660 (UL12Y371F), YK661 (UL12Y371F-repair), YK662 (UL12Y371E), YK665 (UL12G336A/S338A), or YK666 (UL12GA/SA-repair). Exonuclease activity was measured in lysates of the infected-cell cultures at 12 h postinfection. In agreement with previous reports (6–8), Vero cells that were mock infected or infected with YK655 (ΔUL12) or YK665 (UL12G336A/S338A) did not contain a significant amount of exonuclease activity, but there was considerable exonuclease activity in cells infected with wild-type HSV-1(F), YK656 (ΔUL12-repair), or YK666 (UL12GA/SA-repair) (Fig. 6A). In Vero cells infected with YK660 (UL12Y371F), exonuclease activity was significantly lower than that in cells infected with wild-type HSV-1(F) or YK661 (UL12Y371F-repair). The level of exonuclease activity in Vero cells infected with YK660 (UL12Y371F) was about the same as that in cells that were mock infected or infected with YK655 (ΔUL12) or YK665 (UL12G336A/S338A). In contrast, exonuclease activity in Vero cells infected with YK662 (UL12Y371E) was similar to that in cells infected with wild-type HSV-1(F) (Fig. 6A). Immunoblotting showed no pUL12 band in mock- or YK655 (ΔUL12)-infected Vero cell lysates but strong bands with similar amounts of pUL12 in all the other infected-cell lysates (Fig. 6B).
FIG 6.
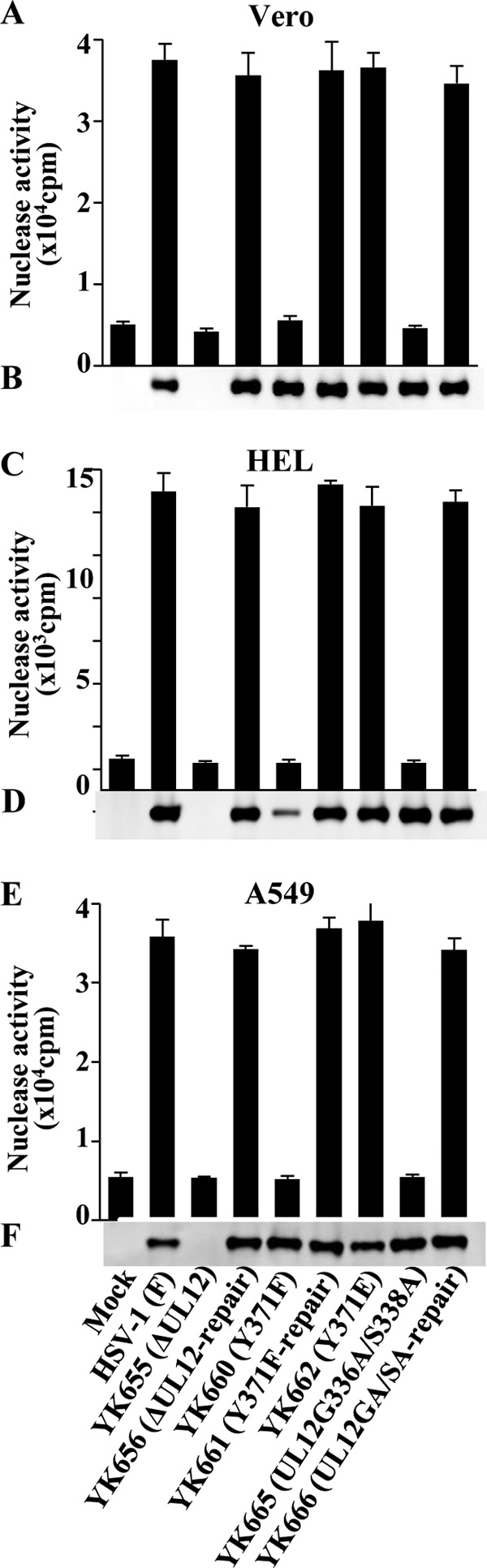
Effects of mutations in pUL12 on exonuclease activity in infected cells. (A, C, and E) Vero, HEL, and A549 cells were either mock infected or infected with either wild-type HSV-1(F), YK655 (ΔUL12), YK656 (ΔUL12-repair), YK660 (UL12Y371F), YK661 (UL12Y371F-repair), YK662 (UL12Y371E), YK665 (UL12G336A/S338A), or YK666 (UL12GA/SA-repair) at an MOI of 3, harvested at 12 h postinfection, lysed, and assayed for exonuclease activity. Each value is the mean ± standard error of the results of triplicate experiments. Data are representative of three independent experiments. (B, D, and F) Immunoblots of the lysates prepared for the assays in panels A, C, and E, respectively. The cell lysates were analyzed by immunoblotting with an anti-pUL12 antibody. Data are representative of three independent experiments.
Results similar to those observed in Vero cells were also found in infected HEL and A549 cells, except that the amount of pUL12 in YK660 (UL12Y371F)-infected HEL cell lysates was less than that in the other infected-cell lysates (Fig. 6C to F). This finding was in agreement with the data presented above (Fig. 5) showing that HEL cells infected with YK660 (UL12Y371F) produced less pUL12 than HEL cells infected with the wild-type virus or the other recombinant viruses. Taken together, these data suggested that phosphorylation of pUL12 at Tyr-371 was critical for the exonuclease activity of pUL12 in HSV-1-infected cells.
Effect of Tyr-371 phosphorylation on the subcellular localization of pUL12.
To examine whether phosphorylation of pUL12 at Tyr-371 regulated the subcellular localization of pUL12 and of ICP8, which interacts with pUL12 (34), in HSV-1-infected cells, Vero, HEL, and A549 cells were infected with either wild-type HSV-1(F), YK660 (UL12Y371F), YK661 (UL12Y371F-repair), or YK662 (UL12Y371E) at an MOI of 10 and were analyzed by immunofluorescence at 12 h postinfection. In agreement with previous reports (1, 35), pUL12 was localized diffusely throughout the nucleus, and ICP8 was detected as numerous punctate dots in the nucleus, in all of the cell types infected with wild-type HSV-1(F) or YK661 (UL12Y371F-repair) (Fig. 7). In contrast, the subcellular localization of pUL12 and ICP8 in Vero cells infected with YK660 (UL12Y371F) was significantly different from that in Vero cells infected with wild-type HSV-1(F) or YK661 (UL12Y371F-repair). In Vero cells infected with YK660 (UL12Y371F), pUL12 was detected as globular structures in the nucleus, and the ICP8 punctate dots appeared to be in aggregates in the nucleus (Fig. 7A). However, the localization of pUL12 and ICP8 in YK662 (UL12Y371E)-infected Vero cells was restored to the pattern in wild-type HSV-1-infected Vero cells (Fig. 7A).
FIG 7.
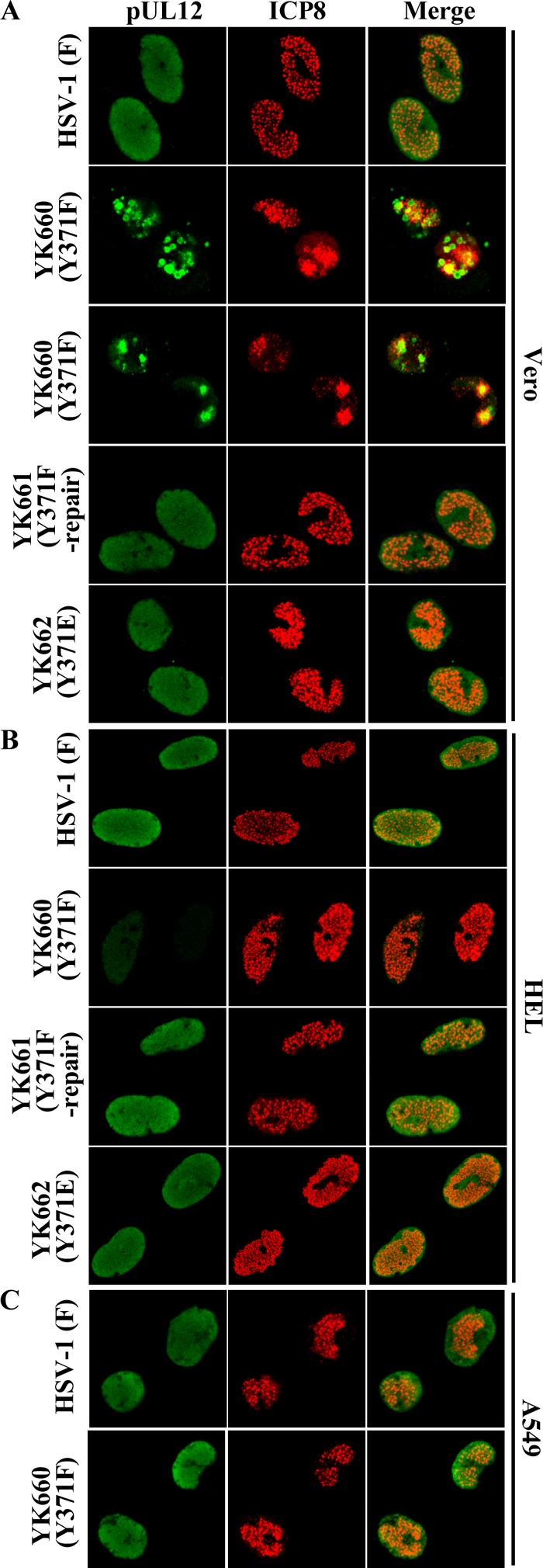
Effects of mutations in pUL12 on the subcellular localization of pUL12 and ICP8 in infected cells. (A and B) Vero (A) and HEL (B) cells were infected with wild-type HSV-1(F), YK660 (UL12Y371F), YK661 (UL12Y371F-repair), or YK662 (UL12Y371E). (C) A549 cells were infected with wild-type HSV-1(F) or YK660 (UL12Y371F). All cells were infected at an MOI of 10, fixed at 12 h postinfection, permeabilized, stained with an anti-pUL12 or anti-ICP8 antibody, and examined by confocal microscopy. Since the number of globular structures of pUL12 and the level of aggregation of ICP8 in the nucleus were different among cells infected with YK660 (UL12Y371F), two fields of these infected cells are shown here as examples of the aberrant localization observed.
In A549 cells infected with YK660 (UL12Y371F), the subcellular localization of pUL12 and ICP8 was similar to that in A549 cells infected with wild-type HSV-1(F) (Fig. 7C). In agreement with our observation above that the Y371F mutation in pUL12 reduced steady-state expression levels of pUL12 in HSV-1-infected cells, the fluorescence intensity of immunostained pUL12 in HEL cells infected with YK660 (UL12Y371F) was lower than that in HEL cells infected with wild-type HSV-1(F) or YK661 (UL12Y371F-repair) (Fig. 7B). However, the subcellular localization of pUL12 in HEL cells infected with YK660 (UL12Y371F) appeared to be similar to that in cells infected with wild-type HSV-1(F) or YK661 (UL12Y371F-repair) (Fig. 7B). In addition, the fluorescence intensity of immunostained ICP8 and its subcellular localization in HEL cells infected with YK660 (UL12Y371F) were similar to those in HEL cells infected with wild-type HSV-1(F) or YK661 (UL12Y371F-repair) (Fig. 7B). The pUL12 nuclease-dead double mutation (UL12 G336A S338A) had no effect on the localization of pUL12 and ICP8 in any of the cell types tested (data not shown). Taken together, these observations suggested that phosphorylation of pUL12 Tyr-371 was required for proper localization of both pUL12 and ICP8 in HSV-1-infected cells in a cell type-dependent manner.
Effect of the phosphorylation of pUL12 Tyr-371 on viral replication.
To examine the effect of the phosphorylation of pUL12 Tyr-371 on viral replication in cell cultures, we analyzed progeny virus yields in Vero, HEL, and A549 cells infected with wild-type HSV-1(F), YK655 (ΔUL12), YK656 (ΔUL12-repair), YK660 (UL12Y371F), YK661 (UL12Y371F-repair), YK662 (UL12Y371E), YK665 (UL12G336A/S338A), or YK666 (UL12GA/SA-repair) at an MOI of 0.01 for 48 h (Fig. 8A to C) or at an MOI of 3 for 24 h (Fig. 8D to F). As shown in Fig. 8A, the progeny virus yield in Vero cells infected with YK655 (ΔUL12) at an MOI of 0.01 was 11,000-fold less than that in Vero cells infected with wild-type HSV-1(F) and 3,700-fold less than that in Vero cells infected with YK656 (ΔUL12-repair). In contrast, the progeny virus yield in Vero cells infected with YK665 (UL12G336A/S338A) at an MOI of 0.01 was 21-fold less than that in Vero cells infected with wild-type HSV-1(F) and 6.7-fold less than that in Vero cells infected with YK666 (UL12GA/SA-repair) (Fig. 8A). Similar results were obtained with Vero cells infected with each of these viruses at an MOI of 3 (Fig. 8D). Thus, in agreement with our previous report (8), although the pUL12 nuclease-dead double mutation (UL12G336A/S338A) consistently reduced viral replication in Vero cells, the double mutation had much less effect than the UL12-null mutation on viral replication in Vero cells. Similarly, the pUL12 nuclease-dead double mutation (UL12 G336A S338A) had much less effect than the UL12-null mutation on viral replication in HEL and A549 cells at MOIs of 0.01 and 3; indeed, the nuclease-dead double mutation barely reduced viral replication in these cells (Fig. 8B, C, E, and F).
FIG 8.

Effects of mutations in pUL12 on progeny virus yields. Vero (A and D), HEL (B and E), and A549 (C and F) cells were infected with either wild-type HSV-1(F), YK655 (ΔUL12), YK656 (ΔUL12-repair), YK660 (UL12Y371F), YK661 (UL12Y371F-repair), YK662 (UL12Y371E), YK665 (UL12G336A/S338A), or YK666 (UL12GA/SA-repair) at an MOI of 0.01 (A to C) or 3 (D to F). Total virus from the cell culture supernatants and infected cells was harvested at 48 h (A to C) or 24 h (D to F) postinfection and was assayed on 6-5 cells. Each data point is the mean ± standard error of results for triplicate samples and is representative of three independent experiments.
In Vero cells infected with YK660 (UL12Y371F) at an MOI of 0.01, the progeny virus yield was 130- or 33-fold lower than that in Vero cells infected with wild-type HSV-1(F) or YK661 (UL12Y371F-repair), respectively (Fig. 8A). The Y371F mutation in pUL12 had more effect than the pUL12 nuclease-dead double mutation (UL12 G336A S338A) on viral replication in Vero cells infected at an MOI of 0.01. The progeny virus titer in Vero cells infected with YK660 (UL12Y371F) was 6.4-fold lower than that in Vero cells infected with YK665 (UL12G336A/S338A) (Fig. 8A).
In results similar to those for Vero cells infected at an MOI of 0.01, the progeny virus yields in HEL cells infected with YK660 (UL12Y371F) at MOIs of 0.01 and 3 were 8.0- to 11-fold lower than those in HEL cells infected with wild-type HSV-1(F) or YK661 (UL12Y371F-repair) and 5.0- and 4.8-fold lower than those in HEL cells infected with YK665 (UL12G336A/S338A), respectively (Fig. 8B and E). In contrast, the progeny virus yield in A549 cells infected with YK660 (UL12Y371F) at an MOI of 0.01 was 7.9- or 3.0-fold lower than that in A549 cells infected with wild-type HSV-1(F) or YK661 (UL12Y371F-repair), respectively, but was similar to that in A549 cells infected with YK665 (UL12G336A/S338A) (Fig. 8C). Similar results were obtained with A549 cells infected with each of these viruses at an MOI of 3 (Fig. 8F).
In Vero cells infected with YK660 (UL12Y371F) at an MOI of 3, the progeny virus yield was 12- or 7.2-fold lower than that in Vero cells infected with wild-type HSV-1(F) or YK661 (UL12Y371F-repair), respectively, but was similar to that in cells infected with YK665 (UL12G336A/S338A) (Fig. 8D).
The progeny virus yields in HEL, Vero, and A549 cells infected at MOIs of 0.01 and 3 with YK662 (UL12Y371E), carrying a phosphomimetic mutation in pUL12 Tyr-371, were similar to those in these cells infected with wild-type HSV-1(F) or the repaired viruses YK656 (ΔUL12-repair), YK661 (UL12Y371F-repair), and YK666 (UL12-GA/SA-repair) (Fig. 8). These results indicated that phosphorylation of pUL12 Tyr-371 was required for efficient HSV-1 replication in cell cultures but that the effect of phosphorylation on viral replication differed with the cell type and MOI. In addition, the Y371F mutation in pUL12 had more effect on viral replication than did the G336A S338A double mutation in pUL12, in a cell type- and MOI-dependent manner.
Effect of the phosphorylation of pUL12 Tyr-371 on cell-cell spread.
To examine the significance of the phosphorylation of pUL12 Tyr-371 for viral cell-cell spread in cell cultures, we analyzed plaque sizes in Vero, HEL, and A549 cells infected with either wild-type HSV-1(F), YK655 (ΔUL12), YK656 (ΔUL12-repair), YK660 (UL12Y371F), YK661 (UL12Y371F-repair), YK662 (UL12Y371E), YK665 (UL12G336A/S338A), or YK666 (UL12GA/SA-repair) using plaque assay conditions. In Vero cells, YK660 (UL12Y371F) produced plaques smaller than those for wild-type HSV-1(F) and YK661 (UL12Y371F-repair), similar in size to those for YK665 (UL12G336A/S338A), and larger than those for YK655 (ΔUL12) (Fig. 9A). The wild-type plaque size was restored in Vero cells infected with YK662 (UL12Y371E), carrying the phosphomimetic mutation in pUL12 Tyr-371 (Fig. 9A).
FIG 9.
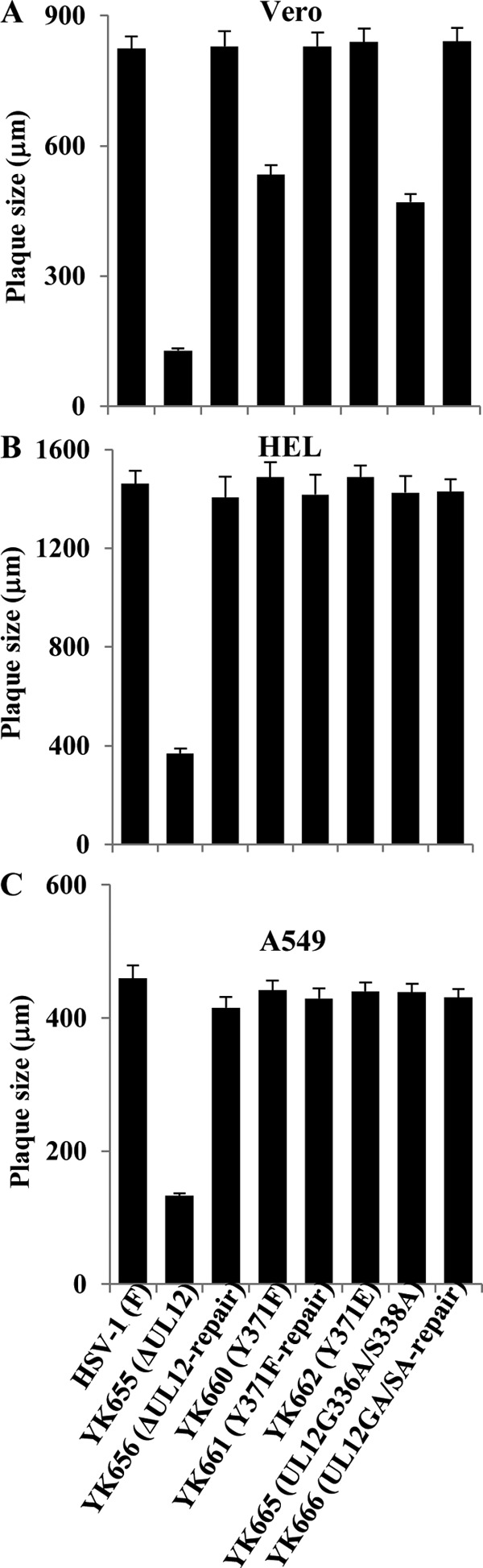
Effects of mutations in pUL12 on virus plaque size. Vero (A), HEL (B), and A549 (C) cells were infected with either wild-type HSV-1(F), YK655 (ΔUL12), YK656 (ΔUL12-repair), YK660 (UL12Y371F), YK661 (UL12Y371F-repair), YK662 (UL12Y371E), YK665 (UL12G336A/S338A), or YK666 (UL12GA/SA-repair) at an MOI of 0.0001, 0.001, or 0.00001, respectively, under plaque assay conditions. The diameters of 25 single plaques for each of the indicated viruses were measured 48 h postinfection. Each data point is the mean ± standard error of the measured plaque sizes and is representative of three independent experiments.
In contrast, YK660 (UL12Y371F) produced plaques similar in size to those produced by wild-type HSV-1(F), YK661 (UL12Y371F-repair), and YK665 (UL12G336A/S338A) in HEL and A549 cells (Fig. 9B and C). The effect of the nuclease-dead double mutation in pUL12 on cell-cell spread in Vero, HEL, and A549 cells was in agreement with our previous report (8). These results indicated that phosphorylation of pUL12 Tyr-371 was required for efficient cell-cell spread in a cell type-dependent manner.
Effect of the phosphorylation of pUL12 Tyr-371 on neurovirulence in mice.
To examine the effect of the phosphorylation of pUL12 Tyr-371 on HSV-1 pathogenesis in vivo, 3-week-old female mice were infected intracerebrally with 10-fold serial dilutions of YK655 (ΔUL12), YK656 (ΔUL12-repair), YK660 (UL12Y371F), YK661 (UL12Y371F-repair), YK662 (UL12Y371E), YK665 (UL12G336A/S338A), or YK666 (UL12GA/SA-repair); mortality was monitored; and LD50 values were calculated (Table 2). In agreement with our previous report (8), the LD50 values of YK655 (ΔUL12) and YK665 (UL12G336A/S338A) were 2,500- and 100-fold greater than those of YK656 (ΔUL12-repair) and YK666 (UL12GA/SA-repair), respectively. The LD50 of YK660 (UL12Y371F) was 200-fold greater than that of YK661 (UL12Y371F-repair) and similar to that of YK665 (UL12G336A/S338A). In addition, the LD50 of YK662 (UL12Y371E), carrying the phosphomimetic mutation in pUL12, was 100-fold lower than that of YK660 (UL12Y371F) and was similar to those of repaired viruses YK656 (ΔUL12-repair), YK666 (UL12GA/SA-repair), and YK661 (UL12Y371F-repair). Similar LD50 values were also obtained for YK660 (UL12Y371F), YK661 (UL12Y371F-repair), and YK662 (UL12Y371E) in repeated experiments (data not shown). These results indicated that phosphorylation of pUL12 Tyr-371 was critical for viral neurovirulence in mice following intracerebral inoculation.
TABLE 2.
LD50 values of recombinant HSV-1 strains in mice following intracerebral inoculation
| Virus | LD50 (PFU)a |
|
|---|---|---|
| Expt 1 | Expt 2 | |
| YK655 (ΔUL12) | 105.2 | |
| YK656 (ΔUL12-repair) | 101.8 | |
| YK665 (UL12G336A/S338A) | 103.5 | 103.5 |
| YK666(UL12GA/SA-repair) | 101.5 | 101.5 |
| YK660 (UL12Y371F) | 103.8 | |
| YK661 (UL12Y371F-repair) | 101.5 | |
| YK662 (UL12Y371E) | 101.8 | |
Three-week-old female ICR mice were infected intracranially with 10-fold serial dilutions of each of the indicated viruses in groups of 3 per dilution; mortality was monitored for 14 days; and LD50 values were calculated.
DISCUSSION
The enzymatic activities of cellular nucleases are often regulated by phosphorylation. The activity of cellular nuclease Mus81–Mms4, which plays an important role in the maintenance of genomic integrity in eukaryotic cells, has been reported to be regulated by phosphorylation in a cell cycle-dependent manner (36). In addition, the activity of Exo1, a cellular 5′-to-3′ exonuclease that regulates mismatch repair, double-strand break (DSB) repair, meiosis, and the response to stalled replication forks and uncapped telomeres, has been shown to be regulated by phosphorylation during the DNA damage checkpoint pathway (37). Although the HSV-1 alkaline nuclease pUL12 has long been known to be a phosphoprotein (1, 2), the biological significance of pUL12 phosphorylation in HSV-1-infected cells remained to be elucidated.
In the present study, LC–MS-MS analysis identified Tyr-371 as one of the phosphorylation sites in pUL12, and Phos-tag SDS-PAGE analysis confirmed this result. Studies with recombinant viruses in which pUL12 Tyr-371 was mutated then showed that the Y371F mutation in pUL12 abolished the phosphorylation of pUL12 Tyr-371 and pUL12 exonuclease activity in HSV-1-infected cells and that a phosphomimetic mutation at pUL12 Tyr-371 restored wild-type pUL12 exonuclease activity. From these observations, we concluded that phosphorylation of pUL12 Tyr-371 was essential for the pUL12 nuclease activity in HSV-1-infected cells. We noted that Tyr-371 is conserved in pUL12 homologs in herpesviruses in all Herpesviridae subfamilies (5, 13), suggesting that the exonuclease activity of herpesvirus pUL12 homologs may be regulated by phosphorylation of the conserved tyrosine residues. In support of this hypothesis as well as our conclusion, the crystal structures of the pUL12 homologs of Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV) revealed that the tyrosines that correspond to HSV-1 pUL12 Tyr-371 were located in linker regions close to the catalytic sites (38, 39). Therefore, it is reasonable that structural change mediated by phosphorylation of these tyrosines affects the catalytic activities of the pUL12 homologs.
Our results appeared to contradict a previous report by Henderson et al. (13) that the Y371F mutation in pUL12, which was generated in that study by in vitro translation using rabbit reticulocyte lysates, had no effect on the exonuclease activity of pUL12. This discrepancy may be due to the lack of a protein kinase(s) or kinase activity to phosphorylate pUL12 Tyr-371 in rabbit reticulocyte lysates. In support of this possibility, it has been noted (13) that in vitro-translated pUL12 was consistently observed to migrate slightly faster in denaturing gels than pUL12 from lysates of HSV-1-infected baby hamster kidney cells, probably due to the lack of phosphorylation in the in vitro-translated pUL12.
We reported recently that pUL12 nuclease activities were required for efficient viral replication and cell-cell spread in cell cultures in a cell type-dependent manner and were critical for viral neurovirulence in mice following intracerebral inoculation (8). In agreement with that report, we have shown here that the Y371F mutation in pUL12, which abolished its exonuclease activity in HSV-1-infected cells, had effects on viral replication, cell-cell spread in infected-cell cultures, and viral neurovirulence in mice following intracerebral inoculation similar to those of the nuclease-dead double mutation in pUL12. However, the effect of the Y371F mutation on viral replication was sometimes greater than that of the nuclease-dead double mutation in a cell type- and MOI-dependent manner. Furthermore, we showed that the reductions in viral replication, cell-cell spread in cell cultures, and viral neurovirulence in mice mediated by the Y371F mutation in pUL12 were reversed by a phosphomimetic mutation at pUL12 residue 371. From these observations, it seems reasonable to conclude that phosphorylation of pUL12 Tyr-371 promoted HSV-1 replication, cell-cell spread in cell cultures, and HSV-1 neurovirulence in mice mainly by upregulating the enzymatic activity of pUL12.
We reported recently that a yet-to-be-defined pUL12 function(s), unrelated to its nuclease activities, played a major role in viral replication in cell cultures (8). In this study, we found that the Y371F mutation in pUL12 consistently reduced viral replication in Vero and HEL cells infected at an MOI of 0.01, and in HEL cells infected at an MOI of 3, severalfold more than the nuclease-dead double mutation in pUL12. In contrast, the Y371F mutation had effects similar to those of the nuclease-dead double mutation on viral replication in Vero and A549 cells at an MOI of 3 and in A549 cells at an MOI of 0.01. These results suggested that phosphorylation of pUL12 Tyr-371 regulated both the nuclease activity of pUL12 and a pUL12 function(s) unrelated to its nuclease activity in a cell type- and MOI-dependent manner. We have also shown that the Y371F mutation led to aberrant subcellular localization of pUL12 and ICP8, which interacts with pUL12 (34), in Vero cells and reduced steady-state expression levels of UL12 in HEL cells. However, the phosphomimetic mutation at pUL12 residue 371 restored wild-type localization of pUL12 and ICP8 in Vero cells and steady-state expression of UL12 in HEL cells, suggesting that pUL12 phosphorylation at residue 371 was required for proper subcellular localization of pUL12 and ICP8 and for the expression of UL12 in a cell type-dependent manner. These cell type-dependent effects of phosphorylation of pUL12 Tyr-371 may be required for pUL12 to efficiently express a function(s) unrelated to its nuclease activities, which may promote viral replication in cell cultures. In contrast, since the Y371F mutation had effects on viral cell-cell spread in cell cultures and on viral neurovirulence in mice similar to those of the nuclease-dead double mutation, the cell type-dependent effects of pUL12 Tyr-371 phosphorylation appeared to play no role in these processes.
The HSV-1 UL12 gene encodes both full-length UL12 and UL12.5, an amino-terminally truncated protein that initiates at UL12 codon 127 (4, 40). Although pUL12 is localized in the nucleus, pUL12.5 is localized predominantly in mitochondria and can trigger mitochondrial DNA degradation, but the biological significance of mitochondrial DNA degradation in HSV-1-infected cells remains to be elucidated (41, 42). When expressed in a baculovirus expression system, pUL12.5 has pUL12 nuclease and strand exchange activities in vitro (4, 43). Since pUL12.5 contains the pUL12 Tyr-371 residue (Fig. 1), this Tyr residue may also be phosphorylated in pUL12.5, and the activities and/or function(s) of pUL12.5 may be regulated by this phosphorylation in HSV-1-infected cells. It has been assumed that the role of pUL12.5 in viral replication in cell cultures is negligible, based on observations that pUL12.5 was expressed much less than pUL12 in HSV-1-infected cells (4). Also, a recombinant virus expressing pUL12.5 at the wild-type level, but incapable of expressing pUL12, showed levels of nuclease activity, viral replication, and cell-cell spread in cell cultures almost identical to those of the UL12-null mutant virus (4). These observations suggested that the phenotype of the pUL12 Y371F mutation in cell cultures observed in this study resulted mainly from the loss of phosphorylation of pUL12, not from the loss of phosphorylation of pUL12.5. In contrast, the role of pUL12.5 in viral pathogenesis in vivo has not been elucidated thus far. Therefore, we cannot eliminate the possibility that the reduced viral neurovirulence in mice due to the Y371F mutation in pUL12 resulted from the loss of phosphorylation of both pUL12 and pUL12.5.
In conclusion, the data presented here have shown that the exonuclease activity of pUL12 was regulated by phosphorylation of the tyrosine residue that is conserved in herpesviruses and that this regulation played an important role in viral replication and pathogenesis. Thus far, studies of most herpesvirus enzymes have concentrated on the downstream effects of the enzymes. The present study showed that regulation of HSV-1 pUL12 by phosphorylation was critical for viral pathogenesis in vivo, indicating that in order to understand the overall features of a viral enzyme, data both on the mechanism of regulation of the viral enzyme in infected cells and on the downstream effects of enzyme regulation are needed. We are currently carrying out experiments to identify the cellular protein kinase(s) that phosphorylates pUL12 Tyr-371 in infected cells, so as to further clarify the mechanism of phosphorylation-mediated regulation of pUL12. Studies to investigate whether the conserved tyrosines in herpesvirus pUL12 homologs are phosphorylated in infected cells and whether this phosphorylation regulates the nuclease activities of pUL12 homologs, viral replication, and pathogenesis are also of interest. Since, as shown in the present study, the loss of phosphorylation of pUL12 Tyr-371 significantly reduced viral neurovirulence in vivo, the tyrosine kinase responsible for the phosphorylation of pUL12 Tyr-371 may be a novel therapeutic target for HSV and other herpesvirus infections, if the effects of phosphorylation of the conserved tyrosines in pUL12 homologs in other herpesviruses on viral replication and virulence are also conserved.
ACKNOWLEDGMENTS
We thank Sandra Weller for providing 6-5 cells. We thank Tomoko Ando and Shihoko Koyama for excellent technical assistance.
This study was supported by the Funding Program for Next Generation World-Leading Researchers, Grants for Scientific Research from the Japan Society for the Promotion of Science (JSPS), a contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases (J-GRID), a grant for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan, and grants from the Takeda Science Foundation and the Naito Foundation. H.F. was supported by research fellowships from JSPS for Young Scientists.
Footnotes
Published ahead of print 2 July 2014
REFERENCES
- 1.Banks LM, Halliburton IW, Purifoy DJ, Killington RA, Powell KL. 1985. Studies on the herpes simplex virus alkaline nuclease: detection of type-common and type-specific epitopes on the enzyme. J. Gen. Virol. 66(Part 1):1–14. 10.1099/0022-1317-66-1-1 [DOI] [PubMed] [Google Scholar]
- 2.Banks L, Purifoy DJ, Hurst PF, Killington RA, Powell KL. 1983. Herpes simplex virus non-structural proteins. IV. Purification of the virus-induced deoxyribonuclease and characterization of the enzyme using monoclonal antibodies. J. Gen. Virol. 64(Part 10):2249–2260 [DOI] [PubMed] [Google Scholar]
- 3.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897 In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B. (ed), Fields virology, 6th ed. Lippincott-Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 4.Martinez R, Shao L, Bronstein JC, Weber PC, Weller SK. 1996. The product of a 1.9-kb mRNA which overlaps the HSV-1 alkaline nuclease gene (UL12) cannot relieve the growth defects of a null mutant. Virology 215:152–164. 10.1006/viro.1996.0018 [DOI] [PubMed] [Google Scholar]
- 5.Goldstein JN, Weller SK. 1998. The exonuclease activity of HSV-1 UL12 is required for in vivo function. Virology 244:442–457. 10.1006/viro.1998.9129 [DOI] [PubMed] [Google Scholar]
- 6.Shao L, Rapp LM, Weller SK. 1993. Herpes simplex virus 1 alkaline nuclease is required for efficient egress of capsids from the nucleus. Virology 196:146–162. 10.1006/viro.1993.1463 [DOI] [PubMed] [Google Scholar]
- 7.Weller SK, Seghatoleslami MR, Shao L, Rowse D, Carmichael EP. 1990. The herpes simplex virus type 1 alkaline nuclease is not essential for viral DNA synthesis: isolation and characterization of a lacZ insertion mutant. J. Gen. Virol. 71(Part 12):2941–2952. 10.1099/0022-1317-71-12-2941 [DOI] [PubMed] [Google Scholar]
- 8.Fujii H, Mugitani M, Koyanagi N, Liu Z, Tsuda S, Arii J, Kato A, Kawaguchi Y. 2014. Role of the nuclease activities encoded by herpes simplex virus 1 UL12 in viral replication and neurovirulence. J. Virol. 88:2359–2364. 10.1128/JVI.03621-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reuven NB, Staire AE, Myers RS, Weller SK. 2003. The herpes simplex virus type 1 alkaline nuclease and single-stranded DNA binding protein mediate strand exchange in vitro. J. Virol. 77:7425–7433. 10.1128/JVI.77.13.7425-7433.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reuven NB, Willcox S, Griffith JD, Weller SK. 2004. Catalysis of strand exchange by the HSV-1 UL12 and ICP8 proteins: potent ICP8 recombinase activity is revealed upon resection of dsDNA substrate by nuclease. J. Mol. Biol. 342:57–71. 10.1016/j.jmb.2004.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schumacher AJ, Mohni KN, Kan Y, Hendrickson EA, Stark JM, Weller SK. 2012. The HSV-1 exonuclease, UL12, stimulates recombination by a single strand annealing mechanism. PLoS Pathog. 8:e1002862. 10.1371/journal.ppat.1002862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porter IM, Stow ND. 2004. Virus particles produced by the herpes simplex virus type 1 alkaline nuclease null mutant ambUL12 contain abnormal genomes. J. Gen. Virol. 85:583–591. 10.1099/vir.0.19657-0 [DOI] [PubMed] [Google Scholar]
- 13.Henderson JO, Ball-Goodrich LJ, Parris DS. 1998. Structure-function analysis of the herpes simplex virus type 1 UL12 gene: correlation of deoxyribonuclease activity in vitro with replication function. Virology 243:247–259. 10.1006/viro.1998.9054 [DOI] [PubMed] [Google Scholar]
- 14.Sagou K, Imai T, Sagara H, Uema M, Kawaguchi Y. 2009. Regulation of the catalytic activity of herpes simplex virus 1 protein kinase Us3 by autophosphorylation and its role in pathogenesis. J. Virol. 83:5773–5783. 10.1128/JVI.00103-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arii J, Uema M, Morimoto T, Sagara H, Akashi H, Ono E, Arase H, Kawaguchi Y. 2009. Entry of herpes simplex virus 1 and other alphaherpesviruses via the paired immunoglobulin-like type 2 receptor alpha. J. Virol. 83:4520–4527. 10.1128/JVI.02601-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382–1391. 10.1128/JVI.77.2.1382-1391.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J. Virol. 82:5198–5211. 10.1128/JVI.02681-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ichimura T, Yamamura H, Sasamoto K, Tominaga Y, Taoka M, Kakiuchi K, Shinkawa T, Takahashi N, Shimada S, Isobe T. 2005. 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J. Biol. Chem. 280:13187–13194. 10.1074/jbc.M412884200 [DOI] [PubMed] [Google Scholar]
- 19.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J. Virol. 71:7328–7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maruzuru Y, Fujii H, Oyama M, Kozuka-Hata H, Kato A, Kawaguchi Y. 2013. Roles of p53 in herpes simplex virus 1 replication. J. Virol. 87:9323–9332. 10.1128/JVI.01581-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. 1995. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. U. S. A. 92:7297–7301. 10.1073/pnas.92.16.7297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. 10.1038/nature09420 [DOI] [PubMed] [Google Scholar]
- 23.Savitski MM, Lemeer S, Boesche M, Lang M, Mathieson T, Bantscheff M, Kuster B. 2011. Confident phosphorylation site localization using the Mascot Delta Score. Mol. Cell. Proteomics 10:M110.003830. 10.1074/mcp.M110.003830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nozawa N, Kawaguchi Y, Tanaka M, Kato A, Kato A, Kimura H, Nishiyama Y. 2005. Herpes simplex virus type 1 UL51 protein is involved in maturation and egress of virus particles. J. Virol. 79:6947–6956. 10.1128/JVI.79.11.6947-6956.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato A, Tsuda S, Liu Z, Kozuka-Hata H, Oyama M, Kawaguchi Y. 2014. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral dUTPase and regulates its catalytic activity in infected cells. J. Virol. 88:655–666. 10.1128/JVI.02710-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. 2006. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell. Proteomics 5:749–757. 10.1074/mcp.T500024-MCP200 [DOI] [PubMed] [Google Scholar]
- 27.Kato A, Yamamoto M, Ohno T, Kodaira H, Nishiyama Y, Kawaguchi Y. 2005. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J. Virol. 79:9325–9331. 10.1128/JVI.79.14.9325-9331.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J. Virol. 82:6172–6189. 10.1128/JVI.00044-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sagou K, Uema M, Kawaguchi Y. 2010. Nucleolin is required for efficient nuclear egress of herpes simplex virus type 1 nucleocapsids. J. Virol. 84:2110–2121. 10.1128/JVI.02007-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bell C, Desjardins M, Thibault P, Radtke K. 2013. Proteomics analysis of herpes simplex virus type 1-infected cells reveals dynamic changes of viral protein expression, ubiquitylation, and phosphorylation. J. Proteome Res. 12:1820–1829. 10.1021/pr301157j [DOI] [PubMed] [Google Scholar]
- 31.Kato A, Shindo K, Maruzuru Y, Kawaguchi Y. 2014. Phosphorylation of a herpes simplex virus 1 dUTPase by a viral protein kinase, Us3, dictates viral pathogenicity in the central nervous system but not at the periphery. J. Virol. 88:2775–2785. 10.1128/JVI.03300-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kato A, Arii J, Shiratori I, Akashi H, Arase H, Kawaguchi Y. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J. Virol. 83:250–261. 10.1128/JVI.01451-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mou F, Wills E, Baines JD. 2009. Phosphorylation of the UL31 protein of herpes simplex virus 1 by the US3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J. Virol. 83:5181–5191. 10.1128/JVI.00090-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vaughan PJ, Banks LM, Purifoy DJ, Powell KL. 1984. Interactions between herpes simplex virus DNA-binding proteins. J. Gen. Virol. 65(Part 11):2033–2041. 10.1099/0022-1317-65-11-2033 [DOI] [PubMed] [Google Scholar]
- 35.Quinlan MP, Chen LB, Knipe DM. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:857–868. 10.1016/0092-8674(84)90035-7 [DOI] [PubMed] [Google Scholar]
- 36.Gallo-Fernandez M, Saugar I, Ortiz-Bazan MA, Vazquez MV, Tercero JA. 2012. Cell cycle-dependent regulation of the nuclease activity of Mus81-Eme1/Mms4. Nucleic Acids Res. 40:8325–8335. 10.1093/nar/gks599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morin I, Ngo HP, Greenall A, Zubko MK, Morrice N, Lydall D. 2008. Checkpoint-dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J. 27:2400–2410. 10.1038/emboj.2008.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dahlroth SL, Gurmu D, Schmitzberger F, Engman H, Haas J, Erlandsen H, Nordlund P. 2009. Crystal structure of the shutoff and exonuclease protein from the oncogenic Kaposi's sarcoma-associated herpesvirus. FEBS J. 276:6636–6645. 10.1111/j.1742-4658.2009.07374.x [DOI] [PubMed] [Google Scholar]
- 39.Buisson M, Geoui T, Flot D, Tarbouriech N, Ressing ME, Wiertz EJ, Burmeister WP. 2009. A bridge crosses the active-site canyon of the Epstein-Barr virus nuclease with DNase and RNase activities. J. Mol. Biol. 391:717–728. 10.1016/j.jmb.2009.06.034 [DOI] [PubMed] [Google Scholar]
- 40.Costa RH, Draper KG, Banks L, Powell KL, Cohen G, Eisenberg R, Wagner EK. 1983. High-resolution characterization of herpes simplex virus type 1 transcripts encoding alkaline exonuclease and a 50,000-dalton protein tentatively identified as a capsid protein. J. Virol. 48:591–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saffran HA, Pare JM, Corcoran JA, Weller SK, Smiley JR. 2007. Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep. 8:188–193. 10.1038/sj.embor.7400878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duguay BA, Saffran HA, Ponomarev A, Duley SA, Eaton HE, Smiley JR. 2014. Elimination of mitochondrial DNA is not required for herpes simplex virus 1 replication. J. Virol. 88:2967–2976. 10.1128/JVI.03129-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reuven NB, Antoku S, Weller SK. 2004. The UL12.5 gene product of herpes simplex virus type 1 exhibits nuclease and strand exchange activities but does not localize to the nucleus. J. Virol. 78:4599–4608. 10.1128/JVI.78.9.4599-4608.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]