

ABSTRACT
Immunization with modified vaccinia virus Ankara (MVA) can rapidly protect mice against lethal ectromelia virus (ECTV) infection, serving as an experimental model for severe systemic infections. Importantly, this early protective capacity of MVA vaccination completely depends on virus-specific cytotoxic CD8+ T cell responses. We used MVA vaccination in the mousepox challenge model using ECTV infection to investigate the previously unknown factors required to elicit rapid protective T cell immunity in normal C57BL/6 mice and in mice lacking the interferon alpha/beta receptor (IFNAR−/−). We found a minimal dose of 105 PFU of MVA vaccine fully sufficient to allow robust protection against lethal mousepox, as assessed by the absence of disease symptoms and failure to detect ECTV in organs from vaccinated animals. Moreover, MVA immunization at low dosage also protected IFNAR−/− mice, indicating efficient activation of cellular immunity even in the absence of type I interferon signaling. When monitoring for virus-specific CD8+ T cell responses in mice vaccinated with the minimal protective dose of MVA, we found significantly enhanced levels of antigen-specific T cells in animals that were MVA vaccinated and ECTV challenged compared to mice that were only vaccinated. The initial priming of naive CD8+ T cells by MVA immunization appears to be highly efficient and, even at low doses, mediates a rapid in vivo burst of pathogen-specific T cells upon challenge. Our findings define striking requirements for protective emergency immunization against severe systemic infections with orthopoxviruses.
IMPORTANCE We demonstrate that single-shot low-dose immunizations with vaccinia virus MVA can rapidly induce T cell-mediated protective immunity against lethal orthopoxvirus infections. Our data provide new evidence for an efficient protective capacity of vaccination with replication-deficient MVA. These data are of important practical relevance for public health, as the effectiveness of a safety-tested, next-generation smallpox vaccine based on MVA is still debated. Furthermore, producing sufficient amounts of vaccine is expected to be a major challenge should an outbreak occur. Moreover, prevention of other infections may require rapidly protective immunization; hence, MVA could be an extremely useful vaccine for delivering heterologous T cell antigens, particularly for infectious diseases that fit a scenario of emergency vaccination.
INTRODUCTION
Severe human infections with recently emerging pathogens, such as avian influenza virus H7N9 or the Middle East respiratory syndrome coronavirus (MERS-CoV) (1, 2), demonstrate the need for public health strategies that rapidly contain potentially dangerous emerging infectious diseases. Thus, developing innovative vaccination principles that will be ready for use in an immediate public health response are essential. Emergency vaccines should include early induction of protective immunity and the capacity to elicit diverse antigen-specific immune responses. However, our understanding of the immunological principles of successful emergency vaccination is limited.
Eradication of human smallpox was achieved by massive prophylactic use of live vaccinia virus (VACV) more than 30 years ago (3). The smallpox vaccine was applied not only during outbreaks but also postexposure, which was generally believed to be at least partially protective (for a recent review, see reference 4). However, the efficacy of postexposure vaccination is poorly defined, and the immune correlates of rapidly protective immunization against smallpox remain largely unclear.
The modified vaccinia virus Ankara (MVA), a replication-deficient and safety-tested VACV (5, 6), is already licensed as a replacement smallpox vaccine in Europe and has been actively investigated as a nonreplicating multipurpose viral vector vaccine against various infections and cancer diseases (7–10). Thus, MVA is a promising platform to develop candidate vaccines inducing strong innate and adaptive immune responses. Immunization with MVA proved highly efficacious in different animal models and elicited antigen-specific humoral as well as cellular immunity (11, 12). Moreover, MVA vaccination can fully protect even when administered shortly before or after systemic infection of mice or macaques with pathogenic orthopoxviruses (13–15). Such early protective capacity is highly attractive for public health preparedness and might also be applicable for other MVA-based emergency vaccines. However, it is still not well understood how MVA can trigger rapid activation of protective immunity.
The orthopoxvirus (OPV) ectromelia virus (ECTV) is a natural mouse pathogen inducing mousepox, a lethal disease in mice. After initial respiratory infection, the virus spreads via lymph and blood circulation to internal organs, resulting in a severe systemic disease (for reviews, see references 16 and 17). The genetic similarity of ECTV to variola virus (VARV), the causative agent of human smallpox, along with their common disease progression and the ease of using mice as laboratory animals, has led to the development of ECTV models as surrogates for human smallpox, once one of the most serious infectious diseases in humans (18).
MVA immunization can fully protect mice against mousepox, even when applied in a time window very close to a lethal infection with ECTV (13, 19). Thus, the immune mechanisms mediating protection in this mouse model may be comparable to those activated after vaccinations of humans against smallpox.
Using the MVA vaccination-ECTV challenge model, we recently demonstrated that orthopoxvirus-specific CD8 T cell responses are essential to rapidly induce protective immunity against lethal systemic mousepox (20). Here, the requirement of the cytolytic protein perforin for protective MVA vaccination suggests that cytotoxic T cells play a key role in rapidly containing the fatal ECTV infection.
It is thought that to induce specific antibody response levels comparable to those elicited by replication-competent VACV, high doses and repeated applications of nonreplicating MVA vaccines are required. It was recommended that MVA immunizations be used at 100-fold-higher doses, in a two-shot regimen, to equal immune responses induced by percutaneous vaccination with the Dryvax smallpox vaccine (15). Therefore, a standard dosage of 108 PFU MVA was tested in most studies, including those reporting protective emergency vaccination (13, 14, 19–21). Such high dosage is also recommended in the recent marketing approval of MVA as a next-generation smallpox vaccine (22).
Here we demonstrate that a 1,000-fold-lower dose of MVA vaccine is sufficient to protect C57BL/6 mice and even immunodeficient mice lacking the interferon alpha/beta receptor. Importantly, protective low-dose vaccination against lethal mousepox required induction of CD8+ T cell responses. Moreover, low-dose MVA immunization seemed to allow efficient initial activation of antigen-specific CD8+ T cells followed by a marked expansion of these T cells in response to ECTV infection.
MATERIALS AND METHODS
Cells and viruses.
Monkey MA-104 cells (ATCC CRL-2378.1) and primary chicken embryo fibroblasts (CEF) were used. Plaque-purified ectromelia virus (ECTV) strain Moscow (ATCC VR-1374; kindly provided by Mark L. Buller, St. Louis University School of Medicine, St. Louis, MO, USA) was propagated on MA-104 cells. Modified vaccinia virus Ankara (MVA) (clonal isolate F6) was propagated on CEF. Viral titers were determined by plaque assay and titrated, with values reported in PFU.
Mice.
Female C57BL/6N mice (6 to 10 weeks old) were purchased from Charles River Laboratories (Sulzfeld, Germany). Type I interferon receptor-deficient (IFNAR−/−) mice have been 20-fold backcrossed with C57BL/6N mice and have a deficient type I IFN system. For experimental work, mice were housed in an Isocage unit (Tecniplast, Germany) and had free access to food and water. All animal experiments were handled in compliance with the German regulations for animal experimentation (Animal Welfare Act).
Immunization experiments.
Intramuscular (i.m.) vaccination was performed by injection of 50 μl of virus suspension containing 105 or 108 PFU of MVA or phosphate-buffered saline (PBS) into the left hind leg. For intranasal infection, mice were anesthetized by intraperitoneal (i.p.) injection with 1 mg ketamine and 0.04 mg xylazine per 10 g body weight. Intranasal infection was performed by instillation of 20 μl of virus suspension containing 200 PFU (∼3 50% lethal doses [LD50]) ECTV. Signs of illness, weight loss, and survival were monitored daily for at least 3 weeks. In all experiments, inoculations of corresponding amounts of PBS were used as controls (mock vaccine).
Depletion of specific subsets of immune cells.
Mice were depleted of CD8+ T cells by i.p. administration of mouse monoclonal antibodies purchased from Harlan Bioproducts, Indianapolis, IN, USA. CD8+ T cell depletion was performed by administration of 100 μg anti-CD8 clone 2.43 antibody on days −2 and −1 prior to immunization on day 0. Successful depletion of immune cells was confirmed by flow-cytometric analysis of blood and spleen cells from antibody-treated animals.
Flow cytometry.
Approximately 106 cells were stained in 50 μl PBS supplemented with 3% fetal calf serum (FCS) using monoclonal antibodies obtained from Biolegend. T cells were detected using phycoerythrin (PE)-labeled CD3, PE-Cy7-labeled CD4, and fluorescein isothiocyanate (FITC)-labeled CD8 antibodies. To detect antigen-specific CD8+ T cells, CD8+ T cells were analyzed with allophycocyanin (APC)-labeled multimers (Dextramer; Immudex, Denmark) containing the VACV peptide B8R20–27 (TSYKFESV), an antigenic determinant present in MVA and ECTV. To ensure specificity of staining, all staining tests contained negative controls from mice that had been mock vaccinated/infected with PBS. Stained cells were analyzed with MACS Quant VYB and MACSQuantify software (Miltenyi Biotec, Bergisch-Gladbach, Germany).
Analysis of antigen-specific CD8+ T cells by enzyme-linked immunospot assay (ELISPOT).
Mice were sacrificed 8 days postimmunization. A cell suspension was prepared by homogenizing the spleens through 200-μm mesh sieves, and red blood cells were removed by adding red cell lysis buffer (Sigma). After centrifugation, the cell pellet was resolved in RPMI medium supplemented with 10% fetal calf serum, 2 mM l-glutamine, and 100 IU/ml penicillin-streptomycin.
Interferon gamma (IFN-γ)-secreting CD8+ T cells were analyzed by using the ELISPOTPLUS kit for mouse IFN-γ (MABTECH, Germany) following the manufacturer's instructions. ELISPOT plates were preincubated overnight with the antibody solution and then incubated with the cell suspension that had been stimulated with the virus-specific peptide B8R20–27 (TSYKFESV). The spots were counted and analyzed by using an automated ELISPOT plate reader and software following the manufacturer's instructions (A.EL.VIS Eli.Scan software; A.EL.VIS, Hanover, Germany).
Histology.
Sections of lungs, livers, and spleens of sacrificed mice were fixed in formaldehyde (4%) for 24 h and subsequently embedded in paraffin. Sections of 4 μm were stained with hematoxylin and eosin before being evaluated by light microscopy. Primary antibody for immunohistochemistry was a rabbit anti-VACV diluted 1:2,000. Lungs of PBS-inoculated mice served as a negative controls. To exclude false-positive reactions of the secondary antibody, an irrelevant primary antibody (polyclonal rabbit anti-Escherichia coli; no. B0357; Dako, Hamburg, Germany) combined with lung material of an infected IFNAR−/− mouse served as additional negative control. After deparaffinization, sections were blocked with hydrogen peroxide followed by diluted normal goat serum (30 min). Primary antibody incubation was 60 min at room temperature. Secondary antibody (biotinylated goat anti-rabbit-Ig; no. BA-1000; Vector, Burlingame, CA, USA) incubation was carried out for 45 min, followed by incubation with ABC (no. PK-6100; Vector); hydrogen peroxide served as the substrate and diaminobenzidine (DAB) as the chromogen (no. 4170; Biotrend, Cologne, Germany). Tissues were counterstained with hematoxylin, dehydrated, and covered with glass coverslips.
Determination of ECTV loads in mouse organs.
Organs (lungs, livers, and spleens) were removed under aseptic conditions from sacrificed or dead mice. The organs were frozen and subsequently thawed, weighed, and homogenized using 0.1 g of organ material with 1 ml PBS in a microtube (Retsch TissueLyser MM 300; Qiagen GmbH, Hilden, Germany). Tubes were centrifuged for 1 min at 1,500 rpm and 4°C. Supernatants were taken and stored in −80°C. Viral titers in organ supernatants were determined by plaque assay and indicated in PFU per 1 g organ material.
Measurement of cytokines in mouse sera.
Cytokine serum levels were analyzed by enzyme-linked immunosorbent assay (ELISA). Interleukin 12 (IL-12) was measured using the IL-12p70 ELISA (Biolegend, Fell, Germany). For detection of IFN-α in sera of vaccinated mice, we used the VeriKine mouse interferon alpha ELISA kit (from PBL Biomedical Laboratories; distributed by R&D Systems Europe Ltd., Wiesbaden, Germany). All assays were performed according to the manufacturer's instructions and repeated three times.
Statistical analysis.
Statistical comparison of different groups of mice was analyzed by one-factorial analysis of variance (ANOVA) for the area under the percentage-of-initial-weight curves (AUC).The differences between vaccination groups were analyzed with a one-factorial analysis of variance model. For multiple comparisons, P values were adjusted with the Bonferroni method. CD8+ T cell responses were compared by t test. The statistical evaluation was performed with GraphPad Prism for Windows (GraphPad Prism Software, USA).
RESULTS
Low doses of MVA vaccine induce OPV-specific CD8+ T cell responses.
We recently demonstrated that CD8+ T cell-mediated protection from lethal mousepox can be achieved by administering standard doses of 108 PFU MVA 2 days before a lethal respiratory ECTV infection (20). Here, to determine the T cell response corresponding to this protective immunization, we measured OPV-specific CD8+ T cells by ELISPOT at day 8 postimmunization (Fig. 1).
FIG 1.
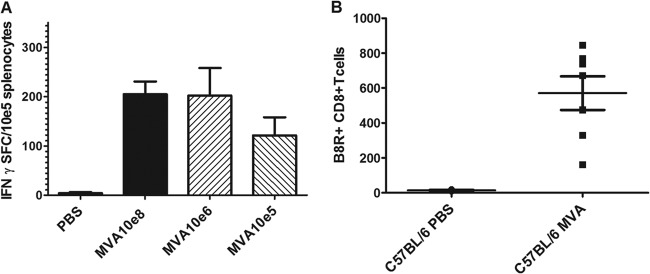
Virus-specific CD8+ T cell response induced by low-dose MVA immunization. (A) Groups of C57BL/6 mice (n = 5) were immunized intramuscularly with 108, 106, or 105 PFU MVA or mock vaccinated (PBS). At 8 days postvaccination, splenocytes were prepared, and B8R20–27-specific IFN-γ-producing CD8+ T cells were measured by ELISPOT. Data are representative of two similar experiments. (B) Total numbers of B8R+ CD8+ T cells in blood from C57BL/6 mice (7 per MVA-immunized group, 3 per mock-vaccinated group) on day 6 after immunization with 105 PFU MVA or mock vaccination.
Unexpectedly, immunization of C57BL/6 mice with various doses of MVA revealed that 100-fold less MVA (106 PFU) vaccine induced levels of IFN-γ-producing T cells comparable to those elicited by the standard dose of 108 PFU MVA (Fig. 1A). Inoculations with only 105 PFU MVA still resulted in clearly detectable amounts of virus-specific CD8+ T cells (Fig. 1A). Moreover, immunizations with 105 PFU MVA were sufficient to elicit detectable levels of B8R20–27-binding CD8+ T cells in the blood of vaccinated animals as early as 6 days postvaccination (Fig. 1B). These data suggested that MVA vaccination results in an efficient and rapid induction of OPV-specific CD8+ T cells, even when 100- to 1,000-fold-smaller amounts of MVA are used.
Low-dose MVA immunization efficiently protects against lethal mousepox.
Since vaccination with 105 PFU MVA resulted in activating substantial numbers of virus-specific CD8+ T cells, we tested whether low doses of MVA vaccine could protect C57BL/6 mice against a lethal infection with ECTV. We intramuscularly vaccinated C57BL/6 mice with 10-fold-increasing amounts of vaccine, ranging from 102 to 108 PFU MVA. Two days later, the mice were intranasally infected with 200 PFU ECTV and monitored for signs of disease and survival.
Confirming data from previous studies (13, 20), all animals receiving 108 PFU MVA were fully protected against the challenge infection, showing no symptoms of disease and showing steadily increasing body weights during the observation period (Fig. 2). In contrast, immunizations using only 102 or 103 PFU MVA failed to show any protective capacity (data not shown). All mice in these groups developed systemic mousepox disease, starting to show progressive body weight loss from 9 dpi, and died or had to be euthanized within 13 days after challenge. This course of disease in mice vaccinated with 102 and 103 PFU MVA was similar to the fate of unvaccinated control mice.
FIG 2.
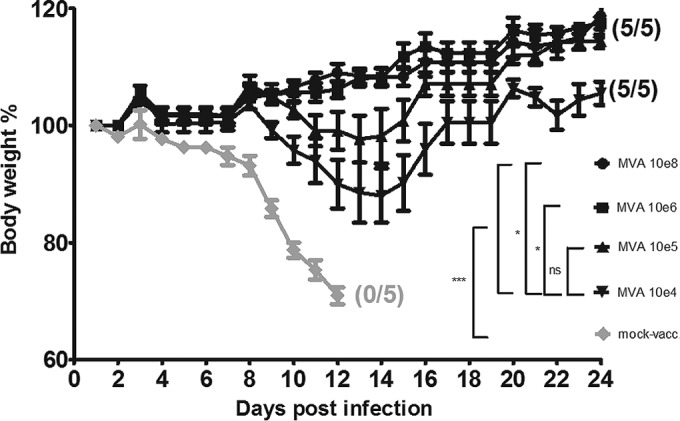
Protective capacity of low-dose MVA immunization against a lethal mousepox challenge infection. C57BL/6 mice were challenged with ECTV 2 days after immunization with MVA vaccine (104 to 108 PFU) or PBS (mock-vaccinated animals, used as controls). In all experiments, weight loss of individual mice was monitored daily (5 per group). Error bars indicate standard errors of the means (SEMs), and the numbers of surviving/total animals are given in parentheses. *, P < 0.05; ***, P < 0.001. Data are representative of two or three experiments.
Vaccinations with 104 PFU MVA did not prevent the onset of morbidity, as characterized by weight loss of up to 15% of original body weight (Fig. 2), reduced motility, accelerated respiration, or conjunctivitis. Disease symptoms peaked at about 12 to 14 dpi. However, all mice in this group recovered rapidly, fully regaining their initial body weights by day 18 and surviving the infection.
Immunizations with doses equal to or higher than 105 PFU MVA were sufficient to prevent any overt disease symptoms, apparently providing robust protection against the respiratory challenge infection (Fig. 2). C57BL/6 mice in this group demonstrated only minimal weight loss (<5%) or delayed increase in body weights on days 10 to 16 following ECTV infection. These data show that compared to the standard dosage of 108 PFU MVA, much smaller amounts of vaccine were sufficient to rapidly induce protective immunity.
CD8+ T cells are essential for the protective capacity of low-dose MVA immunization.
Our previous studies had demonstrated a key role for T cell immunity in rapidly protective immunization against OPV (20). Here, we asked whether that protective immunization with 105 PFU MVA also required the presence of CD8+ T cells. The answer seemed to be yes, since all vaccinated mice depleted of CD8+ T cells succumbed to mousepox after challenge with ECTV (Fig. 3A). Indeed, CD8+ T cell depletion in the MVA-vaccinated C57BL/6 mice resulted in accelerated disease progression, with symptoms and mortalities occurring about 2 days earlier than in mock-vaccinated wild-type controls. As before, the vaccinated wild-type mice were effectively protected from disease and death. Thus, low-dose immunization with 105 PFU MVA also essentially depends on CD8+ T cells for protective immunity against a lethal ECTV infection.
FIG 3.
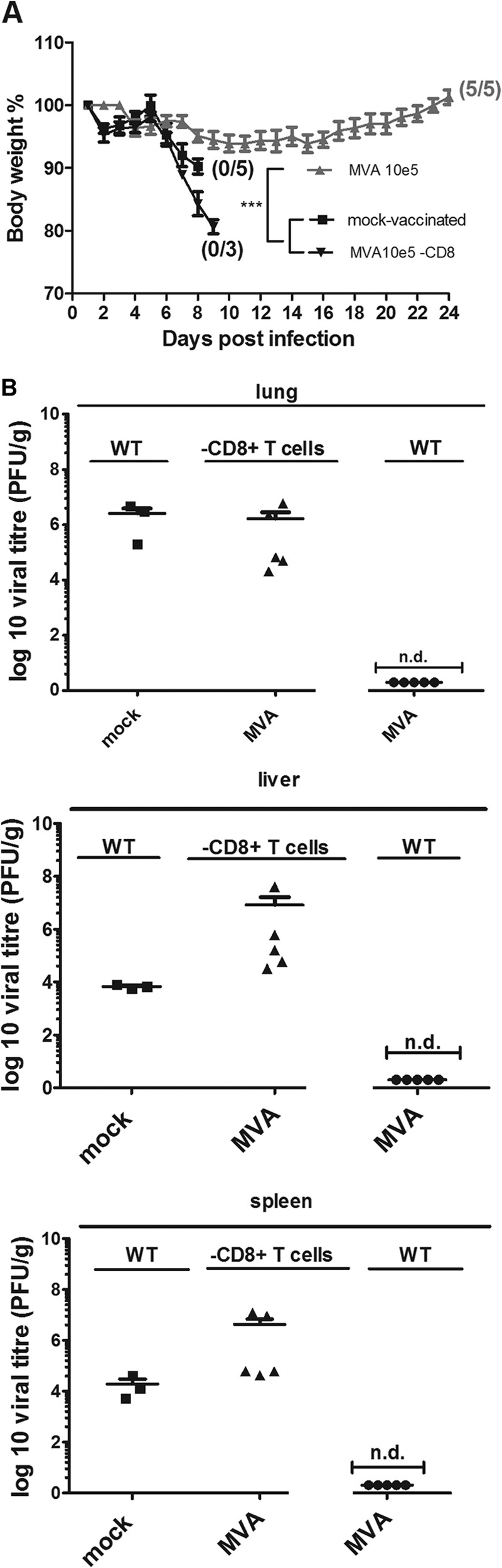
Protective capacity of low-dose MVA immunization is lost in the absence of CD8+ T cells. (A) C57BL/6 mice (WT) and mice depleted of CD8+ T cells were challenged with ECTV 2 days after immunization with MVA (105 PFU) or PBS (mock-vaccinated mice) (5 per group). Error bars indicate SEMs, and the numbers of surviving/total animals are given in parentheses. ***, P < 0.001. (B) Virus titers in livers and spleens after lethal ECTV infection. At the time of death or at the end of the experiments (day 21 for wt MVA-vaccinated mice), spleens and livers were removed and homogenized, and the amount of virus was determined by plaque assays (3 to 5 animals per group). Error bars indicate SEMs, and data are representative of at least two independent experiments. n.d., not detectable.
To determine whether immunization with 105 PFU MVA can clear the challenge virus, we determined ECTV loads in the lungs, livers, and spleens of vaccinated and mock-vaccinated mice at times of death or 21 days postchallenge (Fig. 3B). In the organs from MVA-vaccinated and surviving mice, we failed to detect infectious ECTV, suggesting complete elimination of the challenge virus. In contrast, we found high viral loads in the lungs, livers, and spleens of mock-vaccinated mice (obtained at the day of death). These virus titers ranged from 104 to 106 PFU ECTV per gram of tissue, as is typical for the systemic spread of ECTV infection in C57BL/6 mice. Supporting our observations of accelerated disease, we found clearly increased amounts of ECTV (>107 PFU per g tissue) in lungs, livers, and spleens from mice depleted of CD8+ T cells. Notably, histological analysis and immunohistochemistry of lung, liver, and spleen samples from ECTV-infected and mock-vaccinated C57BL/6 mice confirmed the systemic spread of virus to internal organs, as demonstrated by the detection of necrotic lesions in the epithelia of the lungs and focal necrosis in livers and spleens (Fig. 4). We found more pronounced tissue damage in the livers and spleens than in the lungs as the primary site of infection. In the lungs, pathological changes were restricted to single areas in the bronchioles and blood vessels, suggesting lymphohematogenous dissemination of the virus.
FIG 4.

Histological analysis and immunohistochemistry of organs from ECTV-infected C57BL/6 mice at day 7 postinfection. Lungs, livers, and spleens were removed, and sections of the organs were routinely stained with hematoxylin and eosin (HE). Bars, 50 μm (A, C, and F) and 100 μm (B, D, and E). (A to C) Micrographs of representative tissues from a mock-vaccinated (PBS) mouse. (A) Lung with necrosis of bronchiolar epithelial cells (arrowhead); (B) liver with hepatocyte necrosis (arrowheads); (C) spleen with areas of red and white pulp necrosis. (D to F) Sections of tissues were immunostained (IHC) with polyclonal rabbit antibody raised against VACV Lister virions to detect ECTV antigen. Micrographs show representative areas of lung (D), liver (E), and spleen (F) from a mock-vaccinated C57BL/6 mouse.
Low-dose MVA immunization protects IFNAR−/− mice from mousepox.
In previous work investigating emergency vaccination with 108 PFU MVA, we could also protect immunodeficient mice that lack the type I interferon receptor (IFNAR−/− mice) (13). Here, we examined the capacity of IFNAR−/− mice to rapidly mount protective CD8+ T cells upon vaccination with the 1,000-fold-lower dose of MVA. Remarkably, we detected reasonable levels of OPV-specific CD8+ T cells specifically recognizing the peptide epitope B8R20–27 (TSYKFESV) in the peripheral blood 6 days postimmunization (Fig. 5A) and in the spleen 8 days postimmunization (Fig. 5B). We found slightly reduced levels of TSYKFESV-binding T cells in the blood of IFNAR−/− mice compared to C57BL/6 mice. In the spleens, however, low-dose MVA vaccination of IFNAR−/− mice resulted in IFN-γ-positive CD8+ T cell responses similar to those elicited in wild-type C57BL/6 mice. When we analyzed sera of MVA-vaccinated animals for the presence of proinflammatory cytokine IL-12 and type I interferon (IFN), we detected significant amounts of IL-12 in IFNAR−/− but not in C57BL/6 mice, whereas IFN-α was induced to high levels in C57BL/6 mice only (Fig. 5C and D).
FIG 5.

MVA immunization in mice lacking the type I interferon receptor (IFNAR−/− mice). (A and B) Induction of virus-specific CD8+ T cells. IFNAR−/− mice were inoculated intramuscularly with 105 PFU MVA or PBS. (A) On day 6 after immunization, total numbers of B8R+ CD8+ T cells in blood from IFNAR−/− mice (n = 5) were determined by fluorescence-activated cell sorting (FACS) analysis. (B) At 8 days postvaccination, splenocytes were prepared, and B8R20–27-specific IFN-γ-producing CD8+ T cells were measured by ELISPOT. Data are representative of two similar experiments. (C and D) Induction of IL-12 or IFN-α in sera of MVA-vaccinated mice. (C) Serum levels of IL-12 in IFNAR−/− mice or C57BL/6 mice at 18 h postimmunization. The asterisk indicates the minimum detectable concentration of IL-12(p70) (4 pg/ml). (D) Serum levels of IFN-α in IFNAR−/− mice or C57BL/6 mice at 12 h after immunization with MVA. n.d., not detectable. (E) Protective capacity of low-dose (105 PFU) MVA vaccination in IFNAR−/− mice. Animals were infected with ECTV 2 days after vaccination with MVA or PBS (mock-vaccinated controls), and weight loss of individual mice was monitored daily (3 to 5 per group). Error bars indicate SEMs, and the numbers of surviving/total animals are given in parentheses. ***, P < 0.001.
Immunizations of IFNAR−/− mice with 105 PFU MVA also fully protected against lethal ECTV infection; the MVA-vaccinated IFNAR−/− mice, very similar to immunized wild-type C57BL/6 mice, showed no signs of disease or weight loss. In contrast, mock-vaccinated IFNAR−/− mice all developed systemic mousepox and succumbed to the infection within 8 days after challenge (Fig. 5E).
To further evaluate the level of protection induced in IFNAR−/− mice, we performed histological analyses of lungs, livers, and spleens of MVA-vaccinated and mock-vaccinated mice (Fig. 6). Organs from mock-vaccinated mice showed significant tissue damage following ECTV infection. In the lungs, we detected clear necrotic lesions located primarily in the epithelial cell layers of the bronchi and bronchioli. More sporadically, we found lesions in the blood vessels surrounding the bronchioli. In livers and spleens, we detected patterns of more extensive tissue damage with multifocal necrotic lesions. Immunohistochemical analysis of the tissues revealed multiple foci of infected cells in the spleens and livers from mock-vaccinated animals (Fig. 6B and C). In contrast, the lungs of mock-vaccinated animals contained less abundant and more isolated areas of ECTV-infected cells (Fig. 6A). Immunohistochemical staining of organs from MVA-vaccinated IFNAR−/− mice failed to detect ECTV infected cells, indicating complete clearance of the virus (Fig. 6D to F).
FIG 6.

Immunohistochemical analysis of organs from ECTV-infected and mock-vaccinated (PBS) or MVA (105 PFU)-vaccinated IFNAR−/− mice. At day 7 (PBS) or at day 12 (MVA) postinfection, sections of tissues were immunostained (IHC) with polyclonal rabbit antibody raised against VACV Lister virions to detect ECTV antigen. Bars, 50 μm. Micrographs show representative areas of lung, liver, and spleen from a mock-vaccinated IFNAR−/− mouse (A, B, and C), or lung, liver, and spleen from an MVA-vaccinated mouse (D, E, and F).
To also assess the necessity of T cells for protective vaccination in the IFNAR−/− mouse model, we depleted CD8+ T cells from these mice and vaccinated the animals with 105 PFU MVA 2 days before the lethal challenge infection with ECTV (Fig. 7). Nondepleted and vaccinated control mice were again fully protected. In contrast, all IFNAR−/− mice depleted of CD8+ T cells succumbed to mousepox within 9 days postinfection despite prior MVA immunization (Fig. 7A).
FIG 7.
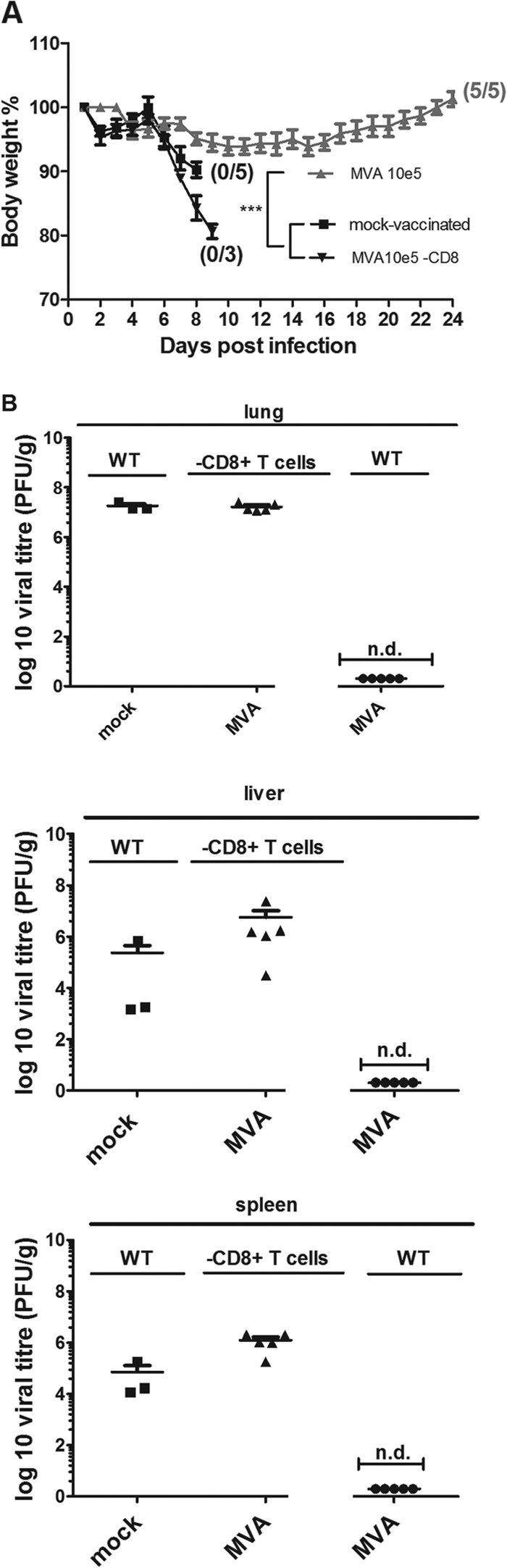
Protection of IFNAR−/− mice requires CD8+ T cell-mediated immunity after low-dose MVA vaccination. (A) IFNAR−/− mice and IFNAR−/− mice depleted of CD8+ T cells (IFNAR−/− CD8+) were vaccinated or mock vaccinated (IFNAR−/− mice only) with 105 PFU MVA. Weight loss of individual mice was monitored daily (3 to 5 per group). Error bars indicate SEMs, and the numbers of surviving/total animals are given in parentheses. ***, P < 0.001. (B) ECTV titers in lungs, livers and spleens of WT or CD8+ cell-depleted IFNAR−/− mice after challenge infection. At the time of death or at the end of the experiments (day 21 p.i. for WT MVA-vaccinated mice), organs were removed and homogenized, and the amount of virus was determined by plaque assay (3 to 5 animals per group). Error bars indicate SEMs, and data are representative of at least two independent experiments. n.d., not detectable.
These results clearly demonstrate that CD8+ T cells are also essential to allow MVA-mediated protection of IFNAR−/− mice. When monitoring virus loads in the organs of mice at times of death (mock-vaccinated or CD8+ T cell-depleted, MVA-vaccinated animals) or 21 days postchallenge (MVA-vaccinated animals), we failed to detect ECTV in the lungs, livers, and spleens of MVA-vaccinated IFNAR−/− mice but found large amounts of virus in the organs from unvaccinated or CD8+ T cell-depleted, MVA-vaccinated IFNAR−/− mice (Fig. 7B). In general, we found moderately increased levels of virus in mock-vaccinated IFNAR−/− mice compared to unvaccinated C57BL/6 mice (Fig. 3B). Again, CD8+ T cell depletion resulted in an increase of ECTV loads in liver and spleen.
MVA-primed CD8+ T cell responses are boosted upon ECTV challenge.
Our data so far showed that protective vaccination by a single application of 105 PFU MVA is associated with rapid cellular immune responses in wild-type C57BL/6 and IFNAR−/− mice. This suggests a scenario of primary activation of virus-specific CD8+ T cells by MVA immunization, followed by powerful T cell expansion, leading to rapid immune control of the closely related ECTV. Apparently, in C57BL/6 (and C57BL/6-derived IFNAR−/−) mice, both MVA and ECTV induce a Kb-restricted immunodominant CD8+ T cell response directed to the conserved B8R20–27 epitope (23).
We monitored this T cell specificity to investigate a possible effect of the ECTV infection on the developing CD8+ T cell immunity. Indeed, analyzing IFN-γ-producing CD8+ T cells by ELISPOT showed that vaccinated and ECTV-challenged C57BL/6 or IFNAR−/− mice produced significantly more B8R20–27 epitope-specific CD8+ T cells than mice inoculated with only MVA (Fig. 8A and B). This was confirmed by analyzing B8R20–27 epitope-binding CD8+ T cells following multimer staining and fluorescence-activated cell sorting (Fig. 8C and D). Obviously, ECTV infection in a time window very close to the immunization strongly amplifies the MVA-induced virus-specific T cell responses.
FIG 8.

MVA-primed CD8+ T cell responses are efficiently boosted upon ECTV challenge infection. (A and B) Virus-specific CD8+ T cells in C57BL/6 (A) and IFNAR−/− (B) mice after MVA immunization or MVA immunization and ECTV challenge infection, as measured by IFN-γ ELISPOT at day 8 after vaccination. (C and D) FACS analysis of B8R+ multimer-binding CD8+ T cells after MVA vaccination or vaccination and ECTV infection in C57BL/6 (C) and IFNAR−/− (D) mice, measured on day 8 after MVA immunization. Data are representative of three similar experiments (5 mice per group). **, P < 0.01.
DISCUSSION
Here, we detected potent activation of virus-specific CD8+ T cell responses in C57BL/6 and interferon receptor-deficient (IFNAR−/−) mice after low-dose i.m. immunization with 105 PFU MVA. Our ELISPOT analyses clearly demonstrate that single-shot vaccinations with 100- to 1,000-fold less MVA than usual induce levels of antigen-specific CD8+ T cells comparable to those elicited by the standard dose of 108 PFU Moreover, low-dose (105 PFU) MVA immunization protected mice against a lethal mousepox challenge with ECTV.
Possibly, in contrast to humoral responses, the activation of MVA-specific CD8+ T cells is highly efficient. This is not completely unexpected, since previous studies demonstrated that low-dose MVA immunizations of HLA-A*0201 transgenic mice can elicit substantial numbers of epitope-specific T cells. However, the possible contribution of the T cell response to MVA-mediated protection of these mice against a lethal challenge with VACV Western Reserve remained unclear (24).
Mousepox infection is an excellent surrogate model for human smallpox because very small amounts of ECTV can efficiently spread and cause fatal systemic disease following inoculation via the upper respiratory route (13, 20). Histology and immunohistochemistry confirmed the generalized nature of the infection (Fig. 4). We found more pronounced tissue damage in the livers and spleens than in the lungs as the primary site of infection. In the lungs, pathological changes were restricted to single areas in the bronchioles and blood vessels, suggesting lymphohematogenous dissemination of the virus. Using immunohistochemistry, we further identified ECTV-positive cells, suggesting phagocytes as potential target cells supporting systemic spreading (Fig. 4).
When we tested whether low-dose inoculations of MVA vaccine could provide rapid protection of C57BL/6 mice against a lethal intranasal challenge with ECTV after intramuscular immunizations with doses ranging from 102 to 108 PFU MVA, 105 PFU MVA was sufficient to protect against the onset of clinical disease and death, indicating vaccine efficacy following challenge. Moreover, the absence of detectable ECTV in the organs of all vaccinated animals suggests that low-dose vaccination can completely eliminate the challenge virus.
The question was whether this protection was due to the vaccine-induced CD8 T cells that were induced with low-dose MVA immunization (20). Upon testing of C57BL/6 mice depleted of CD8 T cells, vaccinated animals remained entirely unprotected, and their organs contained even higher viral loads than those of the unprotected mock-vaccinated control mice. The latter observation may indicate the efficacy of a developing CD8 T cell response for some partial control of the respiratory ECTV infection in naive C57BL/6 mice.
Interestingly, the relevance of cytotoxic T cells for early control of primary ECTV replication in C57BL/6 mice is well documented in the footpad (f.p.) infection model (25). The distinctive feature of the f.p. model is that only C57BL/6-derived mice with deficiencies in various immune compartments, including T cells, will develop severe disease and succumb to ECTV infections. In contrast to the respiratory ECTV infection model, however, f.p.-infected immunocompetent wild-type (wt) C57BL/6 mice do not develop fatal mousepox unless very high doses of ECTV are used for challenge infection (18).
MVA activates a broad range of different innate immune signaling pathways (26–31), probably since it lacks many immune evasion proteins encoded by other VACVs (32, 33). In particular, the induction of type I IFN in various MVA-infected cells is well described (27, 31, 34, 35), and type I IFNs are essential for C57BL/6 mice to resist primary ECTV f.p. infection (25, 36, 37). Curiously, we also found protection of IFNAR−/− mice lacking the type I interferon receptor following immunization with only 105 PFU MVA vaccine. Importantly, as with fully immunocompetent C57BL/6 mice, lesions of ECTV infection were not detected in organs of interferon-deficient mice vaccinated with MVA. In sharp contrast, we found large amounts of ECTV in organs of mock-vaccinated IFNAR−/− mice. Histological analysis revealed severe necrotic lesions in the livers and spleens of unvaccinated IFNAR−/− mice, concurring with the rapid disease course upon ECTV infection observed previously (13). In CD8+ T cell-depleted, MVA-vaccinated animals, we also observed high virus loads and comparable, severe histological changes in the organs. These findings clearly demonstrate a particular protective capacity of MVA-induced CD8+ T cell immunity in IFNAR−/− mice. This was somewhat unexpected, since it is well established that proper activation of CD8+ T cells is supported by type I IFN as the signal (38, 39). Thus, other innate immune factors induced upon MVA immunization are likely involved in activating T cell responses.
Cytokines such as IL-2 and IL-12 have been reported to provide the inflammatory signals to activate CD8+ T cells in vivo (39). Recent studies with IL-12-deficient mice showed that IL-2 activities contribute to the accumulation of memory CD8+ T cells (38, 40, 41), whereas IL-12 and IFN-α, as the signal 3 cytokines, are essential to generate an efficient effector T cell response (42, 43). Thus, we hypothesize that IL-12 may regulate the induction of CD8+ T cell effector function in the IFNAR−/− mice. Indeed, we found that MVA-vaccinated IFNAR−/− mice produced IL-12, while immunized C57BL/6 mice produced IFN-α but not detectable IL-12 (Fig. 5C and D). This agrees with previous studies showing that high levels of IFN-I actively inhibit production of IL-12 (34). Furthermore, expansion and survival of CD8+ T cells during VACV infection was found to depend less critically on type I IFNs (44). Therefore, we believe that IL-12 may replace IFN-I signaling for efficient T cell activation and clonal expansion in IFNAR-deficient mice (45). This assumption is in agreement with the recent finding of Rubio and coworkers that NF-κB activation in ECTV-infected mice can compensate for deficiencies in the type I IFN signaling pathway (46).
In previous work, we had demonstrated the essential need for the direct cytotoxic effector function of CD8+ T cells to allow rapid protection by intranasal immunization with 108 PFU MVA (20). We confirmed this requirement for rapidly protective low-dose (105 PFU) MVA immunization by the intramuscular route again using C57BL/6 mice lacking the cytolytic effector molecule perforin (Prf−/− mice) (data not shown). MVA- or mock-vaccinated and challenged Prf−/− mice showed very comparable courses of disease, and all succumbed to the ECTV infection within 14 days p.i. These data appears to fit well with recent work in the ECTV f.p. infection model showing the essential importance of cytolytic killing for protection mediated by virus-specific memory CD8+ T cells (47). Moreover, this study also demonstrates the critical requirement for production of the antiviral cytokine IFN-γ. Thus, we believe that, while not yet formally shown, IFN-γ responses could also contribute significantly to the rapid protection achieved by low-dose MVA immunization.
Finally, the mousepox infection also seemed to substantially influence the antiviral CD8+ T cell response elicited by MVA immunization just 2 days before challenge. Monitoring for B8R20–27-specific CD8+ T cells revealed significantly enhanced levels of IFN-γ-producing or multimer-binding T cells in animals after vaccination plus ECTV challenge, compared to mice that were only vaccinated. B8R20–27 is the immunodominant determinant of the orthopoxvirus-specific CD8+ T cell response in C57BL/6 mice (23), although other T cell specificities could also contribute to the protective immunity we observed (48, 49). The protective capacity of only B8R20–27-specific T cells against ECTV infection is well established (23) and underlines the general relevance of this CD8+ T cell specificity. The low-dose MVA immunization may be sufficient for primary activation of virus-specific CD8+ T cells, which are efficiently expanded following exposure of ECTV. Once activated by encountering the pathogen, antigen-specific CD8+ T cells undergo an intensive clonal expansion in response to the infection (50–53). Hence, following primary MVA vaccination, the ECTV challenge seems to provide the prolonged specific antigen exposure required for optimal activation of CD8+ T cells, in addition to costimulation and signal 3 signaling (43, 54–56).
Taken together, our findings define important requirements for protective emergency immunization against severe systemic infections with orthopoxviruses. Our results reveal the previously unrecognized potential of rapidly developing CD8+ T cell immunity elicited by low doses of MVA vaccine. This is of considerable practical relevance for public health, since producing sufficient amounts of vaccine is considered a major challenge should an outbreak occur. Moreover, many other infectious diseases may require emergency immunization, and we believe that more attention should be paid to vaccines potentially able to induce protective T cell immunity. Since MVA is a proven vector for efficiently delivering heterologous antigens, it could be a useful tool for generating emergency vaccinations targeting newly emerging pathogens.
ACKNOWLEDGMENTS
We thank Ursula Klostermeier for expert help in animal studies.
This work was supported by grants from the German Centre for Infection Research (DZIF; TTU 01.802) and from the European Commission (VECTORIE, FLUNIVAC, grant agreement numbers HEALTH-F3-2010-261466 and FP7-HEALTH-2013-602604). TWINCORE is a joint venture between the Helmholtz Centre for Infection Research, Braunschweig, Germany, and the Hannover Medical School, Hanover, Germany.
Footnotes
Published ahead of print 9 July 2014
REFERENCES
- 1.Gao H-N, Lu H-Z, Cao B, Du B, Shang H, Gan J-H, Lu S-H, Yang Y-D, Fang Q, Shen Y-Z, Xi X-M, Gu Q, Zhou X-M, Qu H-P, Yan Z, Li F-M, Zhao W, Gao Z-C, Wang G-F, Ruan L-X, Wang W-H, Ye J, Cao H-F, Li X-W, Zhang W-H, Fang X-C, He J, Liang W-F, Xie J, Zeng M, Wu X-Z, Li J, Xia Q, Jin Z-C, Chen Q, Tang C, Zhang Z-Y, Hou B-M, Feng Z-X, Sheng J-F, Zhong N-S, Li L-J. 2013. Clinical findings in 111 cases of influenza A (H7N9) virus infection. N. Engl. J. Med. 368:2277–2285. 10.1056/NEJMoa1305584 [DOI] [PubMed] [Google Scholar]
- 2.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus ADME, Fouchier RAM. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367:1814–1820. 10.1056/NEJMoa1211721 [DOI] [PubMed] [Google Scholar]
- 3.Fenner F. 1988. Smallpox and its eradication. World Health Organization, Geneva, Switzerland [Google Scholar]
- 4.Keckler MS, Reynolds MG, Damon IK, Karem KL. 2013. The effects of post-exposure smallpox vaccination on clinical disease presentation: addressing the data gaps between historical epidemiology and modern surrogate model data. Vaccine 31:5192–5201. 10.1016/j.vaccine.2013.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramírez JC, Gherardi MM, Esteban M. 2000. Biology of attenuated modified vaccinia virus Ankara recombinant vector in mice: virus fate and activation of B- and T-cell immune responses in comparison with the Western Reserve strain and advantages as a vaccine. J. Virol. 74:923–933. 10.1128/JVI.74.2.923-933.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stittelaar KJ, Kuiken T, de Swart RL, van Amerongen G, Vos HW, Niesters HGM, van Schalkwijk P, van der Kwast T, Wyatt LS, Moss B, Osterhaus ADME. 2001. Safety of modified vaccinia virus Ankara (MVA) in immune-suppressed macaques. Vaccine 19:3700–3709. 10.1016/S0264-410X(01)00075-5 [DOI] [PubMed] [Google Scholar]
- 7.Gilbert SC. 2013. Clinical development of modified vaccinia virus Ankara vaccines. Vaccine 31:4241–4246. 10.1016/j.vaccine.2013.03.020 [DOI] [PubMed] [Google Scholar]
- 8.Harrop R, John J, Carroll MW. 2006. Recombinant viral vectors: cancer vaccines. Adv. Drug Deliv. Rev. 58:931–947. 10.1016/j.addr.2006.05.005 [DOI] [PubMed] [Google Scholar]
- 9.Volz A, Sutter G. 2013. Protective efficacy of Modified vaccinia virus Ankara in preclinical studies. Vaccine 31:4235–4240. 10.1016/j.vaccine.2013.03.016 [DOI] [PubMed] [Google Scholar]
- 10.Gómez CE, Nájera JL, Domingo-Gil E, Ochoa-Callejero L, González-Aseguinolaza G, Esteban M. 2007. Virus distribution of the attenuated MVA and NYVAC poxvirus strains in mice. J. Gen. Virol. 88:2473–2478. 10.1099/vir.0.83018-0 [DOI] [PubMed] [Google Scholar]
- 11.Hirsch VM, Fuerst TR, Sutter G, Carroll MW, Yang LC, Goldstein S, Piatak M, Elkins WR, Alvord WG, Montefiori DC, Moss B, Lifson JD. 1996. Patterns of viral replication correlate with outcome in simian immunodeficiency virus (SIV)-infected macaques: effect of prior immunization with a trivalent SIV vaccine in modified vaccinia virus Ankara. J. Virol. 70:3741–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sutter G, Wyatt LS, Foley PL, Bennink JR, Moss B. 1994. A recombinant vector derived from the host range-restricted and highly attenuated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine 12:1032–1040. 10.1016/0264-410X(94)90341-7 [DOI] [PubMed] [Google Scholar]
- 13.Paran N, Suezer Y, Lustig S, Israely T, Schwantes A, Melamed S, Katz L, Preuß T, Hanschmann K-M, Kalinke U, Erez N, Levin R, Velan B, Löwer J, Shafferman A, Sutter G. 2009. Postexposure immunization with modified vaccinia virus Ankara or conventional Lister vaccine provides solid protection in a murine model of human smallpox. J. Infect. Dis. 199:39–48. 10.1086/595565 [DOI] [PubMed] [Google Scholar]
- 14.Staib C, Suezer Y, Kisling S, Kalinke U, Sutter G. 2006. Short-term, but not post-exposure, protection against lethal orthopoxvirus challenge after immunization with modified vaccinia virus Ankara. J. Gen. Virol. 87:2917–2921. 10.1099/vir.0.82068-0 [DOI] [PubMed] [Google Scholar]
- 15.Wyatt LS, Earl PL, Eller LA, Moss B. 2004. Highly attenuated smallpox vaccine protects mice with and without immune deficiencies against pathogenic vaccinia virus challenge. Proc. Natl. Acad. Sci. U. S. A. 101:4590–4595. 10.1073/pnas.0401165101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esteban D, Parker S, Schriewer J, Hartzler H, Buller RM. 2012. Mousepox, a small animal model of smallpox, p 177–198 In Isaacs SN. (ed), Vaccinia virus and poxvirology, vol 890 Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 17.Parker S, Siddiqui AM, Painter G, Schriewer J, Buller RM. 2010. Ectromelia virus infections of mice as a model to support the licensure of anti-orthopoxvirus therapeutics. Viruses 9:1918–1932. 10.3390/v2091918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parker S, Siddiqui AM, Oberle C, Hembrador E, Lanier R, Painter G, Robertson A, Buller RM. 2009. Mousepox in the C57BL/6 strain provides an improved model for evaluating anti-poxvirus therapies. Virology 385:11–21. 10.1016/j.virol.2008.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samuelsson C, Hausmann J, Lauterbach H, Schmidt M, Akira S, Wagner H, Chaplin P, Suter M, O'Keeffe M, Hochrein H. 2008. Survival of lethal poxvirus infection in mice depends on TLR9, and therapeutic vaccination provides protection. J. Clin. Invest. 118:1776–1784. 10.1172/JCI33940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kremer M, Suezer Y, Volz A, Frenz T, Majzoub M, Hanschmann K-M, Lehmann MH, Kalinke U, Sutter G. 2012. Critical role of perforin-dependent CD8+ T cell immunity for rapid protective vaccination in a murine model for human smallpox. PLoS Pathog. 8:e1002557. 10.1371/journal.ppat.1002557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Earl PL, Americo JL, Wyatt LS, Espenshade O, Bassler J, Gong K, Lin S, Peters E, Rhodes L, Spano YE, Silvera PM, Moss B. 2008. Rapid protection in a monkeypox model by a single injection of a replication-deficient vaccinia virus. Proc. Natl. Acad. Sci. U. S. A. 105:10889–10894. 10.1073/pnas.0804985105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.European Medicines Agency. 2013. Imvanex: modifiziertes Vacciniavirus Ankara, lebend. EMA/490157/2013. European Medicines Agency, London, United Kingdom [Google Scholar]
- 23.Tscharke DC, Karupiah G, Zhou J, Palmore T, Irvine KR, Haeryfar SMM, Williams S, Sidney J, Sette A, Bennink JR, Yewdell JW. 2005. Identification of poxvirus CD8+ T cell determinants to enable rational design and characterization of smallpox vaccines. J. Exp. Med. 201:95–104. 10.1084/jem.20041912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drexler I, Staib C, Kastenmüller W, Stevanović S, Schmidt B, Lemonnier FA, Rammensee H-G, Busch DH, Bernhard H, Erfle V, Sutter G. 2003. Identification of vaccinia virus epitope-specific HLA-A*0201-restricted T cells and comparative analysis of smallpox vaccines. Proc. Natl. Acad. Sci. U. S. A. 100:217–222. 10.1073/pnas.262668999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karupiah G, Buller RM, Van Rooijen N, Duarte CJ, Chen J. 1996. Different roles for CD4+ and CD8+ T lymphocytes and macrophage subsets in the control of a generalized virus infection. J. Virol. 70:8301–8309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Förster RWG, Mayr A. 1994. Highly attenuated poxviruses induce functional priming of neutrophils in vitro. Arch. Virol. 136:219–226. 10.1007/BF01538831 [DOI] [PubMed] [Google Scholar]
- 27.Büttner MCC, Lehner KH, Wertz K. 1995. Interferon induction in peripheral blood mononuclear leukocytes of man and farm animals by poxvirus vector candidates and some poxvirus constructs. Vet. Immunol. Immunopathol. 46:237–250. 10.1016/0165-2427(94)05357-X [DOI] [PubMed] [Google Scholar]
- 28.Delaloye J, Roger T, Steiner-Tardivel Q-G, Le Roy D, Knaup Reymond M, Akira S, Petrilli V, Gomez CE, Perdiguero B, Tschopp J, Pantaleo G, Esteban M, Calandra T. 2009. Innate immune sensing of modified vaccinia virus Ankara (MVA) is mediated by TLR2-TLR6, MDA-5 and the NALP3 inflammasome. PLoS Pathog. 5:e1000480. 10.1371/journal.ppat.1000480 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Lehmann MH, Kastenmuller W, Kandemir JD, Brandt F, Suezer Y, Sutter G. 2009. Modified vaccinia virus Ankara triggers chemotaxis of monocytes and early respiratory immigration of leukocytes by induction of CCL2 expression. J. Virol. 83:2540–2552. 10.1128/JVI.01884-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lynch HE, Ray CA, Oie KL, Pollara JJ, Petty ITD, Sadler AJ, Williams BRG, Pickup DJ. 2009. Modified vaccinia virus Ankara can activate NF-κB transcription factors through a double-stranded RNA-activated protein kinase (PKR)-dependent pathway during the early phase of virus replication. Virology 391:177–186. 10.1016/j.virol.2009.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waibler Z, Anzaghe M, Ludwig H, Akira S, Weiss S, Sutter G, Kalinke U. 2007. Modified Vaccinia virus Ankara induces Toll-like receptor-independent type I interferon responses. J. Virol. 81:12102–12110. 10.1128/JVI.01190-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antoine G, Scheiflinger F, Dorner F, Falkner FG. 1998. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology 244:365–396. 10.1006/viro.1998.9123 [DOI] [PubMed] [Google Scholar]
- 33.Blanchard TJ, Alcami A, Andrea P, Smith GL. 1998. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: implications for use as a human vaccine. J. Gen. Virol. 79:1159–1167 [DOI] [PubMed] [Google Scholar]
- 34.Frenz T, Waibler Z, Hofmann J, Hamdorf M, Lantermann M, Reizis B, Tovey MG, Aichele P, Sutter G, Kalinke U. 2010. Concomitant type I IFN receptor-triggering of T cells and of DC is required to promote maximal modified vaccinia virus Ankara-induced T-cell expansion. Eur. J. Immunol. 40:2769–2777. 10.1002/eji.201040453 [DOI] [PubMed] [Google Scholar]
- 35.Hornemann S, Harlin O, Staib C, Kisling S, Erfle V, Kaspers B, Häcker G, Sutter G. 2003. Replication of modified vaccinia virus Ankara in primary chicken embryo fibroblasts requires expression of the interferon resistance gene E3L. J. Virol. 77:8394–8407. 10.1128/JVI.77.15.8394-8407.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karupiah G. 1998. Type 1 and type 2 cytokines in antiviral defense. Vet. Immunol. Immunopathol. 63:105–109. 10.1016/S0165-2427(98)00086-5 [DOI] [PubMed] [Google Scholar]
- 37.Karupiah G, Fredrickson TN, Holmes KL, Khairallah LH, Buller RM. 1993. Importance of interferons in recovery from mousepox. J. Virol. 67:4214–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bachmann MF, Beerli RR, Agnellini P, Wolint P, Schwarz K, Oxenius A. 2006. Long-lived memory CD8+ T cells are programmed by prolonged antigen exposure and low levels of cellular activation. Eur. J. Immunol. 36:842–854. 10.1002/eji.200535730 [DOI] [PubMed] [Google Scholar]
- 39.Valenzuela J, Schmidt C, Mescher M. 2002. The roles of IL-12 in providing a third signal for clonal expansion of naive CD8 T cells. J. Immunol. 169:6842–6849. 10.4049/jimmunol.169.12.6842 [DOI] [PubMed] [Google Scholar]
- 40.D'Souza WN, Schluns KS, Masopust D, Lefrançois L. 2002. Essential role for IL-2 in the regulation of antiviral extralymphoid CD8 T cell responses. J. Immunol. 168:5566–5572. 10.4049/jimmunol.168.11.5566 [DOI] [PubMed] [Google Scholar]
- 41.Williams MA, Tyznik AJ, Bevan MJ. 2006. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature 441:890–893. 10.1038/nature04790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Curtsinger JM, Lins DC, Mescher MF. 2003. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J. Exp. Med. 197:1141–1151. 10.1084/jem.20021910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mescher MF, Curtsinger JM, Agarwal P, Casey KA, Gerner M, Hammerbeck CD, Popescu F, Xiao Z. 2006. Signals required for programming effector and memory development by CD8+ T cells. Immunol. Rev. 211:81–92. 10.1111/j.0105-2896.2006.00382.x [DOI] [PubMed] [Google Scholar]
- 44.Keppler SJ, Rosenits K, Koegl T, Vucikuja S, Aichele P. 2012. Signal 3 cytokines as modulators of primary immune responses during infections: the interplay of type I IFN and IL-12 in CD8 T cell responses. PLoS One 7:e40865. 10.1371/journal.pone.0040865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thompson LJ, Kolumam GA, Thomas S, Murali-Krishna K. 2006. Innate inflammatory signals induced by various pathogens differentially dictate the IFN-I dependence of CD8 T cells for clonal expansion and memory formation. J. Immunol. 177:1746–1754. 10.4049/jimmunol.177.3.1746 [DOI] [PubMed] [Google Scholar]
- 46.Rubio D, Xu R-H, Remakus S, Krouse TE, Truckenmiller ME, Thapa RJ, Balachandran S, Alcamí A, Norbury CC, Sigal LJ. 2013. Crosstalk between the type 1 interferon and nuclear factor kappa B pathways confers resistance to a lethal virus infection. Cell Host Microbe 13:701–710. 10.1016/j.chom.2013.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Remakus S, Rubio D, Lev A, Ma X, Fang M, Xu R-H, Sigal LJ. 2013. Memory CD8+ T cells can outsource IFN-γ production but not cytolytic killing for antiviral protection. Cell Host Microbe 13:546–557. 10.1016/j.chom.2013.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Croft NP, Smith SA, Wong YC, Tan CT, Dudek NL, Flesch IEA, Lin LCW, Tscharke DC, Purcell AW. 2013. Kinetics of antigen expression and epitope presentation during virus infection. PLoS Pathog. 9:e1003129. 10.1371/journal.ppat.1003129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin LCW, Flesch IEA, Tscharke DC. 2013. Immunodomination during peripheral vaccinia virus infection. PLoS Pathog. 9:e1003329. 10.1371/journal.ppat.1003329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Badovinac VP, Porter BB, Harty JT. 2002. Programmed contraction of CD8+ T cells after infection. Nat. Immunol. 3:619–626. 10.1038/nrm880 [DOI] [PubMed] [Google Scholar]
- 51.Harty JT, Badovinac VP. 2002. Influence of effector molecules on the CD8+ T cell response to infection. Curr. Opin. Immunol. 14:360–365. 10.1016/S0952-7915(02)00333-3 [DOI] [PubMed] [Google Scholar]
- 52.Prlic M, Williams MA, Bevan MJ. 2007. Requirements for CD8 T-cell priming, memory generation and maintenance. Curr. Opin. Immunol. 19:315–319. 10.1016/j.coi.2007.04.010 [DOI] [PubMed] [Google Scholar]
- 53.van Heijst JWJ, Gerlach C, Swart E, Sie D, Nunes-Alves C, Kerkhoven RM, Arens R, Correia-Neves M, Schepers K, Schumacher TNM. 2009. Recruitment of antigen-specific CD8+ T Cells in response to infection is markedly efficient. Science 325:1265–1269. 10.1126/science.1175455 [DOI] [PubMed] [Google Scholar]
- 54.Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL, Mescher MF. 2009. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J. Immunol. 183:1695–1704. 10.4049/jimmunol.0900592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bevan MJ, Fink PJ. 2001. The CD8 response on autopilot. Nat. Immunol. 2:381–382. 10.1038/87676 [DOI] [PubMed] [Google Scholar]
- 56.van Stipdonk MJB, Hardenberg G, Bijker MS, Lemmens EE, Droin NM, Green DR, Schoenberger SP. 2003. Dynamic programming of CD8+ T lymphocyte responses. Nat. Immunol. 4:361–365. 10.1038/ni912 [DOI] [PubMed] [Google Scholar]